
Backbone and side chain-linker tunability among dithiocarbamate, ester and amide in sequence-defined oligomers: synthesis and structure–property–function relationship†
Anna
Jose
and
Mintu
Porel
 *
*
Discipline of Chemistry, Indian Institute of Technology Palakkad, Environmental Sciences and Sustainable Engineering Center, Kerala-678577, India. E-mail: mintu@iitpkd.ac.in
First published on 15th February 2022
Abstract
Structural diversity and tunable properties achieved by a defined monomeric sequence are the trademarks of a sequence-defined polymer (SDP). Herein, we report a modular synthetic platform where, in addition to the provision of changing the side chain, the backbone functional groups of the SDP can also be tuned among three key functional groups, namely dithiocarbamate (DTC), ester and amide. An efficient synthetic strategy has been developed to incorporate DTC into the SDP backbone without the need for separate design of the monomer. This unique strategy with the potential to vary both the backbone as well as the side chain is, to the best of our knowledge, the first report in the field of man-made SDPs. Three different classes of sequence-defined oligomers (SDOs) have been synthesized: (i) SDO1 with DTC alone in the backbone; (ii) SDO2 with DTC and ester in the backbone; and (iii) SDO3 with DTC and ester in the backbone and amide in the side-chain linkage. The structure–activity relationships of the three SDOs were investigated, which is important for their biomedical and material applications. This led to the conclusion that SDO1 with DTC functionality was stable under conditions of hydrolysis, heat and acid treatment, whereas SDO2 (DTC and ester) and SDO3 (DTC, ester and amide) are not stable under those conditions. On the other hand, for applications related to toxic Hg2+ removal, SDO3, which contained all three functional groups (DTC, ester and amide), was the most efficient, followed by SDO2 (DTC and ester) with medium efficiency and SDO1 (DTC) with the lowest efficiency. The investigation of the protein binding affinity with serum albumin concluded that all three SDOs are transportable via serum proteins. Taken together, this is a proof-of-concept to explore SDOs with tunable backbones and side-chains to modulate their properties and employ the best suited SDO for any given application.
Introduction
The surge in demand for functional polymers in interdisciplinary fields goes hand-in-hand with the ceaseless progress that polymer chemistry has made to date. Advanced polymers with diverse functionalities, properties and architectures are vital. This calls for mild and efficient strategies for the conversion of functional monomers to advanced materials that are efficient, high yielding and amenable to large-scale purification. However, the inability to control the monomeric sequence in the conventional polymers leads to poor structural diversity and property tunability, indicating the pressing need for advancements in this field. Taking inspiration from natural sequence-defined polymers (SDPs), like nucleic acids and peptides, materials are being designed by the sequential addition of specific monomers at preordained positions. This results in synthetic SDPs1–9 with pendant group functionality, desirable chain length and defined monomeric sequence. However, all of the reports on the synthesis of SDPs focus on the same backbone with different hanging residues for a specific SDP class. In contrast, this report describes strategies for the synthesis of SDPs harbouring (a) the same backbone with different side chain residues and (b) different backbones with the same side chain residues. As a proof-of-principle, three important functional groups have been selected for this study: dithiocarbamate, amide and ester. The effect of these diverse functional groups in modulating the properties of the SDP was studied by choosing one application each in the material and biomedical field.Amide linkages are the basis of biological systems and are regarded as nature's connectors.10 The pervasiveness of this fundamental functional entity in proteins and peptides is seldom overlooked, leading to a delusion that the synthetic challenges to its formation are minimal. However, the use of expensive coupling reagents11 for the synthesis of amide linkages and the associated generation of harmful waste is one among the many factors that urge researchers to explore new methodologies for amide synthesis. Excellent mechanical strength, high temperature resistance, polarity, conformational diversity and tolerance to extreme oxidizing and reducing environments makes them a potential entity in the synthesis of biologically active compounds and synthetic materials. Their application, however, is restricted due to their infusibility and limited solubility in organic solvents. The incorporation of ester linkages is performed to improve the solubility of polyamides. Polymers with biodegradability find applications in packaging, materials and medicine. For example, polyesters undergo non-enzymatic hydrolysis resulting in the cleavage of the ester linkages. They also suffer from stability issues under acidic environments. However, polyamides, due to the formation of strong hydrogen bonds between individual chains, take a longer time to degrade. Thus, the above duo is applied for biomedical applications where both thermomechanical aspects and degradability cannot be jeopardized.12,13
The properties of a polymer, therefore, come by virtue of the careful addition of monomers that carry within them the properties that will be reflected on the macromolecular scale. For this, defined monomeric sequence, desirable chain length, and varied backbone and side-chain functionality is crucial as they impart structural diversity and tunable properties to the polymer. These are the hallmarks of a sequence-defined polymer (SDP) that have resulted in a shift of focus from the synthesis of conventional polymers to non-natural SDPs. Hence, we herein put forth strategies for the synthesis of different SDP scaffolds that are different from one another in terms of the functional moieties that each of them possesses, in addition to the scope of attaching different hanging groups via custom synthesis of the monomer. The different functional groups that are taken into consideration for the design are dithiocarbamate (DTC), amide and ester linkages. Maintaining DTC as a fixed functional entity, we attempted to synthesize three different sequence-defined oligomers (SDOs), namely (i) SDO1: with DTC alone in the backbone, (ii) SDO2: with DTC and ester in the backbone and (iii) SDO3: with DTC and ester in the backbone and amide in the side-chain linkage. All three SDO classes have provision to incorporate a wide range of side-chain functional groups. The rationale behind keeping DTC constant in all of the SDO scaffolds is that DTC is an excellent candidate for both material (e.g., heavy metal sensor5 and vulcanization accelerators in rubber industries14) and biomedical (e.g., antileishmanial agents,15 anti-acute myelogenous leukaemia agents,16 antitrypanosomatids,17 anticancer18) applications. This strategy for the synthesis of SDOs thus has a unique advantage of changing both the backbone as well as the side chain. Further comparison of the properties of these polymers will aid the use of these SDOs for various applications.
