
Manipulation of N-heterocyclic carbene reactivity with practical oriented electric fields†
Mitchell T.
Blyth
 a and
Michelle L.
Coote
*b
a and
Michelle L.
Coote
*b
aResearch School of Chemistry, Australian National University, Canberra, Australian Capital Territory 2601, Australia
bInstitute for Nanoscale Science and Technology, College of Science and Engineering, Flinders University, Bedford Park, South Australia 5042, Australia. E-mail: Michelle.coote@flinders.edu.au
First published on 8th December 2022
Abstract
Using density functional theory (DFT) calculations, we demonstrate that the organocatalytic properties of NHCs, such as their nucleophilicity, electrophilicity and singlet triplet gaps, are predictably influenced by electric fields. These electric fields can be delivered in practical systems using charged functional groups to provide designed local electric fields, and their effects are strong enough to be synthetically relevant even in relatively polar solvents. We also show that these electrostatically enhanced NHCs elicit dramatic changes in the energetics of key transition states of a model benzoin condensation in various solvents, which can be tuned by the sign of the applied charge and the solvent polarity. Based on these findings, we suggest that NHCs are plausible candidates for electrostatic catalysts, and that electric field effects should be considered when designing NHC frameworks.
Introduction
In the past decade, research into the use of electric fields to catalyse and control chemical reactions, has exploded.1–3 This interest has been motivated by an increasing awareness of the ubiquity of electric field-mediated transformations in enzymes4,5 and organic chemistry,6–15 as well as their unique advantages, namely that electric fields are orthogonal to other modes of activation and can exhibit highly directional effects. Designed local electrostatic fields (D-LEFs) might take the form of embedded charges or dipoles, such as those delivered by metal ions or charged functional groups,6,16–20 while oriented external electric fields (OEEFs, or EEF) might be realised by the use of scanning tunnelling microscopy,21 by orientation of bulk solvent in an electric field,22,23 or at interfaces.24,25 These studies have demonstrated that appropriately oriented electrostatic fields can operate as an experimentally relevant mechanism of control over a myriad of chemical processes, even in polar solvents where such effects might be expected to be attenuated.22In the present work, we examine the effect of electric fields on the chemistry of N-heterocyclic carbenes (NHCs). NHCs, once considered laboratory curiosities, are today compounds of huge practical importance, with significant applications in organocatalysis,26 materials chemistry,27 and as transition metal ligands.28 Given the ubiquity of NHCs, the ability to manipulate their chemistry with (switchable) electric fields represents a significant opportunity to affect the outcomes of many existing transformations. A growing number of studies have shown that the electrostatic environment can significantly affect reactions involving carbenes or closely related compounds. For instance, Kanan and co-workers have elegantly demonstrated that interfacial electric fields could modify the selectivity of an intramolecular reaction involving a putative carbene intermediate and catalysed by Rh porphyrins.24 We have shown that electrostatic field effects dominate the outcomes of a decarboxylation reaction catalysed by the protonated analogue of a NHC.29 Additionally, Maji and Wheeler have demonstrated the key importance of electrostatic interactions in determining the stereoselectivity of several NHC-catalysed kinetic resolutions,30 and Wang and co-workers have very recently shown the importance of subtle electrostatic effects when designing optimal NHC ligands for palladium-catalysed aryl-nitro bond activations.31
Inspired by these studies, in the present work we aim to systematically investigate the scope and utility of using electric fields to control carbene properties. We also examine whether electric fields differentially affect the singlet and triplet states of NHCs in a directional manner (Fig. 1). To this end, we use computational chemistry to investigate the electrophilicity, nucleophilicity, and relative spin-state stability of representative NHCs in the presence of both external and local electric fields, before demonstrating the utility of the observed, electrostatically induced, changes to a model NHC-catalysed organic transformation, the benzoin condensation of benzaldehyde. In this way, we aim to expand upon the existing synthetic scope and utility of NHCs as organocatalysts by harnessing electrostatic fields as a complementary and orthogonal tool to fine-tune the properties of carbenes in chemical reactions.
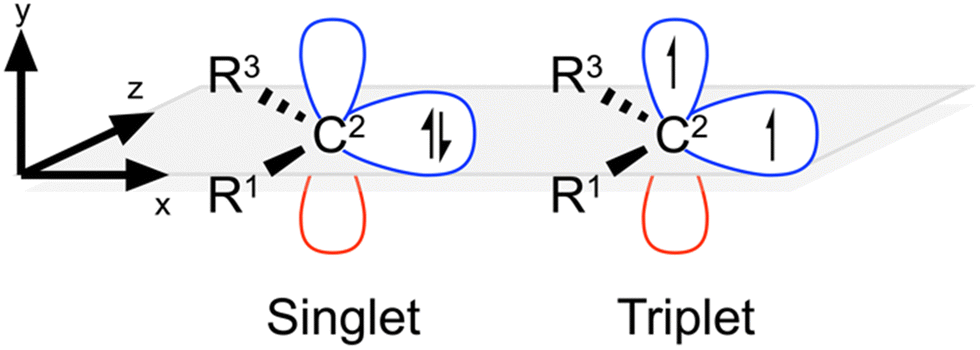 | ||
| Fig. 1 Generalised plane-aligned singlet & triplet carbenes. | ||
Computational methods
Density Functional Theory (DFT) and Time-Dependent DFT (TD-DFT) calculations were performed using the Gaussian 16 electronic structure package.32 Geometries were optimized with the M06-2X exchange correlation functional33 with the aug-cc-pVTZ basis set, except geometries in the benzoin condensation model reactions, for which the cc-pVDZ basis set was used.34,35 M06-2X has previously been used successfully to study electrostatic field effects,10,16–19,29 demonstrated many times to provide accurate structural and thermochemical data on carbenes.29,36 Solvent corrections were obtained with the SMD continuum solvent model,37 and the so-called direct method, in which ideal gas partition functions are applied to solution-phase geometries and frequencies, was employed with a phase correction term, to determine Gibbs free energies in solution.38 All energies given are from the conformationally searched, global minimum energy structures in both gas and solvent phases, unless otherwise indicated. Intrinsic reaction coordinate (IRC) calculations were used to verify the nature and connectivity of key transition states. Selected single-point energy calculations were performed for benchmarking purposes at the CCSD(T)/Def2TZVP//M06-2X/Def2TZVP level of theory.Nucleophilicity was estimated via the relationship N = EHOMO(Nu) − EHOMO(TCE), where tetracyanoethylene (TCE) was chosen as a reference (corrected for solvent where appropriate).39 Global electrophilicity (ω) was estimated via the Global Electrophilicity Index,40,41ω = χ2/2η, where χ = −0.5(EHOMO + ELUMO) is the Mulliken electronegativity (the negative of which is the chemical potential used in the original derivation by Parr40) and η = (ELUMO − EHOMO) is the chemical hardness.42N, ω, χ, and η are reported in eV by convention. NBO7 was used to perform Natural Resonance Theory (NRT) calculations.43 In our NRT calculations, a delocalisation list threshold (NRTE2) of 10 kcal was found to yield both a reasonable number of resonance structures while retaining the correct molecular symmetry.
