
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methane monooxygenases; physiology, biochemistry and structure
Yasuyoshi
Sakai
 *a,
Hiroya
Yurimoto
a and
Seigo
Shima
*b
*a,
Hiroya
Yurimoto
a and
Seigo
Shima
*b
aDivision of Applied Life Sciences, Graduate School of Agriculture, Kyoto University, Kitashirakawa-Oiwake, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: sakai.yasuyoshi.8x@kyoto-u.ac.jp; Fax: +81 75 753 6454; Tel: +81 75 753 6385
bMax Planck Institute for Terrestrial Microbiology, Karl-von-Frisch-Straße 10, 35043 Marburg, Germany. E-mail: shima@mpi-marburg-mpg.de; Fax: +49 6421 178199; Tel: +49 6421 178100
First published on 29th September 2023
Abstract
Methane-utilizing bacteria (methanotrophs), which inhabit various environments such as wetlands, rice paddies and the surface of aquatic plants are known to convert methane to methanol using methane monooxygenase enzymes (MMOs). There are two distinct types of MMOs: the copper-containing membrane-bound enzyme (pMMO) and the iron-containing soluble enzyme (sMMO). Since MMOs catalyze methane oxidation at ambient temperature and pressure, they are potential biocatalysts for industrial methanol production from methane. Understanding the mechanism of the MMO-catalyzed reaction is crucial to develop a new biocatalyst. The catalytic mechanism of MMO has been extensively studied in aspects of enzyme kinetics, protein structure, spectroscopy, biomimetic chemistry and computation. Based on these studies, the catalytic mechanism of sMMO is relatively well understood, and a rigid catalytic mechanism is proposed. On the other hand, the studies of pMMO are still at their early stages and there are debates as to which of several copper sites are the true active sites. In this review, we describe our current knowledge of MMOs including the metabolism, regulation of expression, their enzymatic properties, and the structural and biochemical features. We summarize recent structural and biochemical studies of pMMO and discuss the future directions to develop efficient and robust biocatalysts.
 Yasuyoshi Sakai | Yasuyoshi Sakai is a Professor at Kyoto University, Japan. He received his Ph.D. degree from Kyoto University, a young scholar fellowship from JSPS, and was appointed as an Assistant Professor in 1988. In 1994, he became an Associate Professor, and a Professor at Kyoto University in 2005 –present. His research has been dedicated to molecular and cell biology and biotechnology of the methylotrophic yeast, in which he revealed the molecular mechanism of methanol-induced gene expression, organelle degradation via autophagy, and established the gene expression system. He received Japan Society for Bioscience, Biotechnology, and Agrochemistry (JSBBA) award in 2022. |
 Hiroya Yurimoto | Hiroya Yurimoto is an Associate Professor at Kyoto University, Japan. He received his Ph.D. degree from Kyoto University. Then he became an Assistant Professor at Kyoto University in 1996, and an Associate Professor in 2007 –present. His research interests include molecular basis of metabolism, cellular physiology, and gene regulation of C1-microorganisms which can utilize methane and methanol as the sole carbon source, and their application in production of useful chemicals and proteins. |
 Seigo Shima | Seigo Shima started his career as a scientist at the Central Research Institute of Electric Power Industry (Japan) in 1985. He received his Ph.D. in 1991 from the University of Tokyo. In 1993, he received a fellowship from Alexander von Humboldt Foundation to study at Philipps-Universität Marburg, Germany. In 1995, he was appointed as a Group Leader at the Department of Biochemistry of Max-Planck-Institute for Terrestrial Microbiology in Marburg. He is currently a department-independent Research Group Reader at Max-Planck-Institute for Terrestrial Microbiology in Marburg. His current research focuses on the catalytic mechanisms of the enzymes involved in methanogenesis. |
1. Introduction
Since the shale gas revolution, interest in using methane, the main component of natural gas, not only as a fuel but also as a carbon source for the production of useful materials has increased worldwide. In addition to being found as a natural resource, methane can be produced from biomass (methane fermentation) or CO2 (methanation).1,2 Along with the progress in the technological and conceptional development of “carbon neutral methane” production, there are great expectations for methane as a carbon and energy source in a decarbonized society.3 In the production of useful compounds from methane, it is desirable to convert it into methanol, which is used as a raw material for various chemical products.4 However, there are no feasible industrial processes for producing methanol by direct oxidation of methane. At present, methanol is produced through an indirect reaction using synthesis gas (syngas), which contains mainly CO and H2, produced from natural gas or coal at high temperatures, pressures and energy inputs.5,6 Therefore, the development of novel catalysts for the direct oxidation of methane to methanol under ambient conditions is highly desirable especially for application in biotechnology.In nature, there is a group of microorganisms (C1 microorganisms) that utilize C1 compounds such as methane and methanol as carbon and energy sources.7,8 C1 microorganisms are ubiquitously found in various environments and contribute to drive the global carbon cycle (methane cycle) between the two major greenhouse gases methane and CO2.9,10 In the methane cycle, methane is produced by methanogenic archaea in anaerobic environments such as wetlands, paddy fields and the rumen of cattle. The annual emission of methane is estimated to be 560 Tg,11,12 however, this value may be underestimated because it has recently been reported that methane is produced from methyl radicals generated by the action of reactive oxygen species in the cells of plants and other organisms even in an aerobic environment.13,14 More than 80% of the methane released into the atmosphere is oxidized by hydroxyl radicals, and most of the rest is consumed by methane-utilizing bacteria (methanotrophs) before being released into the atmosphere.9 Methane monooxygenases (MMOs) of methanotrophs are responsible for the oxidation of methane to methanol under ambient temperature and pressure.15–19 Besides MMOs, ammonia monooxygenase and butane monooxygenase have been reported to be able to oxidize methane.20,21 MMOs are the sole enzymes responsible for the physiological oxidation of methane to methanol and the use of methanotrophs or MMOs has been thought to be promising for direct oxidation of methane to methanol and the production of useful compounds from methane since the 1970s.22 However, due to problems with the large-scale preparation of MMOs or cells of methanotrophs and the low stability of MMOs, they have not been used in industrial applications yet.
In this review, after overviewing the physiology of methanotrophs and general properties of MMOs, we describe the current understanding of the structural features and reaction mechanisms of pMMO. Finally, attempts to develop efficient and robust biocatalysts for methane oxidation and future perspectives are described.
2. Methanotrophs and methane metabolism
2.1 Physiology and classification of methanotrophs
Methanotrophs inhabit both aerobic and anaerobic environments, but we focus on aerobic methanotrophs in this review. Anaerobic methane oxidation has been described elsewhere.10,23,24Fig. 1 summarizes the general methane metabolism in aerobic methanotrophs. The first reaction of methane oxidation in aerobic methanotrophic bacteria is catalyzed by two types of methane monooxygenases, namely a membrane-bound copper-containing particulate methane monooxygenase (pMMO) and a cytosolic soluble methane monooxygenase (sMMO).25 pMMO is located in the characteristic intracellular membrane structure in most methanotrophic bacteria, but sMMO is found only in the cytosol of some methanotrophic bacteria.26 Methanol is oxidized to formaldehyde by pyrroloquinoline quinone (PQQ)-dependent methanol dehydrogenases (MDHs).15 In addition to the conventional calcium-dependent MDH (MxaFI), the lanthanide-dependent MDH (XoxF) has recently emerged as an important enzyme for methanol metabolism in natural environments.27–29 Formaldehyde is further metabolized by the oxidation pathway to obtain energy and the assimilation pathway to become cellular components. There are two major types of formaldehyde assimilation pathways, the ribulose monophosphate (RuMP) pathway and the serine pathway.7,15 Some methanotrophs possess the Calvin-Benson-Bassham (CBB) cycle for the fixation of CO2.30,31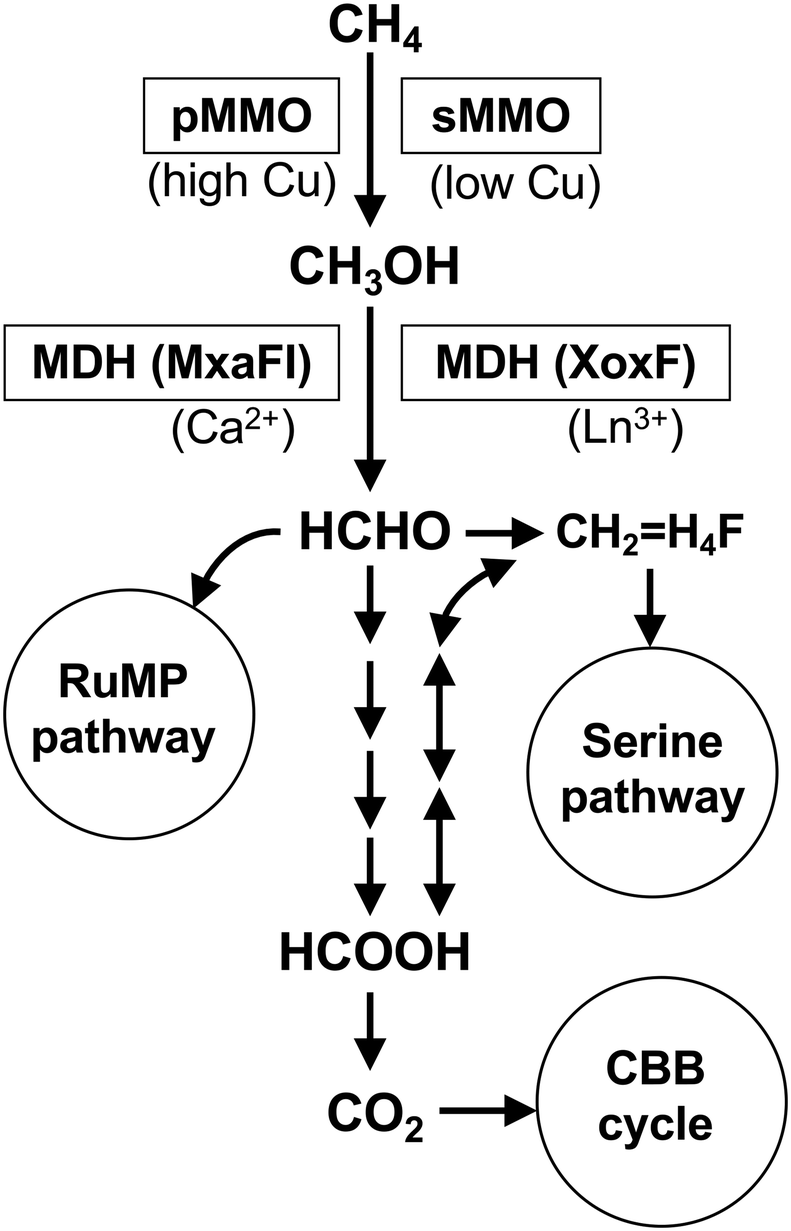 | ||