Results and discussion
In the present study, we developed a synthetic strategy for SDOs that does not involve separate effort for the design and synthesis of the monomers. The iterative addition of readily available inexpensive substrates was employed to prepare the SDO. The synthesis starts with a secondary amine (1, Scheme 1) reacting with carbon disulphide (CS2) and tert-butyl (2-bromoethyl)carbamate (the detailed synthetic procedure is in Scheme S1 in the ESI† and the 1H-NMR spectrum is in Fig. S22†) to form 2 with DTC functionality (Scheme 1). This was then reacted with an alkyl bromide in the presence of K2CO3 in acetonitrile (ACN) solvent to form 3 (tert-butyloxycarbonyl (Boc)-protected 2-mer, Scheme 1), which was further treated with 6 M HCl in methanol to yield the Boc-deprotected 2-mer, 4 (R1 and R2 are the two tunable pendant groups, Scheme 1). Subsequently, the steps 1, 2 and 3 were repeated to generate n-mer SDO1, 5. As a proof-of-concept, N,N-diethyl amine, 6, was reacted with CS2 and tert-butyl (2-bromoethyl)carbamate at room temperature in PEG-200 to form 7 (Fig. 1). This reaction between the secondary amine and CS2 involves the generation of an in situ thiol, which further undergoes a nucleophilic substitution of the terminal bromide of tert-butyl (2-bromoethyl)carbamate to form 7 (Scheme S2†). The quantitative conversion occurred in 30 min. The reaction mixture was washed with water, and the pure product was extracted in ethyl acetate. This compound was utilized for the next step without any further purification. In the next step, 7 was reacted with butyl bromide in the presence of K2CO3 as a base and acetonitrile as a solvent to form 8 (Scheme S3†). Quantitative conversion occurred in 12 hours and the reaction mixture was filtered to remove K2CO3. The reaction mixture was extracted with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/ethyl acetate mixture. The reaction mixture was purified via column chromatography and the pure product was taken for the next reaction. In the third step, 8 was treated with 6 M HCl to deprotect the amine group, which yielded 2-mer, 9 in a quantitative yield (Scheme S4†). High-performance liquid chromatography (HPLC) traces and LC-MS spectra of the products formed in each step are shown alongside each other in Fig. 1. 7 and 8 showed the expected [M + H]+ peaks at 293.25 and 349.25 Da, respectively, in the LC-MS spectrum (Fig. 1). 9, the Boc-deprotected 2-mer, showed the expected [M + H]+ peaks at 249.10 Da in LC-MS. Similarly, the reaction proceeded with propyl bromide and benzyl bromide under identical conditions to yield the 4-mer. The 3-mer (12) and 4-mer (14) were characterized by their [M + H]+ at 410.22 and 719.20 Da, respectively. The retention time of the HPLC traces indicates that the hydrophobicity of the SDO increases as the chain length increases (for example from 6 to 7) and the hydrophobicity decreases during the formation of 8, as it has a free secondary amine. The sequence was validated via a tandem-MS (MS/MS) experiment on the [M + H]+ ion of the 4-mer (Fig. 2). From the fragmentation, it was identified that the cleavage of SDO1a occurs between the β-carbon of the DTC sulfur and the nitrogen of the next DTC unit. An [M-100]+ fragment was also obtained, which is due to the cleavage of the Boc group. All the fragment ions from the MS/MS spectrum confirm the precise sequence of the diethyl-butyl-propyl-benzyl-groups in SDO1a. The final SDO was also characterized by 1H NMR (Fig. S25†).
:1 water/ethyl acetate mixture. The reaction mixture was purified via column chromatography and the pure product was taken for the next reaction. In the third step, 8 was treated with 6 M HCl to deprotect the amine group, which yielded 2-mer, 9 in a quantitative yield (Scheme S4†). High-performance liquid chromatography (HPLC) traces and LC-MS spectra of the products formed in each step are shown alongside each other in Fig. 1. 7 and 8 showed the expected [M + H]+ peaks at 293.25 and 349.25 Da, respectively, in the LC-MS spectrum (Fig. 1). 9, the Boc-deprotected 2-mer, showed the expected [M + H]+ peaks at 249.10 Da in LC-MS. Similarly, the reaction proceeded with propyl bromide and benzyl bromide under identical conditions to yield the 4-mer. The 3-mer (12) and 4-mer (14) were characterized by their [M + H]+ at 410.22 and 719.20 Da, respectively. The retention time of the HPLC traces indicates that the hydrophobicity of the SDO increases as the chain length increases (for example from 6 to 7) and the hydrophobicity decreases during the formation of 8, as it has a free secondary amine. The sequence was validated via a tandem-MS (MS/MS) experiment on the [M + H]+ ion of the 4-mer (Fig. 2). From the fragmentation, it was identified that the cleavage of SDO1a occurs between the β-carbon of the DTC sulfur and the nitrogen of the next DTC unit. An [M-100]+ fragment was also obtained, which is due to the cleavage of the Boc group. All the fragment ions from the MS/MS spectrum confirm the precise sequence of the diethyl-butyl-propyl-benzyl-groups in SDO1a. The final SDO was also characterized by 1H NMR (Fig. S25†).
