
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical subcomponent self-assembly of covalent triple-stranded complexes with 3d–4f vertices: luminescence and magnetic properties†
Zhi
Liu
ab,
Fan
Yin
ab,
Jian
Yang
b,
Xiao-Qing
Guo
b,
Li-Peng
Zhou
 b,
Chong-Bin
Tian
*ab and
Qing-Fu
Sun
*ab
b,
Chong-Bin
Tian
*ab and
Qing-Fu
Sun
*ab
aCollege of Chemistry, Fuzhou University, Fuzhou 350108, People's Republic of China. E-mail: tianchongbin@fjirsm.ac.cn; qfsun@fjirsm.ac.cn
bFujian College, University of Chinese Academy of Sciences, Fuzhou 350002, People's Republic of China
First published on 22nd May 2023
Abstract
Well-defined 3d–4f heterometallic supramolecular architectures have attracted attention because of their applications in the field of luminescence and magnetism. However, covalent metallo-supramolecular discrete complexes, decorated with hetero-metallic vertices, have never been reported because of the difficulties in design and control. Herein, we report a series of covalent metallo-supramolecular discrete complexes with 3d–4f vertices synthesized by hierarchical subcomponent self-assembly of tris(2-aminoethyl)amine, 2,6-diformyl-p-cresol, and lanthanide ions (Ln) with different amines and transition metal ions. The programmable self-assembly process results in the formation of triple-stranded hetero-metallic covalent organic discrete complexes, namely 3a–3c-(Ln, Zn) (Ln = SmIII, EuIII, DyIII, YbIII and LuIII) and 3a′-(Dy, Co), which are characterized by nuclear magnetic resonance (NMR) analysis, electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS), and single-crystal X-ray analysis. Photophysical investigations disclose that the organic skeleton of 3a-(Ln, Zn) exhibits an excellent sensitizing ability toward SmIII, EuIII, and YbIII ions, displaying characteristic luminescence emission in both the visible and near-infrared (NIR) regions. AC susceptibility measurements of 3a′-(Dy, Co) reveal the frequency-independent performance under zero dc field, suggesting the absence of slow relaxation of magnetization. This work offers a new approach for the fabrication of discrete metallic covalent architectures with 3d–4f vertices.
Introduction
Over the past few decades, coordination-driven self-assembly, as a powerful synthesis technique, has been employed by many chemists in the construction of various supramolecular architectures.1–7 By referring to the template synthesis of Busch,8 Nitschke extended the synthesis technique and proposed the versatile “subcomponent self-assembly” strategy.9 In this strategy, the metal–ligand coordination bonds and covalent dynamic imine bonds (formed by condensation of an amine and an aldehyde) are in situ formed.A myriad of diverse metallo-supramolecular compounds with sophisticated structures and fascinating functions, including, but not limited to, macrocycles,10–12 helicates,13–15 grids,16,17 catenanes,18 rotaxanes,19 and metal–organic polyhedra,20–23 have been successfully fabricated by the subcomponent self-assembly approach. Besides these types of structures, there is another type of structure composed of a discrete covalent organic skeleton and metal ions, namely discrete covalent metallo-supramolecular architectures, in which the metal ions are located at the vertices or panels of the covalent organic skeleton. This type of structure is particularly appealing, since the introduction of metal ions not only can template the synthesis of covalent organic cages or covalent organic macrocycles24–26 that cannot be acquired by traditional direct imine condensation,27–32 but also endow the structure embedded by metal ions with special functions.22,23,33–35 Therefore, templated metal ions play a dominant role in determining the structures and functions of discrete covalent metallo-supramolecular complexes. If two different metal ions, such as transition metal ions (3d) and lanthanide ions (4f), are simultaneously introduced into such systems, the intrinsic optical and large single-ion magnetic anisotropy of lanthanides together with remarkable magnetic interactions between 3d and 4f make such 3d–4f based complexes excellent candidates for luminescent materials and single molecular magnets (SMMs).36–40 As far as we are aware, however, the syntheses of covalent metallo-supramolecular complexes were primarily based on single transition metal ions,25,26,30,41–48 very limited to single lanthanide ions.35 In contrast, 3d-4f heterometallic ions have not been explored, largely because of the challenge of designing a suitable ligand that can bridge the gap between the divergent coordination preferences and abilities of 4f and 3d ions.