Fig. 1 General methane metabolism in aerobic methanotrophs. pMMO, particulate methane monooxygenase; sMMO, soluble methane monooxygenase; MDH, methanol dehydrogenase; Ln3+, lanthanide ion; CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H4F, methylenetetrahydrofolate, RuMP, ribulose monophosphate; CBB, Calvin–Benson–Bassham. H4F, methylenetetrahydrofolate, RuMP, ribulose monophosphate; CBB, Calvin–Benson–Bassham. | ||
Most aerobic methanotrophs belong to the α-Proteobacteria, γ-Proteobacteria and Verrucomicrobia.32 Classically, the proteobacterial methanotrophs are divided into type I and type II based on their taxonomy at phylum level, and physiological and morphological traits.7,33 Type I methanotrophs belong to γ-Proteobacteria (families Methylococcaceae and Methylotermaceae) and utilize the RuMP pathway for formaldehyde fixation. Type II methanotrophs belong to α-Proteobacteria (families Methylocystaceae and Beijerinckiaceae) and utilize the serine pathway. A representative methanotroph Methylococcus capsulatus Bath is classified as type X because it has both RuMP and serine pathways.7Verrucomicrobia, which have been discovered relatively recently, are not included in the conventional classification. Verrucomicrobial methanotorphs are extremophiles living in acidic geothermal ecosystems and they possess only pMMO.34 Very recently, Candidatus Mycobacterium methanotorophicum belonging to Actinobacteria has been reported to have methanotrophic traits.35 Methane oxidation in this species is catalyzed by sMMO and formaldehyde fixation by the RuMP pathway.
Almost all methanotrophs are obligate C1 utilizers, i.e., they utilize only methane or methanol, while some species in the genera Methylocella, Methylocapsa, Methylooceanibacter and Methylocystis were found to be facultative methanotrophs that can utilize multi-carbon compounds, such as acetate, ethanol and short-chain alkanes.32 All known Methylocella strains and Methylooceanibacter methanicus R-67171 possess only sMMO, whereas other facultative methanotrophs possess only pMMO or both.32
2.2 Structure of MMO genes and their regulation
pMMO and sMMO have completely different properties with respect to substrate specificity, metal ions involved, and protein structure. pMMO has a trimer of heterotrimers consisting of three subunits, PmoA, PmoB, and PmoC, encoded by the pmoCAB gene operon (Fig. 2).36,37 The structure of pMMO is described in the section 3.4.37 Some methanotrophs of the Methylocystaceae family produce methanobactin, a peptide with high affinity to Cu(I) ion which contributes copper ion trafficking into the bacterial cells.15,38 Another example of the copper collection is a surface-bound protein (MopE) and a secreted form of it (MopE*) in M. capsulatus.39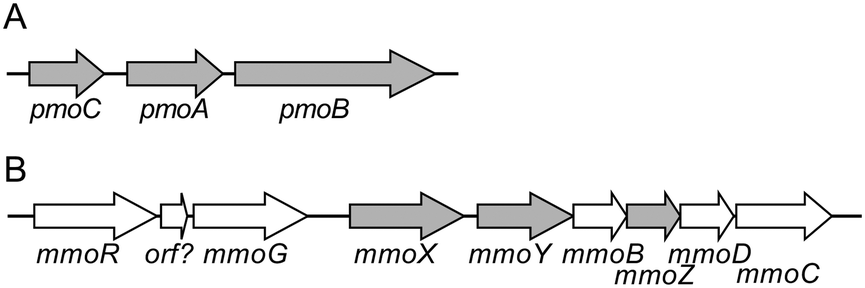 | ||
| Fig. 2 Gene organization of pMMO- and sMMO-encoding operons of M. trichosporium OB3b. (A) The pMMO operon. The pmoA, pmoB, and pmoC encode three subunits of pMMO, PmoA (24 kDa), PmoB (47 kDa), and PmoC (22 kDa). (B) The sMMO operon. The mmoX, mmoY, and mmoZ encode three subunits of MMOH, α (60 kDa), β (45 kDa), and γ (19 kDa). | ||
sMMO has a binuclear iron-active center, and recently, the structure of important chemical species involved in the reaction have also been elucidated.18,40 sMMO consists of a hydroxylase (MMOH) that catalyzes methane hydroxylation, a regulatory protein MMOB, and a reductase MMOR. MMOH is a dimer of heterotrimers encoded by the mmoXYZ genes (Fig. 2).18,36 The α subunit has a binuclear iron active center, where the activation of molecular oxygen and the oxidation of methane take place. MMOR, encoded by the mmoC gene, contains FAD and an iron–sulfur cluster (2Fe-2S). It accepts electrons from NADH, and reduces the active center of the α subunit. MMOB, which is encoded by the mmoB gene, regulates the transfer of electrons.18 Furthermore, the mmo gene cluster contains the mmoG gene, which encodes a chaperone-like protein that mediates protein folding, and the mmoD and mmoR genes, which are thought to be involved in the regulation of gene expression.41,42
In methanotrophs, which have both pMMO and sMMO, the production of pMMO is up-regulated and sMMO is down-regulated at the transcriptional level under copper-ion rich conditions.36 sMMO is expressed when the copper/biomass ratio is low and down-regulated in high copper conditions. This type of regulation is known as the copper switch. The copper switch has been well investigated at the molecular level using a representative methanotroph, Methylosinus tricosporium Ob3B. Several genes, rpoN, mmoR, mmoG and mmoD, were found to play key roles in the regulation of expression of the mmo operon.41,42 Methanobactin may also contribute to the Cu sensing mechanism and the regulatory genes associated with methanobactin, mbnI and mbnR, may play a role in the copper switch.15
sMMO belongs to a large enzyme family of soluble diiron monooxygenases (SDIMOs), which catalyze the oxidation of a wide range of hydrocarbons including short-chain alkanes, alkenes, and aromatic compounds.43,44 SDIMOs consist of six groups based on DNA sequence, gene organization, subunit composition, and substrate specificity. sMMOs are part of group 3. The facultative methanotrophs belonging to Methylocella sp. have a group 5 SDIMO, propane monooxygenase (PrMO), in addition to sMMO, and can utilize propane as a carbon source.44,45 In Methylocella silvestris, the sMMO genes but not the PrMO genes are expressed during growth on methane, whereas both genes are expressed during growth on propane.45
3. Biochemistry of particulate methane monooxygenase
The nature and the catalytic mechanism of sMMO have been extensively studied using biochemistry, spectroscopy, X-ray crystallography and cryo-electron microscopy (cryo-EM), and a plausible catalytic mechanism has been proposed.40 The structure, function and catalytic mechanism of sMMO, has been reviewed elsewhere.18,19,25,46 On the contrary, despite the fact that many active researches have been carried out, the nature of the active site structure and the catalytic mechanism of pMMO are still controversial, although pMMO is a chemically and biotechnologically very important enzyme.16,17 In this section, we shortly review some critical experimental results about pMMO to understand the current status of the study of this enzyme for further development.3.1 Solubilization and purification of pMMO
pMMO is located in the inner cell membrane of methanotrophic bacteria47 and is fractionated into the particulate fraction of the cell extract (Table 1). The occupancy of pMMO in the membrane protein fraction is very high, accounting for ∼80% of the membrane proteins and ∼20% of the total proteins in the cells.47,48 The presence of such a large amount of pMMO in the cells implies a relatively low specific activity of this enzyme compared to the other metabolic enzymes. Comparison of the washed membrane fractions obtained from M. capsulatus showed that the copper-enriched cells contained significantly more copper ions.49| Specific activity (mU mg−1) | Metal ions/heterotrimer | EPR signals | References | ||||
|---|---|---|---|---|---|---|---|
| Whole cell | Membrane fraction | Solubilized | Purified | ||||
| a The enzyme assay was performed at 45 °C by measuring propylene epoxidation with duroquinol, in which the whole cell activity was measured using formate as the reductant. b Total activity. c The enzyme assay was performed at 45 °C using propane, butane oxidation or propene epoxidation. NADH was used as the reductant. d The enzyme assay was performed at 45 °C by measuring epoxidation of propylene with NADH. e The metal was calculated per 200 kDa dimer of heterotrimer. f The enzyme assay was performed at 30 °C by measuring propene epoxidation with duroquinol. g The enzyme assay was performed by measuring propylene epoxidation with NADH or duroquinol. h The enzyme assay was performed by measuring methane oxidation or propene epoxidation with duroquinol. | |||||||
| Methylococcus capsulatus Batha | 60.5 (273![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000)b 000)b |
10.4 (28000)b |
9.86 (19000)b |
11.1 (11000)b |
14.6 ± 1.9 Cu | Type 2 Cu | Zahn et al. 1996 (ref. 55) |
| 2.5 ± 0.7 Fe | High-spin Fe | ||||||
| Broad low-field | |||||||
| Methylococcus capsulatus Bathc | 9.66–12.5 | 2.6–5.1 | 12.4–15 Cu | Type 2 Cu | Nguyen et al.1998 (ref. 54) | ||
| Fe, not detected | Broad-isotropic Cu | ||||||