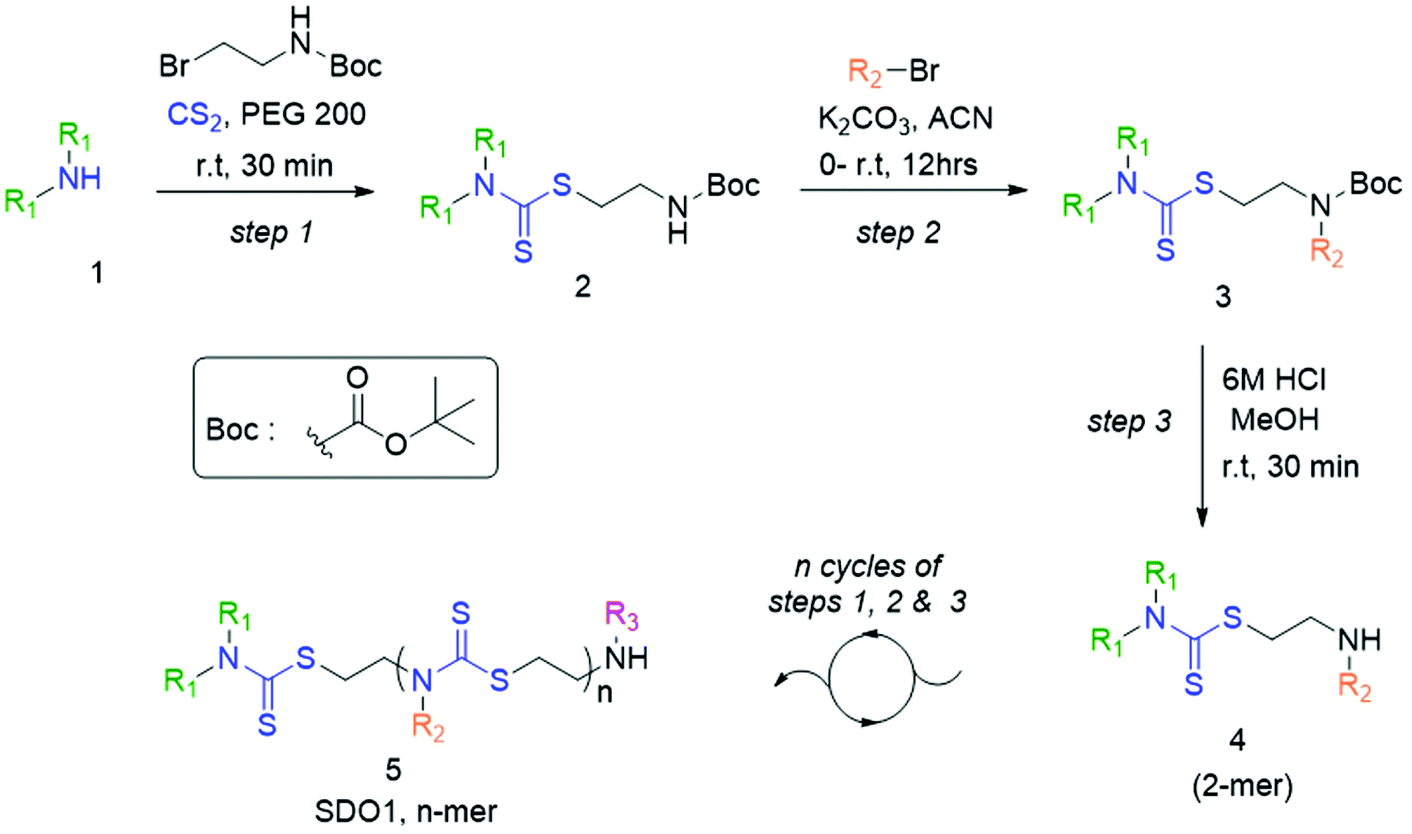 | ||
| Scheme 1 Synthetic strategy for sequence-defined oligomer, SDO1. The equivalents of the substrates taken in each step are indicated as follows: step (1) 1 eq. of 1, 1.2 eq. of tert-butyl (2-bromoethyl)carbamate and 1.2 eq. of CS2; step (2) 1 eq. of 2, 2 eq. of K2CO3 and 4 eq. of alkyl/aryl bromides and step (3) 1 eq. of 3 and 2 eq. of 6 M HCl. | ||
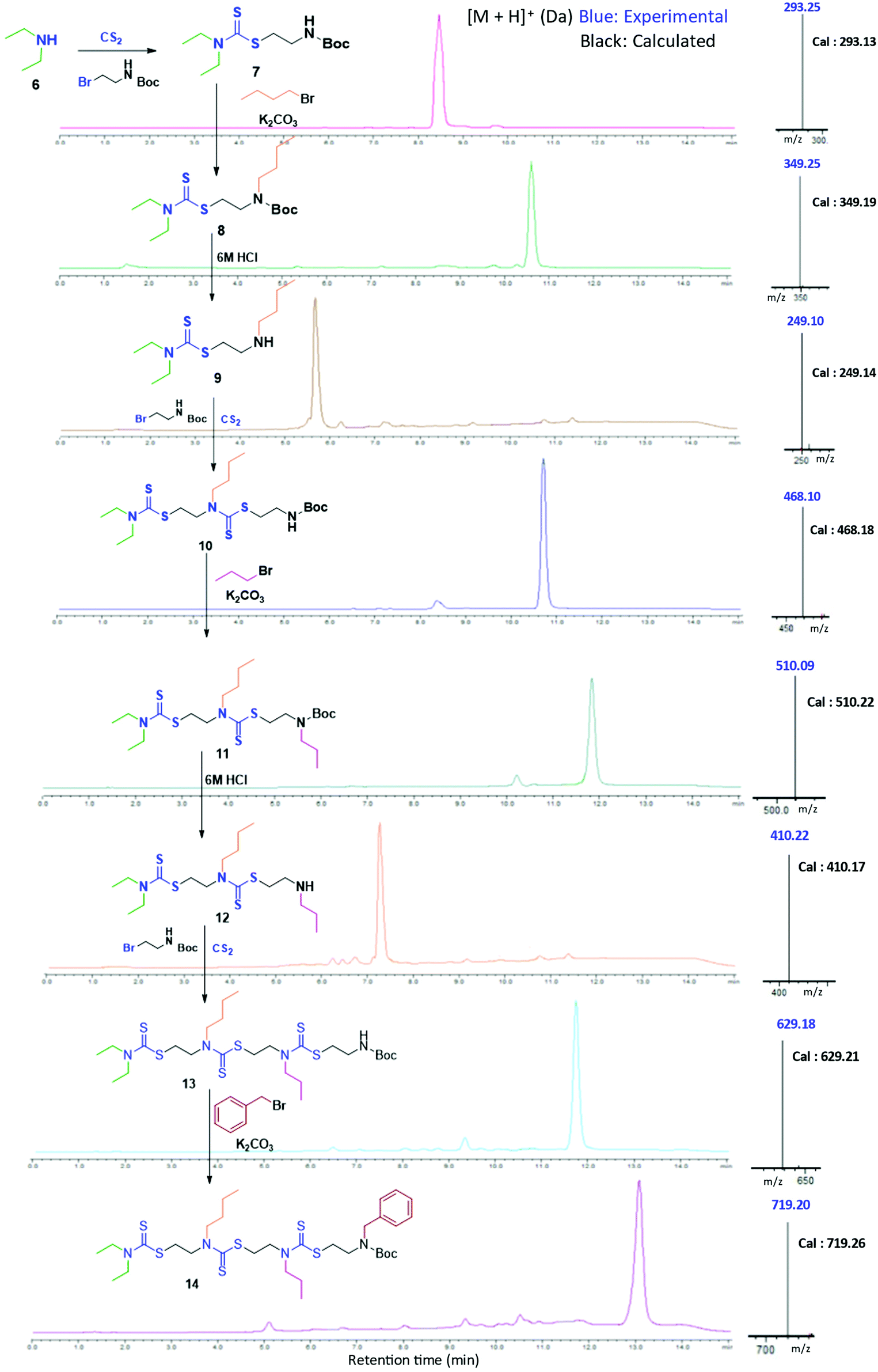 | ||
| Fig. 1 Assembly of 4-mer SDO1a; left – structure, middle – HPLC, and right – [M + H]+ in LC-MS. (Boc represents a C(O)OtBu group.) | ||
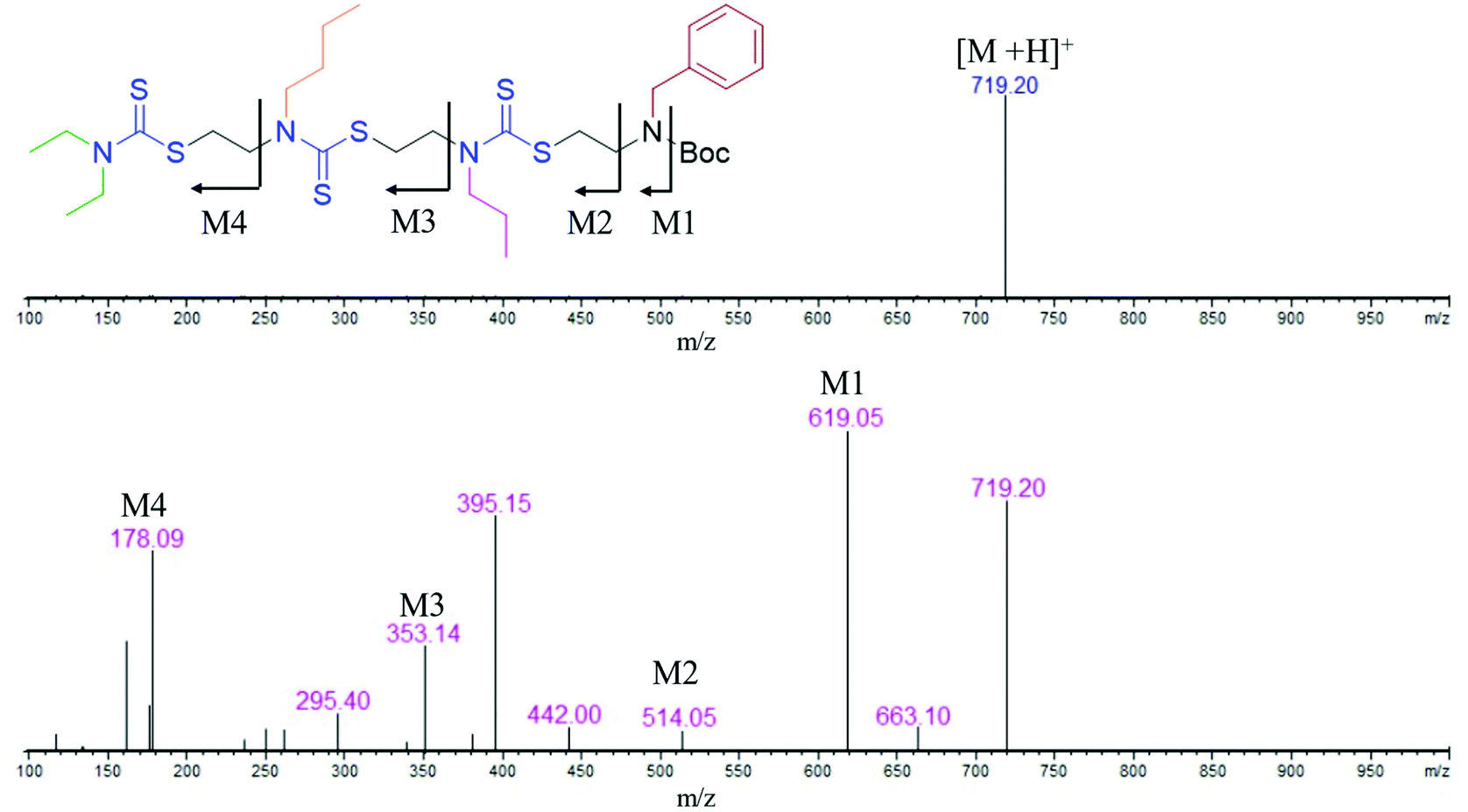 | ||
| Fig. 2 LC-MS (inset: structure, top) and tandem-MS spectrum (bottom) of the 4-mer SDO1a. | ||
Having synthesised this SDO with DTC alone, named as SDO1a, we wanted to explore how the presence of other functional groups in the backbone (in addition to the DTC group) influences the properties of the SDO at the macromolecular level. For this, two different SDOs were also synthesised so as to possess (a) ester and DTC in one SDO, named as SDO2, and (b) ester, amide and DTC in another, named as SDO3. The synthesis of SDO2 starts with the reaction of a secondary amine, here diethyl amine, with chloroacetyl chloride to form the chloroacetylated product (the detailed procedure is shown in the ESI† Schemes S10–S15). This then undergoes a three-component reaction with CS2 and alkyl/aryl ethanolamine (the 1H-NMR spectrum of the monomer used is in Fig. S23†) to form a 2-mer (Scheme S11†). The hydroxy group of the 2-mer was then reacted with chloroacetylchloride and triethylamine as a base (Scheme S12†). The same cycle of reactions was repeated until the formation of the 4-mer of SDO2 (Schemes S13–S15†). SDO3 was synthesized using our earlier reported strategy.6 As the main focus of this study was to investigate the effect of these three key functional groups (DTC, ester and amide) on the properties and applications of the SDO, the