
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA library of Rhodamine6G-based pH-sensitive fluorescent probes with versatile in vivo and in vitro applications†
W. Benton
Swanson
 *a,
Margaret
Durdan‡
bc,
Miranda
Eberle
ad,
Seth
Woodbury
ad,
Ava
Mauser
be,
Jason
Gregory
bf,
Boya
Zhang
bg,
David
Niemann
adf,
Jacob
Herremans
ad,
Peter X.
Ma
aehi,
Joerg
Lahann
befhi,
Megan
Weivoda‡
bgj,
Yuji
Mishina
a and
Colin F.
Greineder
*bgk
*a,
Margaret
Durdan‡
bc,
Miranda
Eberle
ad,
Seth
Woodbury
ad,
Ava
Mauser
be,
Jason
Gregory
bf,
Boya
Zhang
bg,
David
Niemann
adf,
Jacob
Herremans
ad,
Peter X.
Ma
aehi,
Joerg
Lahann
befhi,
Megan
Weivoda‡
bgj,
Yuji
Mishina
a and
Colin F.
Greineder
*bgk
aDepartment of Biologic and Materials Science, School of Dentistry, University of Michigan, 1011 North University Avenue, Ann Arbor, MI 48109, USA. E-mail: wbentons@umich.edu
bBiointerfaces Institute, College of Engineering and Medical School, University of Michigan, Ann Arbor, MI, USA
cCell and Molecular Biology Program, Medical School, University of Michigan, Ann Arbor, MI, USA
dDepartment of Chemistry, College of Literature, Science and the Arts, University of Michigan, Ann Arbor, MI, USA
eDepartment of Biomedical Engineering, College of Engineering and Medical School, University of Michigan, Ann Arbor, MI, USA
fDepartment of Chemical Engineering, College of Engineering, University of Michigan, Ann Arbor, MI, USA
gDepartment of Pharmacology, Medical School, University of Michigan, Ann Arbor, MI, USA
hDepartment of Materials Science and Engineering, College of Engineering, University of Michigan, Ann Arbor, MI, USA
iMacromolecular Science and Engineering Center, College of Engineering, University of Michigan, Ann Arbor, MI, USA
jDepartment of Periodontics and Oral Medicine, School of Dentistry, University of Michigan, Ann Arbor, MI, USA
kDepartment of Emergency Medicine, Medical School, University of Michigan, NCRC 2800 Plymouth Road, Bldg #26, Ann Arbor, MI 48109, USA. E-mail: coling@med.umich.edu
First published on 29th April 2022
Abstract
Acidic pH is critical to the function of the gastrointestinal system, bone-resorbing osteoclasts, and the endolysosomal compartment of nearly every cell in the body. Non-invasive, real-time fluorescence imaging of acidic microenvironments represents a powerful tool for understanding normal cellular biology, defining mechanisms of disease, and monitoring for therapeutic response. While commercially available pH-sensitive fluorescent probes exist, several limitations hinder their widespread use and potential for biologic application. To address this need, we developed a novel library of pH-sensitive probes based on the highly photostable and water-soluble fluorescent molecule, Rhodamine 6G. We demonstrate versatility in terms of both pH sensitivity (i.e., pKa) and chemical functionality, allowing conjugation to small molecules, proteins, nanoparticles, and regenerative biomaterial scaffold matrices. Furthermore, we show preserved pH-sensitive fluorescence following a variety of forms of covalent functionalization and demonstrate three potential applications, both in vitro and in vivo, for intracellular and extracellular pH sensing. Finally, we develop a computation approach for predicting the pH sensitivity of R6G derivatives, which could be used to expand our library and generate probes with novel properties.
Introduction
pH is one of the most important and tightly regulated physiologic parameters, influencing enzymatic activity, ion transport, cellular metabolism, and a host of other processes essential to life.1–3 While typically maintained within a tight range in the bloodstream and tissues, pH well below normal physiologic levels can be found in several locations through the human body – notably the stomach, duodenum, and portions of the colon.4 Apart from the gastrointestinal lumen, acidic microenvironments exist within the endolysosomal compartment of most cells,5,6 as well as in specialized extracellular compartments like osteoclast resorption lacunae.7,8 Notably, acidic pH is essential for proper cellular function in these locations, while in other contexts, such as the tumor microenvironment, it serves as a marker of pathology and potential therapeutic target.9,10 Given the diversity of these areas of study, techniques capable of detecting, visualizing, and monitoring changes in pH on a subcellular scale are of both diagnostic and therapeutic interest.11The traditional means for measuring local pH involves needle microelectrodes painstakingly positioned within target cells or extracellular compartments.12,13 While this approach allows precise determination and real-time monitoring of tissue pH, the limited resolution and inherent disruption of normal tissue architecture drastically limit its utility for in vivo studies. In the last two decades, several alternative and less invasive techniques for sensing pH have been developed, including nuclear magnetic resonance,14,15 photoluminescence,16,17 and fluorescence.18,19 Of these, pH sensing fluorescent probes have emerged as the dominant technology, with dozens of manuscripts and multiple new applications reported each year.18–20 This rapid expansion may be partially attributed to the widespread availability of fluorescent and confocal laser microscopy in biomedical research laboratories, and also reflects the unique capabilities of fluorescent probes as highly sensitive, easily quantified, and spatially informative pH reporters.20–23
Based on a survey of the literature, three commercially available pH-sensitive fluorescent probes account for many reported applications: pHrodo (Invitrogen),24,25 CypHer5E (Cytiva),26 and TPE-Cy (Calbiochem).27 The latter (TPE-Cy), a cell-permeable ratiometric dye, is limited to imaging or quantification of cytosolic pH, while the first two have been formulated as amine reactive esters, allowing modification of macromolecules to expand their number of applications. Labelling of proteins, nucleic acids, phospholipids, and even intact cells has enabled incorporation of these dyes into assays of endocytosis, phagocytosis, and efferocytosis.24–26,28,29 Despite these successes, each of the commercially available probes has limitations, including high cost, limited chemical functionality, and fixed characteristics in terms of fluorescence intensity and pH sensitivity. Together, these limit not only the breadth of research and diagnostic tools which may be developed, but the range of biologic questions to which they can be applied – e.g., determining the prognostic significance of higher vs. lower tumor extracellular pH,9,30 elucidating microenvironmental acidification in stimuli-responsive biomaterials,31–36 or defining the phenotypic differences in osteoclasts with differing levels of lacunar acidification.8,37,38
With this in mind, we sought to develop a library of pH-sensitive probes based on the highly photostable and relatively water-soluble fluorophore, rhodamine 6G, which is widely used as a lasing medium and as a fluorescence tracer.22,23 The overall goals were: 1. to create a family of compounds with tuneable pKa, 2. to begin defining the relationship between this variable and pH-sensitive fluorescence, and 3. to introduce a range of chemical functionalities for coupling to small molecules, proteins, nanoparticles, regenerative scaffold matrices, and other structures with drug delivery and tissue engineering applications. Ultimately, we report examples within each of these domains, with both in vitro and in vivo demonstration of pH-sensitive fluorescence.
Results
Preparation of Rhodamine 6G-based pH sensing fluorophores and spectral properties
Based on the general principle shown in Scheme 1, whereby amide-functionalization of Rhodamine 6G (R6G) enables pH-dependent spirolactam-quinone isomerization, we synthesized Compounds (2) through (9) (shown in Scheme 2A) via nucleophilic addition of various primary amines to the aromatic ester of R6G. Scheme 2B shows, as an example, the synthesis of Compound (3) via reaction of R6G with 1,2-ethylenediamine. The structure of each compound was confirmed using 1H nuclear magnetic resonance (NMR) spectroscopy (S Fig. S1, Methods, ESI†). | ||
| Scheme 1 Functionalization of Rhodamine 6G allows for acid-mediated transition between spirolactam (high pH) and quinone (low pH) form due to facile protonation/deprotonation of the amide substituent, resulting in dynamic “on/off” fluorescent properties. | ||
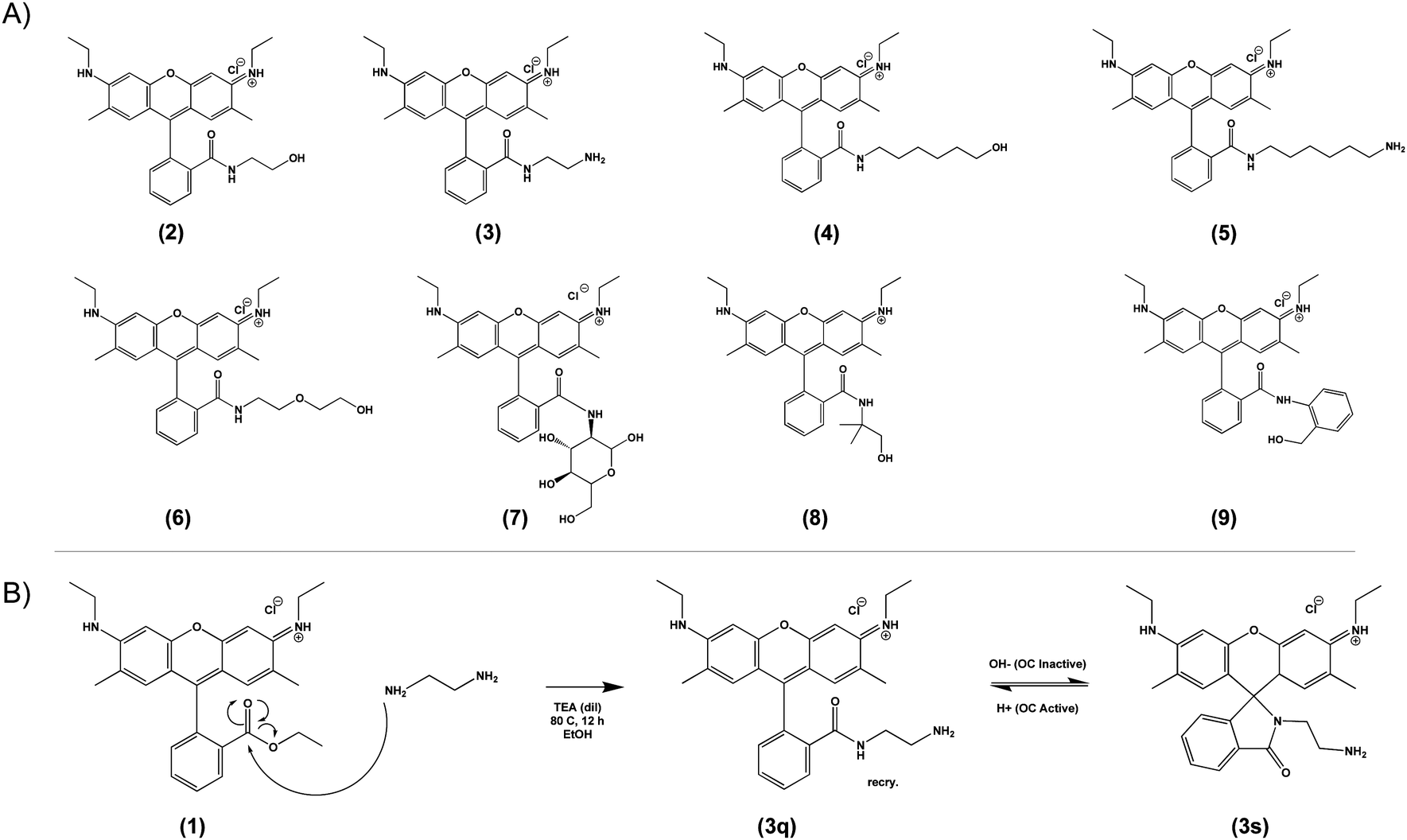 | ||
| Scheme 2 Library of novel amide-functionalized pH-sensitive fluorescent probes (2–9, A) and a general scheme for their synthesis (1, B). | ||
We measured the fluorescence intensity of Compounds (2) through (9) across a range of pHs (Fig. 1A).
