
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Practical considerations for quantitative and reproducible measurements with stimulated Raman scattering microscopy†
Dimitrios
Tsikritsis
 a,
Elizabeth J.
Legge
a and
Natalie A.
Belsey
*ab
a,
Elizabeth J.
Legge
a and
Natalie A.
Belsey
*ab
aChemical and Biological Sciences Department, National Physical Laboratory, Hampton Road, Teddington, Middlesex, TW11 0LW, UK. E-mail: natalie.belsey@npl.co.uk
bDepartment of Chemical & Process Engineering, University of Surrey, Guildford, GU2 7XH, UK
First published on 16th August 2022
Abstract
Stimulated Raman scattering (SRS) microscopy provides rapid label-free 3D chemical imaging with wide-ranging applications including histology, pharmacokinetic studies, and materials characterisation. SRS microscopy has seen a steady increase in utilisation since the early 2000s and has become more accessible due to the increase in availability of facilities, and the development of user-friendly instrumentation. Although some complete SRS systems are now commercially available, many instruments are home-built with highly varied laser sources, optics, and detection mechanisms. Signal intensity is also dependent on the effective spatiotemporal overlap of two (or more) laser beams, thus any drift in alignment can result in variable performance. Currently there is a lack of standard procedures or reference materials for SRS, which has important implications on reproducibility and consistency. These concerns are particularly relevant to the comparison of data from the same instrument at different times and instrument settings as well as between different instruments and laboratories. This tutorial-style review presents the most important practical considerations for sample preparation, instrument set-up, image acquisition and data analysis to obtain reproducible SRS measurements.
Introduction
Optical imaging methods are of critical importance due to their non-destructive nature, meaning that they can be utilised to monitor for example, live cells and chemical or manufacturing processes. Fluorescence imaging has established itself as an invaluable tool in the life sciences, owing to the excellent specificity provided by targeted probe labelling coupled with the high sensitivity afforded by their large absorption cross-sections. It provides highly valuable insight in widespread applications, but suffers from drawbacks such as photobleaching, phototoxicity, and challenging quantification.1 Other drawbacks include potential changes to the sample under investigation due to the presence of tags,2 and possible de-conjugation resulting in uncertainty as to whether the tag is still bound to the target. Fluorescence imaging also necessitates the presence of a fluorophore which is not always possible or appropriate in certain applications, for example at manufacturing lines, and in drug delivery studies such as skin permeation, where the conjugation of a large probe would perturb the physicochemical properties and therefore the behaviour of a small drug molecule. Therefore, there is a need for label-free imaging techniques for which applications span biology,3 chemistry,4 medicine,5 and materials characterisation.6,7Raman spectroscopy (RS) provides a chemical fingerprint of the sample based on the inelastic scattering of light8 without the need for a tag. The frequency difference between the incoming and detected light provides the Raman shift. The Raman shifts for detected peaks can provide information on the types of functional groups present in the sample. Raman signal intensity has a linear relationship with concentration of the chemical species, thus it is possible to elucidate quantitative information from the data.9 Unlike many other chemical characterisation instruments, such as mass spectrometry and photoelectron spectroscopy that require ultra-high vacuum to operate, RS can be performed in ambient environmental conditions and in liquid with minimal requirements for sample preparation. However, there are a few limitations of RS. One of the main shortfalls of RS is its sensitivity, which is poorer than several other analytical methods. This is because only a very small proportion of the incident light is inelastically scattered (Fig. 1) by the sample (typically fewer than 1 in a million photons).9
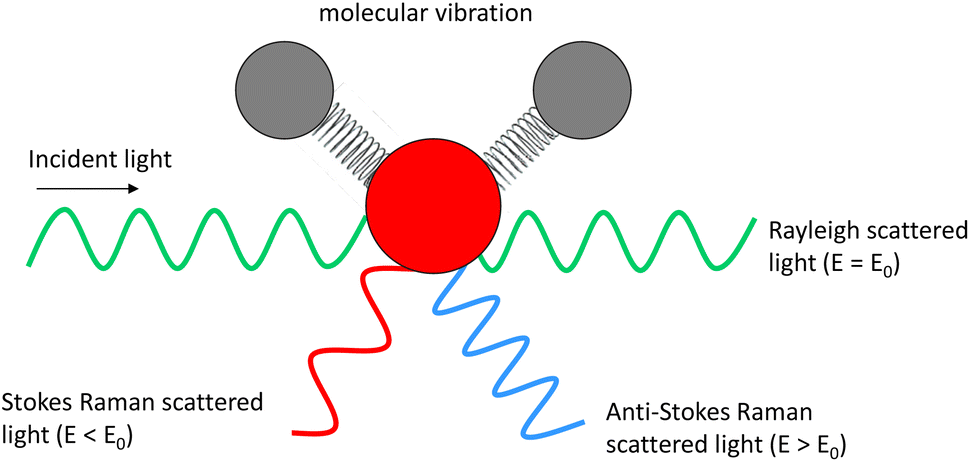 | ||
| Fig. 1 Raman scattering schematic. | ||
There are several Raman-based methods that can offer improvements in sensitivity. Resonant Raman scattering occurs when the incident excitation beam is close in energy to an electronically excited state of the material under examination. The frequency resonance can lead to greatly enhanced intensity of the Raman scattering. A related technique, surface-enhanced Raman scattering (SERS) involves the use of a rough metallic substrate to provide a localised surface plasmon (LSP) which is resonant with the excitation beam and can provide a signal boost of as much as 1011, however despite the increase in sensitivity, quantitative analysis remains a challenge.10 Tip-enhanced Raman spectroscopy (TERS) combines SERS enhancement with scanning probe microscopy (SPM), where LSP resonance occurs at the apex of a metal probe when aligned with the excitation beam, to allow Raman mapping at nanoscale resolution. These enhancement mechanisms should be considered when elucidating quantitative information from RS data, for example the choice of laser source and the presence of metallic nanostructures that could generate LSP resonances, which would locally amplify the detected signal based on their distribution whilst other areas will effectively be silent.
Another disadvantage of RS is the long measurement times when acquiring images. To generate an image with RS, the laser beam must be raster-scanned across a sample, in a stepwise manner, to acquire a spectrum at each pixel. Due to the long acquisition times required to collect sufficient photons for adequate signal to noise, generation of large high-resolution images is extremely time-intensive and is therefore less-well suited to monitoring dynamic processes than laser scanning confocal fluorescence for example.
These long mapping/image acquisition times can be overcome using coherent Raman scattering (CRS) microscopy techniques. This article focuses on the application of stimulated Raman scattering (SRS) microscopy which offers improvements in imaging speed compared to RS. In RS, only one monochromatic laser source is required to excite the sample and the inelastically scattered photons are collected at a different frequency to the incident beam. In SRS and a related CRS technique, coherent anti-Stokes Raman scattering (CARS) microscopy, two laser beams are focused on the sample where the difference in frequency is tuned to match a Raman vibrational mode of interest. This targeted stimulation of the vibrational mode increases the Raman scattering by up to 4 or 5 orders of magnitude and enables rapid imaging11 more than 1000 times faster at a single frequency compared to spontaneous Raman microscopes.12 Both SRS and CARS signals are generated simultaneously by the sample, but they differ in their detection mechanisms. The SRS process results in a loss of intensity in one laser beam and a gain in the other beam. To detect this signal, one of the beams is typically modulated, and the transfer of this modulation pattern to the second beam is detected with high sensitivity using a photodiode detector and lock-in amplifier. In CARS, a new frequency of light is generated, so it can be simply isolated using optical filters and detected with a photomultiplier tube. Although SRS requires a more complicated detection mechanism compared to CARS, it offers some benefits: the signal is linear with chemical concentration (CARS has a quadratic relationship and suffers further complications from a non-coherent background); SRS images are comparatively free from non-resonant background signals such as emitted fluorescence.13
The elucidation of quantitative chemical information provides crucial insight to many applications from drug discovery,14 pharmaceutics,15,16 materials science,17 live cell studies18,19 and medical diagnostics.20 High levels of reproducibility and consistency across Raman instruments is extremely important for many of these applications, for example cancer diagnostics.21 However, there are many parameters affecting the quality and reproducibility of RS and to an even greater extent, SRS data. More generally, there are concerns that the wider scientific community is suffering from a reproducibility crisis,22 for which metrology and standardisation efforts are needed to improve confidence in data.23 There are various levels of reproducibility that need to be met. At a local level, an instrument in a laboratory should be able to generate data with acceptable variability from the same sample on different days and with different operators. This statement implies that there needs to be a stable reference sample from which the day-to-day and operator-to-operator variability in key performance parameters such as intensity, linearity, spatial resolution, and wavelength can be measured. At a wider level, the comparability of results between different instruments and laboratories must be considered and measurements harmonised through agreed procedures, for example for wavelength and intensity calibration.
