DOI:
10.1039/D2PY00600F
(Paper)
Polym. Chem., 2022,
13, 4406-4415
Fully biobased triblock copolymers generated using an unconventional oscillatory plug flow reactor†
Received
9th May 2022
, Accepted 7th July 2022
First published on 14th July 2022
Abstract
Producing polymers in continuous flow offers significant advantages in terms of efficiency, scalability, and safety. Using conventional tubular flow reactors to synthesize polymers comes with some challenges, especially related to maintaining narrow residence time distributions (RTD) when operating with viscous fluids. Laminar flow is typically observed in tubes with restricted dimensions, and significant wall effects result in the broadening of the molar mass distributions. We envisioned that such a limitation can be overcome by the use of an oscillatory flow reactor (OFR). This work describes the use of a novel plate-type OFR to improve continuous-flow polymerization reactions for the first time. The reactor plate is equipped with millimeter-scale cubic pillars, and when combined with a superimposed oscillatory flow regime, promotes turbulent flow to circumvent detrimental wall effects during polymerizations. Additionally, the pulsatile flow intensifies mixing, and careful tuning of the pulsation amplitude and frequency lead to improved (i.e., narrowed) residence time distributions, a crucial parameter when synthesizing complex block polymer scaffolds in continuous flow. This innovative principle of implementing OFRs for improved continuous polymerization reaction is demonstrated with the benchmark ring-opening polymerization of lactide, a well-known renewable monomer. Thorough characterization using the reactor system reveals the relationships between process conditions and molecular attributes, including target molar mass and dispersity (Đ). Further, the OFR enabled the streamlined preparation of a series of block polymers with variable composition and low dispersity in a single experiment by judiciously adjusting the independent inlet feed rates. Finally, the OFR system allows for simple scaling without affecting the critical process parameters. As a result, a multi-gram synthetic protocol was achieved employing a biobased hydroxyl telechelic poly(β-farnesene) macroinitiator in the ring-opening polymerization of lactide to generate fully renewable ABA-type triblock copolymers.
Introduction
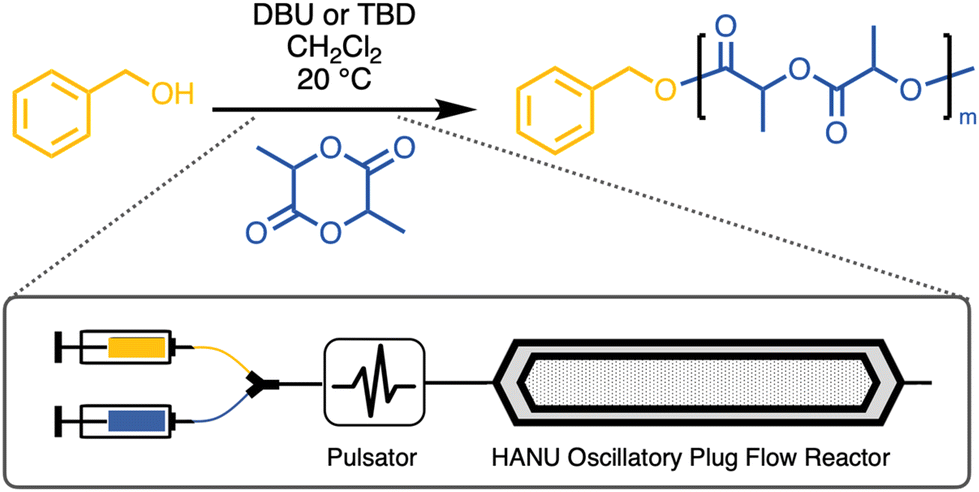
Block polymers are macromolecules with segments of different repeating units that are covalently bound to each other.1 Such segmented copolymers exhibit strongly composition dependent properties related to the adoption of various nanoscale morphologies, the formation of which is driven by the thermodynamic immiscibility between the constituent blocks.2,3 The properties can therefore be tuned by preparing products with different relative block lengths and different block sequences.4,5 However, each individual target composition typically requires a separate synthesis, which can be quite elaborate depending on the architecture.6–9 This is nearly always carried out in batch reactions with each block made sequentially, which is especially necessary with mechanistically incommensurate monomers/repeating units.10–12 Preparing even a limited series of block polymers with different compositions is tedious and resource intensive (e.g., solvents, purification/precipitation). To mitigate this, preparing block polymers in continuous flow reactors has many appealing benefits, as outlined extensively in several recent reviews.13,14 In fact, preparation of block copolymers in flow has been reported using various polymerization methods, using both a macroinitiator approach, sequential monomer addition, or coupling of preformed chains.15–23 This has been demonstrated routinely with acrylates or lactones, typically using conventional tubular reactors or microfluidic devices. Flow polymerizations have received significant attention, with increasingly sophisticated adaptations being made recently to incorporate real-time feedback and machine learning in order to fully automate the process and produce highly customizable polymer constructs.24–26 Despite these gains, the conventional approach of using micrometer- or millimeter sized tubular reactors is still firmly the status quo.27,28 Such reactor geometries severely limit the scale on which materials can be produced. Full characterization of block polymers, especially for applications like thermoplastic elastomers, requires multi-gram synthetic protocols. Recently, a scalable oscillatory flow reactor (OFR) with a planar reaction compartment (15 mL internal capacity) has been used to successfully conduct organic small molecule transformations when processing challenging media (e.g., slurries, viscous solutions).29–32 Polymerizations typically lead to increasing viscosities, which also challenges the integrity of mixing profiles. The novel OFR described in this work (i.e. HANU Flow Reactor, Creaflow B.V.) contains millimeter-sized cubic pillars that enhance turbulent flow, and thus promote intimate mixing. Furthermore, the unique oscillatory plug flow conditions enabled by a pulsator attached in-line to the reactor creates a continuous reaction pathway that does not compromise residence time distributions across a broad range of flow rates.33 This allows a scalable production of materials (i.e., grams per hour) in which the inlet/reactant delivery rates can be adjusted in situ to tailor the product makeup. This is an ideal setup for producing a library of block polymers with variable composition in a “single-pot”, streamlined manner without the need for separate reactors and tedious product isolation protocols. Additionally, control over the polymerization, leading to low dispersities and accurate molar mass targeting, is not compromised by the viscous nature of the reaction medium. Here, we demonstrate the capabilities of the HANU flow reactor by conducting the organo-catalyzed ring-opening transesterification polymerization (ROTEP) of D,L-lactide (LA). Several lactones have been polymerized in a continuous-flow setup recently, but this has been limited to tubular or microfluidic devices.22,34–37 A range of molar masses (Mn) have been targeted, highlighting the limitations and versatility, covering a broad spectrum of flow rates. Dispersity (Đ) of the resulting amorphous polylactide (PLA) was sensitive to the amplitude of the oscillation, but less dependent on the frequency. Finally, a series of ABA-type symmetric block polymers was prepared using optimized conditions in a single-experiment by adjusting the inlet flow rates. We used a biobased hydroxyl-telechelic poly(β-farnesene) as a macroinitiator in the ROTEP of LA monomer, producing a range of compositionally variable block polymers on multi-gram scale. Poly(β-farnesene) is hydrophobic polymer derived from terpenes, a class of highly promising renewable monomers that have wide ranging properties.38–41 Telechelic poly(farnesene) diol (PFD) has already been used in the context of hydrolysis resistant biobased polyurethane precursors.42 Likewise, a related telechelic terpene derivative, poly(myrcene) has been shown to be effective as a midblock by polymerizing LA monomer, exhibiting the utility of structurally similar building blocks in generating triblock copolymers.43 The resulting fully biobased thermoplastics described in this work exhibit a range of physical properties owing to the easily adjustable chemical composition, representing an ideal platform as potential biobased elastomers.44,45 Expanding the technological toolbox to provide convenient access to such a library of biobased block polymers is a highlight.
Results and discussion
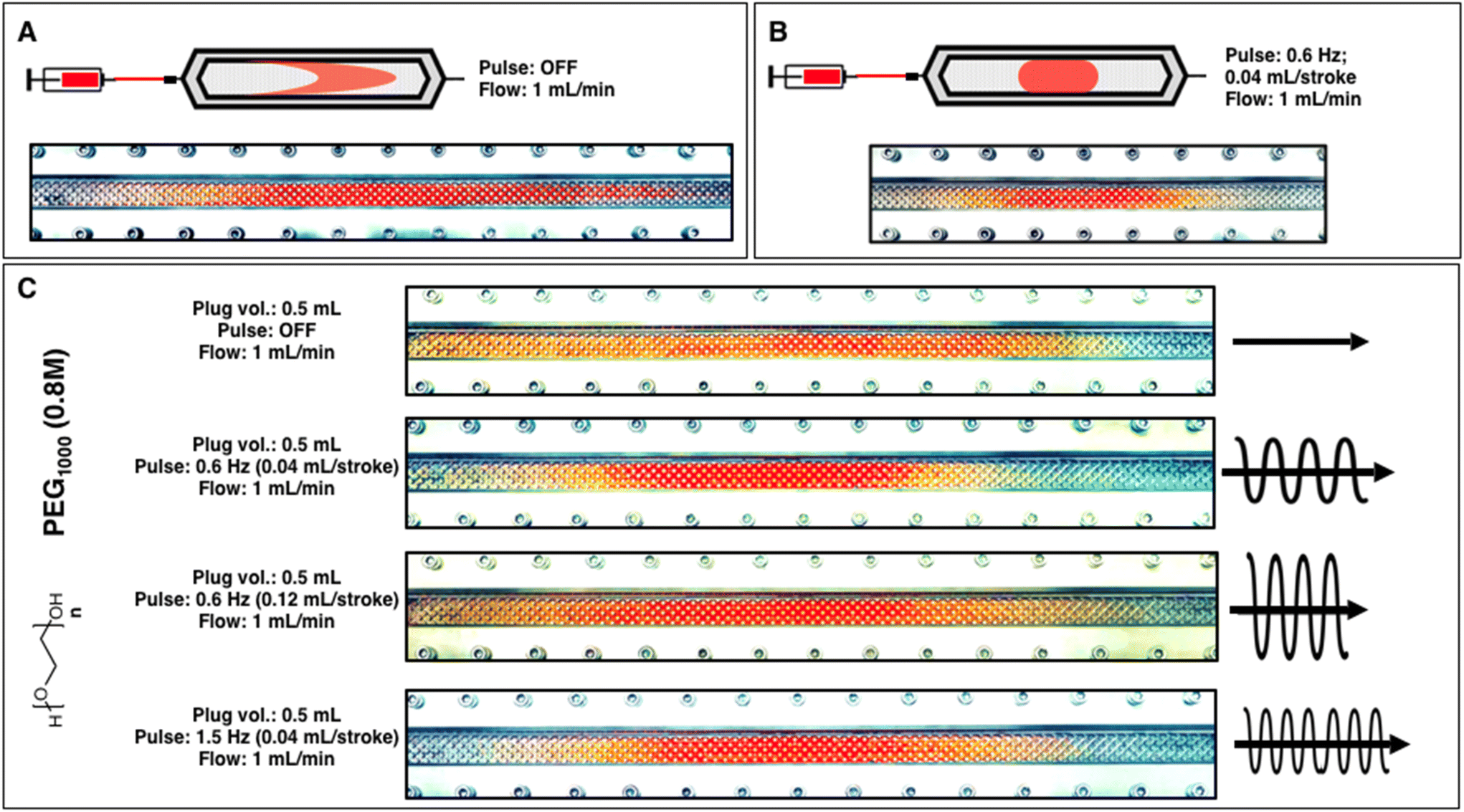
Our initial aim was to interrogate the continuous process conditions for polymerizations by implementing the oscillatory flow reactor (i.e., HANU flow reactor) and establish optimal settings for minimizing the residence time distribution (RTD) in reactions with progressively increasing viscosity (see ESI for a detailed description of the reactor; Fig. S1†).46 The HANU flow reactor has a planar reaction chamber with 15 mL internal capacity. There is a series of regularly spaced stainless steel cube-shaped pillars situated throughout the reaction chamber. The pillars provide a split-and-recombine flow pattern and thus aid in uniform mixing. Further enhancement of the mixing profile is provided by a pulsator unit, which is positioned directly before the reactor inlet. This unit provides superimposed pulsation on the liquid flow. The amplitude and frequency of the pulsation can be independently tuned to optimize the product attributes, which in the case of polymerization is primarily reflected by the molar mass (i.e., conversion) and the molar mass distribution (i.e., dispersity – Đ).47 The dependence of flow profiles on pulsation conditions was initially analyzed qualitatively by visualizing the distribution of a dye as it travels through the reaction chamber. Initial experiments were conducted by adding an organic dye to acetone and then subsequently loading the colored solvent into the injector syringe. A plug volume of 0.5 mL was visually observed for its flow behavior (Fig. 1). In the absence of pulsation (i.e., steady-state flow), a longitudinal spread of the tracer can be observed owing to the laterial velocity gradient present within the reactor channel leading to a distinct laminar flow profile with tailing observed on both sides of the process channel. In contrast, setting the pulsator to a low amplitude setting (5%, equal to 0.04 mL per stroke displacement) and low frequency (0.6 Hz) counteracted the observed tailing effect and led to a more compact, plug flow profile (i.e., narrow RTD) (Fig. 1A and B) and is consistent with previously reported observations for small molecule synthesis.30 In order to take a closer look at the effect of viscosity on the RTD, polyethylene glycol (PEG) with a number average molar mass (Mn) of 1000 g mol−1 was dissolved in acetone to form a 0.8 M solution and subsequently colored using the same organic dye. The resulting polymer solution was significantly more viscous than the acetone solvent. Previous studies point toward the deleterious effect of increasing viscosity on mixing profiles, typically leading to increased RTD and thus higher dispersities.47,48 In this simple experiment in the OFR, improved plug-like flow behavior was visually observed when moderate frequencies (0.6–1.5 Hz) and low amplitudes (≤0.04 mL per stroke) were employed (Fig. 1C). On the other hand, when using higher pulsation amplitudes (>0.08 mL per stroke; 10% amplitude setting) this effect was no longer observed, as intensified mixing promotes a smearing effect because of the increased energy dissipation present. Additionally, it is worth noting that steady-state flow of the PEG solution leads to a longitudinal spread that is significantly more pronounced than that of the pure solvent case, indicating that the lateral velocity gradient increases with higher viscosity fluids due to the more significant wall effects (Fig. 1C).
 |
| | Fig. 1 Photograph of the flow profile using acetone as a medium. (A) A crescent-shaped flow profile is visible as a result of the formation of a lateral velocity gradient within the reactor in the absence of pulsation. (B) A compact flow profile resulting from pulsation leads to a more homogeneous distribution of the medium in both the longetudal and laterial direction. (C) A comparison of higher viscosity flow profiles with respect to increasing pulsation frequency and -amplitude. | |
Typical laminar flow occurs when mixing is limited to diffusion. The combination of static mixers/pillars and pulsation enable rapid mixing in all directions in a relatively short timescale, providing turbulent flow characteristics across a broad range of flow rates. In practice, the turbulent mixing promotes consistent reactions, leading ultimately to polymers with dispersities that are limited by the reaction kinetics, as opposed to being influenced predominantly by mixing profiles. As the pulsation amplitude increases, the longitudinal displacement of the plug per pulse increases and results in the undesirable spread seen in the flow profile analysis (Fig. 1C).
PLA synthesis using constant monomer to initiator ratio
PLA has been produced in various continuous-flow reactor setups, including tubular flow and microfluidic devices.22,34–37 Several of these examples employed highly active organocatalysts that afford low dispersity, controlled polymers with astounding efficiency.49,50 In particular, we have chosen two common organocatalysts to explore in this investigation, namely 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD).
Synthesizing amorphous PLA via ROTEP in flow was performed as a means to evaluate various key reactor parameters such as flow rate, pulsation frequency, and -amplitude on molecular attributes (e.g., monomer conversion, molar mass, and dispersity). Initially, homopolymer PLA was synthesized via ROTEP using benzyl alcohol (BA) as an initiator and DBU as a catalyst, maintaining a constant monomer to initiator ratio ([LA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[BA] = 100) for entries 1–8, while varying the oscillation conditions (Scheme 1; Table 1; Fig. S2†). Importantly, the LA monomer and initiator were placed in the same syringe before injection, while the catalyst solution was prepared in a separate syringe. This translates to an effect on molecular attributes, owing to different mixing efficiencies (vide infra). Conversion was monitored with 1H NMR spectroscopy of the crude samples (Fig. S3†). A systematic approach revealed that high conversions (>80%) were only achieved with flow rates ranging from 1.0–2 mL min−1, whereas both higher flow rates (5 mL min−1) and lower flow rates (0.5 mL min−1) resulted in lower conversions (<60%) at a fixed pulsation setting (0.6 Hz; 0.04 mL per stroke). At fast flow rates, the residence time is too low to allow for sufficient monomer conversion using this particular reactant/catalyst concentrations. Contrarily, at lower flow rates, it is postulated that the effectiveness of the static mixtures are compromised, ultimately resulting in poor mixing between the catalyst and monomer/initiator solutions. This likely results in significantly retarded reaction kinetics compared with moderately faster flow rates. Thus, the optimum flow rate was identified as 1.0 mL min−1 for exploring additional parameters. The dispersity (Đ) was relatively low (<1.2) for all samples, with the exception of 0.5 mL min−1 (entry 1). This further corroborates the hypothesis of poor mixing at very low flow rates, which is ultimately reflected in the moderately higher dispersity.
:[BA] = 100) for entries 1–8, while varying the oscillation conditions (Scheme 1; Table 1; Fig. S2†). Importantly, the LA monomer and initiator were placed in the same syringe before injection, while the catalyst solution was prepared in a separate syringe. This translates to an effect on molecular attributes, owing to different mixing efficiencies (vide infra). Conversion was monitored with 1H NMR spectroscopy of the crude samples (Fig. S3†). A systematic approach revealed that high conversions (>80%) were only achieved with flow rates ranging from 1.0–2 mL min−1, whereas both higher flow rates (5 mL min−1) and lower flow rates (0.5 mL min−1) resulted in lower conversions (<60%) at a fixed pulsation setting (0.6 Hz; 0.04 mL per stroke). At fast flow rates, the residence time is too low to allow for sufficient monomer conversion using this particular reactant/catalyst concentrations. Contrarily, at lower flow rates, it is postulated that the effectiveness of the static mixtures are compromised, ultimately resulting in poor mixing between the catalyst and monomer/initiator solutions. This likely results in significantly retarded reaction kinetics compared with moderately faster flow rates. Thus, the optimum flow rate was identified as 1.0 mL min−1 for exploring additional parameters. The dispersity (Đ) was relatively low (<1.2) for all samples, with the exception of 0.5 mL min−1 (entry 1). This further corroborates the hypothesis of poor mixing at very low flow rates, which is ultimately reflected in the moderately higher dispersity.
 |
| | Scheme 1 Oscillatory plug flow polymerization of lactide using benzyl alcohol initiator. | |
Table 1 Summary of parameter investigation. Selection of the optimal setting was based on high conversions (>90%) with low dispersities (<1.2)
| Entry |
Flow rate (mL min−1) |
Amp. (mL per stroke) |
Freq. (Hz) |
[LA]/[BA] |
M
n,trgta (kg mol−1) |
M
n,exptb (kg mol−1) |
Conv. (%) |
Đ
|
|
Target molar masses are calculated from the LA to BA ratios, assuming 100% monomer conversion.
Experimental molar masses are determined from end-group analysis using 1H NMR spectroscopy.
Dispersity was determined from SEC in chloroform eluent relative to polystyrene standards.
|
| 1 |
0.5 |
0.04 |
0.6 |
100 |
14.4 |
17.5 |
35 |
1.39 |
| 2 |
1.0 |
0.04 |
0.6 |
100 |
14.4 |
11.4 |
95 |
1.16 |
| 3 |
2.0 |
0.04 |
0.6 |
100 |
14.4 |
15.7 |
82 |
1.11 |
| 4 |
5.0 |
0.04 |
0.6 |
100 |
14.4 |
2.4 |
49 |
1.12 |
| 5 |
1.0 |
0 |
0.0 |
100 |
14.4 |
14.2 |
79 |
1.16 |
| 6 |
1.0 |
0.04 |
0.6 |
100 |
14.4 |
14.4 |
84 |
1.15 |
| 7 |
1.0 |
0.08 |
0.6 |
100 |
14.4 |
14.2 |
77 |
1.20 |
| 8 |
1.0 |
0.08 |
1.2 |
100 |
14.4 |
12.2 |
72 |
1.22 |
Similarly, the effect of pulsation frequency on monomer conversion and dispersity was also investigated. The lowest tested frequency (0.6 Hz) combined with a low amplitude (0.04 mL per stroke) provided the highest conversion along with low dispersity (Đ = 1.15). Notably, in the absence of pulsation, the reaction efficiency declined marginally, while the dispersity was not adversely affected. It is postulated that no pulsation has a minimal effect in these circumstances owing primarily to the fact that the initiator and monomer are already pre-mixed before injection into the reactor. The effect of pulsation becomes much more pronounced in experiments that require mixing of initiator and monomer inside the reactor (vide infra, block polymer synthesis). In contrast, increasing the pulsation amplitude above 0.04 mL per stroke (5%) impacts polymerization efficiency, evident in the lower conversion and lower molar mass indicated by size exclusion chromatography (SEC) (Fig. 2, Table 1). Likewise, the dispersity increases very slightly with increased pulsation amplitude (entry 6–8). These results align with the flow profile analysis where a similar trend was observed using a polymer solution with comparable viscosity. Overall, pulsation amplitude contributes more significantly to the product characteristics for the ring-opening polymerization of lactide. Low pulsation amplitude enables efficient mixing without compromising the compact plug flow behavior (i.e., RTD). Notably, in the simple comparison of ROTEP using related organocatalysts in traditional tubular flow reactors, similar conversion and dispersities are achieved within the OFR reactor. The real highlight of the OFR in this context is in scalability. Increasing tube diameter in an effort to accommodate higher flux leads to lower conversion and broader RTD, reflected by higher dispersities.47
 |
| | Fig. 2 Summary SEC chromatograms of different individual parameter variation on molecular weight and distributions. (A) The effect of pulsation amplitude. (B) The effect of pulsation frequency. (C) The effect of flowrate. All polymers had a target molar mass of 14.4 kg mol−1. | |
Poly(lactide) synthesis using variable monomer to initiator ratio
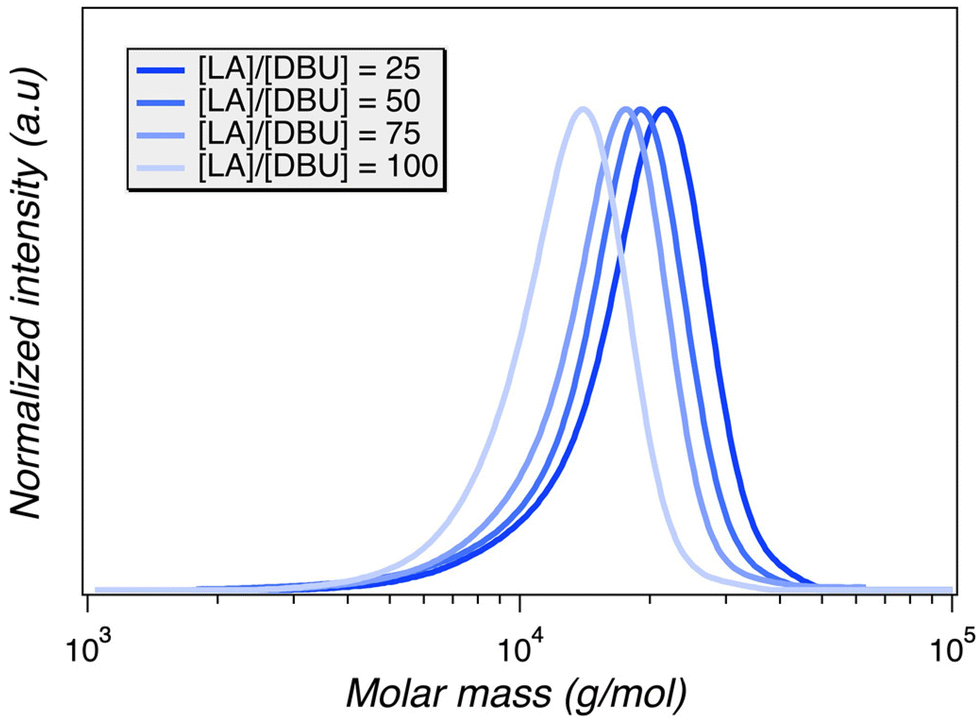
The ability to prepare a series of different polymers in a single stream by varying the individual reactant input rates is one of the most appealing advantages of using flow technology. We set out to vary the monomer to initiator ratio ([LA]:[BA]) in situ, which required multiple syringe injection ports that are independently controlled. Aiming for an operationally simple approach, the number of ports was kept at two. The catalyst was therefore included in the initiator solution. This means that changing the relative monomer and initiator feed rates will accompany a change in catalyst concentration. Therefore, the range of catalyst (i.e., DBU) concentrations that were suitable for achieving desired product targets was initially explored. A preliminary experiment was carried out to estimate a rate constant by varying the lactide to DBU catalyst ratio for various plugs. 1H NMR analysis of monomer conversion revealed a direct relationship with DBU concentrations; more catalyst led to higher monomer conversion over the same residence time (i.e., flow rate). This data was used to roughly estimate a rate constant (Fig. 3, Fig. S4,†Table 2). In this manner, the rate constant enables us to design a system wherein the flow rates are adjusted in situ to reach maximum conversion, taking into account the different target ratios of [LA]:[BA]. The operational conditions are simply tuned to compensate for the variable DBU concentration as the target molar mass is changed, employing the optimal time needed to reach 90% monomer conversion. Importantly, the series of PLA samples was generated in a single experiment, wherein the flow rates of the catalyst solution were merely adjusted relative to monomer flow rates to generate independent plugs corresponding to the indicated [LA] to [DBU] ratios.
 |
| | Fig. 3 SEC chromatograms corresponding to the samples of PLA using various concentrations of DBU catalyst per plug. All samples were generated at fixed flow settings deemed optimal following the flow profile analysis (1 mL min−1; 0.6 Hz; 0.04 mL per stroke). | |
Table 2 Data correlating to the DBU reaction kinetics investigation employing a constant RTD via 1 mL min−1 flow rate, 0.6 Hz frequency, and 0.04 mL per stroke amplitude
| [LA]:[DBU] |
Conv.a (%) |
M
na (kg mol−1) |
Đ
|
|
Conversion and experimental molar masses determined from end-group analysis using 1H NMR spectroscopy.
Dispersity was determined from SEC in chloroform eluent relative to polystyrene standards.
|
| 25 |
95.3 |
20.5 |
1.20 |
| 50 |
94.0 |
13.5 |
1.17 |
| 75 |
76.1 |
14.6 |
1.14 |
| 100 |
42.5 |
9.1 |
1.12 |
This crude kinetic data was used to guide the setup for an experiment in which the target molar mass was adjusted through the LA to BA ratios. One syringe was loaded with LA in CH2Cl2 (1 M) and another smaller syringe was loaded with both initiator (BA) and catalyst (DBU) at a concentration that was determined in order to accommodate a reasonably wide molar mass window without compromising kinetics. However, this design inherently necessitates a change in the overall flow rate to compensate for the lower catalyst concentrations, which is exacerbated in samples with relatively large target molar mass. Decreasing initiator concentration indeed led to an increasing trend in the product molar mass, albeit much less pronounced than expected as indicated by small shifts in the chromatograms (Fig. S5 and Table S1†). In fact, 1H NMR suggested no increase in molar mass between samples for which the ratio of [LA]:[BA] concentration was increased from 200 to 300. Additionally, the obtained molar mass for all samples as measured by 1H NMR was significantly lower than the target molar mass suggesting an acute change in catalytic activity with the different concentrations. Lower concentrations of DBU may lead to retarded reaction kinetics that were not apparent in the initial investigation of a systematic DBU decrease. Operational simplicity is an important factor moving forward, wherein the number of injection ports was strictly limited to two. A critical design criterium is that the initiator and catalyst should be combined in one syringe. In order to access the broadest array of polymer makeup through adjusting the individual feed rates, the catalyst activity should be as high as possible. This decouples the relative feed rates from the final LA monomer conversion. For this reason, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) was investigated in further experiments due to its strong basic properties and high reactivity.49
LFL triblock copolymer synthesis in flow
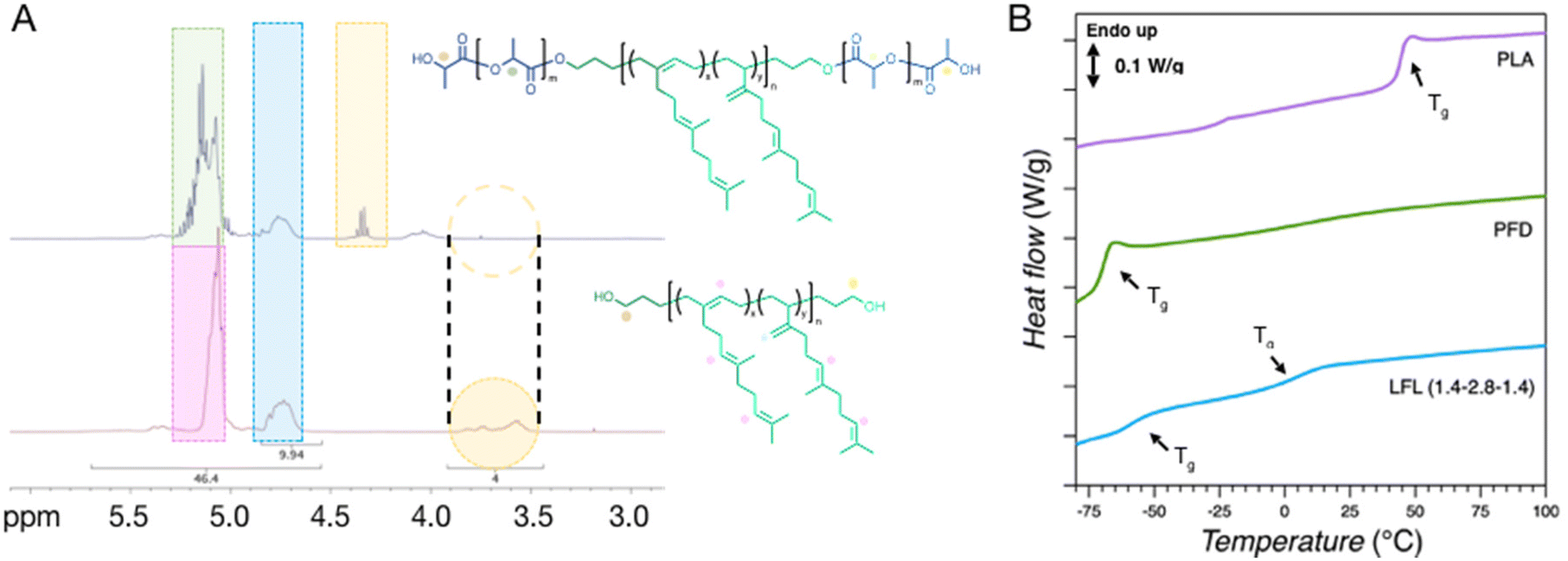
In a further extension of this methodology, telechelic poly(β-farnesene)-diol (PFD) was used as a macroinitiator to generate ABA-type triblock copolymers from the ROTEP of LA. The commercial PFD was provided by Cray Valley (tradename KRASOL F3000) with a molar mass of 2.8 kg mol−1 verified by 1H NMR spectroscopy using end-group analysis (Fig. S6†). Performing the ROTEP using the telechelic PFD ideally leads to symmetric triblock copolymers with final compositions targeted by adjusting the flow rates of PFD relative to LA using two independent syringe feeds. Feed ratios are adjusted by taking into account the anticipated LA monomer conversion. Triblocks are labeled as LFL (X:Y), where X and Y are the relative mass ratios of LA and PFD in the feed, respectively. The reaction setup using PFD was analogous to the previously described experimental protocol using benzyl alcohol, but employing a more active catalyst, TBD (Fig. 4). Initially a 1:1 weight ratio of PLA to PFD was targeted to generate LFL (1:1). A comparison was initially made between DBU and TBD catalysts using various flowrates, unambiguously revealing that TBD resulted in higher conversions across a range of residence times (Fig. S7†). Nearly complete polymerization was even achieved at 5 mL min−1 flow rate with TBD, generating polymers with relatively low dispersities. This flow rate translates to >80 g h−1 of symmetric triblock polymers at a LA concentration of 1 M. Optimized experimental conditions resulted in consistently low dispersities (Đ ∼ 1.3) and relatively high conversion (>90%) for all evaluated flow rates (Table S2†). 1H NMR analysis of the samples using TBD in CHCl3 suggested that the product molar mass was consistent across the range of residence times investigated and experimental values were in good agreement with the target Mn of 5.6 kg mol−1 (Fig. S7 and Table S2†). Additionally, the 1H NMR spectra of the ABA block copolymers suggest that initiation was essentially quantitative from the poly(farnesene)-diol owing to the transition of characteristic end-group signals as the hydroxyl groups are transformed to esters (Fig. 5A).
 |
| | Fig. 4 Schematic overview of LFL triblock polymer synthesis using the HANU flow reactor. | |
 |
| | Fig. 5 (A) 1H NMR analysis comparing commercial PFD and the LFL triblock copolymer. (B) DSC analysis of the synthesized LFL triblock copolymer, as well as PLA and the commercial PFD. Characteristic Tgs are labelled for each sample. | |
The block architecture was further corroborated with differential scanning calorimetry (DSC). Characteristic glass transition temperatures (Tg) corresponding to both the PFD and PLA blocks can be observed in the block copolymer product, suggesting successful incorporation of the PLA substituents onto the poly(farnesene)-diol macroinitiator (Fig. 5B). The Tg corresponding to the PLA blocks is significantly depressed compared with PLA homopolymer. This is consistent with the relatively low molar mass of the PLA blocks and the tethered architecture in the ABA block copolymer, restricting segmental motion.51
LFL series synthesis in flow
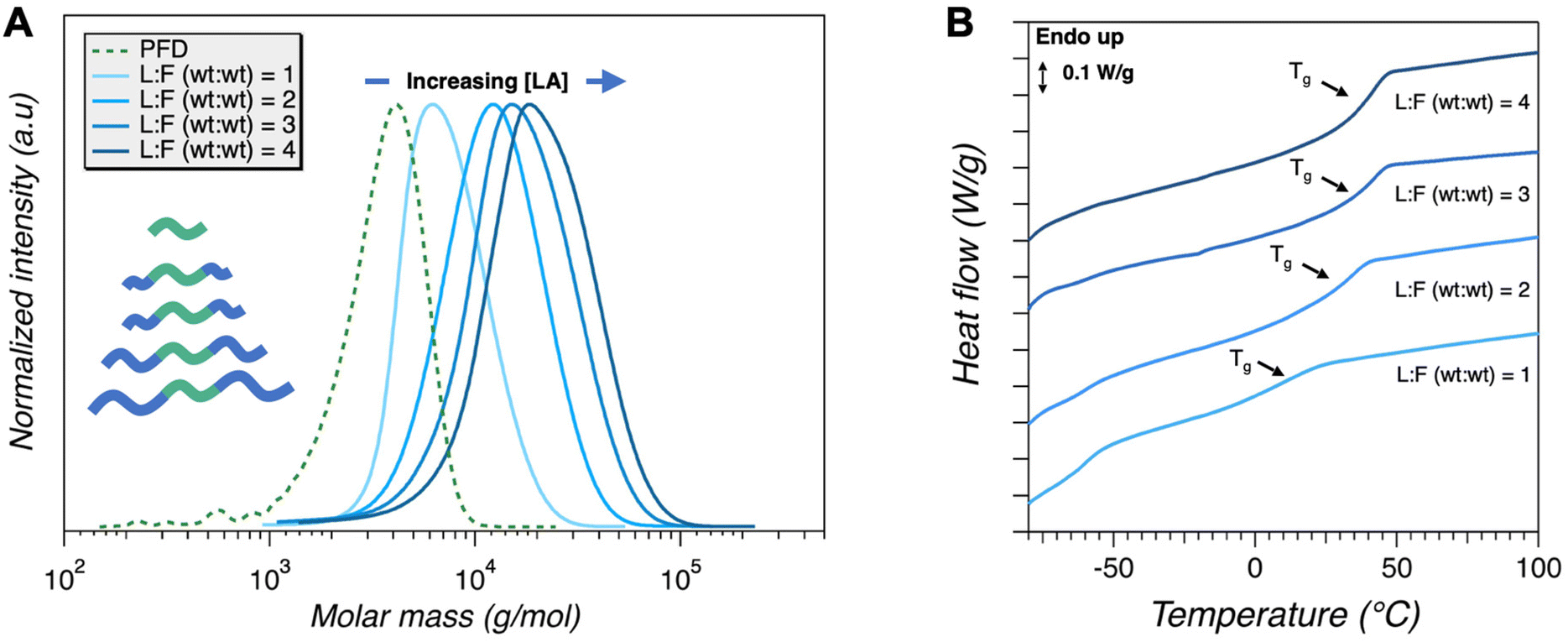
Varying the target composition of the symmetric LFL triblock copolymers in situ was performed similarly to the analogous experiment using benzyl alcohol. However, because the more active TBD catalyst has been employed, the residence time/flow rate compensation is unnecessary with changing monomer concentration. This was because previous results suggest the polymerization reaction was nearly complete in all cases, irrespective of residence time. High conversion (≥94%) was achieved for all target compositions of PLA according to 1H NMR spectroscopy and molar masses (Mn) were in good agreement with theoretical values (Table 3). This was confirmed using 1H NMR analysis showing a gradual increase in corresponding PLA peak intensity (Fig. S8†), as well as SEC, where increased molar mass relative to PFD precursor is apparent as the concentration of LA per plug was increased (Fig. 6A). Surprisingly, the dispersity increased moderately with each consecutive sample, reaching a value of Đ = 1.5 for the last two compositions. The origin of this unexpected result was investigated by performing the experiment again, starting with the highest wt:wt ratio, targeting LFL (4:1). This composition was targeted in four sequentially prepared samples, with increasing pulsation frequencies (0.6 Hz, 1.2 Hz, 1.8 Hz, and 2.4 Hz). The results suggest optimal conversion was achieved for all pulsations whilst the dispersity remained relatively constant and was generally in agreement with the previous experiment (Fig. S9 and Table S3†). These results suggest that the previously observed moderately high dispersity with higher molar masses is not related to the higher lactide concentration in itself, nor is it attributed to different pulsation frequencies. Rather, it is posited that this is an artifact of residual reagents from consecutive plugs. A small volume (<0.5 mL) of each plug inadvertently remains in a small section of tubing in syringe injection port and the T-crossing connected to the HPLC pump. As the ratio of PFD to LA varies across sequential plugs, any residual reagents of a different concentration ratio will contribute to the subsequent sample and thus increase the dispersity. This problem will be overcome in future investigations by briefly flushing the tubing with the desired reagent ratio prior to loading the plug.
 |
| | Fig. 6 (A) SEC analysis and (B) DSC thermograms of LFL triblock copolymers with various compositions. | |
Table 3 Analysis of poly(lactide-b-farnesene-b-lactide) block copolymer synthesis in flow via in situ tuning of wt:wt ratios of lactide to poly(farnesene)-diol. All experiments were performed at 2 mL min−1 using 0.04 mL per stroke pulsation at 0.6 Hz
| [LA]:[PFD] (wt:wt) |
[LA]:[TBD] (mol:mol) |
M
n (theo) (kg mol−1) |
Conv. (%) |
M
n (NMR) (kg mol−1) |
M
n (SEC) (kg mol−1) |
φ
L
|
Đ
|
| 1:1 |
57 |
5.6 |
94 |
5.1 |
6.8 |
0.45 |
1.24 |
| 2:1 |
114 |
8.6 |
95 |
8.1 |
9.6 |
0.65 |
1.34 |
| 3:1 |
170 |
11.5 |
98 |
10.1 |
11.5 |
0.72 |
1.51 |
| 4:1 |
225 |
14.4 |
96 |
14.9 |
15.2 |
0.81 |
1.51 |
Complementary block polymer synthesis targeting identical compositions was performed in the OFR in the absence of any pulsation. Chromatograms from SEC analysis have markedly different distributions (Fig. S10 and Table S4†). The associated dispersities for all samples are significantly larger, consistent with poor mixing compared to the samples prepared with the pulsator on. This is in contrast to the results from homopolymer synthesis, wherein the dispersity was not substantially influenced by pulsation. The block polymer synthetic protocol differs in a critical aspect, with the (macro)initiator and the LA monomer being injected from different syringes. This setup is necessary to enable the streamlined synthesis of a library of compositionally variable samples in a single experiment. However, the independent injection of initiator and monomer also requires intimate mixing to occur immediately upon injection, which is made more challenging with the relatively higher viscosity macroiniator solution of PFD. Thus, the pulsation provides an essential mixing of the reactants in order to achieve uniform polymerization conditions (i.e., low dispersity), which was practically unnecessary during the synthesis of PLA homopolymers.
Finally, DSC was performed on the different triblock copolymers (Fig. 6B). An unambiguous increase of the Tg of the PLA component is observed as L:F (wt:wt) increases (i.e., increasing PLA content). As the length of the PLA chain increases in the LFL triblock copolymer, the free volume of the end groups will increase, thereby inducing a higher Tg as described by the Flory – Fox equation.51 These data are consistent with the different compositions indicated by molecular analysis and the correspondingly varying molar mass of the PLA blocks.
Conclusion
An oscillatory flow reactor (i.e., HANU flow reactor) was used to perform continuous polymerization reactions for the first time. Through initial dye tracer experiments, visual observations were conducted to evaluate the effect of flow pulsation on the flow profile behavior. The obtained data revealed that the implementation of an oscillatory flow in constrained process channel dimensions can have a considerable effect on the polymer product outcome. These first observation will be critical to further assess polymerization studies carried out using this novel flow reactor type system. Providing an overview of the crucial process parameters and the possibility to obtain on-demand control over your polymerization characteristics highlight the added value of continuous polymerization in OFRs. Additionally, we were able to verify the flow profile analysis using the ring opening transesterification polymerization (ROTEP) of D,L-lactide. Follow-up experiments indicated that, when optimized with an efficient organocatalyst, targeted molar masses of ABA-type poly(lactide-b-farnesene-b-lactide) triblock copolymers could be synthesized at relatively high flow rates and high conversions via in situ modulation of the reagent concentrations. A change in various molecular properties was then confirmed using a variety of analytical techniques demonstrating the efficiency and tuneability with which the HANU flow reactor can be utilized to achieve specific polymer architectures for various commercial purposes. We believe that this study has provided the foundation for future polymer research using OFRs in order to realize polymer architectures of increasing complexity with uncompromised efficiency, accuracy, and eventual scalability. Some critical challenges that remain include ensuring more controlled molecular attributes by systematic adjustment of process parameters, reflected foremost in low dispersity. Further, pushing the boundaries toward high molar mass materials, with correspondingly high viscosity remains. And lastly, maintaining the control over molecular attributes while pushing toward high throughput will be a challenge, keeping a keen eye on scale-up.
We think the use of such an OFR is very powerful technology, particularly enabling in the creation of libraries of compositionally variable multi-component or multi-segment polymer constructs. While some optimization is likely necessary as different systems (e.g., different mechanisms, reactant concentrations) are explored in terms of pulsation settings, some universally advantageous conditions are almost certainly identifiable. We are keen to interrogate other systems in order to uncover such features with this versatile polymerization technology.
Author contributions
L. M. P., M. D. H. and H. G. helped design the experimental approach and aided with data interpretation. M. D. H. conducted all of the experiments. All authors contributed to writing and editing the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We greatly appreciate the generous support of Creaflow B.V. for contributing the reactor equipment (i.e. HANU flow reactor), which was utilized to accommodate the polymerization reactions for this project. Partial financial support from Hasselt University under the project Bijzonder Onderzoeksfond (BOF) R-12788 (BOF22K04) is also greatly appreciated.
References
- H. B. Feng, X. Y. Lu, W. Y. Wang, N. G. Kang and J. W. Mays, Polymers, 2017, 9, 494 CrossRef PubMed.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- M. W. Matsen and F. S. Bates, J. Chem. Phys., 1997, 106, 2436–2448 CrossRef CAS.
- G. P. Baeza, J. Polym. Sci., 2021, 59, 2405–2433 CrossRef CAS.
- A. Phatak, L. S. Lim, C. K. Reaves and F. S. Bates, Macromolecules, 2006, 39, 6221–6228 CrossRef CAS.
- G. Fleury and F. S. Bates, Macromolecules, 2009, 42, 3598–3610 CrossRef CAS.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- J. Y. Zhang, D. K. Schneiderrnan, T. Q. Li, M. A. Hillmyer and F. S. Bates, Macromolecules, 2016, 49, 9108–9118 CrossRef CAS.
- Z. L. Li, M. Tang, S. Liang, M. Y. Zhang, G. M. Biesold, Y. J. He, S. M. Hao, W. Choi, Y. J. Liu, J. Peng and Z. Q. Lin, Prog. Polym. Sci., 2021, 116, 101387 CrossRef CAS.
- L. M. Pitet, B. M. Chamberlain, A. W. Hauser and M. A. Hillmyer, Macromolecules, 2010, 43, 8018–8025 CrossRef CAS.
- J. Bolton and J. Rzayev, ACS Macro Lett., 2012, 1, 15–18 CrossRef CAS PubMed.
- M. K. Mahanthappa, F. S. Bates and M. A. Hillmyer, Macromolecules, 2005, 38, 7890–7894 CrossRef CAS.
- N. Zaquen, M. Rubens, N. Corrigan, J. T. Xu, P. B. Zetterlund, C. Boyer and T. Junkers, Prog. Polym. Sci., 2020, 107, 101256 CrossRef CAS.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, ACS Macro Lett., 2020, 9, 123–133 CrossRef CAS PubMed.
- A. Nagaki, A. Miyazaki and J. Yoshida, Macromolecules, 2010, 43, 8424–8429 CrossRef CAS.
- J. Vandenbergh, T. d. m. Ogawa and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2366–2374 CrossRef CAS.
- A. Nagaki, K. Akahori, Y. Takahashi and J. Yoshida, J. Flow Chem., 2014, 4, 168–172 CrossRef.
- J. Vandenbergh, T. Tura, E. Baeten and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1263–1274 CrossRef CAS.
- B. Wenn, M. Conradi, A. D. Carreiras, D. M. Haddleton and T. Junkers, Polym. Chem., 2014, 5, 3053–3060 RSC.
- M. Chen and J. A. Johnson, Chem. Commun., 2015, 51, 6742–6745 RSC.
- N. Corrigan, D. Rosli, J. W. J. Jones, J. T. Xu and C. Boyer, Macromolecules, 2016, 49, 6779–6789 CrossRef CAS.
- W. J. Huang, N. Zhu, Y. H. Liu, J. Wang, J. Zhong, Q. Sun, T. Sun, X. Hu, Z. Fang and K. Guo, Chem. Eng. J., 2019, 356, 592–597 CrossRef CAS.
- N. Zaquen, A. Kadir, A. Iasa, N. Corrigan, T. Junkers, P. B. Zetterlund and C. Boyer, Macromolecules, 2019, 52, 1609–1619 CrossRef CAS.
- M. Rubens, J. H. Vrijsen, J. Laun and T. Junkers, Angew. Chem., Int. Ed., 2019, 58, 3183–3187 CrossRef CAS PubMed.
- C. P. Breen, A. M. K. Nambiar, T. F. Jamison and K. F. Jensen, Trends Chem., 2021, 3, 373–386 CrossRef.
- M. Reis, F. Gusev, N. G. Taylor, S. H. Chung, M. D. Verber, Y. Z. Lee, O. Isayev and F. A. Leibfarth, J. Am. Chem. Soc., 2021, 143, 17677–17689 CrossRef CAS PubMed.
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- Y. H. Su, Y. Song and L. Xiang, Top. Curr. Chem., 2018, 376, 44 CrossRef PubMed.
- W. Debrouwer, W. Kimpe, R. Dangreau, K. Huvaere, H. P. L. Gemoets, M. Mottaghi, S. Kuhn and K. Van Aken, Org. Process Res. Dev., 2020, 24, 2319–2325 CrossRef CAS.
- C. Rosso, S. Gisbertz, J. D. Williams, H. P. L. Gemoets, W. Debrouwer, B. Pieber and C. O. Kappe, React. Chem. Eng., 2020, 5, 597–604 RSC.
- M. Wernik, G. Sipos, B. Buchholcz, F. Darvas, Z. Novak, S. B. Otvos and C. O. Kappe, Green Chem., 2021, 23, 5625–5632 RSC.
- P. Bianchi, J. D. Williams and C. O. Kappe, Green Chem., 2021, 23, 2685–2693 RSC.
- J. R. McDonough, S. Murta, R. Law and A. P. Harvey, Chem. Eng. J., 2019, 358, 643–657 CrossRef CAS.
- N. Zhu, W. Y. Feng, X. Hu, Z. L. Zhang, Z. Fang, K. Zhang, Z. J. Li and K. Guo, Polymer, 2016, 84, 391–397 CrossRef CAS.
- X. Hu, N. Zhu, Z. Fang and K. Guo, React. Chem. Eng., 2017, 2, 20–26 RSC.
- W. Adhami, Y. Bakkour and C. Rolando, Polymer, 2021, 230, 124040 CrossRef CAS.
- C. Bakkali-Hassani, J. P. Hooker, P. J. Voorter, M. Rubens, N. R. Cameron and T. Junkers, Polym. Chem., 2022, 13, 1387–1393 RSC.
- F. Della Monica and A. W. Kleij, Polym. Chem., 2020, 11, 5109–5127 RSC.
- G. John, S. Nagarajan, P. K. Vemula, J. R. Silverman and C. K. S. Pillai, Prog. Polym. Sci., 2019, 92, 158–209 CrossRef CAS.
- C. Wahlen and H. Frey, Macromolecules, 2021, 54, 7323–7336 CrossRef CAS.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS PubMed.
- J. Zhang, J. Chen, M. Yao, Z. Jiang and Y. Ma, J. Appl. Polym. Sci., 2019, 136, 47673 CrossRef.
- C. Zhou, Z. Wei, X. Lei and Y. Li, RSC Adv., 2016, 6, 63508–63514 RSC.
- Z. K. Wang, L. Yuan and C. B. Tang, Acc. Chem. Res., 2017, 50, 1762–1773 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3750 CrossRef CAS.
- Z. R. Zhong, Y. N. Chen, Y. Zhou and M. Chen, Chin. J. Polym. Sci., 2021, 39, 1069–1083 CrossRef CAS.
- M. H. Reis, T. P. Varner and F. A. Leibfarth, Macromolecules, 2019, 52, 3551–3557 CrossRef CAS.
- Y. Zhou, Y. Gu, K. M. Jiang and M. Chen, Macromolecules, 2019, 52, 5611–5617 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS PubMed.
- M. T. Martello and M. A. Hillmyer, Macromolecules, 2011, 44, 8537–8545 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, experimental procedures, and additional characterization data. See DOI: https://doi.org/10.1039/d2py00600f |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Hannes P. L.
Gemoets
b,
Koen
Van Aken
b and
Louis M.
Pitet
a,
Hannes P. L.
Gemoets
b,
Koen
Van Aken
b and
Louis M.
Pitet