Results and discussion
Test set definition and benchmarking
In order to demonstrate the generality of electrostatic field effects upon carbenes and discover those frameworks that are most susceptible to manipulation with electrostatic fields, we investigated the properties of a series of diverse NHCs in the presence of both EEFs and D-LEFs. The test set (Fig. 2) contains NHCs that are broadly representative of several common NHC families,26,44 and were selected for their ability to accommodate D-LEFs in multiple positions and because they exhibited a range of responses to EEFs applied along the Cartesian axes over physically accessible21 field strengths of ±0.003 a.u. in a variety of solvents (see Fig. S3 and S7, ESI†).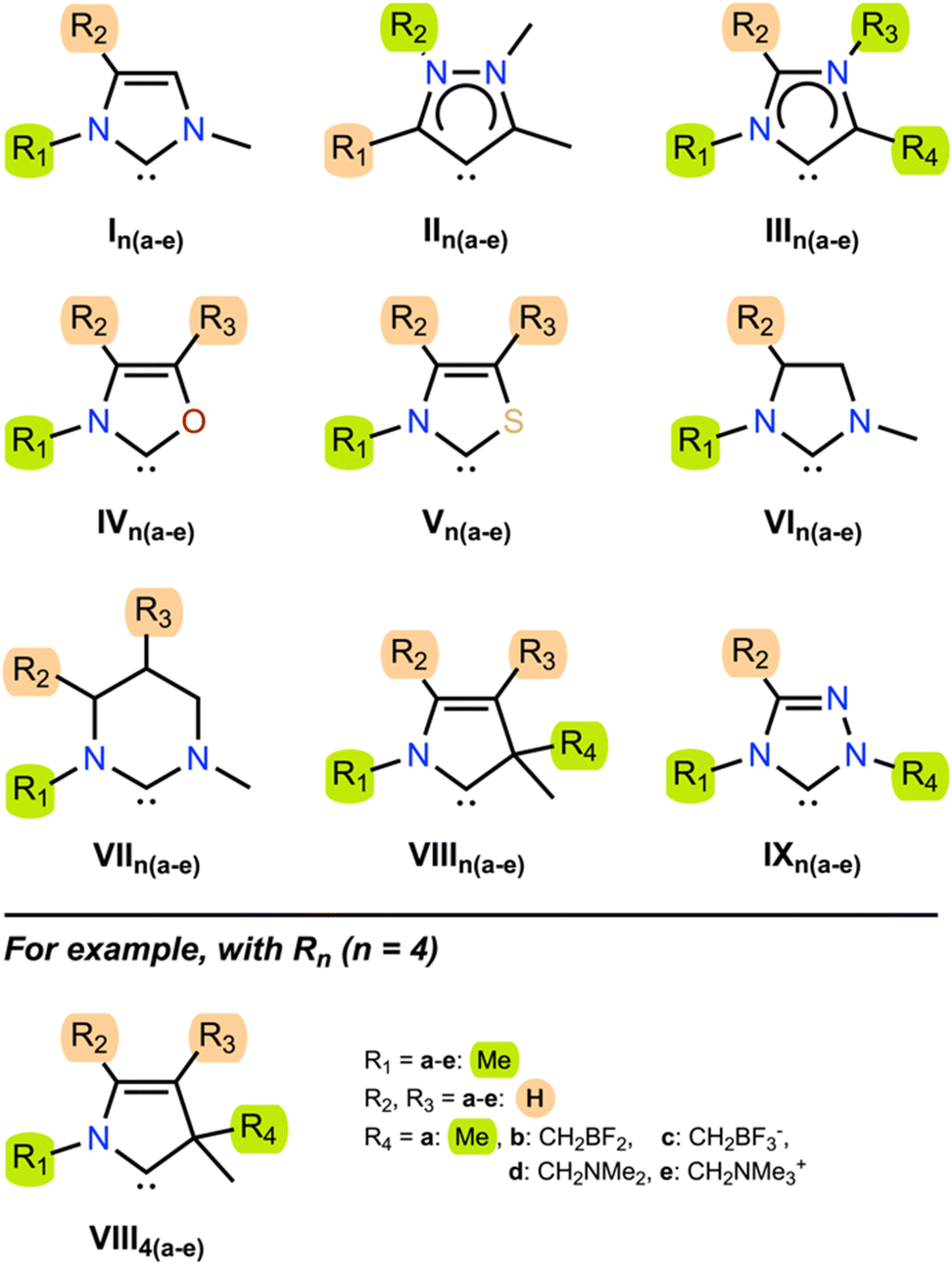 | ||
| Fig. 2 Test set. The test set of 9 NHCs which were investigated in this work. The naming scheme works as follows: for some molecule, the ith (i = a, b, c, d, or e) substitution at position Rn (n = 1, 2, 3, or 4) is either a methyl group (green highlights; if adjacent to the carbenic atom or directly bonded to a heteroatom) or a hydrogen atom (orange highlight; when neither adjacent to the carbenic atom nor bonded directly to a heteroatom) when i = a, and otherwise is substituted with one member of each D-LEF pair (CH2BF2 when i = b, CH2BF3− when i = c, CH2NMe2 when i = d, and CH2NMe3+ when i = e). | ||
The test set contains the imidazolinylidene I1a,45 the “remote” mesoionic carbene II1a (rNHC), the mesoionic carbene III1a (also known as an “abnormal” carbene, aNHC), the chalcogen-substituted oxazolylidene IV1a and thiazolylidene V1a, the unsaturated imidazolylidene VI1a46 and tetrahydropyrimidinylidene VII1a, the cyclic (amino)(alkyl) carbene (CAAC) VIII1a, and the 1,2,4-triazolylidene IX1a. Of these, II1a and III1a were specifically selected for their different mesoionic characters (one being remote-mesoionic), which we believed would be strongly affected and potentially differentiated by an electric field. Likewise, IV1a and V1a were selected to investigate the role of molecular asymmetry and heteroatom identity, if any, in the electric field response along different axes. VI1a and VII1a were selected to permit D-LEFs (vide supra) to adopt out-of-plane conformations (and in the case of VII3b-e, to do so along the central σ mirror plane). Finally, complex derivatives of VIII1a and IV1a are frequently used as asymmetric catalysts, so these models were chosen to investigate whether the addition of D-LEFs to these frameworks might feasibly provide some unique synthetic advantage. Throughout the remainder of this work, we consider the singlet form of the tested carbenes, which we show is always substantially more stable than the corresponding triplet, unless the triplet state is explicitly specified.
The fundamental electronic and steric properties of many of I1a to IX1a in Fig. 2 have been the subject of previous theoretical and experimental work (see for example47–49). Table 1 presents the nucleophilicity parameter (N), global electrophilicity index (GEI), and singlet–triplet gap (ΔGST) of the unsubstituted singlet carbenes I1a–IX1a in Fig. 2, along with corresponding literature results where available. Our computed values of N, GEI, and ΔGST compare favourably with examples from the literature. This is not particularly surprising given each comparison also uses DFT with moderate-to-large basis sets. However, it does provide confidence in our results by virtue of their similarity. We demonstrate later (vide supra) that our results under external electric fields also compare favourably with CCSD(T) benchmarks.
| Structure | N (eV) | GEI (eV) | ΔGST (kJ mol−1) |
|---|---|---|---|
| a Calculations performed at the M06-2X/aug-cc-pVTZ level of theory in the gas phase. b B3LYP/aug-cc-pVTZ//B3LYP/6-31+G*.54 c PBE0/6-31+G*.55 d B3LYP/6-311++G**.56 e B3LYP/6-311G**//B3LYP/6-31G*.57 f B3LYP/aug-cc-pVTZ.58 g PBE/TZVP[SBKJC].59 | |||
| I1a | 3.48 (3.56b) | 0.99 (0.83d) | 349.56 (346.90e) |
| II1a | 5.05 (5.00c) | 0.97 | 171.87 (164.00f) |
| III1a | 4.47 | 0.97 | 257.83 (252.00f) |
| IV1a | 2.85 | 1.12 | 322.24 |
| V1a | 3.15 (3.15b) | 1.08 (1.07d) | 273.23 |
| VI1a | 3.70 | 1.00 | 292.22 (307.52e; 279.07g) |
| VII1a | 4.20 | 0.94 | 250.83 |
| VIII1a | 3.81 (3.83b) | 0.97 | 188.04 (187.03g) |
| IX1a | 3.05 | 1.06 | 355.13 |
According to Pérez and co-workers,50 an electrophilic carbene is one with 1.21 < ω < 2.40 eV, an ambiphilic carbene has ω around 1 eV and a nucleophilic carbene has ω < 1.00 eV. Under these considerations, each of the tested carbenes is considered ambiphilic. The most electrophilic carbene is IV1a, followed by V1a. The least electrophilic carbene is VII1a, though the value of GEI = 0.94 is likely within error of each of II1a, III1a, and VIII1a (GEI = 0.97).
The initial step in most organocatalytic reactions involving NHCs is the addition of the NHC to an electrophile; therefore, the nucleophilicity of the carbene carbon is directly related to its utility as an organocatalyst. The measured nucleophilicity of the tested carbenes I1a–IX1a ranges from 2.85 eV to 5.05 eV, indicating that all except arguably the oxazolylidene are strong nucleophiles. An organic molecule is considered a strong nucleophile if N > 3.00 eV, moderate if 2.00 eV < N < 3.00 eV, and marginal if N < 2.00 eV.39 The most nucleophilic are the mesoionic species II1a and III1a respectively, whereas the least nucleophilic are IV1a and V1a.
The singlet–triplet gap of carbenes is frequently used to approximate their stability towards dimerization, and is a useful, if not independent, measure used to assess the relative stability of spin-states in experiment.51 We find that each NHC has ΔGST ≫ 0 and hence is a stable singlet, which is consistent with the literature. This arises from a combination of conjugative and inductive bonding interactions wherein the π-system of the neighbouring (or remote) nitrogen atom saturates the unoccupied p-orbital on the carbene carbon (π-conjugation), while withdrawing σ electrons from the carbene carbon (σ-induction), thus stabilising the carbene lone pair (which possesses σ-symmetry). The mesoionic carbene II1a has the smallest ΔGST, which is still a relatively large 172 kJ mol−1, followed closely by the CAAC VIII1a with a gap of 188 kJ mol−1. The largest ΔGST values were found for the 1,2,4-triazolylidene IX1a (355 kJ mol−1), followed closely by I1a (350 kJ mol−1), which is to be expected given their saturated backbones and heteroatom substitution.
We also investigated whether EEFs could be used to switch between the singlet- and triplet-states of carbenes at-will. However, as the singlet–triplet gaps of the tested NHCs are of the order of hundreds of kJ mol−1, this was largely unfruitful. The largest change in singlet–triplet gap of 56.9 kJ mol−1 was found for an X-EEF applied to II1a in water, which still yielded a final gap of over 100 kJ mol−1. The relative ordering of the changes in singlet–triplet gap mimics the results presented in Table 1 (Fig. S3, ESI†). We found that the ground states of several other carbenes with known small singlet–triplet gaps52 could be switched between their singlet and triplet states (see Fig. S7, ESI†); however, these switches were generally too small at the CCSD(T)/Def2TZVP//M06-2X/Def2TZVP level to warrant further study with multireference calculations, and their structures offered no realistic opportunity to incorporate DLEFs in the form of e.g., charged functional groups, which would make controlling their orientation with respect to the applied electric field difficult. We note that this issue of alignment could itself be addressed through orientation of the bulk solvent environment itself,22,23 or possibly via orientation within charged metal-organic frameworks,53 but this is beyond the scope of the present work.
Carbenes with designed local electric fields
To investigate the effect of practical electric fields, as delivered by D-LEFs, we have substituted the carbenes in our test set with non-conjugated charged functional groups at all symmetry-unique positions on the carbene backbone and named the resulting molecules according to the rules laid out in Fig. 2. The effects of cationic charges were modelled with the NH2/NH3+ pair, while the effects of anionic charges were modelled with the BF2/BF3− pair. We note that these pairs are intended as model systems only: for example, actual implementations of D-LEFs on NHCs will need to consider the possibility for unfavourable intermolecular carbene-D-LEF interactions, and issues pertaining to solubility. However, given the substantial structural diversity of and existing precedent surrounding the development of novel NHC families, we expect these issues could be rapidly addressed. We restrict ourselves to analysing the effects of D-LEFs upon the singlet carbenes only, as these are the only species likely to be synthetically relevant.Fig. 3 presents the change in nucleophilicity and electrophilicity of I1a (relative to the uncharged D-LEF) upon formation of a positive or negative charge as a function of the sign of the D-LEF charge and the solvent environment. A positive “switch” indicates that the nucleophilicity or electrophilicity increases upon formation of a charge (i.e. by protonation of the amine or addition of fluoride to the BF2 group). Corresponding figures for all nine compounds can be found in the ESI,† Fig. S1 (for GEI) and Fig. S2 (for N), while the raw data can be found in S11. Fig. 4 summarises the average effects of positive and negative D-LEFs on the nucleophilicity and electrophilicity of each compound studied, as averaged over all substitution positions for the D-LEF.
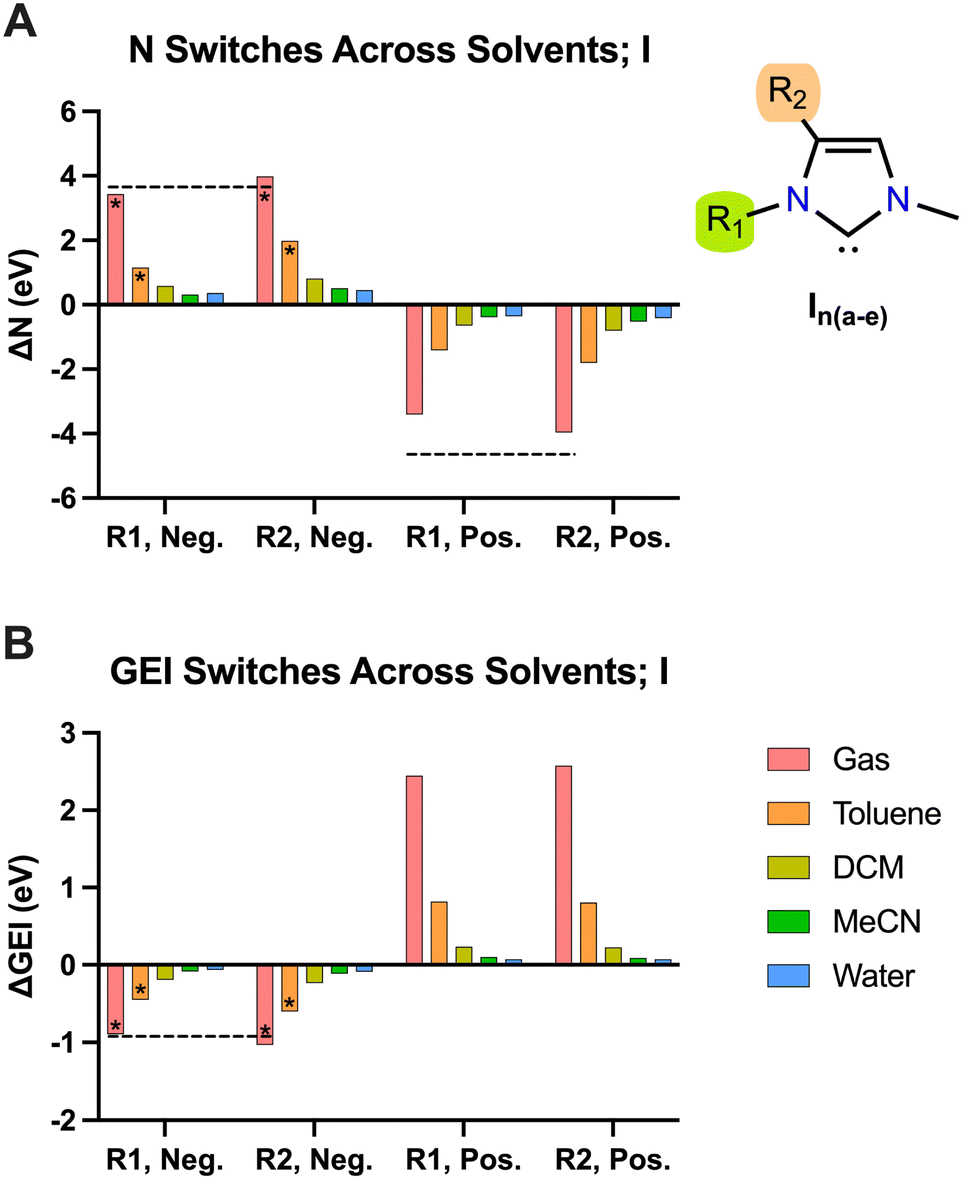 | ||
| Fig. 3 The effect upon the nucleophilicity (A) and electrophilicity (B) of I1a exerted by anionic (Neg.) and cationic (Pos.) D-LEFs in the two tested positions (R1 and R2). Positive values indicate an increase in the measured quantity upon formation of charge. Columns marked with an asterisk were corrected for spurious intramolecular interactions according to the procedure given in Fig. S10 (ESI†). DCM = dichloromethane; MeCN = acetonitrile. For comparison, the corresponding effect of simple point charges in gas is presented as black dotted lines, except for cationic GEI switches. See Fig. S5 (ESI†) for more details. | ||
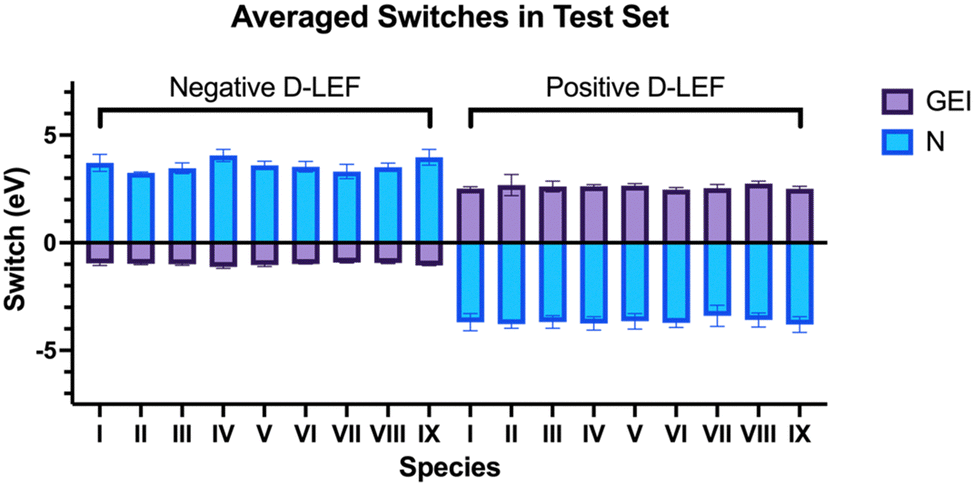 | ||
| Fig. 4 The DLEF-induced changes in electrophilicity and nucleophilicity for each of the carbenes in the test set in the gas-phase, averaged by position. Error bars indicate the standard deviation in values obtained at different positions. | ||
From Fig. 3 and 4, it is seen that D-LEFs affect the nucleophilicity and electrophilicity in a predictable and approximately equal and opposite manner according to the charge of the attached D-LEF, and to an extent that is only subtly affected by the position of the D-LEF. That is, the anionic D-LEF in either the 1- or 2-position on the carbene framework (named per Fig. 2) increases the measured nucleophilicity of the carbene and lowers its electrophilicity, to an extent modulated by the solvent polarity. Conversely, the cationic D-LEF in either position decreases the NHC nucleophilicity and increases its electrophilicity (Fig. 3). This equal-and-opposite behaviour, and the attenuation of the effect in increasingly polar solvents lends credence to the suggestion that these effects are predominately electrostatic, and not operating via conjugative or inductive phenomena. Furthermore, we also demonstrate that these electrostatic switches are induced and approximately reproduced by simple point charges in the case of I1b-e (see Fig. 3 and Fig. S5, ESI,† for further discussion).
In general, the application of D-LEFs yields larger changes in nucleophilicity than electrophilicity (Fig. 4), and this is because the HOMO energies are near-universally more strongly affected by the electric field than the corresponding LUMO energies, which we in turn attribute to the difference in their electronic population, (as the field interacts strongly with charged particles). The electrophilicity also shows a greater disparity between the stabilizing effects of positive D-LEFs and destabilizing effects of negative D-LEFs because the LUMO is more polarisable than the HOMO.
Of the carbenes in the test set, IV and IX generally yielded the greatest changes in nucleophilicity (see Fig. 4 and Fig. S11, ESI†). For these, the average nucleophilicity changes with an anionic D-LEF are between 4.06 eV in gas and 0.32 eV in water, and average electrophilicity changes with an anionic D-LEF are between −1.12 eV in gas to −0.07 eV in water. Large nucleophilicity changes also observed for VIII in dichloromethane, acetonitrile, and water, although the difference between the largest and smallest changes are small. Notably, these changes generally remain significant even in moderately polar solvents. The magnitude of these changes appears to be correlated with the HOMO–LUMO gaps of the unsubstituted carbenes, and uncorrelated with the molecular dipole and with the change in electronic energy under an EEF (Fig. S2, ESI†). Specifically, we found that II and III, as the mesoionic species, naturally yielded the greatest change in electronic energy in our test set under an X-EEF (which is aligned with the molecular dipole), but the properties of these species were not particularly strongly affected by incorporation of a D-LEF.
We were initially surprised to find that the above changes in nucleophilicity and electrophilicity are not strongly affected by the position of the incorporated D-LEF. This behaviour contrasts with many of the systems studied to date (see for example ref. 17 and 18). However, unlike many of these previous systems for which the interaction of the D-LEF can be approximated as a charge–dipole interaction, NRT calculations reveal that the distribution of charge in the tested NHCs is better represented as a quadrupole (Fig. 5), where anionic charge is predominately centred on the carbene carbon and cationic charge distributed on and among the heteroatom(s). This in turn means that an anionic D-LEF in either the 1- or 2-positions will stabilise the carbene framework, (and vice versa for the cationic D-LEF), which should yield favourable changes in subsequent organocatalytic reactivity. We investigate this hypothesis in the following section. The effect of D-LEFs upon the carbene quadrupoles, and their subsequent position-invariance, can be further investigated by comparing the field-independent quadrupoles of each of I2a-e. Upon doing this, we find that the QXX-components of the quadrupoles (and to a lesser extent, the QYY- and QZZ-components; where the D-LEFs are aligned along the X-axis) are affected by the introduction of charges in an approximately equal and opposite manner (Fig. S16, ESI†), relative to the corresponding uncharged species, lending credence to the proposed electrostatic origin of the observed effect upon nucleophilicity and electrophilicity.
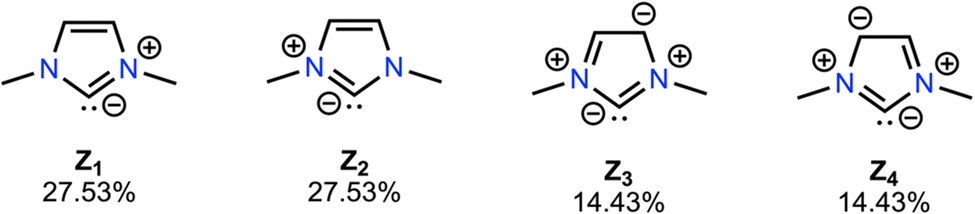 | ||
| Fig. 5 NRT calculations (Fig. S8, ESI†) support the notion that the NHCs in our test set are more accurately represented as quadrupolar carbon ylides (83.92% described by Z1–Z4) rather than as true carbenes, which explains why D-LEF electrostatic switches are not highly position dependent, and why triplet NHCs are not strongly affected by EEFs (Fig. S2, ESI†). | ||
Manipulating an organocatalytic reaction with D-LEFs
We now extend our analysis of electric field effects towards a realistic, multi-step model reaction, the NHC-catalysed benzoin condensation, presented in Fig. 6. In this reaction, the NHC acts as an ambiphilic umpolung catalyst to couple (traditionally) aromatic aldehydes A to yield the corresponding α-hydroxyketone E, though the exact mechanism has been the subject of long-standing debate.60 Under the mechanism presented herein, the zwitterionic primary adduct B is formed via the nucleophilic addition of the NHC catalyst to A, which undergoes a formal 1,2 hydrogen atom shift to yield the diamino enol C, the so-called Breslow intermediate. Then, the azolium hydroxyenolate D is formed by addition of a second equivalent of A, before release of the benzoin product E.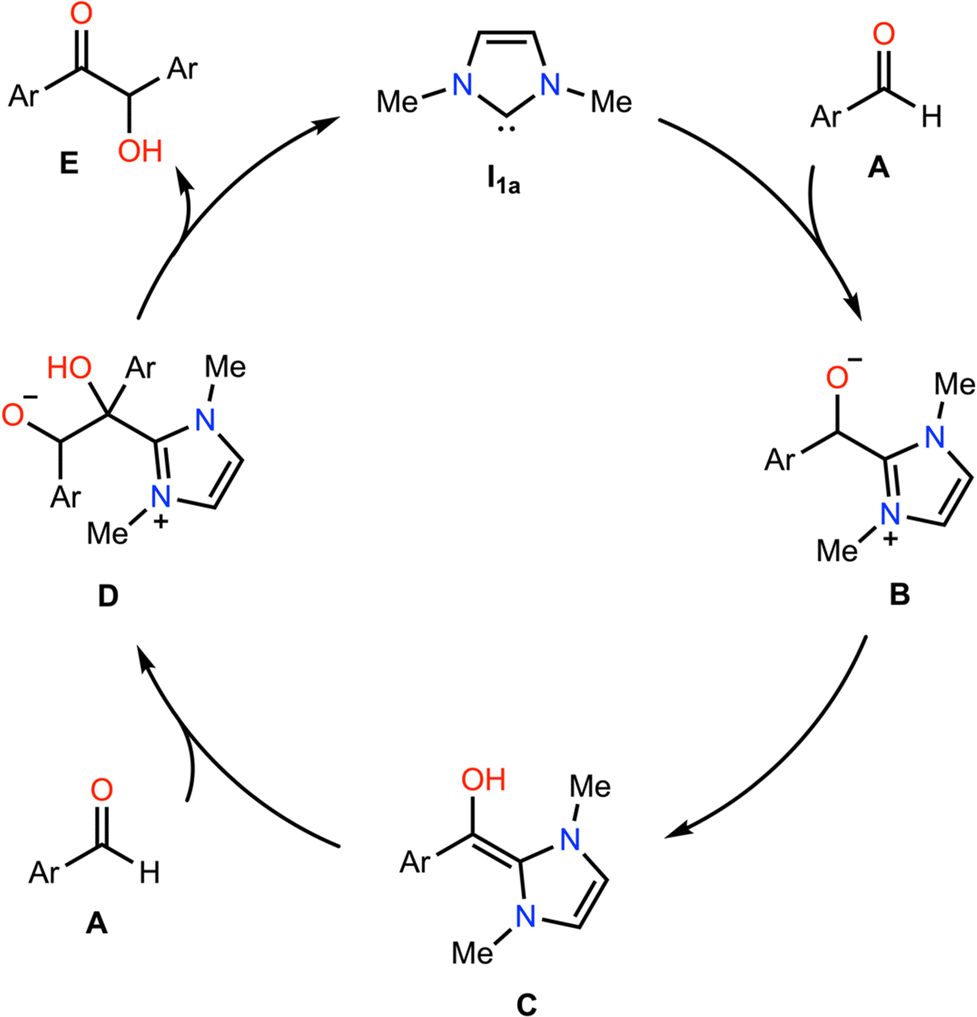 | ||
| Fig. 6 Model NHC-catalysed benzoin condensation of benzaldehyde, Ar = benzyl. | ||
Briefly, we note that the exact process via which C is formed remains an area of active debate; there is reason to believe that C is unlikely to be formed via water-mediated proton-transfer relays,60,61 bimolecular hydrogen-atom translocation,62,63via direct 1,2 hydride shift,64,65 or via radical processes.66 More recently, Gehre and Hollóczki have proposed that C is formed in a single step in the presence of base via the association of A to the azolium salt,67 and Berkessel and co-workers have suggested on the basis of kinetic data that C is formed autocatalytically in C under some combination of different regimes each defined by the relative excesses of the NHC and aldehyde species, and whether the reaction is run under anhydrous conditions.60 As our present focus is upon electric field effects upon carbene organocatalytic reactivity more broadly, we here focus only on electric field effects upon the elementary steps. Therefore, for simplicity and cost considerations, we model the formation of C as occurring via a catalytic proton transfer from B with methanol (rather than autocatalytically with C), which is feasible under the excess-NHC regime proposed by Berkessel and co-workers, and has previously been used successfully to model related benzoin condensations.68 We were unable to obtain the key transition state in the associative mechanism proposed by Gehre and Hollóczki for comparison within reasonable time, at our level of theory.
We begin by assessing whether our model reaction yields reasonable agreement with the literature. We find that, consistent with past research,69–71 the rate-determining step of benzoin condensation is the assisted intramolecular proton transfer (TS2; ΔG‡ = 88.5 kJ mol−1 in toluene, black line in Fig. 7). Consequently, the initial nucleophilic addition of I1a to A to form B is somewhat kinetically unfavourable (ΔΔG = 14.1 kJ mol−1 in SMD toluene in the productive direction). However, the subsequent steps exhibit low barriers, and the overall mechanism is predicted to be slightly exothermic (−6.6 kJ mol−1 in SMD toluene). These observations hold across each of tested solvents: toluene, dichloromethane, and acetonitrile (see Fig. S12, ESI†).
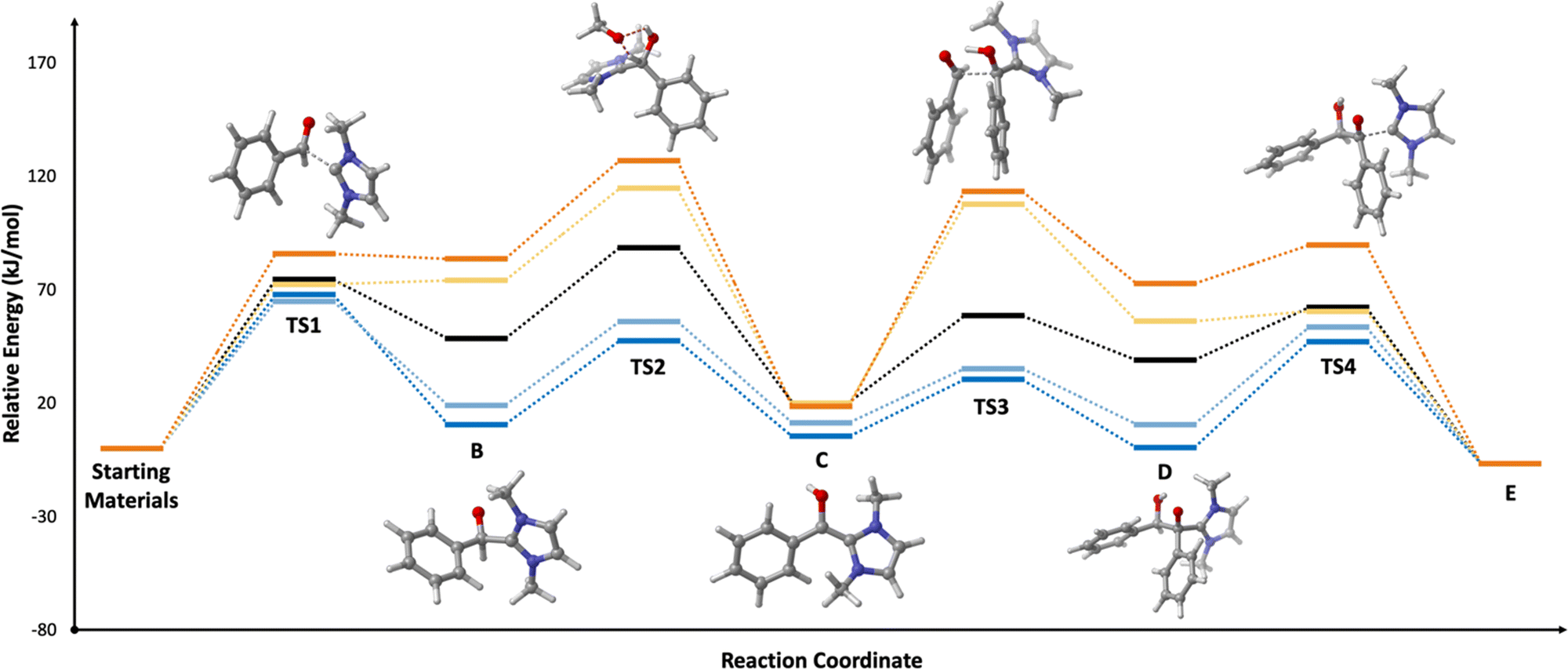 | ||
| Fig. 7 Gibbs free energy (kJ mol−1) at 298.15 K of the benzoin condensation catalysed by I1a (black), I1c (dark blue), I2c (light blue), I1e (yellow), and I2e (orange) at the SMD//M06-2X/cc-pVDZ level of theory in SMD toluene. | ||
Turning now to the effects of D-LEFs upon the benzoin condensation, we were pleased to find that the D-LEFs induced changes that are consistent with a clear electrostatic effect. That is, anionic D-LEFs generally facilitate productive formation of the benzoin product by lowering the energy of key transition states relative to the unsubstituted species (which is to be expected per valence bond theory given the relatively diffuse electronic distribution of transition states), while cationic D-LEFs generally inhibit formation of the product. This outcome is consistent with the results presented in Section B.2. and arises because the anionic D-LEF stabilises the formally cationic carbene framework at many points along the condensation mechanism. Naturally, large effects are also observed for the zwitterionic species, and the energetics of the Breslow intermediate is in turn, relatively unaffected. The magnitude of these effects is, as expected, diminished in increasingly polar solvents, though are broadly consistent with the toluene results presented in Fig. 7 and remain synthetically useful even in acetonitrile. For example, in toluene, an anionic D-LEF in the 1-position lowers the energy of TS1 by 6.6 kJ mol−1, of TS2 by 41.0 kJ mol−1, of TS3 by 28.2 kJ mol−1 and of TS4 by 15.1 kJ mol−1. For the anionic D-LEF in either position, these electrostatically induced changes lower the energy of TS2 to such an extent that the initial addition TS1 is predicted to become competitive with the rate-determining step, even in acetonitrile (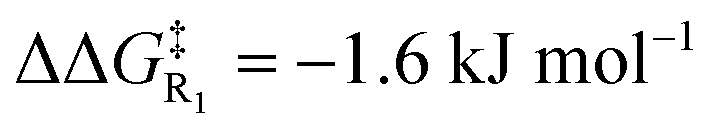 , and