This review presents the most important practical considerations affecting quantitative and reproducible SRS measurements, including sample preparation, instrumentation, image acquisition and data analysis approaches. For a more comprehensive description of the optics and associated mathematics, readers are encouraged to refer to the specialist books and journal publications.24–26
Introduction to optical processes
Irradiation of the sample with a laser beam can result in numerous optical processes. In addition to Rayleigh (elastic) scattering, a small proportion of the light can undergo inelastic scattering. When a single monochromatic laser source is used, spontaneous Raman scattering occurs, whereas when two beams are used with a frequency difference matching a vibrational mode, coherent Raman signals can be generated from SRS and CARS processes. Several other (non-Raman) optical processes may also be taking place, and can also provide complementary information, for example two-photon excited fluorescence (TPEF) and second harmonic generation (SHG).26,27 These different processes are illustrated in the Jablonski diagrams in Fig. 2.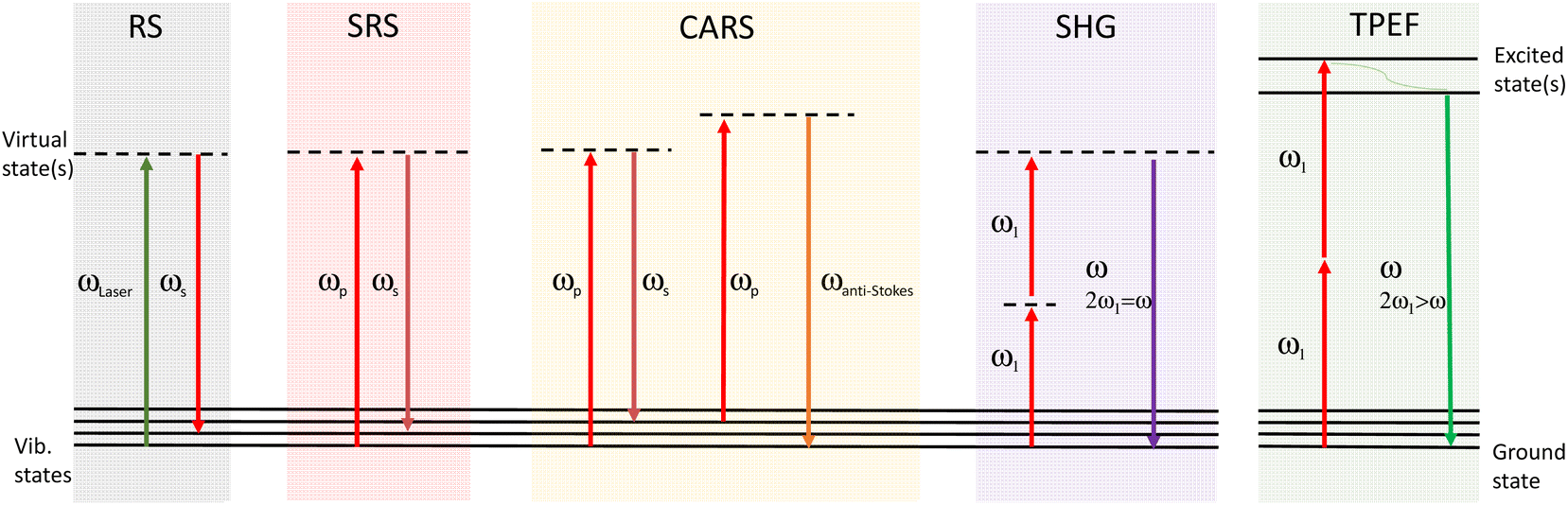 | ||
| Fig. 2 Jablonski diagrams for spontaneous Raman, SRS, CARS, two photon excited fluorescence, and second harmonic generation, where ω is the frequency, and subscript ‘s’ and ‘p’ indicate the Stokes and pump fields respectively. The green wavy line in TPEF represents relaxation. | ||
In RS, a single exciting photon can result in a range of emission energies. For SRS there is a transfer of energy between the pump and Stokes light wavelengths. In CARS, the anti-Stokes signal is generated at a new wavelength. In TPEF the two photons are absorbed by the sample and one fluorescent photon is produced. SHG involves two photons of the same frequency interacting with the sample which combine to produce one photon of double the energy (half the initial wavelength). RS signals are generated in an isotropic distribution, whereas for SRS the signals are propagated more strongly in the forward direction. For this reason, sensitivity of SRS detection tends to be significantly improved in the forward compared to backscattered direction, unless the sample is of significant thickness.28 Therefore, SRS is commonly performed in transmission. The complementary multiphoton modalities such as CARS, TPEF and SHG are typically detected in either backscattered or transmission.
Instrumentation
Since both SRS and CARS signals are generated simultaneously, many microscopes are set up to allow detection of both signals. Similarly, TPEF and SHG signals are also commonly detected in separate channels.A basic CRS instrument is depicted in Fig. 3 and consists of:
1. A dual laser source that provides at least two laser beams of different wavelength with the same repetition rate. The Stokes beam typically has a fixed wavelength, while the pump beam wavelength is tuneable, to target the wavenumbers of interest.
2. An optical parametric oscillator (OPO) enables the tuneability of the pump beam.
3. A modulator that applies a modulation pattern from the frequency generator to the Stokes beam.
4. A frequency generator generates the modulation frequency for the optical modulator.
5. A delay line, which comprises a mechanical stage and mirrors, enables the temporal overlap of the pump and Stokes beams to be optimised.
6. A dichroic mirror spatially combines the pump and the Stokes beams to create the train pulse.
7. An optical scanner (controlled by the image acquisition software) typically consists of a pair of galvo mirrors and the scanning and tube lens. This scanner allows the beam to be rastered laterally across the sample.
8. An objective lens suitable for the range of wavelengths being used, i.e. for most multiphoton imaging, a near infrared transmission-corrected lens.
9. A condenser lens that collects all the forward-propagated SRS signal and the rest of the light at the sample.
10. Filters that remove the modulated Stokes beam.
11. A detector, usually a silicon photodiode.
12. A lock-in amplifier reads the signal from the diode using the frequency generator signal as a carrier wave reference and amplifies the modulated SRS signal.
13. The signal from the lock-in amplifier is read by the PC and the signal is assigned to the appropriate spatial coordinates in a data array.
14. Photomultiplier tubes (PMTs) are typically used for the detection of CARS and other complementary multiphoton signals.
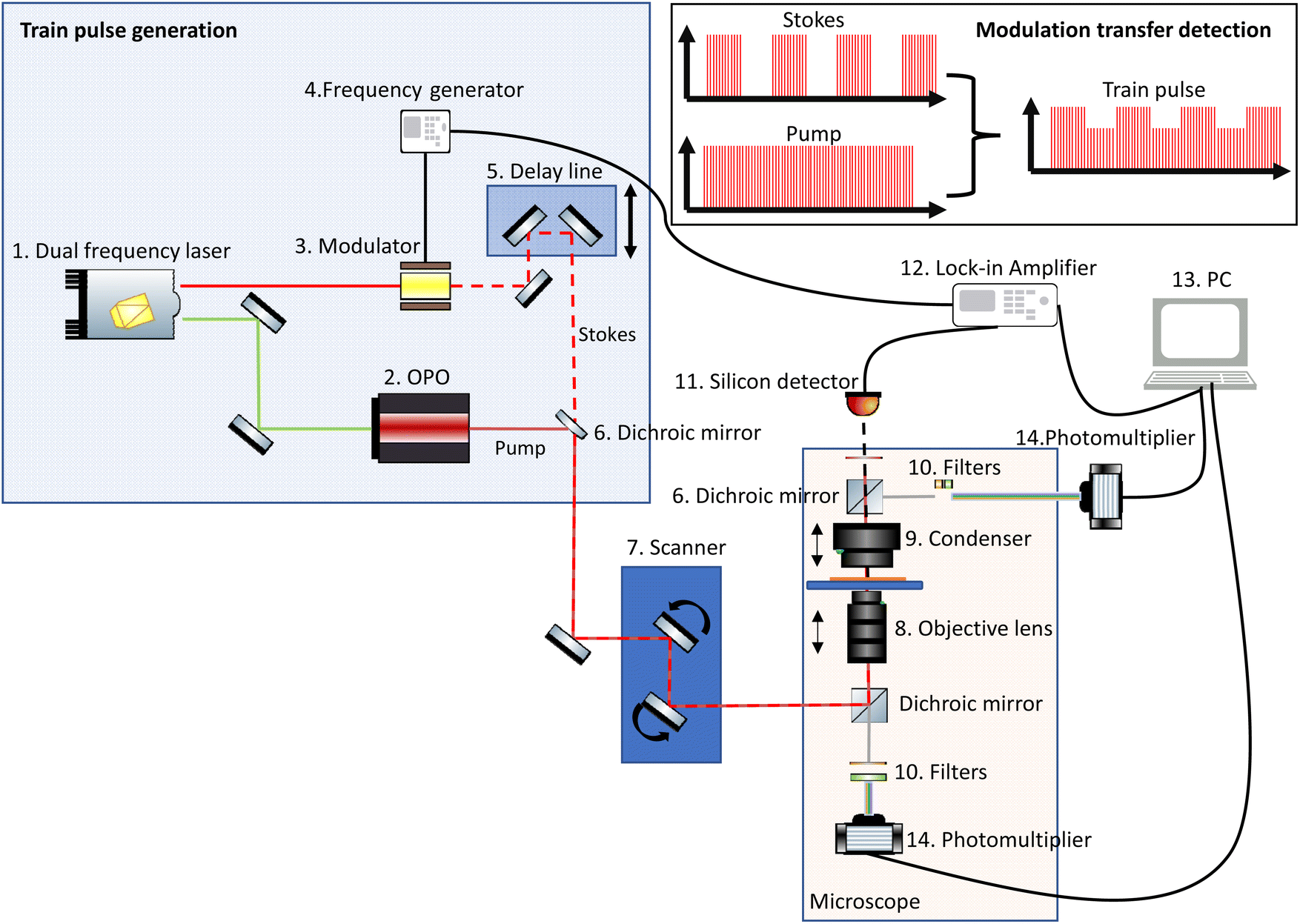 | ||
| Fig. 3 Simplified schematic of a typical CRS system, which often comprises detection mechanisms for both SRS and other multiphoton imaging modalities. | ||
The laser source generates two different wavelength beams with the same repetition frequency. The wavelength of the Stokes beam is kept constant (typically between 1000–1100 nm for most systems). The pump beam passes through the OPO, which facilitates tuning of the laser wavelength to correspond to the required wavelength for the beat frequency. Eqn (1) describes the relation between the pump and Stokes beam wavelengths and the targeted wavenumber stimulated by both SRS and CARS. The 107 factor is included for use of the most practical units: the wavenumber, ṽvib is in cm−1, and the wavelength, λ, is in nm. Eqn (2) is specific to CARS microscopy and shows the relationship between the pump and Stokes beam wavelengths, and the new signal wavelength generated for the corresponding CARS signal.
