
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating the porosity of carbons for improved adsorption of hydrogen, carbon dioxide, and methane: a review
L. Scott
Blankenship
 * and
Robert
Mokaya
*
* and
Robert
Mokaya
*
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: leo.blankenship@nottingham.ac.uk; r.mokaya@nottingham.ac.uk
First published on 12th January 2022
Abstract
Porous carbons provide a low-cost route to a highly stable material for the adsorption of various gases. In particular, activated carbons (ACs) and zeolite templated carbons (ZTCs) show promise in their ability to capture and store environmentally relevant small molecules such as hydrogen, carbon dioxide and methane. Indeed biogas upgrading and methane storage are already partially commercialised. While ZTCs offer precise control over porosity, ACs have the advantage of being easy to synthesise from a wide range of sources. This review examines state-of-the-art techniques to control the porosity of both ACs and ZTCs in order to fine-tune their ability towards the capture and storage of various gases under different pressure and temperature applications.
 L. Scott Blankenship | L. Scott Blankenship is a PhD student in the Mokaya group at the University of Nottingham, and obtained his MSc in the same group in 2016 working on porous carbons from cigarette butts for H2 storage and CO2 capture. His research interests focus on critical evaluation of porosimetric methods for carbons and the ability of these methods to predict uptake of environmentally relevant small molecules. |
 Robert Mokaya | Robert Mokaya received his BSc in Chemistry from the University of Nairobi in 1988 after which he spent a year working for Unilever in Kenya. He was awarded his PhD from the University of Cambridge in 1992. In 1992, he was elected to a Research Fellowship at Trinity College, Cambridge and in 1996 was awarded an EPSRC Advanced Fellowship. He joined the School of Chemistry in Nottingham, as a lecturer in Materials Chemistry in 2000, where he is Professor of Materials Chemistry. His research interests are on the design, synthesis and characterisation of new forms of sustainable porous materials and the study of their structure–property relations. The research involves exploring fundamentally new synthesis methods that are simpler, cheaper, and more efficient and offer valorisation routes to materials with optimised properties for targeted sustainable energy applications. |
1. Introduction
The current climate and ecological emergency threatens all life on planet earth, and is largely caused by incessant anthropogenic emissions of greenhouse gases. The resultant climate change, which is already causing extreme weather events and is threatening mass extinction, is well underway but the full extent of effects of the climate crisis are yet to come.1,2 In order to keep within the temperature limits prescribed by the Paris Agreement,3 it is necessary not only to drastically reduce global net greenhouse gas (GHG) emissions by reducing consumption and transitioning to ‘green’ energy sources but also to capture CO2 and CH4 in the interim.3–6 Although bioremediation of carbon capturing ecosystems has a great role to play in the offsetting of GHG emissions, it is insufficiently expedient to keep up with the reduction in emissions necessitated by both the aforementioned Paris Climate Agreement and the UK's limited goal of achieving net zero emissions by 2050.3,6–8These factors necessitate development of novel, low cost CO2 capture technologies.9 Additionally the global economy must rapidly transition away from fossil fuels as vehicular energy sources and towards cleaner, renewable fuels such as natural gas (CH4) and H2.10–12 Although natural gas is not a carbon neutral fuel, it is produced sustainably from the breakdown of biomass and releases much less CO2 and other pollutants than other fossil fuels.13,14 As for H2, the sole product of its combustion is water making it a carbon-neutral fuel at the point of use.10 The difficulty with storing or capturing CO2, CH4 and H2 is that they all exist in the gas phase under ambient conditions. As a result, the challenge becomes both economic and practical – how these gases can be stored and transported in a cost and space-efficient way.10,14–22 Currently, industrial CO2 capture is achieved principally through reaction with liquid amines in the presence of water to form aqueous ammonium bicarbonates,23,24 while commercial technologies for the storage of natural gas/CH4 and H2 rely on compression or liquefaction.10,14,18,20–22,25 The cost of liquid amine capture is principally a result of its relatively low CO2 capacity as well as energy requirements for regeneration of the amine from the ammonium bicarbonate. Though compression or liquefaction of gaseous fuels does not come with the same regeneration issues present in chemical capture of CO2, it is plagued by the high energy costs of maintaining the high pressures and/or low temperatures required.14,20 In the case of compressed gases the weight of sufficiently robust containers needed to maintain these pressures is very high, leading to energy losses for on-board applications.22
Promising alternative methods of gas storage involve chemical (chemisorption) or physical (physisorption) adsorption onto a suitable solid material. Physisorption has the distinct advantage over chemisorption of facile regeneration of the adsorbate by decreasing pressure or increasing temperature. Microporous adsorbents such as Metal Organic Frameworks (MOFs),26–28 porous polymers,29 Porous Inorganic Membranes,30 and zeolites31–33 present possible storage solutions but have the disadvantage of relatively high cost of production and/or finite lifecycles due to thermal instability or undesirable reactivity with contaminants such as water. On the other hand, porous carbons provide a storage medium that is relatively inexpensive to produce and extremely thermally and chemically stable.18,20,34 Furthermore, the porosity of carbons can be tailored to be selective for specific adsorbates and adsorption conditions.20,35–37
Porous sorbents are actually already beginning to see some commercial use in the capture/storage of small molecules. This is particularly evident for CH4, which is already being used as an on-board fuel.38–42 It is principally stored via compression (CNG) or liquefaction (LNG).38,39 However, adsorbed natural gas (ANG) is now competitive with the CNG and LNG as a result of the much higher energy density than CNG, and is already commercially available.43,44 As pressures required for ANG are significantly lower than those for CNG, ANG infrastructure is less costly.38,40,45,46 Additionally, adsorption of CH4 poses a much lower safety risk relative to compression and improves overall efficiency at point of use.12,46 Due to its low cost, activated carbons are used as the adsorbents in on-board applications as well as at fuelling stations.43,44 Additionally, porous carbons are in commercial use for the upgrading of biogas (typically ∼60% CH4) by selective removal of H2O, CO2 and H2S resulting in improved CH4 concentration of over 98%.44,47,48 The principal problem with porous materials for this application is imperfect selectivity resulting in so-called methane slip, wherein some methane is lost to the sorbent.48–50 In theory this problem can be mitigated by finely tuning pore widths and/or surface chemistry.
Porous carbons fall into four main categories; activated carbons, carbonised frameworks, templated carbons, and carbide-derived carbons.51 Of these, the simplest to produce are activated carbons, which in the case of so-called chemical activation, require simple heating of a carbonised (typically graphitic) material with an activating agent to generate pores. In fact, the initial carbonisation step can be performed simultaneously with the so-called activation step.20,52 The perceived disadvantage of this technique is that the amorphous nature of the product means tuning of the pore size can be difficult. Framework carbonisation involves pyrolysis of an already porous organic material such as an aerogel or other polymer network.53–55 Templating is one of the newer techniques and is performed by depositing a carbon rich material onto some inorganic template (typically silicas or zeolites) with the desired pore size and surface area, followed by heat treatment in order to form a graphenic crystalline structure within the template pores, before removing the template.34,51,56 Finally carbide-derived carbons, which use metal carbides as starting material, allow for tuning of porosity at the atomic level. This is possible as suitable carbides are selected according to their lattice spacing, wherein removal of the metal counterion generates pure carbon with pore channels whose size is determined by the precursor's lattice geometry.57–59 Broadly, porous carbons for small molecule gas adsorption should be microporous and have high surface area and pore volume.18,60–63 Although it is possible to achieve porous carbons with such characteristics via any of the aforementioned routes, by far the most popular methods are activation and templating using zeolites and as such these form the subject of this review. For both of these processes, the challenge is tuning the porosity of resultant carbons to optimise uptake performance for specific adsorbates and/or adsorption conditions; in the case of activated carbons (ACs) this centres on techniques to narrow the pore size distribution (PSD),20 whilst with zeolite templated carbons (ZTCs) improvements come via more improved replication of the template structure.34,56
Gas uptake and molecular selectivity of ZTCs and ACs can also be improved chemically by introducing, as dopants, moieties with affinities for the targeted adsorbate.63–68 This review however, focuses on improvements in physisorption of small gas molecules by porous carbons achieved by tailoring the size, shape, and abundance of pore channels available to the adsorbate. Any reference herein to doping of the carbon structure with heteroatoms is made in the context of the effect on pore structure.
2. Synthesis of porous carbons
Methods for synthesizing porous carbons can be split up into two broad groups; top-down or bottom-up. A top-down method takes an existing carbon or carbonaceous material and develops porosity in it by etching away material; this process is commonly known as activation.20 Conversely, bottom-up methods consist of constructing the carbon structure from its building blocks in such a way that voids or pores are formed, often with the use of a porous template. Such templates include mesoporous silica,69,70 micro-phase separated block copolymers71 or even MOFs,72,73 however perhaps the most well-known microporous templates are zeolites where the product of such a process is known as zeolite-templated carbon (ZTC).34 Bottom-up synthesis can also occur in the absence of a template, for example via the direct carbonization of organic crystals.742.1. Activation of carbon-rich precursors
Activation, here meaning the development of porosity in a carbon precursor, can be achieved by either physical or chemical means. Physical activation is a two-step process whereby the precursor is first pyrolised at 400–900 °C in an inert atmosphere, in order to increase carbon content. Thereafter the sample is exposed to an oxidizing gas at 350–1000 °C in order to develop pores. Chemical activation however, can take place in a single step wherein the carbon precursor is mixed with an activating agent - also known as a porogen (such as alkali metal hydroxides, H3PO4, or ZnCl2), then pyrolised at 450–900 °C.20 Precursors can essentially be any organic matter, and include biomass such as wood,75,76 fruit seeds,35,77 grass,78 and refined biopolymers.79 The degree of activation, i.e. the extent of porosity development, can be improved by pre-carbonisation of the precursor.80–82 Newer forms of carbon such as templated carbons, nanotubes, and carbide derived carbons (CDCs) have also been activated to improve their porosity.83–88It should be noted that physical activation can be achieved without addition of an oxidising gas because volatile components of the precursor are converted to (among other things) oxidising gases on pyrolysis resulting in the development of small micropores. In the case of biomass carbonisation in the absence of a porogen, porosity and structure are maintained in the resulting so-called biochar material.89–91
Activation with CO2 and steam generally result in higher yields than using air as a porogen. This is because the reaction between carbon and oxygen in the air is highly exothermic and leads to a much faster reaction, burning off more of the carbon.20 This does however mean that much lower temperatures are required for activation using air, as compared to CO2 and steam.52,102,103 Steam and CO2 are nonetheless the preferred physical activating agents due to the aforementioned problems with air.20 Steam is generally characterised as the more reactive porogen of the two, requiring lower activation temperatures than CO2.92,104,105 There is however still some disagreement as to what type of porosity is developed by each activating agent.105–108
Chemical activating agents can be divided into two groups according to their activation mechanism; the first group are dehydrating agents such as H3PO4 and ZnCl2 wherein the dehydration of the carbonaceous structure, triggered by the action of the activating agent leads to the formation of cross-linkages.110,111 This in turn triggers condensation of the structure around the activating compound and its hydrates. Pore size is thus limited by size of these compounds. Further, the breadth of the PSD is a function of the variation in molecular sizes of the activating agent and its hydrates. For example, activated carbons produced using ZnCl2 have narrow PSDs as structural condensation occurs around ZnCl2 and its hydrates which are of similar size.112 On the other hand, H3PO4 is converted to molecules with a broader range of sizes such as H4P2O5 and H13P11O34 resulting in a heterogeneous PSD, regardless of activating conditions.113
Despite the ubiquity of KOH as a porogen in the literature, the mechanism of activation is a matter of some debate.20,109,114–121 Nonetheless, there is a broad agreement that pore formation occurs via three principle processes:20,109,114–116,118 (i) chemical activation wherein redox reactions between K compounds and the carbon framework occur; (ii) formation of the physical activating agents CO2 and H2O which induce gasification of carbon to further develop porosity; (iii) the intercalation of metallic K between graphitic sheets. Washing of the sample removes both intercalated K as well as K oxides and carbonates, resulting in a permanently porous carbon. There is inconsistent evidence on the exact nature and validity of processes (i) and (iii). For example, work by Linares-Solano indicates that (i) occurs directly via the oxidation of carbon by KOH (eqn (1)).116,117
| 6KOH + 2C → 2K + 3H2 + 2K2CO3 | (1) |
| 2KOH → K2O + H2O | (2) |
| C + H2O → CO + H2 | (3) |
| CO + H2O → CO2 + H2 | (4) |
Chemical activation using KOH is noted for the distinct advantage of giving a higher degree of control over the PSD as compared with other activating agents.20,79,126,127 Though PSDs for carbons derived using small amounts of KOH or NaOH are typically narrow, significant broadening occurs with increasing MOH/precursor ratio.128–130 For applications in which high porosity materials with narrow PSDs are required, alkali metal carbonates and bicarbonates as well as oxalates show significant promise.131–134 These porogens have been shown to be significantly less corrosive than their hydroxide counterparts, yet produce materials with significant surface area, particularly in the micropore region,135–138 and are discussed in more detail in Section 4.1.1.
While oxidative chemical activation is heavily favoured in the literature due to its ability to produce carbons with extremely high surface areas, it comes with the disadvantage of resulting in significant loss of material. Dehydrating agents are non-destructive thus result in much higher yields. In addition, it has been shown that air, as opposed to an inert gas may be used in such scenarios, for example Fierro et al. produced carbons from lignin activated with H3PO4 at temperatures up to 600 °C.139,140
Pre-carbonisation methods. The porosity of carbons can be improved by subjecting the precursor to an initial carbonisation step before activation. This results in the partial graphitisation of the material and increases the carbon content, accompanied by a reduction in concentration of heteroatoms. This means that fewer oxidising gases are released upon activation resulting in more controlled activation and thus a narrower PSD, less pore collapse and generally higher surface area. Carbonaceous matter can exist in carbonised form such as coal, pitch or tar and can be converted to high surface area activated carbon in a single step.141–143 Alternatively, biomass can be hydrothermally carbonised by heating in water under high pressure to generate carbonaceous matter composed of microspheres which possess a hydrophobic core and hydrophilic shell, of which the latter is more susceptible to chemical activation.144–147 Another route is so-called air- or flash carbonisation, which is achieved by briefly interrupting the inert atmosphere used during pyrolysis by injection of air into the system.35,80,81
2.2. Templating onto porous zeolitic structures
Zeolites are three-dimensional uniform microporous crystalline framework structures. For aluminosilicates, the structure is based on silica networks where some of the Si4+ cations are replaced by Al3+, resulting in an overall negative charge for the framework. Cations such as Na+, H+ or NH4+ reside within the framework pores to balance the charge.154,165,166 More than 40 naturally occurring zeolites are known to exist,167 and while theoretical zeolites number in the thousands168 only ca. 200 have thus far been synthesised.169 The aforementioned uniformity of zeolitic micropores means they are interesting in their own right as gas sorption and separation materials.32,33,170 However, the chemical and thermal stability (under inert conditions) of some zeolites is lower than that of activated carbons. Interest in using zeolites as sacrificial templates for the preparation of porous carbons arises from a desire to improve the structural ordering of highly stable but totally amorphous porous carbon materials. ZTCs provide a route to uniformly porous graphenic microporous solids with porosity and thermochemical stability comparable to that of activated carbon.171 In addition, this regularity in structure leads to greater scope for functionalisation of the carbon surface.34,172These improved characteristics necessitate more precise synthesis conditions. ZTCs can be divided into three types according to how graphenic the overall structure is, which is connected to the degree of replication of the template's pore channels ranging from type-I where near complete template replication is achieved, to type-III where the pore structure bears little resemblance to its template and possesses a high degree of graphene stacking. Type-II ZTCs are formed when the template is partially replicated, but there is still a high degree of graphiticity due to deposition of carbon on the outer surface of the template.34 The structural uniformity of the zeolite can only be conferred on the resultant ZTC (i.e. a type-I ZTC) when the template's pore structure is composed of large 3D channels.173–175 Specifically, pore entrances should contain a minimum of 12 O atoms in the prototypical Si–O–Si ring which forms the zeolite channel opening to allow for uniform pore filling by the carbon monomer without risking pore blocking.34,176 Smaller pores or low dimensional pore networks always result in a disordered, graphitic structure (type-III) more akin to activated carbons. As most natural zeolites have small pore entrances, they typically yield poorly ordered ZTCs. To achieve type-I ZTCs, sufficient carbon monomer must be inserted evenly into the zeolite pores and allowed to polymerise before carbonisation. Thus two heating stages are required to achieve discrete, uniform graphenic structures.34 Carbonisation of the monomer without polymerisation can lead to partial uniformity with some graphene stacking (type-II) with a mixture of graphitic and graphenic ordering, while failure to achieve even distribution of monomer is likely to result in no replication of the template structure. As type-I ZTCs typically have the highest degree of porosity (ABET > 2100 m2 g−1)177 precise selection of zeolite, monomer, and activation conditions is vital for producing a ZTC with adventitious porosity for small molecule physisorption.