Previously, we reported several hetero-metallic cages or macrocycles by hierarchical self-assembly of PdII or PtII metallo-ligands with lanthanide ions.49–53 With our continuing interest in hierarchical self-assembly, herein, we report the hierarchical subcomponent self-assembly of hetero-metallic covalent organic triple-stranded complexes. The subcomponent self-assembly of tris(2-aminoethyl)amine (TREN) and 2,6-diformyl-p-cresol (DFMP) with lanthanide ions (Ln) leads to the formation of metallo-ligand 1-Ln (Scheme 1). The subsequent subcomponent self-assembly of preformed 1-Ln, diamine (2a and 2b), or triamine (2c) with transition metal ions gives rise to a family of heterometallic covalent organic triple-stranded molecules, that is 3a–3c-(Ln, Zn) and 3a′-(Dy, Co), in which the hetero-metal nodes are situated at the vertices of triple-stranded molecules. Photophysical investigations indicate that the organic backbone of 3a-(Ln, Zn) can not only sensitize the visible lanthanide ions (Ln = SmIII and EuIII), but also sensitize NIR-region YbIII ions. Moreover, the magnetic properties of 3a′-(Dy, Co) are also studied.
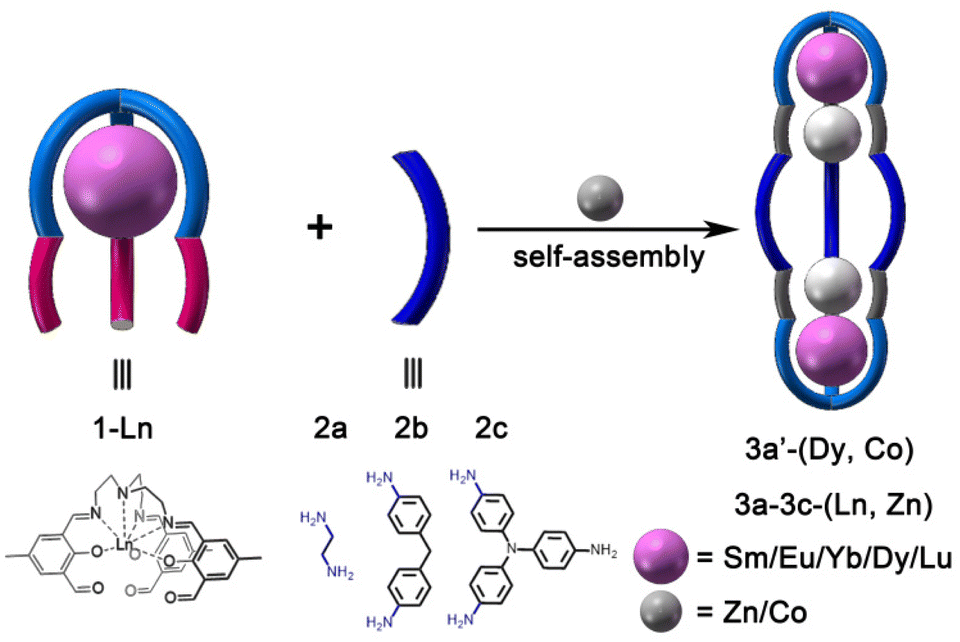 | ||
| Scheme 1 Self-assembly of 3a–3c-(Ln, Zn) and 3a′-(Dy, Co). | ||
Experimental section
Synthesis of subcomponent 1-Ln
Metallo-ligands 1-Dy, 1-Lu and 1-Yb were synthesized according to previously reported literature.54 By substitution of Ln(OTf)3·9H2O with Ln(NO3)3·6H2O, the metallo-ligands 1-Sm and 1-Eu were also prepared with yields of 35% and 30%. 1-Sm: 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H), 7.56 (d, J = 2.2 Hz, 1H), 7.43 (s, 1H), 7.40 (d, J = 2.2 Hz, 1H), 3.73 (s, 2H), 2.63 (s, 2H), 2.23 (s, 3H). ESI-TOF-MS for 1-Sm: calcd for [(1-Sm) + Na]+ 756.1431, found 756.1488; [(1-Sm)2 + Na]+ 1486.3057, found 1486.3075. ESI-TOF-MS for 1-Eu: calcd for [(1-Eu) + Na]+ 757.1508, found 757.1496.Synthesis of complex 3a-(Sm, Zn)
Metallo-ligand 1-Sm (7.32 mg, 0.01 mmol) was mixed with ethylenediamine (0.9 mg, 0.15 mmol) in a mixture of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chloroform and methanol. After heating at 50 °C for about 5 h, the suspension was centrifuged to obtain a precipitate. When the precipitate was dissolved in 600 μL of acetonitrile-d3 in the presence of 1.0 eq. Zn(OTf)2, a homogeneous yellow solution was obtained. 1H NMR (400 MHz, Acetonitrile-d3) δ 8.91 (s, 1H), 7.64 (d, J = 1.7 Hz, 1H), 7.30 (d, J = 1.7 Hz, 1H), 6.94 (s, 1H), 4.71–4.50 (m, 2H), 4.39–4.16 (m, 3H), 3.76–3.65 (m, 1H), 2.30 (s, 3H). ESI-TOF-MS for 3a-(Sm, Zn): the following picked signals are those at the highest intensities. m/z calcd for [3a-(Sm, Zn)(OTf)2]2+ 983.1101, found 983.1085; calcd for [3a-(Sm, Zn)(OTf)1]3+ 605.7559, found 605.7555.