pendant group or side-chain of the SDO was kept constant. The structures of the synthesised SDOs, their HPLC traces and their characterization by mass spectrometry are shown in Fig. 3. The sequence validation via tandem-MS (MS/MS) is shown in Fig. S19–S21† and their characterisation by 1H NMR is shown in Fig. S25–S28.†
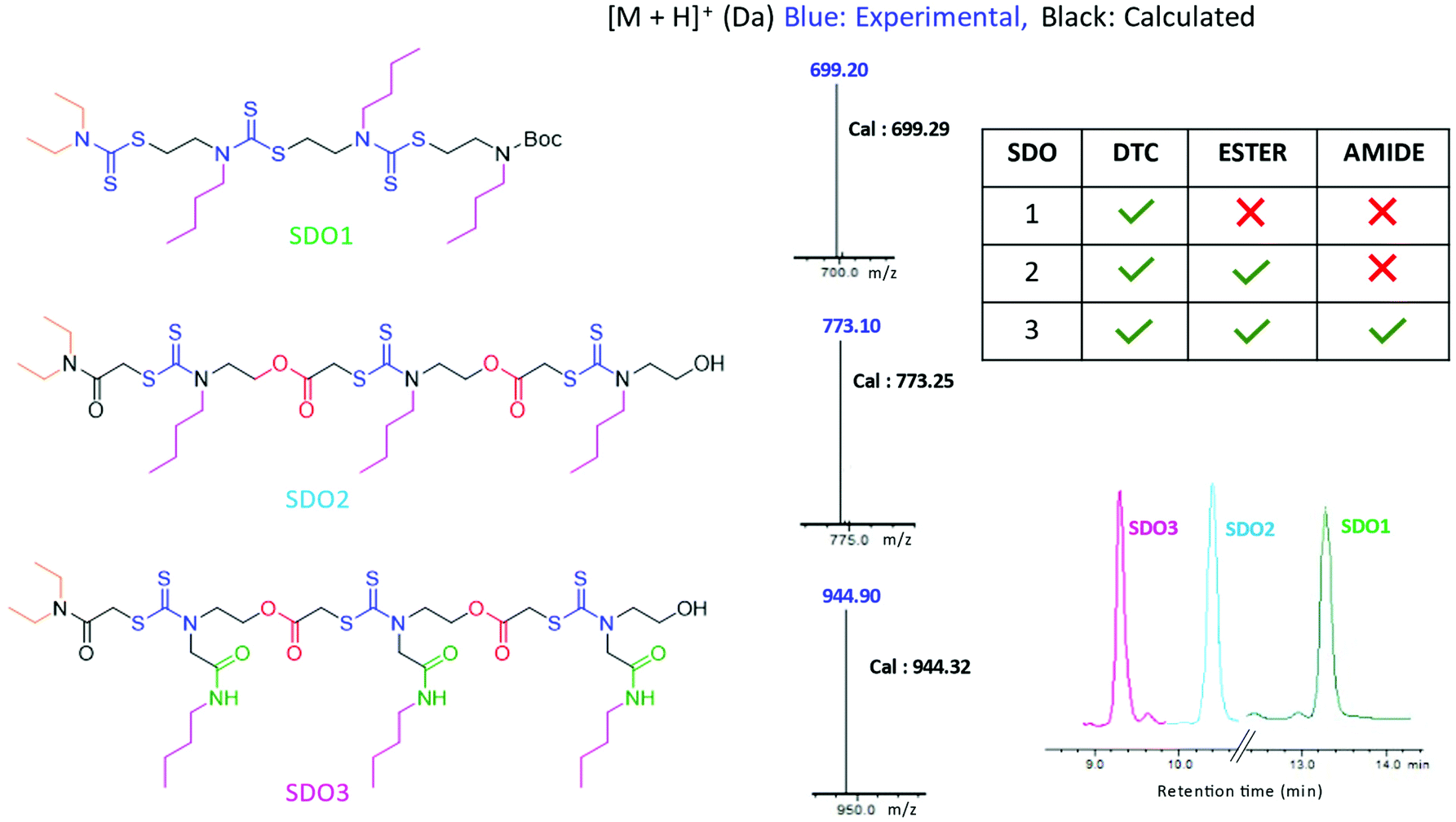 | ||
| Fig. 3 Structures of the SDOs taken for comparison on the left, middle – [M + H]+ in LC-MS and right – HPLC (bottom) and tabular representation of the functional groups present in each SDO (top). | ||
The hydrophobicity of the synthesised SDOs increases in the order SDO3 < SDO2 < SDO1. This is reflected in the HPLC traces of all three SDOs (Fig. 3), as the retention time in HPLC is proportional to the hydrophobicity of the system. This is due to the addition of polar groups (ester and amide) on the backbone, which helps to reduce the hydrophobicity. As it is well-known that acidic conditions have a strong influence on the stability of ester and amide groups,19 acid degradation studies were carried out with 6 M HCl + methanol (1:1) at room temperature. The solutions of SDO1, SDO2 and SDO3 in methanol were mixed with 6 M HCl solution and these were analysed by LCMS after 12 hours. It was found that SDO2 and SDO3, which possess ester and (ester + amide) linkages respectively, were not stable in acidic conditions, whereas SDO1 without ester/amide (DTC alone on the backbone) was stable. The analysis of the acid degradation studies indicated that the fragment ions obtained were due to the cleavage of the ester linkages in the case of SDO2 and that of the ester and amide linkages in the case of SDO3 (Fig. S29–S31 and Schemes S19–S23†). Additionally, the stability of the synthesised SDOs was tested under hydrolysis conditions. For this, methanolic solutions of SDO1, SDO2 and SDO3 were mixed with equal volumes of water and were heated at 70 °C for 12 hours. The LC-MS analysis was done after 