| Methylococcus capsulatus Bathd | 21 (NADH) | n.d. (NADH) | n.d. (NADH) | 4.8 ± 0.8 Cue | Type 2 Cu | Lieberman et al. 2003 (ref. 58) | |
| 16 (duroquinol) | 3.9 (duroquinol) | 17.7 (duroquinol) | 1.5 ± 0.7 Fee | ||||
| Methylosinus trichosporium OB3b (30 °C)f | 2.3 (668)b | 0.24 (35)b | 0.47 (5.7)b | 12.8 Cu | Type 2 Cu | Takeguchi et al. 1998 (ref. 57) | |
| 0.9 Fe | High-spin Fe | ||||||
| Methylosinus trichosporium OB3bg | 2.2 (NADH) | n.d. (NADH) | 2 Cu | Type 2 Cu | Miyaji et al. 2002 (ref. 56) | ||
| 3.8 (duroquinol) | 3.3 (duroquinol) | High-spin Fe | |||||
| Methylocystis sp. Mh | ∼500 (CH4) | ∼250 (methane) | Not reported | 0–10 (methane) | 1–3 Cu | Not tested | Gvozdev et al. 2006 (ref. 60) |
| ∼70 (propene) | 05 (propene) | 0.08–1 Fe | |||||
The activity of pMMO is unstable, and upon when cells are disrupted, much of the methane monooxygenase activity found in the whole cells disappears (Table 1). Nevertheless, it is possible to purify pMMO by tracking the residual enzymatic activity. To purify the membrane protein from the particulate fraction, the membrane protein is solubilized by using detergents. Again, much activity is lost in the solubilization step. The Dalton group reported solubilization of pMMO by using dodecyl-β-D-maltoside as a detergent, which abolished the enzyme activity, but the addition of egg or soybean lipids reactivated the enzyme,50 which suggests that lipids are required to maintain the active structure of pMMO. This finding is supported by reports on the effect of lipid structures (bicelles and nanodiscs) on the pMMO activity (see below).51–54 However, in most of the reports, active pMMO was purified from the detergent-solubilized fraction by column chromatography using detergent-containing buffer solutions.55–58
Tonge et al. first purified pMMO from M. trichosporium OB3b.59 They followed the enzymatic activity by measuring the conversion of methane to methanol using ascorbic acid as an electron donor. However, the size of the proteins in the purified fraction differed from the standard protein composition of the subsequently purified pMMO. Zahn and DiSpirito purified pMMO, which has a typical pMMO subunit composition, from M. capsulatus Bath, in which the total activity from the whole cells was reduced to 10% by disruption of the cells, and subsequent solubilization resulted in a 67% yield.55 In total, 4% of the pMMO activity from whole cells was obtained in the purified fraction (Table 1). The specific activity of the purified enzyme was 11 mU mg−1, where one U of enzyme is defined as the activity of catalyzing 1 μmol propylene epoxidation per minute with duroquinol as the reductant.55 Later, Nguyen et al. also reported the purification of pMMO from the same organism, where the specific activity of the purified enzyme was 2.6–5.1 mU mg−1.54 In addition, Liebermann et al. reported a slightly higher activity (17.7 mU mg−1) of the purified enzyme from the same organism.58 In the case of Methylocystis sp. M, 50% of the cellular activity was recovered in the membrane fraction, but less than a few percent of the enzyme activity was detected in the purified enzyme.60 In the case of M. trichosporium OB3b, less than 1% of the activity in the membrane fraction was obtained in the purified fraction, in which the final specific activity was 0.5 mU mg−1.57 Thus, the yield of isolating pMMO activity from cells is generally very low.
Purified pMMO contains copper ions. However, the amount of copper ions in purified pMMO preparations varies (Table 1). Partial removal of the copper ions by EDTA from purified pMMO from M. trichosporium OB3b and M. capsulatus Bath decreased the enzyme activity and addition of external copper ions reactivated the enzyme.61–64 A stimulating effect of addition of copper ions on the pMMO activity of the membrane fraction of M. capsulatus Bath was reported, in which the Cu(II) electron paramagnetic resonance (EPR) signal increased with MMO activity.49,65 Thus, the copper ions are critical for the enzymatic activity of pMMO but they are only loosely bound to the protein, and addition of external copper ions can restore the copper site by diffusing into the binding sites. This finding suggests that the loosely bound copper ions in pMMO should be simple copper ions complexed by amino-acid residues rather than complicated metal complexes. In addition, these observations suggest that the copper active site is likely exposed to the solvent and not deeply embedded in the protein core. Interestingly, copper ions are also inhibitory at higher concentrations,49 suggesting that external copper ions may bind to the active site copper complex. The reversible binding of copper ions to the pMMO provides some clues to the character of the active site; however, this property makes it difficult to determine the number and character of copper ions in purified pMMO.
3.2 pMMO activity assay
pMMO oxidizes a wide range of substrates including alkanes and alkenes.66 Most of the purification work used propylene epoxidation activity rather than methane oxidation because of the difficulty of detecting the latter reaction. As summarized by Koo et al., many of the purified pMMOs show no enzymatic activity against methane unless reconstituted with lipids.53 The use of the propylene epoxidation assay is based on the hypothesis that methane oxidation and propylene epoxidation are catalyzed by the same active site. If the enzymatic activity of the purified pMMO preparations has not been determined by the methane oxidation assay, the data should be interpreted with caution.The direct physiological reductant of the pMMO reaction has been proposed to be the reduced form of quinone or NADH. NADH is the physiological electron donor of sMMO and this electron carrier should be involved in the reaction sequence of metabolism involving the pMMO reaction. In fact, membrane-fractions and some purified pMMO preparations use NADH as an electron donor (Table 1). Quinol is reproduced from quinone by membrane enzymes involved in the respiratory chain. Choi et al. purified pMMO from M. capsulatus Bath as a complex with type 2 NADH:quinone oxidoreductase.67 This finding supports the hypothesis that quinone is the direct electron carrier for pMMO. However, it was recently shown by cryo-electron tomography that pMMO molecules themselves form higher-order hexagonal arrays in intact cells,47 suggesting that pMMO and type 2 NADH:quinone oxidoreductase do not always form a tight complex. Most enzyme assays use a quinonol analog, duroquinol, but some purified pMMO show activity when using NADH,17 which may be mediated by minute amounts of contaminating type 2 NADH:quinone oxidoreductase.67
The use of duroquinol as a reductant in the pMMO reaction was introduced by Shiemke et al.68 This compound is an analogous to biological quinoles but with relatively higher solubility in water-based solutions. The redox potential at pH 7 of duroquinol is +7 mV.69 Duroquinol is a useful compound for detecting pMMO activity; however, great care must be taken when measuring the enzyme preparation with very low pMMO activity. This is because duroquinol reduces O2 to form H2O2,70 which becomes a hydroxy radical and oxidizes methane.71 The pMMO preparation contains a certain amount of copper ions, which accelerate the formation of H2O2 and the hydroxy radical.72,73 Therefore, the pMMO activity measured in the presence of duroquinol and copper ions shows a certain background of the non-specific methanol-producing activity. This background activity can mislead the enzymatic activity of the pMMO.
To confirm non-enzymatic methane oxidation in the presence of duroquinol or H2O2, we tested the methane oxidation using [13C]-methane as a substrate. We added 5 mg of duroquinol or 500 mM of H2O2 in 250 μl of phosphate-buffered saline pH 7.0 in a 7.7 ml sealed vial, into which 5 mL of [13C]-methane was injected, and incubated the vial at 28 °C or 35 °C for 4 h. We quantified the amount of [13C]-methanol in the reaction mixture by gas-chromatography coupled to mass-spectrometry (GC/MS) analysis. The reaction rates at 35 °C were estimated to be 3.0 × 10−2 μM min−1 (duroquinol) and 6.8 × 10−2 μM min−1 (H2O2). These rates are comparable to the production of methanol by the heterologously-produced soluble-domain of the PmoB subunit (spmoB) reported by Ross et al. (approximately 10 μM methanol production for a 1 h reaction at 45 °C).71 Thus, we confirmed that duroquinol and H2O2 caused non-enzymatic oxidation of methane at ambient temperature in the absence of a biological or artificial catalyst.