, and 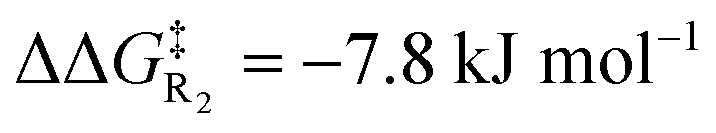 in the forward direction).
in the forward direction).
Critically, this competition between TS1 and TS2 should have both interesting and experimentally-verifiable kinetic implications: for example, all else being equal, it could permit access to a broader substrate scope for which the barrier to the formal 1,2 hydrogen atom shift is otherwise limiting. The relative changes in the energies of TS1 and TS2 can be understood through an analysis of their electronic populations, and how they change. For example, the charges of the relevant heavy atoms are largely unchanged from the starting materials to TS1, which supports the surprising finding that TS1 is only weakly affected by the D-LEF. That TS2 is strongly affected is actually a consequence of the effects of the D-LEF upon the changes in electronic population at the carbene carbon from TS1 to B. Specifically, the carbene carbon is approximately neutral in TS1 and becomes partially cationic in B, such that an anionic D-LEF stabilises the forming charge while a cationic D-LEF destabilises it (Table S308, ESI†).
The electrostatic enhancement from the anionic D-LEF comes with an increased cost for dissociation of the NHC catalyst from the benzoin product in TS4 (in the most extreme case in toluene, 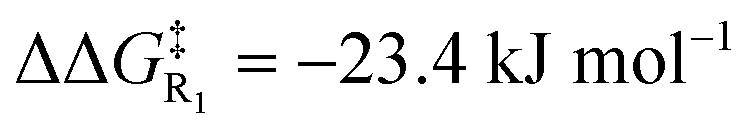 , and
, and 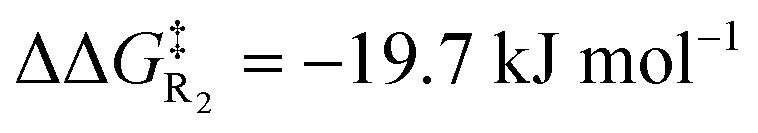 ). This arises because the D-LEF stabilises the zwitterionic form of D, before separating to yield an isolated charge upon release of E. Even this disadvantage, though, yields an overall barrier to release which is still less than the corresponding barrier to initial addition, such that after accounting for the increased population of C, the incorporation of an anionic D-LEF should overall promote the benzoin condensation in a variety of solvents. These various enhancements should be broadly applicable to other NHC-catalysed reactions.
). This arises because the D-LEF stabilises the zwitterionic form of D, before separating to yield an isolated charge upon release of E. Even this disadvantage, though, yields an overall barrier to release which is still less than the corresponding barrier to initial addition, such that after accounting for the increased population of C, the incorporation of an anionic D-LEF should overall promote the benzoin condensation in a variety of solvents. These various enhancements should be broadly applicable to other NHC-catalysed reactions.
Conclusions
We have demonstrated that the catalytic properties of NHCs are, in general, predictably influenced by local electric fields, to an extent that is synthetically relevant even in moderately polar solvents and have applied this understanding to the model benzoin condensation of benzaldehyde under various solvent conditions. Dramatic electrostatic improvements for key transition states were observed, overall promoting the benzoin condensation.Several extensions to this work are clear, and further study in this direction is encouraged. Firstly, given that metal–ligand bonding in transition metal NHC catalysis is highly electrostatic,58,72,73 can the properties of NHCs as transition-metal ligands be similarly modulated in synthetically useful ways by electric fields? Also, given the well-enumerated and 3-dimensional structural diversity of NHC frameworks, incorporating D-LEFs onto these frameworks is highly likely to yield favourable diastereomeric outcomes, and/or favourable rate outcomes for a very broad range of NHC-catalysed organic reactions. For example, during this work we found that the wavelength required for the initial step in the photo-NHC catalysis recently proposed by Hopskinson and co-workers74 could be dramatically influenced by D-LEFs incorporated onto the photoactive benzoyl azolium salt (Fig. S13, ESI†), suggesting that D-LEFs incorporated onto NHCs might represent a general and orthogonal method of modifying the catalytic behaviour of NHCs for both ground- and excited-state chemistry. It is also possible that these electrostatic effects could be highly relevant in the catalytic activity of other high-energy intermediates such as nitrenes and silylenes, and their metal complexes.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
MLC gratefully acknowledges an Australian Research Council Laureate Fellowship (FL170100041), and generous allocations of supercomputing time on the National Facility of the Australian National Computational Infrastructure.Notes and references
- S. Shaik, R. Ramanan, D. Danovich and D. Mandal, Structure and reactivity/selectivity control by oriented-external electric fields, Chem. Soc. Rev., 2018, 47, 5125–5145 RSC
.
- S. Shaik, D. Mandal and R. Ramanan, Oriented electric fields as future smart reagents in chemistry, Nat. Chem., 2016, 8, 1091–1098 CrossRef CAS PubMed
- S. Shaik, D. Danovich, J. Joy, Z. Wang and T. Stuyver, Electric-Field Mediated Chemistry: Uncovering and Exploiting the Potential of (Oriented) Electric Fields to Exert Chemical Catalysis and Reaction Control, J. Am. Chem. Soc., 2020, 142, 12551–12562 CrossRef CAS PubMed
- S. D. Fried, S. Bagchi and S. G. Boxer, Extreme electric fields power catalysis in the active site of ketosteroid isomerase, Science, 2014, 346, 1510–1514 CrossRef CAS PubMed
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Electrostatic Basis for Enzyme Catalysis, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed
-
M. T. Blyth and M. L. Coote, Effects of Electric Fields on Structure and Reactivity, 2021, pp. 119–146 Search PubMed
- C. Q. He, C. C. Lam, P. Yu, Z. Song, M. Chen, Y. Lam, S. Chen and K. N. Houk, Catalytic Effects of Ammonium and Sulfonium Salts and External Electric Fields on Aza-Diels–Alder Reactions, J. Org. Chem., 2020, 85, 2618–2625 CrossRef CAS PubMed
- V. M. Lau, W. C. Pfalzgraff, T. E. Markland and M. W. Kanan, Electrostatic Control of Regioselectivity in Au(I)-Catalyzed Hydroarylation, J. Am. Chem. Soc., 2017, 139, 4035–4041 CrossRef CAS PubMed
- M. P. Frushicheva, S. Mukherjee and A. Warshel, Electrostatic Origin of the Catalytic Effect of a Supramolecular Host Catalyst, J. Phys. Chem. B, 2012, 116, 13353–13360 CrossRef CAS PubMed
- L.-J. Yu and M. L. Coote, Electrostatic Switching between SN1 and SN2 Pathways, J. Phys. Chem. A, 2019, 123, 582–589 CrossRef CAS PubMed
- C. Payne and S. R. Kass, Structural considerations for charge-enhanced Brønsted acid catalysts, J. Phys. Org. Chem., 2020, 33, e4069 CAS
- J. Ma and S. R. Kass, Asymmetric Arylation of 2,2,2-Trifluoroacetophenones Catalyzed by Chiral Electrostatically-Enhanced Phosphoric Acids, Org. Lett., 2018, 20, 2689–2692 CrossRef CAS PubMed
- K. Kang, J. Fuller, A. H. Reath, J. W. Ziller, A. N. Alexandrova and J. Y. Yang, Installation of internal electric fields by non-redox active cations in transition metal complexes, Chem. Sci., 2019, 10, 10135–10142 RSC
- T. Stuyver, R. Ramanan, D. Mallick and S. Shaik, Oriented (Local) Electric Fields Drive the Millionfold Enhancement of the H-Abstraction Catalysis Observed for Synthetic Metalloenzyme Analogues, Angew. Chem., Int. Ed., 2020, 59, 7915–7920 CrossRef CAS PubMed
- M. Akamatsu, N. Sakai and S. Matile, Electric-Field-Assisted Anion–π Catalysis, J. Am. Chem. Soc., 2017, 139, 6558–6561 CrossRef CAS PubMed
- H. M. Aitken and M. L. Coote, Can electrostatic catalysis of Diels–Alder reactions be harnessed with pH-switchable charged functional groups?, Phys. Chem. Chem. Phys., 2018, 20, 10671–10676 RSC
- M. T. Blyth and M. L. Coote, A pH-Switchable Electrostatic Catalyst for the Diels–Alder Reaction: Progress toward Synthetically Viable Electrostatic Catalysis, J. Org. Chem., 2019, 84, 1517–1522 CrossRef PubMed
- M. T. Blyth, B. B. Noble, I. C. Russell and M. L. Coote, Oriented Internal Electrostatic Fields Cooperatively Promote Ground- and Excited-State Reactivity: A Case Study in Photochemical CO2 Capture, J. Am. Chem. Soc., 2020, 142, 606–613 CrossRef CAS PubMed
- N. S. Hill and M. L. Coote, Internal Oriented Electric Fields as a Strategy for Selectively Modifying Photochemical Reactivity, J. Am. Chem. Soc., 2018, 140, 17800–17804 CrossRef CAS PubMed
- G. Gryn’ova and M. L. Coote, Origin and Scope of Long-Range Stabilizing Interactions and Associated SOMO–HOMO Conversion
in Distonic Radical Anions, J. Am. Chem. Soc., 2013, 135, 15392–15403 CrossRef PubMed
- A. C. Aragonès, N. L. Haworth, N. Darwish, S. Ciampi, N. J. Bloomfield, G. G. Wallace, I. Diez-Perez and M. L. Coote, Electrostatic catalysis of a Diels–Alder reaction, Nature, 2016, 531, 88–91 CrossRef PubMed
- M. Belotti, X. Lyu, L. Xu, P. Halat, N. Darwish, D. S. Silvester, C. Goh, E. I. Izgorodina, M. L. Coote and S. Ciampi, Experimental Evidence of Long-Lived Electric Fields of Ionic Liquid Bilayers, J. Am. Chem. Soc., 2021, 143, 17431–17440 CrossRef CAS PubMed
- L. Xu, E. I. Izgorodina and M. L. Coote, Ordered Solvents and Ionic Liquids Can Be Harnessed for Electrostatic Catalysis, J. Am. Chem. Soc., 2020, 142, 12826–12833 CrossRef CAS PubMed
- C. F. Gorin, E. S. Beh, Q. M. Bui, G. R. Dick and M. W. Kanan, Interfacial Electric Field Effects on a Carbene Reaction Catalyzed by Rh Porphyrins, J. Am. Chem. Soc., 2013, 135, 11257–11265 CrossRef CAS PubMed
- A. S. Malkani, J. Li, N. J. Oliveira, M. He, X. Chang, B. Xu and Q. Lu, Understanding the electric and nonelectric field components of the cation effect on the electrochemical CO reduction reaction, Sci. Adv., 2020, 6, eabd2569 CrossRef CAS PubMed
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485–496 CrossRef CAS PubMed
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, N-Heterocyclic Carbenes in Materials Chemistry, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed
- A. Doddi, M. Peters and M. Tamm, N-Heterocyclic Carbene Adducts of Main Group Elements and Their Use as Ligands in Transition Metal Chemistry, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed
- Z. Pei, Q. Qiao, C. Gong, D. Wei and M. L. Coote, Electrostatic effects in N-heterocyclic carbene catalysis: revealing the nature of catalysed decarboxylation, Phys. Chem. Chem. Phys., 2021, 23, 24627–24633 RSC
- R. Maji and S. E. Wheeler, Importance of Electrostatic Effects in the Stereoselectivity of NHC-Catalyzed Kinetic Resolutions, J. Am. Chem. Soc., 2017, 139, 12441–12449 CrossRef CAS PubMed
- Y. Ma, A. A. Hussein and Z. Wang, Boosting Palladium-Catalyzed Aryl–Nitro Bond Activation Reaction by Understanding the Electronic, Electrostatic, and Polarization Effect: A Computational Study from a Basic Understanding to Ligand Design, J. Org. Chem., 2022, 87, 531–539 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC
- D. Gerbig and D. Ley, Computational methods for contemporary carbene chemistry, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 242–272 CAS
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS PubMed
- L. R. Domingo and P. Pérez, The nucleophilicity N index in organic chemistry, Org. Biomol. Chem., 2011, 9, 7168 RSC
- R. G. Parr, L. v Szentpály and S. Liu, Electrophilicity Index, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS
- A. R. Jupp, T. C. Johnstone and D. W. Stephan, Improving the Global Electrophilicity Index (GEI) as a Measure of Lewis Acidity, Inorg. Chem., 2018, 57, 14764–14771 CrossRef CAS PubMed
- R. G. Parr and R. G. Pearson, Absolute hardness: companion parameter to absolute electronegativity, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed
- A. J. Arduengo, J. R. Goerlich and W. J. Marshall, A stable diaminocarbene, J. Am. Chem. Soc., 1995, 117, 11027–11028 CrossRef CAS
- A. J. Arduengo, R. L. Harlow and M. Kline, A stable crystalline carbene, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS
- C. A. Gaggioli, G. Bistoni, G. Ciancaleoni, F. Tarantelli, L. Belpassi and P. Belanzoni, Modulating the Bonding Properties of N-Heterocyclic Carbenes (NHCs): A Systematic Charge-Displacement Analysis, Chem. – Eur. J., 2017, 23, 7558–7569 CrossRef CAS PubMed
- L. Morán-González, J. R.-G. Pedregal, M. Besora and F. Maseras, Understanding the Binding Properties of N-heterocyclic Carbenes through BDE Matrix App, Eur. J. Inorg. Chem., 2022, e202100932 Search PubMed
- D. G. Gusev, Electronic
and Steric Parameters of 76 N-Heterocyclic Carbenes in Ni(CO) 3 (NHC), Organometallics, 2009, 28, 6458–6461 CrossRef CAS
-
P. Pérez, L. R. Domingo, A. Aizman and R. Contreras, Theoretical and Computational Chemistry, Elsevier, 2007, vol. 19, pp. 139–201 Search PubMed
- S. Gronert, J. R. Keeffe and R. A. More O’Ferrall, Stabilities of Carbenes: Independent Measures for Singlets and Triplets, J. Am. Chem. Soc., 2011, 133, 3381–3389 CrossRef CAS PubMed
- I. Alkorta, M. M. Montero-Campillo and J. Elguero, Remote modulation of singlet–triplet gaps in carbenes, Chem. Phys. Lett., 2018, 694, 48–52 CrossRef CAS
- J. P. Dürholt, B. F. Jahromi and R. Schmid, Tuning the Electric Field Response of MOFs by Rotatable Dipolar Linkers, ACS Cent. Sci., 2019, 5, 1440–1448 CrossRef PubMed
- M. Z. Kassaee, F. A. Shakib, M. R. Momeni, M. Ghambarian and S. M. Musavi, Carbenes with Reduced Heteroatom Stabilization: A Computational Approach, J. Org. Chem., 2010, 75, 2539–2545 CrossRef CAS PubMed
- B. Borthakur, B. Silvi, R. D. Dewhurst and A. K. Phukan, Theoretical strategies toward stabilization of singlet remote N-heterocyclic carbenes, J. Comput. Chem., 2016, 37, 1484–1490 CrossRef CAS PubMed
- M. Koohi, M. Z. Kassaee, B. N. Haerizade, M. Ghavami and S. Ashenagar, Substituent effects on cyclonona-3,5,7-trienylidenes: a quest for stable carbenes at density functional theory level, J. Phys. Org. Chem., 2015, 28, 514–526 CrossRef CAS
- M.-J. Cheng and C.-H. Hu, Computational study on the stability of imidazol-2-ylidenes and imidazolin-2-ylidenes, Chem. Phys. Lett., 2001, 349, 477–482 CrossRef CAS
- J. C. Bernhammer, G. Frison and H. V. Huynh, Electronic Structure Trends in N-Heterocyclic Carbenes (NHCs) with Varying Number of Nitrogen Atoms and NHC-Transition-Metal Bond Properties, Chem. – Eur. J., 2013, 19, 12892–12905 CrossRef CAS PubMed
- A. A. Tukov, A. T. Normand and M. S. Nechaev, N-heterocyclic carbenes bearing two, one and no nitrogen atoms at the ylidene carbon: insight from theoretical calculations, Dalton Trans., 2009, 7015 RSC
- A. Wessels, M. Klussmann, M. Breugst, N. E. Schlörer and A. Berkessel, Formation of Breslow Intermediates from N-Heterocyclic Carbenes and Aldehydes Involves Autocatalysis by the Breslow Intermediate, and a Hemiacetal, Angew. Chem., Int. Ed., 2022, 61, e202117682 CAS
- K. M. Melancon and T. R. Cundari, Computational investigations of NHC-backbone configurations for applications in organocatalytic umpolung reactions, Org. Biomol. Chem., 2020, 18, 7437–7447 RSC
- K. J. Hawkes and B. F. Yates, The Mechanism of the Stetter Reaction – A DFT Study, Eur. J. Org. Chem., 2008, 5563–5570 CrossRef CAS
- Y. He and Y. Xue, Theoretical Investigations on the Mechanism of Benzoin Condensation Catalyzed by Pyrido[1,2-a]-2-ethyl[1,2,4]triazol-3-ylidene, J. Phys. Chem. A, 2011, 115, 1408–1417 CrossRef CAS PubMed
- Y. Reddi and R. B. Sunoj, Origin of Stereoselectivity in a Chiral N-Heterocyclic Carbene-Catalyzed Desymmetrization of Substituted Cyclohexyl 1,3-Diketones, Org. Lett., 2012, 14, 2810–2813 CrossRef CAS PubMed
- P. Verma, P. A. Patni and R. B. Sunoj, Mechanistic Insights on N-Heterocyclic Carbene-Catalyzed Annulations: The Role of Base-Assisted Proton Transfers, J. Org. Chem., 2011, 76, 5606–5613 CrossRef CAS PubMed
- M.-H. Hsieh, G.-T. Huang and J.-S. K. Yu, Can the Radical Channel Contribute to the Catalytic Cycle of N-Heterocyclic Carbene in Benzoin Condensation?, J. Org. Chem., 2018, 83, 15202–15209 CrossRef CAS PubMed
- S. Gehrke and O. Hollóczki, Are There Carbenes in N-Heterocyclic Carbene Organocatalysis?, Angew. Chem., Int. Ed., 2017, 56, 16395–16398 CrossRef CAS PubMed
- L. R. Domingo, M. J. Aurell and M. Arnó, Understanding the mechanism of the N-heterocyclic carbene-catalyzed ring-expansion of 4-formyl-β-lactams to succinimide derivatives, Tetrahedron, 2009, 65, 3432–3440 CrossRef CAS
- W. Zhang, Y. Wang, D. Wei, M. Tang and X. Zhu, A DFT study on NHC-catalyzed intramolecular
aldehyde–ketone crossed-benzoin reaction: mechanism, regioselectivity, stereoselectivity, and role of NHC, Org. Biomol. Chem., 2016, 14, 6577–6590 RSC
- S. M. Langdon, M. M. D. Wilde, K. Thai and M. Gravel, Chemoselective N-Heterocyclic Carbene-Catalyzed Cross-Benzoin Reactions: Importance of the Fused Ring in Triazolium Salts, J. Am. Chem. Soc., 2014, 136, 7539–7542 CrossRef CAS PubMed
- T. Liu, S.-M. Han, L.-L. Han, L. Wang, X.-Y. Cui, C.-Y. Du and S. Bi, Theoretical investigation on the chemoselective N-heterocyclic carbene-catalyzed cross-benzoin reactions, Org. Biomol. Chem., 2015, 13, 3654–3661 RSC
- M. A. Celik, C. Dash, V. A. K. Adiraju, A. Das, M. Yousufuddin, G. Frenking and H. V. R. Dias, End-On and Side-On π-Acid Ligand Adducts of Gold(I): Carbonyl, Cyanide, Isocyanide, and Cyclooctyne Gold(I) Complexes Supported by N-Heterocyclic Carbenes and Phosphines, Inorg. Chem., 2013, 52, 729–742 CrossRef CAS PubMed
- C. Boehme and G. Frenking, N-Heterocyclic Carbene, Silylene, and Germylene Complexes of MCl (M = Cu, Ag, Au). A Theoretical Study, Organometallics, 1998, 17, 5801–5809 CrossRef CAS
- A. Mavroskoufis, K. Rajes, P. Golz, A. Agrawal, V. Ruß, J. P. Götze and M. N. Hopkinson, N-Heterocyclic Carbene Catalyzed Photoenolization/Diels–Alder Reaction of Acid Fluorides, Angew. Chem., Int. Ed., 2020, 59, 3190–3194 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: All computational data, benchmarks, energies and geometries. See DOI: https://doi.org/10.1039/d2cp04507a |
| This journal is © the Owner Societies 2023 |