DOI:
10.1039/D2TB00325B
(Paper)
J. Mater. Chem. B, 2022,
10, 6644-6654
Design of molecularly imprinted hydrogels with thermoresponsive drug binding sites†
Received
12th February 2022
, Accepted 23rd April 2022
First published on 25th April 2022
Abstract
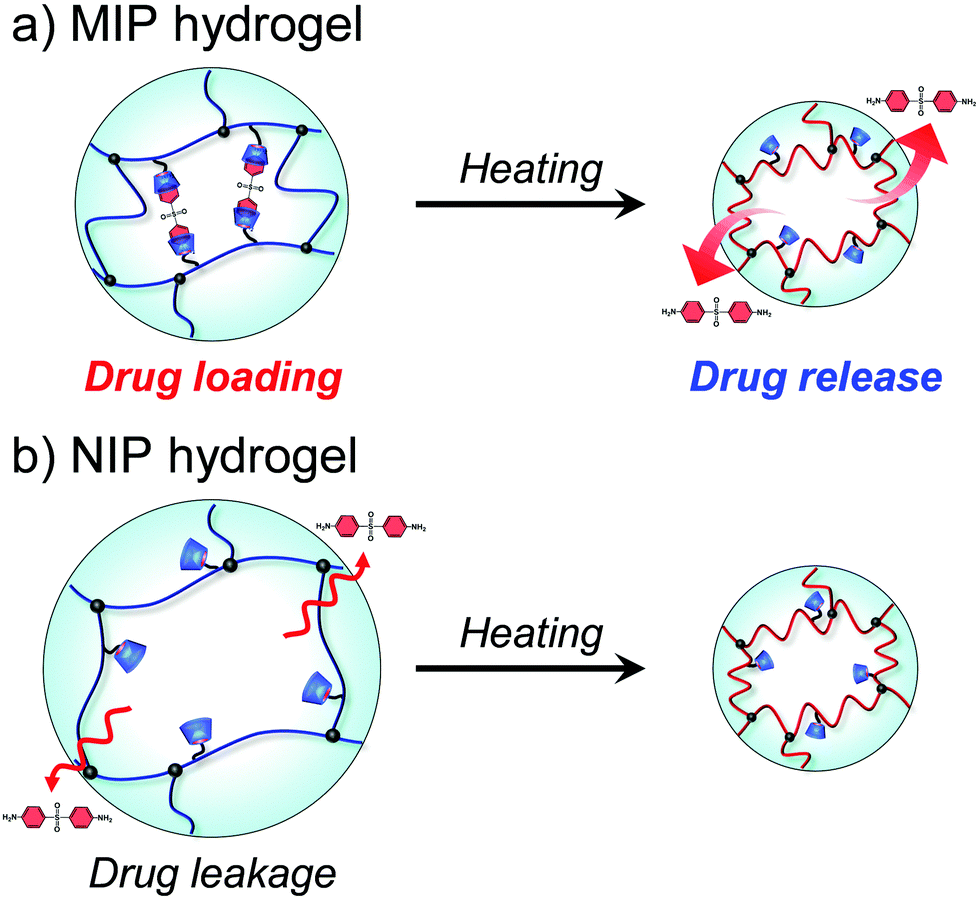
Drug delivery systems (DDS) regulate the spatiotemporal distribution of drugs in vivo to maximize efficacy and minimize side effects. Stimuli-responsive hydrogels, which exhibit a drastic change in volume in response to external stimuli such as temperature and pH, have attracted considerable interest as drug reservoirs for self-regulating DDS, as stimuli-responsive changes in the network size can regulate drug diffusion. However, such hydrogels have the disadvantage of leaking drugs even in the absence of stimulation. Proteins such as hemoglobin have dynamic molecular binding sites that modify their binding capacities by their conformational changes induced when an effector molecule binds to allosteric sites. Such dynamic binding sites are useful for loading drugs into reservoirs because their conformational changes can be used to control drug loading and release. In this study, we prepared thermoresponsive hydrogels with a controlled drug binding capacity to design drug reservoirs capable of both suppressing drug leakage below the transition temperature and accelerating drug release above it. Dynamic molecular binding sites were created by molecular imprinting that used 4,4′-diaminodiphenyl sulfone (dapsone) as the model drug, β-cyclodextrin (CD) as the ligand, and N-isopropylacrylamide as the primary monomer. The molecularly imprinted (MIP) and nonimprinted (NIP) hydrogels with CD ligands, as well as the poly(N-isopropylacrylamide) (PNIPAAm) hydrogels without CD ligands, drastically shrunk above their transition temperature because of the PNIPAAm major chains changing conformation from a hydrophilic random coil to a hydrophobic globule as temperature increased. Because the MIP hydrogel has dynamic molecular binding sites, it absorbs a larger amount of dapsone than the NIP hydrogels in an aqueous solution below the transition temperature. The amount of dapsone adsorbed into the MIP hydrogel significantly decreased with increasing temperatures above 37 °C, despite the fact that the hydrophobic interaction between the polymer chains and dapsone became strong. The decrease in dapsone adsorption capability of the MIP hydrogel is due to a conformational change from a swollen to a shrunken state as temperature increases. The MIP hydrogel suppressed drug leakage below its transition temperature due to the high binding capacity of dynamic binding sites, but accelerated the drug release above its transition temperature due to the collapse of dynamic molecular binding sites, in contrast to the drug release behavior of general PNIPAAm-based hydrogels. Thus, the thermoresponsive MIP hydrogels with dynamic molecular binding sites regulated drug release in response to a change in temperature.
Introduction
Drug delivery systems (DDS) can enhance efficacy and reduce side effects by spatiotemporally regulating the distribution of drugs in the body. There have been many reports on DDS drug carriers and reservoirs, such as liposomes, polymer micelles, nanogels, and hydrogels. In particular, stimuli-responsive hydrogels, which undergo a reversible volume change in response to external stimuli such as temperature and pH, have greatly contributed to the development of the field of DDS.1–10 For example, thermoresponsive hydrogels composed of poly(N-isopropylacrylamide) (PNIPAAm) have been described as DDS reservoirs capable of regulating drug release in response to temperature changes.1,3–6 Due to the lower critical solution temperature (LCST) of PNIPAAm at 32 °C, it is soluble in water below the LCST but insoluble above 32 °C. Thus, thermoresponsive hydrogels prepared with PNIPAAm undergo drastic volume changes in response to changes in temperature, enabling the drug release to be regulated by the diffusion-determined mechanism. These stimuli-responsive hydrogels have a few disadvantages, including drug leakage from the hydrogel even in the absence of stimuli, as hydrogel networks cannot entirely suppress drug diffusion.11,12 This shows that diffusion-determined drug release is limited in self-regulated drug delivery using stimuli-responsive hydrogels. In addition, drugs need to be loaded into hydrogels by strong interactions to suppress their leakage. Therefore, self-regulated drug delivery requires the strategic design of stimuli-responsive hydrogels that efficiently suppress drug leakage in the absence of stimuli such as pH and temperature, yet rapidly release drugs in response to stimuli. This requires fabricating dynamic molecular binding sites with variable binding capacities within stimuli-responsive hydrogels.
Proteins have dynamic binding sites to which a specific molecule is allowed to bind by its complemental interactions. The binding sites are formed within well-defined structures of proteins with folded conformations such as α-helix and β-sheet.13 Folded conformations and structures formed by disulfide bonds, hydrogen bonds, and others are important for proteins to perform their unique functions, such as molecular recognition and enzymatic activities. Protein conformational changes in response to environmental conditions result in either positive or negative changes in their functions. Allosteric regulation of proteins occurs when an effector molecule binds to allosteric sites, inducing conformational changes in proteins and a drastic change in the binding constant.14–16 For example, hemoglobin, an important protein for carrying oxygen, detects and retains oxygen molecules in the presence of excess oxygen.15,16 However, hemoglobin releases oxygen when it undergoes conformational changes in response to a lack of oxygens. These show that proteins regulate the binding capacity of their molecular binding sites by conformational changes in their highly ordered structure. For achieving self-regulated drug delivery, it is useful to create such dynamic binding sites within hydrogels based on bioinspired designs.
Molecular imprinting is a useful method for creating molecular binding sites within synthetic materials by optimizing the position of ligands complementary to a target molecule.17–24 In standard molecular imprinting, after noncovalent interactions arrange functional monomers as ligands around template molecules, they are copolymerized with a large number of cross-linkers, and the template molecule is extracted from the resultant network. Molecular imprinting enables the arrangement of ligands in optimal binding positions for the target molecule, followed by the formation of molecular binding sites. Molecular imprinting has been widely used in preparing synthetic materials for a number of purposes, including separation, concentration, sensing, and detecting. Additionally, a few researchers applied molecular imprinting to design drug reservoirs capable of effectively loading a drug into molecular binding sites.25–31 Drug leakage from the molecularly imprinted hydrogels was successfully suppressed because of the strong binding of the drug to the molecular binding sites. Although such strong bindings can suppress drug leakage from the reservoir, it has been difficult to accelerate drug release in response to stimuli. Thus, strategic design of dynamic binding sites with a variable binding capacity is required to develop smart reservoirs capable of suppressing drug leakage and accelerating release in response to environmental changes such as temperature and pH.
In contrast to conventional molecular imprinting, which uses a large amount of cross-linker, we have prepared molecularly stimuli-responsive hydrogels that change their volume by recognizing a target molecule via molecular imprinting that uses a minute amount of cross-linker and molecular complexes as dynamic cross-links.32–39 For example, tumor-marker-responsive hydrogels that shrank according to the concentration of a tumor marker glycoprotein were prepared by biomolecular imprinting using antibodies and lectins as ligands.32,33 Molecularly imprinted hydrogels with β-cyclodextrin (CD) as a ligand also shrank in response to the target bisphenol A (BPA), which has a high possibility of being an endocrine disrupting chemical, because the formation of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 sandwich-like molecular complex between CD and BPA increased the crosslinking density.35 Furthermore, by simulating dynamic molecular binding sites regulated by the conformational change of proteins, we developed dynamic molecular binding sites within polypeptide hydrogels that changed conformation from random coil to α-helix in response to a change in pH.39 While molecularly imprinted polypeptide hydrogels containing CD ligands and poly(L-lysin) (PLL) major chains have a high binding capacity with the random coil at neutral pH, when the PLL chains undergo a conformational change to a α-helix in response to a basic condition, their binding capacity drastically decreased. Additionally, a few researchers prepared hydrogels with molecularly imprinting using thermoresponsive PNIPAAm as the primary chain.40,41 These results show that when molecular imprinting is combined with stimuli-responsive hydrogels, dynamic molecular binding sites can be formed for effective on–off regulation of drug release.