3. Textural characteristics for gas storage applications
Desirable textural characteristics for gas storage are dependent on the adsorptive, temperature and pressure of interest. Fig. 1 and 2 show how different pressures can affect the optimal textural characteristics of carbons. A summary of relevant properties of common gaseous sorptives is shown in Table 1. In the literature, measurements are commonly taken at 25 °C for H2, CO2, and CH4 while −196 and 0 °C are also typical for H2 and CO2 respectively.14,16,20,212,213 A series of isotherms at various temperatures for a given adsorbate-sample pair may also be reported to allow determination of isosteric heats of adsorption.214 The pressure range used in isotherm measurement may depend on sample porosity; adsorption will typically plateau at relatively low coverage for samples with low surface area thus the upper pressure used need not be very high. Typical reports include data up to ∼40 bar for CH4 and CO2, while H2 uptake is more commonly measured up to ∼100 bar. Of particular interest is the adsorption at 1 bar, however lower pressure such as 0.15 bar are also used for selectivity calculations especially in CO2 uptake studies. Increases in surface area and pore volume generally improve a sample's uptake of an adsorbate by supplying adsorbed molecules with more surface to interact with and greater volume to fill, respectively. This is however limited by pore size, which can differentiate an adsorbent as a good candidate for storing one gas but not another.63,91,215–217 As such, more detailed measures of porosity such as pore size distribution (PSD) and average pore size are used. Furthermore, low pressure adsorption of CO2, CH4 and H2 is dominated by micropores, and thus micropore surface area and volume is typically reported alongside the total values.218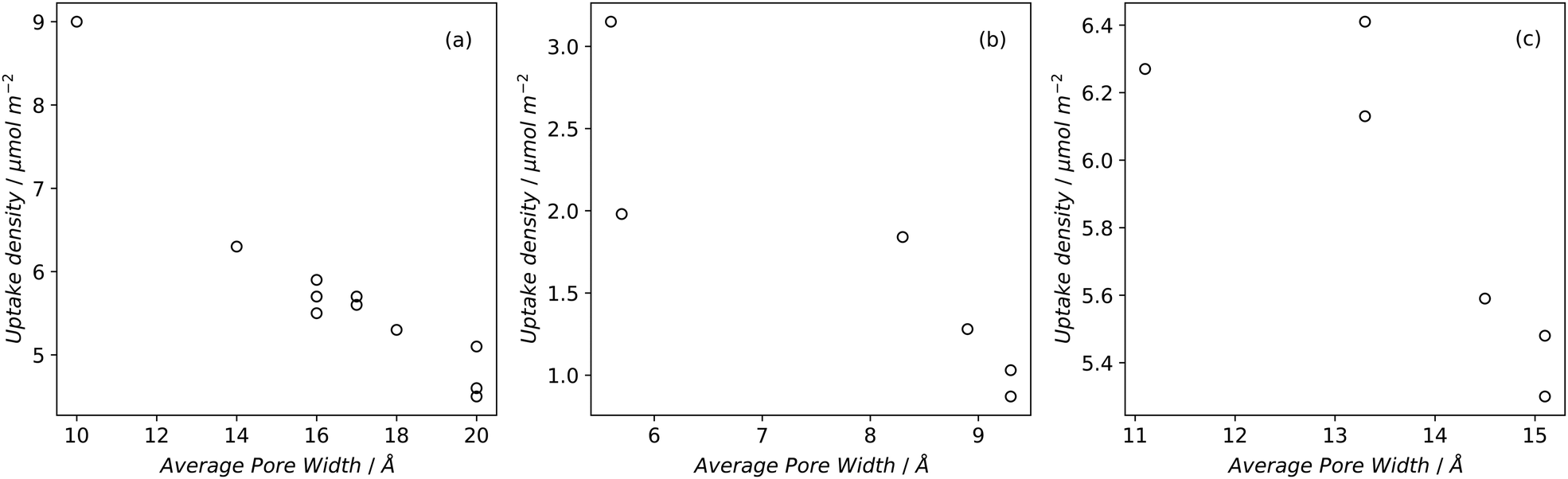 | ||
| Fig. 1 Uptake densities of (a) H2 at 1 bar and −196 °C, (b) CO2 at 1 bar and 25 °C, and (c) CH4 at 65 bar and 25 °C on carbons79,194,219 as a function of average pore size. | ||
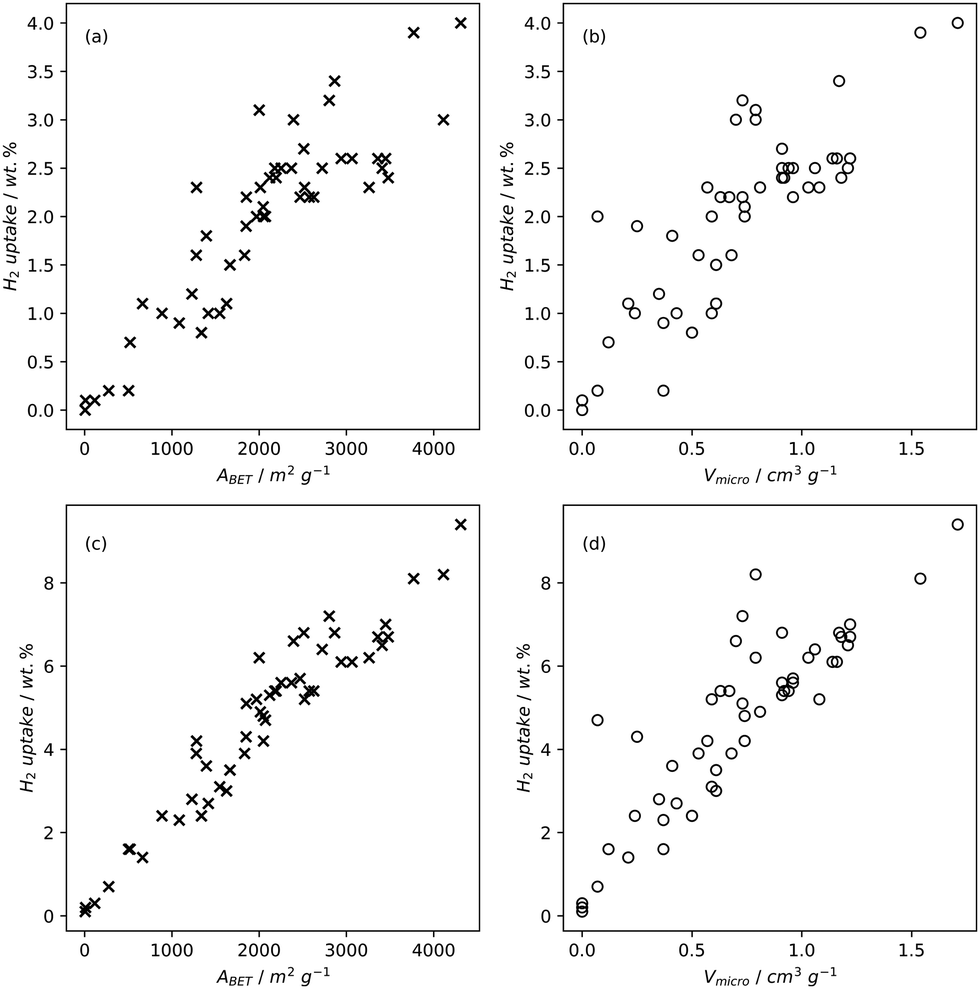 | ||
| Fig. 2 H2 uptake of carbons at −196 °C and 1 bar (a) and (b) or 20 bar (c) and (d) as a function of BET surface area (a) and (c) or micropore volume (b) and (d).79,82,83,196,220,221 | ||
3.1. Pore size
If pore width is less than the diameter of the adsorptive (see Table 1), adsorption will not occur within the pore – indeed even pores with diameters slightly greater than that of the molecule's kinetic diameter (dk) can lead to extremely slow diffusion at low temperatures, meaning that equilibrium may take a long time (hours) to achieve.52,225,226 Additionally, optimum pore size has an upper limit due to the improvements in adsorption from field overlap when pore walls are sufficiently close together.For small molecules like H2, CO2, and CH4 (dk = 2.89, 3.30, and 3.80 Å respectively),154,222,223 this means that highly microporous materials are the most suitable for physisorption (see Fig. 1). Optimal pore size for adsorption of a given molecule is proposed to be that which can hold two layers of the molecule.212,217,227Fig. 1 shows how average pore size affects the uptake density of gases under some standard adsorption conditions. For H2 this means that ultramicropores are the dominant pore structure for storage at atmospheric pressure,60 while optimum pore width is generally agreed upon to be 6 Å.212,216 Maximum hydrogen uptakes can be achieved by samples wherein the PSD is narrow,95 and centred around the optimum pore width; this is true at both cryogenic and ambient temperatures.212,228 There is also evidence that hydrogen and methane preferentially adsorb on carbon slit pores rather than cylindrical pores,229–231 which may give ACs an advantage over ZTCs.
To adsorb CO2 under ambient conditions it is generally agreed that the best carbonaceous sorbents have the majority of their pores smaller than 8 Å, and distributed narrowly62,63,96,97,219,232 (see Fig. 1) but slightly higher and lower limits have also been experimentally demonstrated.233,234 This limit decreases with decreasing pressure – at 0.1 bar, pores of 5 Å have the biggest contribution to adsorption of CO2.97,227 This is particularly applicable for post-combustion industrial capture applications wherein it is necessary to remove CO2 from a mixture of gases (∼75% N2). To achieve the selectivity necessary for such applications, adsorbents require ultramicropores as small as 3.5 Å to prevent the ingress of larger molecules.235,236 At higher pressures the optimum pore size progressively increases into the supermicropore and small mesopore region.62,97,136,234,237 At such pressures, the PSD can broaden without harming overall uptake.66,126,227,238 In fact, high pressure CO2 capture can be harmed by excessive microporosity as overall pore volume needs to be maximized for optimal molecular packing.237
Determination of the optimum pore size for methane storage appears to have yielded less conclusive results than that for H2 and CO2. Computational estimates range from 11 to 15 Å.217,239,240 Through optimization experiments, Biloé et al. propose 15 Å as the optimum,241 whereas other studies seem to suggest that optimal micropore width depends on whether the porous carbon is intended for gravimetric or volumetric storage.
3.2. Surface area
High pressure adsorption is much less dependent on pore size, and available surface area (ABET) begins to dominate as a predictor of gas uptake capacity (Fig. 3e and f). In particular, Bénard and Chahine determined that cryogenic hydrogen storage capacity increases by approximately 1 wt% per 500 m2 g−1.242 However, the degree of influence of ABET on H2 uptake posited in the eponymous Chahine rule has been recently disputed by Kusdhany and Lyth who found via a multivariate analysis that a 500 m2 g−1 increase in surface area corresponds to at most a 0.24 wt% increase in hydrogen uptake.243 Nonetheless high surface area remains an important factor in determining high pressure H2 storage capacity in carbons, with the best performing carbons possessing surface areas approaching 4000 m2 g−1.82,98,178,220,238,244–247 Similarly, in the case of CO2 and CH4 capture and storage, increased surface area is generally associated with improved gravimetric uptake.14,35,131,132,194,248–255 However, depending on the prevailing pressure, CO2 capture is much less dependent on surface area, and having pores of the appropriate size is more important.132,244,252,256 In fact recent work by Jing Cui et al. suggests that excessive volumetric surface area results in poor selectivity of CO2 over N2, a metric which is vital in many industrial applications.257 Furthermore, gravimetric surface area is becoming a less interesting variable for CH4 storage because it does not correlate well to volumetric capacity.18,35,258 As such, metrics like packing density and volumetric surface area (surface area density, see Fig. 1) are often cited in lieu of or as well as the traditional gravimetric value.35,194,259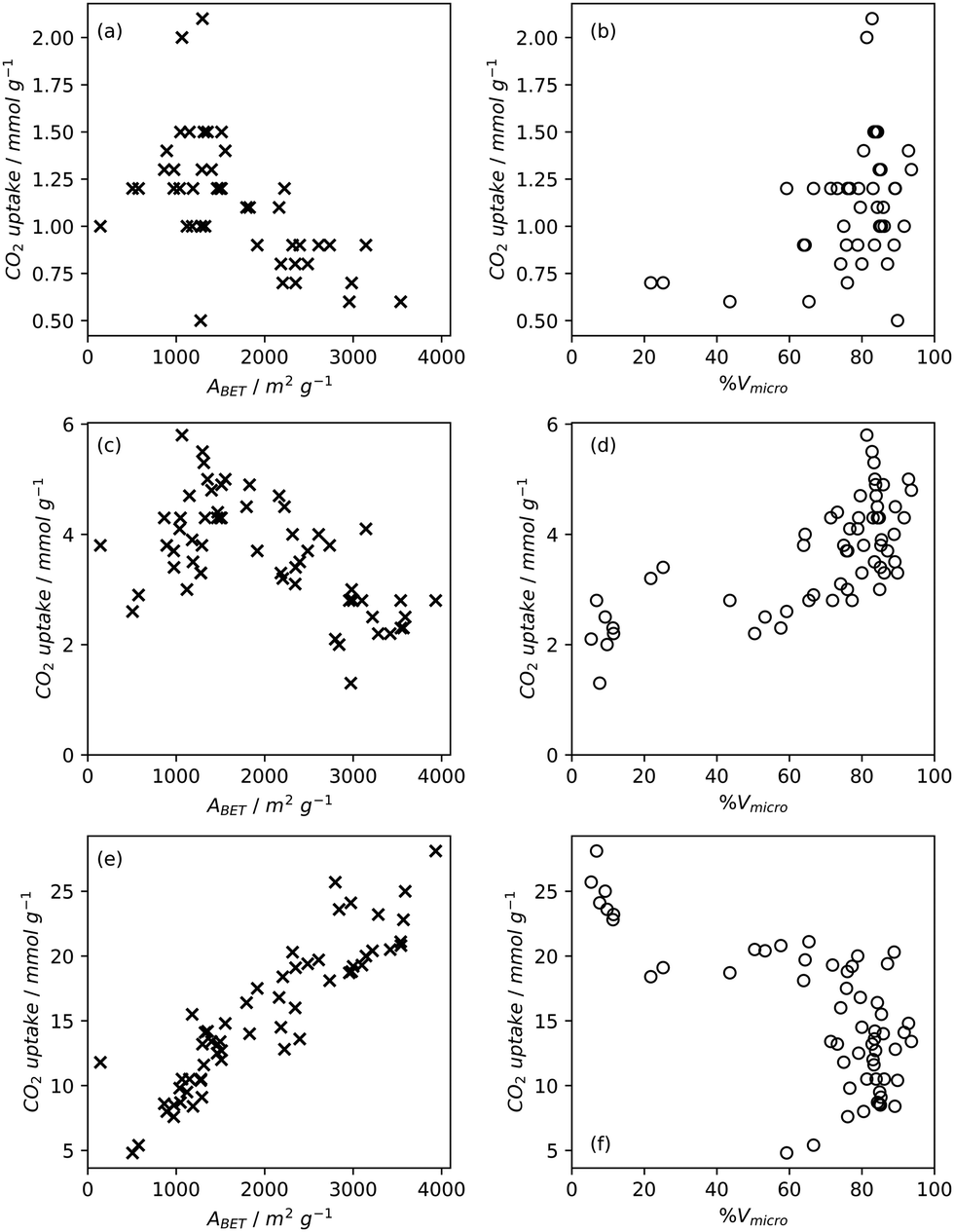 | ||
| Fig. 3 Trends in BET surface area and percentage micropore volume with CO2 uptake capacity of carbons78,80,81,135,237,238,244,248 at 0.15 bar (a) and (b), 1 bar (c) and (d), and 20 bar (e) and (f). | ||
3.3. Pore volume
Pore volume generally correlates to surface area, and thus increases in pore volume are typically associated with increases in gas adsorption capacity. Of course, the strength of the relationship between pore volume and surface area is affected by the size of the pores in question, i.e. an ultramicropore will contribute much less pore volume than a mesopore of identical surface area.20,234,260 If a sample is mostly microporous and has a high total pore volume, this indicates that there are lots of or very deep micropores within the sample. Such a sample is advantageous for small molecule adsorption, especially at lower pressures. As such, (percentage) micropore volume rather than total pore volume is likely a better predictor for H2, CO2 or CH4 uptake (Fig. 2–4).14,18,20,34,68,257,261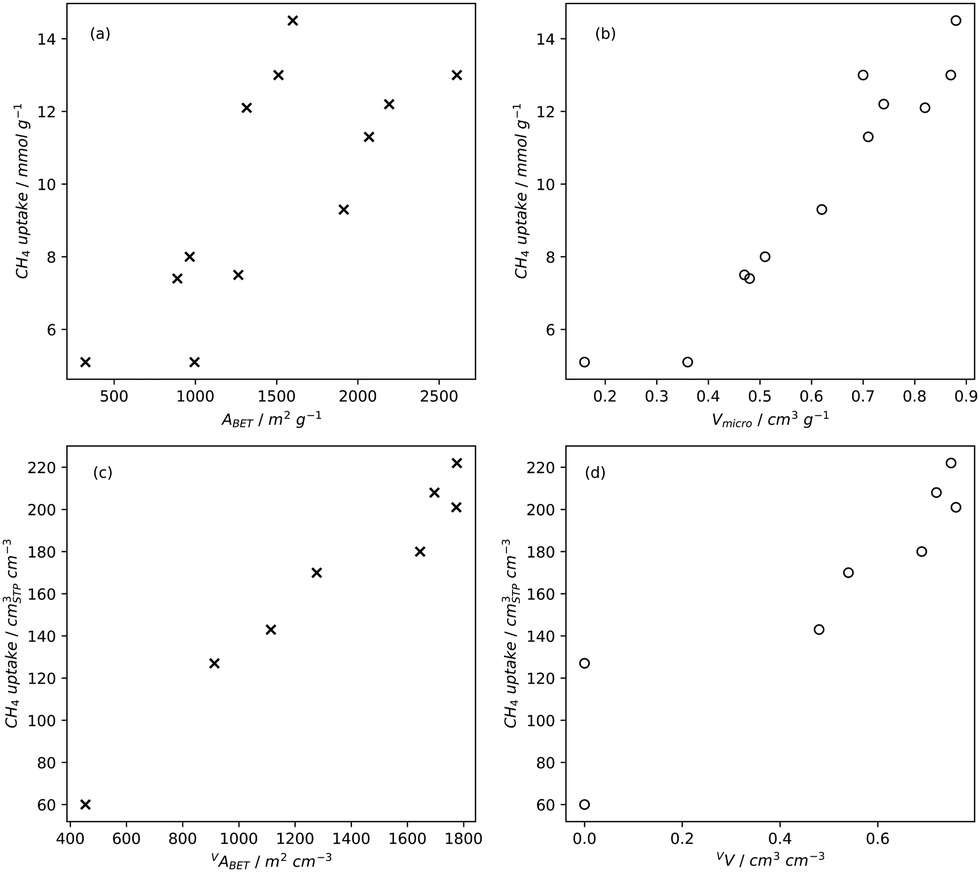 | ||
| Fig. 4 Trends in CH4 uptake with gravimetric (a) and (b) and volumetric (c) and (d) porosity of carbons35,131,258 at 35 bar and 25 °C. | ||
3.4. Measuring porosity
Unlike for crystalline materials, the porosity of ZTCs and ACs cannot be easily determined using diffractive techniques such as XRD. Additionally, due to the size of pores needed for small molecule sorption, use of electron microscopy is not a particularly accurate technique for determining porosity. Thus, sorptometric porosimetry remains the main method for determining specific surface area, pore volume and pore size of carbons.Secondly, nitrogen's relatively high quadrupole moment224 can confound measurements on samples containing polar moieties as is the case for many activated carbons. Polar groups affect the orientation of nitrogen with respect to the surface – this results in the actual cross-sectional area of the molecule differing to that which is used in calculations of textural parameters such as ABET. This is even more problematic for CO2 as it is more polar than N2.224,268 As such, dual gas adsorption techniques may also employ O2 and H2 (at −196 °C) which both have lower quadrupole moments than N2.225,268,269 H2 has the added advantage of being smaller than CO2 thus improving the lower pore size limit to ∼3 Å.225
Classical. Historically, measures of porosity were determined via individual manipulations and/or calculations from the raw isotherm.