12 hours to analyse the fragments ions that were formed. Here also, only SDO1 was stable and the fragment ions formed in the case of SDO2 and SDO3 were due to the cleavage of the ester and amide linkages (Fig. S32–S34 and Schemes S24–S26†). The stability of the SDOs under heating conditions was tested by heating the methanolic solutions of SDO1, SDO2 and SDO3 at 70 °C for 12 hours. The LC-MS analysis after 12 hours (Fig. S35–S37 and Scheme S27, S28†) gave similar results as obtained for degradation under acidic and hydrolysis conditions.
The thermal stability of the synthesised SDOs was tested via TG–DTA analysis, where the SDOs were heated at a rate of 10 °C min−1 under a nitrogen atmosphere (Fig. S38†). It was found that SDO1 was stable up to temperatures of 200 °C without any mass loss, whereas SDO2 and SDO3 were stable up to around 160 °C and 140 °C. Mass loss up to 60–80% was observed after 230 °C.
To explore the effect of the presence or absence of ester/amide groups on the DTC-based SDO in terms of its applications, one example of a material science application and one example of a biomedical application were investigated. The presence of toxic heavy metal ions, especially Hg2+ ions, has been a subject of concern since the advent of the industrial revolution. Methods to mitigate the levels of toxic mercury from environmental samples are extensively delved into, even today. With prior knowledge on the interaction of compounds containing sulfur, nitrogen and oxygen with mercury,20–22 the applicability of the synthesised SDOs towards mercury removal was explored as a material application. Recently, synthetic SDOs have proved their potency for various biomedical applications.23 To investigate the transportability of the synthesised SDOs through serum, the protein binding affinity of the SDOs was determined with a model protein, serum albumin. The results for the Hg2+ removal capacity and protein binding are discussed below individually.