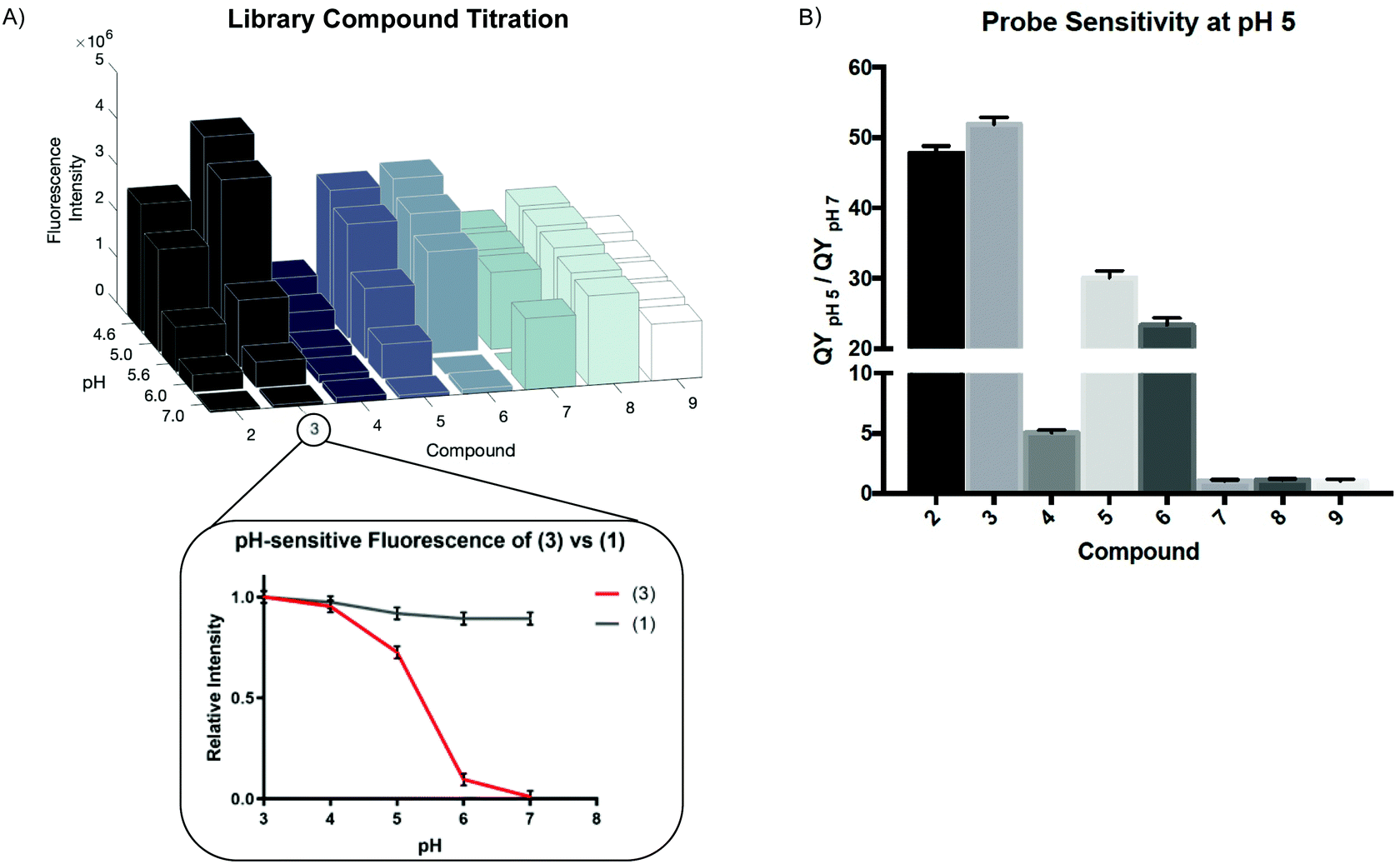 | ||
| Fig. 1 Fluorescence is measured as a function of pH for each compound resulting in titration profiles, used to determine pKa (A). Compared to unmodified starting material (1), library compounds (data for (3) shown) demonstrate an increase in fluorescence as pH decreases (A, inset). The quantum yield of each compound is calculated as the quotient of fluorescence at pH 5 compared to pH 7 (B) to determine sensitivity of our compounds. | ||
While all the compounds demonstrated some pH-dependence, there was significant variation in the ratio of quantum yields at pH 5 and pH 7 (QYpH5/QYpH7, Fig. 1B). pKa is plotted as mean ± standard deviation. Compound (3) had the highest ratio at just over 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The pKa, excitation and emission maxima, and quantum yield ratio of each Compound are shown in Table 1. We demonstrate that absorbance similarly varies as a function of pH (Fig. S2A, ESI†). These compounds are highly photostable and maintain their fluorescent properties after exosure to ambient light and direct irradiation (Fig. S2B and C, ESI†). Finally we demonstrate that these compounds are not cytotoxic at micromolar and low millimolar doses when added to cell culture media (Fig. S2D, ESI†).
:1. The pKa, excitation and emission maxima, and quantum yield ratio of each Compound are shown in Table 1. We demonstrate that absorbance similarly varies as a function of pH (Fig. S2A, ESI†). These compounds are highly photostable and maintain their fluorescent properties after exosure to ambient light and direct irradiation (Fig. S2B and C, ESI†). Finally we demonstrate that these compounds are not cytotoxic at micromolar and low millimolar doses when added to cell culture media (Fig. S2D, ESI†).
| Compound | Base | Ligand | Molecular weight | Excitation (nm) | Emission max (nm) | pKa | (QY pH 5)/(QY pH 7) |
|---|---|---|---|---|---|---|---|
| 2 | R6G | 2-Aminoethanol | 494.032 | 533 | 554 | 5.55 | 47.78 |
| 3 | R6G | 1,2-Ethanediamine | 493.048 | 533 | 558 | 5.36 | 51.91 |
| 4 | R6G | 6-Aminohexanol | 550.14 | 533 | 553 | 5.67 | 5.02 |
| 5 | R6G | 1,6-Hexanediamine | 549.156 | 533 | 554 | 5.87 | 30.05 |
| 6 | R6G | Aminoethoxyethanol | 612.12 | 533 | 557 | 5.70 | 23.32 |
| 7 | R6G | Glucosamine | 538.085 | 533 | 552 | 5.89 | 1.03 |
| 8 | R6G | 2-Amino-2-methyl-1-propanol | 522.086 | 533 | 553 | 5.65 | 1.10 |
| 9 | R6G | 2-Aminobenzylalcohol | 556.103 | 533 | 561 | 5.55 | 1.06 |
To determine the effect of the rhodamine base on the fluorescence properties of the pH-sensitive derivatives, we prepared analogues of two of the most pH-sensitive R6G derivatives, Compounds (3) and (5), except using rhodamine B (RhB) as a starting material (Fig. 2A). In this case, nucleophilic addition of 1,2-ethanediamine and 1,6-hexanediamine to the aromatic carboxylic acid of RhB resulted in Compounds (10) and (11). While the RhB derivatives retained some pH sensitivity, they demonstrated both significantly decreased quantum efficiency at equimolar concentration (Fig. 2B and C) and lower QYpH5/QYpH7 ratios (Fig. 2D). Similarly, we prepared compounds (12) and (13) from rhodamine 110 by nucleophilic addition of 1,2-ethanediamine and 1,6-hexanediamine, respectively (Fig. 2E). In this case, neither Compound (12) nor (13) demonstrated any pH sensitivity (Fig. 2F).
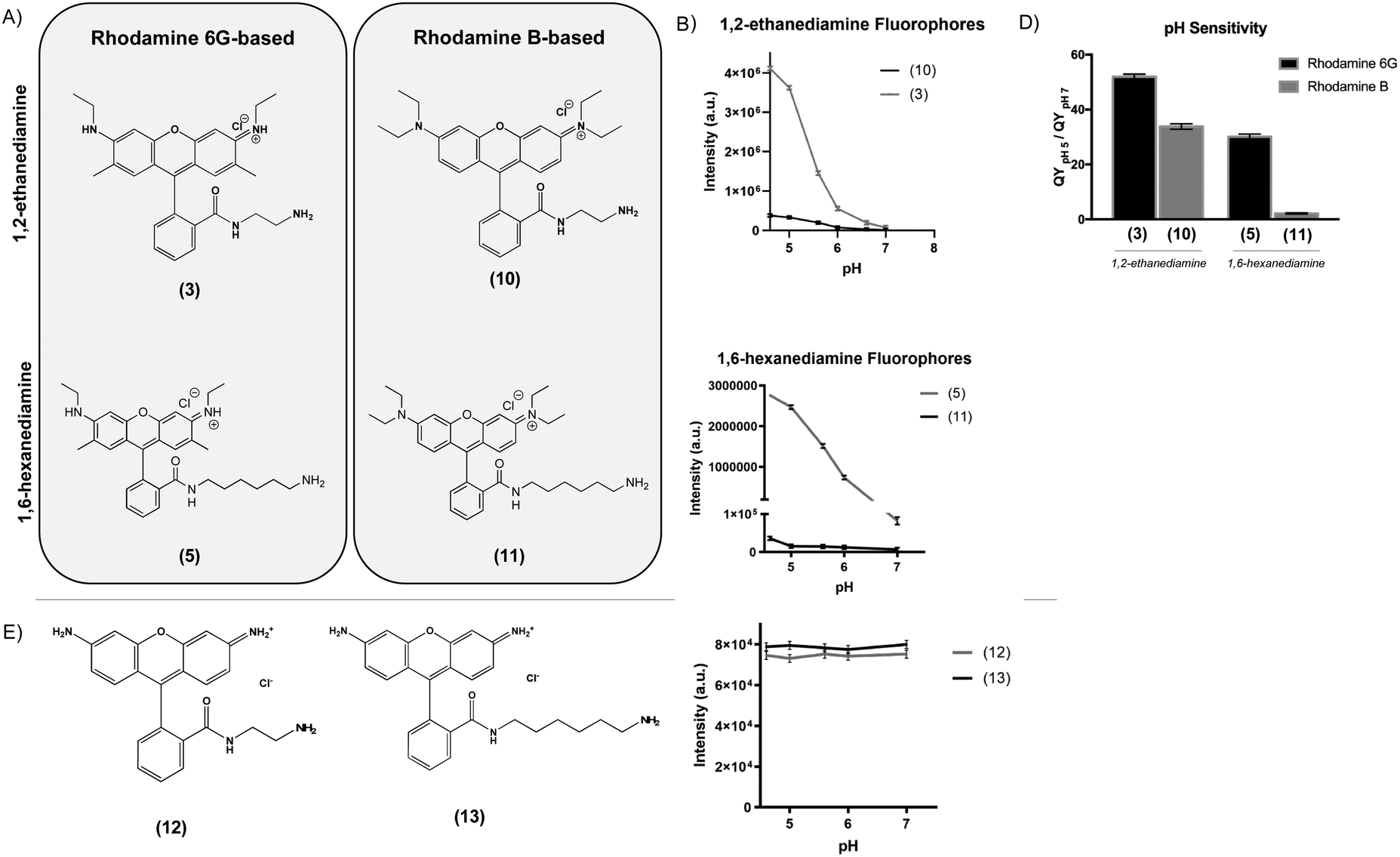 | ||
| Fig. 2 1,2-Ethanediamine derived pH-sensitive fluorophores (A) show similar fluorescence profiles as a function of pH (B). 1,6-Hexanediamine derived pH-sensitive fluorophores also show a similar trend in their relative titration curves (C). Fluorescent intensity at pH 5 and pH 7 for compounds Rhodamine 6G-based (3) and (5) are significantly greater compared to Rhodamine B-based (10) and (11) (D). Rhodamine 110-based compounds (12) and (13) do not show pH-dependent fluorescence (E and F). | ||
Computational model of Gibbs free energy change
We next sought to develop a computational model to explain the pH-dependent fluorescence of Compounds (2) through (11), based on the free energy change of the spirolactam (s, or deprotonated) to quinone (q, or protonated) transition (s → q). We hypothesized that the energetically favourability of the s → q transition would correlate with both the presence of pH sensitivity and the pKa of each dye. Minimum energies were calculated for geometry-optimized structures by convergence using the Universal Force Field (UFF).39,40 For example, this calculation indicated an energetically favourable s → q transition for Compound (3) (Fig. 3A), with an ΔEtrans, or free energy change of transition, of −124.963 kJ mol−1. The calculated ΔEtrans for Compounds (2) through (13) is shown in Fig. 3B. Consistent with our hypothesis, the first ten compounds, which demonstrated pH sensitive fluorescence, all had negative (i.e., energetically favourable) calculated ΔEtrans. In contrast, compounds (12) and (13), based on Rhodamine 110 rather than Rhodamine 6G or Rhodamine B demonstrate a positive, unfavourable, ΔEtrans which agrees with experimental evidence where no fluorescence pH-sensitivity was observed (Fig. 3B). Calculated ΔEtrans (kJ mol−1) was plotted for each compound against its experimentally measured pKa and found strong correlation (Fig. 3C, R2 = 0.89). Given this result, we next used the ΔEtrans calculation to predict the pH sensitivity of several hypothetical compounds of interest. Starting with Compound (3), we created hypothetical Compounds (14) through (18), produced by reaction of R6G with alkane diamines with varying aliphatic chain length. As shown in Fig. 3D, ΔEtrans was affected by the length of the alkane chain, whereas corresponding ethyleneglycol diamine nucleophiles had no effect on the free energy change of transition.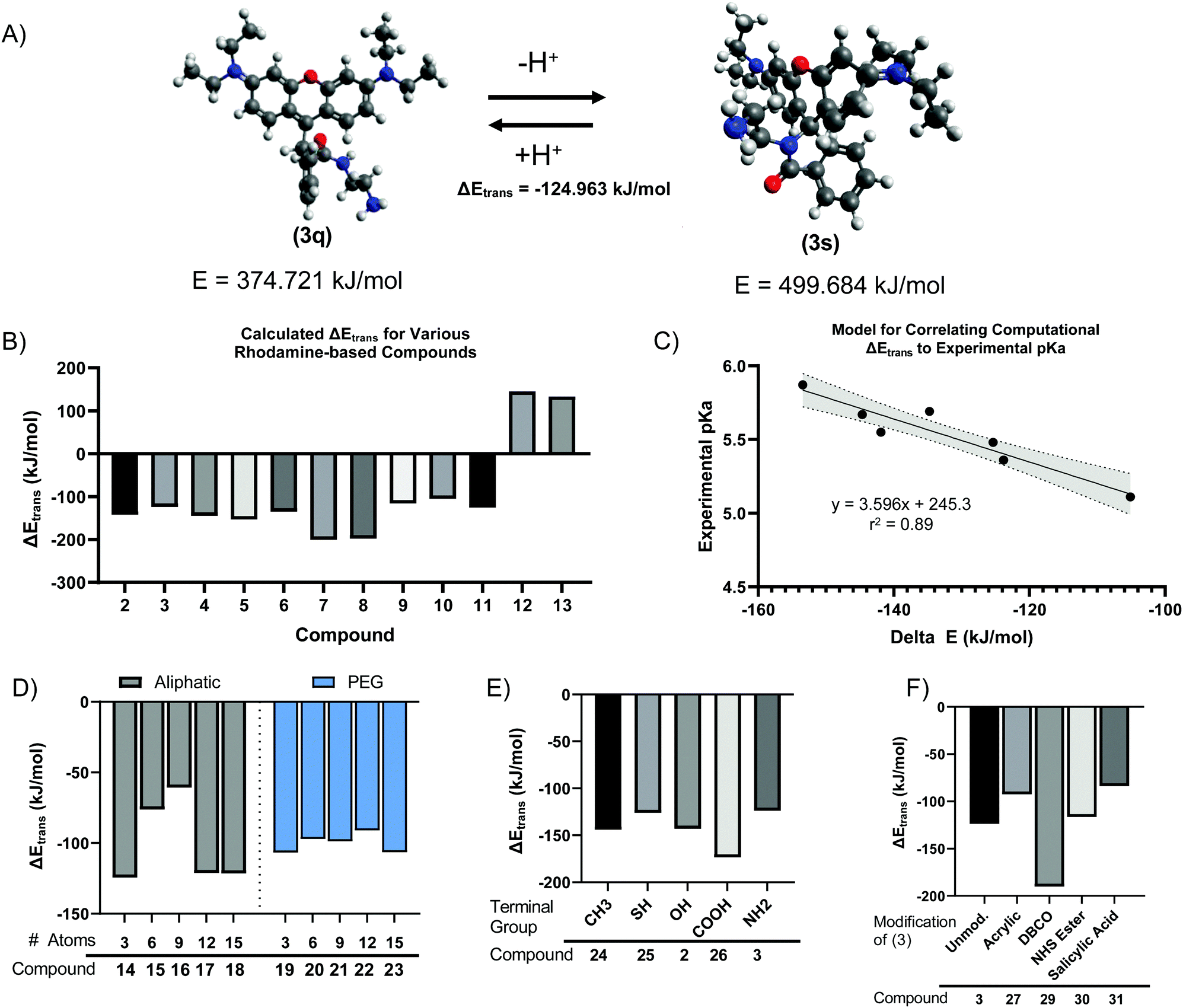 | ||
