
Mechanochemical reactivity of a multimodal 2H-bis-naphthopyran mechanophore†
Skylar K.
Osler
 ,
Molly E.
McFadden
,
Tian
Zeng
and
Maxwell J.
Robb
*
,
Molly E.
McFadden
,
Tian
Zeng
and
Maxwell J.
Robb
*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: mrobb@caltech.edu
First published on 10th May 2023
Abstract
Multimodal mechanophores that react under mechanical force to produce discrete product states with uniquely coupled absorption properties are interesting targets for the design of force-sensing polymers. Herein, we investigate the reactivity of a 2H-bis-naphthopyran mechanophore that generates thermally persistent mono-merocyanine and bis-merocyanine products upon mechanical activation in solution using ultrasonication, distinct from the thermally reversible products generated photochemically. We demonstrate that a force-mediated ester C(O)–O bond scission reaction following ring opening establishes an intramolecular hydrogen bond, locking one merocyanine subunit in the open form. Model compound studies suggest that this locked subunit confers remarkable thermal stability to bis-merocyanine isomers possessing a trans exocyclic alkene on the other subunit, implicating the formation of an unusual trans merocyanine isomer as the product of mechanochemical activation. Density functional theory calculations unexpectedly predict a thermally reversible retro-cyclization reaction of the bis-merocyanine species that could explain the mechanochemical generation of the unusual trans merocyanine isomer.
Introduction
Naphthopyrans are a class of molecular switches that undergo a ring-opening reaction upon photochemical or mechanochemical activation to generate a highly colored merocyanine dye. While the photochemical reactivity of naphthopyran has been studied extensively,1 the mechanochemical reactivity of naphthopyran has only recently been investigated within the nascent field of polymer mechanochemistry.2–9 Mechanical force is transduced to naphthopyrans covalently incorporated into polymers by the application of stress to bulk materials,2–5 or by solution-phase ultrasonication,6–9 leading to merocyanine formation. Naphthopyran is thus a versatile mechanochromic force probe capable of reporting on stress and/or strain colorimetrically. Interestingly, the mechanochemical reactivity of naphthopyran mechanophores often diverges from their photochemical behavior.6,7,9Extended UV photoirradiation of naphthopyrans generates a mixture of merocyanine isomers with cis and trans alkene geometry at the exocyclic olefin (Scheme 1a).10–19 Merocyanines with trans exocyclic olefin geometry are more thermally stable, while the cis isomers readily revert to the naphthopyran under ambient conditions. In contrast to the photochemical reactions, the thermal reversion behavior of most mechanochemically generated naphthopyran-derived merocyanines suggests that only the isomer with cis stereochemistry of the exocyclic alkene is produced using force.2–8 Recently, our group discovered a 2H-naphthopyran mechanophore scaffold that generates a persistent merocyanine dye upon mechanical activation (Scheme 1b).7 Rather than cis–trans isomerization, a unique intramolecular hydrogen bonding mechanism is implicated in the persistent color generation whereby the scission of a C(O)–O ester bond following pyran ring opening establishes a β-hydroxy ketone that prevents merocyanine reversion.
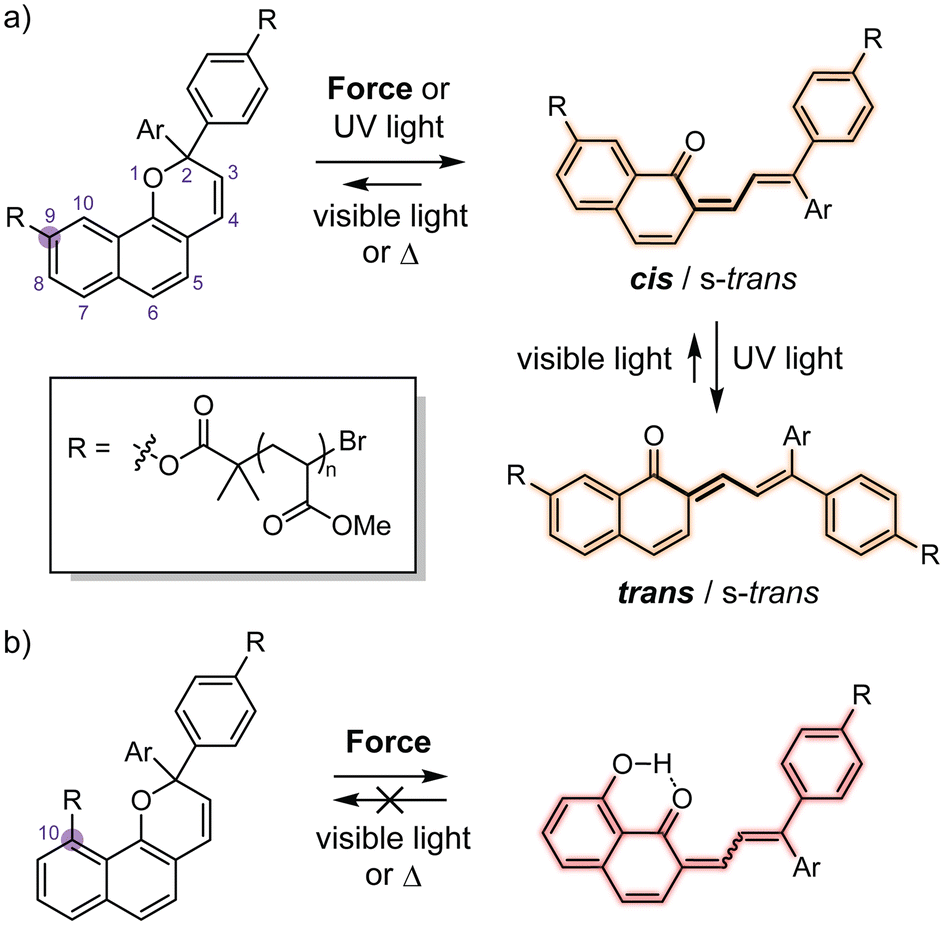 | ||
| Scheme 1 Reactions of 2H-naphthopyran-containing poly(methyl acrylate) polymers and the structures of the merocyanine dyes produced upon mechanochemical or photochemical activation with polymer attachment via an ester linkage at either the (a) 9-position, or (b) 10-position of the naphthopyran scaffold. | ||
Mechanochromic mechanophores possessing multiple reactive sites are theoretically capable of a more complex colorimetric response, accessing distinct states under mechanical force that are coupled to unique changes in optical properties.20 For example, a bis-naphthopyran mechanophore previously developed in our group displays force-dependent multicolor mechanochromism due to a force-modified dynamic equilibrium that is established between two different merocyanine states.6 In contrast to the photochemical reaction, which proceeds via sequential ring-opening reactions of each pyran unit,21–24 mechanical activation was found to simultaneously open both pyran rings to generate the bis-merocyanine species. Because the mono-merocyanine product is formed via thermal reversion of the bis-merocyanine species, increasing the amount of force applied to the mechanophore increases the rate of bis-merocyanine formation relative to mono-merocyanine and shifts the relative distribution of the two distinctly colored dyes. Naphthodipyran9 and diketopyrrolopyrrole25,26 mechanophores also display multicolor mechanochromism under force; however, the dyes produced from these mechanophores are typically either transient and revert to their original state after stress is removed, or require a trapping agent for persistent color generation. In bulk materials, spiropyran27,28 and rhodamine29 mechanophores have been shown to generate uniquely colored states under active tension and after stress relaxation. Notably, mechanophores that display multicolor mechanochromism and generate persistent coloration remain limited.26,30
We were interested in exploring the mechanochromism of a bis-naphthopyran mechanophore potentially capable of generating persistent coloration via the same intramolecular hydrogen bonding mechanism associated with the 2H-naphthopyran mechanophore described above (Scheme 2). In analogy to our previously studied 3H-bis-naphthopyran mechanophore,6 we imagined that the two pyran rings of a 2H-bis-naphthopyran (2H-BNP) mechanophore would open simultaneously upon ultrasound-induced mechanical activation to generate bis-merocyanine 2H-BNPoo (open–open), followed by rapid scission of a C(O)–O ester bond on one of the merocyanine units. This bond cleavage would position the mechanophore at the end of the polymer chain, precluding further mechanochemical reaction.31 The merocyanine unit with an intact ester linkage would be expected to undergo a thermal ring-closing reaction to produce persistent mono-merocyanine 2H-BNPoc (open–closed). Here, we study the mechanochemical reactivity of this 2H-BNP mechanophore in dilute polymer solutions using ultrasonication. While mechanical activation results in the anticipated ring-opening reactions accompanied by ester cleavage, the reactivity is unexpectedly complex. A multifaceted experimental and computational approach provides insights into the unusual reactivity of this 2H-BNP mechanophore.
 | ||
| Scheme 2 The anticipated mechanochemical reactivity of 2H-BNP wherein mechanical force induces a dual ring-opening reaction accompanied by ester scission leading to a transient bis-merocyanine intermediate (2H-BNPoo), which thermally reverts to form a stable mono-merocyanine dye (2H-BNPoc). The actual reactivity is more complex. | ||
Results and discussion
We investigated the mechanochemical reactivity of the 2H-BNP mechanophore through solution-phase ultrasonication experiments.31,32 Polymer attachment at the 10-position of each naphthopyran subunit is critical to form the persistent hydrogen-bonded merocyanine dye.7 Polymer 2H-BNP109 (Mn = 109 kDa, Đ = 1.17) was synthesized by a Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction between bis-alkyne 1a and two equivalents of azide-terminated poly(methyl acrylate) (PMA) polymer PMA-Azide (Mn = 59 kDa, Đ = 1.10) (Fig. 1a, see the ESI† for details).33 Characterization by GPC-MALS with in-line UV absorption detection confirmed that the 2H-BNP mechanophore was incorporated near the chain-center where mechanical force is maximized (Fig. 1b).31,34,35 | ||
| Fig. 1 (a) Synthesis of a poly(methyl acrylate) polymer (109 kDa, Đ = 1.17) containing a chain-centered 2H-bis-naphthopyran mechanophore unit via CuAAC. (b) Characterization by GPC-MALS with differential refractive index (dRI) and in-line UV detection (345 nm) confirms 2H-bis-naphthopyran functionalization near the chain center. | ||
To investigate the mechanochemical activation of the 2H-BNP mechanophore, a solution of 2H-BNP109 (2 mg mL−1 in THF containing 30 mM BHT) maintained at ∼20 °C was subjected to continuous ultrasonication (20 kHz, 8.2 W cm−2) for 12 h (see the ESI† for details). Generation of colored merocyanine products was monitored by synchronous UV-vis absorption spectroscopy using a continuous flow setup.6,36 Ultrasound-induced mechanochemical activation of 2H-BNP109 produces an absorption spectrum with two local maxima at 510 and 550 nm (Fig. 2a). The absorption spectrum of the mechanochemical reaction products remains essentially unchanged 16 h after the cessation of ultrasound, indicating that mechanical activation produces a thermally persistent merocyanine species. Extended irradiation with intense visible light (565 nm, 150 min) causes a slight attenuation of the absorption features, but does not induce significant bleaching (Fig. S1†), consistent with the formation of a permanent merocyanine dye locked via an intramolecular H-bonding interaction.7 To our surprise, however, even the longer wavelength absorption features attributed to the bis-merocyanine are largely persistent under these conditions, indicating the presence of an unusually stable bis-merocyanine species. Notably, the simultaneous cleavage of both C(O)–O ester linkages is unlikely. Thus, this persistent bis-merocyanine species likely contains a single intact ester group on one merocyanine subunit, consistent with the structure of 2H-BNPoo shown in Scheme 2. The slight attenuation of the absorption spectrum after irradiation with visible light is consistent with trans-to-cis isomerization of the exocyclic alkene in the merocyanine subunit containing the intramolecular H-bond (Fig. S2 and S3, see the ESI† for additional details).12 Control experiments demonstrate that the absorption spectrum generated from photochemical activation of 2H-BNP109 is distinct from the spectrum obtained during sonication experiments (Fig. 2b and Fig. S4†). Furthermore, in contrast to the products of mechanochemical activation, the colored species produced under UV light undergo significant reversion under ambient conditions and completely revert to the colorless state upon irradiation with visible light. Together, these data suggest that the thermally persistent species produced upon ultrasound-induced mechanochemical activation of 2H-BNP109 are neither the expected mono-merocyanine locked by an intramolecular H-bond, nor the bis-merocyanine product with two intact ester linkages.
 | ||
| Fig. 2 (a) Ultrasound-induced mechanochemical activation (12 h, 20 °C) of 2H-BNP109 generates a product mixture with absorption maxima at approximately 510 and 550 nm. After incubation at 20 °C for 16 h in the dark, the spectrum remains essentially unchanged. Irradiation with 565 nm light (150 min) results in minimal bleaching. (b) Photoirradiation of 2H-BNP109 with 365 nm UV light (40 min, 20 °C) generates a product mixture with absorption maxima at ∼490 and 575 nm. Some attenuation of the absorbance is observed after 16 h in the dark (20 °C). Irradiation with 565 nm light (180 min) results in nearly complete bleaching. Insets show time-dependent absorbance at the specified wavelengths. | ||
To identify the persistent mechanochemical product(s) of 2H-BNP109 activation, small molecule model compound M1 was synthesized and characterized by 1H NMR and UV-vis absorption spectroscopy (Fig. 3). Consistent with prior studies of a related 2H-naphthopyran mechanophore,7 deprotection of the 10-hydroxyl group on the bis-naphthopyran precursor results in spontaneous thermal equilibration to the mono-merocyanine form. Characteristic proton resonances in the 13–14 ppm region of the 1H NMR spectrum of M1 are attributed to the hydrogen-bonded OH proton (Ha) on various merocyanine stereoisomers (Fig. 3a). These diagnostic signals are also observed in the 1H NMR spectrum of 2H-BNP109 after ultrasonication. The intact pyran ring on the naphthopyran subunit of M1 is identified by a doublet at 6.40 ppm (Hb, J = 9.5 Hz). This resonance is also observed in the 1H NMR spectrum of 2H-BNP109 after ultrasound-induced mechanical activation, along with a pair of overlapping doublets at 6.14 and 6.15 ppm (J = 9.6 Hz) corresponding to pyran protons of residual unreacted 2H-BNP109. While signatures of the mono-merocyanine species with a single β-hydroxy ketone motif are clearly visible in the 1H NMR spectrum of 2H-BNP109 after ultrasonication, further analysis by NMR spectroscopy is obscured by overlapping peaks in the aromatic region. The UV-vis absorption spectrum of M1 contains a single peak with a maximum at 530 nm, which is notably distinct from the spectrum obtained after mechanochemical activation of 2H-BNP109 (Fig. 3b). The absorption spectrum acquired after sonication of 2H-BNP109 has an absorption edge that is bathochromically shifted from that of M1, further suggesting that the sonication product mixture contains a persistent bis-merocyanine species with longer wavelength absorption in addition to mono-merocyanine isomers.
 | ||
| Fig. 3 (a) Characteristic resonances in the 1H NMR (THF-d8, 400 MHz) spectrum of model compound M1 are also observed in the product mixture generated from ultrasound-mediated mechanical activation of 2H-BNP109. (b) The absorption spectrum generated upon ultrasonication of 2H-BNP109 is bathochromically shifted compared to the absorption spectrum of M1. | ||
Further structural elucidation of the mechanochemical product mixture was achieved by photochemical activation of model compound M1 to generate the bis-merocyanine species. A solution of M1 (0.01 mg mL−1 in THF) was irradiated with UV light (365 nm, 8 min) at 20 °C using the same analytical flow setup as above (Fig. 4a). Immediately following photoirradiation, the absorption spectrum exhibits two local maxima at 505 and 580 nm. This extended UV irradiation generates a substantial amount of bis-merocyanine, resulting in significant absorption in the longer wavelength region. However, after an extended period under ambient conditions in the dark (16 h, 20 °C), the absorption spectrum evolves to ultimately match that obtained from ultrasonication of polymer 2H-BNP109 (Fig. 4a, inset). Partial retention of the longer wavelength absorption features suggests that photoirradiation of M1 generates a mixture of thermally transient and thermally persistent bis-merocyanine isomers (Fig. 4b). We identify the thermally transient bis-merocyanine species as having cis exocyclic alkene geometry on the merocyanine unit with an intact ester linkage at the 10-position (cis-2H-BNPoo).18 The thermally persistent bis-merocyanine species is attributed to isomers with trans exocyclic alkene geometry on the merocyanine subunit with an intact ester linkage at the 10-position (trans-2H-BNPoo). Increasing exposure of M1 to UV light results in a greater proportion of thermally persistent trans-2H-BNPoo, consistent with the expected photochemical cis-to-trans isomerization of the exocyclic alkene (Fig. S5†).18,19 Visible light irradiation is known to accelerate trans-to-cis isomerization, which must occur before electrocyclization to regenerate the naphthopyran.11,12,18,19 However, photochemical activation of M1 produces products that do not bleach appreciably under visible light, similar to the mechanochemically-generated products of 2H-BNP109 (Fig. S6†). On the other hand, complete reversion of photochemically produced merocyanines is possible using visible light when both ester linkages at the 10-position are intact (Fig. 2b). These results suggest that the unique persistence of trans-2H-BNPoo relies on having one merocyanine subunit locked in the open form via the intramolecular hydrogen bond.
 | ||
| Fig. 4 (a) Photoirradiation of M1 with UV light (345 nm, 8 min) results in a bathochromic shift in absorption with maxima at 505 and 580 nm. Upon cessation of photoirradiation and 16 h in the dark (20 °C), the spectrum evolves to closely match the final absorption spectrum obtained upon ultrasonication of 2H-BNP109 (inset). (b) Reaction scheme illustrating the formation of a mixture of thermally transient and persistent bis-merocyanine isomers with cis and trans exocyclic alkene geometry on the ester-containing subunit upon exposure of M1 to UV light. | ||
The results presented above indicate that the photochemical activation of M1 and the mechanochemical activation of 2H-BNP109 generate similar product mixtures. An important implication is that the mechanochemical ring-opening reaction of the 2H-BNP mechanophore produces a mixture of bis-merocyanine isomers with both cis and trans geometry of the exocyclic alkene on the merocyanine subunit with an intact ester linkage at the 10-position. For typical naphthopyran-derived merocyanines, thermal cis-to-trans isomerization of the exocyclic alkene is rare.16,19 As discussed above, the mechanochemical generation of merocyanines with a trans exocyclic olefin is unusual from naphthopyran mechanophores. Consistent with the mechanical activation of 2H-BNP109, sonication experiments performed on analogous polymers with lower (2H-BNP44: Mn = 44 kDa, Đ = 1.25) and higher (2H-BNP317: Mn = 317 kDa, Đ = 1.17) molar mass result in product mixtures with similar absorption maxima at 510 and 550 nm, suggesting that the ratio of mechanically-generated cis and trans merocyanine products is not force-dependent (Fig. S7 and S8†).37 Notably, low-temperature sonication of 2H-BNP317 results in the detectable accumulation of a thermally transient bis-merocyanine species due to the increased reaction rate,36 confirming that mechanical activation of the 2H-BNP mechanophore does generate cis-2H-BNPoo in addition to the thermally persistent trans isomers analogous to the photochemical pathway illustrated in Fig. 4b (Fig. S9†).
Density functional theory (DFT) calculations using the constrained geometries simulate external force (CoGEF) method38,39 performed on a truncated model offer an intriguing explanation for the unique mechanochemical generation of merocyanine isomers with trans configuration of the exocyclic alkene in the ester-containing subunit (Fig. 5). Molecular elongation first results in the expected ring-opening reactions to form the bis-merocyanine product. Upon further extension, however, a retro-cyclization reaction is predicted to occur simultaneously on both merocyanine subunits to generate two sets of ketene and allene moieties. Electrocyclic ketene-forming reactions have been reported under thermal40 and photochemical41,42 conditions, providing some precedent for this unusual transformation. If such a retro-cyclization reaction occurs in practice, the species would be predicted to undergo rapid thermal cyclization42 upon chain relaxation to regenerate a mixture of merocyanines with cis and trans exocyclic alkene geometry. Notably, this unusual retro-cyclization reaction is not predicted by CoGEF for a 2H-BNP model with the ester linkages at the 9-position of the two naphthopyran subunits (Fig. S10†). Consistent with these computational predictions, ultrasonication experiments performed on a polymer containing a chain-centered 2H-BNP mechanophore connected at the 9-position via ester linkages on each subunit does not generate any thermally persistent merocyanine isomers (Fig. S11†).
 | ||
| Fig. 5 Density functional theory (DFT) calculations using the constrained geometries simulate external force (CoGEF) method performed on a truncated model of 2H-BNP predict an unusual retro-cyclization reaction upon extended mechanical elongation to generate two sets of ketene and allene motifs. Structures are shown that correspond to the computed products of pyran ring opening (B and C) and the unusual retro-cyclization reaction (D) on the CoGEF profile. Calculations were performed at the B3LYP/6-31G* level of theory. The purple carbon atoms of the terminal methyl groups define the distance constraint. | ||
Conclusions
In summary, we have investigated the mechanochemical reactivity of a 2H-bis-naphthopyran (2H-BNP) mechanophore. Instead of the expected force-dependent multicolor mechanochromism, which is predicated on the thermal reversion of the bis-merocyanine product,6 we find that ultrasound-induced mechanochemical activation of 2H-BNP results in an unexpectedly persistent bis-merocyanine species. Scission of a C(O)–O ester bond at the 10-position on one naphthopyran subunit establishes an intramolecular hydrogen bond that locks one merocyanine subunit open. In contrast to previously reported naphthopyran mechanophores that appear to exclusively produce the cis merocyanine product upon ring opening, mechanical activation of the 2H-BNP mechanophore generates a mixture of merocyanine isomers with cis and trans geometry of the exocyclic alkene on the merocyanine subunit with an intact ester linkage. Supported by small molecule model studies, the bis-merocyanine isomers with a trans exocyclic alkene on the ester-containing subunit are thermally persistent and resist bleaching under visible light irradiation. Density functional theory calculations implicate an unusual, thermally reversible mechanochemical retro-cyclization reaction of the bis-merocyanine species that is plausibly responsible for the formation of the trans merocyanine isomer, rather than a direct mechanically facilitated cis-to-trans isomerization. Efforts within our group are currently underway to experimentally probe the possibility of this unusual transformation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Caltech and an NSF CAREER award (CHE-2145791) is gratefully acknowledged. S. K. O. was supported by an Institute Fellowship from Caltech. M. E. M. was supported by an NSF Graduate Research Fellowship (DGE-1745301) and a Barbara J. Burger Fellowship. We thank the Center for Catalysis and Chemical Synthesis of the Beckman institute at Caltech for access to equipment. M. J. R. is an Alfred P. Sloan Research Fellow and a Camille Dreyfus Teacher-Scholar.References
- J. D. Hepworth and B. M. Heron, in Functional Dyes, Elsevier, 2006, pp. 85–135 Search PubMed.
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS PubMed.
- G. Kim, V. M. Lau, A. J. Halmes, M. L. Oelze, J. S. Moore and K. C. Li, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10214–10222 CrossRef CAS PubMed.
- T. Kosuge, X. Zhu, V. M. Lau, D. Aoki, T. J. Martinez, J. S. Moore and H. Otsuka, J. Am. Chem. Soc., 2019, 141, 1898–1902 CrossRef CAS PubMed.
- B. A. Versaw, M. E. McFadden, C. C. Husic and M. J. Robb, Chem. Sci., 2020, 11, 4525–4530 RSC.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS PubMed.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2021, 143, 7925–7929 CrossRef CAS PubMed.
- S. K. Osler, M. E. McFadden and M. J. Robb, J. Polym. Sci., 2021, 59, 2537–2544 CrossRef CAS.
- M. E. McFadden, S. K. Osler, Y. Sun and M. J. Robb, J. Am. Chem. Soc., 2022, 22391–22396 CrossRef CAS PubMed.
- S. Delbaere, B. Luccioni-Houze, C. Bochu, Y. Teral, M. Campredon and G. Vermeersch, J. Chem. Soc., Perkin Trans. 2, 1998, 1153–1158 RSC.
- G. Ottavi, G. Favaro and V. Malatesta, J. Photochem. Photobiol., A, 1998, 115, 123–128 CrossRef CAS.
- S. Jockusch, N. J. Turro and F. R. Blackburn, J. Phys. Chem. A, 2002, 106, 9236–9241 CrossRef CAS.
- J. C. Arnall-Culliford, Y. Teral, P. Sgarabotto, M. Campredon and G. Giusti, J. Photochem. Photobiol., A, 2003, 159, 7–16 CrossRef CAS.
- S. Delbaere and G. Vermeersch, J. Photochem. Photobiol., A, 2003, 159, 227–232 CrossRef CAS.
- S. Delbaere, J.-C. Micheau and G. Vermeersch, J. Org. Chem., 2003, 68, 8968–8973 CrossRef CAS PubMed.
- S. Delbaere, J.-C. Micheau, M. Frigoli and G. Vermeersch, J. Org. Chem., 2005, 70, 5302–5304 CrossRef CAS PubMed.
- S. Aiken, K. Booth, C. D. Gabbutt, B. M. Heron, C. R. Rice, A. Charaf-Eddin and D. Jacquemin, Chem. Commun., 2014, 50, 7900–7903 RSC.
- S. Brazevic, S. Nizinski, R. Szabla, M. F. Rode and G. Burdzinski, Phys. Chem. Chem. Phys., 2019, 21, 11861–11870 RSC.
- S. Brazevic, M. Baranowski, M. Sikorski, M. F. Rode and G. Burdziński, ChemPhysChem, 2020, 21, 1402–1407 CrossRef CAS PubMed.
- D. Kim, M. S. Kwon and C. W. Lee, Polym. Chem., 2022, 13, 5177–5187 RSC.
- W. Zhao and E. M. Carreira, J. Am. Chem. Soc., 2002, 124, 1582–1583 CrossRef CAS PubMed.
- W. Zhao and E. M. Carreira, Org. Lett., 2006, 8, 99–102 CrossRef CAS PubMed.
- W. Zhao and E. M. Carreira, Chem. – Eur. J., 2007, 13, 2671–2685 CrossRef CAS PubMed.
- X. Lu, Q. Dong, X. Dong and W. Zhao, Tetrahedron, 2015, 71, 4061–4069 CrossRef CAS.
- M. Raisch, W. Maftuhin, M. Walter and M. Sommer, Nat. Commun., 2021, 12, 4243 CrossRef CAS PubMed.
- R. Hertel, W. Maftuhin, M. Walter and M. Sommer, J. Am. Chem. Soc., 2022, 144, 21897–21907 CrossRef CAS PubMed.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS PubMed.
- T. A. Kim, M. J. Robb, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2018, 51, 9177–9183 CrossRef CAS.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS PubMed.
- W. He, Y. Yuan, M. Wu, X. Li, Y. Shen, Z. Qu and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202218785 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- P. A. May and J. S. Moore, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC.
- R. Nixon and G. De Bo, Nat. Chem., 2020, 12, 826–831 CrossRef CAS PubMed.
- K. L. Berkowski, S. L. Potisek, C. R. Hickenboth and J. S. Moore, Macromolecules, 2005, 38, 8975–8978 CrossRef CAS.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- P. A. May, N. F. Munaretto, M. B. Hamoy, M. J. Robb and J. S. Moore, ACS Macro Lett., 2016, 5, 177–180 CrossRef CAS PubMed.
- J. A. Odell and A. Keller, J. Polym. Sci., Part B: Polym. Phys., 1986, 24, 1889–1916 CrossRef CAS.
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- H. R. Smallman, J. A. Leitch, T. McBride, S. V. Ley and D. L. Browne, Tetrahedron, 2021, 93, 132305 CrossRef CAS.
- R. Boch, J. C. Bradley, T. Durst and J. C. Scaiano, Tetrahedron Lett., 1994, 35, 19–22 CrossRef CAS.
- J. K. Parker and S. R. Davis, J. Am. Chem. Soc., 1999, 121, 4271–4277 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00344b |
| This journal is © The Royal Society of Chemistry 2023 |