The application of the synthesised SDOs towards Hg2+ ion removal was tested by adding the different SDOs (4 mM) to Hg2+ solution (12 mM). It was found that a precipitate was formed upon the addition of SDO3 to the Hg2+ solution. Mild precipitation was found in the case of SDO2 and there was slight turbidity in the solution for SDO1 (Fig. S39†). To confirm the efficiency of mercury removal, the supernatant solution was analysed by ICP-MS (inductively coupled plasma-mass spectrometry). The concentrations of Hg2+ in the supernatant for all three SDOs obtained after ICP-MS are tabulated in Table 1. This shows that the mercury removal efficiency was obtained in the order SDO3 > SDO2 > SDO1. The self-assembled structures formed due to the non-covalent interactions between Hg2+ and DTC groups present in each SDO are the basis for Hg2+-mediated precipitation. Also, in addition to the DTC functionality, the presence of a greater number of carbonyl groups from the ester and amide linkages, neighbouring to DTC, is helpful in increasing the binding efficiency to Hg2+ ions in the case of SDO3. This indicates that SDO3 with more carbonyl groups in it, favours higher Hg2+ binding. SDO2 has fewer adjacent carbonyl groups as compared to SDO3, but more than SDO1, and thus has an intermediate binding with Hg2+. This concludes that in addition to the DTC group, the presence of amide and ester linkages is important for the removal of Hg2+ ions.24–26 It was also found that the precipitation was exclusive for Hg2+ ions. The precipitates obtained from the Hg2+ removal studies were characterised using scanning electron microscopy/energy dispersive X-ray (SEM/EDAX) analysis. The SEM images (Fig. S40 and S41†) and the elemental analysis by EDAX also confirmed the presence of mercury in the precipitate.
| SDO | Hg2+ concentration in the supernatant (ppm) | Removal efficiency from ICP-MS (±5%) | |
|---|---|---|---|
| Initial | Final | ||
| SDO1 | 240 | 148.4 | 38.1% |
| SDO2 | 240 | 84.71 | 64.7% |
| SDO3 | 240 | 21.69 | 90.9% |
Serum albumin is the most abundant protein in blood and is the carrier for drug molecules.27,28 Being an analogue of human serum albumin (HSA), the interactions of synthetic molecules with bovine serum albumin (BSA) are extensively studied in the medicinal field to examine their pharmacokinetics.29,30 To determine the protein binding ability of the synthesised SDOs, they were treated with BSA and the fluorescence spectra were recorded. For this, a 10 μM solution of BSA solution in phosphate buffer was taken and was titrated with different SDO solutions (0.2 to 30 μM). The fluorescence spectra were recorded after each addition. It was found that the addition of SDOs resulted in the quenching of the fluorescence of BSA, indicating that the SDOs were interacting with the protein. The binding constant values for the titration with SDO1, SDO2 and SDO3 were calculated with Stern–Volmer plots (Fig. 4, Fig. S42 andS43†) and they were 2.05 × 105 M−1, 1.26 × 105 M−1 and 1.17 × 105 M−1, respectively (Table 2). These values are comparable with the reported systems29,31 and hence the synthesised SDOs can be considered to be potential candidates for biomedical applications. Also, to see the mode of interaction of these diverse SDOs with BSA, molecular docking studies were performed and the obtained receptor–ligand interactions are represented in the 2D image (Fig. S44–S46†). It was found that both the hydrophobic side chains and the main link motifs are interacting with the model protein efficiently via van der Waals interactions, hydrogen bonds and pi–sulphur interactions.
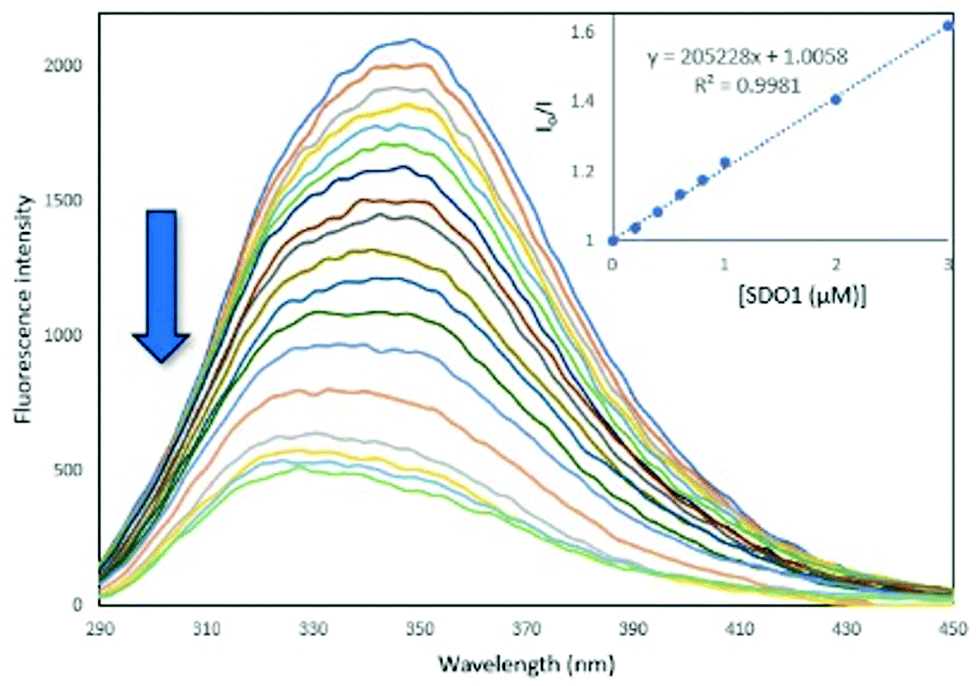 | ||
| Fig. 4 The fluorescence titration of SDO1 (0.2–30 μM) solution with BSA (10 μM) in phosphate buffer at an excitation wavelength of 280 nm at 25 °C. | ||
| SDO | Binding constant (M−1) |
|---|---|
| SDO1 | 2.05 × 105 |
| SDO2 | 1.26 × 105 |
| SDO3 | 1.17 × 105 |
In this manner, the comparative study on the three diverse SDOs efficiently proves that different functional moieties play an integral role for varied applications. The presence of amide and ester linkages in SDO2 and SDO3 makes them unstable in acidic/heating/hydrolysis environments, whereas the absence of those in SDO1 renders it more stable. On the other hand, as a removal system of Hg2+ ions, the efficiency of the SDOs increases in the order of SDO1 < SDO2 < SDO3. All of the SDOs (SDO1, SDO2 and SDO3) proved their biomedical value via binding with serum albumin proteins. This indicates that the precise use of SDOs with fitting functional groups is vital for applications in diverse domains.