| Fig. 3 Ball and stick models of minimum energy geometry-optimized structures of rhodamine 6G (1) modified by 1,2-ethanolamine (3) in its quinone (3q) and spirolactam (3s) forms (A). Calculated free energies predict pH-mediated fluorescent properties because of a favorable transition to the quinone state when protonated (B) which correlate with experimental pKa values (C, shaded are represents 95% confidence interval of the regression). Computational interrogation of theoretical compounds 14–23 to investigate aliphatic chain length-dependence of the energy difference between the spirolactam and quinone forms (D, 14–18) and effect of ethylene glycol substitution of the same length (D, 19–23). Amide derivates of (1) with various terminal functional groups are energetically favorable (E). Amide derivatives, such as (3), can be further modified favorably to impart specific chemical functionality (F) while maintaining energetically favorable pH-dependent fluorescence. | ||
We next created analogues of Compound (2) and (3) with varying terminal functional groups. Fig. 3E shows the predicted ΔEtrans of hypothetical Compounds (24) through (31), with a wide range of terminal functionalities imparted by the ligand. Similarly, we created compounds based on Compound (3) where the terminal primary amine acts as a nucleophile to impart additional chemical functionality (Fig. 3F). Based on the negative free energy change, all these compounds would be expected to demonstrate s → q transition and have the potential to demonstrate pH sensitive fluorescence. It is worth noting that an energetically favorable s → q transition may be necessary for pH sensitive fluorescence, while at the same time not sufficient. Indeed, S Fig. S3 (ESI†) shows a plot of ΔEtrans against the observed QYpH5/QYpH7 ratio for Compounds (2) through (11), with no apparent correlation between the two variables. It stands to reason that pH sensitive fluorescence may require an energetically favourable ΔEtrans – as otherwise the fluorophore will not demonstrate s → q transition – but that other yet unidentified parameters determine the extent to which the quinone form fluoresces.
Protein functionalization with (3) for live cell imaging of endosomal uptake
We next tested the ability of Compound (3) to confer pH-sensitive fluorescence to biomacromolecules. Specifically, we hypothesized that the free primary amine would enable straightforward modification of proteins without loss of pH-sensitivity or fluorescent properties. Using human serum albumin (HSA) as a model protein, we activated carboxylate functional groups (aspartate, glutamate, N-linked glycosylation sites) via 1-ethyl-3-(3-dimethylaminopropyl) carboiimide-HCl (EDC) and N-hydroxysuccinimide (NHS), resulting in (33), which was in turn reacted with an excess of (3), yielding (34) (Scheme 3). The degree of labelling (DOL) was 1.05 ± 0.07, based on the calculation:where the concentration of (3) was measured via absorbance at 533 nm, and the concentration of HSA was determined by Lowry protein assay.41
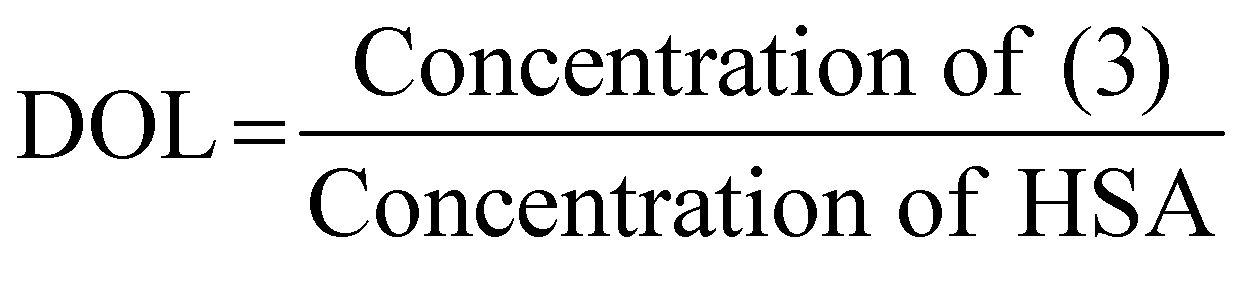
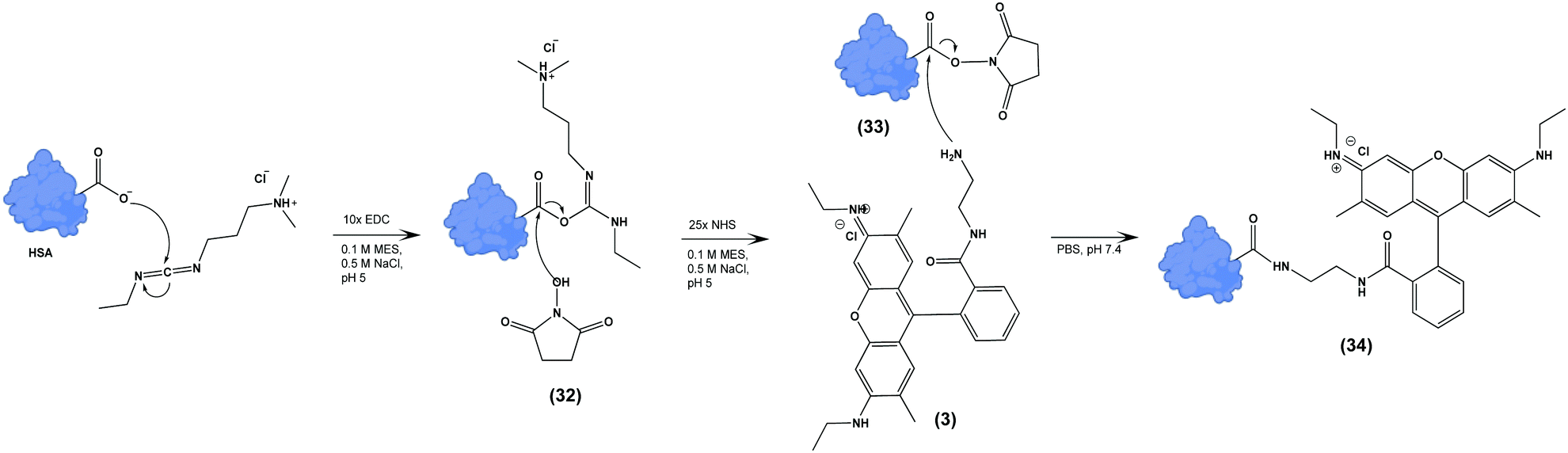 | ||
| Scheme 3 Terminal carboxylate residues of human serum albumin (HSA) are functionalized with (3) by EDC/NHS yielding pH-sensitive fluorescent HSA (34). | ||
To demonstrate the potential utility of (34), we prepared pH-sensing HSA protein nanoparticles (HSA-PNPs) via electrohydrodynamic (EHD) co-jetting.42–44 HSA-PNPs are a biocompatible and highly scalable drug delivery system, which has demonstrated promising results in translational animal models of disease, including induction of siRNA knockdown and prolonged survival in mice with intracranial glioblastoma tumors.42 While the ability of these particles to induce gene knockdown in target cells suggests escape of the particles from the acidified endolysosome following cell uptake, direct visualization of this process has never been achieved. To accomplish this, we formulated HSA-PNPs with 5%, 10%, and 50% pH-sensitive (34). Similarly, HSA-PNPs with AlexaFluor647 (AF647)-HSA were prepared as a fluorescent, but non-pH-sensitive control. All formulations were found to be relatively uniform in terms of size and surface charge, based on scanning electron microscopy (Fig. 4A), dynamic light scattering (DLS), and zeta potential (Fig. 4B and S Fig. S4A, ESI†). (34)-HSA-PNPs showed marked pH-sensitivity, mirroring that of free (3) (Fig. 4C and S Fig. S4B–D, ESI†), with quantum yield directly proportional to the percent incorporation of (34) (Fig. 4D). In contrast, AF647-HSA-PNPs were fluorescent at both acidic and neutral pH (S Fig. S4E, ESI†).
 | ||
| Fig. 4 Protein nanoparticles (PNP) of (34) (34-HSA PNPs) fabricated by electrohydrodynamic jetting are observed by scanning electron microscopy (A). Dynamic light scattering is measured for each variety of PNP fabricated, demonstrating monodispersity and consistency among formulations (B). Fluorescence spectra are recorded as a function of pH ((C)); quantum yield increases with increasing incorporation of (34) into the nanoparticle (D). Non-pH sensitive AF647-HSA PNPs and pH-sensitive (34-HSA PNPs), functionalized with anti-ICAM antibodies, target receptor-mediated endocytosis in REN ICAM cells, shown schematically, and their efficacy is assessed in a pulse-chase experimental design. Uptake of anti-ICAM1 functionalized (34)-HSA NPs in ICAM overexpressing and wild type (WT) REN cells was observed over time by confocal laser microscopy for 60 minutes (E), where peak acidification was observed at 30 minutes (F). | ||
To increase receptor-mediated endocytosis and enhance visualization of 50% (34)-HSA-PNP in the endolysosomal compartment, we surface modified the particles with antibodies to the cell-adhesion molecule, mouse ICAM-1 (anti-mICAM-1, clone YN1),45 and observed uptake by REN-mICAM cells,46 a human mesothelioma cell line which stably express the surface target. Comparison of these cells to their wild-type counterpart, REN-WT, which do not express mouse ICAM-1, enables straightforward comparison of receptor-mediated and non-receptor-mediated forms of cellular uptake (scheme shown in S Fig. S5A, ESI†).
A pulse chase design was used to synchronize uptake of ICAM-1 targeted HSA-PNPs. Briefly, particles were incubated with cells on ice to enable binding, but not uptake. After washing to remove unbound particles, cells were warmed to 37 °C and imaged at multiple time points (scheme shown in S Fig. S5B, ESI†). As shown in Fig. 4E (top panel), non-pH-sensitive (i.e., AF647), ICAM-1 targeted HSA-PNPs were visualized at all time points on REN-mICAM, but not REN-WT cells, indicating surface binding (time 0) and receptor-mediated endocytosis (subsequent time points). In contrast, the pH-sensitive, ICAM-1 targeted (34)-HSA-PNPs were not visualized at time 0, despite presumably being surface bound. Consistent with this idea, fluorescence was seen at the 15 and 30 minute time points, indicating localization in acidified endolysosomes (Fig. 4F, bottom panel). Moreover, (34)-HSA-PNP fluorescence completely disappeared at later time points (120 and 240 min), while only a modest decrease was seen in the AF647 signal. This suggests release of HSA-PNPs into a non-acidified compartment (e.g., the cytosol) and indicates the potential utility of pH-sensing macromolecules, like (34), in studying the critically important process of endosomal escape. Our fluorescent compounds are highly photostable (S Fig. S2B and C, ESI†) lending to significant utility in this type of study.
Biomaterial functionalization with (3) by free radical polymerization
Given our success with modification of protein, we next sought to incorporate (3) into several commonly used biomaterial constructs used in tissue engineering and controlled drug release. As illustrated in Scheme 4A, we activated the primary amine of (3) with acryloyl chloride to yield (27), an acrylate-functionalized pH-sensitive fluorescent probe, enabling subsequent incorporation into synthetic polymers via free radical polymerization. pH-sensitive fluorescence was not significantly altered by the reaction, with (27) and (3) demonstrating nearly identical spectra (S Fig. S6, ESI†). We then combined (27) with poly (lactic acid) (PLA), a widely-used, FDA-approved synthetic biodegradable polymer, recognized for its biocompatability.47 Briefly, acrylate end-functionalized PLA was synthesized from 2-hydroxyethylmethacrylate initiator (HEMA-PLA), and (27) was incorporated as a monomer with HEMA-PLA (macromonomer) and HEMA in a free radical polymerization, yielding (28), as shown in Scheme 4B.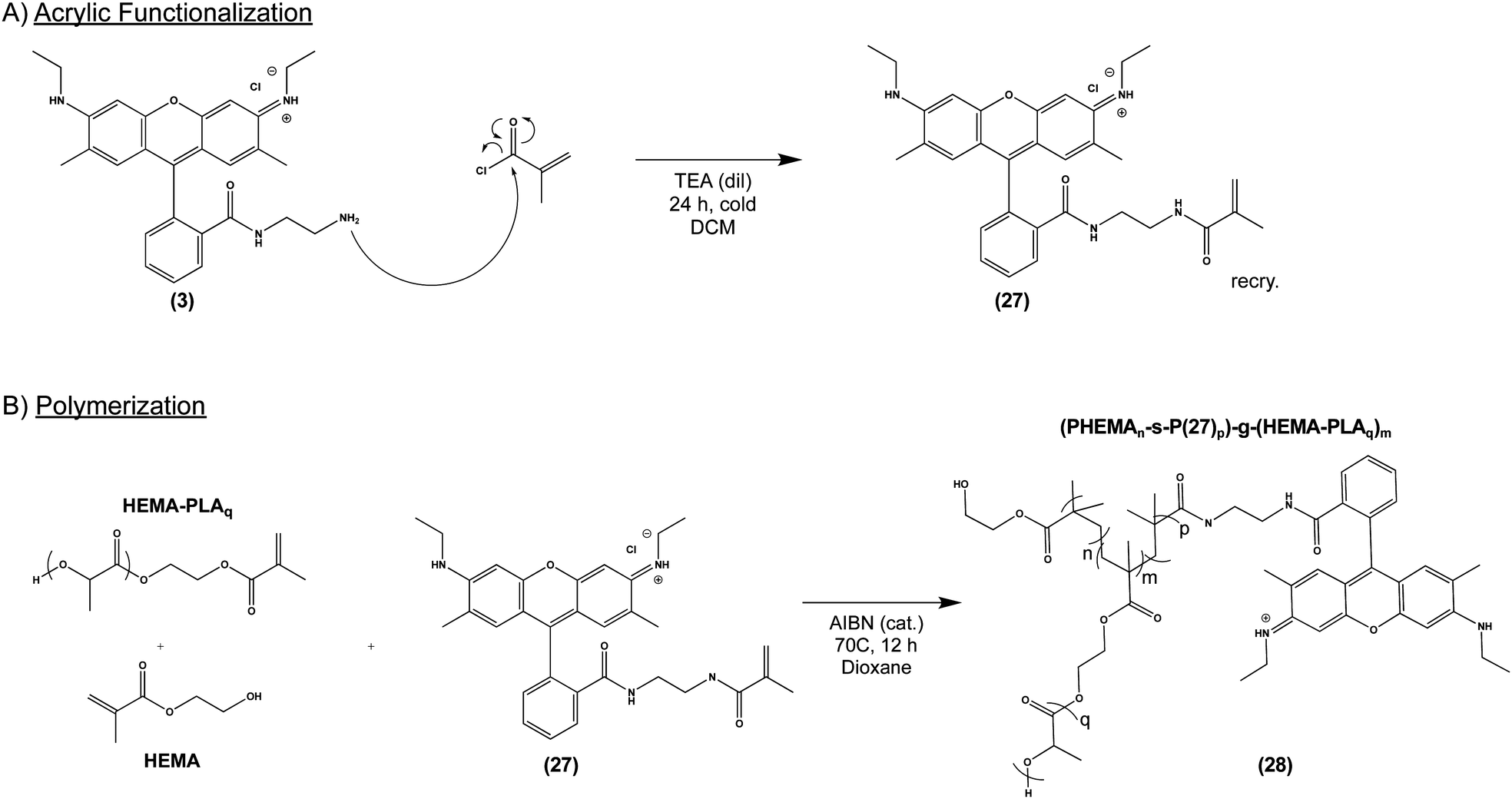 | ||
| Scheme 4 General scheme for synthesis of acrylic-modified pH-sensitive fluorescent probe by nucleophilic addition of acryloyl chloride (top). Synthesis of biodegradable matrix, poly (lactic acid) (PLA), incorporating pH-sensitive probe (27), by radical polymerization (bottom). | ||
Thin polymer films fabricated from (28) demonstrated clear fluorescent signal at pH 5 when visualized by confocal laser microscopy (Fig. 5A) and overall mirrored the pH-sensitivity of (27) and (3) by solid state fluorescence spectroscopy (Fig. 5B). These films from (28) were found to be uniformly smooth, as visualized by scanning electron microscopy (Fig. 5C). In terms of kinetics, the fluorescent signal from (28) increased over the course of 10 minutes in vitro, presumably due to solvent wetting of the film,48 and was maintained up to 1 week with little variation in signal intensity (S Fig. S7, ESI†).
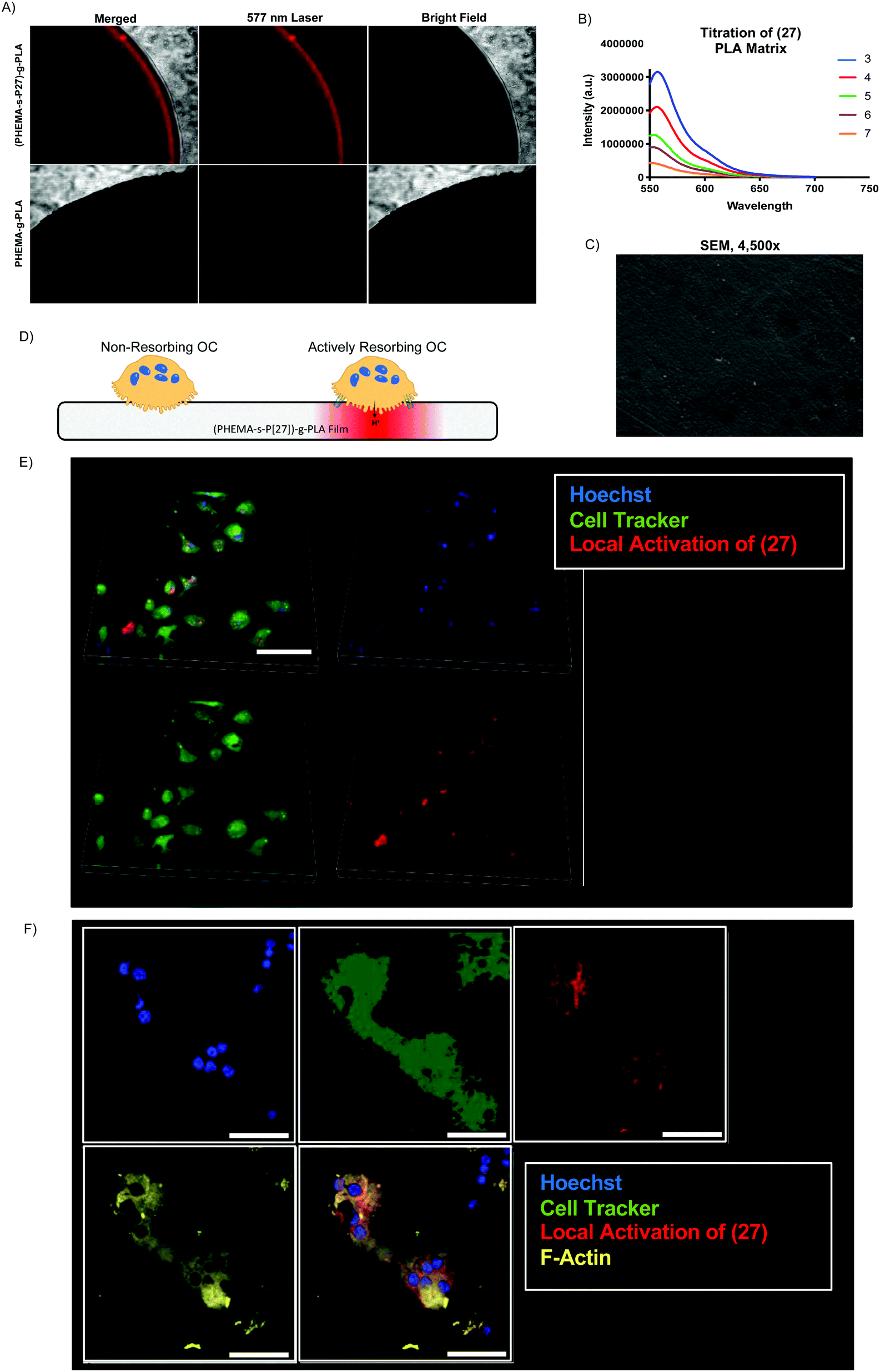 | ||
| Fig. 5 (27)-Functionalized PLA films are incubated in pH 4 solution for one hour and observed by confocal laser microscopy (A, scale = 200 μm). Solid state fluorescence spectra of (27)-functionalized PLA films as a function of pH (B). Thin film surface morphology is observed by SEM (C). Osteoclasts are plated on films to detect local acidification lacunae, shown schematically in (D) and observed by confocal microscopy. Live cell imaging of osteoclasts at low magnification (E, scale = 50 μm) and high magnification (F, scale =30 μm) details local activation of (27) in the polymer matrix (Hoescht = blue; Cell Tracker membrane dye = green; local activation of (27) = red; F-actin = yellow). | ||
To demonstrate the potential utility of the (28)-derived biomaterial, we cultured osteoclasts (OCs) on the surface of the thin film. OCs are bone-resorbing cells which create an actin-sealed, acidified compartment (resorption lacuna) between their cell body and bone surface.49 We hypothesized that culture of OCs on (28)-films would enable direct visualization of this acidic extracellular microenvironment (Fig. 5D). As shown in Fig. 5E and F, OC culture on (28)-films caused local activation in the matrix, demonstrating strong colocalization of the fluorescent signal with the OC cell body and minimal background fluorescence, at both low (Fig. 5E) and high magnification (Fig. 5F) in live cell imaging.
In addition to the (28)-thin films, we fabricated (28) into a second construct, nanofibrous spongy microspheres (NF-SMS) phase inversion emulsion.50 NF-SMS are an injectable tissue engineering scaffold and cell carrier which could be used for a variety of tissue engineering applications including bone, cartilage, and dentin regeneration and non-invasive tumor monitoring, shown in S Fig. S8A (ESI†). NF-SMS from (28) incubated at pH 4 and pH 7 maintain the of pH-sensitive fluorescence of (28), (27), and (3) (S Fig. S8B, ESI†), which can be visualized by confocal laser microscopy (S Fig. S8C, ESI†). Secondly, we fabricated nanospheres ((28)-NPs) by a double emulsion method51 using an ultrasonic probe (S Fig. S8D, ESI†); (28)-NPs maintained pH-sensitive fluorescent properties of (28), (27), and (3) (S Fig. S8E, ESI†).
Affinity targeting functionalization of (3) with a small molecule drug
Finally, we tested the ability of Compound (3) to confer pH-sensitive fluorescence to sodium alendronate, a bisphosphonate (BP) small molecule osteoporosis drug (Fosamax). Following ingestion and absorption, BPs localize to the bone surface, binding to positively charged hydroxyapatite via their two negatively charged phosphonate groups.52 BPs are released by actively resorbing OCs, allowing them to enter the cells and inhibit a key intracellular enzyme (farnesyl pyrophosphate synthase) needed for bone resorption.53 By covalently modifying alendronate with (3), we sought to directly observe the localization of the drug within acidified OC resorption lacunae on the bone surface in vitro and in vivo. To accomplish this, we imparted (3) with a terminal carboxylic acid by conjugation to succinic anhydride, allowing NHS ester activation and conjugation to the primary amine of alendronate (Scheme 5), producing (36). As in previous instances, these covalent modifications of (3) did not affect its underlying pH-sensitive fluorescence (S Fig. S9, ESI†).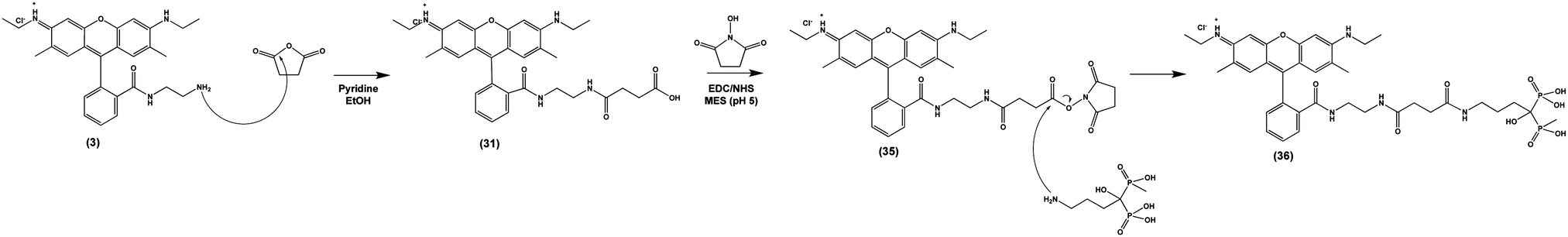 | ||
| Scheme 5 Nucleophilic addition of salicylic acid to (3) yields (31), which is functionalized with a bisphosphonate targeting ligand via EDC/NHS addition, yielding (36). | ||
We incubated bovine bone chips with (36) before inoculating with differentiated primary osteoclasts from mice (Fig. 6A). The fluorescent signal of (36) is readily observed from live cell culture in vitro by confocal laser microscopy (Fig. 6B). (36) was injected into 4 month-old mice at a dose of 0.5 mg kg−1 (Fig. 6C). All mice tolerated this dose well, with no mortality or abnormal clinical conditions noted. After two hours mice were euthanized; femurs were dissected, fixed, and sectioned for histologic analysis without demineralization. Resorptive OCs are identified by Elf97 positive (OC-specific54) staining along the periosteal and endosteal surface of cortical bone at both low magnification and high magnification (Fig. 6D and E). In both cases, Hoeschst stain is used as a fluorescent DNA stain.
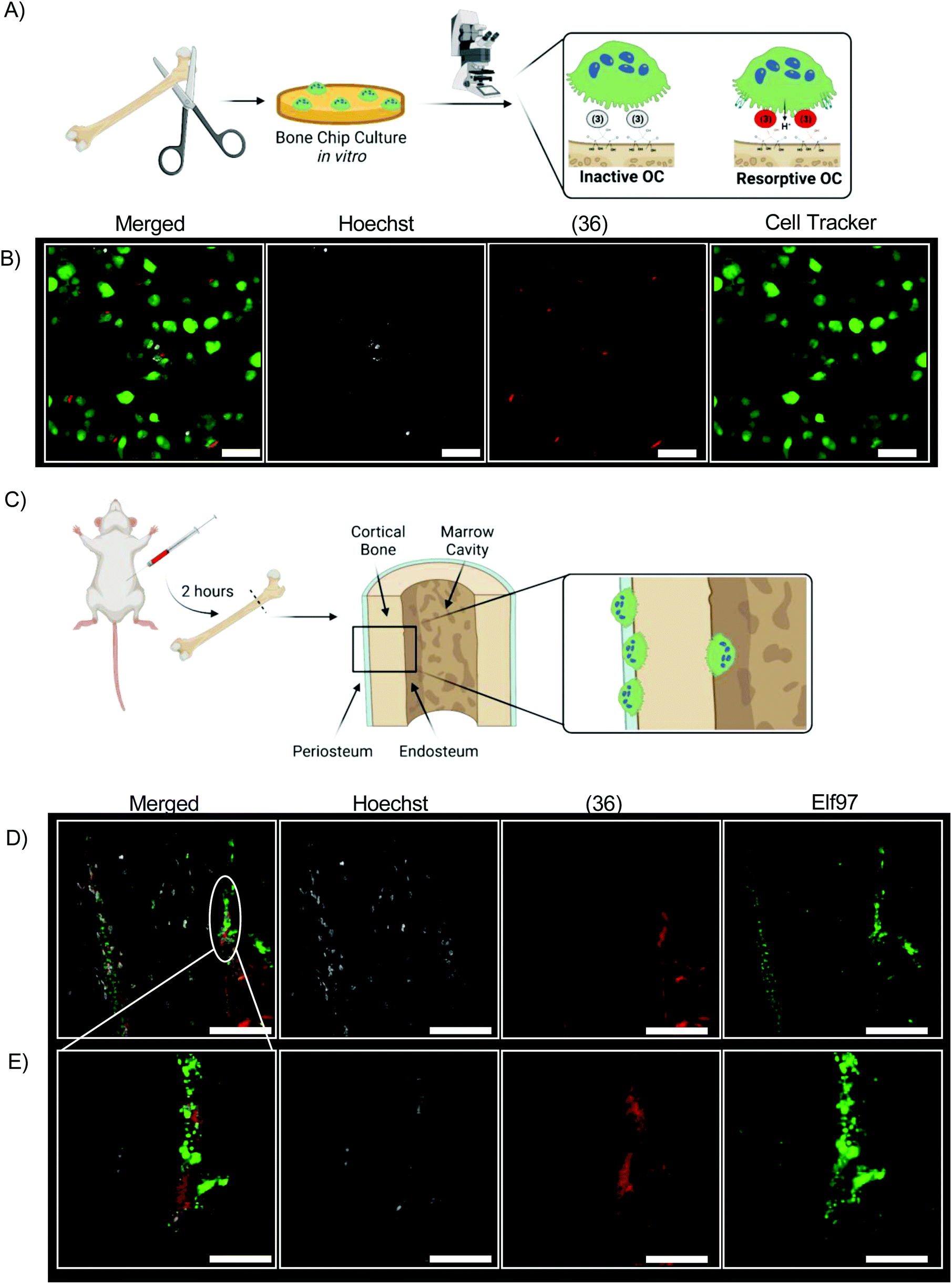 | ||
| Fig. 6 Bisphosphonate targeting anchors (36) to the bone surface while maintaining the fluorescent properties of (3) when fluorescence is observed as a function of pH. To assess the abilities of (36) to detect osteoclast resorption in vitro, primary osteoclasts isolated from mice are cultured on bone chips and imaged by confocal laser microscopy, shown schematically (A). Osteoclasts are cultured on bovine bone chips with (36) for 24 hours and observed by confocal microscopy to determine local acidification (B, scale = 50 μm). In vivo, mice were injected with (36) by a tail vein injection and sacrificed two hours later; femurs were removed for non-decalcified histologic observation, shown schematically (C). In vivo bone resorption is observed by osteoclasts on the cortical bone surface of the femur (D, scale = 150 μm; E, scale = 30 μm) in mice injected with (36). | ||
Discussion
Rhodamine, with its relative photostability, water solubility, and long excitation and emission wavelengths, has long been recognized as a promising substrate for creation of pH-sensitive fluorophores. While most previous efforts have focused on rhodamine B, we chose R6G as the base compound, due to its higher fluorescence quantum yield and straightforward nucleophilic substitution at the aromatic ester. Unlike modification of rhodamine B, which has been reported to require expensive catalysts, multi-step syntheses, and laborious purification strategies,55–60 the pH-sensitive compounds shown in Scheme 2 were synthesized at low cost using methods easily scalable for industrial production, and adaptable to versatile biologic applications.Scheme 1 illustrates our proposed mechanism for the pH-sensitivity of our library of R6G-derivatives – namely spirolactam-quinone (s → q) isomerization following protonation of the amide nitrogen.55,57,59,61 While the s → q transition has never been directly observed via biophysical measurements, its existence is supported by the pH sensitive behaviour of Compounds (2) through (9), and further validated by the strong correlation between the calculated free energy change of s → q transition (ΔEtrans) and the observed pKa of each of these compounds. The lack of pH sensitivity of Compounds (12) and (13), which are predicted to have energetically unfavorable s → q transition, offers yet another layer of support for the proposed mechanism. We demonstrate that the experimental fluorescence response window is about 2 pH units for all compounds reported, as anticipated by the Hendersen-Hasselback equation. We demonstrate a significant advantage of Rhodamine 6G-based (3) and (5) in comparison to Rhodamine B-based (5) and (11) (Fig. 2B) because of its high quantum yield, correlating to enhanced sensitivity to discriminating small pH changes which are of biologic relevance and interest. For situations where small pH variations are critically important, other sophisticated approaches are under development.62 We believe that there is significant synergy between these advanced approaches and our work which highlights the critical importance of preserved pH sensitivity following covalent attachment to various modalities enabling their advanced biologic and biomedical applications.
In contrast to pH sensitivity, the mechanism of pH-dependent fluorescence remains incompletely understood. In the protonated quinone form, a pair of electrons from the amide nitrogen likely contributes to one large, conjugated π system, like the aromatic ester of unmodified R6G. In contrast, in the deprotonated spirolactam form, the aromatic amide nitrogen may borrow electrons from the conjugated system, decentralizing it and preventing fluorescence due to an increased band gap in the energies of the excited and ground states. This mechanism is largely speculative, however, and the wide variation in QYpH5/QYpH7 ratio amongst Compounds (2) through (9) indicates that not all amide substituted R6G-derivatives are fluorescent in the quinone form. Future work will be required to develop and refine a model capable of predicting structures with both pH-sensitive s → q transition and high quantum yield below the pKa.59,63
Beyond these insights, the major contribution of the current manuscript is the development of Compound (3) and subsequent demonstration of its potential applications. The lead compound was chosen not only for its high QYpH5/QYpH7 ratio, but also its readily functionalized terminal primary amine. Importantly, our results indicate that coupling of this amine to a variety of functional groups – i.e., NHS esters (Scheme 3), acrylate (Scheme 4), and succinic anhydride (Scheme 5) – does not impact pH sensitive fluorescence, opening the door to a wide variety of potential biologic applications.
With regards to these applications, our manuscript explores three areas of interest, in which local pH has an important physiologic or pathophysiologic role and demonstrates the feasibility of Compound (3) as a pH-sensing probe for answering relevant biologic questions. The first involves extracellular acidification in the osteoclast (OC) resorption lacunae, a process critical to physiologic bone turnover and regeneration7,8,64,65 Alterations in either the number of actively resorbing OCs or their level of acidification may result in pathology. For example, joint inflammation in patients with rheumatoid arthritis can lead to excessive osteoclast activity and result in bone erosion and structural joint collapse.66,67 Conversely, OC acidification is impaired in the autosomal dominant form of the rare disease, osteopetrosis, and patients develop entrapment of cranial nerves and other painful complications of excessive bone.68,69 Bone anchored, pH-sensing fluorescent probes, such as Compound (36), may be used in vitro or in animal models to quantitate OC acidification and identify promising therapeutic strategies aimed at altering bone resorption.70,71 Similarly, future probes capable of distinguishing differing levels of acidification (e.g., pH 4 vs. pH 5) could help identify osteoclasts with different phenotypes (e.g., pit- and trench-forming OCs) and collagenolytic power.72 Inflammatory osteoclasts with seemingly aggressive tissue destruction phenotypes have been reported,73 and may be responsible in part for bony erosion observed in rheumatoid arthritis,74 delayed bone healing75 and periodontitis.76 However, their difference from homeostatic osteoclasts remains unanswered.77
A second area of interest in which pH plays a central role is that of intracellular drug delivery. For many therapeutic cargoes, avoiding catabolism in the acidified and proteolytic endolysosomal compartment is necessary to achieving requisite concentrations at the intended site of action.78 In fact, “endosomal escape” is often cited as a key feature of novel advanced drug delivery systems, despite limited understanding of the underlying biology.79 To some extent, this knowledge gap results from inadequate methods for visualizing therapeutics within the endolysosome, which typically rely on immunofluorescent colocalization with cellular markers or genetically expressed biosensors80,81–83 In contrast, incorporation of a pH-sensitive probe into the drug carrier, as described for (34)-HSA-PNPs, allows real-time monitoring of trafficking into and out of acidified compartments. Indeed, future application of this technology could help define the physical properties (e.g., size, shape, composition, rigidity) or other factors (e.g., the route of cellular entry) which influence the magnitude and kinetics of endosomal escape. Previous reports of similar technology rely on genetically encoded biosensors, rather than as a synthetic component of the delivery vector itself and serve to demonstrate interest in this application of our technology.82,83
Finally, we explore incorporation of Compound (3) into synthetic biomaterials, which are increasingly used in nanotechnology, tissue engineering, and regenerative medicine.84 PLA and its polyester copolymers are FDA-approved and widely used. Real-time microenvironment monitoring is a major priority for implantable biomaterials, particularly when it can be achieved without causing major modification of the material's chemistry and properties.31,85,86 In theory, this approach could be used to non-invasively image deposition of extracellular matrix or changes in the supply of nutrients or removal of acidic metabolites. While specific biologic applications were not pursued in the current manuscript, we have demonstrated the feasibility of incorporation of Compound (27) into two distinct biomaterial structures. Future directions may include incorporation of pH-sensors in previously reported biomaterials designed to recruit and capture metastatic cancer cells.87 Subcutaneous implantation of these scaffolds has been shown to detect microscopic metastases prior to traditional methods.88 Given the acidity of the tumor microenvironment – even in relatively small tumor spheroids89 – incorporation of Compounds (3) or (27) might enable repeated, non-invasive fluorescent monitoring of early metastasis.
Materials and methods
Materials
Resomer 207S poly(L-lactic acid) was purchased from Evonik. Silica wafers for preparation of thin films were purchased from Corning. Spectrum spectra/por dialysis tubing was purchased from Fisher. All other reagents were purchased from Sigma Aldrich unless specifically mentioned in the below methods. Reagents were used as received unless otherwise specified.Cells and antibodies. RAW 264.7 murine mouse cell line was used for cytotoxicity assessment (Sigma, 91062702). No cytotoxicity was observed in subsequent studies in vitro or in vivo. REN-mICAM and REN wt cells were cultured as previously described. Monoclonal antibody specific for mouse ICAM (clone YN1) was isolated from hybridoma supernatant using Protein G resin. >95% purity was confirmed via SDS-PAGE. Function was validated via binding to REN-mICAM vs. REN wt cells.
Proliferation assay. Cell proliferation was assessed by MTT assay (Sigma, 11465007001), following the manufacturer's protocol. Absorbance was measured using a plate reader.
Primary osteoclast isolation from mice. Long bones were dissected from 4–6 month C57Bl/6 female mice. The epiphyses were removed from both femurs and tibias, and the bone marrow was flushed with 1× PBS. Bone marrow was treated with Red Cell Lysis Buffer (eBioscience, Inc., San Diego, CA, USA), and cells were plated in 10 cm dishes (Corning, Corning, NY, USA) with α-MEM supplemented with 10% (vol/vol) fetal bovine serum (FBS, Hyclone Laboratories, Logan, UT, USA), and 25 ng mL−1 macrophage colony-stimulating factor (M-CSF) for 24 hours. Cells were then plated into 24-well plates at 4 × 105 per well with osteoclast differentiation media (100 ng mL−1 RANKL and 25 ng mL−1 macrophage colony-stimulating factor (M-CSF)) for 72 hours. Cells were lifted using 100 μL per well of 0.02% EDTA (Bio-rad Laboratories) and P100 pipette tips to scrape the bottom of each well. Cells were plated onto either bovine bone chips (Fisher; Immunodiagnostic Systems) or polymer films at 1 × 105 per well in a 96 well plate (Fisher) in osteoclast differentiation media. 24 hours after plating onto bone or films, cells were given an additional media change with osteoclast differentiation media and stained and imaged 12–24 hours later.
All procedures involving animals were performed following a protocol approved by the University of Michigan Institutional Animal Care and Use Committee (IACUC, PRO00009377).
Synthesis of Rhodamine-based pH-sensitive compounds (2–11). In general, the rhodamine base (Rhodamine 6G) was solubilized in pure ethanol (EtOH). The ligand (5× molar equivalents) and triethylamine (TEA, 1× molar equivalent) were added to the solution, dropwise, with mechanical stirring. The reaction mixture was heated to 80 °C for 12 hours to react in a round bottom flask with water-cooled condenser. On cooling, solvent was removed by rotary evaporation and the product was isolated from acetonitrile and collected by suction filtration, then recrystallized. For all compounds, the product is a solid with pink/red color. Products were confirmed by nuclear magnetic resonance (NMR), fluorescence titration, and Fourier-transformed infrared spectroscopy (FTIR). Resulting compounds were stored in a desiccator, protected from light.
Synthesis of acrylate-modified (3): compound (27). (3) Was dissolved with triethylamine (TEA, 3× molar equivalent) in dichloromethane (DCM) at 0 °C. Acryloyl chloride was dissolved in dichloromethane and added dropwise to the solution over the course of one hour. The resulting mixture was left to stir on ice and warm to room temperature, reacting for 24 hours. Solvent was concentrated by rotary evaporation, the product was reconstituted and washed against water to remove biproducts of the reaction, then dried to collect a solid product. (27) is a solid with pink/red color. Each batch of compound was confirmed by NMR spectroscopy and fluorescence titration. Resulting compounds are stored in the dark, in a desiccator. 1H NMR (599 MHz, chloroform-d) δ 7.48 (s, 1H), 7.07 (s, 1H), 6.35 (d, J = 11.3 Hz, 1H), 6.22 (d, J = 17.9 Hz, 2H), 5.99 (s, 1H), 3.32 (s, 1H), 3.26 (s, 11H), 3.22 (t, J = 8.4 Hz, 2H), 3.10 (s, 2H), 3.05 (s, 1H), 1.94 (s, 1H), 1.90 (d, J = 14.0 Hz, 3H), 1.46 (s, 1H), 1.41–1.38 (m, 3H), 1.33 (d, J = 10.0 Hz, 3H), 1.26 (s, 4H).
Synthesis of (28). (PHEMA-s-P27)-g-(HEMA-PLA): HEMA-PLAq was synthesized from 5.760 g lactide and 232 μL (232 mg) 2-hydroxyethyl methacrylate (HEMA) by bulk polymerization in a round bottom flask charged with 115 μL (162 mg) stannous 2-ethylhexanoate. q represents the molar ratio percent of HEMA to PLA in the preparation of HEMA-PLA, typically 5%. The flask is purged with argon for 30 minutes, then sealed and heated to 120 °C in an oil bath with magnetic stirring to melt the reactants. The reaction was allowed to proceed for two hours, then quenched by opening to air. After cooling to room temperature, a solid white material resulted, which was redissolved in a minimum volume of chloroform and precipitated into cold methanol (−20 °C, 5× volume excess), and collected by suction filtration. Identity is confirmed by NMR spectroscopy. 1H NMR (599 MHz, Chloroform-d) δ 5.19 (q, 1H), 1.53 (d, 3H). Minor peaks: 6.45–6.35 (d, 1H), 6.2–6.0 (dd, 1H), 5.9–5.8 (d, 1H).
(PHEMAn-s-P27p)-g-(HEMA-PLAq)m (28) is a graft copolymer prepared by radical polymerization, where m is the molar ratio percent of HEMA-PLA compared to HEMA, n + p represents the total amount of acrylic monomers HEMA and (27), where n + p = 1; q is the molar ratio percent of HEMA to PLA in the preparation of HEMA-PLA. The typical preparation is: (PHEMA90-s-P2710)-g-(HEMA-PLA5)10. 1.22 g HEMA-PLA5, 363 μL (388 mg) HEMA, and 149 mg (27) are dissolved in 5 mL dioxane with 9.8 mg azobisisobutyronitrile catalyst (2% molar ratio of total acrylic monomer, recrystallized from methanol). The mixture is kept at 70 °C for 24 hours, then concentrated by rotary evaporation. The reaction product is precipitated from dioxane into cold methanol (−20 °C, 5× molar excess) and collected by suction filtration. Identity is confirmed by NMR spectroscopy. 1H NMR (599 MHz, chloroform-d) δ 5.19 (q, 1H), 1.53 (d, 3H). Minor peaks: δ 7.48, 7.07, 4.25, 3.88, 3.35–3.05, 2.42, 1.94, 1.46–1.38, 1.22.
Synthesis of (34). 1.5 mL of 25 mg mL−1 human serum albumin (HSA) in 0.1 M MES buffer (0.5 M NaCl, pH 5) was combined with 30 μL 200 mM 1-ethyl-3-(3-dimethylaminopropyl) carboiimide HCl (EDC, 10× molar excess) and 75 μL 200 mM N-hydroxysuccinimide (NHS, 25× molar ratio) in an Eppendorf tube at room temperature, rotating for 30 minutes to yield (33). NHS-functionalized HSA (Sigma) (33) was desalted using a PD-10 column equilibrated with PBS (pH 6.5) to recover 27 mg (33). (3) was dissolved in methanol at 1 mg mL−1; 2 mL of (3) in methanol (8× molar excess) was combined with 2 mL (33) in PBS (pH 6.5) and 8 mL PBS (pH 7.4) and reacted at room temperature for 2 hours, with rotation. The reaction was quenched by the addition of 1 mL 0.1 M sodium bicarbonate (NaHCO3) buffer (pH 8.3). The resulting solution, containing (34), was dialyzed into 1 L PBS using 12–14 kDa molecular weight cutoff dialysis tubing, resulting in 25 mg of (34) after lyophilization. AlexaFluor-647-modified HSA was prepared by the same method as a non-pH sensitive fluorescent control.
Synthesis of (31). 50.0 mg (3) was dissolved in 3 mL ethanol and 10 μL pyridine. Separately, 20.0 mg succinic anhydride were dissolved in ethanol (2× molar equivalents), and added to the solution of (3), dropwise. The reaction was left with magnetic stirring overnight at room temperature. The solvent was evaporated, and the product (31) was redissolved in chloroform. Excess succinic anhydride and biproducts are removed by extraction with 0.1 M NaHCO3. 1H NMR (599 MHz, DMSO-d6) δ 7.79 (s, 1H), 7.49 (s, 2H), 6.96 (s, 1H), 6.27 (d, J = 13.4 Hz, 3H), 6.12 (s, 1H), 5.11 (s, 2H), 3.17 (s, 5H), 3.13 (s, 3H), 3.08 (s, 2H), 3.0–2.8 (m, 4H) 2.78 (s, 1H), 2.75 (s, 4H), 2.51 (s, 1H), 1.90 (s, 2H), 1.87 (s, 3H), 1.82 (s, 1H), 1.22 (d, J = 14.7 Hz, 4H).
Synthesis of (36). 21.67 mg sodium alendronate trihydrate (ALD, 0.066 mmol) was dissolved in MES buffer (pH 5) at 5 mg mL−1. Separately, 15 mg (31) was dissolved in 400 μL ethanol and diluted to a total volume of 2 mL in MES buffer. (31) was added dropwise to the solution containing ALD, slowly, and allowed to equilibrate for 5 minutes. 1.67 mL 200 mM EDC in MEF buffer (15× molar equivalents) and 1.67 mL 200 mM NHS in MEF buffer (15× molar equivalents). The reaction mixture was stirred with a magnetic stir bar for 24 hours at room temperature, and the reaction is quenched by the addition of 250 μL 0.1M NaHCO3 buffer (pH 8.3). The resulting solution containing (36) was dialyzed in 300 Da cutoff dialysis tubing against 500 mL deionized water, then lyophilized to recover (36). 1H NMR (599 MHz, Deuterium Oxide) δ 7.79 (s, 1H), 7.49 (s, 2H), 6.96 (s, 1H), 6.27 (d, J = 13.4 Hz, 3H), 6.12 (s, 1H), 5.11 (s, 2H), 3.17 (s, 5H), 3.13 (s, 3H), 3.10 (m, 2H), 3.08 (s, 2H), 3.0–2.8 (m, 4H) 2.78 (s, 1H), 2.75 (s, 4H), 2.51 (s, 1H), 2.1–2.0 (m, 4H), 1.90 (s, 2H), 1.87 (s, 3H), 1.82 (s, 1H), 1.22 (d, J = 14.7 Hz, 4H). 31P NMR (242 MHz, Deuterium Oxide) δ 17.50.
Fluorescence spectroscopy. Spectra were recorded from 1 cm quartz cuvettes on a Horiba Quanta Master spectrometer with xenon arc lamp and PMT detector. Small molecule probe stock solutions were prepared at 2.0 μmol mL−1 in methanol. Aliquots of stock solution (100 μL) were dissolved in 3.0 mL McIlvaine Buffer (described below) to record spectra (final concentration 6.76 nmol mL−1). Sync Scan spectra were acquired to determine excitation and emission maxima, iteratively. Emission spectra were recorded using maximum excitation wavelength. All spectra were acquired with slit widths set to 1.0 nm and 0.1 s integration time.
Determination of pKa. Fluorescence spectra of compounds prepared in various buffer solutions were acquired in series. Fluorescent intensity at emission maxima was plotted as a function of pH. The first derivative of each plot was calculated and plotted in GraphPad Prism. The peak maxima of the first derivative correspond to the pH at which pH is equal to pKa.
McIlvaine buffer solution. Titrations were performed using a McIlvaine phosphate-citrate buffer.90 Stock solutions of disodium phosphate dihydrate (Na2HPO4·2H2O, 0.20 M) and citric acid (C6H8O7, 0.10 M) were prepared in established ratios to yield buffers in the pH range of 3–8. The pH of buffer solutions is confirmed with a digital pH probe before use.
Nuclear magnetic resonance (NMR) spectroscopy. All compounds were characterized by nuclear magnetic resonance (NMR) spectroscopy to confirm their molecular identity. 1H spectra were recorded with a Bruker 600 MHz NEO600 spectrometer operating at room temperature. Spectra were observed from compounds dissolved in deuterated chloroform (CDCl3). Spectral analysis is carried out in ICONNMR (Bruker) and Mestre Nova (Version 12.0.0–2
000080, Metrelab Research).
Fourier-transformed infrared spectroscopy. All compounds were characterized by Fourier-transformed infrared spectroscopy to confirm the addition and modification of functional groups throughout synthesis. Spectra were recorded directly from a diamond crystal from solid compounds, recording from 4000 cm−1 to 400 cm−1 on a Thermo-Nicolet IS-50 bench top spectrometer.
UV-Vis spectroscopy. Spectra were recorded from 1 cm quartz cuvettes on a Hitachi U-2910 spectrophotometer. Small molecule probe stock solutions were prepared at 2.0 μmol mL−1 in methanol. Aliquots (300 μL) of stock solution were dissolved in McIlvaine Buffer (3 mL, described above) to record spectra.
Assessment of photostability. Small molecule probe stock solutions were prepared at 2.0 μmol mL−1 in methanol. Aliquots of stock solution (100 μL) were dissolved in 3.0 mL McIlvaine Buffer (described above) to record spectra (final concentration 6.76 nmol mL−1). Compounds were either: (a) protected from ambient light with aluminium foil and kept in the dark, (b) exposed to ambient light for 24 hours, or (c) directly irradiated with 560 nm laser irradiation (confocal laser microscope, to mimic biologic application) for 3 hours. Fluorescence spectra were recorded in the same method as previously described.
Confocal laser microscopy. Images were acquired using a Nikon Eclipse C1 Plus confocal microscope (Melville, NY, USA).
Scanning electron microscopy. HSA-PNPs were observed by scanning electron microscopy (NOVA SEM/FIB) with 5 kV acceleration voltage at a working distance of 5 mm. Surface morphologies of biomaterials were observed by scanning electron microscopy (JEOL JSM-7800 FLM) with an accelerating voltage of 5 kV and working distance of 10–15 mm. Prior to observation of all samples, samples were coated with gold using a sputter coater (40–60 seconds, Desk II, Denton Vacuum Inc.).
Computational methods. Theoretical structures and experimentally synthesized structures of chemical entities were drawn in ChemDraw. We performed iterative geometry optimization using a molecular mechanics model and Universal Force Field (UFF) for 500 steps, optimized to the steepest descent, until dE, the change in energy between geometric structures, converges to 10 × 10−7. The minimum energy of the geometry optimized structure was calculated using these parameters (Avogadro, v1.90.0, Avogadro Chemistry).39 The minimum energies of geometry optimized structures of the spirolactam and quinone structures were calculated, and the energy difference is defined as:
| ΔEnergy = Espirolactam − Equinone |
Conclusions
pH is a critical biologic parameter influencing cellular and enzyme activity, tissue turnover, homeostasis, and disease pathology. Development of high-throughput, non-invasive research tools for diverse applications of quantitative fluorescence-based pH sensing are critical for improving our ability to probe the roles of acidic microenvironments in biologic processes. In summary, we have developed a library of pH-sensitive fluorescent probes with remarkable sensitivity and quantum yield. Drugs, proteins, affinity ligands and polymeric biomaterials are synthesized which readily incorporate these probes and maintain their pH-sensitive fluorescent properties. These probes allow for high-throughput, high resolution visualization and analysis of acidic microenvironments in a diverse array of biologic systems including lysosome/endosome acidification and bone resorption activity. We demonstrate that the versatility of these probes extends between both in vitro and in vivo applications. Based on the examples presented and discussed herein, a platform approach to designing pH-sensitive fluorescent probes with modular chemical functionality is a meaningful advancement in developing highly sensitive research tools for biomedical applications.Author contributions
Conceptualization: W. B. S., M. D. D., M. M. W., C. F. G.; data curation: W. B. S., M. D. D., M. E., S. W., A. M., J. G., B. Z., D. N., J. H., M. M. W., C. F. G.; formal analysis: W. B. S., M. D. D., M. E., S. W., A. M., J. G., C. F. G.; funding acquisition: W. B. S., M. D. D., Y. M., A. M., C. F. G., M. M. W.; investigation: W. B. S., M. D. D., M. E., S. W., A. M., J. G., B. Z., D. N., JH, M. M. W., C. F. G.; methodology: W. B. S., M. D. D., S. W., J. G., M. M. W., C. F. G.; project administration: W. B. S., M. D. D., M. M. W., C. F. G., YM; resources: M. D. D., M. M. W., C. F. G., PXM, JL, YM; software: W. B. S., M. E., A. M., J. G., C. F. G.; supervision: J. L., M. M. W., YM, C. F. G.; validation: W. B. S., M. D. D., M. E., S. W., J. G., M. M. W., C. F. G.; visualization: W. B. S., M. D. D., M. M. W., C. F. G.; writing – original draft: W. B. S., M. D. D., S. W., A. M., M. M. W., YM, C. F. G.; writing – review and editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding sources that supported this work: National Institutes of Health (NIH) R01-DE027661 (Y. M., NIDCR), K08-HL130430 (C. F. G., NHLBI), F30-DE029359 (W. B. S., NIDCR), T32-DE07057 (W. B. S., NIDCR), T32-GM007315 (M. D. D., NIGMS), Michigan Integrative Musculoskeletal Health Core Center (MiMHC, P30-AR069620) Pilot Grant (C. F. G., M. W., NIAMS); National Science Foundation (NSF) Graduate Research Fellowship DGE-1256260 (A. M., J. L.). The authors are grateful to resources provided by the University of Michigan Biointerfaces Institute Nanotechnicum (DLS, Zeta potential), University of Michigan Electron Microbeam Analysis Laboratory (SEM), University of Michigan Center for Materials Characterization (College of Engineering, NSF DMR-0320740), and University of Michigan BioNMR Core (funded by the College of Literature, Sciences and Arts, Life Sciences Institute, College of Pharmacy, Medical School and the UM Biosciences Initiative) for equipment assistance.References
- E. Hopkins, T. Sanvictores and S. Sharma, StatPearls, Treasure Island (FL), 2021 Search PubMed.
- S. Grinstein, C. J. Swallow and O. D. Rotstein, Clin. Biochem., 1991, 24, 241–247 CrossRef CAS PubMed.
- M. Dixon, Biochem. J., 1953, 55, 161–170 CrossRef CAS PubMed.
- J. Fallingborg, Dan Med Bull, 1999, 46, 183–196 CAS.
- N. Demaurex, News Physiol. Sci., 2002, 17, 1–5 CAS.
- Y. Chen and E. A. Arriaga, Anal. Chem., 2006, 78, 820–826 CrossRef CAS PubMed.
- A. V. Rousselle and D. Heymann, Bone, 2002, 30, 533–540 CrossRef CAS PubMed.
- F. V. Marcoline, Y. Ishida, J. A. Mindell, S. Nayak and M. Grabe, Bone, 2016, 93, 167–180 CrossRef CAS PubMed.
- C. R. Justus, L. Dong and L. V. Yang, Front. Physiol., 2013, 4, 354 Search PubMed.
- S. Uthaman, K. M. Huh and I. K. Park, Biomater. Res., 2018, 22, 22 CrossRef PubMed.
- H. Hou, Y. Zhao, C. Li, M. Wang, X. Xu and Y. Jin, Sci. Rep., 2017, 7, 1759 CrossRef PubMed.
- D. Ellis and R. C. Thomas, Nature, 1976, 262, 224–225 CrossRef CAS PubMed.
- K. Engin, D. B. Leeper, J. R. Cater, A. J. Thistlethwaite, L. Tupchong and J. D. McFarlane, Int. J. Hyperthermia, 1995, 11, 211–216 CrossRef CAS PubMed.
- D. Malhotra and J. I. Shapiro, Concepts Magn. Reson., 1993, 5, 123–150 CrossRef CAS.
- J. L. Slonczewski, B. P. Rosen, J. R. Alger and R. M. Macnab, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 6271–6275 CrossRef CAS PubMed.
- Q. Zheng, C. Xu, Z. Jiang, M. Zhu, C. Chen and F. Fu, Front. Chem., 2021, 9, 650358 CrossRef CAS PubMed.
- Y. Feng, Y. Liu, C. Su, X. Ji and Z. He, Sens. Actuators, B, 2014, 203, 795–801 CrossRef CAS.
- L. Di Costanzo and B. Panunzi, Molecules, 2021, 26(10), 2952 CrossRef PubMed.
- J.-T. Hou, W. X. Ren, K. Li, J. Seo, A. Sharma, X.-Q. Yu and J. S. Kim, Chem. Soc. Rev., 2017, 46, 2076–2090 RSC.
- G. P. Drummen, Molecules, 2012, 17, 14067–14090 CrossRef CAS PubMed.
- N. El-Hage, M. Rodriguez, S. M. Dever, R. R. Masvekar, D. A. Gewirtz and J. J. Shacka, J. Virol., 2015, 89, 1024–1035 CrossRef PubMed.
- E. R. Simons, Curr. Protoc. Cytom., 2010, Chapter 9, Unit9 31 Search PubMed.
- A. Scott, S. T. Lugg, K. Aldridge, K. E. Lewis, A. Bowden, R. Y. Mahida, F. S. Grudzinska, D. Dosanjh, D. Parekh, R. Foronjy, E. Sapey, B. Naidu and D. R. Thickett, Thorax, 2018, 73, 1161–1169 CrossRef PubMed.
- M. Miksa, H. Komura, R. Wu, K. G. Shah and P. Wang, J. Immunol. Methods, 2009, 342, 71–77 CrossRef CAS PubMed.
- B. Stijlemans, J. Cnops, P. Naniima, A. Vaast, V. Bockstal, P. De Baetselier and S. Magez, PLoS Neglected Trop. Dis., 2015, 9(3), e0003561 CrossRef PubMed.
- S. Chen, Y. Hong, Y. Liu, J. Liu, C. W.-T. Leung, M. Li, R. T.-K. Kwok, E. Zhao, J. W.-Y. Lam, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 4926–4929 CrossRef CAS PubMed.
- A. Beletskii, M. Cooper, P. Sriraman, C. Chiriac, L. Zhao, S. Abbot and L. Yu, Biotechniques, 2005, 39, 894–897 CrossRef CAS PubMed.
- E. K. Lehrman, D. K. Wilton, E. Y. Litvina, C. A. Welsh, S. T. Chang, A. Frouin, A. J. Walker, M. D. Heller, H. Umemori, C. Chen and B. Stevens, Neuron, 2018, 100, 120–134e126 CrossRef CAS PubMed.
- E. E. Bosco, R. J. Christie, R. Carrasco, D. Sabol, J. Zha, K. DaCosta, L. Brown, M. Kennedy, J. Meekin, S. Phipps, J. Ayriss, Q. Du, B. Bezabeh, P. Chowdhury, S. Breen, C. Chen, M. Reed, M. Hinrichs, H. Zhong, Z. Xiao, R. Dixit, R. Herbst and D. A. Tice, Oncotarget, 2018, 9, 22960–22975 CrossRef PubMed.
- P. Swietach, R. D. Vaughan-Jones, A. L. Harris and A. Hulikova, Philos. Trans. R. Soc. London, Ser. B, 2014, 369, 20130099 CrossRef PubMed.
- S. Municoy, M. I. Álvarez Echazú, P. E. Antezana, J. M. Galdopórpora, C. Olivetti, A. M. Mebert, M. L. Foglia, M. V. Tuttolomondo, G. S. Alvarez, J. G. Hardy and M. F. Desimone, Int. J. Mol. Sci., 2020, 21, 4724 CrossRef CAS PubMed.
- M. Rizwan, R. Yahya, A. Hassan, M. Yar, A. D. Azzahari, V. Selvanathan, F. Sonsudin and C. N. Abouloula, Polymers, 2017, 9, 137 CrossRef PubMed.
- L. Palanikumar, S. Al-Hosani, M. Kalmouni, V. P. Nguyen, L. Ali, R. Pasricha, F. N. Barrera and M. Magzoub, Commun. Biol., 2020, 3, 95 CrossRef CAS PubMed.
- J.-O. You, M. Rafat, D. Almeda, N. Maldonado, P. Guo, C. S. Nabzdyk, M. Chun, F. W. LoGerfo, J. W. Hutchinson, L. K. Pradhan-Nabzdyk and D. T. Auguste, Biomaterials, 2015, 57, 22–32 CrossRef CAS PubMed.
- S. Baliga, S. Muglikar and R. Kale, J. Ind. Soc. Periodontol., 2013, 17, 461–465 CrossRef PubMed.
- E. Persi, M. Duran-Frigola, M. Damaghi, W. R. Roush, P. Aloy, J. L. Cleveland, R. J. Gillies and E. Ruppin, Nat. Commun., 2018, 9(1), 2997 CrossRef PubMed.
- R. Baron, L. Neff, D. Louvard and P. J. Courtoy, J. Cell Biol., 1985, 101, 2210–2222 CrossRef CAS PubMed.
- X. G. Borggaard, D. C. Pirapaharan, J.-M. Delaissé and K. Søe, Int. J. Mol. Sci., 2020, 21, 5924 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 2002, 114, 10024–10035 CrossRef.
- O. Lowry, N. Rosebrough, A. L. Farr and R. Randall, J. Biol. Chem., 1951, 193, 265–275 CrossRef CAS.
- J. V. Gregory, P. Kadiyala, R. Doherty, M. Cadena, S. Habeel, E. Ruoslahti, P. R. Lowenstein, M. G. Castro and J. Lahann, Nat. Commun., 2020, 11, 5687 CrossRef CAS PubMed.
- S. Hwang, K. H. Roh, D. W. Lim, G. Wang, C. Uher and J. Lahann, Phys. Chem. Chem. Phys., 2010, 12, 11894–11899 RSC.
- S. Rahmani, S. Ashraf, R. Hartmann, A. F. Dishman, M. V. Zyuzin, C. K.-J. Yu, W. J. Parak and J. Lahann, Bioeng. Transl. Med., 2016, 1, 82–93 CrossRef CAS PubMed.
- K. J. Horley, C. Carpenito, B. Baker and F. Takei, EMBO J., 1989, 8, 2889–2896 CrossRef CAS PubMed.
- C. F. Greineder, A. M. Chacko, S. Zaytsev, B. J. Zern, R. Carnemolla, E. D. Hood, J. Han, B. S. Ding, C. T. Esmon and V. R. Muzykantov, PLoS One, 2013, 8, e80110 CrossRef PubMed.
- W. B. Swanson and P. X. Ma, in Biomaterials Science, An Introduction to Materials in Medicine, ed. G. Zhang, Elsiever, 2020 Search PubMed.
- E. I. Vargha-Butler, E. Kiss, C. N.-C. Lam, Z. Keresztes, E. Kálmán, L. Zhang and A. W. Neumann, Colloid Polym. Sci., 2001, 279, 1160–1168 CrossRef CAS.
- A. V. Rousselle and D. Heymann, Bone, 2002, 30, 533–540 CrossRef CAS PubMed.
- R. Kuang, Z. Zhang, X. Jin, J. Hu, M. J. Gupte, L. Ni and P. X. Ma, Adv. Healthcare Mater., 2015, 4, 1993–2000 CrossRef CAS PubMed.
- R. L. McCall and R. W. Sirianni, J. Vis. Exp., 2013, 51015, DOI:10.3791/51015.
- A. G. Porras, S. D. Holland and B. J. Gertz, Clin. Pharmacokinet., 1999, 36, 315–328 CrossRef CAS PubMed.
- J. Park, V. R. Pandya, S. J. Ezekiel and A. M. Berghuis, Front Chem, 2020, 8, 612728 CrossRef CAS PubMed.
- L. Filgueira, J. Histochem. Cytochem., 2004, 52, 411–414 CrossRef CAS PubMed.
- Z.-Q. Hu, M. Li, M.-D. Liu, W.-M. Zhuang and G.-K. Li, Dyes Pigm., 2013, 96, 71–75 CrossRef CAS.
- S.-L. Shen, X.-P. Chen, X.-F. Zhang, J.-Y. Miao and B.-X. Zhao, J. Mater. Chem. B, 2015, 3, 919–925 RSC.
- G. Bao, K. L. Wong and P. A. Tanner, ChemPlusChem, 2019, 84, 816–820 CrossRef CAS PubMed.
- L. Yuan, W. Lin and Y. Feng, Org. Biomol. Chem., 2011, 9, 1723–1726 RSC.
- S. G. Stratton, G. H. Taumoefolau, G. E. Purnell, M. Rasooly, W. L. Czaplyski and E. J. Harbron, Chem. – Eur. J., 2017, 23, 14064–14072 CrossRef CAS PubMed.
- H. Yang, Y. Imanishi and A. Harata, Anal. Sci., 2015, 31, 1005–1010 CrossRef CAS PubMed.
- Q. A. Best, R. Xu, M. E. McCarroll, L. Wang and D. J. Dyer, Org. Lett., 2010, 12, 3219–3221 CrossRef CAS PubMed.
- W. Ma, L.-a Yan, X. He, T. Qing, Y. Lei, Z. Qiao, D. He, K. Huang and K. Wang, Anal. Chem., 2018, 90, 1889–1896 CrossRef CAS PubMed.
- F.-Y. Lin and A. D. MacKerell, Biomolecular Simulations, 2019, Ch. 2, pp. 21–54 DOI:10.1007/978-1-4939-9608-7_2.
- N. A. Sims, H. A. Morris, R. J. Moore and T. C. Durbridge, Calcif. Tissue Int., 1996, 59, 121–127 CrossRef CAS PubMed.
- N. A. Sims and T. J. Martin, Front. Endocrinol., 2015, 6, 41 Search PubMed.
- K. Sato and H. Takayanagi, Curr. Opin. Rheumatol., 2006, 18, 419–426 CrossRef CAS PubMed.
- S. Karmakar, J. Kay and E. M. Gravallese, Rheum. Dis. Clin. North. Am., 2010, 36, 385–404 CrossRef PubMed.
- A. E. Coudert, M. C. de Vernejoul, M. Muraca and A. Del Fattore, Int. J. Endocrinol., 2015, 2015, 372156 Search PubMed.
- C. Sobacchi, A. Schulz, F. P. Coxon, A. Villa and M. H. Helfrich, Nat. Rev. Endocrinol., 2013, 9, 522–536 CrossRef CAS PubMed.
- F. Cosman, Curr. Osteoporos. Rep., 2014, 12, 385–395 CrossRef PubMed.
- M. Minoshima, J. Kikuta, Y. Omori, S. Seno, R. Suehara, H. Maeda, H. Matsuda, M. Ishii and K. Kikuchi, ACS Cent. Sci., 2019, 5, 1059–1066 CrossRef CAS PubMed.
- D. M.-H. Merrild, D. C. Pirapaharan, C. M. Andreasen, P. Kjærsgaard-Andersen, A. M.-J. Møller, M. Ding, J.-M. Delaissé and K. Søe, Bone Res., 2015, 3, 15032 CrossRef CAS PubMed.
- S. Epsley, S. Tadros, A. Farid, D. Kargilis, S. Mehta and C. S. Rajapakse, Front. Physiol., 2021, 11, 511799 CrossRef PubMed.
- K. Sato and H. Takayanagi, Curr. Opin. Rheumatol., 2006, 18, 419–426 CrossRef CAS PubMed.
- J. M. Wagner, S. V. Schmidt, M. Dadras, J. Huber, C. Wallner, S. Dittfeld, M. Becerikli, H. Jaurich, F. Reinkemeier, M. Drysch, M. Lehnhardt and B. Behr, Life, 2022, 12(2), 134 CrossRef CAS PubMed.
- X. Huang, M. Xie, Y. Xie, F. Mei, X. Lu, X. Li and L. Chen, J. Transl. Med., 2020, 18, 479 CrossRef CAS PubMed.
- D. V. Novack, Arthritis Rheum., 2016, 68, 2834–2836 CrossRef PubMed.
- D. Pei and M. Buyanova, Bioconjugate Chem., 2019, 30, 273–283 CrossRef CAS PubMed.
- S. A. Smith, L. I. Selby, A. P.-R. Johnston and G. K. Such, Bioconjugate Chem., 2018, 30, 263–272 CrossRef PubMed.
- H. Du Rietz, H. Hedlund, S. Wilhelmson, P. Nordenfelt and A. Wittrup, Nat. Commun., 2020, 11, 1809 CrossRef CAS PubMed.
- W. Liang and J. K.-W. Lam, Molecular Regulation of Endocytosis, 2012, Ch. 17 DOI:10.5772/46006.
- M. Serresi, R. Bizzarri, F. Cardarelli and F. Beltram, Anal. Bioanal. Chem., 2008, 393, 1123–1133 CrossRef PubMed.
- S. Padilla-Parra, P. M. Matos, N. Kondo, M. Marin, N. C. Santos and G. B. Melikyan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17627–17632 CrossRef CAS PubMed.
- P. X. Ma, Adv. Drug Delivery Rev., 2008, 60, 184–198 CrossRef CAS PubMed.
- S. Zhuo, F. Zhang, J. Yu, X. Zhang, G. Yang and X. Liu, Molecules, 2020, 25, 5649 CrossRef CAS PubMed.
- F. Li, Y. Liu, Y. Xu, Y. Li, J. Liu, M. Lv, C. Ruan, H. Pan and X. Zhao, ACS Omega, 2020, 5, 19796–19804 CrossRef CAS PubMed.
- S. M. Azarin, J. Yi, R. M. Gower, B. A. Aguado, M. E. Sullivan, A. G. Goodman, E. J. Jiang, S. S. Rao, Y. Ren, S. L. Tucker, V. Backman, J. S. Jeruss and L. D. Shea, Nat. Commun., 2015, 6, 8094 CrossRef PubMed.
- A. H. Morris, S. M. Orbach, G. G. Bushnell, R. S. Oakes, J. S. Jeruss and L. D. Shea, Cancer Res., 2020, 80, 3786–3794 CrossRef CAS PubMed.
- E. V. Zagaynova, I. N. Druzhkova, N. M. Mishina, N. I. Ignatova, V. V. Dudenkova and M. V. Shirmanova, Multi-Parametric Live Cell Microscopy of 3D Tissue Models, 2017, Ch. 7, pp. 105–119 DOI:10.1007/978-3-319-67358-5_7.
- T. C. McIlvaine, J. Biol. Chem., 1921, 49, 183–186 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Methods, Supplementary Figures. See DOI: https://doi.org/10.1039/d2cb00030j |
| ‡ Current address: Division of Hematology, Department of Internal Medicine, Mayo Clinic, Rochester, MN USA. |
| This journal is © The Royal Society of Chemistry 2022 |