:1 sandwich-like molecular complex between CD and BPA increased the crosslinking density.35 Furthermore, by simulating dynamic molecular binding sites regulated by the conformational change of proteins, we developed dynamic molecular binding sites within polypeptide hydrogels that changed conformation from random coil to α-helix in response to a change in pH.39 While molecularly imprinted polypeptide hydrogels containing CD ligands and poly(L-lysin) (PLL) major chains have a high binding capacity with the random coil at neutral pH, when the PLL chains undergo a conformational change to a α-helix in response to a basic condition, their binding capacity drastically decreased. Additionally, a few researchers prepared hydrogels with molecularly imprinting using thermoresponsive PNIPAAm as the primary chain.40,41 These results show that when molecular imprinting is combined with stimuli-responsive hydrogels, dynamic molecular binding sites can be formed for effective on–off regulation of drug release.
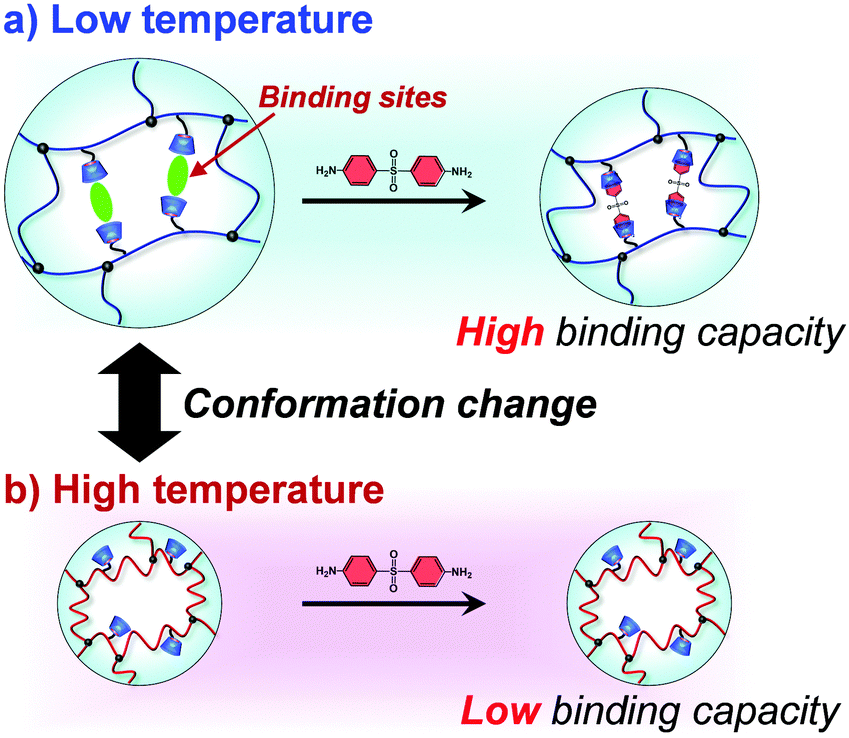
In this study, we designed molecularly imprinted hydrogels that can regulate the binding capacity of a model drug by changing their conformation from a random coil to a globule in response to temperature changes (Fig. 1). The molecularly imprinted (MIP) hydrogels were prepared using the model drug, 4,4′-diaminodiphenyl sulfone (dapsone) as the template molecule, CD as the ligand, and PNIPAAm as the primary chain. The effects of temperature on the conformational change and drug adsorption of the resultant MIP hydrogels were investigated. After loading dapsone into MIP and NIP hydrogels, their release behaviour in response to a temperature change was investigated.
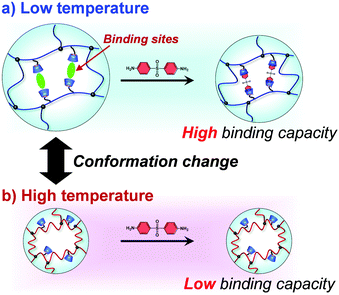 |
| | Fig. 1 Schematic illustration of the regulation of the drug-binding capacity by a conformational change of a thermoresponsive MIP hydrogel with dynamic molecular binding sites at low (a) and high (b) temperatures. | |
Materials and methods
Materials
p-Toluenesulfonyl chloride, p-toluenesulfonic acid monohydrate, dichloromethane, β-cyclodextrin (β-CD), sodium hydroxide (NaOH), hydrochloric acid (HCl), sodium azide (NaN3), triphenylphosphine, N,N-dimethylformamide (DMF), sodium hydrogen carbonate (NaHCO3), benzene, hexane, N-isopropylacrylamide (NIPAAm), N,N′-methylenebisacrylamide (MBAA), ammonium persulfate (APS), and N,N,N′,N′-tetramethylethylenediamine (TEMED) were supplied by FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Additionally, Fujifilm Wako Pure Chemical Corporation supplied 4,4′-diaminodiphenyl sulfone (dapsone), an antibiotic commonly used as an antimycobacterial to treat mycobacterial infections, as a model drug. β-CD and NIPAAm were used after being recrystallized. All aqueous solutions were prepared with ultrapure water (Milli-Q, 18.2 MΩ cm). Other solvents and reagents of analytical grade were bought from commercial sources and were used without further purification.
Synthesis of acryloyl-modified β-CD
Acryloyl-modified β-CD was synthesized via a reaction process with five steps, as shown in Scheme S1 (ESI†). p-Toluenesulfonyl chloride (40.1 g, 210 mmol) and p-toluenesulfonic acid monohydrate (20.1 g, 105 mmol) were dispersed in 250 mL of dichloromethane. The mixture was stirred overnight, and then the unreacted p-toluenesulfonyl chloride was removed by filtration. The filtrate was evaporated, and then the residue was recrystallized from isopropyl ether to yield p-toluenesulfonic anhydride (Ts2O) as a white solid; yield, 22.7 g (66%).
β-CD (27.6 g, 24.4 mmol) and Ts2O (11.6 g, 35.7 mmol) were dispersed in 240 mL of water, and then the suspension was stirred for 2 h. After 10 minutes, 120 mL of NaOH solution (2.5 M) was added to the mixture, and the unreacted Ts2O was filtered off. The filtrate was neutralized by the addition of HCl, affording 6-O-monotosyl-6-deoxy-β-cyclodextrin (TsO-CD), which was collected after cooling at 4 °C for overnight: the yield was 3.98 g (13%).
TsO-CD (3.80 g, 2.95 mmol) and NaN3 (2.49 g, 38.3 mmol) were dissolved in 55 mL of water, and then stirred for 5 h at 80 °C, followed by pouring into acetone to precipitate the 6-deoxy-6-azide-β-cyclodextrin (CD-N3) as a white powder. The resulting CD-N3 was dried in vacuo; the yield was 3.20 g (94%).
CD-N3 (2.50 g, 2.16 mmol) and triphenylphosphine (0.65 g, 2.48 mmol) were dissolved in 4.3 mL of N,N-dimethylformamide (DMF), and then the mixture was stirred overnight. 5.43 mL of water and 5.00 mL of DMF were added to the mixture, and then stirred for 2 h at 90 °C, followed by pouring into acetone to precipitate the 6-deoxy-6-amino-β-cyclodextrin (CD-NH2) as a white powder. The resulting CD-NH2 was dried in vacuo; the yield was 2.34 g (96%).
CD-NH2 (1.01 g, 0.89 mmol) was dissolved in 15.0 mL of NaHCO3 aq. (pH 11). In an ice bath, acryloyl chloride (4.03 g, 44.2 mmol) was added to the solution of CD-NH2. The solution was stirred overnight, and then the mixture was poured into acetone. The precipitate was dried in vacuo to yield acryloyl-6-amino-6-deoxy-β-cyclodextrin (acryloyl-CD) as a white powder; the yield was 0.90 g (85%).
The chemical structures of all products were confirmed by proton nuclear magnetic resonance spectroscopy (1H NMR) and a Fourier-transform infrared spectrometer (FT-IR) (Spectrum 100: PerkinElmer, Inc., MA, USA) (Fig. S1–S5, ESI†). 1H NMR spectra were recorded using a GSX-400 NMR instrument (JEOL, Tokyo, Japan) operating at 400 MHz with deuterated dimethyl sulfoxide (DMSO-d6) and D2O as a solvent and tetramethylsilane (TMS) as an internal standard.
Preparation of molecularly imprinted hydrogels with CD ligands
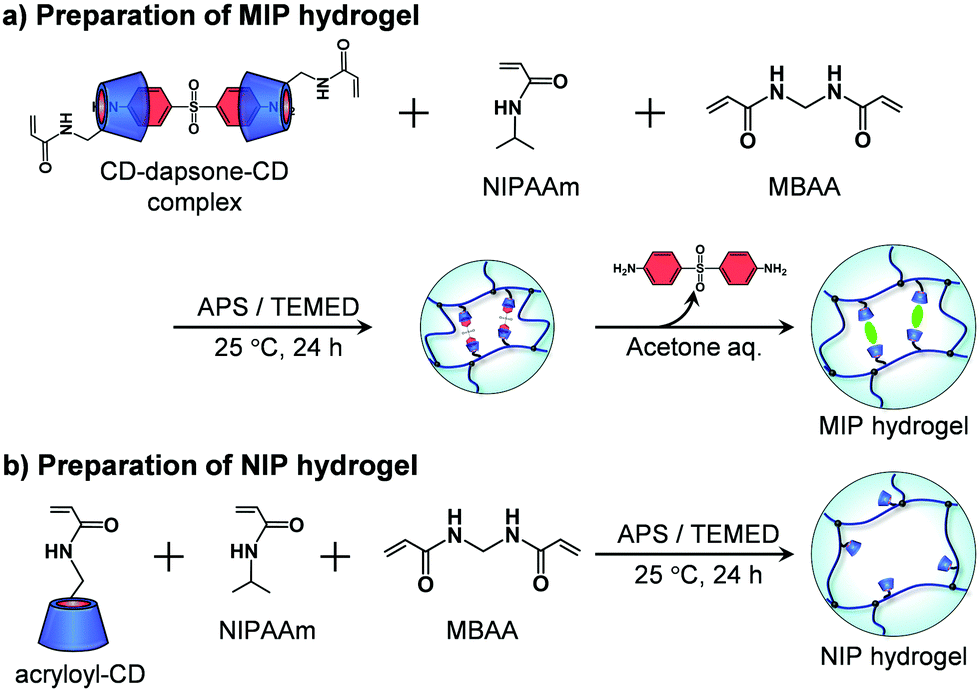
A MIP hydrogel was synthesized by molecular imprinting using 4,4′-diaminodiphenyl sulfone (dapsone) as the template drug molecule and CD as the ligand (Fig. 2a). Acryloyl-CD (5.94 mg, 5.00 μmol), dapsone (0.740 mg, 2.50 μmol), NIPAAm (11.3 mg, 100 μmol), and MBAA (0.509 mg, 3.30 μmol) were dissolved in 134 μL of water and then incubated at room temperature for one day to form sandwich-like CD-dapsone-CD complexes. After adding 33 μL of 0.8 M aqueous TEMED solution and 33 μL of 0.1 M aqueous APS solution as redox initiators to the resulting monomer solution, the solution was injected into a cylindrical glass mold with an inner diameter of 5 mm and an inner depth of 5 mm, and polymerization was performed at a temperature of 25 °C for 24 h. After removing the hydrogels from the glass mold, they were immersed in a 30% (v/v) aqueous acetone solution for three days to remove template dapsone and unreacted monomers from their networks. The hydrogels were then immersed in water to remove acetone until their swelling ratio reached equilibrium in water.