3.3 Inhibitors of pMMO
The inhibitors of pMMO can be classified into three types; (i) analogs of the substrate or product of pMMO: acetylene,55,74,75 duroquinol,76 duroquinone,77 and oxygen54,55 and H2O2;78 (ii) chelating or metal-binding reagents: azide,55 cyanide,55,68,79 EDTA,68 and nitrite;71 and (iii) metal ions: copper,49 zinc.80 Acetylene and acetylenic compounds react with O2 and reducing equivalents and irreversibly inhibit pMMO by binding the reaction product to the protein.75 Isotope labeling with [14C]-acetylene showed that the reaction product localized to the 26–27 kDa band on SDS-PAGE, the size of which corresponds to the PmoA subunit of pMMO.55,75 On the other hand, Pham et al. reported that the acetylene-reaction product is bound to the residue of PmoC.74 Because the reaction product of acetylene, ketene, can move from the active site to another position in the protein, exact localization of the active site is not easy to determine.74The inhibitory effect of chelating or metal-binding inhibitors suggest that the active site of pMMO is composed of metal ions. Treatment with cyanide or EDTA partially removed copper ions from pMMO and decreased the enzyme activity,61–64 indicating that the copper active site is located in the hydrophilic region of pMMO. Zinc is found in the crystal structure when zinc compounds are present in the crystallization solution. The zinc ion is located at the probable copper binding site; therefore, zinc could inhibit the MMO activity by displacing the copper. The zinc-substituted metal-binding site is identified in the CuC site of the PmoC subunit (see below), which suggests that the CuC site may be the active site of pMMO.71,80 Inhibition by copper ions suggests that external copper ions may interact with the endogenous copper site, which could form an artificial multinuclear copper cluster. Oxygen affects the activity and oxidation states of pMMO; therefore, most of purification experiments were performed under anoxic conditions.54,55,61,76
3.4 Protein structure of pMMO
To discuss the structure and function of the metal centers of pMMO, it is essential to have three-dimensional coordination information. The first protein crystal structure was obtained by the Rosenzweig group using pMMO purified from M. capsulatus Bath.37 The 2.8 Å crystal structure showed a conformation of a trimer of the heterotrimer composed of the 24 kDa PmoA, 47 kDa PmoB, and 22 kDa PmoC (Fig. 3). The PmoA and PmoC subunits are mainly composed of transmembrane helices, which is typical for membrane anchoring proteins. PmoB is mainly located in the cytosolic region and is anchored to the membrane by a hydrophobic domain. There are electron densities corresponding to a mononuclear metal site (CuA) and a dinuclear metal site (CuB) in PmoB, and a mononuclear metal site in PmoC. X-ray anomalous Fourier maps indicated that the metals at the metal binding sites are copper in PmoB and zinc in PmoC. The zinc-binding site in PmoC could be a site for the copper-binding site in the native form of the enzyme; therefore, this metal site is referred to as the CuC site.37 A zinc ion could be incorporated from the crystallization solution and replaced by the native copper ion.79–81 Analysis of the purified pMMO indicated the presence of 2 to 15 copper ions and 2 iron ions, but no further binding sites for copper and iron ions were detected in the X-ray crystal structure.37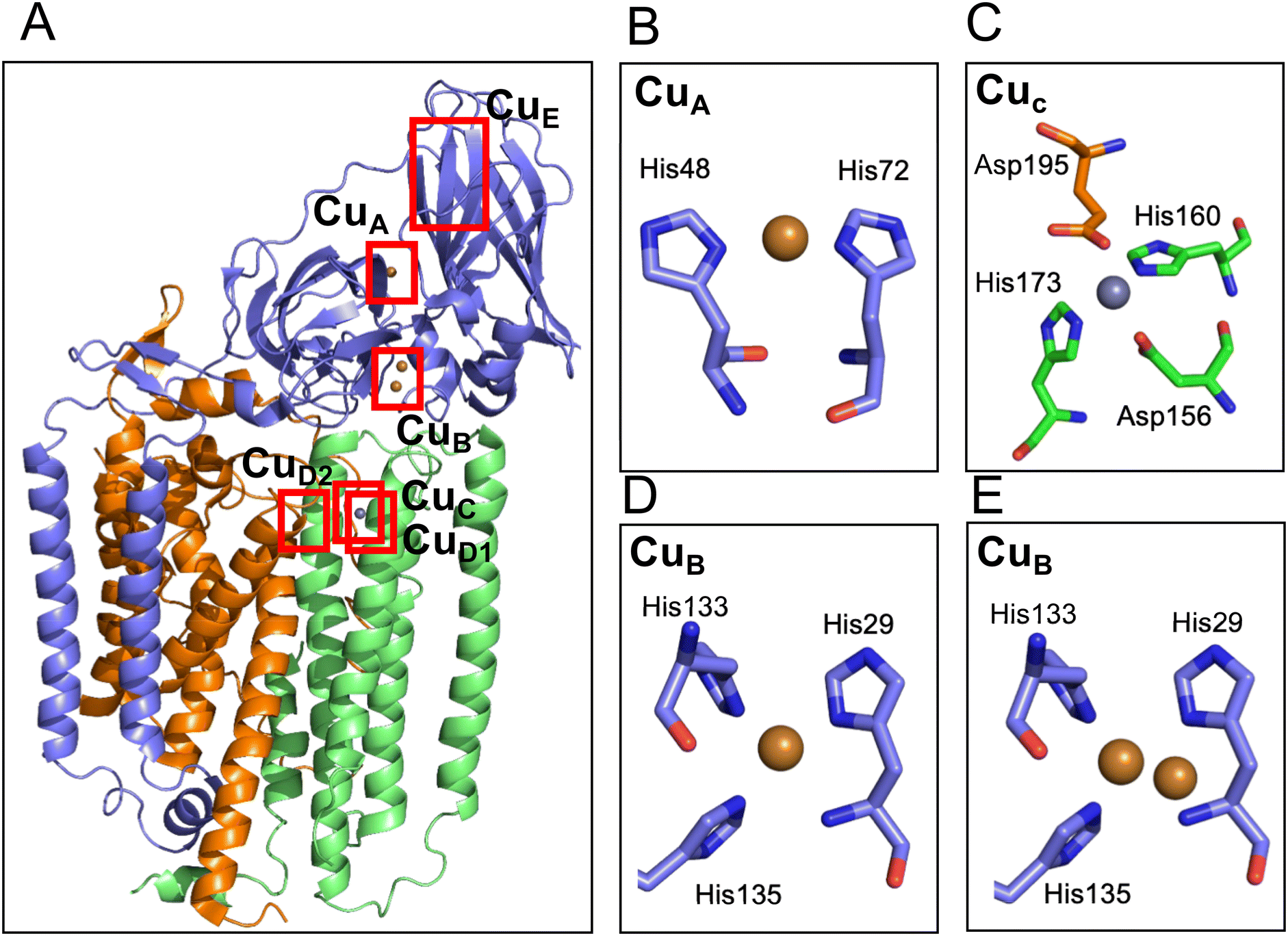 | ||
| Fig. 3 Cartoon model of the structure of pMMO and the metal sites. (A) Overall structure of pMMO from M. capsulatus in heterotrimer (PDB code 1YEW). The cartoon models of the PmoA, PmoB, and PmoC are shown in orange, blue, and green, respectively. The modeled copper and zinc ions are shown as brown and gray spheres, respectively. The ligand structure of CuA, CuC and two types of CuB are shown in the panels B–E. The CuE site was modeled only in one of the cryo-electron microscopy structure (Table 2) but not detected in the crystal structure; therefore, the CuE site metals are not depicted in this panel. (B) CuA site of pMMO from M. capsulatus (PDB code 1YEW). (C) CuC site of pMMO from M. capsulatus (1YEW). Glu195 is tentatively assigned to bind 2.9 Å away from the Zn site. (D and E) Mononuclear (D) and dinuclear (E) models of the CuB site in pMMO from Methylocystis sp. M (PDB code 3RFR). | ||
In addition to pMMO from M. capsulatus Bath, pMMOs from M. trichosporium OB3b,81Methylocystis sp. M,79Methylocystis sp. Rockwell,80 and Methylomicrobium alcaliphilum 20Z51 were analyzed by X-ray crystallography. The metal-binding sites of the pMMO structures found in pMMO from M. capsulatus Bath are not fully conserved among the different pMMOs (Table 2). The CuB site in PmoB of the pMMO structure in M. alcaliphilum 20Z and Methylocystis sp. Rockwell was modeled as a mononuclear copper site, and that of Methylocystis sp. M was modeled as a mixture of mononuclear and dinuclear copper sites (Fig. 3D and E). The CuA mononuclear copper site was not reproduced in other crystal structures of pMMO (Table 2), although the density was observed in the cryo-EM structures.53,82 At the CuC mononuclear metal site of PmoC, a copper ion was identified in the structure of M. trichosporium OB3b and Methylocystis sp. Rockwell, and zinc was observed in the structure from Methylocystis sp. M. In the crystal structure of pMMO from M. alcaliphilum 20Z, 60% of the structure of PmoC was disordered and the CuC metal site was not observed.51 The purified samples for crystallization show no or very low activity, which is one of the reason for the difficulty in studying the structure–function relationship of the metal site.53
| Microorganism | Resolution | Modeled metals in the structure of the pMMO subunits and the coordinating residues | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| PmoB | PmoB | PmoC | PmoC | PmoA | PmoB | |||
| CuA | CuB | CuC | CuD1 | CuD2 | CuE | |||
| a Distance between two Cu ions by EXAFS measurement. b A Cu–Cu site is modeled in one of three pmoB subunits, in which the other two PmoB subunits were modeled with a single copper ion. c The crystal structure showed single a Cu ion but EXAFS showed a Cu–Cu cluster at a distance of 2.48 Å. d Most of the PmoC structure is disordered. e The methyl carbon of Met42 is modeled to coordinate to a Cu ion in the distance of 1.9 Å. f The modeled mononuclear Cu is coordinated only to a single E316, in which the other proposed coordination residues are too distant to ligate; Tyr330 (3.2 Å from the modeled Cu), Arg323 (3.6 Å) and Arg400 (4.0 Å).82 | ||||||||
| Crystal structure | ||||||||
| Methylococcus capsulatus Bath (1YEW) | 2.8 Å | Cu | Cu–Cu (2.57 Å)a | Zn | No metal | No metal | No metal | Lieberman et al. 2005 (ref. 37) |
| H48H72 | H33H137H139 | D156H160H173EA195 | ||||||
| Methylosinus trichosporium OB3b (3CHX) | 3.9 Å | No metal | Cu–Cu (2.52 Å)a | Cu | No metal | No metal | No metal | Hakemian et al. 2008 (ref. 81) |
| H40H144H146 | D129H133H146E200 | |||||||
| Methylocystis sp. M (3RFR) | 2.68 Å | No metal | Cu–Cu or Cub | Zn | No metal | No metal | No metal | Smith et al. 2011 (ref. 79) |
| H29H133H135 (2.52–2.64 Å)a | D129H133E146 | |||||||
| Methylocystis sp. Rockwell (4PHZ) | 2.59 Å | No metal | Cu | Cu | No metal | No metal | No metal | Sirajuddin et al. 2014 (ref. 80) |
| H29H133H135 (2.48 Å)c | D129H133H146 | |||||||