(i) Surface area: Stephen Brunauer, Paul Hugh Emmett, and Edward Teller expanded Langmuir theory to account for multilayer adsorption, which occurs at higher pressures and temperatures. The eponymous BET surface area, ABET, is calculated by first determining the total quantity of gas adsorbed, Q, and from this calculating Qm, the quantity of gas in the monolayer using the following equation;
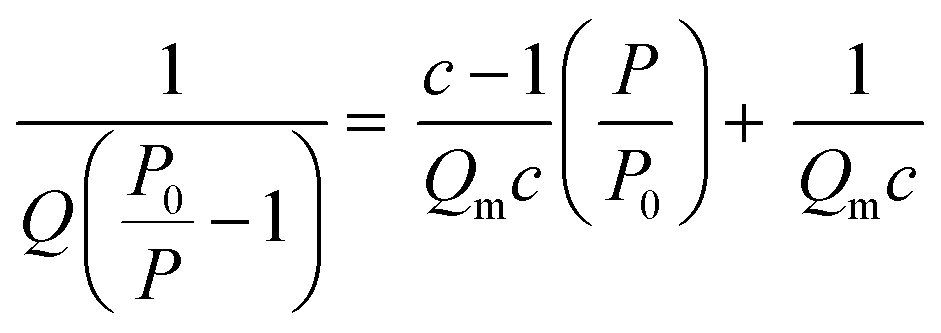
A measured isotherm can be transformed in accordance with the BET equation allowing for the determination of Qm from a linear portion of the plot, selected according to the Rouquerol criteria.270,271 Thus ABET is determined using the adsorption cross-section, σ, and mass of the adsorbate, a;
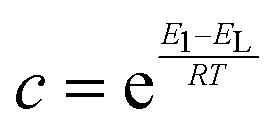
Despite its ubiquity, the theoretical backing of ABET does not robustly describe the surface area of all materials, particularly in the case of materials having very small pores as so-called multilayer adsorption is not relevant here.218,271 In addition, the model does not account for surface chemical or energetic heterogeneity, and as such σ may differ at different points on the surface.218,225,272 As a result ABET should not be considered a ‘true’ value for the surface area of porous carbons. Nevertheless if applied consistently and correctly, (i.e. selecting the relative pressure range as ascribed by the aforementioned Rouquerol criteria),218,271ABET provides the ‘apparent’ surface area for microporous materials, which is a useful metric for comparing the porosity of different, related samples. This is in contrast to the much simpler typical criteria used to select the pressure region for determination of ABET in mesoporous, macroporous or nonporous materials, wherein the range is typically between 0.05 and 0.30.
It should be noted however that manual application of the Rouquerol criteria can be extremely inconsistent when applied by different people. In a round-robin study, the Fairen-Rodriguez group found that the manually determined ABET from the same isotherm could differ by more than 7000 m2 g−1. Furthermore, even when applied correctly the Rouquerol method can yield multiple acceptable pressure regions for an isotherm. They therefore propose an algorithmic approach to determine of the optimal Rouquerol range and thus the valid BET area.273
(ii) Pore volume: is typically measured using the quantity of gas adsorbed at the isothermal plateau and at relative pressure approaching 1, as by this point the pores are considered to be fully filled by the adsorbate. As this method only uses one point on the isotherm, it is known as the single-point pore volume calculation.218 This method is not applicable to all isotherms, as some do not exhibit a plateau.
(iii) Microporosity: there are various classical methods used for this, the first of these being the t-plot method which relies upon comparison of the experimental isotherm with that of a standard adsorbent under the same analytical conditions.274 This of course relies on an appropriate standard, which is not always available. Furthermore this method may give inaccurate micropore volumes for samples that contain significant mesoporosity,275 in some cases yielding negative values for microporosity.276 Further developments include the αS, Dubinin–Astakhov (DA) and Dubinin–Radushkevich (DR) plots.274,277 These methods utilise a transformation of the isotherm to yield a roughly linear plot. The slopes and intercepts of such plots can then be used to calculate micropore volume and surface area.
(iv) Pore size: methods are available for determination of a pore size distribution such as H–K (Horváth–Kawazoe) and BJH (Barrett, Joyner and Halenda) from an N2 isotherm in the micropore and mesopore region, respectively. The former relies on the assumption that pores of some width fill at a certain relative pressure, which in turn is calculated using molecular potentials. The latter method uses the isotherm's desorption branch to relate the amount of N2 removed from pores at decreasing relative pressure to pore width.278,279 Whatever the method, PSDs can be displayed in cumulative or differential form and according to either surface area or pore volume. Once the PSD is achieved, determination of an average pore width is trivial.
Density functional theory. Classical models for pore structure determination rely on parameters including (but not limited to) the monolayer capacity of the adsorbent, as well as the adsorbate–adsorbent interaction. Additionally, they make use of potentially false assumptions such as that the adsorbate behaves as a two-dimensional ideal gas (in the case of the Horvath–Kawazoe model). Conversely, Density Functional Theory (DFT), when applied to porosity, makes use of statistical modelling of adsorbate–adsorbate and adsorbate–adsorbent interactions specific to a system determined by pore size, pore geometry, nature of the adsorptive and temperature. A set of theoretical isotherms is generated according to the specific variables of the system under examination, where each individual, theoretical isotherm is calculated for an idealized adsorbent having a single pore width. This library of calculated isotherms is known as the kernel,
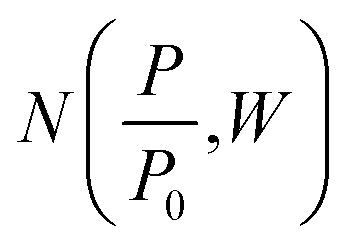 which can be used in conjunction with the experimental isotherm,
which can be used in conjunction with the experimental isotherm, 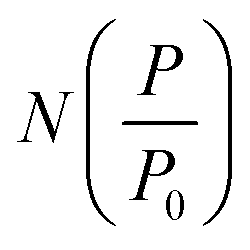 to yield a pore size distribution as a function of pore width, f(W);274
to yield a pore size distribution as a function of pore width, f(W);274This data can be displayed in terms of differential or cumulative pore volume and surface area, and as such can be used to determine textural quantities traditionally calculated via classical methods.215,266 Recent reports indicate that gas uptake may be better predicted using porosity determined according to DFT rather than classical methods.280,281 DFT methods have also been employed in recent years to combine isotherms determined using two separate gases to yield a single PSD.91,225
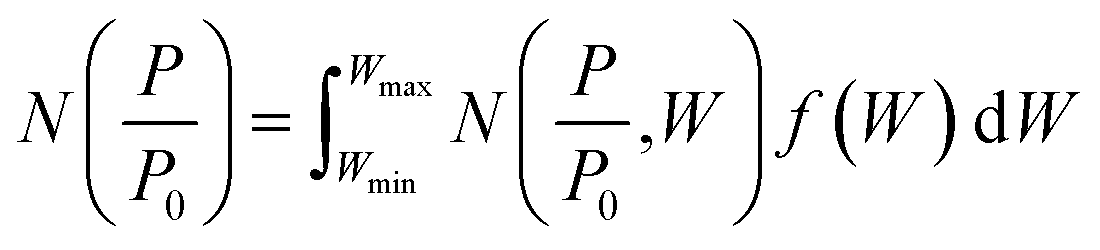
It is useful to compare measures of porosity derived using DFT methods to those using classical methods. In terms of pore volume, Jagiello et al. reported that both total and micropore volume of a set of biochars and activated carbons as determined using the single-point and DR methods respectively, were essentially the same as those found using DFT.91 This was shown to also be true for micropore volumes.282 Villarroel-Rocha et al. found broad agreement between values of micropore volume in microporous materials derived using the DR, t-plot, αS and DFT methods.275 However, this is not true in the application of these methods to all isotherms; DFT and t-plot methods show greater disagreement the further the isotherm deviates from type-I.275,276
3.5. Grain density
While gravimetric measures of porosity have historically been the metric associated with gas uptake increasingly, high volumetric capacity is desired in applications such as methane storage,127,260,283 as well as CO2 capture.174,284,285 Various studies have shown that grain density is a good predictor of H2, CH4 and CO2 capacity, provided that this is balanced with appropriate pore size as well as high surface area and pore volume.35,81,143,178,182,194,255,284,286 Grain density, d may be calculated according towhere ρs is the skeletal density determined via helium pycnometry and VT is the total pore volume.35 Alternatively it can be measured directly by packing the sample at high pressure.127 Determination of grain density allows for the derivation of volumetric equivalents to surface area and pore volume, which are typically calculated gravimetrically. These can be used as further metrics to assess volumetric gas uptake (see Fig. 4).
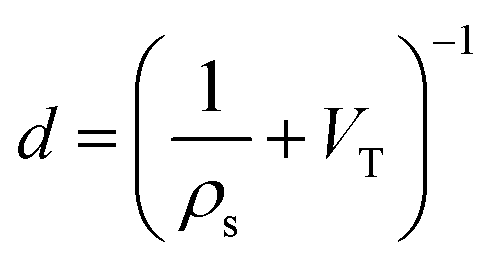
4. Controlling porosity
While the achievement of reasonable porosity in carbons for small gas molecule adsorption is relatively well established, various methods for fine control over various textural parameters are under constant development. Of particular recent interest in the literature are methods to improve volumetric as opposed to gravimetric porosity so as to optimise volumetric capacity for applications such as on-board storage.4.1. Activated carbons
| Porogen | Precursora | A BET | V t | %Vmicd | S | U | Ref. |
|---|---|---|---|---|---|---|---|
| a Precursor description. b BET surface area (m2 g−1). c Total pore volume (cm3 g−1). d Percent microporosity by volume. e Adsorbate measured (values in brackets are pressure (bar) followed by temperature (°C)). f Uptake in mmol g−1 except for H2, which is wt%. g Precursor was converted to hydrochar prior to activation. | |||||||
| CO2 | Graphite oxide | 908 | 3.08 | 3 | CO2 (1, 0) | 1.6 | 249 |
| KOH | Cigarette buttsg | 4310 | 2.09 | 82 | H2 (20, −196) | 9.4 | 82 |
| K2CO3 | Polyacrylonitrile | 1250 | 0.64 | 89 | CO2 (1, 30) | 2.4 | 252 |
| KHCO3 | Glucoseg | 2210 | 0.97 | 84 | 138 | ||
| Potassium oxalate | Sawdustg | 1470 | 0.71 | 73 | CO2 (1, 25) | 4.4 | 135 |
| NaOH | Polyacrylonitrile | 1020 | 0.57 | 89 | CO2 (1, 30) | 2.2 | 252 |
| NaNH2 | Polyacrylonitrile | 833 | 0.36 | 94 | CO2 (1, 30) | 1.8 | 252 |
| H3PO4 | Coconut shell | 304 | CH4 (1, 10) | 0.3 | 287 | ||
The success of group 1 metal hydroxides as activating agents for highly microporous carbons has led to the exploitation of other hydroxides, as well as salts of potassium and sodium as porogens.232,252,288,296,297 Theoretically a gentler activating agent, Ca(OH)2-activation yields carbons with much lower surface areas and a lower degree of microporosity than KOH under identical conditions.288 Taylor found that careful selection of conditions may produce carbons with pore volumes exceeding 1 cm3 g−1, with the majority of this coming from mesopores.296 Although microporosity is low, activation at 800 °C yields carbons whose micropores are dominated by pores around 8 Å, thus achieving moderate CO2 capacity of up to 2.3 mmol g−1 at 1 bar and 25 °C.296 Similarly, Singh et al. reported that the activation of polyacrylonitrile with NaNH2 yields carbons with low overall porosity development, but a higher concentration of pores centred at 7 Å when compared to carbons activated with NaOH under similar conditions. This indicates that when using NaNH2 as an activating agent much of the porosity is likely formed via Na intercalation, rather than redox reactions – in contrast to what happen with NaOH as activating agent.252
While KOH has been identified as a uniquely superior activating agent, its corrosive nature qualifies it as an aggressive reagent in activated carbon synthesis, thus resulting in low yields. As the majority of the microporosity comes about via the intercalation of potassium ions into graphitic layers as opposed to corrosive action of the hydroxide counterion, gentler anions such as carbonate,232,298 and bicarbonate,138,299 have been shown to give improved yields, resulting in high surface area carbons with tuneable porosity. For example, Sevilla and Fuertes produced KHCO3-activated carbons from glucose hydrochar with surface areas of 2000 m2 g−1 with more than 80% microporosity. Porosity development is lower than that for an equivalent amount of KOH under similar conditions because the initial pore formation process – the oxidation of C by K2CO3 – requires temperatures in excess of 700 °C, while KOH can oxidise C at much lower temperatures.138 On the other hand, Xia and co-workers use of an acetate counterion has shown that a contributing factor to pore formation may be hydrogen bonding between polar moieties within the biomass and the activating agents.297
Another promising ‘gentle’ potassium activating agent is potassium oxalate (PO).131,135–137,300,301 The activation mechanism proceeds principally via gasification of K2CO3 at or below 800 °C, resulting in carbons that are almost exclusively microporous. Therefore activating with PO allows a high degree of control over sample porosity – Aljumialy and Mokaya found that pore volume of sawdust-derived carbons could be reliably increased within the micropore region by increasing activation temperature. Interestingly this improvement in overall porosity had little effect on pore sizes. When the activation temperature is increased to 900 °C, oxidative etching of the carbon surface begins,135,136 which results in mesopore formation without the collapse of the previously formed micropores, producing carbons with a hierarchical pore network optimised for moderate pressure (20 bar) CO2 storage (18 mmol g−1 excess uptake). The highly microporous (>90% microporosity) carbons produced at lower temperatures achieve CO2 capacity of 4.3 mmol g−1 at room temperature and ambient pressure.135 Lee et al. achieved similarly high degrees of microporosity (approaching 100%) in the PO activation of carbonized corn husks (CCH). Quite unusually, increasing quantities of PO lead to greater development of ultramicropores. Methane capacity was optimised (7.75 mmol g−1 at 35 bar and 25 °C) at PO/CCH ratio of 3 as a result of highest surface area as well as hierarchical pore structure including ultramicropores.131
Mixing method. Chemical activating agents may be combined with the precursor material either through physical mixing35,135,255,302 or by impregnation with a dissolved agent.75,288,290,295,303 The latter technique, so-called solution impregnation, is followed by a drying step to remove the solvent prior to activation. The perceived advantage of solution impregnation over solid mixing is that the activating agent is more evenly distributed, thus resulting in more consistent porosity throughout the material. Nevertheless, PSDs of carbons derived using the physical mixing technique show narrow PSDs indicating that an impregnation and drying step may not strictly be necessary.80,82,135 There is, unfortunately, a dearth of literature comparing the porosity of carbons derived using the two techniques. However, in 2018 Boujibar et al. reported activated carbons synthesised using KOH and NaOH via both solution impregnation and physical mixing. In terms of surface area, the KOH-activated samples showed improvements of 20% when physical mixing was employed but these gains reduced the proportion of microporosity. Conversely, surface area of NaOH-activated samples were 25% greater using solution impregnation over physical mixing, though the former samples had a much broader PSD. Despite the differences in textural properties attained through these two mixing techniques, CO2 uptake at 25 °C and 1 bar does not significantly differ between the two sample sets, implying that both impregnation and solid mixing can be effectively used to balance overall porosity development and level of microporosity for gas adsorption.292
Combined activating agents. While chemical activating agents provide highly microporous samples which are suited to H2 storage and low pressure CO2 capture, other applications such as high pressure CO2 capture require so-called hierarchical or multimodal pore structures wherein most or all microporosity is retained but further porosity is generated in the mesopore region. Caturla et al. achieved this in 1991 by subjecting a ZnCl2-activated carbon to physical activation using CO2.112 Using both a chemical and physical activating agent has become known as physicochemical activation. Since then other researchers have produced activated carbons via a simultaneous physicochemical method, wherein during the pyrolysis of a mixture of precursor and chemical activating agent the flow of inert gas is interrupted with a physical activating agent.152,287,304,305 In fact, Hu and Srinivasan reported that simultaneous physicochemical activation using ZnCl2 and CO2 is more effective for formation of mesopores than an equivalent sequential activation.304 KOH and H3PO4 have also been used as the chemical porogen in conjunction with CO2 although it appears that using KOH makes the material more resistant to mesopore development.305,306Table 3 gives examples of the porosity of carbons derived through single and two step-activations.