:1 chloroform and methanol. After heating at 50 °C for about 5 h, the suspension was centrifuged to obtain a precipitate. When the precipitate was dissolved in 600 μL of acetonitrile-d3 in the presence of 1.0 eq. Zn(OTf)2, a homogeneous yellow solution was obtained. 1H NMR (400 MHz, Acetonitrile-d3) δ 8.91 (s, 1H), 7.64 (d, J = 1.7 Hz, 1H), 7.30 (d, J = 1.7 Hz, 1H), 6.94 (s, 1H), 4.71–4.50 (m, 2H), 4.39–4.16 (m, 3H), 3.76–3.65 (m, 1H), 2.30 (s, 3H). ESI-TOF-MS for 3a-(Sm, Zn): the following picked signals are those at the highest intensities. m/z calcd for [3a-(Sm, Zn)(OTf)2]2+ 983.1101, found 983.1085; calcd for [3a-(Sm, Zn)(OTf)1]3+ 605.7559, found 605.7555.
The synthesis procedures of 3a′-(Dy, Co), 3b-(Ln, Zn) and 3c-(Ln, Zn) were similar to that of 3a-(Sm, Zn) by changing the amine or metal source (see the ESI† for details).
Results and discussion
As shown in Scheme 1, 3a′-(Dy, Co) and 3a–3c-(Ln, Zn) (Ln = SmIII, EuIII, DyIII, YbIII and LuIII) were synthesized through subcomponent self-assembly of 1-Ln with different amines in the presence of transition metal ions. As a representative example, the subcomponent self-assembly of 3a-(Sm, Zn) was investigated. The 1H NMR spectrum reveals the generation of a sole species with a high degree of symmetry other than oligomeric species (Fig. 1B). Compared with metallo-ligand 1-Sm, all singles stemming from the formed species are shifted, in agreement with the coordination to Zn2+ ions. Moreover, 1H diffusion ordered spectroscopy (DOSY) displays that all signals are located at the same diffusion band (Fig. 1C), further confirming the single species in solution. Using the Stokes–Einstein equation, the diffusion coefficients and dynamic radius were estimated to be 1.0 × 10−9 m2 s−1 and 6.33 Å, respectively. As displayed in Fig. 1D, electrospray ionization time-of-flight mass spectroscopy (ESI-TOF-MS) again suggests the formation of the expected hetero-metallic covalent organic triple-stranded molecule with the formula of 3a-(Sm, Zn)·(OTf)4. Two main peaks (m/z = 605.7555 and 982.1086) observed in the ESI-TOF-MS spectrum can be assigned to [3a-(Sm, Zn)·OTf]3+ and [3a-(Sm, Zn)·(OTf)2]2+, respectively. The assignments of these peaks are also confirmed by the careful comparison of the experimental data with the simulated isotopic distributions (Fig. 1D, inset). After changing 2a with 2b or 2c, the ESI-TOF-MS confirms the formation of 3b-(Sm, Zn) or 3c-(Sm, Zn) (Fig. 1E and F). | ||
| Fig. 1 1H NMR (400 MHz, 298 K) spectra of 1-Sm in DMSO-d6 solution (A) and 3a-(Sm, Zn) in CD3CN solution (B). 1H DOSY NMR (400 MHz, 298 K, CD3CN) spectrum of complex 3a-(Sm, Zn) (C). ESI-TOF-MS spectra of (D) 3a-(Sm, Zn), (E) 3b-(Sm, Zn) and (F) 3c-(Sm, Zn). | ||
The formations of other covalent organic triple-stranded molecules were also identified by various characterization methods including NMR (1H, COSY, DOSY), ESI-TOF-MS and single-crystal X-ray diffraction studies, which are highly consistent with the expected stoichiometry.