| Methylomicrobium alcaliphilum 20Z (6CXH) | 2.7 Å | No metal | Cu | No metald | No metal | No metal | No metal | Ro et al. 2018 (ref. 51) |
| H33H137H139 | ||||||||
| Cryo-EM structure | ||||||||
| Methylococcus capsulatus Bath (7EV9) | 2.6 Å | Cu | Cu–Cu | No metal | No metal | Cu–Cue | Cu–Cu | Chang et al. 2021 (ref. 82) |
| H48H72 | H33H137H139 | H38M42E100 | T281N306D395 | |||||
| Cuf | ||||||||
| E316 | ||||||||
| Methylococcus capuslatus Bath (7S4H) | 2.14 Å | Cu | Cu | No metal | Cu | No metal | No metal | Koo et al. 2022 (ref. 53) |
| H48H72 | H33H137H139 | N227H231 H245 | ||||||
To overcome the instability of pMMO, the Rosenzweig group reported the stabilization of the purified pMMO by reconstitution of a solubilized membrane protein in a lipidic environment using bicelles and nanodiscs, and successfully increased the specific activity of the pMMO preparation.51,52 The active reconstituted pMMO in lipid nanodiscs was used for the structural analysis by cryo-EM.53 As shown in Table 2, the 2.14–2.46 Å cryo-EM structure of the nanodisc-stabilized pMMO from M. capsulatus Bath revealed the presence of a new metal site in PmoC, which is proposed to be a mononuclear copper site. We refer to this metal site as CuD1 to distinguish it from a copper D site in PmoA proposed by the Chan group, which is referred to as CuD2 in this review. The protein region at the CuD1 site in PmoC was stabilized by embedding into lipid nanodiscs. The authors proposed that the CuD1 site is the actual methane oxidation active site, which was not visible in the structural analysis of inactive pMMO without stabilization by lipids. The Chan group also reported a 2.5 Å cryo-EM structure of pMMO from M. capsulatus Bath, which was solubilized using the detergent n-dodecyl-β-D-maltoside.82 In the cryo-EM density, they modeled several copper ions at the CuD2 site in PmoA and at the CuE sites in PmoB (Table 2). In the detergent-solubilized pMMO structure, the CuD1 site was not observed. On the contrary, the CuD2 and CuE sites were not observed in the nanodisc-stabilized structure. The quality of the cryo-EM density maps appears to be too low to be used for the discussion of the metal ligand. The presence of the metal ions at the sites must be confirmed by anomalous scattering of the crystal structure and/or careful X-ray absorption spectroscopy (XAS) analysis combined with mutation analysis.
3.5 Copper complex structures based on spectroscopy analyses
However, the majority of experimental results contradict the hypothesis of the presence of a trinuclear copper site. All structural analyses do not show the presence of the trinuclear copper center (see Table 2). In addition to the data of purified pMMO shown in Table 1, the EPR studies on the samples of M. capsulatus,71,91,92M. trichosporium OB3b,81Methylocystis sp. M,79,80M. alcaliphilum 20Z,51Methylomicrobium album BG8,93,94 did not show the presence of the trinuclear copper center. Takeguchi et al. discussed an EPR signal attributable to the trinuclear clusters in the purified pMMO from the M. trichosporium OB3b fraction,57 but in a later study, the same group concluded the absence of such an EPR signal using pMMO from the same organism with an improved purification method.56
3.6 Oxidation states of the copper sites
Most of the EPR analysis of the pMMO samples indicated the presence of Cu(II) sites (Table 1). The Cu(II) signal decreased upon reduction with dithionite, indicating that the copper site is redox active and reduced to Cu(I).49,54,58,92 X-ray absorption near edge (XANE) analysis indicates that the copper in purified pMMO is a mixture of Cu(I) and Cu(II).37,51,79,81 However, recent work by the DeBeer group pointed out that X-ray beam causes reduction of the copper sites.98 To minimize the radiation effects, the analyses were performed with lower intensity beam and the authors concluded that most of the copper ion species in pMMO are Cu(II) and the copper sites in pMMO are only mononuclear. The effect of background copper ions is one reason for the misinterpretation of the previous EXAFS data that led to modeling dinuclear copper sites.983.7 Characterization of the engineered PmoB subunit
The crystal structure of pMMO from M. capsulatus Bath indicated that the cytosolic subunit PmoB contains a mononuclear CuA and dinuclear CuB copper site.37 A genetically engineered PmoB subunit (spmoB) was heterologously produced in E. coli. The hydrophobic membrane-anchoring domain of PmoB was genetically removed in the artificial spmoB protein by conjugation of the soluble regions with a peptide linker.99 Reconstitution of soluble spmoB from the inclusion bodies in the presence of copper ions produced an active form of psmoB, which exhibited methane mooxygenase activity; epoxidation of propylene to propylene oxide and oxidation of methane to methanol. Mutation of possible copper-binding residues at the CuB abolished the enzyme activity but mutation of the residues at the CuA site left some residual activity. The authors concluded that the CuB site is crucial for the methane monooxygenase activity of this protein.99 In the same report, EXAFS revealed the coordination structure of the copper site of spmoB, which is similar to that in pMMO.O2 or H2O2 reacts with the reduced form of pMMO and spomB to change the UV-visible spectrum of pMMO, but the mutants lacking the CuB site did not show the reaction, indicating that the PmoB subunit is responsible for the reaction with the oxidants.100 In addition, the oxidation states of the copper center of spmoB were studied using quantitative EPR analysis.101 Kim et al. constructed a fusion protein of a PmoB soluble fragment conjugated to ferritin, which increased the solubility of the PmoB fragment.102 The engineered PmoB fusion protein exhibited methane monooxygenase activity, supporting the previous spmoB studies.102
Later, however, an improved form of spmoB was constructed that had increased solubility by conjugating soluble protein regions to spmoB.16,71 The new form of spmoB does not catalyze the methane monooxygenase activity. Based on these results, the authors discussed that the observed methane monooxygenase activity of the original spmoB in Balasubramanian et al.99 is a background reaction dependent on duroquinol.71 However, if the observed methane monooxygenase activity is due to a non-enzymatic reaction, then the results of the mutation experiments at the CuB ligation site in the previously reported spmoB are contradictry.99 Even if the PmoB analogues cannot directly catalyze the methane monooxygenase activity, it cannot be excluded that under certain conditions, spomB catalyzes the formation of the reactive oxygen species, that in turn mediate the oxidation of methane to methanol.
Chan et al. reported the construction of an engineered PmoB conjugated with a maltose-binding protein.103 This engineered protein did not show methane monooxigenase activity but it catalyzed H2O2 production. Miyaji et al. also reported the H2O2-producing activity of pMMO from Methylosinus trichosporium OB3b.78 These data suggest that PmoB might be able to produce H2O2. The reactive oxygen species produced can catalyze the formation of methanol from methane. However, this interpretation should be carefully considered due to the presence of the H2O2 formation reactions mediated by duroquinol as discussed in the section 3.2 of this review.
3.8 Proposed catalytic mechanisms
According to the progress of the biochemical and biophysical analyses of pMMO, the proposed catalytic-site structures are changed from the trinuclear/dinuclear copper center to the mononuclear copper one.53,71,96 Some catalytic mechanisms are proposed based on the predicted mononuclear copper site structures of pMMO, and the reactivity has been studied using computational methods. Yoshizawa and Shiota reported that the mononuclear copper CuA site is reactive to cleave the C–H bond of methane, in which CuIII-oxo species is proposed as the active species for the reaction.104 Morris reported the mechanism at the CuC site of pMMO, in which the CuIII-oxo species is proposed as the active site species.105 Peng et al. also reported a catalytic mechanism using the CuC site with duroquinol as a part of the catalytic cycle. In this mechanism, the CuII–O anion-radical reacts with methane.106 Thus, theoretical study supported the catalytic function of the mononuclear copper sites. Theoretical studies using the coordination of the CuD1 site in the cryo-EM structure are expected.Concluding remarks
The structure and function of the active sites of pMMO are still controversial. The main reason for this uncertainty is the unstable nature of pMMO. Despite extensive efforts to stabilize the enzyme, the specific activity remains very low and not highly reproducible. Although careful spectroscopic and structural work has been done, it is still unclear whether the data reflect the native enzyme, a damaged enzyme, or due to impurities and/or background. To further advance research in this field, it is essential to have access to highly active and stable enzyme samples as purified enzymes. Application of new methods like the one using bicelles and nanodiscs can contribute to further our understanding. A high-resolution structural model of the enzyme should be constructed from the highly active and stable enzyme samples using crystallography and/or cryo-EM methods. Spectroscopic analysis should be carefully performed based on the new structural models. It is also necessary to construct a heterologous and/or homologous expression system for this enzyme for mutational analysis of pMMO to identify the active site. Recently, cell-free biosynthesis of pMMO was reported, but it yielded an inactive enzyme.107It is not feasible to use cells of methanotrophs or MMOs for the conversion of methane to methanol on an industrial scale due to difficulties with large-scale and high-cell-density cultivation of methanotrophs as well as with large-scale production of MMO proteins. It is also difficult to express MMOs in a fully or highly active form in heterologous hosts such as E. coli.25,102 If MMOs can be heterologously expressed in methanol-utilizing bacteria and yeasts, such microbes could be used as a methane-oxidizing cell catalyst to directly produce useful compounds from methane. In these cells, methanol generated from methane can be rapidly metabolized and converted into useful compounds. The rapid consumption of methanol can also avoid product inhibition of MMOs by methanol.
Recently, we have succeeded in constructing methanol sensor cells in the methanol-utilizing yeast and bacterium in which the fluorescent proteins are expressed under the control of methanol-induced gene promoters.108,109 Using the yeast methanol sensor, we have demonstrated that high-throughput screening of methanol-generating enzymes, e.g. pectin methylesterase, could be conducted.108 Highly-functional novel methane-oxidizing biocatalysts will be developed by using these methanol sensor cells as expression hosts.
While the use of advanced technologies is expected, perhaps, we may need to go back to more basic research to identify stable enzymes by exploring new microorganisms. Considering the industrial importance of this enzyme, it is hoped that more researchers will enter this field and conduct active research.
Author contributions
Y. S., H. Y. and S. S. co-wrote the paper.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank Dr. Tomoyuki Takeya for his help in evaluation of non-enzymatic methane oxidation. This research work was supported by Japan Science and Technology Agency (CREST program JPMJCR to Y. S. and Mirai program JPMJMI22E8 to H. Y.) and Max Planck Society (S. S.). Open Access funding provided by the Max Planck Society.References
- R. Munõz, L. Meier, I. Diaz and D. Jeison, Rev. Environ. Sci. Bio/Technol., 2015, 14, 727–759 CrossRef.
- B. Miao, S. S. K. Ma, X. Wang, H. B. Su and S. H. Chan, Catal. Sci. Technol., 2016, 6, 4048–4058 RSC.
- A. Schoedel, Z. Ji and O. M. Yaghi, Nat. Energy, 2016, 1, 16034 CrossRef CAS.
- M. J. da Silva, Fuel Process. Technol., 2016, 145, 42–61 CrossRef.
- A. A. Latimer, A. Kakekhani, A. R. Kulkarni and J. K. Norskov, ACS Catal., 2018, 8, 6894–6907 CrossRef CAS.
- H. T. Luk, C. Mondelli, D. C. Ferre, J. A. Stewart and J. Perez-Ramirez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- R. S. Hanson and T. E. Hanson, Microbiol. Rev., 1996, 60, 439–471 CrossRef CAS PubMed.
- L. Chistoserdova, M. G. Kalyuzhnaya and M. E. Lidstrom, Annu. Rev. Microbiol., 2009, 63, 477–499 CrossRef CAS PubMed.
- R. Conrad, Environ. Microbiol. Rep., 2009, 1, 285–292 CrossRef CAS PubMed.
- C. Knief, Curr. Issues Mol. Biol., 2019, 33, 23–56 CrossRef PubMed.
- S. Kirschke, P. Bousquet, P. Ciais, M. Saunois, J. G. Canadell, E. J. Dlugokencky, P. Bergamaschi, D. Bergmann, D. R. Blake, L. Bruhwiler, P. Cameron-Smith, S. Castaldi, F. Chevallier, L. Feng, A. Fraser, M. Heimann, E. L. Hodson, S. Houweling, B. Josse, P. J. Fraser, P. B. Krummel, J. F. Lamarque, R. L. Langenfelds, C. Le Quere, V. Naik, S. O'Doherty, P. I. Palmer, I. Pison, D. Plummer, B. Poulter, R. G. Prinn, M. Rigby, B. Ringeval, M. Santini, M. Schmidt, D. T. Shindell, I. J. Simpson, R. Spahni, L. P. Steele, S. A. Strode, K. Sudo, S. Szopa, G. R. van der Werf, A. Voulgarakis, M. van Weele, R. F. Weiss, J. E. Williams and G. Zeng, Nat. Geosci., 2013, 6, 813–823 CrossRef CAS.
- M. Saunois, P. Bousquet, B. Poulter, A. Peregon, P. Ciais, J. G. Canadell, E. J. Dlugokencky, G. Etiope, D. Bastviken, S. Houweling, G. Janssens-Maenhout, F. N. Tubiello, S. Castaldi, R. B. Jackson, M. Alexe, V. K. Arora, D. J. Beerling, P. Bergamaschi, D. R. Blake, G. Brailsford, V. Brovkin, L. Bruhwiler, C. Crevoisier, P. Crill, K. Covey, C. Curry, C. Frankenberg, N. Gedney, L. Hoglund-Isaksson, M. Ishizawa, A. Ito, F. Joos, H. S. Kim, T. Kleinen, P. Krummel, J. F. Lamarque, R. Langenfelds, R. Locatelli, T. Machida, S. Maksyutov, K. C. McDonald, J. Marshall, J. R. Melton, I. Morino, V. Naik, S. O'Doherty, F. J. W. Parmentier, P. K. Patra, C. H. Peng, S. S. Peng, G. P. Peters, I. Pison, C. Prigent, R. Prinn, M. Ramonet, W. J. Riley, M. Saito, M. Santini, R. Schroeder, I. J. Simpson, R. Spahni, P. Steele, A. Takizawa, B. F. Thornton, H. Q. Tian, Y. Tohjima, N. Viovy, A. Voulgarakis, M. van Weele, G. R. van der Werf, R. Weiss, C. Wiedinmyer, D. J. Wilton, A. Wiltshire, D. Worthy, D. Wunch, X. Y. Xu, Y. Yoshida, B. Zhang, Z. Zhang and Q. Zhu, Earth Syst. Sci. Data, 2016, 8, 697–751 CrossRef.
- F. Keppler, J. T. G. Hamilton, M. Brass and T. Rockmann, Nature, 2006, 439, 187–191 CrossRef CAS PubMed.
- L. Ernst, B. Steinfeld, U. Barayeu, T. Klintzsch, M. Kurth, D. Grimm, T. P. Dick, J. G. Rebelein, I. B. Bischofs and F. Keppler, Nature, 2022, 603, 482–487 CrossRef CAS PubMed.
- J. D. Semrau, A. A. DiSpirito, W. Y. Gu and S. Yoon, Appl. Environ. Microbiol., 2018, 84, e02289-17 CrossRef PubMed.
- C. W. Koo and A. C. Rosenzweig, Chem. Soc. Rev., 2021, 50, 3424–3436 RSC.
- S. I. Chan, V. C. C. Wang, P. P. Y. Chen and S. S. F. Yu, J. Chin. Chem. Soc., 2022, 69, 1147–1158 CrossRef CAS.
- R. Banerjee, J. C. Jones and J. D. Lipscomb, Annu. Rev. Biochem., 2019, 88, 409–431 CrossRef CAS PubMed.
- W. X. Wang, A. D. Liang and S. J. Lippard, Acc. Chem. Res., 2015, 48, 2632–2639 CrossRef CAS PubMed.
- M. R. Hyman, I. B. Murton and D. J. Arp, Appl. Environ. Microbiol., 1988, 54, 3187–3190 CrossRef CAS PubMed.
- K. H. Halsey, L. A. Sayavedra-Soto, P. J. Bottomley and D. J. Arp, J. Bacteriol., 2006, 188, 4962–4969 CrossRef CAS PubMed.
- I. Y. Hwang, S. H. Lee, Y. S. Choi, S. J. Park, J. G. Na, I. S. Chang, C. Kim, H. C. Kim, Y. H. Kim, J. W. Lee and E. Y. Lee, J. Microbiol. Biotechnol., 2014, 24, 1597–1605 CrossRef CAS PubMed.
- M. M. Cui, A. Z. Ma, H. Y. Qi, X. L. Zhuang and G. Q. Zhuang, Microbiology, 2015, 4, 1–11 CAS.
- W. T. Yang, L. D. Shen and Y. N. Bai, Environ. Res., 2023, 219, 115174 CrossRef CAS PubMed.
- M. O. Ross and A. C. Rosenzweig, J. Biol. Inorg. Chem., 2017, 22, 307–319 CrossRef CAS PubMed.
- J. D. Semrau, A. A. DiSpirito and S. Yoon, FEMS Microbiol. Rev., 2010, 34, 496–531 CrossRef CAS PubMed.
- Y. Hibi, K. Asai, H. Arafuka, M. Hamajima, T. Iwama and K. Kawai, J. Biosci. Bioeng., 2011, 111, 547–549 CrossRef CAS PubMed.
- T. Nakagawa, R. Mitsui, A. Tani, K. Sasa, S. Tashiro, T. Iwama, T. Hayakawa and K. Kawai, PLoS One, 2012, 7, e50480 CrossRef CAS PubMed.
- L. Chistoserdova and M. G. Kalyuzhnaya, Trends Microbiol., 2018, 26, 703–714 CrossRef CAS PubMed.
- M. C. F. van Teeseling, A. Pol, H. R. Harhangi, S. van der Zwart, M. S. M. Jetten and H. J. M. O. den Camp, Appl. Environ. Microbiol., 2014, 80, 6782–6791 CrossRef PubMed.
- A. F. Khadem, A. Pol, A. Wieczorek, S. S. Mohammadi, K. J. Francoijs, H. G. Stunnenberg, M. S. M. Jetten and H. J. M. Op den Camp, J. Bacteriol., 2011, 193, 4438–4446 CrossRef CAS PubMed.
- M. F. Ul Haque, H. J. Xu, J. C. Murrell and A. Crombie, Microbiology, 2020, 166, 894–908 CrossRef CAS PubMed.
- S. Guerrero-Cruz, A. Vaksmaa, M. A. Horn, H. Niemann, M. Pijuan and A. Ho, Front. Microbiol., 2021, 12, 678057 CrossRef PubMed.
- H. J. M. Op den Camp, T. Islam, M. B. Stott, H. R. Harhangi, A. Hynes, S. Schouten, M. S. M. Jetten, N. K. Birkeland, A. Pol and P. F. Dunfield, Environ. Microbiol. Rep., 2009, 1, 293–306 CrossRef CAS PubMed.
- R. J. M. Van Spanning, Q. Guan, C. Melkonian, J. Gallant, L. Polerecky, J. F. Flot, B. W. Brandt, M. Braster, P. I. Espinoza, J. W. Aerts, M. M. Meima-Franke, S. R. Piersma, C. M. Bunduc, R. Ummels, A. Pain, E. J. Fleming, N. N. Van der Wel, V. D. Gherman, S. M. Sarbu, P. L. E. Bodelier and W. Bitter, Nat. Microbiol., 2022, 7, 2089–2100 CrossRef CAS PubMed.
- J. C. Murrell, B. Gilbert and I. R. McDonald, Arch. Microbiol., 2000, 173, 325–332 CrossRef CAS PubMed.
- R. L. Lieberman and A. C. Rosenzweig, Nature, 2005, 434, 177–182 CrossRef CAS PubMed.
- A. El Ghazouani, A. Basle, S. J. Firbank, C. W. Knapp, J. Gray, D. W. Graham and C. Dennison, Inorg. Chem., 2011, 50, 1378–1391 CrossRef CAS PubMed.
- C. S. Kang-Yun, X. J. Liang, P. Dershwitz, W. Y. Gu, A. Schepers, A. Flatley, J. Lichtmannegger, H. Zischka, L. J. Zhang, X. Lu, B. H. Gu, J. C. Ledesma, D. J. Pelger, A. A. DiSpirito and J. D. Semrau, ISME J., 2022, 16, 211–220 CrossRef CAS PubMed.
- R. Banerjee, Y. Proshlyakov, J. D. Lipscomb and D. A. Proshlyakov, Nature, 2015, 518, 431–434 CrossRef CAS PubMed.
- G. P. Stafford, J. Scanlan, I. R. McDonald and J. C. Murrell, Microbiology, 2003, 149, 1771–1784 CrossRef CAS PubMed.
- J. D. Semrau, S. Jagadevan, A. A. DiSpirito, A. Khalifa, J. Scanlan, B. H. Bergman, B. C. Freemeier, B. S. Baral, N. L. Bandow, A. Vorobev, D. H. Haft, S. Vuilleumier and J. C. Murrell, Environ. Microbiol., 2013, 15, 3077–3086 CrossRef CAS PubMed.
- J. G. Leahy, P. J. Batchelor and S. M. Morcomb, FEMS Microbiol. Rev., 2003, 27, 449–479 CrossRef CAS PubMed.
- N. V. Coleman, N. B. Bui and A. J. Holmes, Environ. Microbiol., 2006, 8, 1228–1239 CrossRef CAS PubMed.
- A. T. Crombie and J. C. Murrell, Nature, 2014, 510, 148–151 CrossRef CAS PubMed.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- Y. A. Zhu, C. W. Koo, C. K. Cassidy, M. C. Spink, T. Ni, L. C. Zanetti-Domingues, B. Bateman, M. L. Martin-Fernandez, J. Shen, Y. W. Sheng, Y. Song, Z. Y. Yang, A. C. Rosenzweig and P. J. Zhang, Nat. Commun., 2022, 13, 5221 CrossRef CAS PubMed.
- S. S. F. Yu, K. H. C. Chen, M. Y. H. Tseng, Y. S. Wang, C. F. Tseng, Y. J. Chen, D. S. Huang and S. I. Chan, J. Bacteriol., 2003, 185, 5915–5924 CrossRef CAS PubMed.
- H. H. T. Nguyen, A. K. Shiemke, S. J. Jacobs, B. J. Hales, M. E. Lidstrom and S. I. Chan, J. Biol. Chem., 1994, 269, 14995–15005 CrossRef CAS PubMed.
- D. Drummond, S. Smith and H. Dalton, Eur. J. Biochem., 1989, 182, 667–671 CrossRef PubMed.
- S. Y. Ro, M. O. Ross, Y. W. Deng, S. Batelu, T. J. Lawton, J. D. Hurley, T. L. Stemmler, B. M. Hoffman and A. C. Rosenzweig, J. Biol. Chem., 2018, 293, 10457–10465 CrossRef PubMed.
- S. Y. Ro, L. F. Schachner, C. W. Koo, R. Purohit, J. P. Remis, G. E. Kenney, B. W. Liauw, P. M. Thomas, S. M. Patrie, N. L. Kelleher and A. C. Rosenzweig, Nat. Commun., 2019, 10, 2675 CrossRef PubMed.
- C. W. Koo, F. J. Tucci, Y. He and A. C. Rosenzweig, Science, 2022, 375, 1287–1291 CrossRef CAS PubMed.
- H. H. T. Nguyen, S. J. Elliott, J. H. K. Yip and S. I. Chan, J. Biol. Chem., 1998, 273, 7957–7966 CrossRef CAS PubMed.
- J. A. Zahn and A. A. DiSpirito, J. Bacteriol., 1996, 178, 1018–1029 CrossRef CAS PubMed.
- A. Miyaji, T. Kamachi and I. Okura, Biotechnol. Lett., 2002, 24, 1883–1887 CrossRef CAS.
- M. Takeguchi, K. Miyakawa and I. Okura, J. Mol. Catal. A: Chem., 1998, 132, 145–153 CrossRef CAS.
- R. L. Lieberman, D. B. Shrestha, P. E. Doan, B. M. Hoffman, T. L. Stemmler and A. C. Rosenzweig, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3820–3825 CrossRef CAS PubMed.
- G. M. Tonge, D. E. F. Harrison and I. J. Higgins, Biochem. J., 1977, 161, 333–344 CrossRef CAS PubMed.
- R. I. Gvozdev, I. A. Tukhvatullin and L. V. Tumanova, Biol. Bull., 2008, 35, 161–169 CrossRef CAS.
- M. Takeguchi, K. Miyakawa and I. Okura, J. Mol. Catal. A: Chem., 1999, 137, 161–168 CrossRef CAS.
- A. Miyaji, M. Nitta and T. Baba, J. Biotechnol., 2019, 306S, 100001 CrossRef PubMed.
- M. Takeguchi and I. Okura, Catal. Surv. Jpn., 2000, 4, 51–63 CrossRef CAS.
- S. A. Cook and A. K. Shiemke, J. Inorg. Biochem., 1996, 63, 273–284 CrossRef CAS.
- J. D. Semrau, D. Zolandz, M. E. Lidstrom and S. I. Chan, J. Inorg. Biochem., 1995, 58, 235–244 CrossRef CAS PubMed.