| Precursor | Porogena | A BET | V t | %Vmicf | S | U | Ref. |
|---|---|---|---|---|---|---|---|
| a First and second steps. Activation temperature (°C) in parenthesis, S = steam, M = melamine, PO = potassium oxalate, NaA = sodium alginate. b Simultaneous activation. c Sequential activation. d BET surface area (m2 g−1). e Total pore volume (cm3 g−1). f Percentage microporosity by volume. g Adsorbate gas (CH4, CO2 or H2); values in parenthesis are measurement temperature (°C) and pressure (bar), respectively. h Gravimetric uptake in mmol g−1, except for H2, which is wt%. i Precursor was pyrolysed before porogenesis. j Precursor was hydrothermally carbonised before porogenesis. | |||||||
| Coal | ZnCl2 (500) | 1530 | 0.69 | 61 | 152 | ||
| ZnCl2 (500) and CO2 (950)b | 1820 | 0.97 | 44 | ||||
| Coconut shell | S (unknown) | 875 | CH4 (10, 1) | 0.2 | 287 | ||
| S, then KOH (600)c | 961 | 0.3 | |||||
| Coconut shell | S (unknown) | 875 | CH4 (10, 1) | 0.2 | 287 | ||
| S, then H3PO4 (500)c | 304 | 0.3 | |||||
| Oil palm shell | H3PO4 (450) | 615 | 0.28 | 93 | CH4 (30, 1) | 0.7 | 306 |
| H3PO4 (450), then CO2 (855)c | 642 | 0.28 | 93 | 1.1 | |||
| Neem woodi | KOH (800) | 764 | 0.44 | 81 | 290 | ||
| KOH (800) and S (800)b | 963 | 0.55 | 87 | ||||
| Starchj | KOH (800) | 3000 | 1.4 | 77 | 307 | ||
| KOH (800) and M (800)b | 3280 | 2.4 | 45 | ||||
| Sodium alginate | HNO3 (750), | 444 | 0.21 | CO2 (25, 1) | 2.0 | 308 | |
| HNO3 (750) and H3PO4 (750)b | 1740 | 1.6 | 4.5 | ||||
| Cellulosej | KOH (700) | 1280 | 0.68 | 84 | H2 (−196, 20) | 4.2 | |
| KOH (700) then KOH (700)c | 2470 | 1.18 | 81 | 5.7 | |||
| Sawdust | PO (800) | 1270 | 0.52 | 96 | 136 | ||
| PO (800) and M (800)b | 3090 | 1.8 | 66 | ||||
| Melon seedsi | KOH (600) | 1140 | 0.41 | 100 | H2 (−196, 1) | 1.4 | 309 |
| KOH (600) and NaA (600)b | 2310 | 0.89 | 96 | 2.6 | |||
Physicochemical activation is not limited to broadening of PSDs; for example Song et al. explored the sequential physicochemical activation of coconut shell charcoal using H3PO4 and steam and found that steam treatment at 800 °C produced carbons with twice the surface area of those activated with H3PO4 alone. In contrast to earlier carbons produced using ZnCl2 and CO2, the majority of new porosity development was in the micropore region, with 80% of surface area from pores of width 4.5–6.5 Å. This sample achieved 58% higher gravimetric CH4 uptake at 1 bar and 10 °C compared to a conventionally activated equivalent.287 Adlak et al. reported similar improvements in both overall surface area and microporosity for carbons activated using KOH and steam simultaneously. This could be a result of the dissolution of K compounds in steam, which results in greater mobility of the activating agent through the sample and thus further activation via intercalation.290
The porosity of carbon can also be modulated by adding a so-called mediator to the precursor-activating agent mixture. The mediator is a species that is not in itself an activating agent, but produces activating species as it breaks down during pyrolysis, which can then work in conjunction with the activating agent to produce multimodal porosity. An early example of a mediator is the use of melamine by Fuertes and Sevilla in the KOH-activation of hydrochars. Samples synthesised using melamine had higher surface areas than their conventionally activated counterparts, which came about through the development of mesopores. These mesopores purportedly originate from gasification by volatile nitrogen compounds released upon the decomposition of melamine.307 Indeed, there are other notable instances of volatiles from nitrogenous compounds having an activating effect.221,308 Further work revealed that surface areas above 3200 m2 g−1 could be achieved by this method, with optimal porosity for cryogenic H2 storage at 20 bar, and room temperature CO2 capture at 40 bar, achieving uptakes of 7 wt% and 21 mmol g−1, respectively.244 By swapping KOH for the gentler PO, CO2 uptake at 40 bar was improved to almost 40 mmol g−1 due to a broad, continuous PSD spanning the ultramicropore and small mesopore region.136 More recently Hu et al. reported the use of sodium alginate as a mediator in conjunction with KOH to prevent the over-activation of carbons derived from a variety of biochars.309 The pyrolysis of sodium alginate had previously been shown to produce non-porous carbons.308 On pyrolysis the mediator broke down to produce Na2O which appears to have restricted porosity development and limited the PSD to the micropore region accompanied by further development of ultramicropores. As a result, H2 storage capacity at 1 bar and −196 °C was improved by up to 87% from 1.37 wt% to 2.56 wt%.309
Self-activation. In recent years, research interest has turned to precursors that can develop porosity via pyrolysis without the assistance of an external activating agent. As previously mentioned, so-called biochar is derived through physical self-activation of biomass to produce microporous carbons,89–91 through the release of oxidising gases such as CO2 and H2O upon biomass pyrolysis.310,311 Under typical conditions, the bulk of these oxidising gases are removed from the sample due to a flow of N2 or Ar before they can develop significant porosity. Xia and Shi found that long, harsh pyrolysis of kenaf core in a closed system allowed the oxidising gases to react more with the carbonised biomass to form pores, resulting in mesoporous carbons with surface areas approaching 2500 m2 g−1. Carbons with hierarchical pore structures were produced when temperature and dwell time were limited to 1000 °C and 10 h respectively.312
Precursors or additives with high nitrogen contents are often employed to produce N-doped carbons for use in CO2 capture. However, nitrogen also has a part to play in porosity development in the mesopore region. Sevilla et al. found that polypyrrole-derived carbons gave higher than expected surface area for the activation conditions.126 Later work confirmed that by increasing the N concentration by use of an additive, the porosity of microporous carbons could be extended into the mesopore region, resulting in higher H2 and CO2 storage capacity. This effect is presumed to come about due to the gasification of the carbonaceous structure by volatile N-compounds.244,307 Similar effects have also been reported by Ariharan et al. for precursors containing phosphorous.313,314
Self-activation may also have had a part to play in the unusual temperature-porosity relationship of a series of cigarette butt-derived KOH-activated carbons. It is supposed that the unexpectedly high surface area (4300 m2 g−1) of highly microporous sample activated at 600 °C may be a result of metal contaminants within the cigarette butt that assist KOH in the activation process. Such textural properties resulted in unprecedented hydrogen storage capacity of 8.1 wt% at −196 °C and 20 bar.82 This idea of activation by contaminants is supported by a 2018 study by Longxin Li et al. wherein carbons produced via the simultaneous physicochemical activation of demineralised coal achieved lower surface areas than and equivalent carbon from untreated coal.152
Organic salt carbonisation. Organic salts are an interesting class of so-called self-activating precursors for activated carbon production. To achieve self-activation, the cation must be a metal that can take part in chemical activation. Furthermore, the anion must be both sufficiently rich in carbon, and stable enough that it does not fully degrade before its cation can cause it to undergo activation. For example, while PO is interesting as a so-called gentle porogen, the oxalate (C2O42−) anion degrades to CO32− before activation can be achieved, meaning that no residual carbon remains.133 More bulky precursors such as K, Na, Ca, Mg, Fe, and Zn salts of citric acid have been successfully carbonised to yield carbons with surface areas of at least 500 m2 g−1.315–320 Other notable examples include K, Na, and Ca gluconate,315,321–323 Na and Ca alginate,315 and potassium tartrate.324 K salts of polymers such as poly(acrylamide-co-acrylic acid),325 Na salts of several forms of poly(styrene sulfonate),326–328 and even a Zn organic framework329 have been carbonised in order to build on structural voids in the precursor.
Use of organic salts as self-activation precursors means that porosity of the carbons is easily tuneable simply by changing the anion or cation. For example, Sevilla and Fuertes found that while pyrolysis at 800 °C of potassium gluconate or citrate yields relatively high surface area and highly microporous carbons, the equivalent Ca salts produce a greater degree of mesoporosity in the resultant carbon.315 Pyrolysis of Mg, Zn, and Fe salts also results in highly mesoporous carbons.318–320 Indeed, mesoporous carbons derived from magnesium citrate and zinc glycolate have pore volumes in excess of 2.5 cm3 g−1.318,330 The pore structure of carbons derived from Na salts often lie somewhere in the middle, typically having hierarchical (microporous plus meso- or macroporous) PSDs.321,326–328,331 The identity of the cation can also affect pore shape, for example carbonisation of sodium gluconate produces unusually long, narrow slit-shaped pores, whereas pores in calcium gluconate derived carbons have a more cellular structure.315,332 On the other hand, anion composition and structure seems to have a greater effect on large scale morphological features rather than the fine pore structure.133 For example, while salts of smaller anions like gluconate and citrate can be used to produce regular, sheet-like structures under appropriate conditions, polymeric salts typically yield carbons with a much lower degree of structural regularity.315,321,322,326,327,333,334
Despite the easily tuneable porosity of organic salt-derived carbons, reports on their application as gas storage media are minimal. By far the dominant application of such materials appears to be as electrodes for supercapacitors,315,316,319,320,330,334,335 however their use as (electro)catalysts,319,322,329 as well as ion storage318,331 and battery326,336 materials is well documented. This may be due to the fact that although microporous carbons can easily be formed from organic salts, the surface areas of such materials are typically quite low (<1500 m2 g−1). Of course some gas storage/capture applications require more hierarchical micro-mesoporous pore structure, but surface areas of such carbons still seem to be largely limited to no more than 2000 m2 g−1. A notable exception to this are some carbons synthesised by Sevilla et al. from potassium citrate, which when carbonised with urea at 750–800 °C yields a carbon with surface area of up to 3350 m2 g−1 and pore volume >2.0 cm3 g−1 which is relatively evenly divided between meso- and micropores.335 Such a material would be an interesting candidate for CO2 pressure swing adsorption (PSA) applications but unfortunately this wasn’t examined by the authors. The sole instance of an organic salt-derived carbon being employed in gas storage/capture is that of potassium hydrogen phthalate (KHP). Uniquely narrow PSDs were achieved by researchers in the Mokaya group through gentle pyrolysis conditions producing carbons with a single micropore width. This is presumably the result of mechanistic differences when K+ (as opposed to the harsher KOH) is the primary activating agent – that is, activation proceeds solely via intercalation of potassium ions into the pores. At temperatures above 800 °C, the PSD begins to broaden resulting in higher surface area samples with CO2 capacities up to 4.5 mmol g−1 under ambient conditions.296,337
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :precursor weight ratio to 2 to 4, it was possible to retain much of the microporosity whilst doubling the surface area to over 2000 m2 g−1. The authors attributed the lack of so-called over-activation to the high activation resistance of carbonised date seeds. Porosity of carbons could be controlled via temperature or KOH:precursor ratio towards high gravimetric or volumetric methane capacity (25 °C and 35 bar) up to 11.4 mmol g−1 and 196 cm3STP cm−3, respectively.35
:precursor weight ratio to 2 to 4, it was possible to retain much of the microporosity whilst doubling the surface area to over 2000 m2 g−1. The authors attributed the lack of so-called over-activation to the high activation resistance of carbonised date seeds. Porosity of carbons could be controlled via temperature or KOH:precursor ratio towards high gravimetric or volumetric methane capacity (25 °C and 35 bar) up to 11.4 mmol g−1 and 196 cm3STP cm−3, respectively.35
The O/C ratio of a precursor can also be modulated according to pre-carbonisation conditions. For example, Zhen Zhang et al. synthesised a set of precursors by calcining glucose-derived hydrochar in air at temperatures between 200 and 300 °C, as well as calcining pure glucose at 300 °C, and also examined non-calcined hydrochar. These precursors differed most apparently due to the abundance of COOH moieties present on their surface (so-called ion exchange capacity) which varied from 0.22 to 3.92 mmol g−1. This corresponds to overall O/C ratios in the range 0.37–0.45. The authors posit that the presence of COOH is significant for activation via aqueous solution impregnation with KOH, as the carboxyl group undergoes an ion-exchange reaction to form COOK. The closely bound K+ can thus act as a more effective activating agent than if it were present in the form of KOH. It was found that the content of ultramicropores was optimised for the activated carbon derived from hydrochar calcined at 300 °C, having an ion exchange capacity of 3.92 (O/C ratio 0.45). This sample showed the highest ambient (25 °C, 1 bar) CO2 uptake of 4.3 mmol g−1.251
This does not however mean that a pre-carbonisation or calcination step automatically improves a precursor's ability to form carbons with favourable porosity for small molecule adsorption. Balahmar et al. found that pre-treatment of precursors by hydrothermal carbonisation had either marginal or negative effects on the porosity of KOH-activated carbons derived from a variety of biomasses.339 This may be a function of decreasing O/C ratio and thus increasing resistance to activation.
A simple way to change precursor composition is simply to mix two precursors with significantly different compositions. An example of this is the mixing of polypyrrole (PPY) with sawdust hydrochar (H) or raw sawdust (SD) which have an O/C ratio of 0.672, 0.483 and 0.773, respectively (see Fig. 5). Researchers in the Mokaya group determined that a greater degree of control over porosity could be achieved by activating the mixed precursor than by activating any of the three alone.98,135,221,237 For samples activated with KOH, porosity was mainly developed in the mesopore region. Increasing concentrations of H in a PPY–H mixture results in decreased mesoporosity, which is attributed to the decrease in O/C ratio; more specifically the mesoporosity is a function of the amount of PPY in the mixture whereas the mesopore size is increased with increasing PPY:H ratio.98 However, the same does not apply to carbons synthesised by from mixtures of PPY and SD, wherein increased O/C ratio actually results in higher a higher degree of microporosity and lower porosity over all. This is attributed to stabilising interactions between aromatic moieties on PPY and SD which confer resistance to activation.98 On the other hand, using a gentler activating agent (potassium oxalate) results in more hierarchical meso–microporous carbons, with optimised pore structure for high pressure CO2 capture.135 The appropriate selection of Ppy:H ratio, activating agent and activation conditions can be used to produce carbons with a variety of porosities and textural characteristics suited to applications ranging from low pressure CO2 capture to high pressure H2 storage.98,135
 | ||
| Fig. 5 Variation of pore volume and micropore percentage (by volume) with precursor O/C ratio as controlled by mixing in different quantities the precursors polypyrrole (PPY), sawdust (SD) and sawdust hydrochar (H). Adapted from work by Balahmar et al. O/C ratio. All samples activated at 800 °C for 1 h.98 O/C ratio determined using weighted sums of the O/C contents of PPY, H, and SD. | ||
| Precursor | T | d | A BET | V | v A BET | v V | Pore sizeg | g U | v U | S | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Activation temperature (°C). b Packing density (g cm−3). c BET surface area (m2 g−1). d Total pore volume (cm3 g−1). e Volumetric BET surface area (m2 cm−3). f Volumetric pore volume (cm3 cm−3). g Pore size taken from centre of most prominent peak in PSD. h Gravimetric uptake, in mmol g−1 for CO2 and wt% for H2. i Volumetric uptake (g dm−3). j Adsorbate gas (CO2 or H2); values in parenthesis are pressure (bar) and temperature (°C). | |||||||||||
| Sawdust | 600 | 0.94 | 866 | 0.46 | 814 | 0.43 | 6.8 | 4.3 | 178 | CO2 (1, 25) | 248 |
| 0.95 | 1066 | 0.59 | 1013 | 0.56 | 5.9 | 5.8 | 242 | ||||
| Polypyrrole | 600 | 0.92 | 976 | 0.47 | 898 | 0.43 | 6.8 | 3.4 | 136 | CO2 (1, 25) | 238 |
| 0.85 | 1297 | 0.64 | 1102 | 0.54 | 5.9 | 5.5 | 206 | ||||
| Polypyrrole | 800 | 0.32 | 3589 | 2.71 | 1148 | 0.87 | 28 | 45 | 638 | CO2 (50, 25) | 237 |
| 0.38 | 3945 | 2.92 | 1499 | 1.11 | 34 | 54 | 928 | ||||
| Flash-carbonised wood | 700 | 0.89 | 1280 | 0.65 | 1139 | 0.58 | 6.5 | 2.2 | 129 | CO2 (1, 25) | 81 |
| 0.78 | 2315 | 1.08 | 1806 | 0.84 | 7.0 | 4.0 | 137 | ||||
| Waste tyres | 800 | 775 | 0.41 | 5.8 | 1.1 | H2 (1, −196) | 340 | ||||
In 2018 Rambau et al. reported mesoporous compactivated carbons from used tyre-derived char, however surface areas of the reported carbons do not approach those in other studies under similar conditions.340 This may be a result of the char's relatively high carbon content and associated resistance to activation. An alternative explanation is that the compaction pressure was limited to 10 MPa whereas other studies used up to 740 MPa. Despite the clear advantages of compactivation for carbons for gas storage/capture applications there has been very little published on the topic in the past few years. As such, there is as yet no information on the effect of compaction pressure and duration on the porosity of synthesised carbons.
4.2. Zeolite templated carbons
While the synthesis of ZTCs continues to be a popular area of research,34,172,193,341–344 articles reporting their application in the adsorption of CO2, H2 or CH4 have dwindled in the past three years. In fact since 2018 there exist only nine journal articles on the subject of capture/storage of these molecules in ZTCs.194,256,257,345–350 Furthermore, of these papers two are based on simulations of hypothetical optimized ZTCs.257,345 While these provide insights into what an optimal ZTC structure might be for these applications, they do not provide novel methods for modulating ZTC porosity.257,345 This may be due to a consensus within the community that the more facile synthesis of ACs leads to materials whose pore geometry is more suitable for small molecule sorption. Nonetheless the templating approach provides interesting and unique opportunities for modulation of porosity for use in storage of H2 as well as CO2 capture. In the case of CH4 however, ZTCs have severe limitations on their volumetric storage capacity meaning that they are less feasible as ANG sorbents,34,240,351 although there has been a recent noteworthy attempt to improve volumetric porosity of ZTCs to this end.194| Template | Carbonisationa | A BET | V | H2 uptake conditionsd | H2 uptakee wt% | Ref. | |
|---|---|---|---|---|---|---|---|
| Step 1 | Step 2 | ||||||
| a Carbon source, followed by carbonisation method and, in parenthesis the carbonisation technique (LI or CVD) and temperature (°C); FA = furfuryl alcohol, ET = ethylene, PP = polypropylene, AN = acetonitrile. b BET surface area (m2 g−1). c Pore volume (cm3 g−1); values in parenthesis are micropore volume. d Temperature (°C) and pressure (bar) at which H2 uptake was measured. e H2 uptake capacity (wt%). f No total pore volume reported, thus sum of Vmicro and Vmeso is used. | |||||||
| NaY | FA (LI, 700) | PP (CVD, 900) | 3590 | −196, 24 | 5.5 | 245 | |
| 13X | FA (LI, 700) | ET (CVD, 900) | 3330 | 1.66 (1.18) | −196, 20 | 7.3 | 178 |
| Y | ET (CVD, 900) | 2000 | 1.11 (0.83) | −196, 1 | 1.8 | 190 | |
| Y | ET (CVD, 900) | ET (CVD, 900) | 2160 | 1.26 (0.92) | −196, 1 | 1.9 | 190 |
| Y | FA (LI, 900) | 860 | 0.93 (0.24) | −196, 1 | 0.7 | 190 | |
| Y | FA (LI, 900) | ET (CVD, 900) | 1940 | 1.18 (0.73) | −196, 1 | 1.6 | 190 |
| Y | Sucrose (LI, 800) | 930 | 0.97 (0.30) | 30, 1 | 0.5 | 346 | |
| Y | Sucrose (LI, 800) | 1250 | 0.95 (0.09) | 30, 1 | 1.7 | 352 | |
| Y | Sucrose/AN (LI, 800) | 1670 | 2.28 (0.55) | 31, 1 | 3.9 | 352 | |
| 13X | FA (LI, 700) | Tyre pyrolysis vapour (CVD, 900) | 3250 | 1.72 (1.61) | −196, 1 | 2.5 | 348 |
| 13X | FA (LI, 700) | ET (CVD, 900) | 3340 | 1.88 (1.72) | −196, 1 | 1.9 | 348 |
| Clinoptilolite | 1:5 glucose/urea (LI, 900) |
66 | 0.32 (0.00) | 350 | |||
| Clinoptilolite | 2:5 glucose/urea (LI, 900) |
151 | 0.36 (0.03) | 350 | |||
| Y | ET (CVD, 790) | 1930 | 0.97f (0.73) | 193 | |||
| Y, surfactant templated | ET (CVD, 790) | 2140 | 1.59f (0.74) | 193 | |||
In recent years attempts have been made to use alternative carbon sources such as sucrose. In order to be used for LI the disaccharide must be dissolved. Wijiyanti et al. produced a ZTC with moderate surface area (932 m2 g−1) and pore volume (0.97 cm3 g−1) with hierarchical porosity using sucrose dissolved in H2SO4, resulting in a material which allows rapid ingress of H2 (at 30–50 °C and 1 bar) into the porous structure.346 While the bulk of the literature has focused on using pristine carbon sources in synthesis of ZTCs, Musyoka et al. found that a ‘dirty’ source, namely the pyrolysis gases from used tyres can be used as a CVD vapour (see Table 5). The vapour is composed primarily of small aromatic molecules. When performed following LI templation with furfuryl alcohol on zeolite-13X, the resulting carbons exhibited higher porosity (3254 m2 g−1 and 91% microporosity) compared to those derived using ethylene in the second (CVD) step. Indeed, carbons produced using tyre pyrolysis vapour as a CVD source exhibit a narrower PSD than those conventionally-produced ZTCs. High H2 storage capacity of 2.5 wt% was achieved at −196 °C and 1 bar.348 Although H2 adsorption was not measured above atmospheric pressure, both the ZTC textural characteristics and the trajectory of the reported isotherm indicate this carbon may be suited to higher pressure applications.