Attempts to solve the crystal structures of 3a–3c-(Sm, Zn) failed, as we cannot get single crystals of 3a–3c-(Sm, Zn). However, upon replacing SmIII with LuIII, the single crystals of 3a–3c-(Lu, Zn), which were the isostructural compounds of 3a–3c-(Sm, Zn), were successfully obtained by slow vapor diffusion of ether (diethyl ether for 3a-(Lu, Zn), isopropyl ether for 3b-(Lu, Zn) and methyl tertiary-butyl ether for 3c-(Lu, Zn)) into the acetonitrile solutions of the corresponding assemblies. Single-crystal X-ray diffraction analysis shows that 3a–3c-(Lu, Zn) are the hetero-metallic triple-stranded covalent organic molecules, where hetero-metallic Lu–Zn nodes are located at the vertices of the molecules (Fig. 2). The refinement parameters and crystallographic data for 3a–3c-(Lu, Zn) are shown in Tables S1 and S2.† The important bond distances (Å) and angles (°) for 3a–3c-(Lu, Zn) are listed in Tables S3–S5.† Both 3a-(Lu, Zn) and 3b-(Lu, Zn) crystallized in the monoclinic C2/c space group, while 3c-(Lu–Zn) crystallized in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. To the best of our knowledge, these complexes represent the first example of covalent organic metallo-supramolecular molecules with 3d–4f hetero-metallic vertices.
space group. To the best of our knowledge, these complexes represent the first example of covalent organic metallo-supramolecular molecules with 3d–4f hetero-metallic vertices.
 | ||
| Fig. 2 X-Ray crystal structures of (A) 3a-(Lu, Zn), (B) 3b-(Lu, Zn) and (C) 3c-(Lu, Zn) where the free counter-ions and solvent molecules are omitted for clarity. (C, gray; H, white; N, blue; O, red; Lu, magenta; Zn, dark gray; S, yellow; F, cyan). | ||
Considering the very similar structure of 3a–3c-(Lu, Zn), only the structural description of 3a-(Lu, Zn) will be given here in detail. As shown in Fig. 2A, the 3a-(Lu, Zn) molecule consists of two LuIII ions, two ZnIII ions, one triple-stranded covalent organic backbone, one coordinated OTf− anion and one coordinated H2O molecule. The three strands of the covalent organic backbone wrap around the four metal centers (two LuIII and two ZnIII ions) and form the heterometallic covalent organic triple-stranded assembly. Both the Lu1 and Lu2 atoms are eight-coordinated, adopting a triangular dodecahedron (TDD-8) coordination geometry (Table S9†) with different coordination environments: the Lu1 atom is ligated by four nitrogen atoms (N1, N2, N3 and N4) from the TREN unit, three oxygen atoms (O2, O3 and O4) from the phenolic groups of the covalent organic backbone and one oxygen atom (O1) from the coordinated water molecule, while the Lu2 atom is surrounded by four nitrogen atoms (N11, N12, N13 and N14) from the TREN unit, three oxygen atoms (O5, O6 and O7) from the phenol groups and one oxygen atom (O8) from the coordinated OTf− anion (Fig. S58†). The bond distance of Dy–Ophenol is shorter than the others, owing to the strong coordination ability of the deprotonated phenolic oxygen atoms toward the Lu atoms. The bond length between the lanthanide center and the tertiary amine N atom of TREN (Lu–N1 or Lu–N11) is the longest among the Lu–O/N bond lengths, which can be ascribed to the limitation of the conformation of TREN. Unlike the Lu atoms, two Zn atoms are six-coordinated and coordinated by three nitrogen atoms from the three covalent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N groups and three oxygen atoms from three phenol groups belonging to the three strands of the covalent organic backbone, forming a OC-6 coordination geometry (Table S10†). The phenolic oxygen atoms in the covalent organic backbone adopt the μ2-OR coordination model, i.e., the LuIII ion and ZnII ion are connected together by phenolic oxygen and produce the hetero-metallic nodes, in which the average Lu–O–Zn bond angle is 94.28° and the average Lu–Zn distance is 3.23 Å. The distance between the tertiary amine N atoms of TREN (N1–N11) is 16.62 Å in 3a-(Lu, Zn), which is shorter than those in 3b-(Lu, Zn) (23.03 Å) and 3c-(Lu, Zn) (23.13 Å).