Conclusion
An efficient strategy for the synthesis of a sequence-defined oligomer (SDO) was established via the iterative addition of commercially available inexpensive substrates, without the need for monomer design. The precise sequence of the oligomer was validated by tandem MS spectra. The properties of this new SDO (DTC backbone) were compared with two other SDOs, one having ester and DTC linkages and the other having ester, amide and DTC linkages. The comparison of HPLC traces of all three SDOs indicates the increase in hydrophobicity from SDO3 < SDO2 < SDO1. The stability of the synthesised SDOs was compared in acidic medium, hydrolysis and heating conditions. In all cases, SDO1 with DTC functionality alone showed excellent stability. However, for material applications like toxic Hg2+ removal, SDO3, which contained all three functional groups (DTC, ester and amide), was the most efficient followed by SDO2 (DTC and ester) with medium efficiency and SDO1 (DTC) with the least efficiency. Protein binding was studied and all three SDOs showed excellent binding, indicating their promising value for biomedical applications. Therefore, this study puts forth the importance of various functional groups and their role in regulating properties at the macromolecular level and how that can be modulated to cater for diverse applications in the material and biomedical fields. Currently we are working on different applications of the best suited SDO from this library.Experimental section
Materials and methods
All chemicals were purchased from Sigma-Aldrich, Alfa Aesar, and Spectrochem. Solvents were of analytical grade and were used directly without any further purification.Instrumentation
1H NMR spectra were recorded on INOVA-400 spectrometers. The chemical shifts are reported in units of ppm relative to tetramethylsilane. Liquid chromatography-mass spectrometry (LC-MS) experiments were carried out on a Shimadzu LC-MS-8045 with a Sprite TARGA C18 column (40 × 2.1 mm, 5 μm) monitoring at 210 and 254 nm in positive mode for mass detection. The solvents used for LC-MS were water with 0.1% acetic acid (solvent A) and acetonitrile with 0.1% acetic acid (solvent B). High performance liquid chromatography (HPLC) was done on a Shimadzu Nexera X2 instrument with a Shim-pack C-18 column (20 × 250 mm, 5 μm) connected to a diode array detector. The solvents used for HPLC were the same as that for LC-MS. Fluorescence was recorded on a PerkinElmer FL 6500. All fluorescence spectra were recorded at 25 °C with an excitation wavelength of 280 nm and excitation and emission slit widths of 5 nm. SEM images were obtained using a Carl Zeiss Gemini SEM 300 scanning electron microscope with 2000000× magnification and a resolution of 0.8 nm at 15 kV, and 1.3 nm at 1 kV. The quantitative analysis of mercury was performed using inductively coupled plasma-mass spectrometry (ICP-MS) (Thermo Fisher iCAP RQ ICP-MS).
Synthesis procedures
:1 water/ethyl acetate. The ethyl acetate layer, which contained the pure product, was passed through a Na2SO4 bed and the solvent was evaporated to isolate 7. This was reacted with butyl bromide in the presence of K2CO3 as a base and acetonitrile as a solvent to form 8 (Scheme S3†). Quantitative conversion occurred in 12 hours and the reaction mixture was filtered to remove K2CO3. The reaction mixture was extracted with 1:1 water/ethyl acetate. The product was obtained with 97% purity and was directly used for the next reaction. In the third step, 8 was deprotected by treating with 6 M HCl in methanol solvent to yield 9 in quantitative yields (Scheme S4†). The product 9 was then reacted with CS2 (1.5 mmol) and tert-butyl (2-bromoethyl)carbamate (1 mmol) in PEG 200 solvent at room temperature for 30 min to form 10 (Scheme S5†). After extraction with 1:1 ethyl acetate + water, the product was obtained in the organic layer with 70% purity. The expected product was purified by column chromatography and the product eluted in a 20% ethyl acetate + 80% hexane mixture. The pure product 10 was used for the next reaction. Similarly, the above-described steps were repeated one after the other with propyl bromide and benzyl bromide to obtain the 4-mer with an overall yield of 60%.
:1 ethyl acetate + hexane solvent. The hydroxy group of the 2-mer (1 mmol) was then reacted with chloroacetylchloride (1.5 mmol) and triethylamine (2 mmol, base) in the presence of DCM to obtain the chloroacetylated product (Scheme S12†). In the next step, the chloroacetylated product underwent a three-component reaction to form the 3-mer (Scheme S13†). The 3-mer reaction mixture was also column purified and the pure product eluted in 70% ethyl acetate + 30% hexane mixture. The same cycle of reactions was repeated until the formation of the 4-mer of SDO2. Column purification of the 4-mer mixture was also done and the product eluted in 90% ethyl acetate + 10% hexane mixture. The overall yield of SDO2 was 68%.
:1 water:ethyl acetate was added into the reaction mixture, and the 2-mer was extracted into the ethyl acetate layer and was employed for the next step without column chromatography purification. Next, the chloro derivative of the 2-mer was prepared via the reaction of the 2-mer (1 mmol) and chloroacetyl chloride (2 mmol) in the presence of triethylamine (2 mmol) in dichloromethane (5 mL) at room temperature (step 2). The reaction was completed in 20 min, and thereafter, the excess chloroacetyl chloride was quenched with sodium bicarbonate solution. The reaction mixture was extracted with 1:1 water:DCM and the product was obtained in the organic layer. The solvent, DCM, was removed under reduced pressure. This product was taken for the next reaction. The same cycle of reactions was repeated for the synthesis of 4-mer SDO3. The 4-mer was purified by column chromatography and the pure product eluted in 90% ethyl acetate + 10% hexane mixture. The overall yield of 4-mer SDO3 was 71%.