The introduction of nitrogen into the structure of ZTCs is meant to improve adsorption of small molecules due to improved adsorbate-adsorbent interactions – typically this is performed by using a nitrogen-rich carbon source.179,253,353,354 Such methods also tend to have the side-effect of changing the porosity of the ZTC. For example, Dewi and Widiastuti found that mixing small amounts of acetonitrile into a sucrose solution prior to its use as an LI carbon source on zeolite-Y resulted in a ZTC with increased surface area (1250 to 1670 m2 g−1) and increased microporosity from 5 to 24%. However, with increasing concentrations of the nitrogen dopant, overall surface area falls to 867 m2 g−1 along with absolute microporosity.352 Similarly, Rangel-Sequeda et al. have recently shown that increasing the amount of urea in a urea-glucose mixture resulted in higher surface area carbons when the mixture was used as an LI agent on the natural zeolite (clinoptilolite) template.350 While the former did measure H2 storage capacity and the latter CO2 capture, it is impossible to disentangle the effects of improved porosity and chemical doping on the improved uptakes.
Template modification. A major stumbling block with the use of microporous zeolites as hard templates for carbons is that if the structure is replicated well, the PSD will invariably be unimodal. This lack of pore hierarchy can limit the gas storage/capture capacity of such ZTCs due to slow ingress into pores. Similarly, problems can arise in the synthesis of ZTCs if the carbon source cannot easily enter the narrow channels.355,356 Mesopores can be created in zeolites (while retaining microporosity) by the use of surfactant-templating wherein the material is steamed in order to partially remove aluminium from the framework357 thus yielding hierarchical templates. Aumond et al. achieved ZTCs with approximately equal micropore and mesopore volume by CVD using ethylene on a surfactant-templated zeolite-Y. The product showed an increase in total pore volume from 0.97 to 1.59 cm3 g−1 relative to a ZTC derived by templating on non-treated zeolite.193 Although not measured, such a material is an interesting candidate for CO2 pressure swing adsorption applications.
Densification. Densification, also known as compaction or pelletisation is a process used to improve the volumetric storage of small molecules on carbons by compressing the skeletal structure i.e. increasing the packing density. As early as 2005, Celzard and Fierro found that increasing the apparent density of AC powders resulted in an approximately proportional improvement in volumetric methane capacity at 35 bar and 20 °C. Increases in apparent density did however lead to reductions in gravimetric uptake, thus densification pressure should be optimised.360 Early attempts to increase density of ZTCs utilised so-called hot densification wherein pressures up to 147 MPa and temperatures up to 300 °C were used in order to convert the diffuse ZTC powder into a dense, compact pellet.258,361 While studying the hot densification (300 °C) of a ZTC derived from zeolite-Y (Na-form), Hou et al. found that although gravimetric surface area decreased with increasing compaction pressure, the opposite trend was found when surface area is measured volumetrically.361 Similarly Guan et al. found that hot densification of a ZTC halved gravimetric measures of porosity, while doubling their volumetric counterparts. As a result, volumetric CH4 storage capacity (at 35 bar and 25 °C) was improved from 60 to 127 cm3STP cm−3 for the pristine and compacted ZTC, respectively.258 Gravimetric porosity generally decreases following densification, but to varying degrees depending on the compaction conditions and nature of the ZTC.178,182,362 However, a report from Almasoudi et al. indicates that if compaction pressure is sufficiently low (370 MPa), gravimetric surface area can increase alongside volumetric porosity resulting in improved gravimetric and volumetric H2 capacity.363 As for pore size, compaction generally leads to the development of further microporosity and/or shifting of the PSD and average pore size towards the ultramicropores region as pores contract under pressure,178,361,363 which may result in improved low pressure gas uptake. Fig. 6 illustrates the effect of densification pressure on various measures of porosity.
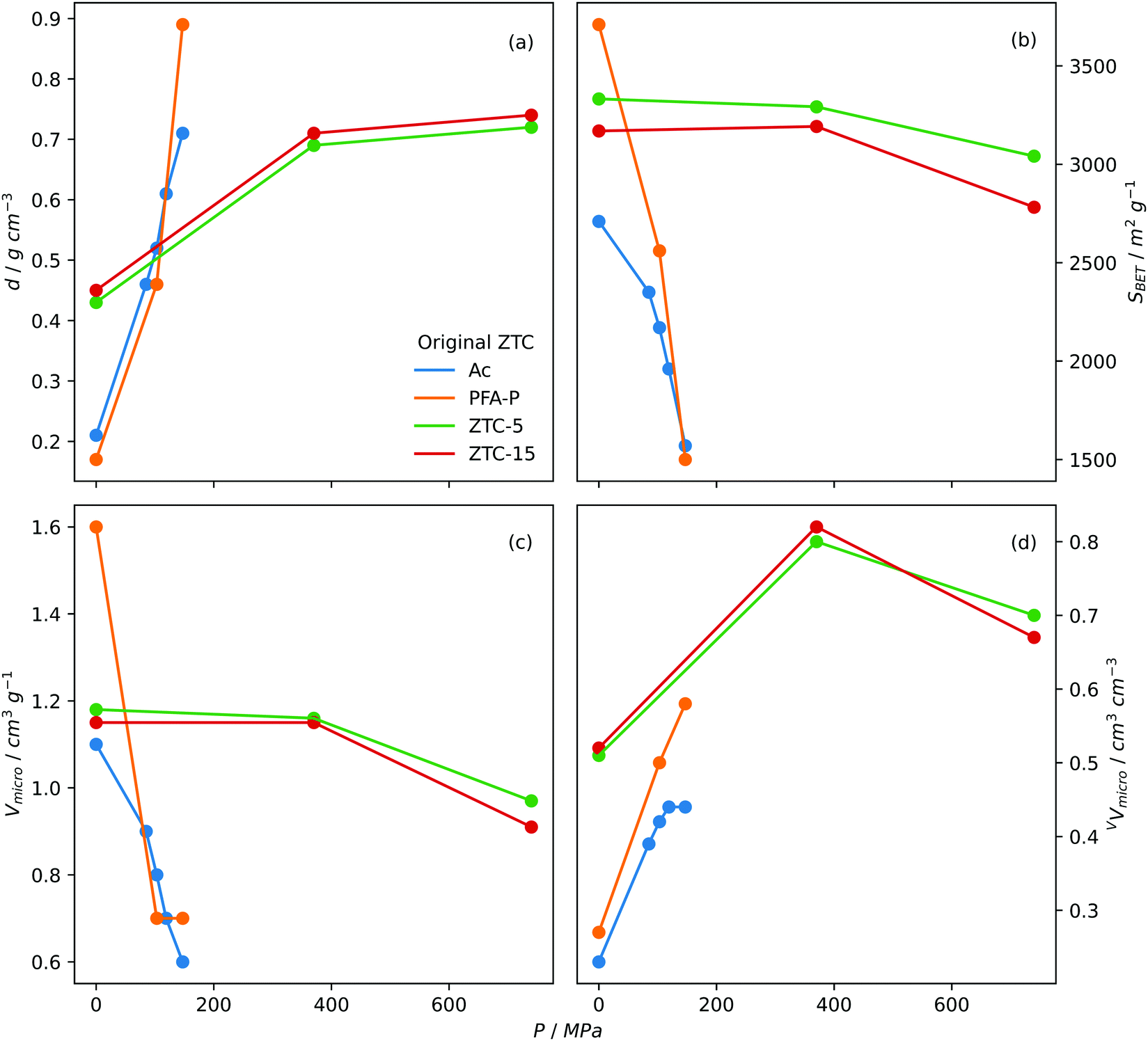 | ||
| Fig. 6 Trends in (a) packing density, (b) BET surface area, (c) gravimetric micropore volume and (d) volumetric micropore volume of densified ZTCs with densification pressure. Ac and PFA-P both templated on Na-zeolite Y with Ac synthesized via acetylene CVD and PFA-P via polyfurfuryl alcohol LI followed by propylene CVD. Densification performed at 300 °C.361 ZTC-5 and ZTC-15 synthesized via polyfurfuryl alcohol LI followed by ethylene CVD on zeolite 13X. 5 and 15 refer to the ramp rate used in the first carbonisation step.178 | ||
More recently, work by Balahmar et al. compared the result of compacting a ZTC to the compaction of the zeolite scaffold prior to templation (so-called pre-compaction) on the textural characteristics and H2 and CO2 storage capacities of the resultant ZTCs. Materials were synthesised on zeolite-13X or -Y templates using CVD of acetonitrile. It was found that pre-compaction actually increased gravimetric porosity in terms of both surface area and pore volume as compared with the conventional ZTC. Furthermore, packing density increased by over 50% from 0.53 to 0.82 g cm−3. This resulted in simultaneous improvement to gravimetric (11.6 to 13.1 mmol g−1) and volumetric (271 to 473 v/v) CO2 uptake at moderate pressure (20 bar and 25 °C). Conversely, post-compaction had almost no effect on gravimetric storage and showed only mild improvements volumetrically.182 A recent paper by Gabe et al. detailed the optimisation of densification conditions for a ZTC mixed with reduced graphene oxide. The authors found that volumetric H2 storage was doubled at all pressures from 0 to 100 bar, when the zeolite-Y-derived carbon was subjected to compaction at 150 °C and 145 MPa. The densified composite also has the advantage of being extremely mechanically robust.364
Activation. The porosity of templated carbons can be expanded via chemical or physical activation; for example the porosity of mesoporous carbons formed when using a silica template can be expanded into the micropore region on activation with KOH or CO2.61,365,366 With the advent of microporous templated carbons based on zeolites, researchers have attempted activation to both improve absolute, overall microporosity and extend porosity into the mesopore region. The activating agent of choice has become KOH as unlike CO2 it can improve overall porosity without destroying the intricate network of micropores already present in the ZTC.83,88,367 Inevitably, activation of ZTCs leads to destruction of much of the crystal structure meaning that the narrow PSDs which ZTCs are prized for are not present in activated-ZTCs,34 although work by Anggarini et al. suggests that increased ordering can be achieved via activation with ZnCl2.368 Relatively aggressive KOH-activation (700–800 °C, 4
:1 w/w) leads to doubling or even tripling of overall surface area according to work done in the Mokaya group. At the same time much, but not all of the original microporosity is retained.83,88,199 However, it is not yet clear to what extent the initial porosity of the ZTC influences that of the activated ZTC. For example, KOH activated carbons with surface areas around 3000 m2 g−1 can be produced from ZTCs with initial surface area as low as 367 m2 g−1 and as high as 1670 m2 g−1.83,367 In fact, in one case ZTCs with the highest initial porosity show the smallest improvements following activation.88 This unclear relationship is probably a result of entangled variables such as ZTC structural type and elemental composition.34,77,135 The benefit of activation of ZTCs is somewhat questionable as the highest surface area achieved through this technique is only around 3350 m2 g−1, significantly less than the record for conventionally activated carbons82 or indeed non-activated ZTCs.369
Nevertheless, activation of ZTCs can lead to nearly doubling of cryogenic hydrogen storage capacity at 20 bar.83,88 On the other hand improvements are not nearly as significant for applications wherein pore size, rather than overall surface area or porosity is the more significant factor in determining gas uptake. For example, Almasoudi et al. showed that despite improvements in overall surface area upon KOH-activation of a carbon derived from the templation of furfuryl alcohol on a zeolitic imidazole framework, ambient gravimetric CO2 capacity could not be improved by more than 23%. Even more striking are accompanying decreases (up to 78%) in CO2 uptake density. This is attributed to significant destruction of micropores during the activation process.199 More recently Susanti and Widiastuti reported KOH-activation of a zeolite-Y templated carbon (ZTC-Y) synthesised via LI. They report that improvement in CO2 (ambient pressure, 30 °C) capture capacity is optimised at a KOH:ZTC-Y ratio of 1.5 from 0.24 mmol g−1 to 0.62 mmol g−1. Interestingly, activation of ZTC-Y is accompanied by a reduction in total surface area and simultaneous decrease in average pore width. This lends further credence to the vital role of pore size in such applications.256
5. Summary and outlook
Experimental control over the porosity of ZTCs and ACs is now well established, with a variety of routes to high surface area carbons possessing either unimodal or hierarchical porosities. As a result, it is possible to achieve carbons with consistently appropriate porosity for good gravimetric uptake of gases under a variety of conditions. For example, there are multiple routes to the narrow PSDs with high surface area useful in H2 storage. For cyclic CO2 capture applications, hierarchical carbons with moderate porosity can be formed both via the templation or activation routes. Paradoxically, of the three sorptives examined in this review, CH4 while the most commercially developed has seen the lowest degree of development in so far as determination of optimal porosity for sorption on carbons. As such, works on porosity modulation for this particular application are limited.Materials with high volumetric capacity are significantly under-investigated, a metric which is particularly relevant to CH4 storage. While there have been some significant and interesting methods used to improve packing density and volumetric porosity of carbons via post-synthetic methods or by altering experimental conditions, work in this area is fairly limited and seems to be restricted to a handful of research groups. In particular, techniques such as compaction and densification have showed a lot of promise but the full effects of the balance of variables such as compaction time, pressure and temperature are yet to be examined – in stark contrast to well-established techniques like KOH-activation.
ACs show more promise than ZTCs as, despite their amorphous structure, it is at least as facile to control their porosity for any given application. Furthermore, conditions and reagents for AC synthesis typically require less precision than those needed for ZTCs resulting in reduced costs and the ability to use abundant and low value recycled materials. Besides for many applications, the exquisitely uniform porosity found in ZTCs is not necessary, and indeed the dearth of literature surrounding CH4 adsorption on ZTCs reflects this. Their greatest utility appears to be in hydrogen storage where extremely homogenous pore widths are required. Further work is needed to determine whether good ZTC carbon sources can be generated from waste materials, and to determine the simplest routes to good ZTC porosity.
Author contributions
L. Scott Blankenship: Investigation, Visualisation, Writing – original draft. Robert Mokaya: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to the EPSRC (Low-Dimensional Materials & Interfaces DTP) for a studentship for LSB. RM thanks the Royal Society for a Royal Society Wolfson Research Merit Award.Notes and references
- V. Ramanathan and Y. Xu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8055–8062 CrossRef CAS PubMed.
- V. Masson-Delmotte, P. Zhai, H.-O. Pörtner, D. C. Roberts, J. Skea, P. R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan and R. Pidcock, Global warming of 1.5 °C: Summary for policy makers, 2018 Search PubMed.
- C.-F. Schleussner, J. Rogelj, M. Schaeffer, T. Lissner, R. Licker, E. M. Fischer, R. Knutti, A. Levermann, K. Frieler and W. Hare, Nat. Clim. Change, 2016, 6, 827–835 CrossRef.
- T. Kuramochi, A. Ramírez, W. Turkenburg and A. Faaij, Prog. Energy Combust. Sci., 2012, 38, 87–112 CrossRef CAS.
- C.-C. Lin, R.-X. He and W.-Y. Liu, Energies, 2018, 11, 3432 CrossRef.
- G. P. Peters, R. M. Andrew, J. G. Canadell, S. Fuss, R. B. Jackson, J. I. Korsbakken, C. Le Quéré and N. Nakicenovic, Nat. Clim. Change, 2017, 7, 118–122 CrossRef.
- R. Clémençon, J. Environ. Dev., 2016, 25, 3–24 CrossRef.
- UK becomes first major economy to pass net zero emissions law, https://www.gov.uk/government/news/uk-becomes-first-major-economy-to-pass-net-zero-emissions-law, accessed 23rd of March, 2021.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- U. Eberle, B. Müller and R. Von Helmolt, Energy Environ. Sci., 2012, 5, 8780–8798 RSC.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- K. V. Kumar, K. Preuss, M.-M. Titirici and F. Rodríguez-Reinoso, Chem. Rev., 2017, 117, 1796–1825 CrossRef CAS PubMed.
- I. Angelidaki, L. Treu, P. Tsapekos, G. Luo, S. Campanaro, H. Wenzel and P. G. Kougias, Biotechnol. Adv., 2018, 36, 452–466 CrossRef CAS PubMed.
- T. A. Makal, J.-R. Li, W. Lu and H.-C. Zhou, Chem. Soc. Rev., 2012, 41, 7761–7779 RSC.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 RSC.
- M. Cozier, Greenhouse Gases: Sci. Technol., 2019, 9, 1084–1086 CrossRef.