N groups and three oxygen atoms from three phenol groups belonging to the three strands of the covalent organic backbone, forming a OC-6 coordination geometry (Table S10†). The phenolic oxygen atoms in the covalent organic backbone adopt the μ2-OR coordination model, i.e., the LuIII ion and ZnII ion are connected together by phenolic oxygen and produce the hetero-metallic nodes, in which the average Lu–O–Zn bond angle is 94.28° and the average Lu–Zn distance is 3.23 Å. The distance between the tertiary amine N atoms of TREN (N1–N11) is 16.62 Å in 3a-(Lu, Zn), which is shorter than those in 3b-(Lu, Zn) (23.03 Å) and 3c-(Lu, Zn) (23.13 Å).
It should be noted that the covalent organic tetrahedron may be obtained if the tritopic triamine subcomponent was used, as observed in the previous reports.35,42,44 However, in our work, only the discrete covalent organic triple-stranded compound was observed when the ditopic diamine subcomponent was replaced with the tritopic triamine subcomponent. In 3c-(Lu, Zn), the three coordination atoms closest to the panel backbone are three nitrogen atoms originating from three imine bonds (CN), while the corresponding atoms in the reported examples are three pyridine nitrogen atoms. This difference should be responsible for the formation of a triple-stranded compound other than the tetrahedron, due to the dihedral angle generated by the plane of three coordinated nitrogen atoms closest to the panel backbone and one benzene ring plane of the tritopic subcomponent in 3c-(Lu, Zn) is about 98°, which is much smaller than the corresponding angle (≈120°) in the tetrahedron (Fig. S62†).
Photophysical properties
The UV-vis absorption and luminescence properties of 3a–3c-(Sm, Zn) were measured in solution at room temperature. Upon excitation at 386 nm, the organic skeleton of 3a-(Sm, Zn) can well sensitize SmIII, while the organic backbone of 3c-(Sm, Zn) cannot sensitize SmIII (Fig. 3a). Five characteristic bands at 561, 609, 645, 705 and 787 nm can be clearly found for complex 3a-(Sm, Zn), corresponding to the transitions from the emitting 4G5/2 to the 6HJ (J = 5/2–13/2) states. To gain insight into the differences observed in the emission spectra, the phosphorescence spectra of complexes 3a–3c-(Gd, Zn) were measured at 77 K (Fig. S54†), from which the triplet state energy levels of the organic skeletons were determined to be 21053 cm−1, 20167 cm−1 and 17605 cm−1 for 3a-(Gd, Zn), 3b-(Gd, Zn) and 3c-(Gd, Zn), respectively. The organic skeleton triplet state energy level of 3c-(Gd, Zn) is smaller than those of 3a-(Gd, Zn) and 3b-(Gd, Zn), and can be comparable with the energy level of 4G5/2 of SmIII ions, meaning that the organic skeleton of 3c-(Gd, Zn) cannot sensitize SmIII ions. Thus, we infer that the difference in the organic skeleton triplet state energy level should be responsible for the distinct emission spectra of 3a–3c-(Sm, Zn).
 | ||
| Fig. 3 (A) Emission spectrum of 3a–3c-(Sm, Zn) (1.0 × 10−5 M in CH3CN) at room temperature. (B) Emission spectrum of 3a-(Sm, Zn), 3a-(Eu, Zn) and 3a-(Yb, Zn) (1.0 × 10−5 M in CH3CN) at room temperature. | ||
Besides the SmIII ion, the organic skeleton of 3a-(Sm, Zn) can also sensitize other lanthanide ions, such as EuIII ions and NIR-emissive YbIII ions. As shown in Fig. 3b, complex 3a-(Eu, Zn) exhibits apparent peaks at 592, 612, 650 and 700 nm, corresponding to the 5D0 to 7FJ (J = 0–4) transition of the EuIII ion. For 3a-(Yb, Zn), it displays a well-resolved emission band at 977 nm, which can be assigned to the transition from the 2F5/2 to the 2F7/2 state. It is worth pointing out that the luminescence of the organic backbone (emission at about 456 nm, Fig. S41†) was not observed for complexes 3a-(Eu, Zn) and 3a-(Yb, Zn), revealing the complete energy transfer from the organic skeleton to the lanthanide ions. In contrast, 3a-(Sm, Zn) has a significant amount of the residual ligand emitted at about 456 nm, suggesting the incomplete organic skeleton to lanthanide energy transfer.34
Magnetic properties
Given that CoII and DyIII ions are the large magnetic anisotropy ions, and that once the super-exchange ferromagnetic interaction occurs between CoII and DyIII, quantum tunneling of magnetization (QTM) can be mitigated,55,56 we prepared compound 3a′-(Dy, Co) and characterized it through single-crystal X-ray diffraction, ESI-TOF-MS, TG, PXRD and elemental analysis (see the ESI†). The M–H curves (2, 3 and 5 K) and AC magnetic susceptibility were measured to testify the possible single molecular magnets properties of 3a′-(Dy, Co). As shown in Fig. 4, the observed χMT value at room temperature is 33.50 cm3 K mol−1, which is larger than the theoretical value of 32.09 cm3 K mol−1 expected for two isolated CoII ions (S = 3/2 and g = 2.0) and two isolated DyIII ions (J = 15/2 and g = 4/3). Such a disparity should be attributed to the significant orbital contribution of the high-spin octahedral CoII ions.57–59 The χMT products remain nearly constant up to 100 K, below which they decrease slowly and reach the minimum value of 27.66 cm3 K mol−1 at 2 K. The decreases below 100 K are likely to be caused by the depopulation of the Stark levels of DyIII and the spin–orbital effect of CoII. Moreover, the possible weak antiferromagnetic interactions between DyIII and CoII may also make some contributions to the decreases of χMT below 100 K, as observed in other heterometallic CoII–DyIII compounds.60 The magnetization isotherm curves display a rapid increase at low fields and then a gradual increase at higher fields without reaching saturation even at 8 T, which is very common behavior for such compounds and indicative of the presence of anisotropy in this compound. As shown in Fig. S56,† no out-of-phase AC signals were found for 3a′-(Dy, Co), suggesting the absence of slow magnetization relaxation behaviour in 3a′-(Dy, Co). To further investigate the possible field-induced SMM behavior, field dependent x′′m data were collected to acquire an optimum dc field (Fig. S57†). From Fig. S57,† we can see that no maximum was found in the x′′m–H curve, indicating the absence of an optimum dc field. This phenomenon can be attributed to the relatively small energy barriers for the reverse of the magnetic moment in 3a′-(Dy, Co).61 | ||
| Fig. 4 Temperature-dependent χMT plots for 3a′-(Dy, Co). Insert: Field dependences of magnetization between 0 and 80 kOe and at temperatures of 2.0, 3.0, and 5.0 K. | ||
Conclusions
In summary, a series of hetero-metallic covalent organic triple-stranded molecules with 3d–4f vertices have been synthesized through the hierarchical subcomponent self-assembly approach. These compounds are the first examples of covalent organic metallo-supramolecular backbones decorated with 3d–4f heteronuclear vertices. Besides, 3d–4f vertices of these complexes bring photophysical and magnetic properties to the assemblies. The luminescence investigations revealed that the organic backbone fabricated by using ethylenediamine can sensitize the luminescence emissions of lanthanide ions (SmIII, EuIII and YbIII) in both the visible and NIR domains. The AC magnetic susceptibility measurements revealed the absence of slow magnetization relaxation behaviour in compound 3a′-(Dy, Co). This work confirms that the hierarchical subcomponent self-assembly is a potential route to fabricate discrete hetero-metallic covalent organic architectures.Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grants 21971237, 21825107 and 22171264), the National Key Research and Development Program of China (Grant 2021YFA1500400), and the Science Foundation of Fujian Province (Grants 2021J02016 and 2022J01507).References
- X. Z. Li, C. B. Tian and Q. F. Sun, Chem. Rev., 2022, 122, 6374 CrossRef CAS PubMed.
- C. B. Tian and Q. F. Sun, Chem. – Eur. J., 2023, 29, e202300195 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- Y. Domoto and M. Fujita, Coord. Chem. Rev., 2022, 466, 214605 CrossRef CAS.
- M. Yoshizawa and L. Catti, Acc. Chem. Res., 2019, 52, 2392 CrossRef CAS PubMed.
- W. Liu, P. J. Das, H. M. Colquhoun and J. F. Stoddart, CCS Chem., 2022, 4, 755 CrossRef CAS.
- D. Chu, W. Gong, H. Jiang, X. Tang, Y. Cui and Y. Liu, CCS Chem., 2021, 3, 1692 Search PubMed.
- T. J. Hubin and D. H. Busch, Coord. Chem. Rev., 2000, 200, 5 CrossRef.
- J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103 CrossRef CAS PubMed.
- Q. Zhang, F. Chen, X. Shen, T. He, H. Qiu, S. Yin and P. J. Stang, ACS Macro Lett., 2021, 10, 873 CrossRef CAS PubMed.
- J. Dong, C. Tan, K. Zhang, Y. Liu, P. J. Low, J. Jiang and Y. Cui, J. Am. Chem. Soc., 2017, 139, 1554 CrossRef CAS PubMed.
- E. F. V. Dry, J. K. Clegg, B. Breiner, D. E. Whitaker, R. Stefak and J. R. Nitschke, Chem. Commun., 2011, 47, 6021 RSC.
- J. Jiao, J. Dong, Y. Li and Y. Cui, Angew. Chem., 2021, 133, 16704 CrossRef.
- J. R. Nitschke, D. Schultz, G. Bernardinelli and D. Gerard, J. Am. Chem. Soc., 2004, 126, 16538 CrossRef CAS PubMed.
- L. Wu, M. Tang, L. Jiang, Y. Chen, L. Bian, J. Liu, S. Wang, Y. Liang and Z. Liu, Nat. Synth., 2022, 2, 17 CrossRef.
- J. R. Nitschke, M. Hutin and G. Bernardinelli, Angew. Chem., Int. Ed., 2004, 43, 6724 CrossRef CAS PubMed.
- X. L. Li, Z. Ma, J. Wu, Q. Zhou and J. Tang, Dalton Trans., 2022, 51, 17579 RSC.
- M. Hutin, C. A. Schalley, G. Bernardinelli and J. R. Nitschke, Chem. – Eur. J., 2006, 12, 4069 CrossRef CAS PubMed.
- C. Browne, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2014, 53, 10701 CrossRef CAS PubMed.
- W. Xue, T. K. Ronson, Z. Lu and J. R. Nitschke, J. Am. Chem. Soc., 2022, 144, 6136 CrossRef CAS PubMed.
- J. P. Carpenter, T. K. Ronson, F. J. Rizzuto, T. Heliot, P. Grice and J. R. Nitschke, J. Am. Chem. Soc., 2022, 144, 8467 CrossRef CAS PubMed.
- G. Wu, Y. Chen, S. Fang, L. Tong, L. Shen, C. Ge, Y. Pan, X. Shi and H. Li, Angew. Chem., Int. Ed., 2021, 60, 16594 CrossRef CAS PubMed.
- X. C. Zhou, L. X. Wu, X. Z. Wang, Y. L. Lai, Y. Y. Ge, J. Su, X. P. Zhou and D. Li, Inorg. Chem., 2022, 61, 5196 CrossRef CAS PubMed.
- R. Lavendomme, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 12147 CrossRef CAS PubMed.
- R. Modak, B. Mondal, P. Howlader and P. S. Mukherjee, Chem. Commun., 2019, 55, 6711 RSC.
- J. H. Zhang, H. P. Wang, L. Y. Zhang, S. C. Wei, Z. W. Wei, M. Pan and C. Y. Su, Chem. Sci., 2020, 11, 8885 RSC.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- Y. Chen, Y. Lei, L. Tong and H. Li, Chem. – Eur. J., 2022, 28, e202102910 CAS.
- P. S. Mukherjee and D. Chakraborty, Chem. Commun., 2022, 58, 5558 RSC.
- J. Mosquera, S. Zarra and J. R. Nitschke, Angew. Chem., Int. Ed., 2014, 53, 1556 CrossRef CAS PubMed.
- H. Li, H. Zhang, A. D. Lammer, M. Wang, X. Li, V. M. Lynch and J. L. Sessler, Nat. Chem., 2015, 7, 1003, DOI:10.1038/nchem.2392.
- F. Wang, C. Bucher, Q. He, A. Jana and J. L. Sessler, Acc. Chem. Res., 2022, 55, 1646 CrossRef CAS PubMed.
- Z. W. Li, X. Wang, L. Q. Wei, I. Ivanovic-Burmazovic and G. F. Liu, J. Am. Chem. Soc., 2020, 142, 7283 CrossRef CAS PubMed.
- F. F. Chang, F. D. Feng, J. Geng and W. Huang, Chem. Commun., 2021, 57, 9220 RSC.
- S. L. Han, J. Yang, D. Tripathy, X. Q. Guo, S. J. Hu, X. Z. Li, L. X. Cai, L. P. Zhou and Q. F. Sun, Inorg. Chem., 2020, 59, 14023 CrossRef CAS PubMed.
- H. Yan, C. M. Wang, P. Chen, Y. Q. Zhang and W. B. Sun, Dalton Trans., 2022, 51, 6918 RSC.
- X. Liu, X. Yang, Y. Ma, J. Liu, D. Shi and D. Schipper, Chin. Chem. Lett., 2021, 32, 569 CrossRef CAS.
- Y. Peng, H. Kaemmerer and A. Powell, Chem. – Eur. J., 2021, 27, 15044 CrossRef PubMed.
- H. S. Wang, K. Zhang, Y. Song and Z. Q. Pan, Inorg. Chim. Acta, 2021, 521, 120318 CrossRef CAS.
- C. L. Yin, Z. B. Hu, Q. Q. Long, H. S. Wang, J. Li, Y. Song, Z. C. Zhang, Y. Q. Zhang and Z. Q. Pan, Dalton Trans., 2019, 48, 512 RSC.
- P. D. Frischmann, V. Kunz and F. Wurthner, Angew. Chem., Int. Ed., 2015, 54, 7285 CrossRef CAS PubMed.
- A. M. Castilla, N. Ousaka, R. A. Bilbeisi, E. Valeri, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 17999 CrossRef CAS PubMed.
- E. G. Percastegui, J. Mosquera, T. K. Ronson, A. J. Plajer, M. Kieffer and J. R. Nitschke, Chem. Sci., 2019, 10, 2006 RSC.
- A. M. Castilla, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 2342 CrossRef CAS PubMed.
- J. Holub, A. Santoro, M.-A. Stadler and J.-M. Lehn, Inorg. Chem. Front., 2021, 8, 5054 RSC.
- M. Urbani and T. Torres, Chem. – Eur. J., 2020, 26, 1683 CrossRef CAS PubMed.
- L. Meng, Y.-F. Deng, S. Liu, Z. Zheng and Y.-Z. Zhang, Sci. China: Chem., 2021, 64, 1340 CrossRef CAS.
- E. T. Luis, H. Iranmanesh, K. S. A. Arachchige, W. A. Donald, G. Quach, E. G. Moore and J. E. Beves, Inorg. Chem., 2018, 57, 8476 CrossRef CAS PubMed.
- Z. Wang, L. P. Zhou, T. H. Zhao, L. X. Cai, X. Q. Guo, P. F. Duan and Q. F. Sun, Inorg. Chem., 2018, 57, 7982 CrossRef CAS PubMed.
- Q. Y. Zhu, L. P. Zhou and Q. F. Sun, Dalton Trans., 2019, 48, 4479 RSC.
- Q. Y. Zhu, L. P. Zhou, L. X. Cai, X. Z. Li, J. Zhou and Q. F. Sun, Chem. Commun., 2020, 56, 2861 RSC.
- Q. Y. Zhu, L. P. Zhou, L. X. Cai, S. J. Hu, X. Z. Li and Q. F. Sun, Inorg. Chem., 2022, 61, 16814 CrossRef CAS PubMed.
- G. L. Zhang, L. P. Zhou, D. Q. Yuan and Q. F. Sun, Angew. Chem., Int. Ed., 2015, 54, 9844 CrossRef CAS PubMed.
- C. D. Buch, S. H. Hansen, C. M. Tram, D. Mitcov and S. Piligkos, Inorg. Chem., 2020, 59, 16328 CrossRef CAS PubMed.
- J. Wang, M. Feng, M. N. Akhtar and M.-L. Tong, Coord. Chem. Rev., 2019, 387, 129 CrossRef CAS.
- Y. Peng and A. K. Powell, Coord. Chem. Rev., 2021, 426, 213490 CrossRef CAS.
- C. B. Tian, Y. H. Han, Z. Z. He and S. W. Du, Chem. – Eur. J., 2017, 23, 767 CrossRef CAS PubMed.
- Q. Yang, G. L. Wang, Y. Q. Zhang and J. Tang, Dalton Trans., 2022, 51, 13928 RSC.
- N. Ahmed and K. Uddin Ansari, Dalton Trans., 2022, 51, 4122 RSC.
- E. Moreno Pineda, N. F. Chilton, F. Tuna, R. E. Winpenny and E. J. McInnes, Inorg. Chem., 2015, 54, 5930 CrossRef CAS PubMed.
- H. S. Wang, Q. Q. Long, Z. B. Hu, L. Yue, F. J. Yang, C. L. Yin, Z. Q. Pan, Y. Q. Zhang and Y. Song, Dalton Trans., 2019, 48, 13472 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2256435–2256438. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01139a |
| This journal is © The Royal Society of Chemistry 2023 |