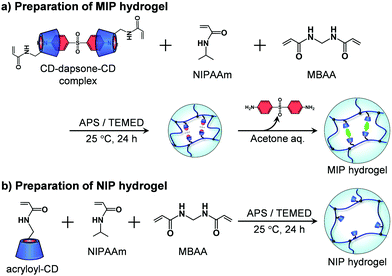 |
| | Fig. 2 Preparation of the MIP and NIP hydrogels by the copolymerization of acryloyl-CD, NIPAAm and MBAA. | |
The preparation method was also used to produce an NIP hydrogel without the use of the template dapsone (Fig. 2b). Acryloyl-CD (5.94 mg, 5.00 μmol), NIPAAm (11.3 mg, 100 μmol), and MBAA (0.509 mg, 3.30 μmol) were dissolved in 134 μL of water. After adding 33 μL of 0.8 M aqueous TEMED solution and 33 μL of 0.1 M aqueous APS solution as redox initiators to the resultant monomer solution, the solution was injected into a glass mold with an inner diameter of 5 mm, and polymerization was performed at a temperature of 25 °C for 24 h. After removing the hydrogels from the glass mold, they were immersed in a 30% aqueous acetone solution for three days to remove unreacted monomers. The hydrogels were then immersed in water to remove acetone until their swelling ratio reached equilibrium in water. Additionally, a PNIPAAm hydrogel was prepared by copolymerizing NIPAAm and MBAA without using acryloyl-CD or dapsone as a template.
FT-IR measurements
The chemical structures of the MIP hydrogels were examined by the KBr method with a Fourier-transform infrared spectrometer (FT-IR) (Spectrum 100: PerkinElmer, Inc., MA, USA) after drying the hydrogel at 70 °C. All the spectra represent an average of 32 scans taken in the wavenumber range of 4000–800 cm−1.
Transmittance measurements
The thermoresponsive behavior of the MIP, NIP, and PNIPAAm hydrogels was evaluated by measuring their transmittance as a function of temperature. First, their hydrogels were prepared using molds composed of quartz glass. The transmittance of the hydrogels at various temperatures was measured at 650 nm with a UV-visible spectrometer (UV-1550: Shimadzu, Kyoto, Japan).
Measurements of the swelling ratio of hydrogels
The MIP, NIP, and PNIPAAm hydrogels were kept immersed in water until equilibrium was reached at various temperatures. Afterward, the hydrogels were transferred and kept immersed in water at various temperatures. The swelling ratio (V/V25) of hydrogels was determined from their diameters in water by eqn (1):| | 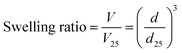 | (1) |
where d is the diameter of the hydrogels swollen in water at each temperature, and d25 is the diameter of the hydrogels swollen in water at 25 °C. The diameters of hydrogels were measured with a microscope (BX51, Olympus, Tokyo, Japan) equipped with a digital camera (DP-21, Olympus, Tokyo, Japan).
Similarly, the relative swelling ratio of the MIP and NIP hydrogels in an aqueous dapsone solution was measured as a function of the dapsone concentration. The MIP and NIP hydrogels were kept immersed in an aqueous dapsone solution until equilibrium was reached at 25 °C. The relative swelling ratio (V/V0) of hydrogels was determined from their diameters in the aqueous dapsone solution by eqn (2):
| |  | (2) |
where
d is the diameter of the hydrogels swollen in an aqueous dapsone solution at 25 °C and
d0 is the diameters of hydrogels swollen in water without dapsone at 25 °C. The diameters of hydrogels were measured with a microscope (BX51, Olympus, Tokyo, Japan) equipped with a digital camera (DP-21, Olympus, Tokyo, Japan). The relative swelling ratio of the hydrogels shows their shrinkage in response to dapsone.
Measurements of the cross-linking density of hydrogels
Compression modulus tests were performed on the MIP, NIP, and PNIPAAm hydrogels using a tensile tester (Shimadzu Co., Ltd; EZ Test/CE) for the measurements of their cross-linking density in an aqueous dapsone solution with a dapsone concentration of 0.035 mmol L−1. The swollen MIP, NIP, and PNIPAAm hydrogels were compressed by the crosshead of the apparatus, and then the relationship between the compressive stress and strain of the hydrogels was recorded. The compressive modulus can be obtained by eqn (3) from the compressive stress and strain of the hydrogels. Additionally, the cross-linking density of the hydrogels can be calculated by eqn (4):32,35where σ is the compressive stress (Pa), G is the compressive modulus (Pa), R is gas constant, T is the absolute temperature (K), α is the ratio of the thickness of the hydrogel before and after the compression, νe is the effective cross-linking density (mol L−1), ϕ is the volume fraction of the polymer during network formation, which is obtained by the swelling measurements.
Measurements of dapsone adsorption into hydrogels
Adsorptions of dapsone as a model drug into the MIP, NIP and PNIPAAm hydrogels were estimated by measuring the concentration of dapsone after the immersion of the hydrogels in water containing dapsone at a concentration of 0.035 mM at various temperatures. The absorbances of the aqueous dapsone solution were measured in the wavelength range of 225–350 nm using UV-vis spectrometer (UV-1550: Shimadzu, Kyoto, Japan) against a blank containing pure water at each temperature. The amounts of dapsone adsorbed into the hydrogels were determined using eqn (5):| | 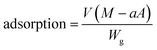 | (5) |
where V is the volume of an aqueous dapsone solution (L), M is the initial dapsone concentration (mg L−1), a is the slope of the standard curve of the aqueous dapsone solution, A is the absorbance of the supernatant of the aqueous dapsone solution, and Wg is the weight of the dried gel (mg).
Release of dapsone from dapsone-loaded hydrogels
Dapsone as a model drug was loaded into the MIP and NIP hydrogels by immersing the hydrogels in an aqueous dapsone solution with a dapsone concentration of 0.035 mM. Then, the resulting hydrogels in which dapsone was loaded were rinsed to wash away dapsone physically adsorbed on the hydrogel surface until dapsone leaked out of the hydrogels in water at 25 °C was not detected with a UV-vis spectrometer (UV-1550, Shimadzu, Kyoto, Japan). The dapsone-loaded hydrogels were transferred and kept immersed in water at a temperature of 40 °C. The amount of dapsone released from the hydrogels was determined by measuring the dapsone concentration after the immersion of the hydrogel in water at various temperatures. The absorbances of the aqueous dapsone solution at each temperature were measured in the wavelength range of 225–350 nm using a UV-vis spectrometer (UV-1550, Shimadzu, Kyoto, Japan) against a blank containing water at each temperature. The release rate of dapsone from the hydrogels was calculated by eqn (6).| | 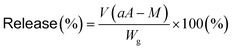 | (6) |
where V is the volume of water (L), a is the slope of the standard curve of the aqueous dapsone solution, A is the absorbance of the supernatant of the aqueous dapsone solution, M is the initial dapsone concentration (mg L−1), and Wg is the weight of the dried gel (mg).
Results and discussion
Preparation of molecularly imprinted hydrogels with CD ligands
In general, standard molecular imprinting uses ligands such as carboxyl and amino groups. In addition, a few researchers have demonstrated that CDs are effective ligands for molecular binding sites in molecular imprinting.22,23,28 We have designed molecularly imprinted hydrogels using lectins, antibodies, and CDs as ligands with high selectivity and strong affinity for target molecules.35–37,39 In this study, molecular imprinting was combined with thermoresponsive polymers that underwent conformational changes from an expanded random coil to a collapsed globule as the temperature increased. First, acryloyl-CD was synthesized by introducing an acryloyl group to the hydroxy group of β-CD, as shown in Scheme S1 (ESI†). As β-CD is a macrocyclic ring composed of seven glucose subunits, it has seven primary and fourteen secondary hydroxyl groups. If two or more acryloyl groups were introduced to a β-CD molecule, the resulting acryloyl-CD works as a chemical cross-linker during the preparation of the molecularly imprinted hydrogels. This results in the formation of rigid binding sites with fixed binding capacity. As a result, we tuned the synthetic condition for introducing only an acryloyl group into a β-CD molecule. As shown in Fig. S1–S5 (ESI†), the acryloyl-CD with an acryloyl group was successfully synthesized by the procedure described in Scheme S1 (ESI†). The 1H NMR analysis showed a molar ratio of 0.98 for the acryloyl group introduced into an acryloyl-CD molecule. Following the forming of sandwich-like CD-dapsone-CD complexes, MIP hydrogels were prepared using the resultant acryloyl-CD as a ligand monomer by copolymerizing it with NIPAAm and MBAA. In our previous study, we demonstrated that molecularly imprinted poly(acrylamide) hydrogels prepared with 5 mol% CD ligands have a high binding capacity. As a result, the concentration of acryloyl-CD in the feed used for preparing the MIP and NIP hydrogels was set at 5 mol% in this study. The introduction of CD into the MIP hydrogel was confirmed by FT-IR measurements. The FT-IR spectra of the PNIPAAm hydrogel, β-CD, and MIP hydrogel after freeze-drying are shown in Fig. S6 (ESI†). The FT-IR spectra of β-CD and the MIP hydrogels show peaks at 1050 cm−1, which are assigned to the antisymmetric glycosidic vibration, but not those of PNIPAAm hydrogels. Additionally, the broad band at 3400 cm−1 in the FT-IR spectra of β-CD and MIP hydrogel is caused by O–H vibrations. Thus, FT-IR measurements of the MIP hydrogel demonstrate that the networks contain CDs as ligands.