Removal of Hg2+via the precipitation method
The removal efficiency of the synthesised compounds was tested using ICP-MS. For this, stock solutions of the SDOs (4 mM) were made in methanol and Hg2+ (12 mM) was made in a 1:1 water methanol system. Equal volumes of SDO solution were mixed with Hg2+ solution. The solution was mixed thoroughly and allowed to settle. The formed precipitate was separated from the liquid, and the supernatant was then analysed for any remaining mercury content using ICP-MS.
BSA binding studies
Stock solutions of SDO1, SDO2, and SDO3 were created at 10 × 10−6 M in DMSO, and were diluted to obtain solutions of different concentrations for fluorescence analysis in a quartz cuvette. A stock solution of BSA (100 × 10−6 M) was made in phosphate buffer. The excitation wavelength was 280 nm, and the SDOs exhibited emission with wavelength maxima at 350 nm. The titration of each SDO with BSA was performed and the fluorescence spectra were recorded after each addition.Author contributions
M. P. conceived the tunable backbone and side-chain concept for the synthesis of the SDOs. M. P. and A. J. conceived the molecular design and synthetic protocols. A. J. carried out all experiments including synthesis, characterization, and other studies. A. J. and M. P. analysed the data. A. J. and M. P. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely acknowledge the Indian Institute of Technology Palakkad, India and the Ramanujan Fellowship, Science and Engineering Research Board, Department of Science and Technology, India, Scheme for Transformational and Advanced Research in Sciences, Ministry of Education and Technology, India and the Core Research Grant, Science and Engineering Research Board, Department of Science and Technology, India for the financial support. The Central Instrumentation Facility (CIF) at the Indian Institute of Technology Palakkad and Indian Institute of Technology Madras are acknowledged for the analytical instrumentation support.References
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- M. A. R. Meier and C. Barner-Kowollik, Adv. Mater., 2019, 31, 1806027 CrossRef PubMed.
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- A. Jose, P. Nanjan and M. Porel, Polym. Chem., 2021, 12, 5201–5208 RSC.
- P. Nanjan, A. Jose, L. Thurakkal and M. Porel, Macromolecules, 2020, 53, 11019–11026 CrossRef CAS.
- P. Nanjan and M. Porel, Polym. Chem., 2019, 10, 5406–5424 RSC.
- B. van Genabeek, B. A. G. Lamers, C. J. Hawker, E. W. Meijer, W. R. Gutekunst and B. V. K. J. Schmidt, J. Polym. Sci., 2021, 59, 373–403 CrossRef CAS.
- R. Aksakal, C. Mertens, M. Soete, N. Badi and F. Du Prez, Adv. Sci., 2021, 8, 2004038 CrossRef CAS PubMed.
- A. Greenberg, C. M. Breneman and J. F. Liebman, The amide linkage: Structural significance in chemistry, biochemistry, and materials science, John Wiley & Sons, 2002 Search PubMed.
- E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 2020, 4641–4651 CrossRef CAS.
- A. C. Fonseca, M. H. Gil and P. N. Simoes, Prog. Polym. Sci., 2014, 39, 1291–1311 CrossRef CAS.
- K. Ghosal, M. S. Latha and S. Thomas, Eur. Polym. J., 2014, 60, 58–68 CrossRef CAS.
- H.-W. Engels, H.-J. Weidenhaupt, M. Pieroth, W. Hofmann, K.-H. Menting, T. Mergenhagen, R. Schmoll and S. Uhrlandt, Ullmann's Encycl. Ind. Chem., 2011 Search PubMed.
- D. S. Pal, D. K. Mondal and R. Datta, Antimicrob. Agents Chemother., 2015, 59, 2144–2152 CrossRef CAS PubMed.
- Y. Ding, Z. Yang, W. Ge, B. Kuang, J. Xu, J. Yang, Y. Chen and Q. Zhang, J. Enzyme Inhib. Med. Chem., 2018, 33, 1376–1391 CrossRef CAS PubMed.
- J. W. de Freitas Oliveira, H. A. O. Rocha, W. M. T. Q. de Medeiros and M. S. Silva, Molecules, 2019, 24, 2806 CrossRef PubMed.
- P. Gaspari, T. Banerjee, W. P. Malachowski, A. J. Muller, G. C. Prendergast, J. DuHadaway, S. Bennett and A. M. Donovan, J. Med. Chem., 2006, 49, 684–692 CrossRef CAS PubMed.
- K. Yates and R. A. McClelland, J. Am. Chem. Soc., 1967, 89, 2686–2692 CrossRef CAS.
- C. Tunsu and B. Wickman, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- U. Haldar and H. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 13685–13693 CrossRef CAS PubMed.
- P. J. Knerr, M. C. Branco, R. Nagarkar, D. J. Pochan and J. P. Schneider, J. Mater. Chem., 2012, 22, 1352–1357 RSC.
- D. Zhang, E. N. Atochina-Vasserman, D. S. Maurya, M. Liu, Q. Xiao, J. Lu, G. Lauri, N. Ona, E. K. Reagan, H. Ni, D. Weissman and V. Percec, J. Am. Chem. Soc., 2021, 143, 17975–17982 CrossRef CAS PubMed.
- J. Hatai, S. Pal, G. P. Jose and S. Bandyopadhyay, Inorg. Chem., 2012, 51, 10129–10135 CrossRef CAS PubMed.
- J. Hatai, S. Pal, G. P. Jose, T. Sengupta and S. Bandyopadhyay, RSC Adv., 2012, 2, 7033–7036 RSC.
- S. Pal, J. Hatai, M. Samanta, A. Shaurya and S. Bandyopadhyay, Org. Biomol. Chem., 2014, 12, 1072–1078 RSC.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- O. A. Chaves, V. A. da Silva, C. M. R. Sant'Anna, A. B. B. Ferreira, T. A. N. Ribeiro, M. G. de Carvalho, D. Cesarin-Sobrinho and J. C. Netto-Ferreira, J. Mol. Struct., 2017, 1128, 606–611 CrossRef CAS.
- S. Jovanović, K. Obrenčević, Ž. D. Bugarčić, I. Popović, J. Žakula and B. Petrović, Dalton Trans., 2016, 45, 12444–12457 RSC.
- A. Ravindran, A. Singh, A. M. Raichur, N. Chandrasekaran and A. Mukherjee, Colloids Surf., B, 2010, 76, 32–37 CrossRef CAS PubMed.
- D. S. Raja, N. S. P. Bhuvanesh and K. Natarajan, Eur. J. Med. Chem., 2011, 46, 4584–4594 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01586a |
| This journal is © The Royal Society of Chemistry 2022 |