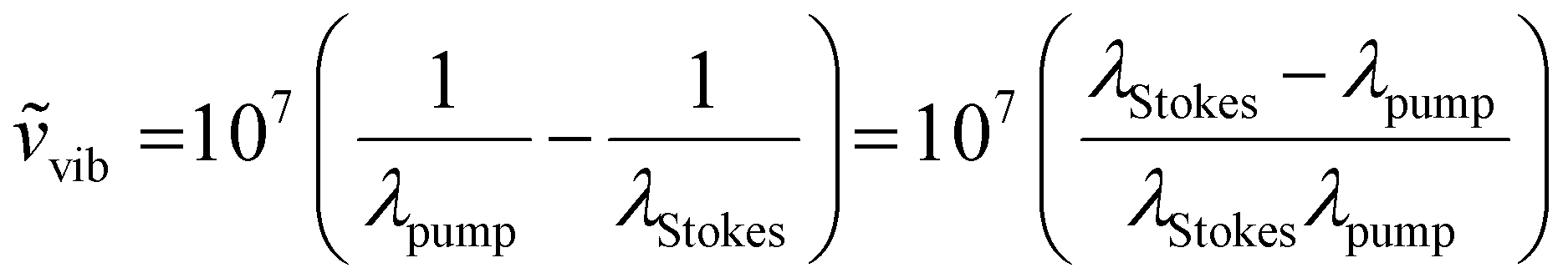 | (1) |
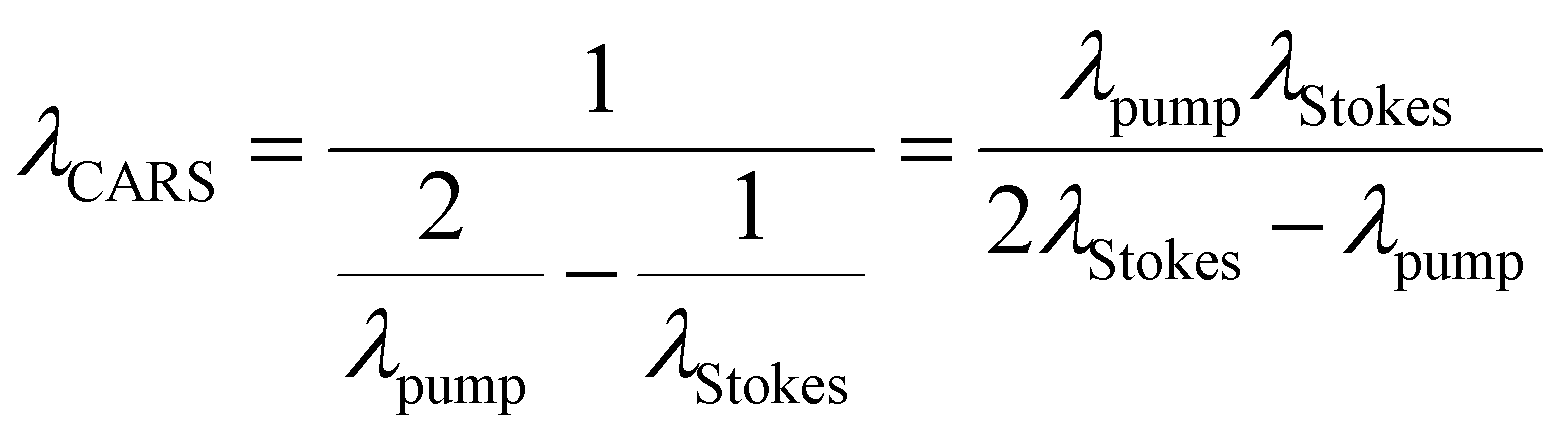 | (2) |
SRS is commonly performed as stimulated Raman loss (SRL) rather than stimulated Raman gain (SRG) for several reasons. SRL requires more power from the longer wavelength (Stokes) beam and less power from the pump beam for excitation of the sample, thus resulting in lower power densities (and photodamage) for the same signal to noise. It is also easier to align modulators in the fixed path of the Stokes beam than in the tuneable pump beam. In addition, silicon photodiodes tend to have better responsivities for the pump compared to the Stokes beam.24 To perform SRL, the Stokes beam is modulated (via electro-optical or acousto-optical modulators) to generate a train-pulse. To overlap the beam pulses in the time domain, the modulated Stokes beam passes through a delay line. This is a mechanical stage that increases or reduces the length of the light path so that when both beams combine spatially, they can be adjusted to overlap the time domain of the pulses. The combined beam travels through the scanning module to the objective lens, where it is focused on to the sample. The forward propagated light from the sample is collected with a condenser lens. It is important to use a condenser with a higher numerical aperture (NA) than the objective lens in order to minimize spurious signals caused by cross phase modulation.24 Typically, the Stokes beam is filtered out from the collected light and the resulting modulated SRS signal is collected with a silicon diode. The silicon diode is connected to a lock-in amplifier that discriminates the modulation transfer signal. This signal is amplified and read by the software.
Many SRS systems are custom-made, but complete commercially available instruments are now available. Custom-made systems are often constructed to be adaptable and usually permit flexibility in the beam path and individual components, whereas integrated commercial systems tend to be more enclosed to optimise instrument footprint and improve safety. Both types of instrumentation should be evaluated in relation to measurement robustness, comparability, and repeatability. To achieve reproducible and meaningful SRS microscopy measurements, instrument settings require careful optimisation. We provide below a summary of the main hardware experimental considerations SRS users should be aware of.
Laser system
In SRS systems used for biological applications, a femtosecond or a picosecond dual laser source is usually employed. Femtosecond sources allow for more sensitive SHG and TPEF imaging due to the higher peak intensity. However, femtosecond sources produce poorer SRS spectral resolution due to the larger bandwidth of shorter pulses, compared to picosecond lasers. Picosecond sources also impart less photodamage to sensitive samples.24 Typically, the Stokes beam is in the near infra-red range (1000 nm to 1100 nm), and the second beam is usually generated from this source by frequency-doubling, for example 1064 nm Stokes beams are used to generate 532 nm. The wavelength of the frequency-doubled beam is then tuned using an OPO, generating wavelength ranges between ∼750 nm to ∼1100 nm, which corresponds to approximately 600 cm−1 to 3500 cm−1, depending on the wavelength of the Stokes beam. These wavelengths are selected to enable improved optical penetration relative to the visible region, due to lower absorption and scattering by the tissue.The laser power can be controlled directly from the laser source and some systems offer the possibility to reduce the power, using for example a polariser which is controlled by the microscope software. It should be noted that the laser power reaching the sample is not the same as at the laser source. Due to the losses along the optical pathway and depending on the employed optical elements, a fraction of the initial laser power may reach the sample. For this reason, it is good practice to measure the laser power of both laser beams at the sample prior to use and during troubleshooting. Monitoring the power over time can help identify when the optical alignment is deteriorating, as well as identifying other problems in the optical path, for example accumulated dust (especially on an open table system) or damaged optics.
An important factor affecting the laser source stability is the consistency of the temperature (both internal and external to the laser). For this reason, the laser systems are coupled to a chiller unit which maintains the stability of the internal laser temperature. Even very small changes in the chiller temperature can significantly affect the laser intensity, so good chiller maintenance is extremely important. The OPO is extremely sensitive to environmental factors such as temperature and vibrations and requires a stable environment for optimal operation and fine tuning. Alignment can be challenging and time-consuming. Care should be taken where the microscope tower and OPO are placed on the same floating table due to the risk of OPO misalignment caused by large vertical displacements of the table when using the microscope tower.
For any CRS method it is critical to maintain the spatial and the temporal overlap of the laser beams. The sensitivity of the technique is entirely dependent on this overlap which can drift with time. Therefore, it is prudent to conduct regular checks of the beam overlap and monitor performance using well-characterised reference samples of known/expected signal to noise and keep records. By stipulating a set of consistent acquisition parameters, it is then possible to ‘quantify’ the health of the instrument response based on the necessary detector gain settings to reach a certain intensity value.
For long time-course experiments, or those particularly sensitive to laser-intensity fluctuations, the laser power should be monitored, for example with a beam-splitter and detector and the data used to correct the acquired images accordingly. Laser noise is an important issue that limits the sensitivity of this technique. A comprehensive tutorial review on laser noise for SRS microscopy has been published by Audier et al.29
Detectors
The SRS signal is most often measured using a silicon detector.24 As is the case for RS, different detectors can exhibit very different wavelength-dependent quantum efficiencies and therefore sensitivities across the wavelength ranges used. An example of a sensitivity response of a silicon photodiode commonly used for SRS detection is presented in Fig. 4.30,31 Detector sensitivity is highly wavelength-dependent, and the response curve can be significantly different for different models of photodiodes. Therefore, signal intensity vs. wavenumber for the same sample could look quite different when analysed on different instruments.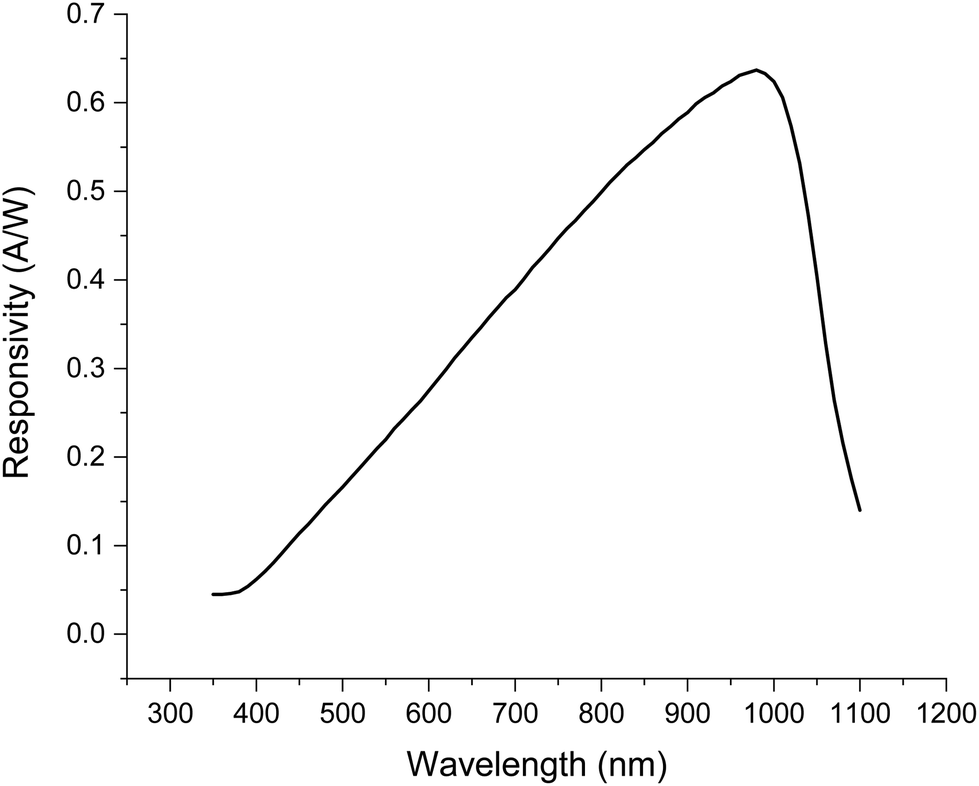 | ||
| Fig. 4 Typical SRS detector responsivity vs. wavelength (FDS1010 silicon photodiode, Thorlabs).30 Typical wavelengths used are 1000–1100 nm for the Stokes and 700–900 nm for the pump beams. | ||
For RS, intensity calibration reference materials have been developed by the National Institute of Standards and Technology (NIST) and documentary standards are available describing these procedures.32,33 However, the calibration reference standards produced by NIST work are based on emitted fluorescence which is not detected by SRS. There remains a need for calibration samples to measure SRS detector responses. Information on the relative intensities of reference materials such as polystyrene can provide some useful insight. These can be compared to the ASTM E1840-96 standard which lists the peak positions and relative intensities.34Fig. 5 shows RS spectra acquired with different excitation sources (left) and an SRS spectrum (right) acquired from a sample of polystyrene sheet. The spectra have been normalised to the highest intensity peak but are not corrected for relative intensity. Large differences in relative intensity across the spectra are observed between different excitation sources in RS and with SRS. The most common reason for these discrepancies is differences in detector responsivity (such as that shown for SRS in Fig. 4) and similarly, reduced CCD response at longer wavelengths in spontaneous Raman spectroscopy. In addition, differences in the OPO output power across the wavelength range used can also strongly impact SRS data.
 | ||
| Fig. 5 Uncalibrated spectra obtained from a polystyrene sheet using RS with a range of excitation sources (left), compared to SRS (right). The spectra have been normalised to the highest intensity peak but have not been corrected for wavelength-dependent sensitivity. The SRS spectrum was acquired with 0.2 nm steps of the pump laser wavelength (2 ps pulse width). | ||
An additional consideration is the detector driving voltage and the gain used which are adjustable. These parameters should be carefully recorded and kept consistent when direct comparison of data sets is required, e.g. in dosed vs. control samples. However, due to the difference in sensitivity across the Raman spectrum, and different signal intensities between samples, it may not be possible to find a gain setting that works well for all targeted wavelengths. One solution is to group the wavenumbers of interest into lower and higher signal intensities, ensure settings are consistent within each group, and that the wavenumbers requiring direct comparison, such as an on-resonance vs. off-resonance control, are acquired within the same group with consistent settings.
Signal to noise will vary between detectors and optimisation to minimise electronic noise may be necessary, especially when targeting weak signals.29,31
Lock-in amplifier
For comparable SRS measurements, uniform settings of the lock-in amplifier are required. The main settings include:• Phase
• Time constant
• Sensitivity
• Channel offset
It is not useful to attempt to prescribe the ‘best’ settings because these will be instrument and sample dependent. However, for sets of experimental work where the data needs to be directly comparable, such as time-dependent studies or comparing treated vs. untreated samples, it is critical to keep these settings consistent. Altering the sensitivity setting will result in incomparable data sets. For complete/built-in systems these settings are usually pre-set and the user can only alter the phase as this is sample dependent. More detailed information can be found in a publication by Zada et al., who describe the effects of various lock-in amplifier settings on SRS signal and image formation, and their freely available simulation programme for SRS imaging.35
Image formation: sample vs. beam scanning
Most SRS microscopes utilise galvo-mirrors for scanning the beam across the sample. One important consideration is that the transmission of the galvo scanners may not be uniform across the whole wavelength range used, which can cause a slight relative signal loss at certain wavelength ranges. Instead of scanning the laser beams across the sample, some systems have a fixed laser path and instead the sample stage is moved. Most piezo stage scanning systems are more restricted in terms of the maximum field of view and image acquisition is typically slower compared to laser-scanning systems.The uniformity of the signal intensity across the field of view should be checked using a reference sample. Samples with distinctive patterns such as grids can also be useful to check there are no distortions in the resulting images (see ESI Fig. S1† for an example).
Filters
Since many instruments are set up to perform multiple imaging modalities, a series of different filters may be incorporated throughout the optical path, particularly when other multiphoton techniques are being performed in parallel. Careful selection and knowledge of the properties of these filters is required to ensure they do not result in accidental partial selectivity of a wavelength-dependent proportion of the signal leading to biased results. Filters should be regularly cleaned following the manufacturers guidelines and inspected before use for scratches or defects.Lenses
The incident light is focused onto the sample using an objective lens, and the forward propagating light is usually collected in transmission using a condenser lens, however for thick, scattering samples SRS can also be detected in a backscattering geometry.36 High NA lenses are required for the best spatial resolution and result in better sensitivity since they collect a greater proportion of the light. It should be noted that the pairing of a condenser with a lower NA than the objective lens can introduce spurious signals from cross-phase modulation arising from the Kerr effect,24,37 so this should be avoided. It is also extremely important that the condenser position is laterally aligned with the objective lens, and that the height is optimally focused, as this strongly impacts the signal intensity.As for other optical microscopy techniques such as two photon fluorescence microscopy, spatial resolution is diffraction-limited. Depending on the choice of lenses, the spatial resolution can reach 500 nm laterally, and approximately one micron in the axial direction. However, efforts are underway to overcome the diffraction limit, and further discussion can be found in the future perspectives section.
The potential maximum depth or 3D volume that can be analysed will depend on the maximum working distances of the lenses. Typically, higher NA lenses have shorter working distances, and thus a trade-off is often required in choosing the lenses that will give the optimum quality imaging within the constraints of the necessary working distance prescribed by the experimental requirements. The best performing lenses tend to require oil or water-immersion media. This can introduce practical challenges, such as ensuring that the immersion oil does not contaminate the sample or any water-immersion lenses. Retaining sufficient water on aqueous immersion lenses for lengthy experiments can be challenging due to the rate of evaporation. Practical solutions include water pumps to top up the lens water, and the use of synthetic media which mimics the refractive index of water but is less volatile and is retained in sufficient volume for many hours.
SRS workflow and experimental planning
SRS generates images quickly, but for single wavenumber at-a-time systems the generation of spectra is quite slow. In contrast RS generates spectra quickly for a single point but takes a long time to generate images. Therefore, it is practical to acquire individual ingredient or component spectra with RS to inform the choice of wavenumbers to target for SRS imaging. However, care should be taken to check the position of the peak maxima by tuning in small increments a few fractions of a nm either side of the expected peak position. Small changes in peak position could be caused by differences in the chemical micro-environment between the reference compounds compared to the complex mixture, in addition to differences in RS compared to SRS instrument wavenumber position calibration. Performing SRS imaging slightly off the peak maxima will result in significant reduction in the signal to noise. A more complete calibration assessment of the system can be performed using reference materials such as polystyrene, for which the peak positions (and relative intensities, as described earlier) can be compared to those published in documentary standards.34Once the calibration between the RS and SRS systems have been checked, RS spectra can be used to inform the choice of Raman shift to tune the SRS system to facilitate rapid imaging at that wavenumber. Fig. 6 shows example RS spectra of water and petrolatum (left), and the Raman shifts selected for SRS imaging of a formulation containing both these ingredients, which have been overlaid to form a composite SRS image (right).
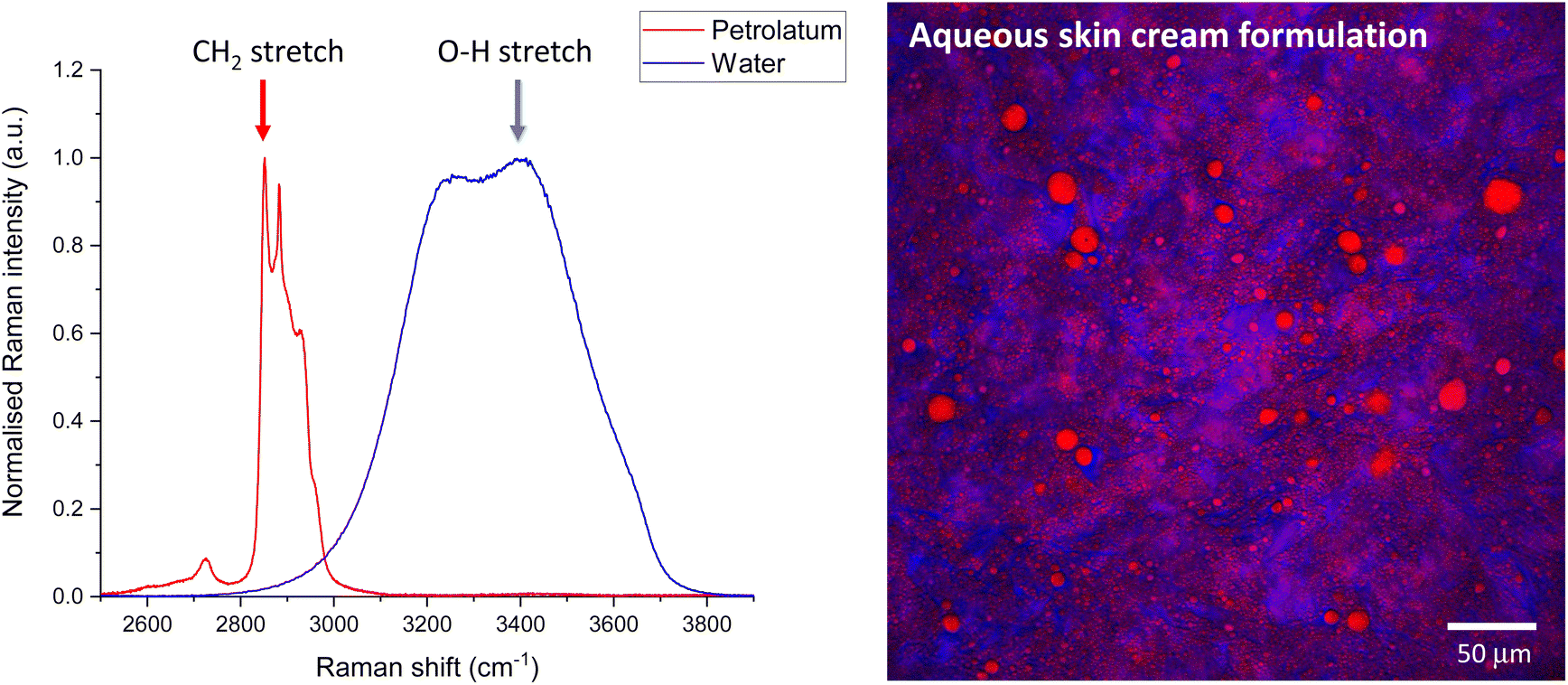 | ||
| Fig. 6 RS to SRS workflow. Step (1) Acquire RS spectra of the individual ingredients. Step (2) Select wavenumbers of peaks corresponding to the components of interest, ideally positions which do not overlap with other components, in addition to at least one off-resonance wavenumber as a control. Step (3) tune the SRS system to the corresponding wavenumbers to acquire contrast for each peak in turn. | ||
When choosing peaks for SRS imaging based on RS spectra, there are several important considerations: the first is a possible difference in detector sensitivity across the wavenumber ranges. Typically, lower wavenumber peaks in the fingerprint region may look relatively more intense in RS compared to SRS due to differences in detector quantum efficiency and instrument sensitivity with wavelength.
Secondly, in addition to performing imaging at the selected wavenumbers of interest, it is necessary to also acquire control images at nearby ‘off-resonance’ wavenumbers where no Raman signal is expected. Although SRS has a minimal non-resonant background there can be some non-Raman contributions to the signal such as two photon absorption, photothermal lensing and cross-phase modulation.38 These contributions are typically observed across a broad wavenumber range compared to the sharp Raman bands, and thus can be identified by tuning away from the Raman bands. The selection of the off-resonance wavenumbers is of great importance. For the reasons described above, it is best to select a wavenumber as close as practically possible to the on-resonance contrast wavenumber of interest. This can prove challenging in complex samples for which there are many peaks with few areas of baseline-level signal. Due to the issues of differing instrument sensitivity across the full range of wavenumbers, it may be necessary to select multiple off-resonance wavenumbers to complement the on-resonance wavenumbers throughout the range of the wavenumbers probed. This is particularly critical when spurious signals are present and must be subtracted prior to the extraction of quantitative information through image analysis.
When considering image acquisition parameters, most variables will affect the signal, noise, and parasitic signals measured. It is best practice to optimise all parameters for a representative sample, and then apply the same settings consistently across samples for which comparable data is required. The instrument response may change over time, so for direct comparisons between samples, they should be measured as close together in time as is practically possible. Consider also monitoring the fluctuations in laser power (see Instrumentation section).
Optimising the detector gain settings can be challenging, due to the large range of signal to noise in SRS data (especially during hyperspectral imaging, see later). It is important to ensure that the detector is not saturated at any point during the data acquisition. This can be easily checked using a look up table (LUT) which has a different colour scheme for the highest and lowest values of the bitmap, for example the ‘HiLo’ LUT in software such as ImageJ or Fiji. Saturated pixels provide limited quantitative information because the true signal magnitude is truncated. Generally for this reason, it is most practical to start with the sample and wavenumbers that will give the highest signal first, for example a dosed sample; optimise all settings for that sample, and then keep everything perfectly consistent to repeat the acquisition on the control/placebo treated sample, and at wavenumbers that exhibit lower intensity signals.
Sample considerations
SRS microscopy can be applied to a wide range of sample types and formats. This section contains some useful tips for sample preparation and considerations for elucidation of quantitative information.Sample thickness and optical properties
As for all optical microscopies, scattering and absorption of the beam by turbid samples occurs as a function of depth, leading to an attenuation in the signal intensity. In addition, the focal volume also increases with depth, particularly in the axial direction,39 and the spatial resolution worsens, as is the case for RS.40 Therefore, the overall sample thickness and optical properties ultimately impact the magnitude and quality of the detected signals.28 To compare between samples such as tissue sections, for example, the sections should be cut to the same nominal thickness and mounted in a consistent manner. When performing 3D analyses, signal attenuation with sample depth due to these losses must be corrected for. Please refer to the data analysis section for more information.Factors that affect the efficiency of the light transmission through the sample include the presence of optical clearing agents that change the refractive index of the sample. These may be intentionally applied to reduce scattering to increase signal intensity allowing deeper measurements,41,42 or they may be an unintentional consequence, for example excipients in a topical drug formulation which in addition to providing a vehicle for drug delivery, also affect the optical properties of the tissue. Common examples of molecules with optical clearing properties include urea, triton-x100, propylene glycol and glucose.41,42 Refractive index changes for example in layered samples, or naturally occurring RI heterogeneity in tissues impart large effects on light scattering and thus the signals detected.40 Van der Kolk et al.43 have shown using finite-difference time-domain simulations that even modest differences in the refractive index (within a range consistent with biological samples) can lead to signal enhancements of up to an order of magnitude, in addition to perturbations of a feature's perceived position by up to 1 μm.
Sample preparation
When elucidating chemical distributions through SRS imaging, careful consideration must be given to the sample preparation methods used prior to analysis. Ideally tissue should be fresh or fresh-frozen with minimum processing. Tissue or cells that have been chemically fixed and embedded may have an altered local biochemical profile. Similarly, the use of optimum cutting temperature or other chemical mounting or embedding media may alter the chemical composition and may interfere with the chemical species under investigation. Performing SRS on tissue embedded in paraffin, requires its prior removal. This process, however, can also strip lipids from the local microenvironment of the sample.When performing quantitative measurements, the potential impact of the chemical microenvironment should be considered. For example, the presence of species which may result in changes to hydrogen bonding that could impact signal intensity or peak position. Depending on the nature of the sample and the information sought, it can be beneficial to ensure optimum hydration by the inclusion of some water or phosphate buffered saline soaked filter paper in contact with the edge of the sample, as this will improve heat dissipation and permit a slightly higher laser power to be used without causing detectable damage.
Sample mounting and environmental control
Due to the short working distance of most high NA lenses used for SRS imaging, samples are commonly mounted on (or between two) thin glass coverslips. Environmental sample control can be challenging due to the short working distance requirements, since many commercially available microscopy environmental chambers will be too thick to image through. Alternative solutions include temperature-control of a larger enclosure that also incorporates the lenses (although this can negatively impact the optics).The thin glass coverslips can be prone to flexing during imaging, for example over time with temperature changes including the warmth of the incident beam, and from surface tension of lens immersion media, particularly when using more viscous oils. A practical solution is to clamp the coverslip(s) between a securing rigid frame to prevent axial movement drift. When mounting samples between glass coverslips, ensure the sample makes good contact with both slides to avoid creation of airgaps. This would introduce a change in the refractive index leading to signal losses and potentially spurious background contributions. Sample hydration can be an important factor in many applications. Consider the extreme case of two tissue sections, cut to the same thickness, but one is dehydrated, while the other remains fully hydrated: loss of water from the tissue will essentially reduce the thickness of the tissue slice, and consequently concentrate the remaining molecular species within the probe volume. Effective air-tight sealing of sections between coverslips, coupled with low laser power can help to minimise sample dehydration (which may also affect the refractive index and thermal properties). Maintaining good hydration also helps dissipate heat generated during imaging and reduced sample damage. Sample movement can cause quite complex data analysis challenges, for example trying to overlay images acquired consecutively at different wavenumbers. The issue of dehydration of tissue sections during imaging leading to shrinkage between frames is common. Similarly, many samples undergo motion for example droplets within emulsions or even live organisms such as cells and small creatures. In some cases, movement can be reduced by cooling the sample, but often this is not possible or feasible; since many lenses are water-immersion, for example, freezing the sample would also freeze the lens immersion water. Alternative solutions are to increase the image acquisition speed such that the movement is no longer on a comparable timescale or apply techniques such as image registration to create the composite image (see data section).
Chemical sensitivity and specificity
A common misconception is that SRS offers improved sensitivity of detection relative to RS. While it is true that the stimulated excitation increases scattering efficiencies relative to spontaneous Raman by up to 4 or 5 orders of magnitude, the technical limitations defining the limit of detection (LOD) are fundamentally very different: in RS the LOD is predominantly limited by the number of generated photons, whereas in SRS the LOD is predominantly limited by the shot noise of the laser. As is the case for RS, the limit of detection in SRS varies strongly with the Raman cross section for the specific functional group. For example, the limit of detection using SRS microscopy for retinol was measured at 50 μM, whereas for methanol it was 5 mM (targeting their vibrational modes at 1595 cm−1 and 2840 cm−1 respectively, corresponding to pump beam wavelengths of 909.6 nm and 817.1 nm respectively, with a 1064 nm Stokes beam).13There have been few direct comparisons between the sensitivity of SRS relative to RS, and of course the exact values will depend on laser power and acquisition parameters. However, there are values reported in the literature for both RS and SRS detection of methanol, for which the LOD was measured to be in the range of 0.99 mM to 4.2 mM with confocal RS,44 and 5 mM with SRS.13 However, it should be noted that these LODs were determined using different vibrational modes (C–O stretching at 1019 cm−1 for RS and CH2 stretching at 2840 cm−1 for SRS).
Raman provides a ‘chemical fingerprint’ through the characteristic vibrational modes of molecules. However, due to overlapping peaks and similarity in peak positions the specificity of Raman is not as good as some analytical methods such as mass spectrometry. This means that for highly complex samples, it may sometimes be impossible to separately identify the molecule of interest from other species in a complex sample matrix e.g. tissue or formulated products that may contain multiple different ingredients. One solution is to increase the chemical specificity of the target molecule using a functional group modification or tag. For example, deuteration is an effective way to shift a C–H bond vibrational signal into the ‘silent’ region of the Raman spectrum (1800–2800 cm−1; where typically there are few endogenous signals in most biological samples). The drawbacks include high cost of the reagents or requirement for chemical synthesis, and that the technique would no longer be non-invasive since samples would not be analysed in their native state.
There are also many researchers developing Raman tags which contain functional groups offering improved chemical specificity such as alkyne moieties which are detected in the biological silent region.45–47 These can also be designed to offer improvements in sensitivity as well as specificity. Although one of the main advantages of SRS is the offer of label-free imaging, such labels are typically less perturbing than large fluorescence probes and do not photobleach.
Where chemical specificity is a challenge, but tagging is not possible, advanced data methods such as spectral unmixing may provide a means of unravelling signals from overlapping spectral components (see the Data section for further information).
Sample damage
When selecting the laser power and image acquisition parameters, care must be taken to ensure the sample is not damaged during the analysis. The type of laser system used will also be a contributing factor, including the wavelengths, pulse length, and the average vs. peak power. Different chemical species and sample types will vary in their threshold for damaging laser exposure. Typical factors include the level of hydration or means of heat dissipation. Although there will always be a trade-off between signal to noise and sample damage, there are some parameters that can be optimised. For example, the total number of pixels in the image: reducing the pixel number in an image reduces the total exposure time (for example, a 256 × 256 image requires a quarter of the total laser exposure compared to a 512 × 512 image). Similarly, line or frame averaging accumulations can be applied instead of increasing the laser power to improve the signal to noise.Typical signs of sample damage include changes to the image over repeat image frames at the same wavenumber. In particular, the appearance of bright spots which increase in intensity and size over time. These signals typically remain when imaging at off-resonance wavenumbers. Additional care should be taken when the sample contains strong absorbers such as pigments or metallic particles: not only do these sample types absorb more power from the laser and therefore induce local damage, they also tend to exhibit high levels of parasitic signals in the resultant SRS images, owing to processes such as two photon absorption and photothermal lensing.48
However, these signals can be useful, for example allowing the visualisation of metal particle distribution (although without any chemical specificity). An example is presented in Fig. 7, which shows a section of rat skin that contains metal particles.
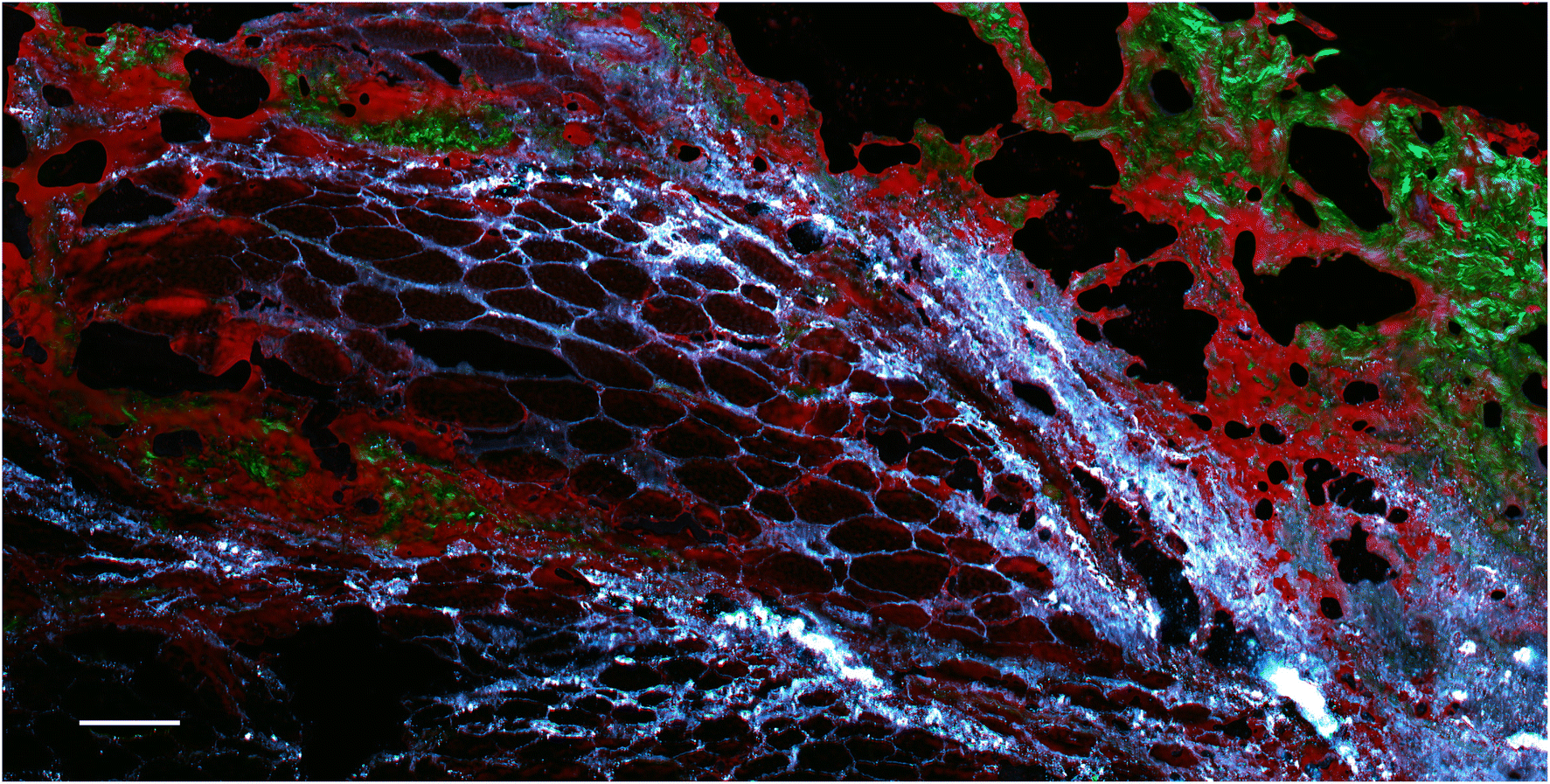 | ||
| Fig. 7 SRS and SHG microscopy composite image of a rat skin section containing metallic particles. SRS contrast for CH2 stretching at 2850 cm−1 is shown in red, and off-resonance signals at 2770 cm−1 are overlaid in cyan hot, revealing the distribution of the metal particles based on their strong signals due to absorption and photothermal lensing. Collagen (green) is visualised using SHG. The scale bar represents 100 μm. | ||
Data considerations
Data management and record keeping
Performing SRS microscopy involves many degrees of freedom in terms of instrumentation and image acquisition settings. Although commercial microscopes often save most of the image acquisition parameters in the metadata, these are not comprehensive. For example, the choice of condenser lens, and immersion media, details of the sample preparations and observations/measurements relating to the laser power, its fluctuation, spatial and temporal overlap of the beams, environmental conditions, and any modifications to the beam path should also be recorded in detail (refer to the checklist in the ESI†).Image processing
Several image processing steps may be required to generate the useful image outputs. Raw SRS images usually contain some spurious signal contributions, for example from two photon absorption or photothermal lensing. These are particularly significant in samples containing strong absorbers such as dyes, pigments and metal particles. If quantitative information is to be extracted from images, these spurious signals must first be removed. This can be achieved by subtracting the spurious signals captured in an ‘off-resonance’ image from the corresponding ‘on-resonance’ image in a pixel-by pixel manner, for example using the ‘Image calculator’ plugin in FIJI/ImageJ/ICY software.49 However, extreme caution should be exercised. Firstly, as discussed previously, the off-resonance wavelength should be as close as possible to the on-resonance image. However, this can prove challenging, especially in the fingerprint region of complex samples, where there may be large numbers of peaks clustered together. Secondly, if the on- and off-resonance images are acquired sequentially, there could be small translational drifts in the sample position, for example if the sample is motile, or is dehydrating on the timescale of the measurement. If such movement has occurred, image registration can be performed to correct for this. This requires some common features, either native to the sample or fiducial markers, between the images to be registered. Images from other modalities acquired simultaneously in separate channels such as TPEF or SHG can also provide information for more robust registration. Image registration may also need to be applied to create image composites comprising different wavenumber contrasts which have been acquired sequentially. Finally, images requiring direct comparison (such as a time series, or a treated vs. untreated control sample) should be displayed with the same image brightness and contrast settings.Generation of SRS spectra from hyperspectral image stacks
SRS spectra can be generated by acquiring SRS images sequentially at very small increments in wavelength of the pump beam (such as 0.1 nm, depending on the spectral resolution of the system) to generate a stack of images that span a wavenumber range of interest. Alternatively, hyperspectral datasets can be acquired more rapidly using spectral focusing, which involves a chirped femtosecond laser source.50 Once this stack of images has been acquired, regions of interest (ROIs) can be selected using image analysis software. From these ROIs, SRS spectra are generated by plotting integrated ROI SRS signal intensity against the wavelength of the pump beam or wavenumber difference between the pump and Stokes beam. Sometimes it can be helpful to generate a maximum intensity projection of all the images in the wavelength stack, to visualize all the features in one single image and ensure no important features have been missed. An example is displayed in Fig. 8.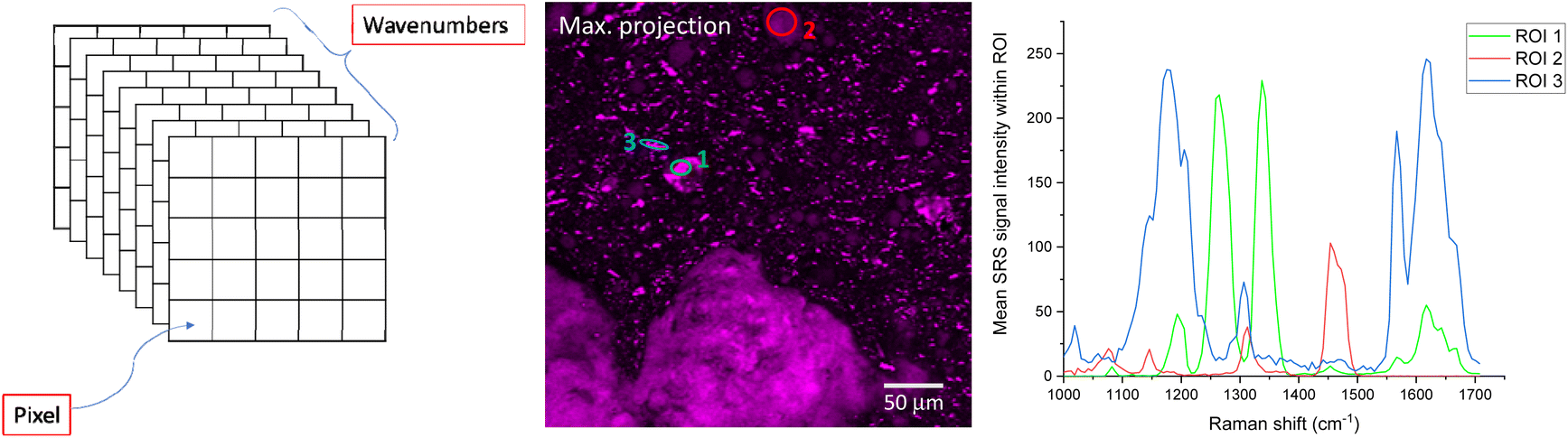 | ||
| Fig. 8 Hyperspectral SRS imaging schematic (left); maximum intensity projection of a hyperspectral SRS stack acquired by recording SRS images of the sample with 0.1 nm increments in wavelength of the pump beam (middle); resulting SRS spectra generated from the ROIs indicated (right). | ||
Care must be taken when generating SRS spectra from hyperspectral image stacks, as there are several potential pitfalls. If there is any movement of the sample during the acquisition, features of interest can move within the field of view throughout the stack, so these translations must be corrected for before selecting a ROI to plot the SRS intensity vs. wavenumber. Correcting each image that represents contrast at a single wavenumber can be difficult, particularly for complex samples where each image corresponds to different chemical components.
Hyperspectral data and multivariate data approaches
SRS generates spectra for which the Raman signals should be consistent with those acquired using RS. However, they can have different non-Raman signal contributions: in the case of RS, a fluorescence background may be present, whereas SRS images do not contain emitted fluorescence contributions, but may contain spurious signals from processes such as two-photon absorption, photothermal lensing or cross-phase modulation.In RS the fluorescence background typically is removed using a polynomial fitting method or other approaches such as small window moving average or wavelets such as Mexican hat (Ricker).51–53 Data pre-treatment include cosmic ray spike removal, denoising, normalisation, data centering and binning etc. It should be noted that this may have a significant effect on any multivariate analysis that may be performed subsequently.54
For SRS, when the sample exhibits no spurious signals, the minimum signal value can be subtracted for each wavenumber if the field of view contains a region without Raman signals, acting as a local control. When spurious signals such as absorption artefacts are present across multiple wavenumbers including off-resonance, they should first be removed. Care is required when spurious signals present themselves in only part of the spectral window, as this can be related to local photothermal damage during hyperspectral scanning or long laser exposures.
A few multivariate data analysis approaches for hyperspectral SRS imaging are briefly described which are also utilised for RS.55 Multivariate data approaches can be broadly separated into 2 categories: methods that don't require previous spectral knowledge (unsupervised), and methods that require some previous knowledge (supervised). Either approach may be appropriate depending on the nature of the data and the information sought. Unsupervised techniques include:
• Non-negative matrix factorization (NMF);56
• Principal component analysis (PCA);56
• Vertex component analysis (VCA);56,57
• N-finder;58
• Factorization into spectra and concentrations of chemical components (FSC3);59
• Spectral phasor analysis.60,61
The most commonly used supervised technique for chemometric purposes is multivariate curve resolution (MCR) which can retrieve the spectral profiles of the components of interest within the spectral volume, in other words resolve the spectral volume to determine the spectral components.62 NMF and MCR are based on similar concepts, the main difference being that NMF creates the components in a volumetric matrix without prior knowledge, whereas for MCR the volumetric matrix is resolved into pre-existing components.
These methods may be used with clustering methods such as K-means, agglomerative hierarchical cluster analysis.63 The specific technique selection is driven by the nature of the sample and the quality of the spectral information as well as the desired application. When the sample does not contain significant spurious signals and has not undergone physical changes/movements then hyperspectral methods can be applied directly.
Quantification
In some applications such as drug delivery studies it is desirable to convert the gray scale intensity values in SRS images into an absolute chemical concentration. This is non-trivial and requires very careful design of a range of calibration samples of known concentration. The samples should have consistent optical properties and thickness compared to the sample under investigation. By preparing samples that have been spiked with known concentrations (resulting in a homogeneous chemical distribution through the sample matrix), a traditional calibration curve can be generated by imaging them in turn with identical acquisition parameters and plotting the known chemical concentrations vs. their respective average SRS signal intensities, i.e. the average gray scale values for a defined region.Similarly, quantification of particles (number per unit area or volume) may be possible.64,65 However, this presents considerable challenges, especially for sub-spatial resolution-sized particles. Care must be taken when analysing particles in liquids which are likely to move during the measurement, in particular, those which may be affected by optical trapping.66 In addition, thermal and other potentially spurious signal contributions may be present.67
As described earlier, when acquiring 3D image stacks there is a loss of signal with depth into the sample due to scattering and absorption and depending on the nature of the sample, potentially other phenomena. Where quantitative information is sought, this attenuation must be corrected for. There are many publications that address optical scattering and correction methods;28,68 however, accurate correction depends on many sample-dependent factors and can be quite complex, especially for inhomogeneous sample types. One practical solution to validate the corrected result, is to compare a Z-stack of a 3D sample with the X–Y image of a physical cross-section of the sample, prepared for example using a microtome or cryostat. In this manner, all depths of the sample can be imaged within the sample optical plane and the signals compared to those in the 3D image stack (Fig. 9). In this way, a correction factor can be assessed and then applied to the 3D samples of similar properties.
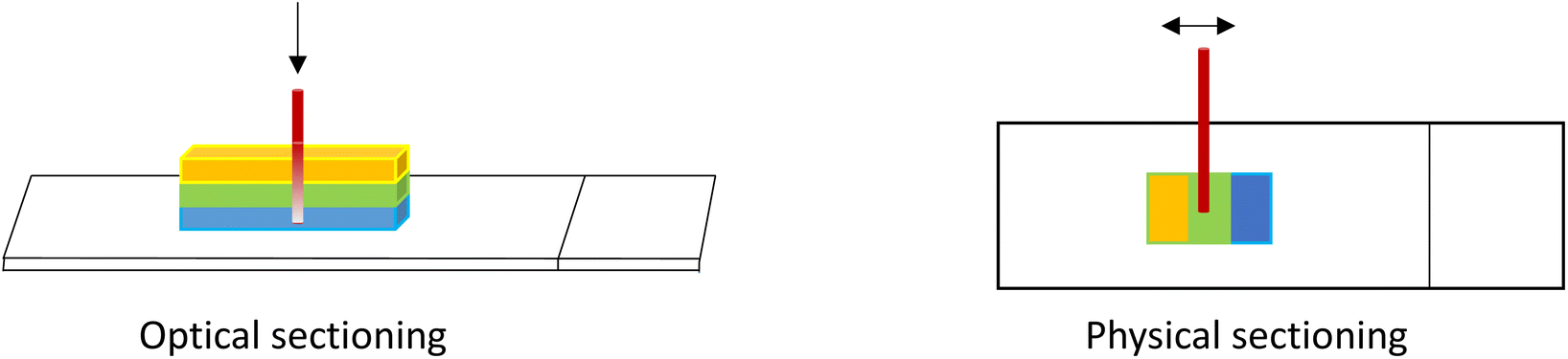 | ||
| Fig. 9 Illustration of ‘optical sectioning’ of a 3D sample which suffers from signal attenuation with depth (left); vs. a physical cross-section of a sample for which all depths can be analysed in the same optical plane (right). | ||
Conclusions and future perspectives
Coherent Raman techniques are finding utility in diverse fields, although the biomedical sector remains a core application. The first diagnostic clinical tools based on SRS are under development5,69 and commercially integrated solutions are now available,70,71 broadening the accessibility to a wider pool of researchers and medical professionals, compared to experimental optical set ups.While SRS becomes more widely employed, new challenges and technology developments are continuously being proposed;72 such as simultaneous stimulated Raman gain and loss detection for eliminating the need for a separate off-resonance image.73 Other important technologies include broadband CRS to enable faster data collection.74 Challenges yet to be solved include much needed improvements in sensitivity, faster detectors with lower noise, and more stable laser sources. Efforts towards super resolution SRS microscopy are also underway,75 employing a range of different mechanisms including saturated SRS,76 near-resonance enhanced visible SRS,77 and stimulated Raman excited fluorescence.78
As SRS transitions into a mainstream analytical and clinical method, standardisation of acquisition protocols and data formats allowing the transferability and comparability of data between different platforms is necessary. This will require the development of reference samples and procedures to ensure data quality and reproducibility, identified as the number one priority at an SRS workshop held at the National Physical Laboratory in November 2021. It will also facilitate greater data utility in studies where comparison to other imaging methods is required, such as mass spectrometry, fluorescence, super-resolution, and electron microscopy. As described earlier, one solution to the sensitivity limit is the development of Raman-active tags; by sacrificing the label-free nature of the approach, better specific sensitivities are accessible. For example, Tipping et al. reported up to two orders of magnitude improvement in Raman activity with increased conjugation of their biorthogonal tags.46
New technological and data treatment methods will facilitate imaging at a greater penetration depth.28 In addition, new approaches for chemometrics will be utilised to maximise the chemical information available, and in real-time. These factors, together with the fact that it provides complementary information to other multiphoton methods, makes CRS a promising tool for in vivo diagnostics as it can be easily corroborated with traditional methods but while providing additional molecular information through robust chemometrics approaches.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Dr Alex Shard and Dr Caterina Minelli for helpful discussions and revision of the manuscript. In addition, the authors would like to acknowledge and thank all the participants of the SRS microscopy workshop held at NPL during November 2021 for the detailed discussions on future standardisation needs. This work was funded by the UK Department of Business, Energy and Industrial Strategy through the projects NMS/ID74 and NMS/ST18 of the UK National Measurement System. This work was also supported by the Community for Analytical Measurement Science through a 2020 CAMS Fellowship Award to Natalie Belsey funded by the Analytical Chemistry Trust Fund.References
- J. B. Pawley, Handbook Of Biological Confocal Microscopy, Springer, Boston, MA, 3rd edn, 2006 Search PubMed.
- V. Marx, It's free imaging—label-free, that is, Nat. Methods, 2019, 16(12), 1209–1212 CrossRef CAS PubMed.
- F.-K. Lu, et al., Label-free DNA imaging in vivo with stimulated Raman scattering microscopy, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(37), 11624 CrossRef CAS PubMed.
- B. Figueroa, et al., Detecting and Quantifying Microscale Chemical Reactions in Pharmaceutical Tablets by Stimulated Raman Scattering Microscopy, Anal. Chem., 2019, 91(10), 6894–6901 CrossRef CAS PubMed.
- D. A. Orringer, et al., Rapid intraoperative histology of unprocessed surgical specimens via fibre-laser-based stimulated Raman scattering microscopy, Nat. Biomed. Eng., 2017, 1(2), 0027 CrossRef PubMed.
- J. Ao, et al., Rapid, 3D Chemical Profiling of Individual Atmospheric Aerosols with Stimulated Raman Scattering Microscopy, Small Methods, 2020, 4(2), 1900600 CrossRef CAS.
- L. M. Malard, et al., Studying 2D materials with advanced Raman spectroscopy: CARS, SRS and TERS, Phys. Chem. Chem. Phys., 2021, 23(41), 23428–23444 RSC.
- C. V. Raman and K. S. Krishnan, A New Type of Secondary Radiation, Nature, 1928, 121(3048), 501–502 CrossRef CAS.
- R. L. McCreery, Raman Spectroscopy for Chemical Analysis, John Wiley & Sons, Inc, 2000 Search PubMed.
- S. Fornasaro, et al., Surface Enhanced Raman Spectroscopy for Quantitative Analysis: Results of a Large-Scale European Multi-Instrument Interlaboratory Study, Anal. Chem., 2020, 92(5), 4053–4064 CrossRef CAS PubMed.
- W. Min, et al., Coherent Nonlinear Optical Imaging: Beyond Fluorescence Microscopy, Annu. Rev. Phys. Chem., 2011, 62(1), 507–530 CrossRef CAS PubMed.
- D. Zhang, et al., Fast Vibrational Imaging of Single Cells and Tissues by Stimulated Raman Scattering Microscopy, Acc. Chem. Res., 2014, 47(8), 2282–2290 CrossRef CAS PubMed.
- C. W. Freudiger, et al., Label-Free Biomedical Imaging with High Sensitivity by Stimulated Raman Scattering Microscopy, Science, 2008, 322(5909), 1857–1861 CrossRef CAS PubMed.
- W. J. Tipping, et al., Stimulated Raman scattering microscopy: an emerging tool for drug discovery, Chem. Soc. Rev., 2016, 45(8), 2075–2089 RSC.
- M. N. Slipchenko, et al., Vibrational imaging of tablets by epi-detected stimulated Raman scattering microscopy, Analyst, 2010, 135(10), 2613–2619 RSC.
- N. A. Belsey, et al., Evaluation of drug delivery to intact and porated skin by coherent Raman scattering and fluorescence microscopies, J. Controlled Release, 2014, 174, 37–42 CrossRef CAS PubMed.
- J. Ling, et al., Vibrational Imaging and Quantification of Two-Dimensional Hexagonal Boron Nitride with Stimulated Raman Scattering, ACS Nano, 2019, 13(12), 14033–14040 CrossRef CAS PubMed.
- H. J. Lee and J.-X. Cheng, Imaging chemistry inside living cells by stimulated Raman scattering microscopy, Methods, 2017, 128, 119–128 CrossRef CAS PubMed.
- D. Tsikritsis, et al., Label-free identification and characterization of living human primary and secondary tumour cells, Analyst, 2015, 140(15), 5162–5168 RSC.
- R. Mittal, et al., Evaluation of stimulated raman scattering microscopy for identifying squamous cell carcinoma in human skin, Lasers Surg. Med., 2013, 45(8), 496–502 Search PubMed.
- S. Guo, et al., Comparability of Raman Spectroscopic Configurations: A Large Scale Cross-Laboratory Study, Anal. Chem., 2020, 92(24), 15745–15756 CrossRef CAS PubMed.
- Reality check on reproducibility, Nature, 2016, 533, 437 Search PubMed.
- M. Sené, I. Gilmore and J.-T. Janssen, Metrology is key to reproducing results, Nature, 2017, 547(7664), 397–399 CrossRef PubMed.
- J-X. Cheng and X. S. Xie, Coherent Raman Scattering Microscopy, CRC Press, Boca Raton, 2012 Search PubMed.
- J-X Cheng, W. Min, Y. Ozeki and D. Polli, Stimulated Raman Scattering Microscopy Techniques and Applications, Elsevier, 2022 Search PubMed.
- B. R. Masters and P. T. C. So, Handbook of Biological Nonlinear Optical Microscopy, Oxford University Press Inc, 2008 Search PubMed.
- S. Yue, M. N. Slipchenko and J. X. Cheng, Multimodal nonlinear optical microscopy, Laser Photonics Rev., 2011, 5(4), 496–512 CrossRef PubMed.
- A. H. Hill, B. Manifold and D. Fu, Tissue imaging depth limit of stimulated Raman scattering microscopy, Biomed. Opt. Express, 2020, 11(2), 762–774 CrossRef CAS PubMed.
- X. Audier, et al., Noise in stimulated Raman scattering measurement: From basics to practice, APL Photonics, 2020, 5(1), 011101 CrossRef CAS.
- Thorlabs. 22-March-2022; Available from: https://www.thorlabs.com/thorproduct.cfm?partnumber=FDS1010.
- J.-X. Cheng, et al., Stimulated Raman scattering microscopy techniques and applications, Elsevier, 2022 Search PubMed.
- S. J. Choquette, et al., Relative Intensity Correction of Raman Spectrometers: NIST SRMs 2241 through 2243 for 785 nm, 532 nm, and 488 nm/514.5 nm Excitation, Appl. Spectrosc., 2007, 61(2), 117–129 CrossRef CAS PubMed.
- ASTM, E2911–13 Standard Guide for Relative Intensity Correction of Raman Spectrometers, ASTM, 2013 Search PubMed.
- Standard Guide for Raman Shift Standards for Spectrometer Calibration, ASTM, 1996 (2014) Search PubMed.
- L. Zada, et al., Stimulated Raman scattering simulation for imaging optimization, J. Eur. Opt. Soc.-Rapid Publications, 2021, 17(1), 10 CrossRef.
- M. J. B. Moester, et al., Stimulated Raman scattering microscopy with long wavelengths for improved imaging depth, J. Raman Spectrosc., 2019, 50(9), 1321–1328 CrossRef CAS.
- K. Ekvall, et al., Cross phase modulation artifact in liquid phase transient absorption spectroscopy, J. Appl. Phys., 2000, 87(5), 2340–2352 CrossRef CAS.
- P. Berto, E. R. Andresen and H. Rigneault, Background-Free Stimulated Raman Spectroscopy and Microscopy, Phys. Rev. Lett., 2014, 112(5), 053905 CrossRef PubMed.
- A.-G. Alba, et al., Biological imaging with coherent Raman scattering microscopy: a tutorial, J. Biomed. Opt., 2014, 19(7), 1–14 Search PubMed.
- N. J. Everall, Confocal Raman Microscopy: Why the Depth Resolution and Spatial Accuracy Can Be Much Worse Than You Think, Appl. Spectrosc., 2000, 54(10), 1515–1520 CrossRef CAS.
- M. Wei, et al., Volumetric chemical imaging by clearing-enhanced stimulated Raman scattering microscopy, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(14), 6608 CrossRef CAS PubMed.
- J. Li, et al., Volumetric stimulated Raman scattering imaging of cleared tissues towards three-dimensional chemical histopathology, Biomed. Opt. Express, 2019, 10(8), 4329–4339 CrossRef CAS PubMed.
- J. van der Kolk, A. C. Lesina and L. Ramunno, Effects of refractive index mismatch on SRS and CARS microscopy, Opt. Express, 2016, 24(22), 25752–25766 CrossRef PubMed.
- B. Guven, et al., A Novel Method for Detection of Ethanol and Methanol in Distilled Alcoholic Beverages Using Raman Spectroscopy, 2012 Search PubMed.
- L. Wei, et al., Live-cell imaging of alkyne-tagged small biomolecules by stimulated Raman scattering, Nat. Methods, 2014, 11(4), 410–412 CrossRef CAS PubMed.
- W. J. Tipping, et al., Imaging drug uptake by bioorthogonal stimulated Raman scattering microscopy, Chem. Sci., 2017, 8(8), 5606–5615 RSC.
- S. Vanden-Hehir, et al., Alkyne-Tagged PLGA Allows Direct Visualization of Nanoparticles In Vitro and Ex Vivo by Stimulated Raman Scattering Microscopy, Biomacromolecules, 2019, 20(10), 4008–4014 CrossRef CAS PubMed.
- S. Berciaud, et al., Photothermal heterodyne imaging of individual metallic nanoparticles: Theory versus experiment, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(4), 045424 CrossRef.
- Image Calculator plugin. 26/12/2021; Available from: https://imagej.nih.gov/ij/docs/menus/process.html#calculator.
- B. Manifold, B. Figueroa and D. Fu, Hyperspectral SRS imaging via spectral focusing, in Stimulated Raman Scattering Microscopy Techniques and Applications, ed. J.-X. Cheng, et al., Elsevier, 2022, pp. 69–79 Search PubMed.
- H. G. Schulze, et al., A Small-Window Moving Average-Based Fully Automated Baseline Estimation Method for Raman Spectra, Appl. Spectrosc., 2012, 66(7), 757–764 CrossRef CAS PubMed.
- Z.-M. Zhang, et al., An intelligent background-correction algorithm for highly fluorescent samples in Raman spectroscopy, J. Raman Spectrosc., 2010, 41(6), 659–669 CrossRef CAS.
- G. Schulze, et al., Investigation of Selected Baseline Removal Techniques as Candidates for Automated Implementation, Appl. Spectrosc., 2005, 59(5), 545–574 CrossRef CAS PubMed.
- R. Gautam, et al., Review of multidimensional data processing approaches for Raman and infrared spectroscopy, EPJ Tech. Instrum., 2015, 2(1), 8 CrossRef.
- I. C. Ragupathy, V. Schweikhard and A. Zumbusch, Multivariate analysis of hyperspectral stimulated Raman scattering microscopy images, J. Raman Spectrosc., 2021, 52(9), 1630–1642 CrossRef.
- B. Prats-Mateu, et al., Multivariate unmixing approaches on Raman images of plant cell walls: new insights or overinterpretation of results?, Plant Methods, 2018, 14(1), 52 CrossRef PubMed.
- A. Alfonso-García, et al., A machine learning framework to analyze hyperspectral stimulated Raman scattering microscopy images of expressed human meibum, J. Raman Spectrosc., 2017, 48(6), 803–812 CrossRef PubMed.
- E. W. Michael, N-FINDR: an algorithm for fast autonomous spectral end-member determination in hyperspectral data, in Proc. SPIE, 1999 Search PubMed.
- F. Masia, et al., Hyperspectral image analysis for CARS, SRS, and Raman data, J. Raman Spectrosc., 2015, 46(8), 727–734 CrossRef CAS PubMed.
- F. Fereidouni, A. N. Bader and H. C. Gerritsen, Spectral phasor analysis allows rapid and reliable unmixing of fluorescence microscopy spectral images, Opt. Express, 2012, 20(12), 12729–12741 CrossRef CAS PubMed.
- D. Fu and X. S. Xie, Reliable Cell Segmentation Based on Spectral Phasor Analysis of Hyperspectral Stimulated Raman Scattering Imaging Data, Anal. Chem., 2014, 86(9), 4115–4119 CrossRef CAS PubMed.
- D. Zhang, et al., Quantitative Vibrational Imaging by Hyperspectral Stimulated Raman Scattering Microscopy and Multivariate Curve Resolution Analysis, Anal. Chem., 2013, 85(1), 98–106 CrossRef CAS PubMed.
- T. Bocklitz, et al., Invited Article: Comparison of hyperspectral coherent Raman scattering microscopies for biomedical applications, APL Photonics, 2018, 3(9), 092404 CrossRef.
- L. Zada, et al., Fast microplastics identification with stimulated Raman scattering microscopy, J. Raman Spectrosc., 2018, 49(7), 1136–1144 CrossRef CAS.
- C. Zhang, et al., Stimulated Raman scattering flow cytometry for label-free single-particle analysis, Optica, 2017, 4(1), 103–109 CrossRef CAS.
- K. C. Neuman and S. M. Block, Optical trapping, Rev. Sci. Instrum., 2004, 75(9), 2787–2809 CrossRef CAS PubMed.
- A. Lombardini, et al., Background-suppressed SRS fingerprint imaging with a fully integrated system using a single optical parametric oscillator, Opt. Express, 2020, 28(10), 14490–14502 CrossRef CAS PubMed.
- F. Helmchen and W. Denk, Deep tissue two-photon microscopy, Nat. Methods, 2005, 2(12), 932–940 CrossRef CAS PubMed.
- Invenio. 1st March 2022; Available from: https://www.invenio-imaging.com/.
- Microsystems, L. 1st March 2022; Available from: https://www.leica-microsystems.com/products/confocal-microscopes/p/stellaris-8-crs/.
- Tech, L. 1st March 2022; Available from: https://lightcore.tech/.
- C. Zhang and J.-X. Cheng, Perspective: Coherent Raman scattering microscopy, the future is bright, APL Photonics, 2018, 3(9), 090901 CrossRef.
- S. Heuke, et al., Simultaneous stimulated Raman gain and loss detection (SRGAL), Opt. Express, 2020, 28(20), 29619–29630 CrossRef CAS PubMed.
- A. De la Cadena, Novel approaches to broadband coherent Raman imaging, in, Proc. SPIE, 2021 Search PubMed.
- C. T. Graefe, et al., Far-Field Super-Resolution Vibrational Spectroscopy, Anal. Chem., 2019, 91(14), 8723–8731 CrossRef CAS PubMed.
- L. Gong, et al., Saturated Stimulated-Raman-Scattering Microscopy for Far-Field Superresolution Vibrational Imaging, Phys. Rev. Appl., 2019, 11(3), 034041 CrossRef CAS.
- Y. Bi, et al., Near-resonance enhanced label-free stimulated Raman scattering microscopy with spatial resolution near 130 nm, Light: Sci. Appl., 2018, 7(1), 81 CrossRef PubMed.
- H. Xiong, et al., Super-resolution vibrational microscopy by stimulated Raman excited fluorescence, Light: Sci. Appl., 2021, 10(1), 87 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an00817c |
| This journal is © The Royal Society of Chemistry 2022 |