Thermoresponsive behaviour of MIP and NIP hydrogels
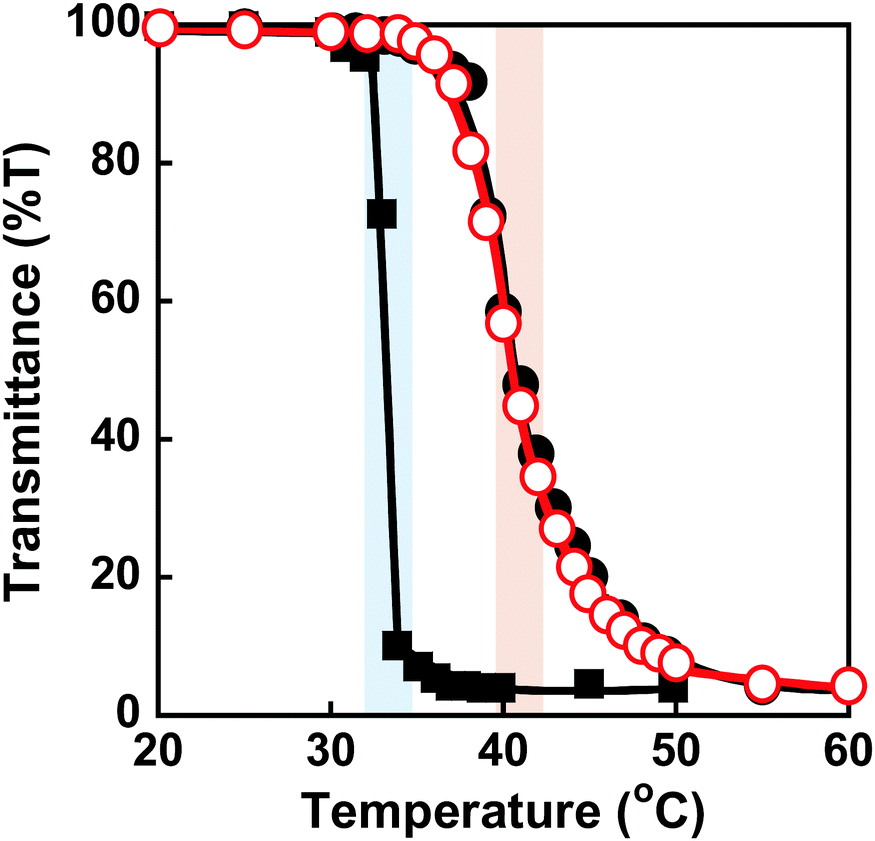
PNIPAAm is a thermoresponsive polymer with a clear LCST at 32 °C.42 PNIPAAm-based hydrogels undergo a drastic change in volume in response to temperature changes.1,3–6 Their drastic change from a swollen to a shrunken state as the temperature increases is known as the volume phase transition of the hydrogels. This is induced by a drastic change of PNIPAAm chains from a hydrophilic random coil to a hydrophobic globule. We evaluated the thermoresponsive behaviour of the MIP and NIP hydrogels by measuring their transmittance and swelling ratio in water at various temperatures. The transmittance of the MIP, NIP, and PNIPAAm hydrogels in water as a function of temperature is shown in Fig. 3. At 32 °C, the transmittance of the PNIPAAm hydrogel sharply decreased with increasing temperature. The transmittance of the MIP and NIP hydrogels decreased at around 37 °C, which is close to body temperature. The decrease in transmittance of their hydrogels is caused by a drastic change in the PNIPAAm-based networks from a hydrophilic random coil to a hydrophobic globule at a temperature close to the LCST of the PNIPAAm chains. The MIP and NIP hydrogels have a higher transition temperature than the PNIPAAm hydrogel. In addition, the MIP and NIP hydrogels’ transmittance changes were less abrupt than those of the PNIPAAm hydrogel. In general, introducing hydrophilic and hydrophobic components to PNIPAAm increases and decreases its LCST, respectively.42 Additionally, it is known that the thermoresponsiveness of PNIPAAm decreases with the introduction of hydrophilic components. Thus, the fact that the MIP and NIP hydrogels showed higher transition temperatures and a little lower thermoresponsiveness than the PNIPAAm hydrogel means that the MIP and NIP hydrogels had hydrophilic CD ligands covalently attached to the PNIPAAm networks. Notably, because the MIP and NIP hydrogels showed the same thermoresponsive behaviour, we can conclude that the amount of CD introduced in the MIP hydrogel was almost the same as that incorporated into the NIP hydrogel.
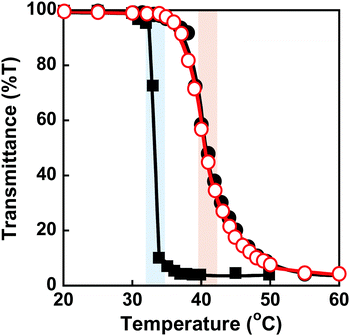 |
| | Fig. 3 Effect of temperature on the transmittance (650 nm) of the MIP ( ), NIP (●) and PNIPAAm (■) hydrogels in water. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively. ), NIP (●) and PNIPAAm (■) hydrogels in water. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively. | |
Conformational changes in polymer chains of hydrogel networks are directly correlated with the swelling ratio of the hydrogel. To evaluate the conformational change of the networks, we investigated the swelling ratios of the MIP, NIP, and PNIPAAm hydrogels as a function of temperature. The relationship between temperature and swelling ratio of the MIP and NIP hydrogels in water is shown in Fig. 4. Similarly to the PNIPAAm hydrogel, the swelling ratio of the MIP and NIP hydrogels decreased dramatically at 37 °C with increasing temperature. It should be noted that the transition temperature of the MIP and NIP hydrogels is about 37 °C, which is a little higher than that of the PNIPAAm hydrogel. These results are consistent with the temperature dependence of the transmittance of their hydrogels shown in Fig. 3. Notably, the transition temperature of the MIP hydrogel is close to body temperature, showing that it could be used as a smart reservoir to regulate drug release in response to temperature changes.
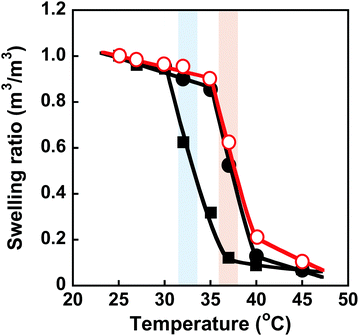 |
| | Fig. 4 Effect of temperature on the swelling ratio of the MIP ( ), NIP (●) and PNIPAAm (■) hydrogels in water. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively. ), NIP (●) and PNIPAAm (■) hydrogels in water. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively. | |
Adsorption behaviour of a model drug into MIP and NIP hydrogels
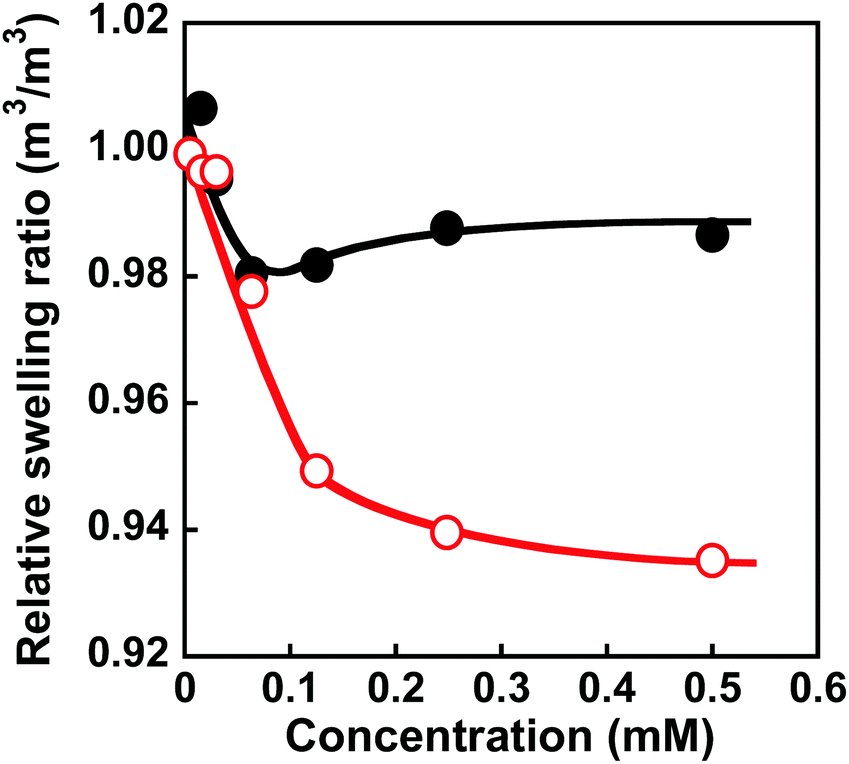
Molecular imprinting enables us to easily create molecular binding sites within chemically cross-linked networks. In this study, we prepared the MIP hydrogel by molecular imprinting, which used CD as a ligand for template dapsone, PNIPAAm as a thermoresponsive primary chain, and a negligible amount of cross-linker, in contrast to standard molecular imprinting, which requires a large amount of cross-linker. As the MIP and NIP hydrogels were prepared by network formation at 25 °C, their dapsone adsorption in aqueous dapsone solutions was measured at this temperature. The relationship between the dapsone concentration and the amount of dapsone adsorbed into the MIP and NIP hydrogels in aqueous dapsone solutions at 25 °C is shown in Fig. 5. The amount of dapsone adsorbed into their hydrogels sharply increased as the dapsone concentration in aqueous solutions increased. The MIP hydrogel adsorbed a greater amount of dapsone than the NIP hydrogel in the aqueous solutions at 25 °C. This is because molecular imprinting was used to establish molecular binding sites for dapsone within the MIP hydrogel. The molecular imprinting enables the arrangement of CDs as ligands at optimal positions for forming sandwich-like CD-dapsone-CD complexes, followed by the formation of molecular binding sites for dapsone, as shown in Fig. 6. In addition, we measured the swelling ratios of the MIP and NIP hydrogels as a function of dapsone concentration to determine the changes in the cross-linked structures induced by the formation of the CD and dapsone complex. The relationship between the dapsone concentration and the swelling ratio of MIP and NIP hydrogels in aqueous dapsone solutions at 25 °C is shown in Fig. 7. The swelling ratio of the MIP hydrogel decreased more effectively than that of the NIP hydrogel with increasing the dapsone concentration in aqueous solutions. The compressive modulus was also used to determine the crosslinking density of the MIP, NIP, and PNIPAAm hydrogels in an aqueous solution with and without dapsone. As shown in Table S1 (ESI†), whereas the cross-linking density of the NIP and PNIPAAm hydrogels changed little by the presence of dapsone, that of the MIP hydrogel increased effectively in an aqueous dapsone solution. The enhanced cross-linking density of the MIP hydrogel in an aqueous dapsone solution shows the formation of the CD-dapsone-CD complexes acting as cross-links. Two CDs are likely to form a sandwich-like CD-dapsone-CD complex with a dapsone molecule due to the presence of two aromatic groups in dapsone. Additionally, our previous publications demonstrated that when CD ligands form sandwich-like CD-target-CD complexes, molecularly imprinted hydrogels with CD ligands shrink in response to the presence of a target molecule due to increased cross-linking densities.35,36,39 Thus, the dapsone concentration-dependent shrinkage of the MIP hydrogel at 25 °C is induced by the formation of dynamic cross-links based on the CD-dapsone-CD complexes. The fact that the MIP hydrogel shrinks more than the NIP hydrogel demonstrates that CDs are efficiently arranged into appropriate sites to form the CD-dapsone-CD complexes by molecular imprinting, as shown in Fig. 6. Unlike the MIP hydrogel, the swelling ratio of the NIP hydrogel increased a little in an aqueous solution with a dapsone concentration of more than 0.2 mM. In an aqueous solution with a high dapsone concentration, the sandwich-like CD-dapsone-CD complexes in the NIP hydrogels are dissociated by the inhibition of excess amount of dapsone because of a small ratio of the complex to dapsone or weak bindings. In addition, when the CDs form not CD-dapsone-CD complexes but CD-dapsone complexes, an amino group of dapsone is not included within CD, followed by an increase in the osmotic pressure. Therefore, a little increase in the swelling ratio of the NIP hydrogel in an aqueous solution with high dapsone concentration might be attributed to a decrease in the cross-linking density and an increase in the osmotic pressure. As a result, we conclude that molecular binding sites for dapsone can be successfully created within the PNIPAAm networks by molecular imprinting, although the slightly cross-linked hydrogel prepared using a minute amount of chemical cross-linker is highly swollen in an aqueous solution at 25 °C, in contrast to general molecularly imprinted polymers with highly cross-linked structures.
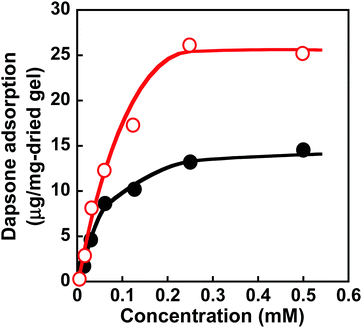 |
| | Fig. 5 Relationship between the dapsone concentration and the amount of dapsone adsorbed into the MIP ( ) and NIP (●) hydrogels in aqueous dapsone solutions at 25 °C. ) and NIP (●) hydrogels in aqueous dapsone solutions at 25 °C. | |
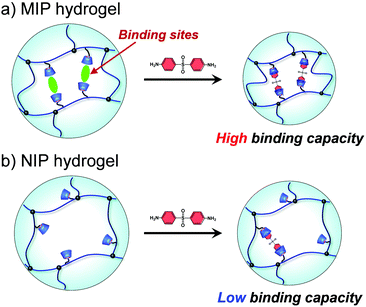 |
| | Fig. 6 Schematic illustration of the dapsone adsorption into the MIP and NIP hydrogels. | |
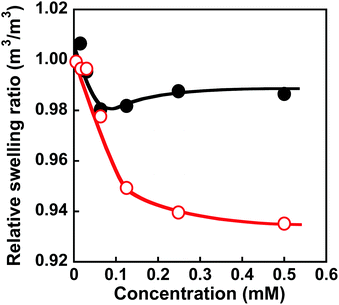 |
| | Fig. 7 Relationship between the dapsone concentration and relative swelling ratio of the MIP ( ) and NIP (●) hydrogels in aqueous dapsone solution at 25 °C. ) and NIP (●) hydrogels in aqueous dapsone solution at 25 °C. | |
We determined the amount of dapsone adsorbed into the MIP, NIP, and PNIPAAm hydrogels as a function of time in an aqueous solution containing 0.035 mmol L−1 of dapsone at various temperatures. As shown in Fig. S7 (ESI†), the amount of dapsone adsorbed into their hydrogels gradually increased with increasing time. The MIP, NIP, and PNIPAAm hydrogels demonstrate different profiles of dapsone adsorption at each temperature. The amount of dapsone adsorbed into the MIP and NIP hydrogels was much more than the amount adsorbed into the PNIPAAm hydrogel at each temperature. This is because the former hydrogels had CDs as ligands for the inclusion complexes formed by the two aromatic groups of a dapsone molecule, whereas the latter hydrogel lacked CDs. Notably, the MIP hydrogels can adsorb more dapsone than the NIP hydrogel at each temperature. These results showed that the MIP hydrogels had molecular binding sites for dapsone in an aqueous solution at each temperature. The relationship between temperature and the amount of dapsone adsorbed into the MIP, NIP, and PNIPAAm hydrogels in an aqueous dapsone solution containing 0.035 mmol L−1 is shown in Fig. 8. The amount of dapsone adsorbed into the PNIPAAm hydrogel without CD ligands increased a little at 32 °C, which is the transition temperature based on the LCST of PNIPAAm. As the PNIPAAm chain undergoes a drastic change from a hydrophilic random coil to a hydrophobic globule at its LCST, a small amount of dapsone is nonspecifically adsorbed onto the hydrophobic PNIPAAm chain by hydrophobic interaction at a temperature above the LCST. In contrast to the PNIPAAm hydrogel, dapsone adsorption into the MIP and NIP hydrogels showed negative temperature dependence, despite their networks consisting of PNIPAAm chains. It should be noted that when the temperature was increased from 25 °C to 45 °C, dapsone adsorption into the MIP hydrogel remarkably decreased by up to one-third. Although the MIP hydrogel adsorbed three times as much dapsone as the NIP hydrogel at 25 °C, the amount of dapsone adsorbed into the MIP hydrogel decreased to approximately that of the NIP hydrogel at 45 °C. Due to the presence of molecular binding sites for dapsone created by molecular imprinting in water at 25 °C, the MIP hydrogel can adsorb a large amount of dapsone compared to the NIP hydrogel at 25 °C. When the temperature rises from 25 °C to 45 °C, the molecular binding sites within the MIP hydrogel collapse due to conformational change of the PNIPAAm chains from a hydrophilic random coil to a hydrophobic globule above their transition temperature of approximately 37 °C, close to body temperature (Fig. 3 and 4). Thus, the amount of dapsone adsorbed into the MIP hydrogel approaches that into the NIP hydrogel above the transition temperature. This shows that specific binding of dapsone to molecular binding sites is more predominant in the dapsone adsorption into the MIP hydrogel than hydrophobic interaction between dapsone and PNIPAAm chains. Additionally, a few researchers created molecular binding sites within weakly cross-linked hydrogels that had been swollen in an aqueous medium.32–41 Furthermore, thermoresponsive PNIPAAm-based hydrogels can be used to separate hydrophobic chemicals, since the temperature-dependent conversion of the PNIPAAm chains to hydrophobicity enhances their hydrophobic interaction with hydrophobic molecules.42,43 These PNIPAAm-based hydrogels preferentially adsorb hydrophobic molecules by hydrophobic interactions above their transition temperature but do not adsorb below their transition temperature. This shows that a decrease in temperature triggers the release of the hydrophobic molecule from the general PNIPAAm-based hydrogels. As a result, applications of PNIPAAm-based hydrogels as self-regulated drug reservoirs are limited to the regulation of drug release using a diffusion-determined mechanism. In contrast to the general PNIPAAm-based hydrogels, the thermoresponsive MIP hydrogel with CD ligands adsorbs dapsone by its specific binding to dynamic molecular binding sites below the transition temperature, but its dapsone adsorption decreases effectively above the transition temperature. As a result, we conclude that the MIP hydrogel has dynamic molecular binding sites for dapsone that can be regulated in response to temperature variations in the conformation of the PNIPAAm networks. Because these dynamic molecular binding sites are different from the PNIPAAm-based hydrogels that were previously reported, they can be useful in designing self-regulated drug reservoirs that can suppress drug leakage in the absence of stimuli but accelerate drug release in response to stimuli.
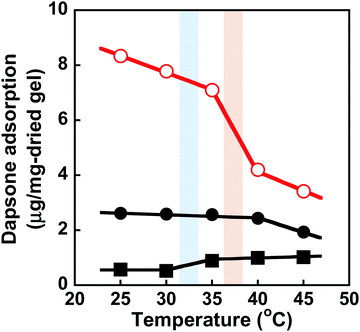 |
| | Fig. 8 Relationship between temperature and the amount of dapsone adsorbed into the MIP ( ), NIP (●) and PNIPAAm hydrogels (■) in an aqueous solution at various temperatures. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively. ), NIP (●) and PNIPAAm hydrogels (■) in an aqueous solution at various temperatures. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively. | |
Drug release behaviour from MIP and NIP hydrogels in response to temperature
After the preparation of the MIP hydrogels, dapsone was loaded into the hydrogel by its adsorption in an aqueous dapsone solution at 25 °C. After that, the dapsone-loaded hydrogels were immersed in water at 25 °C for three hours to wash away the dapsone adsorbed on their surface. The process for loading dapsone within the MIP hydrogel is inefficient because dapsone rebinds with the dynamic binding site after the removal of template dapsone. As an efficient method, we can load dapsone within MIP hydrogels by molecular imprinting without the process for the removal of the template dapsone from the networks, unlike standard molecular imprinting. By such pseudo molecular imprinting without the process for removal of the template molecule, loading efficiency can be improved. However, in this study, to clarify the effect of the dynamic drug binding sites, we loaded dapsone within the MIP hydrogels by the rebinding after the formation of dynamic binding sites by removal of the template dapsone in molecular imprinting.
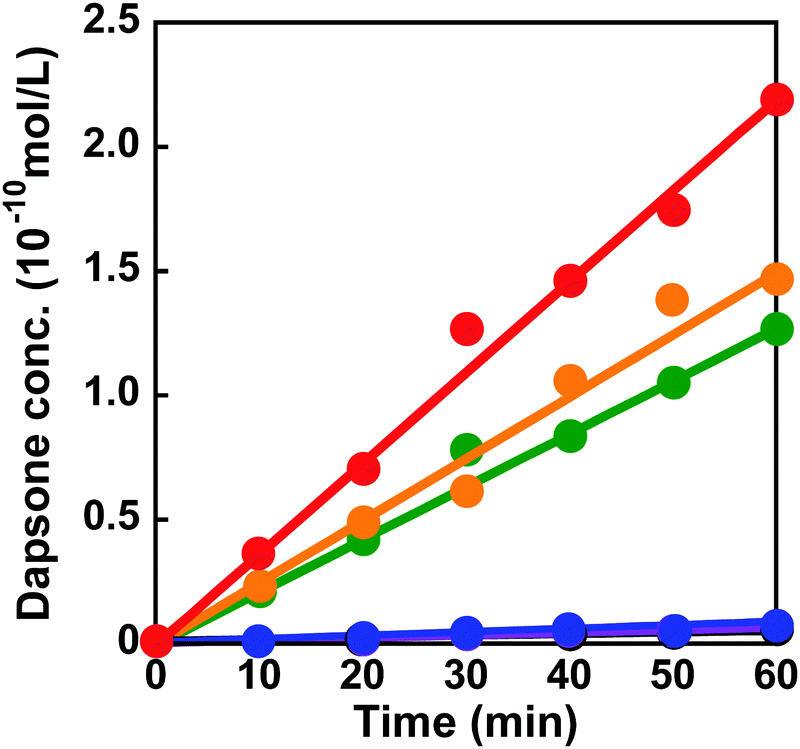
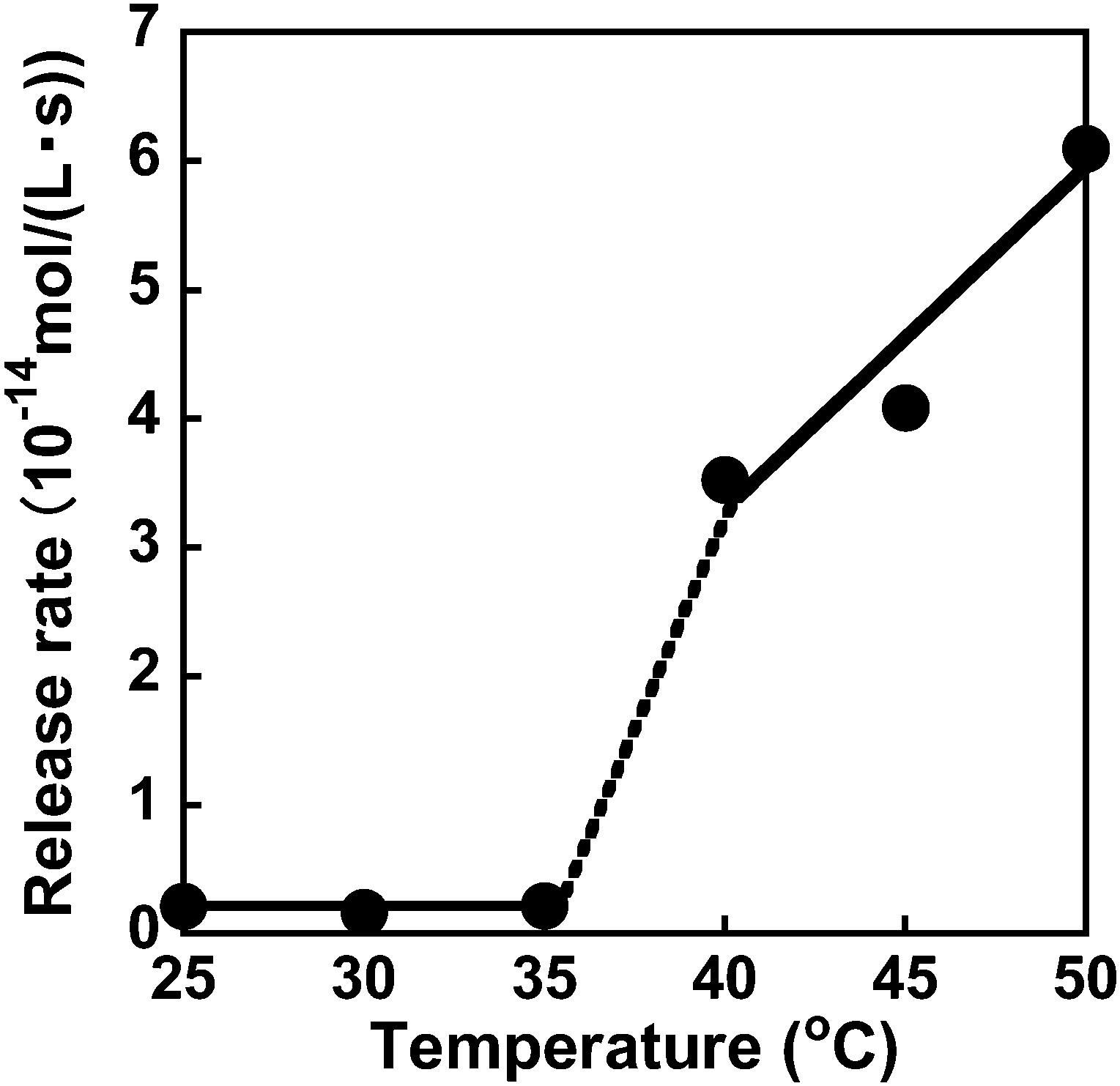
The dapsone release from the resultant dapsone-loaded MIP hydrogel was investigated in water at various temperatures. Fig. 9 shows initial release profiles of dapsone from the dapsone-loaded MIP hydrogel at various temperatures. The release behaviour of dapsone from the dapsone-loaded MIP hydrogel was strongly influenced by temperature. The dapsone-loaded MIP hydrogel suppressed the dapsone release at a temperature below 35 °C which is near its transition temperature, but accelerated it above 40 °C. From the slope of the initial release profile as a function of time shown in Fig. 9, we determined the release rate of dapsone from the MIP hydrogel. The effect of temperature on the release rate of dapsone from the dapsone-loaded MIP hydrogel is shown in Fig. 10. Whereas the release rate of dapsone from the dapsone-loaded MIP hydrogel was very low below its transition temperature, the release rate increased dramatically above its transition temperature. Notably, the temperature at which the dapsone release rate increased discontinuously coincides with that at which the MIP hydrogel dramatically shrank with increasing temperature. Below the transition temperature, dapsone binds to the dynamic molecular binding sites of the MIP hydrogel, enabling the effective suppression of the dapsone leakage (Fig. 11a). Above the transition temperature, however, because the dynamic molecular binding sites are collapsed by conformational changes from a hydrophilic random coil to a hydrophobic globule above the transition temperature, the MIP hydrogel accelerates the dapsone release rapidly. It should be noted that the transition temperature at which the dapsone release rate changes dramatically is near body temperature. These results indicate that the MIP hydrogel with dynamic molecular binding sites can regulate the drug release in response to a change in temperature near body temperature.
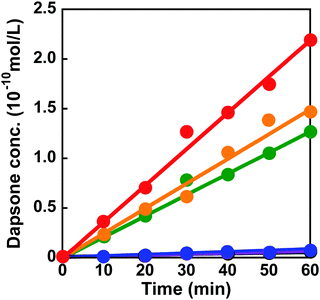 |
| | Fig. 9 Release profiles of dapsone from the dapsone-loaded MIP hydrogel in water at 25 (black), 30 (blue), 35 (purple), 40 (green), 45 (orange) and 50 °C (red). | |
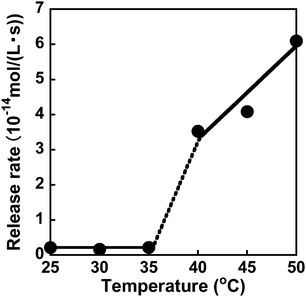 |
| | Fig. 10 Effect of temperature on the release rate of dapsone from the dapsone-loaded hydrogel in water at various temperatures. | |
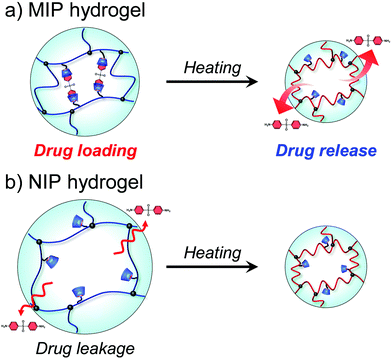 |
| | Fig. 11 Schematic illustration of drug release from MIP and NIP hydrogels with increasing temperature. | |
To discuss the on–off regulation of the drug release, we also investigated the profiles of the dapsone release from MIP and NIP hydrogels in water when the temperature is switched from 25 °C to 45 °C after seven hours (Fig. S8, ESI†). Before switching to 45 °C, dapsone leaked out of the NIP hydrogel at 25 °C, but dapsone leakage from the MIP hydrogel was effectively suppressed. The MIP hydrogel in water at 25 °C had molecular binding sites for dapsone, in which CDs were arranged at optimal positions for forming the sandwich-like CD-dapsone-CD complex because it was prepared by molecular imprinting at 25 °C (Fig. 11). However, it was difficult for CDs to form the CD-dapsone-CD complexes in the NIP hydrogels as CDs were arranged at random within the network in the NIP hydrogel. As dapsone strongly bound to the molecular binding sites of the MIP hydrogel at 25 °C, its leakage from the MIP hydrogel at 25 °C was more effectively suppressed than that from the NIP hydrogel. As soon as the temperature was switched to 45 °C, the MIP hydrogel accelerated the release of dapsone; whereas the NIP hydrogel adsorbed dapsone. Switching temperature from 25 °C to 45 °C resulted in drastic shrinkage of MIP and NIP hydrogels because PNIPAAm chains underwent a conformational change from a hydrophilic random coil to a hydrophobic globule at the transition temperature of 37 °C, close to body temperature, as shown in Fig. 3 and 4. The conformational change results in the collapse of the dynamic molecular binding sites within the MIP hydrogel above the transition temperature. As a result of decreased binding capacity caused by the collapse of the molecular binding sites, dapsone was rapidly released from the MIP hydrogel as the temperature increased above 37 °C. In general, PNIPAAm-based hydrogels tend to suppress drug release above the transition temperature due to the shrunken networks inhibiting the diffusion of the drugs.1,3–6 In contrast to previously reported PNIPAAm-based hydrogels, the MIP hydrogel can successfully suppress drug leakage below the transition temperature while rapidly releasing drugs above the transition temperature. Thus, because thermoresponsive MIP hydrogels with CD ligands have dynamic molecular binding sites with variable drug binding capacity, they can be used as self-regulating drug reservoir capable of both suppressing drug leakage in the absence of stimuli and accelerating drug release in the presence of stimuli. This study demonstrates how molecular imprinting with CD ligands and a thermoresponsive hydrogel enables the creation of dynamic molecular binding sites similar to those found in proteins. This paper focuses on the formation of dynamic drug binding sites within thermoresponsive hydrogels because it is the first report on the combination of molecular imprinting with CD ligands and thermoresponsive polymers. However, in designing stimuli-responsive drug carriers for clinical applications, the formation of nanogels with a diameter of 10–200 nm and the introduction of biocompatibility are required. Detailed studies on the preparation of biocompatible nanogels with dynamic drug binding sites by combining molecular imprinting using CD ligands with thermoresponsive polymers are under investigation for designing anticancer drug carriers for potential applications. Even though the thermoresponsive MIP hydrogels require additional research to determine their potential applications, they are expected to become important reservoirs for self-regulated DDS in the future.
Conclusions
A thermoresponsive hydrogel with dynamic molecular binding sites was prepared by molecular imprinting using a model drug as the template and CD as the ligand. Combining molecular imprinting with thermoresponsive polymer networks enabled us to develop dynamic binding sites with variable binding capacity. An increase in temperature above 37 °C, close to body temperature, resulted in a significant shrinkage of the MIP hydrogel, followed by the collapse of the dynamic molecular binding site. In contrast to the dapsone adsorption into the PNIPAAm hydrogel by hydrophobic interaction, adsorption into the MIP hydrogel was significantly decreased drastically when the temperature was increased above its transition temperature. The MIP hydrogel efficiently suppressed drug leakage below its transition temperature but rapidly accelerated drug release above it due to conformational change in the thermoresponsive networks’ binding capacity. In general, thermoresponsive hydrogels release drugs below their transition temperatures and suppress drug leakage above their transition temperatures by shrinking networks. However, thermoresponsive hydrogels with dynamic molecular binding sites created by molecular imprinting have the opposite effect on drug release in response to temperature changes. Even though the thermoresponsive MIP hydrogels with dynamic molecular binding sites for drugs still require additional research to determine their applications, they are expected to become important reservoirs for self-regulated DDS in the future.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP20H04539 and No. JP20H05236 from the Japan Society for the Promotion of Science (JSPS), and by research grants from the Canon Foundation.
Notes and references
- A. S. Hoffman, Hydrogels for biomedical applications, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS PubMed.
- J. L. Druly and D. J. Mooney, Hydrogels for tissue engineering: Scaffold design variables and applications, Biomaterials, 2003, 24, 4337–4351 CrossRef.
- Z. M. O. Rzaev, S. Dinçer and E. Pişkin, Functional copolymers of N-isopropylacrylamide for bioengineering applications, Prog. Polym. Sci., 2007, 32, 534–595 CrossRef CAS.
- L. Klouda and A. G. Mikos, Thermoresponsive hydrogels in biomedical applications, Eur. J. Pharm. Biopharm., 2008, 68, 34–45 CrossRef CAS PubMed.
- Y. Qiu and K. Park, Environment-sensitive hydrogels for drug delivery, Adv. Drug Delivery Rev., 2012, 64, 49–60 CrossRef.
- R. Yoshida and K. Sakai, Drug release profiles in the shrinking process of thermoresponsive poly(N-isopropylacrylamide-co-alkyl methacrylate) gels, Ind. Eng. Chem. Res., 1992, 31, 2339–2345 CrossRef CAS.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Biodegradable block copolymers as injectable drug-delivery systems, Nature, 1997, 388, 860 CrossRef CAS PubMed.
- T. Miyata, T. Uragami and K. Nakamae, Biomolecule-sensitive hydrogels, Adv. Drug Delivery Rev., 2002, 54, 79–98 CrossRef CAS PubMed.
- T. Miyata, Preparation of smart soft materials using molecular complexes, Polym. J., 2010, 42, 277–289 CrossRef CAS.
- T. Miyata, N. Asami and T. Uragami, A reversibly antigen-responsive hydrogel, Nature, 1999, 399, 766–769 CrossRef CAS PubMed.
- N. T. Zaman, Y.-Y. Yang and J. Y. Ying, Stimuli-responsive polymers for the targeted delivery of paclitaxel to hepatocytes, Nano Today, 2010, 5, 9–14 CrossRef CAS.
- R. Liu and A. Poma, Advances in molecularly imprinted polymers as drug delivery systems, Molecules, 2021, 26, 3589 CrossRef CAS PubMed.
- V. S. Pande, A. Y. Grosberg and T. Tanaka, Heteropolymer freezing and design: Towards physical models of protein folding, Rev. Mod. Phys., 2000, 72, 259 CrossRef CAS.
- P. Cohen, The structure and regulation of protein phosphatases, Annu. Rev. Biochem., 1989, 58, 453–508 CrossRef CAS PubMed.
- J. Monod, J. Wyman and J. P. Changeux, On the nature of allosteric transitions: A plausible model, J. Mol. Biol., 1965, 12, 88–118 CrossRef CAS PubMed.
- E. Antonini, Interrelationship between structure and function in hemoglobin and myoglobin, Physiol. Rev., 1965, 45, 123–170 CrossRef CAS PubMed.
- K. Mosbach, Molecular imprinting, Trends Biochem. Sci., 1994, 19, 9–14 CrossRef CAS PubMed.
- K. Shea, Molecular imprinting of synthetic network polymers: The de novo synthesis of macromolecular binding and catalytic sites, Trends Polym. Sci., 1994, 2, 166–173 CAS.
- G. Wulff, Molecular imprinting in cross-linked materials with the aid of molecular templates-A way towards artificial antibodies, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS.
- K. Haupt and K. Mosbach, Molecular imprinted polymers and their use in biomimtic sensors, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed.
- G. Wulff, Enzyme-like catalysis by molecularly imprinted polymers, Chem. Rev., 2002, 102, 1–27 CrossRef CAS PubMed.
- M. Komiyama, T. Mori and K. Ariga, Molecular imprinting: Materials nanoarchitectonics with molecular information, Bull. Chem. Soc. Jpn., 2018, 91, 1075–1111 CrossRef CAS.
- H. Asamura, T. Hashiya and M. Komiyama, Tailor-made receptors by molecular imprinting, Adv. Mater., 2000, 12, 1019–1030 CrossRef.
- J. J. BelBruno, Molecularly imprinted polymers, Chem. Rev., 2019, 119, 94–119 CrossRef CAS PubMed.
- R. Langer and N. A. Peppas, Advances in biomaterials, drug delivery, and bionanotechnology, AIChE J., 2003, 49, 2990–3006 CrossRef CAS.
- C. Alvarez-Lorenzo and A. Concheiro, Molecularly imprinted polymers for drug delivery, J. Chromatogr. B: Anal. Technol.
Biomed. Life Sci., 2004, 804, 231–245 CrossRef CAS PubMed.
- H. Hiratani, Y. Mizutani and C. Alvarez-Lorenzo, Controlling drug release from imprinted hydrogels by modifying the characteristics of the imprinted cavities, Macromol. Biosci., 2005, 5, 728–733 CrossRef CAS PubMed.
- B. Sellergren and C. J. Allender, Molecularly imprinted polymers: a bridge to advanced drug delivery, Adv. Drug Delivery Rev., 2005, 57, 1733–1741 CrossRef CAS PubMed.
- D. R. Kryscio and N. A. Peppas, Mimicking biological delivery through feedback-controlled drug release systems based on molecular imprinting, AIChE J., 2009, 55, 1311–1324 CrossRef CAS PubMed.
- S. A. Zaidi, Molecular imprinted polymers as drug delivery vehicles, Drug Delivery, 2016, 23, 2262–2271 CrossRef CAS PubMed.
- S. A. Zaidi, Molecular imprinting: A useful approach for drug delivery, Mater. Sci. Energy Technol., 2020, 3, 72–77 CAS.
- T. Miyata, M. Jige, T. Nakaminami and T. Uragami, Tumor marker-responsive behavior of gels prepared by biomolecular imprinting, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1190–1193 CrossRef CAS PubMed.
- T. Miyata, T. Hayashi, Y. Kuriu and T. Uragami, Responsive behavior of tumor-marker-imprinted hydrogels using macromolecular cross-linkers, J. Mol. Recognit., 2012, 25, 336–343 CrossRef CAS PubMed.
- Y. Kuriu, A. Kawamura, T. Uragami and T. Miyata, Formation of molecularly imprinted hydrogel thin layers with lectin recognition sites on SPR sensor chips by atom transfer radical polymerization, Chem. Lett., 2014, 43, 825–827 CrossRef CAS.
- A. Kawamura, T. Kiguchi, T. Nishihata, T. Uragami and T. Miyata, Target molecule-responsive hydrogels designed via molecular imprinting using bisphenol A as a template, Chem. Commun., 2014, 50, 11101–11103 RSC.
- Y. Shiraki, K. Tsuruta, J. Morimoto, C. Ohba, A. Kawamura, R. Yoshida, R. Kawano, T. Uragami and T. Miyata, Preparation of molecule-responsive microsized hydrogels via photopolymerization for smart microchannel microvalves, Macromol. Rapid Commun., 2015, 36, 515–519 CrossRef CAS PubMed.
- K. Matsumoto, B. D. B. Tiu, A. Kawamura, R. C. Advincula and T. Miyata, QCM sensing of bisphenol A using molecularly imprinted hydrogel/conducting polymer matrix, Polym. J., 2016, 48, 525–532 CrossRef CAS.
- R. Naraprawatphong, G. Kawanaka, M. Hayashi, A. Kawamura and T. Miyata, Development of protein–recognition SPR devices by combination of SI-ATRP with biomolecular imprinting using protein ligands, J. Mol. Imprinting, 2016, 4, 21–30 CAS.
- K. Matsumoto, A. Kawamura and T. Miyata, Conformationally regulated molecular binding and release of molecularly imprinted polypeptide hydrogels that undergo helix–coil transition, Macromolecules, 2017, 50, 2136–2144 CrossRef CAS.
- C. Alvarez-Lorenzo, O. Guney, T. Oya, Y. Sakai, M. Kobayashi, T. Enoki, Y. Takeoka, T. Ishibashi, K. Kuroda, K. Tanaka, G. Wang and A. Y. Grosberg, Reversible adsorption of calcium ions by imprinted temperature sensitive gels, J. Chem. Phys., 2001, 114, 2812–2816 CrossRef CAS.
- C. Alvarez-Lorenzo, H. Hiratani, K. Tanaka, K. Stancil, A. Y. Grosberg and T. Tanaka, Simultaneous multiple-point adsorption of aluminum ions and charged molecules a polyampholyte thermosensitive gel: Controlling frustrations in a heteropolymer gel, Langmuir, 2001, 17, 3616–3622 CrossRef CAS.
- H. G. Schild, poly(N-isopropylacrylamide): experiment, theory and application, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- K. L. Wang, J. H. Burban and E. L. Cussler, Hydrogels as separation agents, Adv. Polym. Sci., 1993, 110, 67–79 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Yoshinori
Takashima
ab,
Yoshinori
Takashima
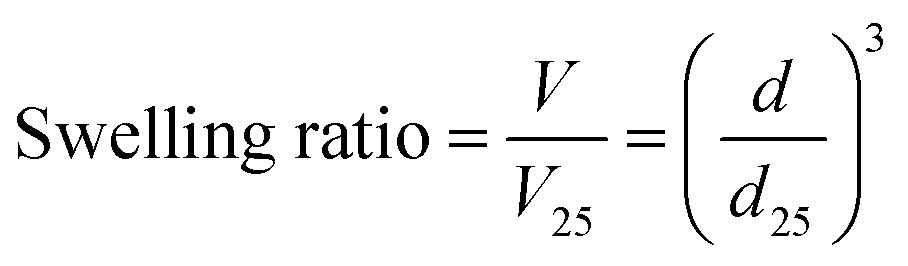

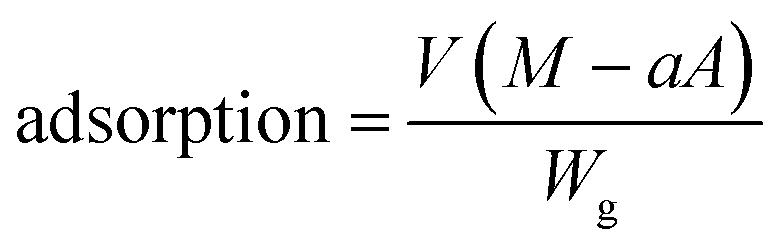
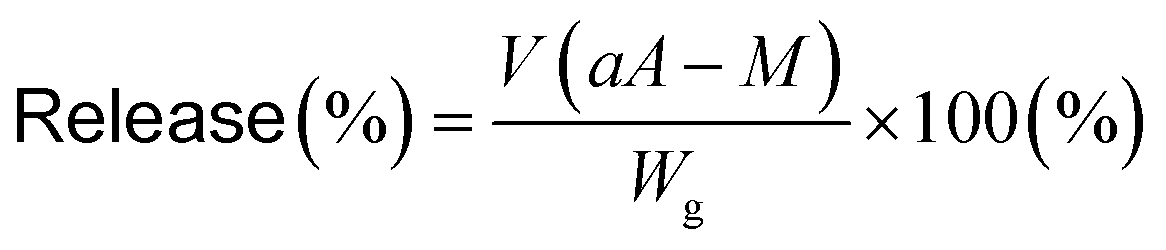
 ), NIP (●) and PNIPAAm (■) hydrogels in water. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively.
), NIP (●) and PNIPAAm (■) hydrogels in water. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively.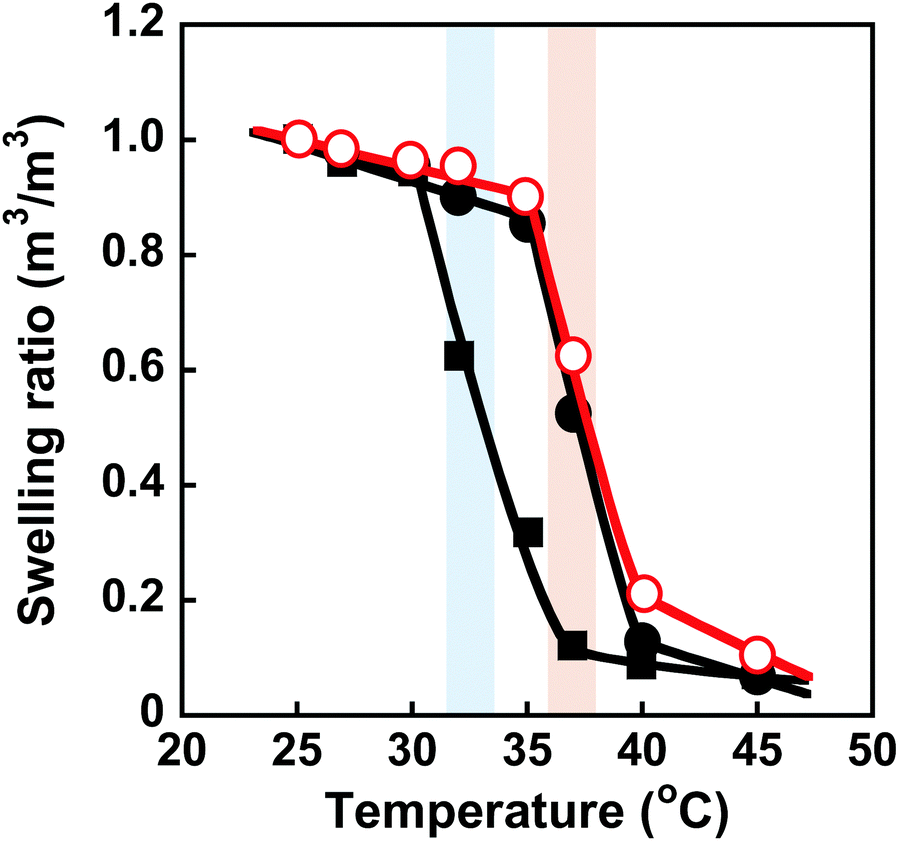
 ), NIP (●) and PNIPAAm (■) hydrogels in water. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively.
), NIP (●) and PNIPAAm (■) hydrogels in water. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively.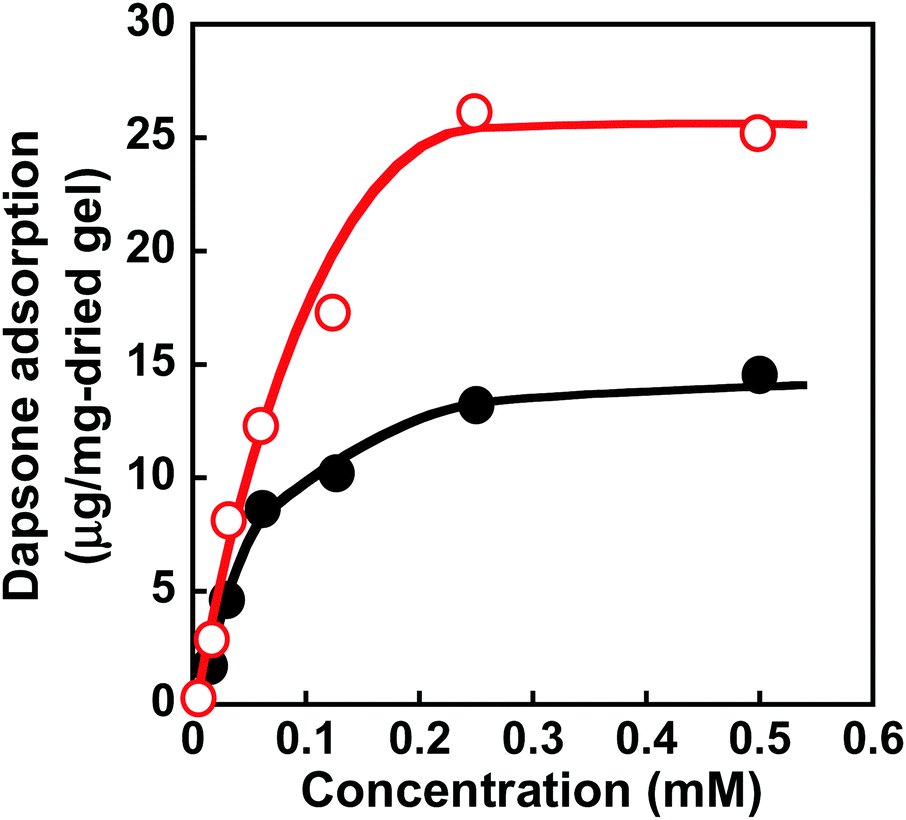
 ) and NIP (●) hydrogels in aqueous dapsone solutions at 25 °C.
) and NIP (●) hydrogels in aqueous dapsone solutions at 25 °C.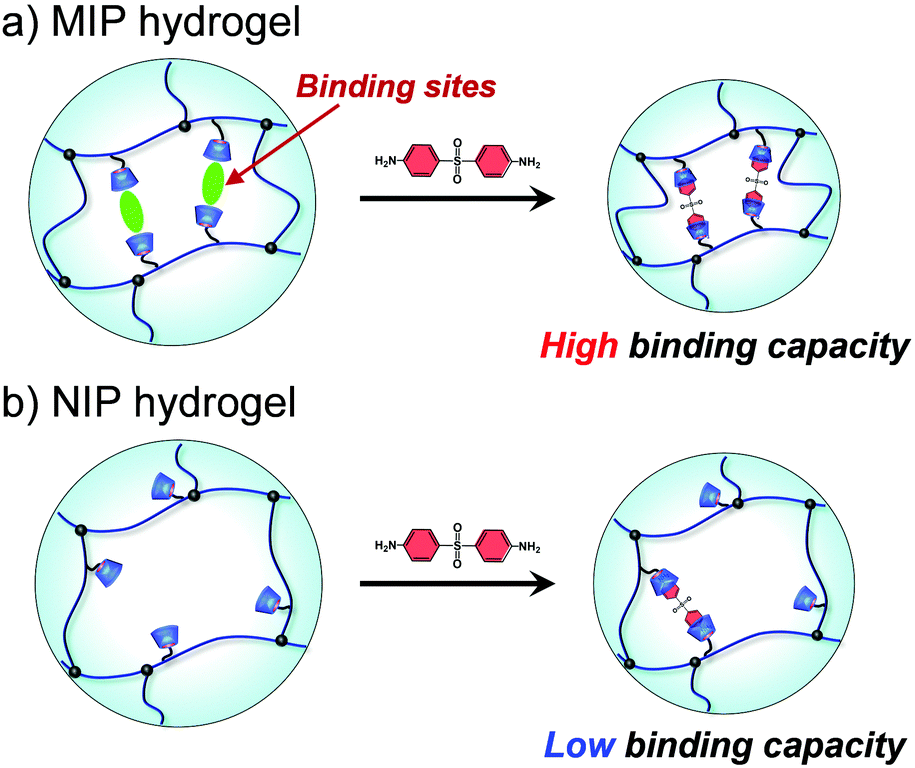
 ) and NIP (●) hydrogels in aqueous dapsone solution at 25 °C.
) and NIP (●) hydrogels in aqueous dapsone solution at 25 °C.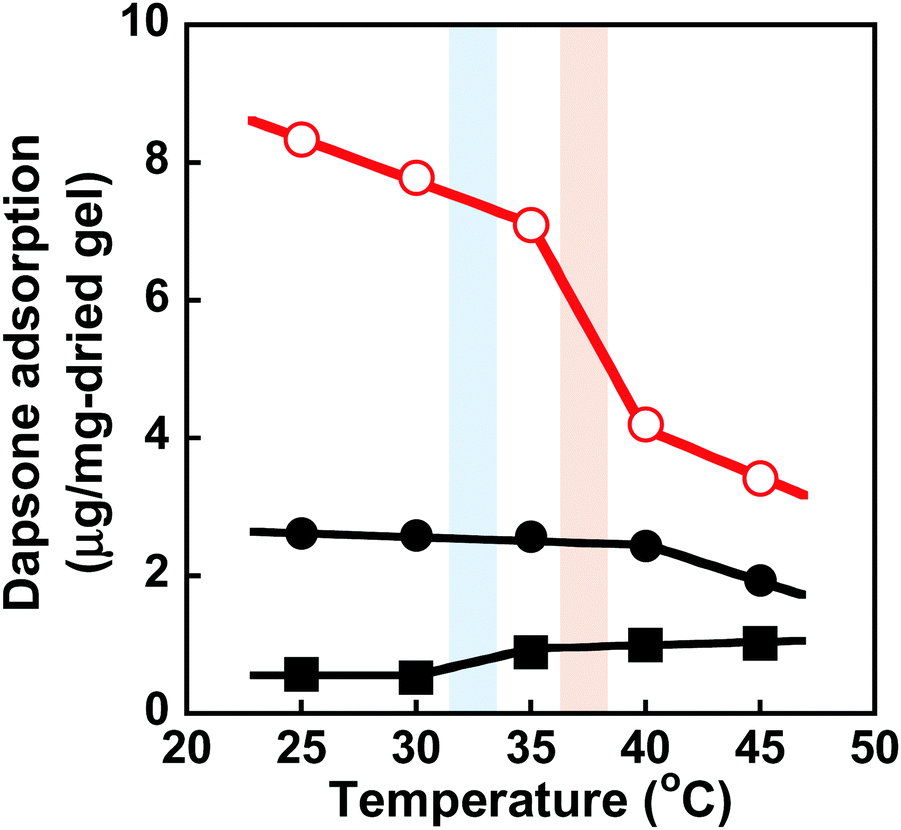
 ), NIP (●) and PNIPAAm hydrogels (■) in an aqueous solution at various temperatures. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively.
), NIP (●) and PNIPAAm hydrogels (■) in an aqueous solution at various temperatures. Blue and red bars indicate the transition temperature of PNIPAAm and MIP & NIP hydrogels, respectively.