- K. J. Burrows, A. Cornish, D. Scott and I. J. Higgins, J. Gen. Microbiol., 1984, 130, 3327–3333 CAS.
- D. W. Choi, R. C. Kunz, E. S. Boyd, J. D. Semrau, W. E. Antholine, J. I. Han, J. A. Zahn, J. M. Boyd, A. M. de la Mora and A. A. DiSpirito, J. Bacteriol., 2003, 185, 5755–5764 CrossRef CAS PubMed.
- A. K. Shiemke, S. A. Cook, T. Miley and P. Singleton, Arch. Biochem. Biophys., 1995, 321, 421–428 CrossRef CAS PubMed.
- I. M. Ibrahim, H. Wu, R. Ezhov, G. E. Kayanja, S. D. Zakharov, Y. Y. Du, W. A. Tao, Y. Pushkar, W. A. Cramer and S. Puthiyaveetil, Commun. Biol., 2020, 3, 13 CrossRef PubMed.
- E. Cadenas, A. Boveris, C. I. Ragan and A. O. M. Stoppani, Arch. Biochem. Biophys., 1977, 180, 248–257 CrossRef CAS PubMed.
- M. O. Ross, F. MacMillan, J. Z. Wang, A. Nisthal, T. J. Lawton, B. D. Olafson, S. L. Mayo, A. C. Rosenzweig and B. M. Hoffman, Science, 2019, 364, 566–570 CrossRef CAS PubMed.
- M. R. Gunther, P. M. Hanna, R. P. Mason and M. S. Cohen, Arch. Biochem. Biophys., 1995, 316, 515–522 CrossRef CAS PubMed.
- B. Halliwell and J. M. C. Gutteridge, Biochem. J., 1984, 219, 1–14 CrossRef CAS PubMed.
- M. D. Pham, Y. P. Lin, Q. V. Vuong, P. Nagababu, B. T. A. Chang, K. Y. Ng, C. H. Chen, C. C. Han, C. H. Chen, M. S. Li, S. S. F. Yu and S. I. Chan, Biochim. Biophys. Acta, 2015, 1854, 1842–1852 CrossRef CAS PubMed.
- S. D. Prior and H. Dalton, FEMS Microbiol. Lett., 1985, 29, 105–109 CrossRef CAS.
- V. I. Vasil'ev, T. V. Tikhonova, R. I. Gvozdev, I. A. Tukhvatullin and V. O. Popov, Biochemistry, 2006, 71, 1329–1335 Search PubMed.
- K. H. C. Chen, H. H. Wu, S. F. Ke, Y. T. Rao, C. M. Tu, Y. P. Chen, K. H. Kuei, Y. S. Chen, V. C. C. Wang, W. C. Kao and S. I. Chan, J. Inorg. Biochem., 2012, 111, 10–17 CrossRef CAS PubMed.
- A. Miyaji, M. Suzuki, T. Baba, T. Kamachi and I. Okura, J. Mol. Catal. B: Enzym., 2009, 57, 211–215 CrossRef CAS.
- S. M. Smith, S. Rawat, J. Telser, B. M. Hoffman, T. L. Stemmler and A. C. Rosenzweig, Biochemistry, 2011, 50, 10231–10240 CrossRef CAS PubMed.
- S. Sirajuddin, D. Barupala, S. Helling, K. Marcus, T. L. Stemmler and A. C. Rosenzweig, J. Biol. Chem., 2014, 289, 21782–21794 CrossRef PubMed.
- A. S. Hakemian, K. C. Kondapalli, J. Telser, B. M. Hoffman, T. L. Stemmler and A. C. Rosenzweig, Biochemistry, 2008, 47, 6793–6801 CrossRef CAS PubMed.
- W. H. Chang, H. H. Lin, I. K. Tsai, S. H. Huang, S. C. Chung, I. P. Tu, S. S. F. Yu and S. I. Chan, J. Am. Chem. Soc., 2021, 143, 9922–9932 CrossRef CAS PubMed.
- P. P. Y. Chen and S. I. Chan, J. Inorg. Biochem., 2006, 100, 801–809 CrossRef CAS PubMed.
- C. H. Yeh, S. S. F. Yu, S. I. Chan and J. C. Jiang, ChemistrySelect, 2018, 3, 5113–5122 CrossRef CAS.
- S. I. Chan, V. C. C. Wang, J. C. H. Lai, S. S. F. Yu, P. P. Y. Chen, K. H. C. Chen, C. L. Chen and M. K. Chan, Angew. Chem., Int. Ed., 2007, 46, 1992–1994 CrossRef CAS PubMed.
- S. Maji, J. C. M. Lee, Y. J. Lu, C. L. Chen, M. C. Hung, P. P. Y. Chen, S. S. F. Yu and S. I. Chan, Chem. – Eur. J., 2012, 18, 3955–3968 CrossRef CAS PubMed.
- P. Nagababu, S. Maji, M. P. Kumar, P. P. Y. Chen, S. S. F. Yu and S. I. Chan, Adv. Synth. Catal., 2012, 354, 3275–3282 CrossRef CAS.
- S. I. Chan, C. Y. C. Chien, C. S. C. Yu, P. Nagababu, S. Maji and P. P. Y. Chen, J. Catal., 2012, 293, 186–194 CrossRef CAS.
- Y. H. Chen, C. Q. Wu, P. H. Sung, S. I. Chan and P. P. Y. Chen, ChemCatChem, 2020, 12, 3088–3096 CrossRef CAS.
- P. P. Y. Chen, R. B. G. Yang, J. C. M. Lee and S. I. Chan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14570–14575 CrossRef CAS PubMed.
- R. L. Lieberman and A. C. Rosenzweig, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 147–164 CrossRef CAS PubMed.
- R. L. Lieberman, K. C. Kondapalli, D. B. Shrestha, A. S. Hakemian, S. M. Smith, J. Telser, J. Kuzelka, R. Gupta, A. S. Borovik, S. J. Lippard, B. M. Hoffman, A. C. Rosenzweig and T. L. Stemmler, Inorg. Chem., 2006, 45, 8372–8381 CrossRef CAS PubMed.
- H. Yuan, M. L. P. Collins and W. E. Antholine, J. Am. Chem. Soc., 1997, 119, 5073–5074 CrossRef CAS.
- H. Yuan, M. L. P. Collins and W. E. Antholine, Biophys. J., 1999, 76, 2223–2229 CrossRef CAS PubMed.
- L. L. Cao, O. Caldararu, A. C. Rosenzweig and U. Ryde, Angew. Chem., Int. Ed., 2018, 57, 162–166 CrossRef CAS PubMed.
- R. J. Jodts, M. O. Ross, C. W. Koo, P. E. Doan, A. C. Rosenzweig and B. M. Hoffman, J. Am. Chem. Soc., 2021, 143, 15358–15368 CrossRef CAS PubMed.
- E. F. Liew, D. C. Tong, N. V. Coleman and A. J. Holmes, Microbiology, 2014, 160, 1267–1277 CrossRef CAS PubMed.
- G. E. Cutsail, M. O. Ross, A. C. Rosenzweig and S. DeBeer, Chem. Sci., 2021, 12, 6194–6209 RSC.
- R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115–119 CrossRef CAS PubMed.
- M. A. Culpepper, G. E. Cutsail, B. M. Hoffman and A. C. Rosenzweig, J. Am. Chem. Soc., 2012, 134, 7640–7643 CrossRef CAS PubMed.
- M. A. Culpepper, G. E. Cutsail, W. A. Gunderson, B. M. Hoffman and A. C. Rosenzweig, J. Am. Chem. Soc., 2014, 136, 11767–11775 CrossRef CAS PubMed.
- H. J. Kim, J. Huh, Y. W. Kwon, D. Park, Y. Yu, Y. E. Jang, B. R. Lee, E. Jo, E. J. Lee, Y. Heo, W. Lee and J. Lee, Nat. Catal., 2019, 2, 342–353 CrossRef CAS.
- Y. J. Lu, M. C. Hung, B. T. A. Chang, T. L. Lee, Z. H. Lin, I. K. Tsai, Y. S. Chen, C. S. Chang, Y. F. Tsai, K. H. C. Chen, S. I. Chan and S. S. F. Yu, J. Inorg. Biochem., 2019, 196, 110691 CrossRef CAS PubMed.
- K. Yoshizawa and Y. Shiota, J. Am. Chem. Soc., 2006, 128, 9873–9881 CrossRef CAS PubMed.
- R. H. Morris, Inorg. Chim. Acta, 2020, 503, 119441 CrossRef CAS.
- W. Peng, X. Y. Qu, S. Shaik and B. J. Wang, Nat. Catal., 2021, 4, 266–273 CrossRef CAS.
- C. W. Koo, J. M. Hershewe, M. C. Jewett and A. C. Rosenzweig, ACS Synth. Biol., 2022, 11, 4009–4017 CrossRef CAS PubMed.
- T. Takeya, H. Yurimoto and Y. Sakai, Appl. Microbiol. Biotechnol., 2018, 102, 7017–7027 CrossRef CAS PubMed.
- V. C. Casaroli, I. Orita, S. Katayama, H. Yurimoto, Y. Sakai and T. Fukui, J. Biosci. Bioeng., 2021, 132, 247–252 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |