
Sequence-regulated vinyl polymers via iterative atom transfer radical additions and acyclic diene metathesis polymerization†
Masato
Miyajima
a,
Kotaro
Satoh
 ab and
Masami
Kamigaito
*a
ab and
Masami
Kamigaito
*a
aDepartment of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan
bDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1-H120 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: kamigait@chembio.nagoya-u.ac.jp
First published on 7th December 2020
Abstract
A new strategy for the synthesis of sequence-regulated vinyl polymers based on a combination of iterative atom transfer radical additions (ATRAs) of vinyl monomers and acyclic diene metathesis (ADMET) polymerization was developed. A molecularly defined vinyl monomer sequence at the single monomer unit level was constructed in telechelic diene sequence-regulated oligomers by iterative metal-catalyzed ATRAs of vinyl monomers to dichloride followed by allylation of both carbon–chloride terminals. ADMET polymerization of the obtained telechelic diene oligomer with a symmetrical monomer sequence resulted in polymers with an embedded vinyl monomer sequence that was repeatedly connected by an olefin formed by olefin metathesis polymerization. The subsequent hydrogenation of the main-chain olefins led to polymers that mimicked sequence-regulated vinyl polymer structures consisting of saturated C–C main chains. In particular, this study enabled the synthesis of sequence-regulated vinyl polymer mimics that have perfect and periodic styrene–acrylate–styrene–ethylene–methylene sequences, no statistical distributions of the comonomer compositions and sequences in polymer chains, relatively high molecular weights, and only carbon–carbon single bonds in the main chain. The thermal properties of the sequence-regulated vinyl polymer mimics were evaluated and compared with those of hypothetical random copolymers of styrene, acrylate, and ethylene units with the same comonomer contents.
Introduction
A molecularly defined structure with controlled monomer sequences is one of the ultimate targets for synthetic polymers. An example of such desirable structures is natural macromolecules, for example, proteins, which have highly controlled monomer sequences that give them special properties and functions. Many approaches to monomer sequence control in various synthetic polymers have been proposed and investigated.1–10For vinyl polymers, one of the major families of synthetic polymers, precision monomer sequence control, particularly at the single monomer unit level, is extremely challenging. Vinyl polymers are usually prepared by chain-growth polymerizations,11 in which the propagation of comonomers statistically occurs according to the monomer reactivity ratios. Statistically governed copolymerization inevitably results in nonhomogeneous distributions of comonomer units among the resulting polymer chains. The comonomer sequence and distribution differ from chain to chain and are molecularly undefined and uncontrolled. Although there are several pairs of monomers, which undergo alternating radical copolymerization with a high probability of cross-propagation, a sequence error is statistically inevitable.12 Furthermore, longer monomer sequence control of more than three monomer units is obviously harder to achieve.13 Therefore, synthesizing sequence-regulated vinyl polymers with a long monomer sequence and high sequence perfection at the single monomer unit level has been one of the most challenging feats in polymer synthesis.
We have proposed a novel approach to synthesize sequence-regulated vinyl polymers based on iterative step-by-step single unit monomer additions of vinyl monomers, followed by an appropriate polymerization of the obtained sequence-regulated oligomers.14–18 Although other researchers have used single unit monomer addition or insertion to control the sequence of vinyl monomers, there have been limitations in the sequence lengths due to the use of iterative approaches, and separating discrete oligomers becomes difficult as the sequence length increases.19–32 We prepared a series of sequence-regulated oligomers by iterative metal-catalyzed atom transfer radical additions (ATRAs) of vinyl monomers to a dichloride and then put unconjugated vinyl groups by selective allylation at one chain end while keeping another chloride intact at the other chain end. The obtained ab-type heterotelechelic sequence-regulated oligomers, which have unconjugated vinyl (a) and halide (b) groups at their terminals, were then polymerized via metal-catalyzed step-growth radical polymerization to produce polymers with embedded vinyl monomer sequences linked by an additional monomer unit, which was equivalent to the vinyl chloride unit and was formed via metal-catalyzed radical addition between the unconjugated vinyl group and chloride. Step-growth polymerization also works for aa- and bb-type homotelechelic sequence-regulated oligomers, which possess two vinyl and two halide groups at both terminals, respectively.15 Although the vinyl monomer sequences in the resulting polymers can vary from 4 to 11 vinyl monomer unit lengths in which various vinyl monomers, including styrenes, acrylates, acrylamides, acrylonitrile, vinyl chloride, and ethylene, can be embedded, the molecular weights are limited to approximately 2000–3000 in number-average molecular weight (Mn) due to inevitable side reactions and slow step-growth radical polymerizations. A more efficient linking reaction for the sequence-regulated oligomers is required for further developments of this approach based on iterative single unit vinyl monomer additions.
Acyclic diene metathesis (ADMET) polymerization is one of the most efficient step-growth polymerizations of linear telechelic diene monomers.33 Polymerization, especially using ruthenium catalysts, is highly effective for various linear telechelic dienes, resulting in relatively high molecular weight polymers, even though the monomers contain polar functional groups. To place periodic functionality in polymer chains, Wagener and other groups used ADMET polymerization for linear telechelic dienes with various lengths and functional groups, such as alkyl, alcohol, ester, amide, and halogen, in the side chains. They then hydrogenated the resulting olefins in the main chain produced by the olefin metathesis reaction to synthesize a series of linear polyethylene mimics with periodic functional groups.34–38 However, no vinyl monomers have been used as starting materials for telechelic dienes, and no vinyl monomer sequences have been embedded in the resulting polymers.
In this study, we investigated the combination of iterative single unit monomer additions via ATRAs and ADMET polymerization of sequence-regulated telechelic diene oligomers to produce sequence-regulated vinyl polymers with a long monomer sequence, high sequence perfection, relatively high molecular weights, and only C–C main chains (Scheme 1). We thus prepared sequence-regulated telechelic diene oligomers possessing a styrene–acrylate–styrene sequence via iterative ATRAs followed by allylation at both chain ends. We then performed ADMET polymerization of the synthesized telechelic dienes and subsequent hydrogenation of the resulting internal olefins to synthesize sequence-regulated vinyl polymer mimics consisting of periodic styrene–acrylate–styrene–ethylene–methylene sequences. Although we quite recently reported ring-opening polymerization of a sequence-regulated cyclic dimer of the telechelic diene oligomers derived from ring-closing metathesis,39 in this study, we report ADMET polymerization for the first time.
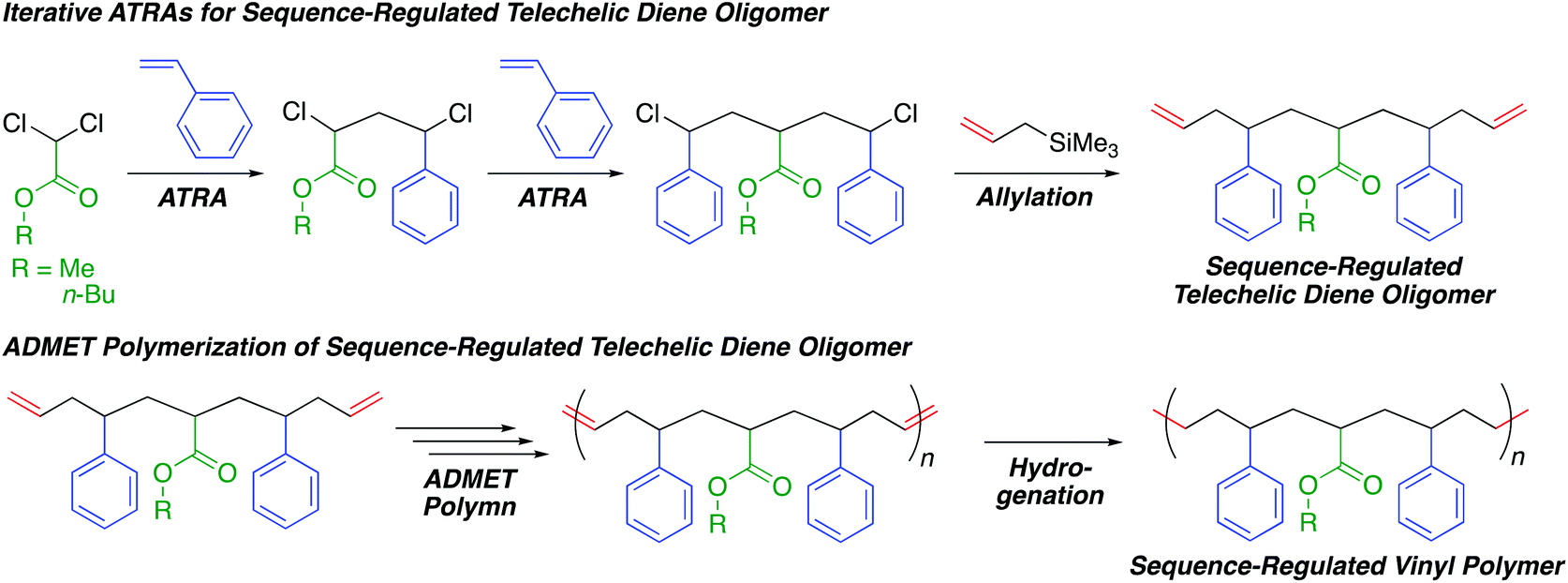 | ||
| Scheme 1 Sequence-regulated vinyl polymers via iterative atom transfer radical additions and acyclic diene metathesis polymerization. | ||
Results and discussion
Synthesis of sequence-regulated telechelic diene oligomers via iterative ATRAs and allylation
We first conducted a single ATRA between styrene and methyl or n-butyl dichloroacetate (ClAMCl or ClABCl), which later became the corresponding alkyl acrylate unit, using CuCl and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) in excess of dichloroacetate over styrene to selectively obtain a dimer (ClSAMCl or ClSABCl) by reducing styrene polymerization. After dimer purification through a silica-gel column and distillation, the second ATRA between styrene and the dimer was performed in the presence of CuCl and another ligand, N,N,N′,N′′,N′′′,N′′′-hexamethyltriethylenetetramine (HMTETA), which is less active than PMDETA but is sufficiently active to a more activated acrylate unit C–Cl bond.40 This ligand choice could contribute to suppressing the unfavorable activation of the less active C–Cl bond of the styrene unit and the more favorable selective formation of the targeted trimer (ClSAMSCl or ClSABSCl) that has symmetrical sequences of styrene–acrylate–styrene units. In addition, the dimer was used in excess of styrene to decrease styrene polymerization, although excess dimer remained after the reaction.The terminal C–Cl bonds of the obtained products were then allylated by allyltrimethylsilane in the presence of TiCl4 to introduce olefins at the terminals. The obtained sequence-regulated telechelic diene oligomers possessing styrene–methyl or butyl acrylate–styrene sequences between the terminal olefins (OSAMSO (M1) or OSABSO (M2), respectively) were purified through a silica-gel column to remove the remaining dimer after selective amidation with ethanolamine to change the polarity. The successful synthesis and purification of M1 and M2 were confirmed by 1H and 13C NMR (Fig. S1 and S2†). The monomer sequences were indeed perfect; however, they were mixtures of diastereomers and enantiomers,39 as expected from the stereo-uncontrolled ATRAs and allylations.
ADMET polymerization of sequence-regulated telechelic diene oligomers
ADMET M1 and M2 polymerization was investigated using a second-generation Grubbs ruthenium catalyst (G2) in bulk at 50 °C under reduced pressure (0.03 MPa) to remove the ethylene generated during the metathesis reaction. Fig. 1A shows time vs. conversion of the terminal olefin plots. Although polymerization proceeded in both cases, the reaction seemed to stop at approximately 70% for M1, which had a shorter alkyl ester group than that of M2. As the polymerization proceeded, particularly in bulk, the viscosity significantly increased, lowering the mobility of the polymer chain and slowing the step-growth ADMET polymerization between the olefin terminals of the resulting polymer chains. As a result, the Mn of the resulting products was low and limited to 1100 for M1 in bulk. In contrast, M2, which had a longer alkyl ester group than that of M1, was polymerized with a higher conversion (93%) due to its higher mobility, resulting in a polymer with an Mn higher than 2000. The Mn increased especially at a later M2 polymerization stage, indicating that the step-growth polymerization proceeded.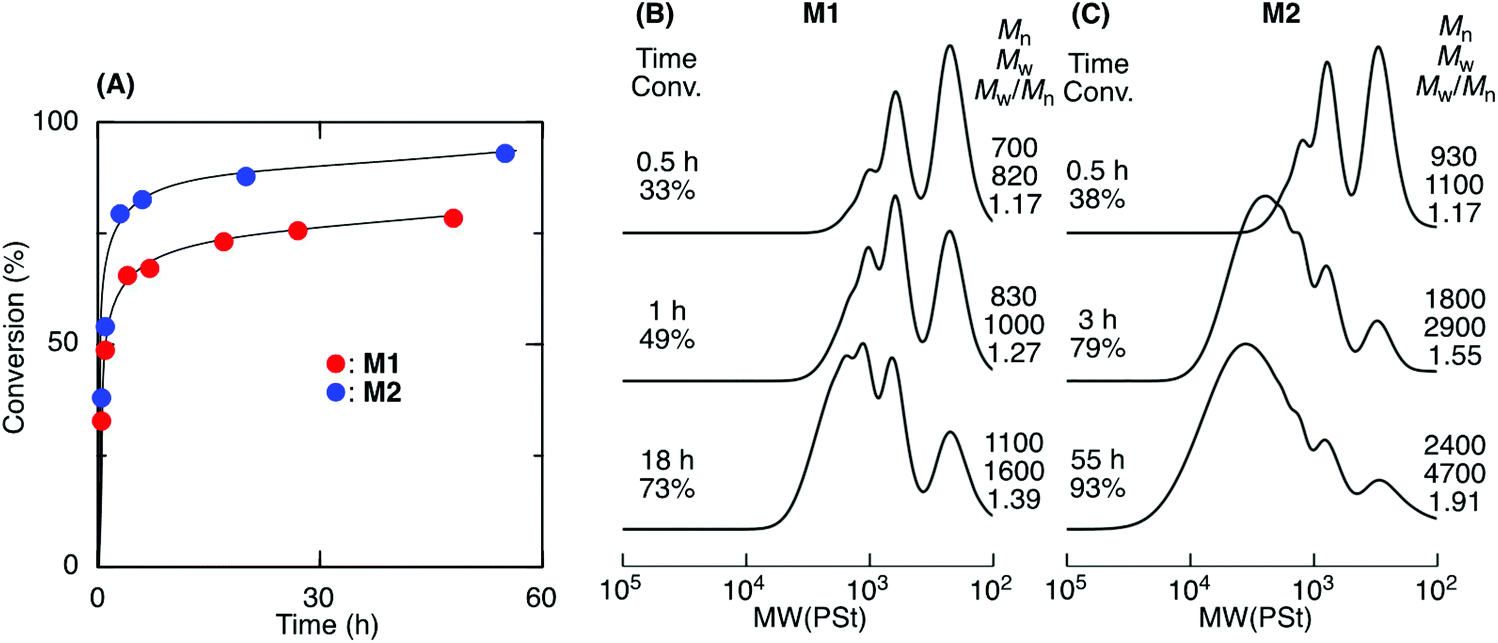 | ||
| Fig. 1 ADMET polymerization of sequence-regulated telechelic diene oligomers (M1 and M2) ([M]0/[G2]0 = 400) in bulk at 50 °C. Time–conversion curves (A) and SEC curves of the products obtained from M1 (B) and M2 (C). | ||
Tetralin (1,2,3,4-tetrahydronaphthalene), which has a relatively high boiling point (208 °C), was used as a solvent to increase the polymer chain mobility in the ADMET M1 and M2 polymerization with G2 under reduced pressure (0.03 MPa) at 50 °C. Both M1 and M2 were consumed in the solution, resulting in polymers with a higher Mn close to 3000 (Fig. 2). The Mn of the products progressively increased particularly at a later stage of the polymerization and was close to the calculated line for the step-growth polymerization based on the assumption that the Mn or DPn (number-average degree of polymerization) increased in inverse proportion to 1 − p, where p means the consumption of the olefin terminal or the extent of reaction. However, a small peak was observed at around 500–800 in the SEC curves that was attributed to the M1 and M2 cyclic dimer formed via dimerization and the subsequent ring-closing metathesis reaction. Selective cyclic dimer formation has been reported during metathesis, particularly at high dilution.39 These results indicated that the sequence-regulated telechelic diene oligomers possessing styrene–acrylate–styrene sequences underwent ADMET polymerization efficiently in the solution under reduced pressure.
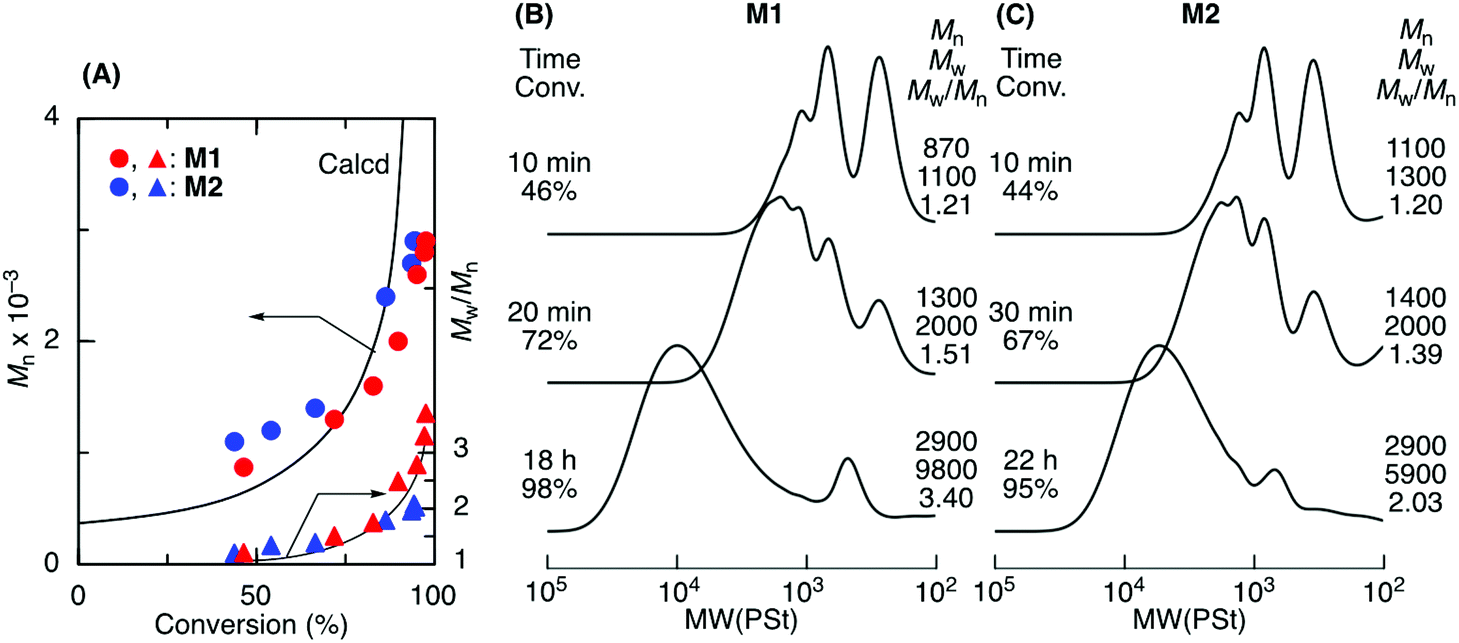 | ||
| Fig. 2 ADMET polymerization of sequence-regulated telechelic diene oligomers (M1 and M2) ([M]0/[G2]0 = 1500/3.75 mM) in tetralin at 50 °C. Mn and Mw/Mnvs. monomer conversion plots (A) and SEC curves the products obtained from M1 (B) and M2 (C). | ||
The obtained polymers were analyzed by 1H and 13C NMR after polymer purification by preparative SEC to remove low-molecular-weight oligomers, including the cyclic dimer (Fig. S3†). When the 1H NMR spectra of the obtained polymers were compared with those of the monomers, the terminal olefins significantly decreased and internal olefins appeared, indicating that the olefin metathesis reaction occurred between the terminal olefins of the telechelic dienes, converting them into internal olefins in the polymer main chains (Fig. 3). However, there were almost no significant changes in the chemical shifts and shapes of the other signals, including main-chain alkyl, side-chain alkyl ester, and phenyl protons, except for a slight broadening of these peaks. These results indicated that the ruthenium catalyst successfully induced ADMET polymerization of the sequence-regulated telechelic diene oligomers without significant side reactions, such as olefin isomerization. In addition, the peak intensity ratios of the main-chain alkyl and side-chain ester and phenyl protons agreed well with the calculated values for the polymers with one acrylate and two styrene units. The 13C NMR spectral changes also showed disappearance of the terminal olefins and appearance of the internal olefins (Figs. S4 and S5†). The methylene carbons adjacent to the internal olefins were observed at two different chemical shifts due to the cis and trans structures.41 The intensity ratios revealed that the internal olefins were rich in the trans structure (trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis = 81:19).
:cis = 81:19).
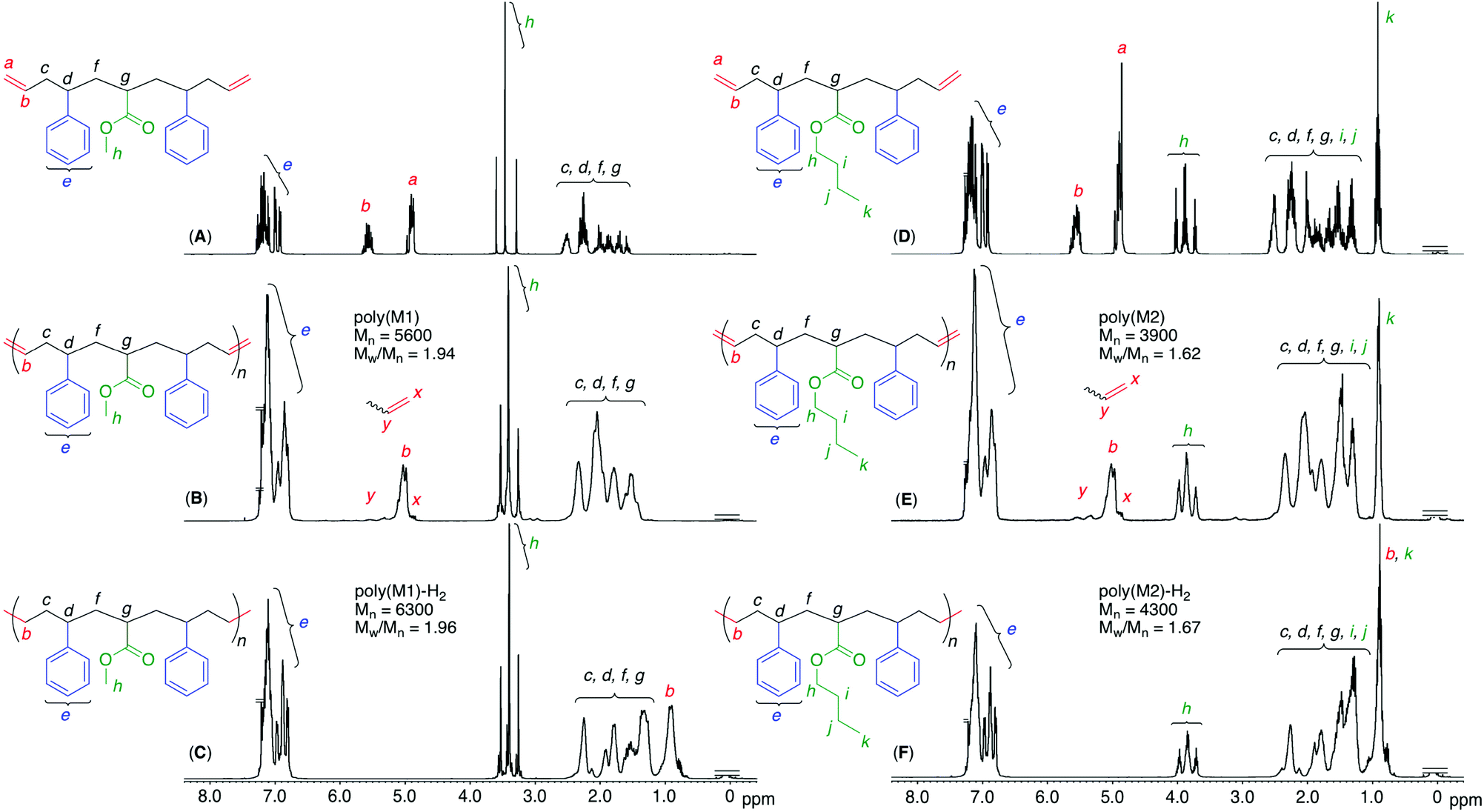 | ||
| Fig. 3 1H NMR spectra (CDCl3, 25 or 55 °C) of sequence-regulated telechelic diene oligomers (M1 and M2) (A and D), sequence-regulated polymers obtained via ADMET polymerization of M1 and M2 (B and E), and the hydrogenated polymers (C and F). | ||
The MALDI-TOF-MS spectrum of poly(M2) consisted of a main series of peaks with almost identical intervals (376.5 Da) equal to the molar mass of the repeating sequence-regulated oligomer unit (Fig. 4). In addition, the peak molar masses were almost the same as those for the sequence-regulated polymers with telechelic olefin terminals produced by M2 ADMET polymerization. A similar result was also obtained for poly(M1) with a methyl ester substituent (Fig. S7†).
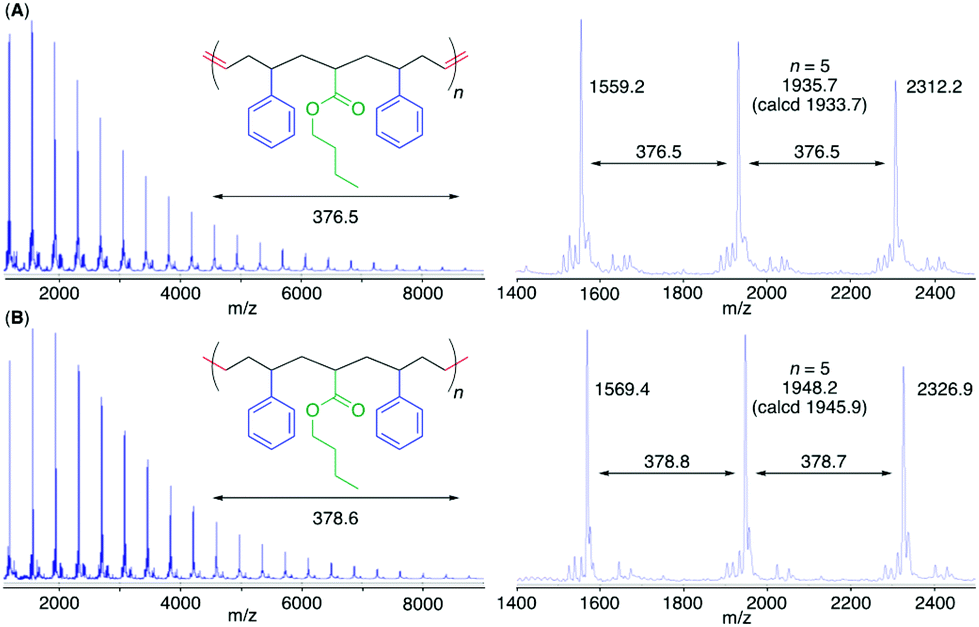 | ||
| Fig. 4 MALDI-TOF-MS spectra of poly(M2) (A) and the hydrogenated poly(M2) (poly(M2)-H2) (B). | ||
As presented in Table S1,† we also investigated various conditions, including different ruthenium catalysts for M1 ADMET polymerization. Although higher molecular weight polymers were obtained by increasing the temperature or extending the reaction time, olefin isomerization became more visible.
These results indicated that sequence-regulated telechelic diene oligomers with the embedded styrene–acrylate–styrene sequence were successfully polymerized by ADMET polymerization, resulting in well-defined sequence-regulated polymers.
Hydrogenation of sequence-regulated polymers to mimic vinyl polymer structures
To further mimic vinyl polymer structures, which generally consist of only C–C single bonds in the main chain, the internal olefins in poly(M1) and poly(M2) were hydrogenated using p-toluenesulfonyl hydrazide in o-xylene at 135 °C.39 The shapes of the polymer SEC curves were almost unchanged after the reaction (Fig. S6†). Although Mn(SEC) based on polystyrene calibration slightly increased, Mn(MALLS) was almost unchanged. These results indicated that no polymer chain scission occurred during the post-polymerization reaction.In the 1H NMR spectra of the obtained products, the complete disappearance of the internal olefin peaks and slight changes in the alkyl proton regions were observed (Fig. 3), indicating quantitative hydrogenation and formation of the fully saturated main chains without any structural changes in the side chains of the styrene and acrylate units. The 13C NMR spectra of the products also showed the disappearance of both olefinic and allylic carbons (Fig. S4 and S5†), also indicating successful hydrogenation. Furthermore, in the MALDI-TOF-MS spectra of the hydrogenated products (poly(M2)-H2), the main series of peaks was separated by 378.6 (Fig. 4), which agreed with the theoretical value of the hydrogenated repeating units, namely, styrene–acrylate–styrene plus ethylene–methylene units, which were linked by single C–C bonds. In addition, there were no statistical distributions of these vinyl monomer units in any of the polymer chains. Specifically, the vinyl polymer chains were homogeneous in terms of not only their monomer compositions but also their monomer sequences. Such vinyl copolymers are generally difficult to prepare in any chain-growth copolymerizations due to the inevitable statistical distributions of comonomers for each polymer chain.
Thermal properties of sequence-regulated vinyl polymers
The thermal properties of a series of the obtained polymers were evaluated before and after hydrogenation using DSC and TGA (Fig. 5A). The glass transition temperatures (Tg) of poly(M1) (Mn = 5600, Mw/Mn = 1.94) and poly(M2) (Mn = 3600, Mw/Mn = 1.62) before hydrogenation were 35 and 0 °C, respectively. The lower Tg of poly(M2) than of that of poly(M1) could be attributed to the longer alkyl side chain of n-butyl acrylate units, which was similar to the relationship between poly(methyl acrylate) and poly(n-butyl acrylate).42 Poly(M1) and poly(M2) hydrogenation led to a slight decrease in the Tg values to 26 and −6 °C, respectively, due to the alkene unit structural change into a more flexible linear alkane unit.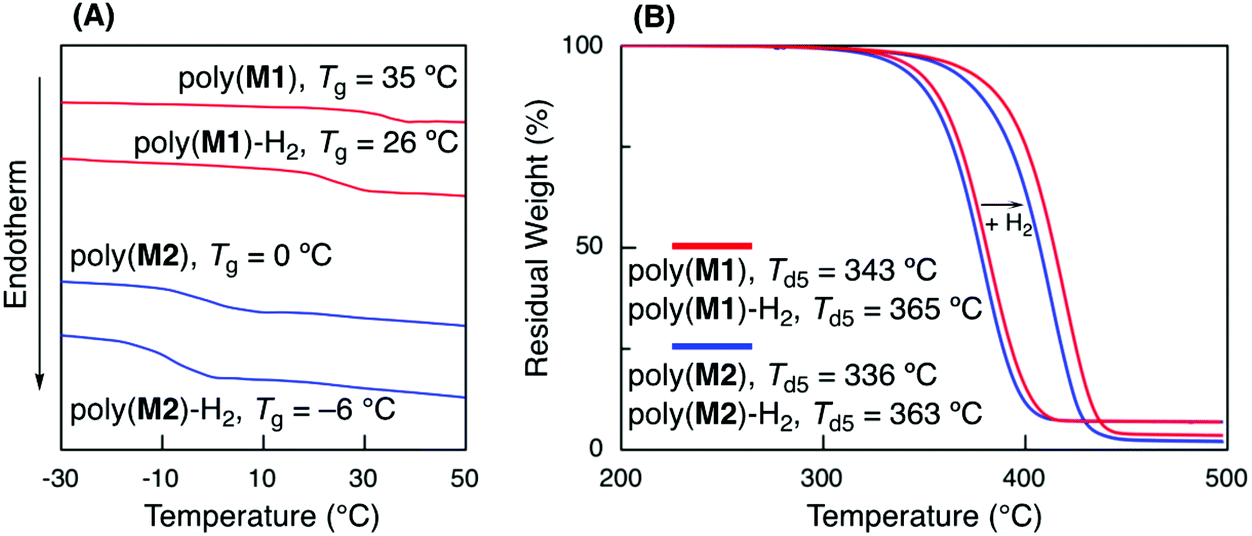 | ||
| Fig. 5 DSC (A) and TGA (B) curves of poly(M1) and poly(M2) before and after hydrogenation. | ||
The Tg values of the sequence-regulated copolymers were compared with those of hypothetical random copolymers of styrene, acrylate, and ethylene, although random copolymers have never been synthesized. In general, the Tg of random copolymers can be estimated from the Tg of each homopolymer (Tg,i) and its weight ratio (wi) in the copolymers according to the Fox equation (1/Tg = w1/Tg,1 + w2/Tg,2 + w3/Tg,3).43 The Tg values of hypothetical random copolymers with the same weight ratios of each monomer unit were thus estimated using typical Tg values of each homopolymer with a high molecular weight (polystyrene: 100 °C, poly(methyl acrylate): 10 °C, poly(n-butyl acrylate): −54 °C, linear polyethylene: −125 °C)40 and the same weight ratios as those of poly(M1)-H2 (polystyrene:poly(methyl acrylate):polyethylene = 0.619:0.256:0.125) and poly(M2)-H2 (polystyrene:poly(n-butyl acrylate):polyethylene = 0.550:0.338:0.111), respectively. The calculated values based on the Fox equation were 20 and −8 °C for the hypothetical random copolymers with methyl and n-butyl acrylate units, respectively.
Since the molecular weights of the obtained sequence-regulated polymers were not high enough to compare to those of the hypothetical random copolymers, the Tg values of sequence-regulated hydrogenated poly(M1) and poly(M2) with infinite molecular weights were then estimated using the Flory–Fox equation (Tg = T∞g − K/Mn).43 The hydrogenated polymers were separated into several fractions with different molecular weights by preparative SEC, and the Tg was measured for each fraction. The calculated T∞g values of poly(M1)-H2 and poly(M2)-H2 were 41 and 10 °C (Fig. 6), respectively, which were both higher than the Tg values of the hypothetical random copolymers (20 and −8 °C) by ca. 20 °C. The differences can be attributed to the effects of monomer sequences, although they are not conclusive.
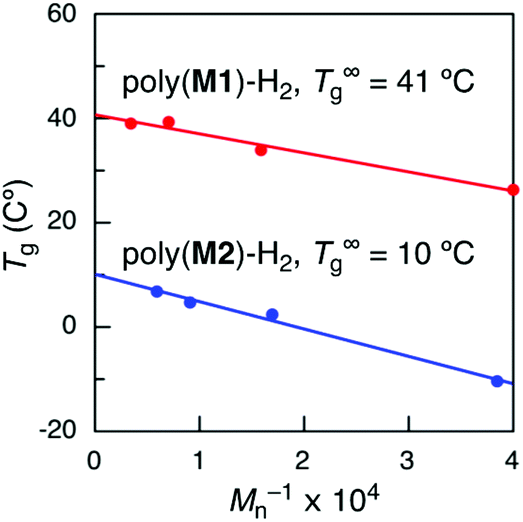 | ||
| Fig. 6 T g values of hydrogenated poly(M1) (poly(M1)-H2) and poly(M2) (poly(M2)-H2) with various molecular weights and T∞g values estimated using the Flory–Fox equation. | ||
The hydrogenation of internal olefins increased the thermal stability of the polymers. After hydrogenation, the 5% degradation temperatures (Td5) increased from 343 to 365 °C for poly(M1) and 336 to 363 °C for poly(M2) (Fig. 5B). These values were almost comparable to those of general vinyl polymers, such as polystyrene and polyacrylate, suggesting the formation of thermally stable vinyl polymers.
Conclusions
A novel synthetic strategy based on a combination of iterative ATRAs of vinyl monomers and ADMET polymerization was developed for the synthesis of molecularly defined copolymers with controlled vinyl monomer sequences at the single monomer unit level. This study enabled the synthesis of sequence-regulated vinyl polymer mimics with perfect and periodic styrene–acrylate–styrene–ethylene–methylene sequences, no statistical distributions of the comonomer compositions and sequences, relatively high molecular weights, and only carbon–carbon single bonds in the main chain. Such molecularly defined vinyl copolymers of styrene, acrylate, and ethylene units with homogeneous distributions in all the polymer chains have not previously been accessible via chain-growth vinyl monomer copolymerization due to the statistical monomer distributions as well as the different monomer reactivity ratios. The thermal properties of the sequence-regulated copolymers could be tuned by the comonomer units, compositions, and sequences, although the sequence effects were not clarified in detail. This strategy can be applied for functionalized vinyl monomers since transition metal-catalyzed ATRAs and ruthenium-catalyzed ADMET polymerization are tolerant of various polar functional groups. This strategy will enable the synthesis of sequence-regulated vinyl polymers with periodic functional groups in vinyl monomer units. The precision synthesis and the detailed analysis of the sequence-regulated copolymers will clarify the effects of monomer sequences in vinyl copolymers and will contribute to further developments of next-generation vinyl polymers with high performance and novel functions.Experimental section
Materials
Styrene (Kishida, 99.5%), methyl dichloroacetate (ClAMCl) (Tokyo Chemical Industry, >99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (Aldrich, >99%), N,N,N′,N′′,N′′′,N′′′-hexamethyltriethylenetetramine (HMTETA) (Aldrich, 97%), allyltrimethylsilane (Tokyo Chemical Industry, >98%), and 1,2,3,4-tetrahydronaphthalene (Wako, 97%) were distilled over calcium hydride under reduced or atmospheric pressure before use. CuCl (Aldrich, 99.99%), a second-generation Grubbs catalyst (G2) ([1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(phenylmethylene)-(tricyclohexylphosphino)ruthenium, Aldrich), a second-generation Hoveyda–Grubbs catalyst (HG2) ([1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(o-isopropoxyphenylmethylene)ruthenium, Aldrich), and a third-generation Grubbs catalyst (G3) ([1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(phenylmethylene)bis(3-bromopyridine)ruthenium, Aldrich) were used as received and handled in a glovebox (MBRAUN LABmaster box) under a moisture- and oxygen-free argon atmosphere (O2, <1 ppm). Toluene (Kanto, >99.5%; H2O <10 ppm) and CH2Cl2 (Kanto, >99.5%, H2O <20 ppm) were dried and deoxygenized by passing through columns from Glass Contour Solvent Systems before use. TiCl4 (Kojundo Chemical Laboratory, Co., Ltd, 99.999%) and 2-aminoethanol (Tokyo Kasei, >99%) were used as received and handled under dry nitrogen. p-Toluenesulfonyl hydrazide (Tokyo Chemical Industry, >98%), 2,6-di-tert-butyl-4-methyl phenol (Tokyo Chemical Industry, >99%), and o-xylene (Wako, >98%) were used as received. n-Butyl dichloroacetate (ClABCl) was prepared according to the literature.39Synthesis of OSAMSO (M1)
CuCl (4.98 g 50.3 mmol), PMDETA (10.6 mL, 50.7 mmol), and toluene (7.6 mL) were placed in a 50 mL round-bottom flask under dry argon at room temperature. The mixture was stirred for 1 h at 80 °C and allowed to cool to room temperature. ClAMCl (364 mL, 3.52 mol), styrene (113 mL, 983 mmol), and the CuCl solution were placed in a 500 mL round-bottom flask under dry argon at room temperature ([ClAMCl]0/[styrene]0/[CuCl]0/[PMDETA]0 = 7000/2000/100/100 mM, total volume: 500 mL). The flask was placed in an oil bath at 80 °C under vigorous stirring. The conversion of styrene was determined by measuring the vinyl group of the residual styrene by 1H NMR. After 68 h, the conversion of styrene reached 99% to form ClSAMCl in a high yield (96%). The solution was diluted with n-hexane and ethyl acetate and washed with aqueous HCl three times and then with water three times. The organic layer was evaporated to remove the solvents. The product was purified through a silica-gel column and eluted with n-hexane/ethyl acetate (9/1), followed by distillation (17 Pa, bp 96 °C) to yield a colorless liquid (ClSAMCl) (200 g, 810 mmol, yield = 82% based on styrene).CuCl (1.98 g, 20.0 mmol), HMTETA (5.5 mL, 20.2 mmol), and toluene (13.8 mL) were placed in a 300 mL round-bottom flask under dry argon at room temperature. The mixture was stirred for 1 h at 80 °C and allowed to cool to room temperature. Styrene (46.0 mL, 400 mmol) and ClSAMCl (133 mL, 662 mmol) were added to the solution at room temperature. The flask was placed in an oil bath at 80 °C under vigorous stirring. The conversion of styrene was determined by measuring the vinyl group of the residual styrene by 1H NMR. After 6 h, the conversion of styrene reached 95% to form ClSAMSCl in a high yield (69%). The solution was diluted with n-hexane and ethyl acetate and washed with aqueous HCl three times and then with water three times. The organic layer was evaporated to remove the solvents. The product (ClSAMSCl) was purified through a silica-gel column and eluted with n-hexane/ethyl acetate (4/1) followed by evaporation to yield a pale yellow liquid (183 g) containing ClSAMSCl and unreacted ClSAMCl.
The liquid was then placed into a 1 L round-bottom flask, into which CH2Cl2 (508 mL) and allyltrimethylsilane (213 mL, 1.34 mol) were added at room temperature. TiCl4 (20 mL, 182 mmol) was added dropwise to the solution and cooled to −78 °C. ([RSCl]0/[allyltrimethylsilane]0/[TiCl4]0 = 1.0/1.5/0.2 M, total volume: 893 mL. The concentration of RSCl was the total value of the styrene-chloride end in ClSAMSCl and ClSAMCl.) After the mixture was stirred at −78 °C for 1 h and at room temperature for 15 h, ammonia methanol (200 mL) was added to quench the reaction. The solution was diluted with diethyl ether and washed with aqueous HCl three times and then with water three times. The organic layer was evaporated to remove the solvents. In addition to OSAMSO, the products contained OSAMCl, which was obtained via allylation of the remaining ClSAMCl.
To efficiently remove OSAMCl using column chromatography based on differences in polarity, the acrylate unit adjacent to the C–Cl bond in OSAMCl was selectively amidated by 2-aminoethanol (174 g mixture of the crude products in 54 mL (902 mmol) of 2-aminoethanol) at 0 °C. The reaction was run at 0 °C for 14 h to amidate the remaining OSAMCl without affecting the ester group in OSAMSO. The solution was diluted with n-hexane and ethyl acetate and washed with aqueous HCl three times and then with water three times. The organic layer was evaporated to remove the solvents. The remaining amidated product was nearly completely removed through a silica-gel column and eluted with n-hexane/ethyl acetate (4/1). After evaporation of the solvents, OSAMSO was distilled under reduced pressure (50 Pa, bp 145 °C) to give a colorless liquid (M1) (68.4 g, 189 mmol, total yield = 30%).
Synthesis of OSABSO (M2)
CuCl (2.97 g 30.0 mmol), PMDETA (6.3 mL, 30.2 mmol), and toluene (2.7 mL) were placed in a 50 mL round-bottom flask under dry argon at room temperature. The mixture was stirred for 1 h at 80 °C and allowed to cool to room temperature. ClABCl (219 mL, 1.46 mol), styrene (69.0 mL, 600 mmol), and the CuCl solution were placed in a 500 mL round-bottom flask under dry argon at room temperature ([ClABCl]0/[styrene]0/[CuCl]0/[PMDETA]0 = 4900/2000/100/100 mM, total volume: 300 mL). The flask was placed in an oil bath at 80 °C under vigorous stirring. The conversion of styrene was determined by measuring the vinyl group of the residual styrene by 1H NMR. After 14 h, the conversion of styrene reached 99% to form ClSABCl in a high yield (79%). The solution was diluted with n-hexane and ethyl acetate and washed with aqueous HCl three times and then with water three times. The organic layer was evaporated to remove the solvents. The product was purified through a silica-gel column and eluted with n-hexane/ethyl acetate (9/1), followed by distillation (26 Pa, bp 125 °C) to yield a colorless liquid (ClSABCl) (112 g, 394 mmol, yield = 66% based on styrene).CuCl (1.27 g, 12.8 mmol) and HMTETA (3.5 mL, 13.0 mmol) were placed in a 300 mL round-bottom flask under dry argon at room temperature. The mixture was stirred for 1 h at 80 °C and allowed to cool to room temperature. Styrene (28.8 mL, 250 mmol) and ClSABCl (92.2 ml, 350 mmol) were added to the solution at room temperature. The flask was placed in an oil bath at 80 °C under vigorous stirring. The conversion of styrene was determined by measuring the vinyl group of the residual styrene by 1H NMR. After 4 h, the conversion of styrene reached 98% to form ClSABSCl in a high yield (62%). The solution was diluted with n-hexane and ethyl acetate and washed with aqueous HCl three times and then with water three times. The organic layer was evaporated to remove the solvents. The product (ClSABSCl) was purified through a silica-gel column and eluted with n-hexane/ethyl acetate (9/1), followed by evaporation to yield a pale yellow liquid (129 g) containing ClSABSCl and unreacted ClSABCl.
The liquid was then placed into a 1 L round-bottom flask, into which CH2Cl2 (286 mL) and allyltrimethylsilane (96.4 mL, 607 mmol) were added at room temperature. TiCl4 (11.2 mL, 101 mmol) was added dropwise to the solution cooled to −78 °C. ([RSCl]0/[allyltrimethylsilane]0/[TiCl4]0 = 1.0/1.2/0.2 M, total volume: 552 mL. The concentration of RSCl is the total value of the styrene-chloride end in ClSABSCl and ClSABCl.) After the mixture was stirred at −78 °C for 7 h and at room temperature for 11 h, ammonia methanol (100 mL) was added to quench the reaction. The solution was diluted with diethyl ether (400 mL) and washed with aqueous HCl three times and then with water three times. The organic layer was evaporated to remove the solvents. In addition to OSABSO, the products contained OSABCl, which was obtained via allylation of the remaining ClSABCl.
To efficiently remove OSABCl using column chromatography based on differences in polarity, the acrylate unit adjacent to the C–Cl bond in OSABCl was selectively amidated by 2-aminoethanol (114 g mixture of the crude products in 120 mL (2.0 mol) of 2-aminoethanol) in THF (140 mL) at 0 °C. The reaction was run at 0 °C for 22 h. The solution was diluted with n-hexane and ethyl acetate and washed with aqueous HCl three times and then with water three times. The organic layer was evaporated to remove the solvents. The remaining amidated product was nearly completely removed through a silica-gel column and eluted with n-hexane/ethyl acetate (95/5). After evaporation of the solvents, OSABSO was distilled under reduced pressure (20 Pa, bp 100 °C) to give a colorless liquid (M2) (33.8 g, 83.5 mmol, total yield = 14%).
ADMET polymerization of M1
Polymerization was carried out by the syringe technique under dry argon in a baked two-necked round-bottom flask equipped with a three-way stopcock. A typical example of the polymerization procedure is given below. M1 (3.3 mL, 9.2 mmol), G2 (19.6 mg, 0.023 mmol), and tetralin (2.8 ml) were placed in the flask. Polymerization was initiated by placing the flask in an oil bath at 50 °C and removing the generated ethylene under vacuum (0.03 MPa). At predetermined intervals, the solution was sampled to monitor the conversions and was terminated by adding ethyl vinyl ether. The monomer conversions were determined from the concentration of the residual monomer using 1H NMR (18 h, conversion = 98%). The quenched reaction mixture was evaporated to dryness under reduced pressure to give the polymer (Mn = 2900, Mw/Mn = 3.40).Hydrogenation of the polymer
Hydrogenation was carried out by the syringe technique under dry nitrogen in baked glass tubes equipped with a three-way stopcock. The obtained polymer and 2,6-di-tert-butyl-4-methyl phenol (0.1 molar equivalent) as a stabilizer were dissolved in o-xylene to give a 1 wt% solution. p-Toluenesulfonyl hydrazide (5 molar equivalents with respect to the unsaturated bonds) was added to the polymer solution, and the mixture was refluxed at 135 °C under stirring. p-Toluenesulfonyl hydrazide was added four times every hour. After 6 h, the solution was allowed to cool, and the mixture was washed with 1 M aqueous NaOH three times and with water three times. The solution was evaporated to dryness to produce the hydrogenated polymer.Measurements
1H and 13C NMR spectra were recorded in CDCl3 at 25 °C on a JEOL ESC-400 spectrometer operating at 400 and 100 MHz, respectively. MALDI-TOF-MS was performed on a Bruker autoflex max (linear mode) with dithranol as the ionizing matrix and sodium trifluoroacetate as the ion source. The number-average molecular weight (Mn) and molecular weight distribution (Mw/Mn) of the product polymers were determined by SEC in THF at 40 °C on two polystyrene gel columns [Shodex GPC KF-805L (8.0 mm i.d. × 30 cm) × 2; flow rate 1.0 mL min−1] connected to a JASCO PU-2080 precision pump and a JASCO RI-2031 detector. The columns were calibrated against standard polystyrene samples (Agilent Technologies; Mp = 580–3053000, Mw/Mn = 1.02–1.23). The glass transition temperature (Tg) of the polymers was recorded on a Q200 differential scanning calorimeter (TA Instruments Inc.). The samples were first heated to 120 °C at 10 °C min−1, equilibrated at this temperature for 10 min, and cooled to −60 °C at 10 °C min−1. After being held at this temperature for 5 min, the samples were then reheated to 160 °C at 10 °C min−1. All the Tg values were determined from the second scan after removing the thermal history. Thermogravimetric analysis (TGA) was performed using a Q500 system (TA Instruments Inc.). The samples were first equilibrated at 110 °C for 30 min and then equilibrated at 40 °C before heating to 500 °C at 5 °C min−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by a Grant-in-Aid for Scientific Research (A) (No. JP18H03917) to M. K. by the Japan Society for the Promotion of Science and the Program for Leading Graduate Schools “Integrative Graduate School and Research Program in Green Natural Sciences”.Notes and references
- Sequence-Controlled Polymers, ed. J.-F. Lutz, Wiley-VCH, Weinheim, 2018 Search PubMed.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 628 CrossRef CAS.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS.
- S. C. Sollender, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef.
- S. Martens, J. O. Holloway and F. E. Du Prez, Macromol. Rapid Commun., 2017, 38, 1700469 CrossRef.
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- M. Ouchi and M. Sawamoto, Polym. J., 2018, 50, 83–94 CrossRef CAS.
- S. L. Perry and C. E. Sing, ACS Macro Lett., 2020, 9, 216–225 CrossRef CAS.
- J. Li and J. He, ACS Macro Lett., 2015, 4, 372–376 CrossRef CAS.
- C. Qu, Z. Li and J. He, Polym. Chem., 2018, 9, 3455–3460 RSC.
- Polymer science: A comprehensive reference, Vol. 3, Chain polymerization of vinyl monomers, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012 Search PubMed.
- Alternating Copolymers, ed. J. M. G. Cowie, Plenum Press, New York, 1985 Search PubMed.
- K. Satoh, M. Matsuda, K. Nagai and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 10003–10005 CrossRef CAS.
- K. Satoh, S. Ozawa, M. Mizutani, K. Nagai and M. Kamigaito, Nat. Commun., 2010, 1, 6 CrossRef.
- K. Satoh, K. Ishizuka, T. Hamada, M. Handa, T. Abe, S. Ozawa, M. Miyajima and M. Kamigaito, Macromolecules, 2019, 52, 3327–3341 CrossRef CAS.
- T. Soejima, K. Satoh and M. Kamigaito, ACS Macro Lett., 2015, 4, 745–749 CrossRef CAS.
- T. Soejima, K. Satoh and M. Kamigaito, J. Am. Chem. Soc., 2016, 138, 944–954 CrossRef CAS.
- T. Soejima, K. Satoh and M. Kamigaito, Polym. Chem., 2016, 7, 4833–4841 RSC.
- C.-H. Wang, Z.-Y. Song, X.-X. Deng, L.-J. Zhang, F.-S. Du and Z.-C. Li, Macromol. Rapid Commun., 2014, 35, 474–478 CrossRef CAS.
- Y. Hibi, M. Ouchi and M. Sawamoto, Nat. Commun., 2015, 7, 11064 CrossRef.
- D. Oh, M. Ouchi, T. Nakanishi, H. Ono and M. Sawamoto, ACS Macro Lett., 2016, 5, 745–749 CrossRef CAS.
- D. Oh, M. Sawamoto and M. Ouchi, Polym. Chem., 2019, 10, 1998–2003 RSC.
- J. Xu, J. S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou and C. Boyer, J. Am. Chem. Soc., 2016, 138, 3094–3106 CrossRef CAS.
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., 2017, 129, 8496–8503 CrossRef.
- Z. Huang, B. B. Noble, N. Corrigan, Y. Chu, K. Satoh, D. S. Thomas, C. J. Hawker, G. Moad, M. Kamigaito, M. L. Coote, C. Boyer and J. Xu, J. Am. Chem. Soc., 2018, 140, 13392–13406 Search PubMed.
- Z. Huang, N. Corrigan, S. Lin, C. Boyer and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1947–1955 Search PubMed.
- J. Xu, Macromolecules, 2019, 52, 9068–9093 Search PubMed.
- R. Liu, L. Zhang, Z. Huang and J. Xu, Polym. Chem., 2020, 1, 4557–4567 Search PubMed.
- J. Vandenbergh, G. Reekmans, P. Adriansens and T. Junkers, Chem. Commun., 2013, 49, 10358–10360 Search PubMed.
- J. Vandengergh, G. Reekmans, P. Adriasens and T. Junkers, Chem. Sci., 2015, 6, 5753–5761 Search PubMed.
- J. J. Haven, J. A. De Neve and T. Junkers, ACS Macro Lett., 2017, 6, 743–747 Search PubMed.
- E. Maron, J. H. Swisher, J. J. Haven, T. Y. Meyer, T. Junkers and H. G. Börner, Angew. Chem., Int. Ed., 2019, 58, 10747–10754 Search PubMed.
- L. C. da Silva, G. Rojas, M. D. Schluz and K. B. Wagener, Prog. Polym. Sci., 2017, 69, 79–107 Search PubMed.
- M. D. Watson and K. B. Wagener, Macromolecules, 2000, 33, 5411–5417 Search PubMed.
- S. E. Leman Jr., K. B. Wagener, L. S. Baugh, S. P. Rucker, D. N. Schulz, M. Varma-Nair and E. Berluche, Macromolecules, 2007, 40, 2643–2656 Search PubMed.
- Z.-L. Li, A. Lv, L. Li, X.-X. Deng, L.-J. Zhang, F.-S. Du and Z.-C. Li, Polymer, 2013, 54, 3841–3849 Search PubMed.
- P. R. Bachler and K. B. Wagener, Monatsh. Chem., 2015, 146, 1053–1061 Search PubMed.
- M. D. Schulz and K. B. Wagener, Macromol. Chem. Phys., 2014, 215, 1936–1945 Search PubMed.
- M. Miyajima, K. Satoh, T. Horibe, K. Ishihara and M. Kamigaito, J. Am. Chem. Soc., 2020, 142, 18955–18962 Search PubMed.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 Search PubMed.
- W. J. Nearly and J. G. Kennemur, Macromol. Rapid Commun., 2016, 37, 975–979 Search PubMed.
- Polymer Handbook, ed. J. Brandrup, E. H. Immergut and E. A. Grulke, John Wiley & Sons, New York, 4th edn, 1999 Search PubMed.
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, CRC Press, Boca Barton, 2nd edn, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01564d |
| This journal is © The Royal Society of Chemistry 2021 |