- D. Lozano-Castello, J. Alcaniz-Monge, M. De la Casa-Lillo, D. Cazorla-Amoros and A. Linares-Solano, Fuel, 2002, 81, 1777–1803 CrossRef CAS.
- J. Romanos, S. Sweany, T. Rash, L. Firlej, B. Kuchta, J. Idrobo and P. Pfeifer, Adsorpt. Sci. Technol., 2014, 32, 681–691 CrossRef CAS.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC.
- L. Schlapbach and A. Züttel, Mater. Sustainable Energy, 2011, 265–270 Search PubMed.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS PubMed.
- Z. Liang, K. Fu, R. Idem and P. Tontiwachwuthikul, Chin. J. Chem. Eng., 2016, 24, 278–288 CrossRef CAS.
- K. S. Fisher, K. Searcy, G. T. Rochelle, S. Ziaii and C. Schubert, Advanced Amine Solvent Formulations and Process Integration for Near-term CO2 Capture Success, Trimeric Corporation, 2007 Search PubMed.
- P. Pfeifer, L. Aston, M. Banks, S. Barker, J. Burress, S. Carter, J. Coleman, S. Crockett, C. Faulhaber and J. Flavin, Chaos, 2007, 17, 41108 CrossRef CAS PubMed.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- D. Farrusseng, Metal-organic Frameworks: Applications from Catalysis to Gas Storage, John Wiley & Sons, 2011 Search PubMed.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- J.-Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672 RSC.
- M. Pera-Titus, Chem. Rev., 2014, 114, 1413–1492 CrossRef CAS PubMed.
- R. V. Siriwardane, M.-S. Shen, E. P. Fisher and J. Losch, Energy Fuels, 2005, 19, 1153–1159 CrossRef CAS.
- J. Weitkamp, M. Fritz and S. Ernst, Zeolites as media for hydrogen storage, Elsevier, Montreal, 1992 Search PubMed.
- R. Krishna and J. M. van Baten, J. Membr. Sci., 2010, 360, 323–333 CrossRef CAS.
- H. Nishihara and T. Kyotani, Chem. Commun., 2018, 54, 5648–5673 RSC.
- A. Altwala and R. Mokaya, Energy Environ. Sci., 2020, 13, 2967–2978 RSC.
- C. Xu and M. Stromme, Nanomaterials, 2019, 9, 103 CrossRef PubMed.
- I. Toda, H. Toda, H. Akasaka, S. Ohshio, S. Himeno and H. Saitoh, J. Ceram. Soc. Jpn., 2013, 121, 464–466 CrossRef CAS.
- S. Alhasan, R. Carriveau and D.-K. Ting, Int. J. Environ. Studies, 2016, 73, 343–356 CrossRef CAS.
- iNatGas, The Natural Gas Industry Solutions, https://inatgas.com/, accessed 12th December, 2021.
- Natural Gas Fueling Infrastructure Development, https://afdc.energy.gov/fuels/natural_gas_infrastructure.html, accessed 21st December, 2021.
- G. Basich and T. Dao, CNG Use Expands Among Government Agencies, https://www.government-fleet.com/152834/cng-use-expands-among-government-agencies, accessed 21st December, 2021.
- Shell, Liquefied Natural Gas (LNG), https://www.shell.com/energy-and-innovation/natural-gas/liquefied-natural-gas-lng.html, accessed 21st December, 2021.
- Ingevity, Adsorbed Natural Gas, https://www.ingevity.com/markets/adsorbed-natural-gas/, accessed 21st December, 2021.
- Daigas, Biogas purification and adsorption storage system, https://www.daigasgps.co.jp/en/service/engineering/energy/biogas_stocksystem/, accessed 21st December, 2021.
- M. Smith and J. Gonzales, Costs associated with compressed natural gas vehicle fueling infrastructure, National Renewable Energy Lab. (NREL), Golden, CO (United States), 2014 Search PubMed.
- A. Y. Tsivadze, O. E. Aksyutin, A. G. Ishkov, I. E. Men'shchikov, A. A. Fomkin, A. V. Shkolin, E. V. Khozina and V. A. Grachev, Russ. Chem. Rev., 2018, 87, 950–983 CrossRef CAS.
- P. Greene, Basics of Biogas Upgrading, https://www.biocycle.net/basics-biogas-upgrading/, accessed 21st December, 2021.
- F. Bauer, T. Persson, C. Hulteberg and D. Tamm, Biofuels, Bioprod. Biorefin., 2013, 7, 499–511 CrossRef CAS.
- T. Kvist and N. Aryal, Waste Manage., 2019, 87, 295–300 CrossRef CAS PubMed.
- A. McLeod, B. Jefferson and E. J. McAdam, Water Res., 2013, 47, 3688–3695 CrossRef CAS PubMed.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- F. Rodriguez-Reinoso and M. Molina-Sabio, Carbon, 1992, 30, 1111–1118 CrossRef CAS.
- J. Ozaki, N. Endo, W. Ohizumi, K. Igarashi and M. Nakahara, Carbon, 1997, 35, 1031–1033 CrossRef CAS.
- N. Patel, K. Okabe and A. Oya, Carbon, 2002, 40, 315–320 CrossRef CAS.
- R. Pekala and D. Schaefer, Macromolecules, 1993, 26, 5487–5493 CrossRef CAS.
- H. Nishihara, K. Imai, H. Itoi, K. Nomura, K. Takai and T. Kyotani, Tanso, 2017, 2017, 169–174 CrossRef.
- V. Presser, M. Heon and Y. Gogotsi, Adv. Funct. Mater., 2011, 21, 810–833 CrossRef CAS.
- Y. Korenblit and G. Yushin, in Carbon Nanomaterials, ed. Y. Gogotsi and V. Presser, 2nd edn, 2014, ch. 11, pp. 303–330 Search PubMed.
- W. Gu and G. Yushin, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 424–473 CAS.
- N. Texier-Mandoki, J. Dentzer, T. Piquero, S. Saadallah, P. David and C. Vix-Guterl, Carbon, 2004, 42, 2744–2747 CrossRef CAS.
- E. Masika and R. Mokaya, J. Phys. Chem. C, 2012, 116, 25734–25740 CrossRef CAS.
- C. F. Martín, M. G. Plaza, J. Pis, F. Rubiera, C. Pevida and T. Centeno, Sep. Purif. Technol., 2010, 74, 225–229 CrossRef.
- M. Sevilla, J. B. Parra and A. B. Fuertes, ACS Appl. Mater. Interfaces, 2013, 5, 6360–6368 CrossRef CAS PubMed.
- Z. Yang, Y. Xia, X. Sun and R. Mokaya, J. Phys. Chem. B, 2006, 110, 18424–18431 CrossRef CAS PubMed.
- Y. Li, S. Wang, B. Wang, Y. Wang and J. Wei, Nanomaterials, 2020, 10, 174 CrossRef CAS PubMed.
- B. Adeniran and R. Mokaya, Chem. Mater., 2016, 28, 994–1001 CrossRef CAS.
- S. E. Moradi, Korean J. Chem. Eng., 2014, 31, 1651–1655 CrossRef CAS.
- A. I. Sultana, N. Saha and M. T. Reza, Sustainability, 2021, 13, 1947 CrossRef.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- S. Oh and K. Kim, Chem. Commun., 1999, 2177–2178 Search PubMed.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- L. Radhakrishnan, J. Reboul, S. Furukawa, P. Srinivasu, S. Kitagawa and Y. Yamauchi, Chem. Mater., 2011, 23, 1225–1231 CrossRef CAS.
- H. Nishihara, T. Hirota, K. Matsuura, M. Ohwada, N. Hoshino, T. Akutagawa, T. Higuchi, H. Jinnai, Y. Koseki and H. Kasai, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed.
- M. L. Botome, P. Poletto, J. Junges, D. Perondi, A. Dettmer and M. Godinho, Chem. Eng. J., 2017, 321, 614–621 CrossRef CAS.
- R. Pedicini, S. Maisano, V. Chiodo, G. Conte, A. Policicchio and R. G. Agostino, Int. J. Hydrogen Energy, 2020, 45, 14038–14047 CrossRef CAS.
- A. E. Ogungbenro, D. V. Quang, K. A. Al-Ali, L. F. Vega and M. R. M. Abu-Zahra, J. Environ. Chem. Eng., 2018, 6, 4245–4252 CrossRef CAS.
- H. M. Coromina, D. A. Walsh and R. Mokaya, J. Mater. Chem. A, 2016, 4, 280–289 RSC.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 4, 1400–1410 RSC.
- E. A. Hirst, A. Taylor and R. Mokaya, J. Mater. Chem. A, 2018, 6, 12393–12403 RSC.
- E. Haffner-Staton, N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2016, 4, 13324–13335 RSC.
- L. S. Blankenship and R. Mokaya, Energy Environ. Sci., 2017, 10, 2552–2562 RSC.
- M. Sevilla, N. Alam and R. Mokaya, J. Phys. Chem. C, 2010, 114, 11314–11319 CrossRef CAS.
- M. Sevilla, R. Foulston and R. Mokaya, Energy Environ. Sci., 2010, 3, 223–227 RSC.
- M. Sevilla and R. Mokaya, J. Mater. Chem., 2011, 21, 4727–4732 RSC.
- S.-Y. Lee and S.-J. Park, J. Solid State Chem., 2010, 183, 2951–2956 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, Adv. Mater. Res., 2010, 123, 695–698 Search PubMed.
- A. Almasoudi and R. Mokaya, J. Mater. Chem., 2012, 22, 146–152 RSC.
- A. Downie, A. Crosky and P. Munroe, in Biochar for Environmental Management: Science and Technology, ed. J. Lehman and S. Joseph, 2009, ch. 2, p. 14 Search PubMed.
- K. Weber and P. Quicker, Fuel, 2018, 217, 240–261 CrossRef CAS.
- J. Jagiello, J. Kenvin, A. Celzard and V. Fierro, Carbon, 2019, 144, 206–215 CrossRef CAS.
- J. F. González, J. M. Encinar, C. M. González-García, E. Sabio, A. Ramiro, J. L. Canito and J. Gañán, Appl. Surf. Sci., 2006, 252, 5999–6004 CrossRef.
- J. Ganan, J. Gonzalez, C. Gonzalez-Garcia, A. Ramiro, E. Sabio and S. Roman, Appl. Surf. Sci., 2006, 252, 5988–5992 CrossRef CAS.
- S. Osswald, C. Portet, Y. Gogotsi, G. Laudisio, J. Singer, J. Fischer, V. Sokolov, J. Kukushkina and A. Kravchik, J. Solid State Chem., 2009, 182, 1733–1741 CrossRef CAS.
- R. Ströbel, L. Jörissen, T. Schliermann, V. Trapp, W. Schütz, K. Bohmhammel, G. Wolf and J. Garche, J. Power Sources, 1999, 84, 221–224 CrossRef.
- N. P. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112–116 RSC.
- V. Presser, J. McDonough, S.-H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 RSC.
- N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2019, 7, 17466–17479 RSC.
- M. G. Plaza, S. Garcia, F. Rubiera, J. J. Pis and C. Pevida, Chem. Eng. J., 2010, 163, 41–47 CrossRef CAS.
- H. An, B. Feng and S. Su, Int. J. Greenhouse Gas Control, 2011, 5, 16–25 CrossRef CAS.
- G. Sdanghi, R. L. Canevesi, A. Celzard, M. Thommes and V. Fierro, Carbon, 2020, 6, 46 CAS.
- B. Feng and S. K. Bhatia, Carbon, 2003, 41, 507–523 CrossRef CAS.
- E. A. Dawson, G. Parkes, P. A. Barnes and M. J. Chinn, Carbon, 2003, 41, 571–578 CrossRef CAS.
- S. K. Ryu, H. Jin, D. Gondy, N. Pusset and P. Ehrburger, Carbon, 1993, 31, 841–842 CrossRef CAS.
- F. Rodriguez-Reinoso, M. Molina-Sabio and M. González, Carbon, 1995, 33, 15–23 CrossRef CAS.
- C.-F. Chang, C.-Y. Chang and W.-T. Tsai, J. Colloid Interface Sci., 2000, 232, 45–49 CrossRef CAS PubMed.
- M. M. Johns, W. E. Marshall and C. A. Toles, J. Chem. Technol. Biotechnol., 1999, 74, 1037–1044 CrossRef CAS.
- S. Román, J. F. González, C. M. González-García and F. Zamora, Fuel Process. Technol., 2008, 89, 715–720 CrossRef.
- H. Wang, Q. Gao and J. Hu, J. Am. Chem. Soc., 2009, 131, 7016–7022 CrossRef CAS PubMed.
- M. Jagtoyen and F. Derbyshire, Carbon, 1998, 36, 1085–1097 CrossRef CAS.
- T. K. Das, S. Banerjee, P. Sharma, V. Sudarsan and P. U. Sastry, Int. J. Hydrogen Energy, 2018, 43, 8385–8394 CrossRef CAS.
- F. Caturla, M. Molina-Sabio and F. Rodriguez-Reinoso, Carbon, 1991, 29, 999–1007 CrossRef CAS.
- M. Molina-Sabio and F. Rodrıguez-Reinoso, Colloids Surf., A, 2004, 241, 15–25 CrossRef CAS.
- J. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710–23725 RSC.
- T. Otowa, R. Tanibata and M. Itoh, Gas Sep. Purif., 1993, 7, 241–245 CrossRef CAS.
- D. Lozano-Castello, J. Calo, D. Cazorla-Amoros and A. Linares-Solano, Carbon, 2007, 45, 2529–2536 CrossRef CAS.
- E. Raymundo-Pinero, P. Azaïs, T. Cacciaguerra, D. Cazorla-Amorós, A. Linares-Solano and F. Béguin, Carbon, 2005, 43, 786–795 CrossRef CAS.
- W. Qiao, S.-H. Yoon and I. Mochida, Energy Fuels, 2006, 20, 1680–1684 CrossRef CAS.
- M. Lillo-Ródenas, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2003, 41, 267–275 CrossRef.
- M. Lillo-Ródenas, J. Juan-Juan, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2004, 42, 1371–1375 CrossRef.
- Y. Yamashita and K. Ouchi, Carbon, 1982, 20, 41–45 CrossRef CAS.
- S. Kelemen and C. Mims, Surf. Sci., 1983, 133, 71–88 CrossRef CAS.
- R. Xue and Z. Shen, Carbon, 2003, 41, 1862–1864 CrossRef CAS.
- E. Fuente, R. R. Gil, R. P. Giron, M. A. Lillo-Ródenas, M. A. Montes-Moran, M. J. Martin and A. Linares-Solano, Carbon, 2010, 48, 1032–1037 CrossRef CAS.
- N. Tsubouchi, M. Nishio and Y. Mochizuki, Appl. Surf. Sci., 2016, 371, 301–306 CrossRef CAS.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- D. Lozano-Castello, D. Cazorla-Amoros, A. Linares-Solano and D. F. Quinn, Carbon, 2002, 40, 989–1002 CrossRef CAS.
- W. Zhao, V. Fierro, C. Zlotea, E. Aylon, M. Izquierdo, M. Latroche and A. Celzard, Int. J. Hydrogen Energy, 2011, 36, 11746–11751 CrossRef CAS.
- W. Zhao, V. Fierro, C. Zlotea, E. Aylon, M. Izquierdo, M. Latroche and A. Celzard, Int. J. Hydrogen Energy, 2011, 36, 5431–5434 CrossRef CAS.
- W. Zhao, V. Fierro, N. Fernández-Huerta, M. Izquierdo and A. Celzard, Int. J. Hydrogen Energy, 2012, 37, 14278–14284 CrossRef CAS.
- J.-H. Lee and S.-J. Park, J. Nanosci. Nanotechnol., 2020, 20, 7124–7129 CrossRef CAS PubMed.
- J. Ludwinowicz and M. Jaroniec, Carbon, 2015, 82, 297–303 CrossRef CAS.
- M. Sevilla, N. Díez and A. B. Fuertes, ChemSusChem, 2021, 14, 94–117 CrossRef CAS PubMed.
- J. V. Guerrera, J. N. Burrow, J. E. Eichler, M. Z. Rahman, M. V. Namireddy, K. A. Friedman, S. S. Coffman, D. C. Calabro and C. B. Mullins, Energy Fuels, 2020, 34, 6101–6112 CrossRef CAS.
- A. M. Aljumialy and R. Mokaya, Mater. Adv., 2020, 1, 3267–3280 RSC.
- M. Sevilla, A. S. M. Al-Jumialy, A. B. Fuertes and R. Mokaya, ACS Appl. Mater. Interfaces, 2018, 10, 1623–1633 CrossRef CAS PubMed.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Carbon, 2017, 114, 50–58 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, ChemSusChem, 2016, 9, 1880–1888 CrossRef CAS PubMed.
- V. Fierro, V. Torné-Fernández and A. Celzard, Microporous Mesoporous Mater., 2006, 92, 243–250 CrossRef CAS.
- V. Fierro, V. Torné-Fernández, A. Celzard and D. Montané, J. Hazard. Mater., 2007, 149, 126–133 CrossRef CAS PubMed.
- A. S. Oberoi, J. Andrews, A. L. Chaffee and L. Ciddor, Int. J. Hydrogen Energy, 2016, 41, 23099–23108 CrossRef CAS.
- N. M. Musyoka, M. Wdowin, K. M. Rambau, W. Franus, R. Panek, J. Madej and D. Czarna-Juszkiewicz, Renewable Energy, 2020, 155, 1264–1271 CrossRef CAS.
- J. Liu, X. Liu, Y. Sun, C. Sun, H. Liu, L. A. Stevens, K. Li and C. E. Snape, Adv. Sustainable Syst., 2018, 2, 1700115 CrossRef.
- L.-P. Xiao, Z.-J. Shi, F. Xu and R.-C. Sun, Bioresour. Technol., 2012, 118, 619–623 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Chem. – Eur. J., 2009, 15, 4195–4203 CrossRef CAS PubMed.
- M. Sevilla, J. A. Maciá-Agulló and A. B. Fuertes, Biomass Bioenergy, 2011, 35, 3152–3159 CrossRef CAS.
- M.-M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- W. Ao, J. Fu, X. Mao, Q. Kang, C. Ran, Y. Liu, H. Zhang, Z. Gao, J. Li and G. Liu, Renewable Sustainable Energy Rev., 2018, 92, 958–979 CrossRef CAS.
- M. Fernanda Gonzalez-Navarro, L. Giraldo and J. Carlos Moreno-Pirajan, J. Anal. Appl. Pyrolysis, 2014, 107, 82–86 CrossRef.
- X. Fang, G. Li, J. Li, H. Jin, J. Li, V. Jegatheesan, S. Li, H. Wang and M. Yang, Desalin. Water Treat., 2017, 96, 120–127 CrossRef CAS.
- P. Veerakumar, T. Maiyalagan, B. G. S. Raj, K. Guruprasad, Z. Jiang and K.-C. Lin, Arabian J. Chem., 2020, 13, 2995–3007 CrossRef CAS.
- L. Li, F. Sun, J. Gao, L. Wang, X. Pi and G. Zhao, RSC Adv., 2018, 8, 14488–14499 RSC.
- J. Y. Lin, Science, 2016, 353, 121–122 CrossRef CAS PubMed.
- D. W. Breck, Zeolite molecular sieves, Wiley, New York, 1974 Search PubMed.
- D. Bastani, N. Esmaeili and M. Asadollahi, J. Ind. Eng. Chem., 2013, 19, 375–393 CrossRef CAS.
- H. C. Foley, in Perspectives in Molecular Sieve Science, ed. W. H. Flank and T. E. Whyte, American Chemical Society, 1988, ch. 21, pp. 335–360 Search PubMed.
- P. Serp, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, 2nd edn, 2013, vol. 7, ch. 13, pp. 323–369 Search PubMed.
- J. S. Beck and J. C. Vartuli, Curr. Opin. Solid State Mater. Sci., 1996, 1, 76–87 CrossRef CAS.
- A. Dyer, An introduction to zeolite molecular sieves, Wiley, United States, 1988 Search PubMed.
- H. Marsh and F. Rodriguez-Reinoso, Activated Carbon, Elsevier Science Ltd., Oxford, UK, 2006, pp. 243–321 Search PubMed.
- A. R. Mohamed, M. Mohammadi and G. N. Darzi, Renewable Sustainable Energy Rev., 2010, 14, 1591–1599 CrossRef CAS.
- T. Horikawa, J. i. Hayashi and K. Muroyama, Carbon, 2002, 40, 709–714 CrossRef CAS.
- C. Gomez-de-Salazar, A. Sepúlveda-Escribano and F. Rodrıguez-Reinoso, Carbon, 2000, 38, 1889–1892 CrossRef CAS.
- G. Bello, R. García, R. Arriagada, A. Sepulveda-Escribano and F. Rodrıguez-Reinoso, Microporous Mesoporous Mater., 2002, 56, 139–145 CrossRef CAS.
- T. Maesen, in Introduction to Zeolite Science and Practice, ed. J. Čejka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, Amsterdam, 3rd edn, 2007, ch. 1, pp. 1–12 Search PubMed.
- J. Liu and J. Yu, in Zeolites and Zeolite-Like Materials, ed. B. F. Sels and L. M. Kustov, Elsevier, 2016, ch. 1, pp. 1–25 Search PubMed.
- D. Barthelmy, Minerals Arranged by the New Dana Classification, http://webmineral.com/danaclass.shtml, accessed 16th March, 2021.
- D. J. Earl and M. W. Deem, Ind. Eng. Chem. Res., 2006, 45, 5449–5454 CrossRef CAS.
- C. Baerlocher, L. B. McCusker, W. M. Meier and D. Olson, Database of Zeolite Structures, http://www.iza-structure.org/databases/, accessed March 16th, 2021.
- N. Kosinov, J. Gascon, F. Kapteijn and E. J. Hensen, J. Membr. Sci., 2016, 499, 65–79 CrossRef CAS.
- B. Sakintuna and Y. Yürüm, Ind. Eng. Chem. Res., 2005, 44, 2893–2902 CrossRef CAS.
- J. Miao, Z. L. Lang, T. Y. Xue, Y. Li, Y. W. Li, J. J. Cheng, H. Zhang and Z. K. Tang, Adv. Sci., 2020, 7, 32 Search PubMed.
- J. Rodriguez-Mirasol, T. Cordero, A. L. Radovic and J. Rodriguez, Chem. Mater., 1998, 10, 550–558 CrossRef CAS.
- T. Kyotani, T. Nagai, S. Inoue and A. Tomita, Chem. Mater., 1997, 9, 609–615 CrossRef CAS.
- S. A. Johnson, E. S. Brigham, P. J. Ollivier and T. E. Mallouk, Chem. Mater., 1997, 9, 2448–2458 CrossRef CAS.
- K. Kim, M. Choi and R. Ryoo, Carbon, 2013, 60, 175–185 CrossRef CAS.
- H. Nishihara and T. Kyotani, in Novel Carbon Adsorbents, ed. J. Tascón, Elsevier, London, 2012 Search PubMed.
- E. Masika and R. Mokaya, Energy Environ. Sci., 2014, 7, 427–434 RSC.
- Y. Xia, G. S. Walker, D. M. Grant and R. Mokaya, J. Am. Chem. Soc., 2009, 131, 16493–16499 CrossRef CAS PubMed.
- N. Alam and R. Mokaya, Energy Environ. Sci., 2010, 3, 1773–1781 RSC.
- N. Alam and R. Mokaya, Microporous Mesoporous Mater., 2011, 142, 716–724 CrossRef CAS.
- N. Balahmar, A. M. Lowbridge and R. Mokaya, J. Mater. Chem. A, 2016, 4, 14254–14266 RSC.
- C. Guan, F. Su, X. Zhao and K. Wang, Sep. Purif. Technol., 2008, 64, 124–126 CrossRef CAS.
- J. Shi, W. Li and D. Li, Colloids Surf., A, 2015, 485, 11–17 CrossRef CAS.
- R. M. Freire, A. H. de Morais Batista, A. G. de Souza Filho, J. M. Filho, G. D. Saraiva and A. C. Oliveira, Catal. Lett., 2009, 131, 135–145 CrossRef CAS.
- R. Ruiz-Rosas, M. J. Valero-Romero, D. Salinas-Torres, J. Rodríguez-Mirasol, T. Cordero, E. Morallon and D. Cazorla-Amorós, ChemSusChem, 2014, 7, 1458–1467 CrossRef CAS PubMed.
- M. Valero-Romero, E. Márquez-Franco, J. Bedia, J. Rodríguez-Mirasol and T. Cordero, Microporous Mesoporous Mater., 2014, 196, 68–78 CrossRef CAS.
- M. Seredych and T. J. Bandosz, Langmuir, 2007, 23, 6033–6041 CrossRef CAS PubMed.
- Y. Lv, Z. Wu, Y. Fang, X. Qian, A. M. Asiri, B. Tu and D. Zhao, Appl. Mater., 2014, 2, 113302 CrossRef.
- Y. Xia, Z. Yang and R. Mokaya, Chem. Vap. Deposition, 2010, 16, 322–328 CrossRef CAS.
- T. Kyotani, Z. Ma and A. Tomita, Carbon, 2003, 41, 1451–1459 CrossRef CAS.
- L. Chen, R. K. Singh and P. Webley, Microporous Mesoporous Mater., 2007, 102, 159–170 CrossRef CAS.
- T. Aumond, J. Rousseau, Y. Pouilloux, L. Pinard and A. Sachse, Carbon Trends, 2021, 2, 100014 CrossRef.
- S. Choi, M. A. Alkhabbaz, Y. G. Wang, R. M. Othman and M. Choi, Carbon, 2019, 141, 143–153 CrossRef CAS.
- P. X. Hou, T. Yamazaki, H. Orikasa and T. Kyotani, Carbon, 2005, 43, 2624–2627 CrossRef CAS.
- N. Alam and R. Mokaya, Microporous Mesoporous Mater., 2011, 144, 140–147 CrossRef CAS.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673–1679 CrossRef CAS PubMed.
- A. Pacuła and R. Mokaya, J. Phys. Chem. C, 2008, 112, 2764–2769 CrossRef.
- A. Almasoudi and R. Mokaya, Microporous Mesoporous Mater., 2014, 195, 258–265 CrossRef CAS.
- C. Ducrot-Boisgontier, J. Parmentier, A. Faour, J. L. Patarin and G. D. Pirngruber, Energy Fuels, 2010, 24, 3595–3602 CrossRef CAS.
- Z. Ma, T. Kyotani and A. Tomita, Chem. Commun., 2000, 2365–2366 RSC.
- H.-K. Youn, J. Kim, G. Chandrasekar, H. Jin and W.-S. Ahn, Mater. Lett., 2011, 65, 1772–1774 CrossRef CAS.
- P.-X. Hou, H. Orikasa, T. Yamazaki, K. Matsuoka, A. Tomita, N. Setoyama, Y. Fukushima and T. Kyotani, Chem. Mater., 2005, 17, 5187–5193 CrossRef CAS.
- F. Su, J. Zeng, Y. Yu, L. Lv, J. Y. Lee and X. Zhao, Carbon, 2005, 43, 2366–2373 CrossRef CAS.
- J. Wang, Z. Liu, X. Dong, C.-E. Hsiung, Y. Zhu, L. Liu and Y. Han, J. Mater. Chem. A, 2017, 5, 6860–6865 RSC.
- R. J. Konwar and M. De, Microporous Mesoporous Mater., 2013, 175, 16–24 CrossRef CAS.
- Y. Li, X. Wang, T. Thersleff, G. Svensson and N. Hedin, ACS Omega, 2019, 4, 9889–9895 CrossRef CAS PubMed.
- M.-C. Silaghi, C. Chizallet and P. Raybaud, Microporous Mesoporous Mater., 2014, 191, 82–96 CrossRef CAS.
- K. Kim, T. Lee, Y. Kwon, Y. Seo, J. Song, J. K. Park, H. Lee, J. Y. Park, H. Ihee and S. J. Cho, Nature, 2016, 535, 131–135 CrossRef CAS PubMed.
- G.-H. Moon, A. Bähr and H. Tüysüz, Chem. Mater., 2018, 30, 3779–3788 CrossRef CAS.
- H. Park, S. K. Terhorst, R. K. Bera and R. Ryoo, Carbon, 2019, 155, 570–579 CrossRef CAS.
- M. De la Casa-Lillo, F. Lamari-Darkrim, D. Cazorla-Amoros and A. Linares-Solano, J. Phys. Chem. B, 2002, 106, 10930–10934 CrossRef CAS.
- S. S. Samantaray, S. R. Mangisetti and S. Ramaprabhu, J. Alloys Compd., 2019, 789, 800–804 CrossRef CAS.
- J. B. Condon, Surface Area and Porosity Determination by Phisisorption Measurements and Theory, 2006, ch. 2, pp. 29–53 Search PubMed.
- J. Jagiello and M. Thommes, Carbon, 2004, 42, 1227–1232 CrossRef CAS.
- I. Cabria, M. J. López and J. A. Alonso, Carbon, 2007, 45, 2649–2658 CrossRef CAS.
- K. R. Matranga, A. Stella, A. L. Myers and E. D. Glandt, Sep. Sci. Technol., 1992, 27, 1825–1836 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- J. Li, B. Michalkiewicz, J. Min, C. Ma, X. Chen, J. Gong, E. Mijowska and T. Tang, Chem. Eng. J., 2019, 360, 250–259 CrossRef CAS.
- L. S. Blankenship, N. Balahmar and R. Mokaya, Nat. Commun., 2017, 8, 1–12 CrossRef PubMed.
- M. Sevilla, R. Mokaya and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 2930–2936 RSC.
- D. R. Lide, Handbook of Chemistry and Physics, CRC Press, United States, 88th edn, 2007 Search PubMed.
- B. E. Poling, J. M. Prausnitz and J. P. O’connell, Properties of gases and liquids, McGraw-Hill Education, 2001 Search PubMed.
- C. Graham, D. A. Imrie and R. E. Raab, Mol. Phys., 1998, 93, 49–56 CrossRef CAS.
- J. Jagiello, J. Kenvin, C. O. Ania, J. B. Parra, A. Celzard and V. Fierro, Carbon, 2020, 160, 164–175 CrossRef CAS.
- J. Jagiello, C. Ania, J. B. Parra and C. Cook, Carbon, 2015, 91, 330–337 CrossRef CAS.
- S. Reljic, E. Jardim, C. Cuadrado-Collados, M. Bayona, M. Martinez-Escandell, J. Silvestre-Albero and F. Rodríguez-Reinoso, Porous Materials: Theory and Its Application for Environmental Remediation, 2021, pp. 139–152 Search PubMed.
- M. Jordá-Beneyto, F. Suárez-García, D. Lozano-Castello, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2007, 45, 293–303 CrossRef.
- M. Rzepka, P. Lamp and M. De la Casa-Lillo, J. Phys. Chem. B, 1998, 102, 10894–10898 CrossRef CAS.
- L. Zhou, Y. Zhou and Y. Sun, Int. J. Hydrogen Energy, 2004, 29, 475–479 CrossRef CAS.
- S. Hlushak, Phys. Chem. Chem. Phys., 2018, 20, 872–888 RSC.
- J. Srenscek-Nazzal and K. Kielbasa, Appl. Surf. Sci., 2019, 494, 137–151 CrossRef CAS.
- J. Ludwinowicz and M. Jaroniec, Carbon, 2015, 94, 673–679 CrossRef CAS.
- M. E. Casco, M. Martínez-Escandell, J. Silvestre-Albero and F. Rodríguez-Reinoso, Carbon, 2014, 67, 230–235 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- M. Cox and R. Mokaya, Sustainable Energy Fuels, 2017, 1, 1414–1424 RSC.
- B. Adeniran and R. Mokaya, Nano Energy, 2015, 16, 173–185 CrossRef CAS.
- Z. Tan and K. E. Gubbins, J. Phys. Chem., 1990, 94, 6061–6069 CrossRef CAS.
- C. M. Simon, J. Kim, D. A. Gomez-Gualdron, J. S. Camp, Y. G. Chung, R. L. Martin, R. Mercado, M. W. Deem, D. Gunter and M. Haranczyk, Energy Environ. Sci., 2015, 8, 1190–1199 RSC.
- S. Biloé, V. Goetz and A. Guillot, Carbon, 2002, 40, 1295–1308 CrossRef.
- P. Bénard and R. Chahine, Scr. Mater., 2007, 56, 803–808 CrossRef.
- M. I. M. Kusdhany and S. M. Lyth, Carbon, 2021, 179, 190–201 CrossRef.
- M. Sevilla, W. Sangchoom, N. Balahmar, A. B. Fuertes and R. Mokaya, ACS Sustainable Chem. Eng., 2016, 4, 4710–4716 CrossRef CAS.
- N. P. Stadie, J. J. Vajo, R. W. Cumberland, A. A. Wilson, C. C. Ahn and B. Fultz, Langmuir, 2012, 28, 10057–10063 CrossRef CAS PubMed.
- A. J. Lachawiec Jr and R. T. Yang, Langmuir, 2008, 24, 6159–6165 CrossRef PubMed.
- H. Nishihara, P.-X. Hou, L.-X. Li, M. Ito, M. Uchiyama, T. Kaburagi, A. Ikura, J. Katamura, T. Kawarada, K. Mizuuchi and T. Kyotani, J. Phys. Chem. C, 2009, 113, 3189–3196 CrossRef CAS.
- N. Balahmar, A. C. Mitchell and R. Mokaya, Adv. Energy Mater., 2015, 5, 1500867 CrossRef.
- K. Xia, R. Xiong, Y. Chen, D. Liu, Q. Tian, Q. Gao, B. Han and C. Zhou, Colloids Surf., A, 2021, 126640 CrossRef CAS.
- X. Ma, R. Chen, K. Zhou, Q. Wu, H. Li, Z. Zeng and L. Li, ACS Sustainable Chem. Eng., 2020, 8, 11721–11728 CrossRef CAS.
- Z. Zhang, D. Luo, G. Lui, G. Li, G. Jiang, Z. P. Cano, Y.-P. Deng, X. Du, S. Yin, Y. Chen, M. Zhang, Z. Yan and Z. Chen, Carbon, 2019, 143, 531–541 CrossRef CAS.
- J. Singh, S. Basu and H. Bhunia, J. Taiwan Inst. Chem. Eng., 2019, 102, 438–447 CrossRef CAS.
- L. Wang and R. T. Yang, J. Phys. Chem. C, 2012, 116, 1099–1106 CrossRef CAS.
- J. E. Park, G. B. Lee, S. Y. Hwang, J. H. Kim, B. U. Hong, H. Kim and S. Kim, Appl. Sci., 2018, 8, 1596 CrossRef.
- N. Byamba-Ochir, W. G. Shim, M. S. Balathanigaimani and H. Moon, Appl. Energy, 2017, 190, 257–265 CrossRef CAS.
- I. Susanti and N. Widiastuti, Malaysian J. Fundament. Appl. Sci., 2019, 15, 249–253 CrossRef.
- J. Cui, K. Zhang, X. Zhang and Y. Lee, Microporous Mesoporous Mater., 2020, 295, 109947 CrossRef CAS.
- C. Guan, L. S. Loo, K. Wang and C. Yang, Energy Convers. Manage., 2011, 52, 1258–1262 CrossRef CAS.
- L. Giraldo and J. C. Moreno-Piraján, Mater. Sci. Appl., 2011, 2, 331 CAS.
- I. Men'shchikov, A. Shiryaev, A. Shkolin, V. Vysotskii, E. Khozina and A. Fomkin, Korean J. Chem. Eng., 2021, 38, 276–291 CrossRef.
- Z. Zhang, Z. P. Cano, D. Luo, H. Dou, A. Yu and Z. Chen, J. Mater. Chem. A, 2019, 7, 20985–21003 RSC.
- F. Rodriguez-Reinoso, J. de, D. Lopez-Gonzalez and C. Berenguer, Carbon, 1982, 20, 513–518 CrossRef CAS.
- W. Hao, E. Björkman, M. Lilliestråle and N. Hedin, Appl. Energy, 2013, 112, 526–532 CrossRef CAS.
- J. Jagiello, Langmuir, 1994, 10, 2778–2785 CrossRef CAS.
- J. Garrido, A. Linares-Solano, J. Martin-Martinez, M. Molina-Sabio, F. Rodriguez-Reinoso and R. Torregrosa, Langmuir, 1987, 3, 76–81 CrossRef CAS.
- D. Grau-Marin, J. Silvestre-Albero, E. O. Jardim, J. Jagiello, W. R. Betz and L. E. Peña, Carbon, 2020, 157, 495–505 CrossRef CAS.
- D. Lozano-Castelló, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2004, 42, 1233–1242 CrossRef.
- R. H. Orcutt, J. Chem. Phys., 1963, 39, 605–608 CrossRef CAS.
- V. W. Couling and S. S. Ntombela, Chem. Phys. Lett., 2014, 614, 41–44 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49–56 CrossRef CAS.
- J. Rouquerol, F. Rouquerol, P. Llewellyn, G. Maurin and K. S. Sing, Adsorption by powders and porous solids: principles, methodology and applications, Academic Press, 2013 Search PubMed.
- J. Osterrieth, J. Rampersad, D. G. Madden, N. Rampal, L. Skoric, B. Connolly, M. Allendorf, V. Stavila, J. Snider, R. Ameloot, J. Marreiros, C. O. Ania, D. C. S. Azevedo, E. Vilarrasa-García, B. F. Santos, X.-H. Bu, X. Zang, H. Bunzen, N. Champness, S. L. Griffin, B. Chen, R.-B. Lin, B. Coasne, S. M. Cohen, J. C. Moreton, Y. J. Colon, L. Chen, R. Clowes, F.-X. Coudert, Y. Cui, B. Hou, D. D’Alessandro, P. W. Doheny, M. Dinca, C. Sun, C. Doonan, M. Huxley, J. D. Evans, P. Falcaro, R. Riccò, O. K. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalovic, S. K. Ghosha, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K. Otake, R. P. Lively, S. J. A. DeWitt, P. L. Llewellyn, B. Lotsch, S. T. Emmerling, A. Pütz, C. Martí-Gastaldo, N. Muñoz, J. Garcia-Martinez, N. Linares, D. Maspoch, J. A. Suarez, P. Moghadam, R. Oktavian, R. Morris, P. Wheatley, J. Navarro, C. Petit, D. Danaci, M. Rosseinsky, A. Katsoulidis, M. Schroder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. G. Telfer, S. J. Lee, V. Ting, J. Rowlandson, T. Uemura, T. Iiyuka, M. V. D. Veen, D. Rega, V. Vanspeybroeck, A. Lamaire, S. Rogge, K. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. Yavuz, T. Nguyen, F. Zamora, C. Montoro, H.-C. Zhou, K. Angelo and D. Fairen-Jimenez, unpublished work.
- A. Marczewski, Adsorption Glossary, 2002.
- J. Villarroel-Rocha, D. Barrera, A. A. G. Blanco, M. E. R. Jalil and K. Sapag, Adsorpt. Sci. Technol., 2013, 31, 165–183 CrossRef CAS.
- M. Occelli, J. Olivier, J. Perdigon-Melon and A. Auroux, Langmuir, 2002, 18, 9816–9823 CrossRef CAS.
- M. M. Dubinin and V. A. Astakhov, in Molecular Sieve Zeolites-II, ed. E. M. Flanigen and L. B. Sand, ACS Publications, United States, 1971, ch. 44, pp. 69–85 Search PubMed.
- G. Horváth and K. Kawazoe, J. Chem. Eng. Jpn., 1983, 16, 470–475 CrossRef.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- P. Ramirez-Vidal, R. L. Canevesi, G. Sdanghi, S. Schaefer, G. Maranzana, A. Celzard and V. Fierro, ACS Appl. Mater. Interfaces, 2021, 13, 12562–12574 CrossRef CAS PubMed.
- J. Jagiello and M. Thommes, International Conference on Carbon for Energy Storage and Environment Protection, Orleans, France, 2005 Search PubMed.
- M. Kwiatkowski and D. Kalderis, Carbon Lett., 2020, 30, 325–329 CrossRef.
- T. Tian, Z. Zeng, D. Vulpe, M. E. Casco, G. Divitini, P. A. Midgley, J. Silvestre-Albero, J.-C. Tan, P. Z. Moghadam and D. Fairen-Jimenez, Nat. Mater., 2018, 17, 174–179 CrossRef CAS PubMed.
- J. Marco-Lozar, J. Juan-Juan, F. Suárez-García, D. Cazorla-Amorós and A. Linares-Solano, Int. J. Hydrogen Energy, 2012, 37, 2370–2381 CrossRef CAS.
- J. Marco-Lozar, M. Kunowsky, F. Suarez-Garcia, J. Carruthers and A. Linares-Solano, Energy Environ. Sci., 2012, 5, 9833–9842 RSC.
- J. Juan-Juan, J. Marco-Lozar, F. Suárez-García, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2010, 48, 2906–2909 CrossRef CAS.
- Y. Song, X. Zhou and J. A. Wang, Energy Sources, Part A, 2021, 43, 1337–1347 CrossRef CAS.
- C. Ge, D. Lian, S. Cui, J. Gao and J. Lu, Processes, 2019, 7, 592 CrossRef CAS.
- E. Mehrvarz, A. A. Ghoreyshi and M. Jahanshahi, Front. Chem. Sci. Eng., 2017, 11, 252–265 CrossRef CAS.
- K. Adlak, R. Chandra, V. K. Vijay and K. K. Pant, J. Anal. Appl. Pyrolysis, 2021, 105102 CrossRef CAS.
- P. Wang, G. Zhang, W. Chen, Q. Chen, H. Jiao, L. Liu, X. Wang and X. Deng, ACS Omega, 2020, 5, 23460–23467 CrossRef CAS PubMed.
- O. Boujibar, A. Souikny, F. Ghamouss, O. Achak, M. Dahbi and T. Chafik, J. Environ. Chem. Eng., 2018, 6, 1995–2002 CrossRef CAS.
- E. García-Díez, S. Schaefer, A. Sanchez-Sanchez, A. Celzard, V. Fierro, M. M. Maroto-Valer and S. García, ACS Appl. Mater. Interfaces, 2019, 11, 36789–36799 CrossRef PubMed.
- X. Liu, C. Zhang, Z. Geng and M. Cai, Microporous Mesoporous Mater., 2014, 194, 60–65 CrossRef CAS.
- W. Shi, Q. Zhang, S. Liu, S. Su, B. Chang and B. Yang, J. Colloid Interface Sci., 2021, 600, 670–680 CrossRef CAS PubMed.
- A. Taylor, PhD thesis, University of Nottingham, 2019.
- M. Xia, W. Chen, J. Wu, Y. Chen, H. Yang, X. Chen, D. Zhu and H. Chen, Fuel, 2021, 291, 120185 CrossRef CAS.
- X.-F. Wang, L. Xiong, J.-J. Zhong, L. Jin, J.-L. Yan, B. Mu, Y.-G. Zhang and S.-L. Song, Ind. Eng. Chem. Res., 2020, 59, 18106–18114 CrossRef CAS.
- J. Deng, T. Xiong, F. Xu, M. Li, C. Han, Y. Gong, H. Wang and Y. Wang, Green Chem., 2015, 17, 4053–4060 RSC.
- J.-S. Wei, S. Wan, P. Zhang, H. Ding, X.-B. Chen, H.-M. Xiong, S. Gao and X. Wei, New J. Chem., 2018, 42, 6763–6769 RSC.
- J. Wang, Q. Yang, W. Yang, H. Pei, L. Zhang, T. Zhang, N. Hu, Y. Suo and J. Wang, J. Mater. Chem. A, 2018, 6, 16690–16698 RSC.
- A. Rehman, Y.-J. Heo, G. Nazir and S.-J. Park, Carbon, 2021, 172, 71–82 CrossRef CAS.
- Z. Han, H. Yu, C. Li and S. Zhou, Appl. Surf. Sci., 2021, 544, 148963 CrossRef CAS.
- Z. Hu and M. P. Srinivasan, Microporous Mesoporous Mater., 2001, 43, 267–275 CrossRef CAS.
- Z. Hu, H. Guo, M. Srinivasan and N. Yaming, Sep. Purif. Technol., 2003, 31, 47–52 CrossRef CAS.
- A. Arami-Niya, W. M. A. W. Daud, F. S. Mjalli, F. Abnisa and M. S. Shafeeyan, Chem. Eng. Res. Des., 2012, 90, 776–784 CrossRef CAS.
- A. B. Fuertes and M. Sevilla, Carbon, 2015, 94, 41–52 CrossRef CAS.
- X. Ma, Y. Li, M. Cao and C. Hu, J. Mater. Chem. A, 2014, 2, 4819–4826 RSC.
- W. Hu, W. Zhang, M. Zheng, Y. Xiao, H. Dong, Y. Liang, H. Hu and Y. Liu, Int. J. Hydrogen Energy, 2021, 46, 896–905 CrossRef CAS.
- C. Bommier, R. Xu, W. Wang, X. Wang, D. Wen, J. Lu and X. Ji, Nano Energy, 2015, 13, 709–717 CrossRef CAS.
- H. Yang, R. Yan, H. Chen, D. H. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- C. Xia and S. Q. Shi, Green Chem., 2016, 18, 2063–2071 RSC.
- A. Ariharan, K. Ramesh, R. Vinayagamoorthi, M. S. Rani, B. Viswanathan, S. Ramaprabhu and V. Nandhakumar, J. Energy Storage, 2021, 35, 102185 CrossRef.
- A. Ariharan, B. Viswanathan and V. Nandhakumar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2016, 55, 649–656 Search PubMed.
- M. Sevilla and A. B. Fuertes, J. Mater. Chem. A, 2013, 1, 13738–13741 RSC.
- W. Yang, W. Yang, F. Ding, L. Sang, Z. Ma and G. Shao, Carbon, 2017, 111, 419–427 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, ACS Nano, 2014, 8, 5069–5078 CrossRef CAS PubMed.
- T. Tsumura, A. Arikawa, T. Kinumoto, Y. Arai, T. Morishita, H. Orikasa, M. Inagaki and M. Toyoda, Mater. Chem. Phys., 2014, 147, 1175–1182 CrossRef CAS.
- G. A. Ferrero, A. B. Fuertes, M. Sevilla and M.-M. Titirici, Carbon, 2016, 106, 179–187 CrossRef CAS.
- G. Ferrero, M. Sevilla and A. Fuertes, Carbon, 2015, 88, 239–251 CrossRef CAS.
- A. B. Fuertes and M. Sevilla, ACS Appl. Mater. Interfaces, 2015, 7, 4344–4353 CrossRef CAS PubMed.
- T. Yang, W. Li, M. Su, Y. Liu and M. Liu, New J. Chem., 2020, 44, 7968–7975 RSC.
- A. Fuertes, G. Ferrero and M. Sevilla, J. Mater. Chem. A, 2014, 2, 14439–14448 RSC.
- H. Luo, Y. Yang, X. Zhao, J. Zhang and Y. Chen, Electrochim. Acta, 2015, 169, 13–21 CrossRef CAS.
- D. Puthusseri, V. Aravindan, S. Madhavi and S. Ogale, Energy Environ. Sci., 2014, 7, 728–735 RSC.
- A. D. Roberts, X. Li and H. Zhang, Carbon, 2015, 95, 268–278 CrossRef CAS.
- P. Yadav, A. Banerjee, S. Unni, J. Jog, S. Kurungot and S. Ogale, ChemSusChem, 2012, 5, 2159–2164 CrossRef CAS PubMed.
- D. Hines, A. Bagreev and T. J. Bandosz, Langmuir, 2004, 20, 3388–3397 CrossRef CAS PubMed.
- L. Tong, L.-L. Zhang, Y.-C. Wang, L.-Y. Wan, Q.-Q. Yan, C. Hua, C.-J. Jiao, Z.-Y. Zhou, Y.-W. Ding and B. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 25211–25220 CrossRef CAS PubMed.
- W. Li, F. Zhang, Y. Dou, Z. Wu, H. Liu, X. Qian, D. Gu, Y. Xia, B. Tu and D. Zhao, Adv. Energy Mater., 2011, 1, 382–386 CrossRef CAS.
- B. Yang, J. Chen, S. Lei, R. Guo, H. Li, S. Shi and X. Yan, Adv. Energy Mater., 2018, 8, 1702409 CrossRef.
- Y. Wang, H. Wang, H. Wang, M. Zhang, X. Liang, K. Xia and Y. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 20272–20280 CrossRef CAS PubMed.
- N. Wang, Y. Wang, X. Xu, T. Liao, Y. Du, Z. Bai and S. Dou, ACS Appl. Mater. Interfaces, 2018, 10, 9353–9361 CrossRef CAS PubMed.
- J. K. Guo, J. Liu and L. B. Kong, ChemElectroChem, 2020, 7, 2592–2598 CrossRef CAS.
- M. Sevilla, G. A. Ferrero, N. Diez and A. B. Fuertes, Carbon, 2018, 131, 193–200 CrossRef CAS.
- D. Li, X. Ren, Q. Ai, Q. Sun, L. Zhu, Y. Liu, Z. Liang, R. Peng, P. Si and J. Lou, Adv. Energy Mater., 2018, 8, 1802386 CrossRef.
- B. Adeniran, E. Masika and R. Mokaya, J. Mater. Chem. A, 2014, 2, 14696–14710 RSC.
- J. Deng, T. Xiong, H. Wang, A. Zheng and Y. Wang, ACS Sustainable Chem. Eng., 2016, 4, 3750–3756 CrossRef CAS.
- N. Balahmar, A. S. Al-Jumialy and R. Mokaya, J. Mater. Chem. A, 2017, 5, 12330–12339 RSC.
- K. M. Rambau, N. M. Musyoka, N. Manyala, J. Ren and H. W. Langmi, Mater. Today: Proc., 2018, 5, 10505–10513 CAS.
- E. E. Taylor, K. Garman and N. P. Stadie, Chem. Mater., 2020, 32, 2742–2752 CrossRef CAS.
- R. K. Bera, H. Park, S. H. Ko and R. Ryoo, RSC Adv., 2020, 10, 32290–32295 RSC.
- H. Park, J. Bang, S. W. Han, R. K. Bera, K. Kim and R. Ryoo, Microporous Mesoporous Mater., 2021, 318, 111038 CrossRef CAS.
- R. S. Ambekar, E. F. Oliveira, B. Kushwaha, V. Pal, L. D. Machado, S. M. Sajadi, R. H. Baughman, P. M. Ajayan, A. K. Roy and D. S. Galvao, Addit. Manuf., 2021, 37, 101628 CAS.
- H. Tanaka, T. Seto, H. Nishihara, T. Kyotani and M. T. Miyahara, Carbon, 2021, 175, 609 CrossRef.
- R. Wijiyanti, T. Gunawan, N. S. Nasri, Z. A. Karim, A. F. Ismail and N. Widiastuti, Indones. J. Chem., 2020, 20, 29–42 CrossRef CAS.
- D. Tiwari, H. Bhunia and P. K. Bajpai, J. Indian Chem. Soc., 2018, 95, 247–252 Search PubMed.
- N. M. Musyoka, K. M. Rambau, N. Manyala, J. W. Ren, H. W. Langmi and M. K. Mathe, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2018, 53, 1022–1028 CrossRef CAS PubMed.
- T. Gunawan, R. Wijiyanti and N. Widiastuti, RSC Adv., 2018, 8, 41594–41602 RSC.
- J. Rangel-Sequeda, M. Loredo-Cancino, V. I. A. Maté, J. A. D. Dobladez, D. A. De Haro-Del Rio, J. J. Salazar-Rábago and E. Soto-Regalado, Water, Air, Soil Pollut., 2021, 232, 1–18 CrossRef.
- N. P. Stadie, M. Murialdo, C. C. Ahn and B. Fultz, J. Am. Chem. Soc., 2013, 135, 990–993 CrossRef CAS PubMed.
- S. T. J. Dewi and N. Widiastuti, in 3rd International Seminar on Chemistry: Green Chemistry and Its Role for Sustainability, ed. Y. Kusumawati, S. Fatmawati, A. S. Purnomo, F. Kurniawan and H. Juwono, Amer Inst Physics, Melville, 2018, vol. 2049 Search PubMed.
- C. Xue, H. Zhu, T. Xu, E. Wang, B. Xiao, X. Liu, X. Hao and G. Guan, RSC Adv., 2017, 7, 24195–24203 RSC.
- X. Ma, L. Li, R. Chen, C. Wang, H. Li and H. Li, Chem. – Asian J., 2018, 13, 2069–2076 CrossRef CAS PubMed.
- A. Garsuch, O. Klepel, R. R. Sattler, C. Berger and R. Gläser, Carbon, 2006, 44, 593–596 CrossRef CAS.
- R. Baur and R. Krishna, Catal. Today, 2005, 105, 173–179 CrossRef CAS.
- A. Sachse and J. Garcia-Martinez, Chem. Mater., 2017, 29, 3827–3853 CrossRef CAS.
- P. Annamalai, N. M. Musyoka, J. W. Ren, H. W. Langmi, M. Mathe, D. Bessarabov and L. F. Petrik, Res. Chem. Intermed., 2017, 43, 4095–4102 CrossRef CAS.
- M. Mazaj, M. Bjelica, E. Žagar, N. Z. Logar and S. Kovačič, ChemSusChem, 2020, 13, 2089–2097 CrossRef CAS PubMed.
- A. Celzard and V. Fierro, Energy Fuels, 2005, 19, 573–583 CrossRef CAS.
- P.-X. Hou, H. Orikasa, H. Itoi, H. Nishihara and T. Kyotani, Carbon, 2007, 45, 2011–2016 CrossRef CAS.
- J. Shao, X. Xiao, X. Fan, X. Huang, B. Zhai, S. Li, H. Ge, Q. Wang and L. Chen, Nano Energy, 2015, 15, 244–255 CrossRef CAS.
- A. Almasoudi and R. Mokaya, J. Mater. Chem. A, 2014, 2, 10960–10968 RSC.
- A. Gabe, M. Ouzzine, E. E. Taylor, N. P. Stadie, N. Uchiyama, T. Kanai, Y. Nishina, H. Tanaka, Z.-Z. Pan and T. Kyotani, J. Mater. Chem. A, 2021, 9, 7503–7507 RSC.
- J. Jiang, Q. Gao, K. Xia and J. Hu, Microporous Mesoporous Mater., 2009, 118, 28–34 CrossRef CAS.
- K. Xia, Q. Gao, S. Song, C. Wu, J. Jiang, J. Hu and L. Gao, Int. J. Hydrogen Energy, 2008, 33, 116–123 CrossRef CAS.
- H. Wang, Q. Gao and J. Hu, Microporous Mesoporous Mater., 2010, 131, 89–96 CrossRef CAS.
- U. Anggarini, E. Agustina and N. Widiastuti, Indones. J. Chem., 2015, 15, 315–318 CrossRef CAS.
- K. Matsuoka, Y. Yamagishi, T. Yamazaki, N. Setoyama, A. Tomita and T. Kyotani, Carbon, 2005, 43, 876–879 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |