
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMimicking the extracellular matrix by incorporating functionalized graphene into hybrid hydrogels†
Josué
M. Galindo
 a,
Irene
San-Millán
a,
Carlos A.
Castillo-Sarmiento
b,
Inmaculada
Ballesteros-Yáñez
c,
M. Antonia
Herrero
*a,
Sonia
Merino
*a and
Ester
Vázquez
*a
a,
Irene
San-Millán
a,
Carlos A.
Castillo-Sarmiento
b,
Inmaculada
Ballesteros-Yáñez
c,
M. Antonia
Herrero
*a,
Sonia
Merino
*a and
Ester
Vázquez
*a
aInstituto Regional de Investigación Científica Aplicada (IRICA) and Facultad de Ciencias y Tecnologías Químicas, Universidad de Castilla-La Mancha, 13071 Ciudad Real, Spain. E-mail: Ester.vazquez@uclm.es; Sonia.merino@uclm.es; MariaAntonia.Herrero@uclm.es
bFacultad de Fisioterapia y Enfermería, Universidad de Castilla-La Mancha, 45071, Toledo, Spain
cFacultad de Medicina, Universidad de Castilla-La Mancha, 13071, Ciudad Real, Spain
First published on 21st August 2023
Abstract
The efficient functionalization of graphene with sulfonic groups using a sustainable approach facilitates the interaction of biomolecules with its surface. The inclusion of these graphene sheets inside a photopolymerized acrylamide-based hydrogel provides a 3D scaffold with viscoelastic behaviour closer to that found in natural tissues. Cell-culture experiments and differentiation assays with SH-SY5Y cells showed that these hybrid hydrogels are non-cytotoxic, thus making them potentially useful as scaffold materials mimicking the extracellular environment.
Introduction
The extracellular matrix (ECM) is a complex mixture of proteins and other molecules that provides structural support and biochemical signalling to cells. It is secreted by cells and surrounds them, forming a structural and functional barrier between the cells and their environment. The ECM also guides cell migration and proliferation and plays a role in tissue development and repair.1In vitro cell culture provides a platform to investigate cell physiology and tissue pathophysiology outside the organism. Conventionally, these kinds of cell-culture studies are carried out on 2D substrates, where cells are cultured on a flat surface. This system has contributed to our understanding of molecular biology, stem-cell differentiation and tissue morphogenesis. This arrangement allows cells to exchange nutrients and waste products easily. However, some reports have shown a non-natural behaviour of cells when they are cultured in 2D systems as physiologically they are surrounded by high concentrations of proteins and hormones.2 In 2D culture systems, cells are grown in a monolayer in which only one part of the cell membrane interacts with the ECM. Consequently, the concentration and spatial distribution of nutrients and biomolecules accessible to cells are limited and not representative of the natural matrix. As such, numerous researchers are making efforts to overcome these difficulties by developing 3D systems for cell culture and tissue engineering.2–5
Although commercial 3D cell-culture products are available, there is still room for improvement in this field. The main challenge is to recreate 3D environments to study how cell signalling occurs and to have more accurate models of tissues and organs.6 There are many ways to prepare such environments, although each method has its own set of advantages and disadvantages. As such, numerous researchers are making efforts to overcome these difficulties by developing 3D systems most representative of the physiological conditions, including cell spheroids7 and polymeric scaffolds, most of which can be integrated into organ-on-a-chip (OoC) devices8–11 and 3D bioprinting.12 Also, the use of beads or gels to support cells is a common method,13 whereas another approach involves using the microenvironments already present in tissues, such as pores and channels in the extracellular matrix. However, although they are usually made with natural and biodegradable components, these systems lack a 3D structure and have poor structural stability.14
An alternative approach relies on the use of hydrogels, which are networks of polymer chains that can mimic ECM characteristics, such as flexibility and water content, or which can swell or shrink in response to changes in pH or temperature.15 This allows the hydrogels to change their shape to accommodate the cells. However, hydrogels sometimes present issues related to: (i) cell adhesion, since cells do not have specific receptors to interact with the chemical compounds present in the hydrogel network, and (ii) mechanical behaviour, which strongly influences the regulation of cell functions, their physiology, cell differentiation and cell viability.
In this sense, glycosaminoglycans (GAGs), which are long-chain, unbranched polysaccharides present on the cell surface and are components of the extracellular matrix, are essential as signalling molecules with regulatory effects on protein activity, structural components, and effectors of cellular activity.16,17 Thus, GAGs have been used as biomaterials to develop hydrogel networks for tissue engineering.18 Indeed, their presence in a hydrogel provides a platform that induces better cell adhesion and cell viability and chondrogenesis in vitro and in vivo, represents a lead for anti-HIV compounds that are receiving attention given their therapeutic potential, or can even be used in the field of drug delivery since they can coordinate with cationic drugs.19–27 Indeed, one key member of the GAG family, namely heparin, is also able to coordinate with growth factors, proteins and other kinds of biomolecules. The most important interactions between these systems are ionic ones between positively charged amino acids from proteins and the sulfonate and carboxylate groups in heparin.28,29 As such, heparin derivatives have been extensively used in the design of hydrogels.30–36 However, most of these hydrogels exhibit poor mechanical properties. In addition, given its high anticoagulant effect, heparin may often lead to haemorrhagic complications, thus meaning that heparin-based hydrogel analogues are required.
The mechanical properties of a hydrogel are also key to good cell development.22 Consequently, different strategies have been followed to modulate the swelling capacity of the network or to modify its elasticity, but the viscoelasticity of the system is usually ignored. Viscoelasticity is the ability of a material to exhibit viscous and elastic behaviour simultaneously under constant forces, with polymer chains flowing and reorganizing in order to adapt themselves to the applied stress. Cell spreading, proliferation or differentiation are influenced by the viscoelasticity of the materials since natural extracellular matrices are viscoelastic and exhibit stress relaxation.37 Thus, an ideal viscoelastic environment is crucial for cell adhesion and proliferation. The viscoelasticity of a hydrogel can be tuned by changing the composition of the gel, the structure of the network or the amount or kind of crosslinker. In general, physically crosslinked hydrogels with dynamic interactions show better viscoelastic behaviour.38 In addition, this behaviour can also be modulated by the inclusion of nanomaterials, such as graphene.39 This strategy will help in tuning the pore size, swelling degree or even mechanical properties of the hydrogel.40 Graphene has been widely used in the field of tissue engineering and, in combination with 3D hydrogels, in cell culture.41 Moreover, the functionalization of graphene by the introduction of organic groups changes its properties, which is crucial for the final application thereof.42–44 As such, the combination of graphene, sulfonic groups and a suitable network could allow the development of hydrogels with excellent viscoelastic properties and with potential applications in the field of biomedicine.
In the present study, few-layer graphene (FLG) was functionalized with sulfonic groups using a green methodology based on microwave (MW) irradiation. The inclusion of this nanomaterial in a hydrogel matrix provides a new approach for the preparation of sulfonic-based hydrogels combining the properties of graphene and sulfonic groups. The synthesis of the hydrogel using UV light was also optimised. The role of graphene and functionalized graphene was studied using different characterization techniques, focusing especially on the viscoelastic behaviour. Finally, cell-viability and -differentiation studies were carried out to check the biocompatibility and potential applications of the hydrogels. It is important to highlight that no sulfonate or sulfonic-based hydrogels with viscoelasticity similar to that found in natural tissues have been described in the literature previously. These gels are promising materials as scaffolds for 3D cell culture that mimic the extracellular environment.
Experimental section
Materials and reagents
Reagents were used as purchased from commercial sources without further purification. SP-1 graphite powder was purchased from Bay Carbon, Inc. and glycine was from Acros Organics (98%). Isopentyl nitrite, sulfanilic acid, acrylamide (AM) and N,N′-methylenebisacrylamide (MBA) were purchased from Sigma Aldrich. IRGACURE 2959 was purchased from TCI Chemicals. Dichloromethane was purchased from Scharlab and ethanol was from Labkem. A planetary ball mill (Retsch PM 100) was used to prepare FLG. Milli-Q water was generated using a Simplicity® UV Water Purification System. SH-SY5Y cells were purchased from ATCC. Culture media, plates, fluorophores, antibodies and reagents used for cell viability and immunochemistry experiments were purchased from Thermo Fisher (MA, USA). All-trans retinoic acid (ATRA) was purchased from Sigma-Aldrich and brain-derived neurotrophic factor (BDNF) was purchased from Thermo Fisher Scientific.Preparation of FLG
In a typical experiment, 75 mg of graphite and 250 mg of glycine were ball-milled (planetary ball-mill Retsch PM 100) in a 250 mL stainless steel jar containing 15 stainless steel balls (2 cm in diameter each) at 100 rpm and room temperature for 2 h. The solid obtained was dispersed and sonicated in 100 ml of water. The dispersion was then placed in dialysis tubing (Spectrum Labs, 3.5 kDa MWCO) and dialyzed with water at room temperature, where the solvent was replaced every 2 hours, overnight in order to remove glycine. Finally, the FLG aqueous dispersion was lyophilized to produce the FLG powder.Functionalization of FLG
FLG was functionalized in a CEM DISCOVER S-Class reactor. Thus, FLG (10 mg) and sulfanilic acid (72.74 mg, 0.42 mmol) were placed in a microwave-proof glass vessel with 2 mL of Milli-Q water. After sonication for 30 s, isopentyl nitrite (72.22 μL, 0.56 mmol) was added to the mixture, which was sonicated for 30 s. The reaction mixture was then irradiated for 30 min at 90 °C and 30 W. After cooling to room temperature, the process was repeated twice to increase the functionalization. Finally, the functionalized FLG (f-FLG) was washed by filtration through a Millipore membrane (fluorophore 0.45 μm) with 30 mL of water, 30 mL of EtOH and 10 mL of dichloromethane, and then redispersed in fresh dichloromethane (5 mL), which was subsequently evaporated to yield a black powder.Synthesis of acrylamide-based hydrogels by photopolymerization
Acrylamide hydrogels were prepared by the radical polymerization of acrylamide (2.81 mmol) and the crosslinker N,N′-methylenebisacrylamide (0.0013 mmol) in 1 mL of Milli-Q water. 2-Hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (IRGACURE 2959) (0.013 mmol) was used as a radical photoinitiator. The mixture was sonicated until homogenization; then, polymerization was performed at room temperature by irradiating with UV light (wavelength 365 nm, four LEDs with an irradiation power of 1200 mW each) for 15 min.For mechanical tests, cylindrical silicon-based molds with a diameter of 1.5 cm and a height of 0.7 cm were used, while for cell experiments, hydrogels were synthesized with a thickness of 300 μm using a REODHR-1 Rheometer with a special platform equipped with UV LEDs.
Synthesis of FLG and f-FLG hybrid hydrogels by photopolymerization
The FLG-based and f-FLG-based hydrogels were synthesized following the same procedure but by using an aqueous FLG and f-FLG dispersions (0.5 mg mL−1), respectively, as the solvents.Characterization of the nanomaterials
Thermogravimetric analyses were performed using a TGA Q50 (TA Instruments) at 100 °C for 20 min for an initial isotherm, followed by an increase of 10 °C min−1 up to 800 °C under a N2 atmosphere.Raman spectra were recorded using a Renishaw inVia microspectrometer with a 532 nm point laser. Raman samples were prepared by dropping the powder onto double-sided tape.
Transmission electron microscopy (TEM) was carried out using a JEOL 2100 HRTEM system at an accelerating voltage of 200 kV. Samples were prepared as dilute ethanol dispersions of FLG and f-FLG by sonication for 2 min, and then settled out on holey C grids (EMS). Images were recorded using a digital Gatan Orius camera (2 × 2 MPi) under low-dose conditions (<5 mA). Z-potential was analyzed using a (PONER EQUIPO/MARCA). Samples were prepared as Milli-Q water dispersions of FLG and f-FLG (0.25 mg mL−1).
Characterization of the hydrogels
Swelling studies were carried out by immersing the xerogels in Milli-Q water and a cell culture medium (CCM) consisting of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Eagle's minimum essential medium (EMEM) and F12 medium at room temperature. The samples were weighed at established time intervals, and measurements were finished once the hydrogels reached constant weight. In order to avoid measurement errors due to the weight of water, excess water was removed using a paper filter before each measurement. The swelling degree was calculated using the following equation:
:1 mixture of Eagle's minimum essential medium (EMEM) and F12 medium at room temperature. The samples were weighed at established time intervals, and measurements were finished once the hydrogels reached constant weight. In order to avoid measurement errors due to the weight of water, excess water was removed using a paper filter before each measurement. The swelling degree was calculated using the following equation:where, Wt is the weight of the hydrogels at each time and W0 is the weight of the xerogels.
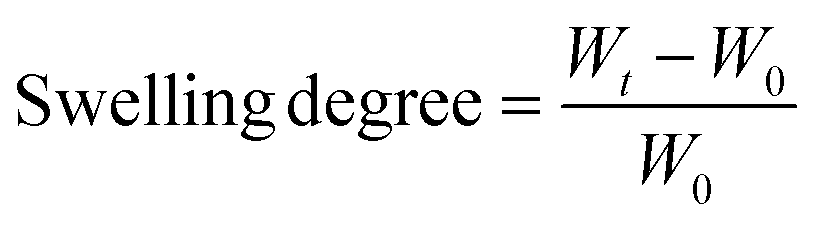
Mechanical testing of the hydrogels was carried out using a REODHR-1 Rheometer at room temperature. Hydrogel disks (×6) were measured at their initial swelling degree (diameter of 1.5 cm and thickness of 0.7 cm). For Young's modulus measurement, hydrogels were compressed in a uniaxial form between two plates at a rate of 100 μm s−1 until 40% compression of the height of the hydrogels; the linear part of the curve was analyzed (from 2% to 10% of strain). For viscoelasticity studies, hydrogel disks were compressed in a uniaxial form until 15% compression of the diameter of the hydrogel also at its initial swelling state at a rate of 16.67 μm s−1. The hydrogels were then maintained under that stress for 10 min; the relaxation of the chains was observed.
The porous structure was analyzed by scanning electron microscopy (SEM) using a Gemini SEM 500 from Zeiss with FEI QUANTA 250 apparatus. Hydrogels were swollen to their maximum swelling state, and then frozen overnight at −4 °C. In order to be sure of the freezing process, samples were also frozen in liquid N2 for a few minutes. The frozen hydrogels were then lyophilized overnight to obtain dry aerogel samples.
SH-SY5Y cell culture
Human neuroblastoma SH-SY5Y cells (ATCC) were maintained in a 1:1 mixture of EMEM and F12 medium, supplemented with 10% fetal bovine serum, 1% L-glutamine, and 1% Pen/Strep, under a humidified atmosphere supplied with 5% CO2 at 37 °C.
Cell-viability assays
Different hydrogels were cut into 1 cm2 squares (p-AM, p-AM_G and p-AM-f_G), transferred into separate wells of a 12-well plate, dried and sterilized by UV radiation for 24 hours. A dilution of 4 × 106 cells per mL in complete medium was prepared and 25 μL of this solution was soaked into each hydrogel. After 30 min of incubation at 37 °C, the complete medium (2 mL) was added to each well and cells were incubated for 2 and 7 days under standard cell culture conditions. After 2 and 7 days, cells were then washed with Hank's balanced salt solution (HBSS) and incubated with 1 μM calcein-AM, 1.5 μM propidium iodide and 1 μM Hoechst 33342 in the complete medium for 15 min at 37 °C. Calcein-AM was used as a cell-permeant dye to determine cell viability (green fluorescence), propidium iodide was used as a non-permeant dye to determine cell death (red fluorescence) and Hoechst 33342 was used to dye all nuclei (blue fluorescence). After washing once with HBSS, cells were maintained in the complete medium before transfer of each hydrogel onto a glass slide to visualize fluorescence.SH-SY5Y cell differentiation
SH-SY5Y differentiation was evaluated using the classical method of Encinas et al.,45 as modified by Shipley et al.46 and adapted to the study of hydrogels. Briefly, cells were treated with 1 μM ATRA for 5 days, after which they were detached with trypsin and a dilution of 4 × 106 cells per mL was prepared. Different hydrogels (p-AM_G and p-AM-f_G) were prepared and sterilised as described in the previous experiment and rehydrated, for at least 24 hours, with the Neurobasal medium complemented with 1× B-27, 1× Pen/Strep, 2 mM Glutamax I and 100 ng mL−1 BDNF (brain-derived neurotrophic factor). Hydrogels in which BDNF was not added were considered as controls. A 25 μL aliquot of the ATRA-treated cell solution was added to each of the hydrogels following the protocol described in the previous experiment, and each well was supplemented with 2 mL of the complemented Neurobasal medium (without BDNF). Cells were cultured for 3 days before analysing the differentiation levels by measuring MAP2 expression.Immunochemistry
MAP2 immunostaining was performed as described previously.47 Briefly, the culture medium was removed and the cells were washed with PBS. The cells were then fixed with 4% formalin for 10 min at room temperature and permeabilized with 0.5% Triton X-100 in PBS for 10 min. They were then washed twice with PBS for 5 min. Blotto buffer was used as the solvent to reduce non-specific binding of primary and secondary antibodies.Blotto buffer contained 50 mg mL−1 BSA and 0.05% Tween-20 in PBS. Thereafter, the cells were incubated for 2 h at 37 °C in Blotto buffer containing rabbit anti-MAP2 (1:1000). After three washes with Blotto buffer (10 min each), cells were exposed to Alexa 488-conjugated goat anti-rabbit IgG (1:1000) for 1 h at 37 °C. Thereafter, the cells were washed three times with PBS, mounted following the standard procedures using ProLong Gold as an antifade reagent, and stored under cold and dark conditions.
Confocal microscopy and image analysis
Images were taken by scanning the hydrogels using a Zeiss LSM 800 confocal microscopy system (Carl Zeiss, Weimar, Germany). Fluorescence images under each set of conditions were taken in duplicate and acquired using the 10× objective (X and Y axes = 638.9 μm) with one excitation laser at 494 nm for calcein-AM, another excitation laser at 613 nm for propidium iodide and another excitation laser at 348 nm for Hoechst 33342 in the viability experiments, and one excitation laser at 488 nm for MAP2 visualization. Confocal z-slices of around 360 μm were used (from 300 to 400 μm) with a 30 μm z-interval. Cell counting was done using ImageJ (Fiji, NIH Image; Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997–2018) by a blind data collection with the Cell Counter plugin. Quantitative variables are presented as a normalized percentage of living cells with respect to the control (p-AM hydrogel). Comparisons between hydrogels were conducted using the Kruskal–Wallis procedure for independent samples and the DMS post hoc test. Three experiments were performed using different cell passages, with two different slices being used for each set of conditions in each experiment.Results and discussion
Preparation and characterization of FLG
FLG was prepared following a previously reported method involving a mechano-chemical exfoliation process in which glycine was used as an exfoliating agent.48 The exfoliated FLG obtained was suspended in water and dialyzed in order to remove glycine. The aqueous FLG dispersion was then lyophilized to obtain a black powder.TGA analysis showed a very stable material with a weight loss of 3% at 600 °C, which corresponds to the small amount of functional groups in the FLG material (Fig. 1, black line).
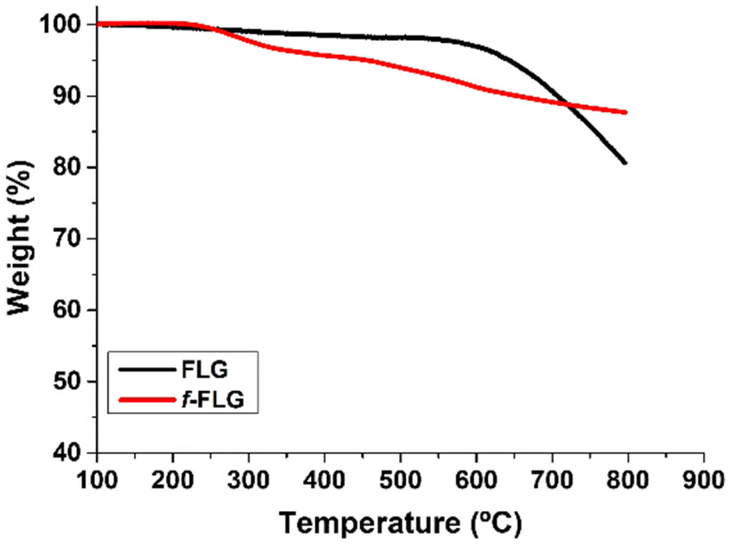 | ||
| Fig. 1 TGA analysis of FLGvs.f-FLG. | ||
This low oxygen content was also confirmed by the elemental analysis (Table 1, entry 1).
| Entry | NM | % C | % H | % O | % N | % S |
|---|---|---|---|---|---|---|
| 1 | FLG | 97.02 ± 0.60 | 0.21 ± 0.04 | 2.08 ± 0.03 | 0.22 ± 0.03 | 0.47 ± 0.03 |
| 2 | f-FLG | 85.90 ± 0.21 | 0.97 ± 0.02 | 10.09 ± 0.03 | 0.56 ± 0.007 | 2.48 ± 0.02 |
The Raman spectrum of FLG shows the most characteristic D, G, D′ and 2D bands at 1350, 1581, 1621 and 2711 cm−1, respectively (Fig. 2, black line), thereby providing information about the FLG structure. The G band, which is the most intense peak, is assigned to the planar sp2-bonded carbon atoms in FLG. The D band shows the disorder-induced mode in the structure of FLG (increasing with Csp3 hybridisation). The intensity ratio of the D and G bands (ID/IG) is used to quantify the number of edge defects in FLG. In this case, ID/IG is 0.36, thus meaning there are very few such defects.49 The D′ band appears in FLG as a shoulder on the G band, and the 2D peak reveals four layers in our FLG material, as calculated according to Paton et al.50 (eqn (1), ESI†).
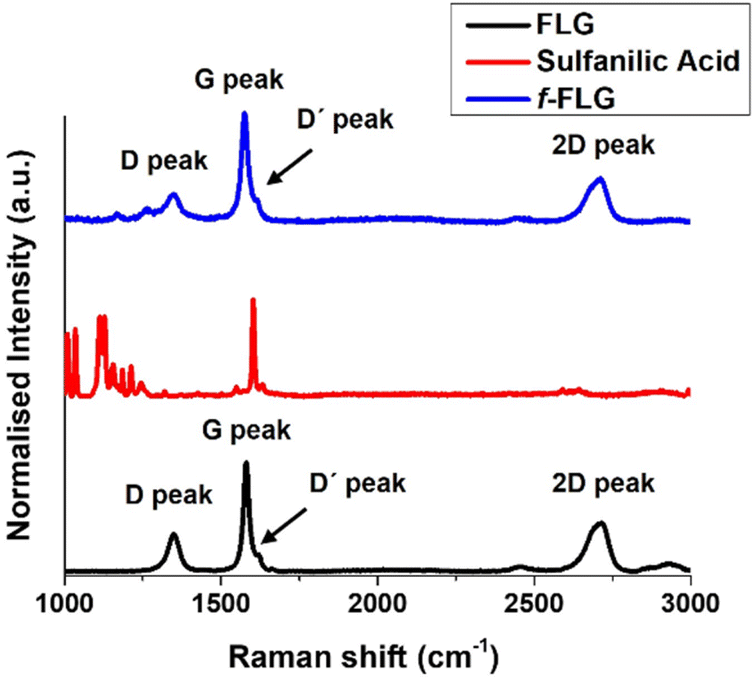 | ||
| Fig. 2 Raman spectra of FLG, sulfanilic acid and f-FLG. | ||
Finally, the TEM analysis of FLG revealed a lateral size distribution of 216 ± 22 nm and confirmed the presence of four layers (Fig. 3a).
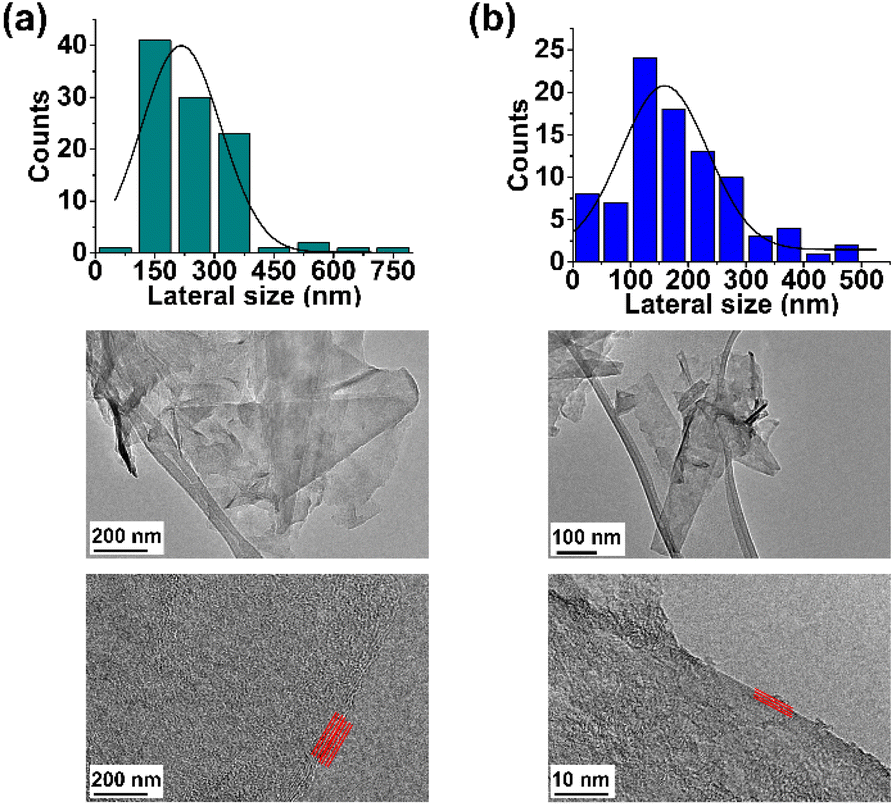 | ||
| Fig. 3 Lateral size distribution and TEM images of (a) FLG and (b) f-FLG. | ||
Functionalization of FLG
The functionalization of pristine graphene is still challenging since this non-reactive material comprises exclusively sp2 carbon atoms with π-electrons delocalized over the entire 2D surface.40 The degree of functionalization of graphene with aryl diazonium salts is heavily influenced by the graphene structure, and some authors have described that only the edges or defects are susceptible to reaction.41In this work, the covalent functionalization of FLG was performed by radical addition using MW irradiation. This methodology has previously been employed with other carbon nanostructures.51–56 In a typical reaction, FLG was dispersed in water, and sulfanilic acid and isopentyl nitrite were added. After sonication, the mixture was irradiated at 90 °C and 30 W for 30 min (Scheme 1). The process was repeated twice to accomplish a certain degree of functionalization. The presence of azo compounds was confirmed using different characterization techniques. The aryl radicals appear to initially react with FLG and then undergo aromatic homolytic substitution after this first modification, as reported previously.57,58
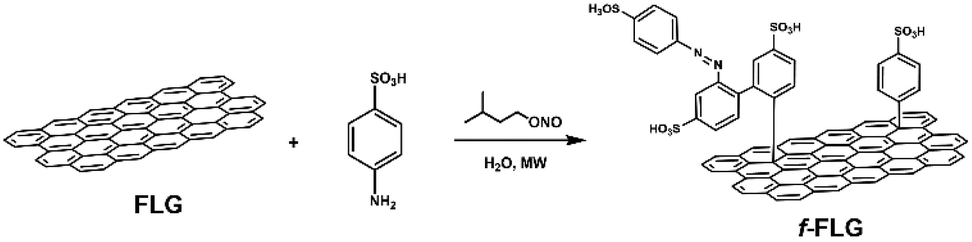 | ||
| Scheme 1 Proposed synthesis scheme for the radical addition of sulfanilic acid to FLG. | ||
The f-FLG obtained was washed thoroughly with water, ethanol and dichloromethane and characterized using Z-potential measurement, TGA, Raman spectroscopy, elemental analysis and TEM.
The Z-Potential of the pristine FLG and f-FLG was analysed, revealing a value of −33.30 and −70.84 mV, respectively (Fig. S3†). These results confirm the successful incorporation of sulfonic groups onto the FLG surface.
TGA showed clear changes after the reaction (Fig. 1, red line). Thus, the FLG starting material showed a weight loss of 3% at 600 °C under a N2 atmosphere, whereas an extra weight loss of 6% at 600 °C was observed after functionalization, thus confirming the success of the reaction.
On the other hand, elemental analysis confirms the presence of 0.78 mmol per gram sulfonic groups (Table 1, entry 2). Elemental analysis also revealed the presence of nitrogen, thereby confirming further reaction of the aryl ring attached to FLG. In addition, the presence of diazonium salts could lead to azo derivatives, as also described previously.57,58
The Raman spectrum shows that the symmetric 2D band at 2711 cm−1 in FLG is retained. Moreover, no clear differences were found between FLG and f-FLG samples due to the minor modifications at the FLG surface. In addition, no increase in the D band was observed, thus confirming the presence of azo derivatives as discussed above (Fig. 2, blue line).
Finally, TEM analysis revealed a lateral size distribution of 159 ± 11 nm (Fig. 3b), similar to that for pristine FLG, thus confirming that the surface of FLG was not heavily influenced by the incorporation of a limited number of organic groups.
Although multiple examples of graphene functionalization via radical addition have been reported, the starting material tends to be graphite59,60 or graphene oxide.61 Furthermore, while the degree of functionalization is very high for GO, fullerenes or SWNTs,52–56 in graphene, only the edges are susceptible to reaction. In this case, pristine FLG with small defects was used and, after functionalization, the FLG structure remained, as shown by the TEM and Raman characterization.
From thermal- to photo-polymerization: synthesis of acrylamide-based hydrogels
Most of the acrylamide-based hydrogels published previously are prepared by thermal radical polymerization, which is very time-consuming. Thus, an optimization process involving photopolymerization under UV light was performed using IRGACURE 2959, which is a highly efficient radical photoinitiator for UV curing systems that is commonly used for unsaturated monomers, especially in aqueous solution.62,63Furthermore, it has been reported that IRGACURE 2959 shows low cellular toxicity and can also be employed in the synthesis of hydrogels for final applications in the field of biomedical or tissue engineering.64 In this case, polymerization took place in water, using AM as the main monomer and MBA as a crosslinker. This reaction resulted in a tough and easily manipulable hydrogel (Scheme 2a). The synthesis of hydrogels by photopolymerization reduces the reaction time, in this case from 1 h to 15 min. In addition, it allows the possibility of using a 3D printer, thereby enhancing the versatility of the hydrogels by creating different shapes or sizes depending on the final application of the hydrogels.65 Furthermore, photopolymerization avoids the precipitation of FLG in the hydrogel structure, thus resulting in highly homogeneous materials.
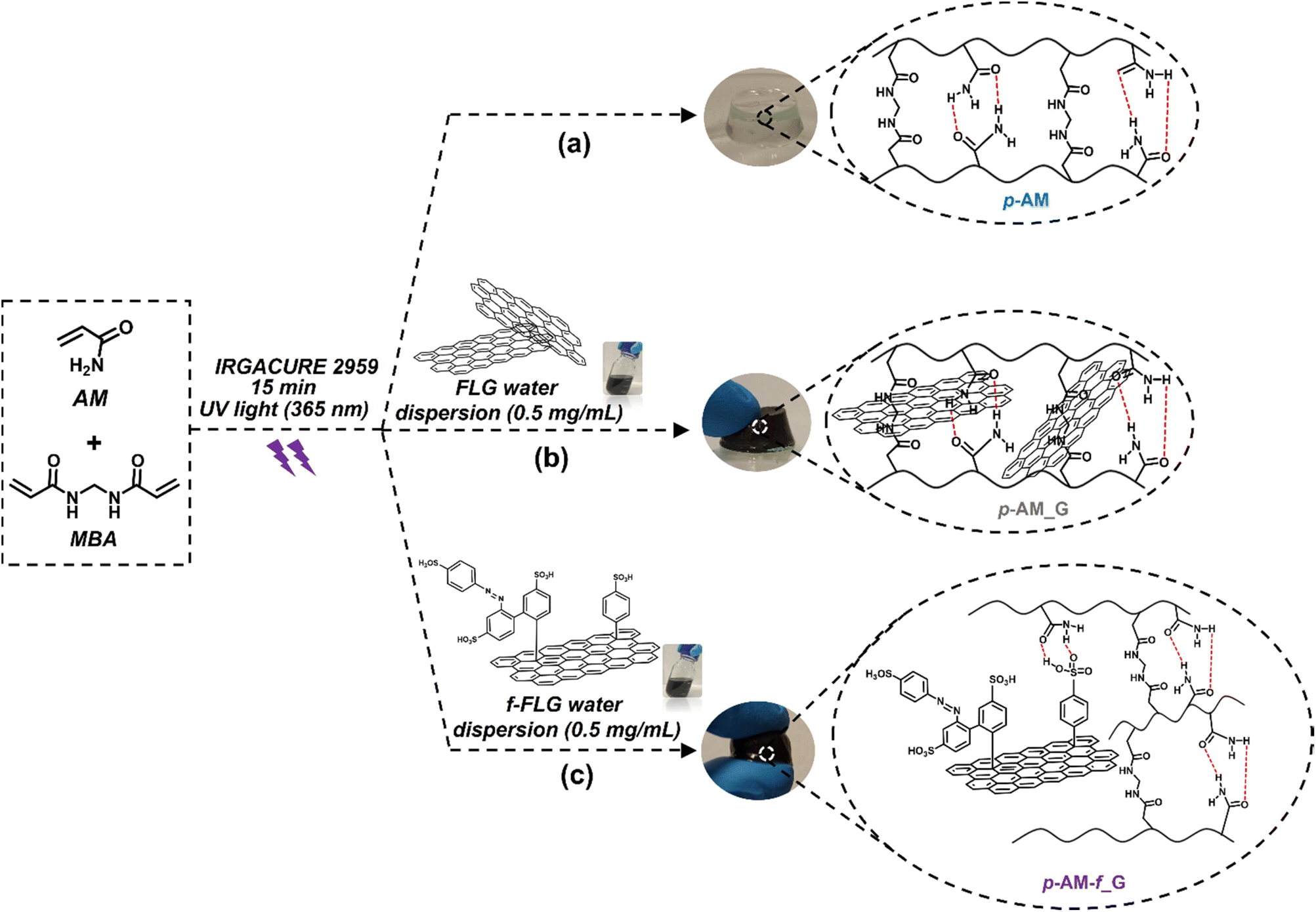 | ||
| Scheme 2 Synthesis scheme and digital images of p-AM hydrogels: (a) blank, (b) FLG- and (c) f-FLG-based hydrogels. | ||
In the case of FLG (p-AM_G)- and f-FLG (p-AM_f-G)-based hydrogels (Scheme 2b and c, respectively), they were prepared by in situ radical polymerization, incorporating them into the synthesis process. A dispersion of 0.5 mg mL−1FLG or f-FLG in water was first prepared, and then AM, MBA and IRGACURE 2959 were added and immediately polymerized under UV light.
The final hydrogels were immersed in 250 mL of Milli-Q water for four days, where water replacement was carried out three times daily to ensure the complete removal of any unreacted monomers or initiator. Subsequently, the hydrogels were dried at 40 °C in an oven until a constant weight was achieved, resulting in the formation of xerogels. The structure and role of the nanomaterials in the polymer network were thoroughly characterized using different techniques.
Characterization of the hydrogels
The swelling degree is a very important parameter when considering the final application of the hydrogels. Thus, each xerogel was immersed in Milli-Q water and the CCM and the swelling degree was determined after 24 h where the weight of the hydrogels remained constant. As shown in Fig. 4, there are no significant differences between the hydrogels, although the swelling degree decreases slightly in the presence of both FLG and f-FLG. Moreover, the swelling degree in the CCM is lower since this medium contains salts and other compounds.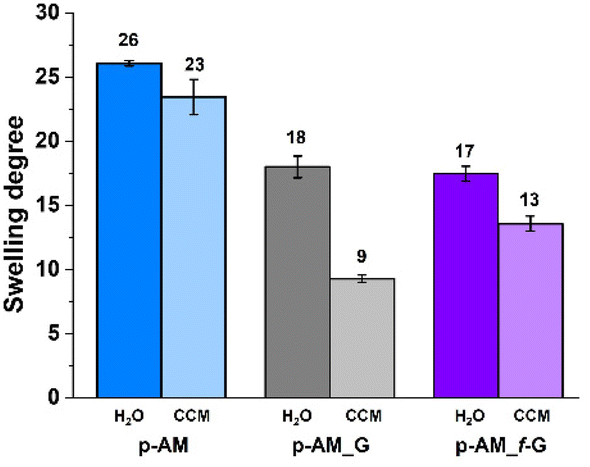 | ||
| Fig. 4 Swelling degree of p-AM hydrogels. | ||
In addition, the microstructure of these hydrogels was analysed by scanning electron microscopy (SEM) (Fig. 5). The presence of FLG and f-FLG in the hydrogel structure gave rise to a heterogeneous pore-size distribution. In the case of f-FLG, a slight decrease in pore size is found since, apart from the π–π interactions between FLG sheets and between these sheets and benzene rings from sulfanilic acid, the sulfonic groups in the network can interact with AM molecules via hydrogen-bond interactions.
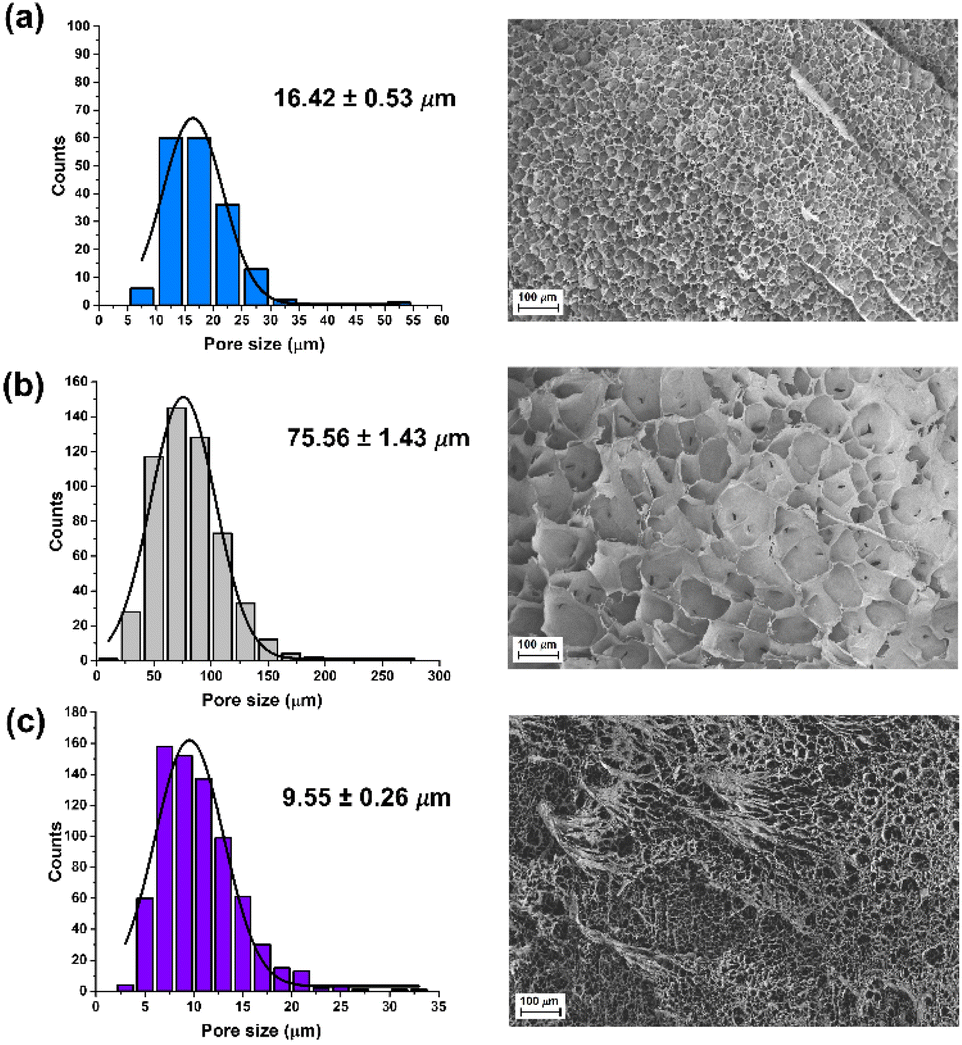 | ||
| Fig. 5 Pore-size distribution and SEM images of (a) blank, (b) FLG and (c) f-FLGp-AM hydrogels. | ||
Mechanical compressive tests were performed (Fig. 6a), and Young's modulus (Fig. 6b) was analysed in order to evaluate the influence of FLG and f-FLG on the hydrogel network. The addition of FLG improves the mechanical strength, thus suggesting that FLG interacts strongly with the network. Despite the increase in pore size in the presence of FLG, the stiffness of the hydrogel also increases, thus resulting in a more rigid material. For p-AM_f-G, no differences are observed in comparison with the blank AM hydrogel, while the elastic modulus decreases with respect to the FLG-based one. In this case, as mentioned above, the presence of dynamic interactions in the network may give rise to a more elastic network and, consequently, a lower Young's modulus.
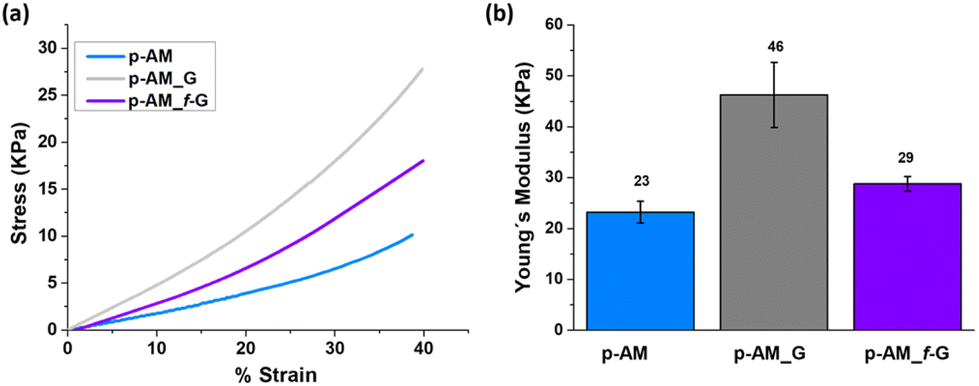 | ||
| Fig. 6 (a) Stress–strain curves and (b) Young's modulus of p-AM hydrogels. | ||
Viscoelastic studies were also performed by compressing hydrogels to 15% of their strain in order to check the stress relaxation of the hydrogel after 10 min of stress (Fig. 7). The inclusion of FLG in the structure did not enhance this property. However, the addition of f-FLG with sulfanilic acid groups improved the viscoelasticity of the material, which exhibited a stress relaxation of 40%. As noted above, hydrogen-bond interactions and π–π stacking between FLG sheets and between FLG and benzene rings from sulfanilic acid can alter the network, thereby making it more elastic. These kinds of interactions are dynamic; in other words, when the hydrogel is under constant stress, the polymer chains reorganise by breaking these links and building new bonds, thus resulting in a material with good stress relaxation. These hydrogels can therefore be modulated owing to the altered connectivity and chain mobility in the polymer network due to the presence of f-FLG with sulfanilic acid moieties. The viscoelasticity values of p-AM_f-G are similar to those found in the literature for human tissues.31
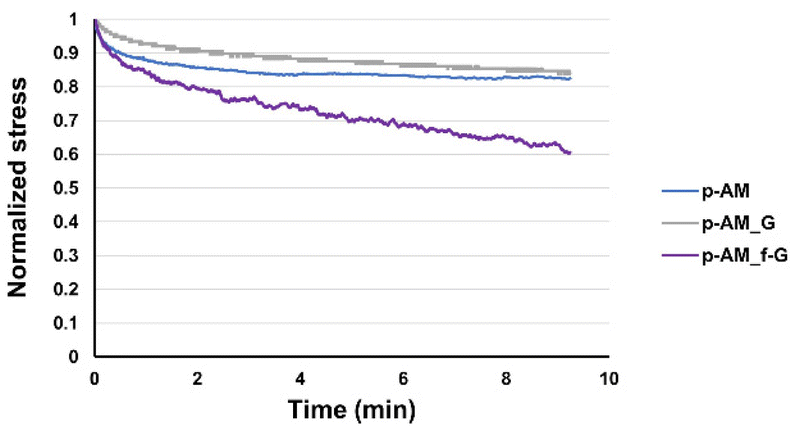 | ||
| Fig. 7 Viscoelastic behaviour of p-AM hydrogels. | ||
Cell-viability assays
Due to the good mechanical and, especially, viscoelastic properties of these new hydrogels with sulfonic f-FLG, cell-viability assays were carried out to evaluate possible cytotoxic effects. Thus, the neuroblastoma cell line SH-SY5Y was seeded onto the three different kinds of hydrogels. This cell line is commonly used in experimental neurobiology and has been extensively employed as a model by our research group because it can differentiate into a neuron-like phenotype. In this case, representative confocal images were taken after 2 and 7 days of cell culture (Fig. 8). | ||
| Fig. 8 Representative confocal fluorescence images of SH-SY5Y cells grown on different scaffolds. The results are presented in the following order: merged image, propidium iodide staining for dead cells in red, calcein-AM staining for living cells in green and Hoechst 33342 for nuclear staining in blue. Scale bar: 100 μm. | ||
The presence of sulfonic groups, together with the viscoelastic environment, promotes cell viability in the early stages of cell culture, with this viability stabilising after 7 days (Fig. 9).
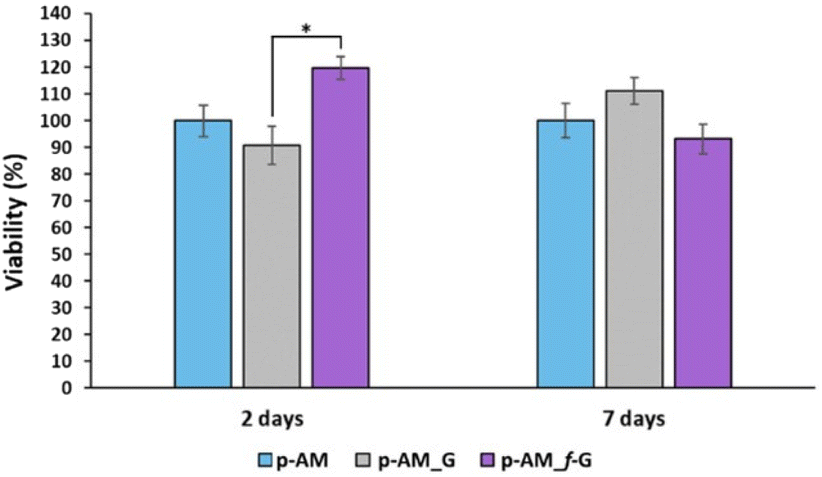 | ||
| Fig. 9 Viability of SH-SY5Y cells after two and seven days of culture on p-AM, p-AM_G and p-AM-f_G hydrogels. The results at each time refer to the corresponding percentage of living cells on the p-AM hydrogel. Differences were obtained using the Kruskal–Wallis procedure for independent samples and the DMS post hoc test (*p < 0.05). | ||
This improved viability observed at short times for the sulfonic-based hydrogel could be due to a combination of several factors. Thus, sulfonic groups can improve the hydrophilicity of the material. This is important because many biological processes take place in aqueous environments and hydrophilicity can improve the ability of the material to interact with cells and biological fluids. Similarly, the presence of sulfonic groups can increase the negative charge on the surface of the material, which can enhance the ability of the material to interact with positively charged molecules and cells in the biological environment. This is particularly important for materials intended for tissue engineering, where a good interaction between cells and the material is crucial for cell adhesion and proliferation. Finally, sulfonic groups can act as anchoring groups for bioactive molecules, such as peptides or proteins, which can enhance the interaction between the material and cells.
Another important scaffolding property of our hydrogel is the viscoelasticity. The viscoelasticity can significantly affect fundamental cellular processes, including differentiation. Extensive research has demonstrated that this property of the extracellular matrix can regulate these processes and promote behaviors that are not observed with purely elastic hydrogels in both two- and three-dimensional culture microenvironments. In addition, these results suggest design guidelines for the next generation of biomaterials, aiming to match tissue and extracellular matrix mechanics for in vitro tissue models and applications in regenerative medicine.66,67
In order to analyse the potential of these materials in SH-SY5Y cell differentiation and therefore their possible application in neuronal regeneration processes, a comparative analysis was performed in which BDNF, a factor that promotes cell differentiation in SH-SY5Y cells after exposure to ATRA, was embedded in p-AM_G and p-AM_f-G (Fig. 10). MAP2 detection is typically used as a hallmark of neuronal differentiation.68 Preliminary experiments suggest that, when cell differentiation takes place on the surface of f-FLG scaffolds, there is an increase in MAP2 expression, that is, an increase in the number of cells that successfully differentiate in comparison with non-functionalized FLG hydrogels. This approach is of great significance as FLG and its derivatives have shown unique and promising properties in various biotechnological applications. The presence of sulfonic groups on FLG may facilitate cell differentiation as a result of potential alterations in surface charge, chemical reactivity, and cellular interactions, which warrants further biological research.
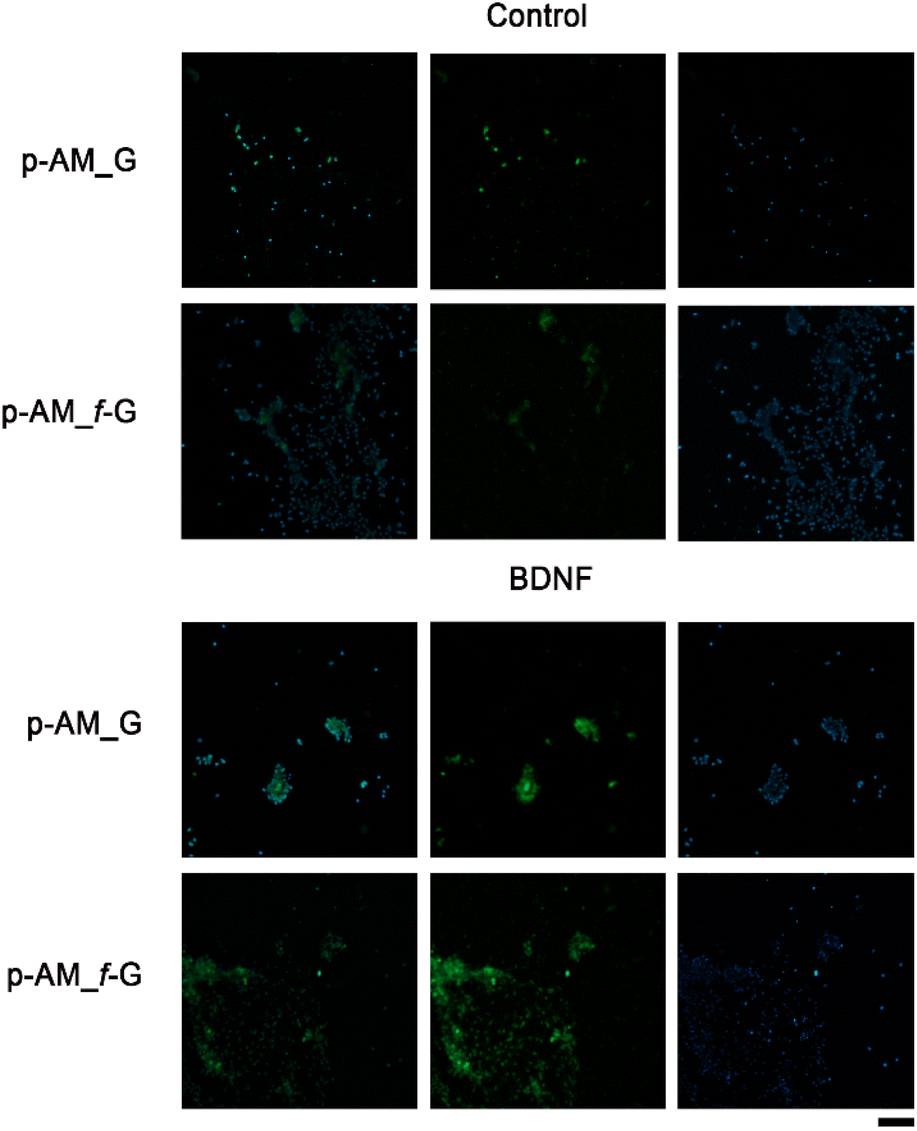 | ||
| Fig. 10 Representative immunochemistry images for microtubule-associated protein 2 (MAP2; a neuronal cytoskeletal marker). Immunochemistry of MAP2 was performed on hydrogels with or without (control) BDNF, seeded with SH-SY5Y cells pre-treated with ATRA. Results are presented in the following order: merged image, MAP2 signal in green and DAPI, for nuclear staining, in blue. Scale bar: 100 μm. | ||
Conclusions
The preparation of synthetic hybrid sulfonic-based hydrogels as soft scaffolds for tissue engineering has been reported. Functionalization of the FLG surface was accomplished using sulfanilic acid, with microwave irradiation as the energy source. Despite the effective functionalization with sulfonic groups on the graphene surface, the FLG structure is preserved. Moreover, the incorporation of this modified 2D nanomaterial into a photopolymerizable AM hydrogel network allows modulation of the macroscopic properties of the 3D system. In fact, the presence of sulfonic-modified-FLG in the network improves the viscoelastic behaviour, which is similar to that found in living tissues. This new hybrid material enhances the cell viability in the early stages of the process and facilitates the differentiation process due to potential alterations in surface charge, chemical reactivity, and cellular interactions, which warrant further biological research.Notes
We encourage the citation of primary research over review articles, where appropriate, in order to give credit to those who first reported a finding. Find out more (https://www.rsc.org/news-events/articles/2020/jun/rsc-signs-dora/) about our commitments to the principles of San Francisco Declaration on Research Assessment (DORA).Author contributions
J. M. G. performed the experiments and wrote the original draft. I. S.-M. performed the biological studies. C. A. C.-S. and I. B.-Y. gave technical suggestions on the biological experiments, co-wrote, and reviewed the manuscript. E. V., S. M. and M. A. H. supervised, reviewed, and edited the manuscript. E. V. and S. M. acquired the financial support for the project leading to this publication. All the authors approved the final manuscript for submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the European Flagship project GA881603 Graphene Core 3, the Spanish government (project PID2020-113080RB-I00), and the Junta de Comunidades de Castilla-La Mancha (projects SBPL4/17/180501/000204 and SBPLY/21/180501/000135/1). This study is a part of the Advanced Materials programme and was supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and by the Junta de Comunidades de Castilla-La Mancha. Josué M. Galindo and I. San-Millán acknowledge the Spanish Ministry of Economy and Competitiveness (MINECO) for their Fellowships (FPI-PRE2018-084047 and FPU19/04293), respectively. Finally, the authors would like to offer special thanks to the instrumentation team at the IRICA center for their valuable help with sample preparation and analysis.Notes and references
- J. Nicolas, S. Magli, L. Rabbachin, S. Sampaolesi, F. Nicotra and L. Russo, Biomacromolecules, 2020, 21(6), 1968–1994 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103(4), 655–663 CrossRef CAS PubMed.
- A. A. Ustyugov, M. M. Chicheva, E. A. Lysikova, E. A. Vikhareva, N. A. Sipyagina, A. N. Malkova, E. A. Straumal, E. V. Bovina, F. S. Senatov, A. I. Salimon, A. V. Maksimkin and S. A. Lermontov, Biomed. Chem.: Res. Methods, 2018, 1(3), e00048 Search PubMed.
- Y. Li and K. A. Kilian, Adv. Healthcare Mater., 2015, 4(18), 2780–2796 CrossRef CAS PubMed.
- K. Da Silva, P. Kumar, Y. E. Choonara, L. C. du Toit and V. Pillay, J. Biomed. Mater. Res., Part A, 2020, 108(12), 2324–2350 CrossRef CAS PubMed.
- F. Pampaloni, E. G. Reynaud and E. H. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8(10), 839–845 CrossRef CAS PubMed.
- H. Otsuka, Gels, 2023, 9(3), 203 CrossRef CAS PubMed.
- T. Ahmed, Biosens. Bioelectron.: X, 2022, 11, 100194 CAS.
- C. M. Leung, P. de Haan, K. Ronaldson-Bouchard, G.-A. Kim, J. Ko, H. S. Rho, Z. Chen, P. Habibovic, N. L. Jeon, S. Takayama, M. L. Shuler, G. Vunjak-Novakovic, O. Frey, E. Verpoorte and Y.-C. Toh, Nat. Rev. Methods Primers, 2022, 2(1), 33 CrossRef CAS.
- J. A. Terrell, C. G. Jones, G. K. M. Kabandana and C. Chen, J. Mater. Chem. B, 2020, 8(31), 6667–6685 RSC.
- D. Vera, M. García-Díaz, N. Torras, M. Álvarez, R. Villa and E. Martinez, ACS Appl. Mater. Interfaces, 2021, 13(12), 13920–13933 CrossRef CAS.
- J. M. Unagolla and A. C. Jayasuriya, Appl. Mater. Today, 2020, 18, 100479 CrossRef PubMed.
- K. Takimoto, T. Arahira and M. Todo, Results Mater., 2022, 16, 100317 CrossRef CAS.
- G. S. Hussey, J. L. Dziki and S. F. Badylak, Nat. Rev. Mater., 2018, 3(7), 159–173 CrossRef CAS.
- E. C. González-Díaz and S. Varghese, Gels, 2016, 2(3), 20 CrossRef PubMed.
- A. Köwitsch, G. Zhou and T. Groth, J. Tissue Eng. Regener. Med., 2018, 12(1), 23–41 CrossRef PubMed.
- J. Rnjak-Kovacina, F. Tang, J. M. Whitelock and M. S. Lord, Adv. Healthcare Mater., 2018, 7(6), 1701042 CrossRef PubMed.
- M. I. Neves, M. Araújo, L. Moroni, R. M. da Silva and C. C. Barrias, Molecules, 2020, 25(4), 978 CrossRef CAS PubMed.
- Y. H. Kim, D. K. Han, K. D. Park and S. H. Kim, Biomaterials, 2003, 24(13), 2213–2223 CrossRef CAS PubMed.
- J. H. Kim, J. G. Kim, D. Kim and Y. H. Kim, J. Appl. Polym. Sci., 2005, 96(1), 56–61 CrossRef CAS.
- K. L. Christman, V. Vázquez-Dorbatt, E. Schopf, C. M. Kolodziej, R. C. Li, R. M. Broyer, Y. Chen and H. D. Maynard, J. Am. Chem. Soc., 2008, 130(49), 16585–16591 CrossRef CAS PubMed.
- H. Joon Kwon, J. Mech. Behav. Biomed. Mater., 2013, 17, 337–346 CrossRef.
- J. Liang, B. B. Karakoçak, J. J. Struckhoff and N. Ravi, Biomacromolecules, 2016, 17(12), 4064–4074 CrossRef CAS PubMed.
- S. Kim, Z.-K. Cui, P. J. Kim, L. Y. Jung and M. Lee, Acta Biomater., 2018, 72, 45–54 CrossRef CAS PubMed.
- M. Suhail, C.-W. Fang, M. U. Minhas and P.-C. Wu, Pharmaceuticals, 2021, 14(4), 350 CrossRef CAS PubMed.
- C.-T. Lee, P.-H. Kung and Y.-D. Lee, Carbohydr. Polym., 2005, 61(3), 348–354 CrossRef CAS.
- P. Mohan, D. Schols, M. Baba and E. De Clercq, Antiviral Res., 1992, 18(2), 139–150 CrossRef CAS PubMed.
- I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41(3), 390–412 CrossRef CAS.
- B. Casu, Structure and Biological Activity of Heparin, in Adv. Carbohydr. Chem. Biochem, ed. R. S. Tipson and D. Horton, Academic Press, 1985, vol. 43, pp. 51–134 Search PubMed.
- D. H. Go, Y. K. Joung, S. Y. Lee, M. C. Lee and K. D. Park, Macromol. Biosci., 2008, 8(12), 1152–1160 CrossRef CAS PubMed.
- S. Nakamura, M. Ishihara, K. Obara, K. Masuoka, T. Ishizuka, Y. Kanatani, B. Takase, T. Matsui, H. Hattori and T. Sato, J. Biomed. Mater. Res., Part A, 2006, 78(2), 364–371 CrossRef.
- M. Fujita, M. Ishihara, M. Shimizu, K. Obara, S. Nakamura, Y. Kanatani, Y. Morimoto, B. Takase, T. Matsui and M. Kikuchi, Wound Repair Regen., 2007, 15(1), 58–65 CrossRef.
- J. Lee, W. I. Choi, G. Tae, Y. H. Kim, S. S. Kang, S. E. Kim, S.-H. Kim, Y. Jung and S. H. Kim, Acta Biomater., 2011, 7(1), 244–257 CrossRef CAS PubMed.
- M. Kim, S. E. Kim, S. S. Kang, Y. H. Kim and G. Tae, Biomaterials, 2011, 32(31), 7883–7896 CrossRef CAS PubMed.
- R. Jin, L. S. M. Teixeira, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, J. Controlled Release, 2011, 152(1), 186–195 CrossRef CAS PubMed.
- M. Kim, B. Hong, J. Lee, S. E. Kim, S. S. Kang, Y. H. Kim and G. Tae, Biomacromolecules, 2012, 13(8), 2287–2298 CrossRef CAS PubMed.
- S.-B. Han, J.-K. Kim, G. Lee and D.-H. Kim, Adv. Biosyst., 2020, 4(11), 2000247 CrossRef PubMed.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H.-p. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15(3), 326–334 CrossRef CAS PubMed.
- Z. Xiong, W. Huang, Q. Liang, Y. Cao, S. Liu, Z. He, R. Zhang, B. Zhang, R. Green, S. Zhang and D. Li, Small Methods, 2022, 6(5), 2200022 CrossRef CAS PubMed.
- C. Martín, A. Martín-Pacheco, A. Naranjo, A. Criado, S. Merino, E. Díez-Barra, M. A. Herrero and E. Vázquez, Nanoscale, 2019, 11(11), 4822–4830 RSC.
- C. Martín, S. Merino, J. M. González-Domínguez, R. Rauti, L. Ballerini, M. Prato and E. Vázquez, Sci. Rep., 2017, 7(1), 10942 CrossRef PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112(11), 6156–6214 CrossRef CAS PubMed.
- I. A. Vacchi, C. Ménard-Moyon and A. Bianco, Phys. Sci. Rev., 2017, 2(1), 20160103 Search PubMed.
- H. Y. Mao, Y. H. Lu, J. D. Lin, S. Zhong, A. T. S. Wee and W. Chen, Prog. Surf. Sci., 2013, 88(2), 132–159 CrossRef CAS.
- M. Encinas, M. Iglesias, Y. Liu, H. Wang, A. Muhaisen, V. Ceña, C. Gallego and J. X. Comella, J. Neurochem., 2000, 75(3), 991–1003 CrossRef CAS PubMed.
- M. M. Shipley, C. A. Mangold and M. L. Szpara, J. Visualized Exp., 2016, 53193 Search PubMed.
- C. A. Castillo, I. Ballesteros-Yáñez, D. A. León-Navarro, J. L. Albasanz and M. Martín, Int. J. Mol. Sci., 2021, 22(12), 6577 CrossRef CAS PubMed.
- V. J. González, A. M. Rodríguez, I. Payo and E. Vázquez, Nanoscale Horiz., 2020, 5(2), 331–335 RSC.
- V. Yokaribas, S. Wagner, D. S. Schneider, P. Friebertshäuser, M. C. Lemme and C.-P. Fritzen, Sensors, 2017, 17(12), 2937 CrossRef PubMed.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13(6), 624–630 CrossRef CAS PubMed.
- E. Vázquez and M. Prato, ACS Nano, 2009, 3(12), 3819–3824 CrossRef.
- J. Liu, M. R. Rodriguez i Zubiri, B. Vigolo, M. Dossot, Y. Fort, J.-J. Ehrhardt and E. McRae, Carbon, 2007, 45(4), 885–891 CrossRef CAS.
- Y. Wang, Z. Iqbal and S. Mitra, Carbon, 2005, 43(5), 1015–1020 CrossRef CAS.
- N. Rubio, M. A. Herrero, A. de la Hoz, M. Meneghetti, M. Prato and E. Vázquez, Org. Biomol. Chem., 2010, 8(8), 1936–1942 RSC.
- F. Langa and P. de la Cruz, Comb. Chem. High Throughput Screening, 2007, 10(9), 766–782 CrossRef CAS PubMed.
- V. Campisciano, S. Riela, R. Noto, M. Gruttadauria and F. Giacalone, RSC Adv., 2014, 4(108), 63200–63207 RSC.
- P. Salice, E. Fabris, C. Sartorio, D. Fenaroli, V. Figà, M. P. Casaletto, S. Cataldo, B. Pignataro and E. Menna, Carbon, 2014, 74, 73–82 CrossRef CAS.
- A. J. Hilmer, T. P. McNicholas, S. Lin, J. Zhang, Q. H. Wang, J. D. Mendenhall, C. Song, D. A. Heller, P. W. Barone, D. Blankschtein and M. S. Strano, Langmuir, 2012, 28(2), 1309–1321 CrossRef CAS PubMed.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H.-P. Steinrück, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3(4), 279–286 CrossRef CAS PubMed.
- Z. Sun, S.-i. Kohama, Z. Zhang, J. R. Lomeda and J. M. Tour, Nano Res., 2010, 3(2), 117–125 CrossRef CAS.
- V. Rebuttini, E. Fazio, S. Santangelo, F. Neri, G. Caputo, C. Martin, T. Brousse, F. Favier and N. Pinna, Chem. – Eur. J., 2015, 21(35), 12465–12474 CrossRef CAS PubMed.
- P. Coimbra, D. Fernandes, P. Ferreira, M. H. Gil and H. C. de Sousa, J. Supercrit. Fluids, 2008, 45(3), 272–281 CrossRef CAS.
- M. Liu, M.-D. Li, J. Xue and D. L. Phillips, J. Phys. Chem. A, 2014, 118(38), 8701–8707 CrossRef CAS PubMed.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11(5), 439–457 CrossRef CAS.
- A. López-Díaz, A. Martín-Pacheco, A. M. Rodríguez, M. A. Herrero, A. S. Vázquez and E. Vázquez, Adv. Funct. Mater., 2020, 30(46), 2004417 CrossRef.
- M. Duran, A. C. M. Luzo, J. G. de Souza, W. J. Favaro, P. Garcia and N. Duran, Curr. Mol. Med., 2017, 17(9), 619–626 CrossRef CAS PubMed.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584(7822), 535–546 CrossRef CAS PubMed.
- L. Hromadkova, D. Bezdekova, J. Pala, S. Schedin-Weiss, L. O. Tjernberg, C. Hoschl and S. V. Ovsepian, Biochim. Biophys. Acta, Mol. Cell Res., 2020, 1867(9), 118737 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02689b |
| This journal is © The Royal Society of Chemistry 2023 |