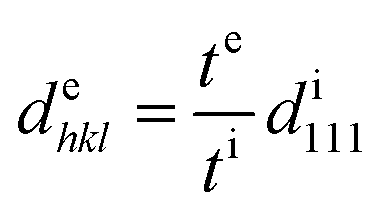DOI:
10.1039/D1MA00133G
(Paper)
Mater. Adv., 2021,
2, 3294-3299
Elastomer-immobilized tunable colloidal photonic crystal films with high optical qualities and high maximum strain†
Received
15th February 2021
, Accepted 8th April 2021
First published on 15th April 2021
Abstract
High-quality elastomer-immobilized colloidal crystal films with low particle volume fractions can be prepared using hydrogel-immobilized single-crystalline colloidal crystal films processed by shear flow. The solvent-free elastomer film demonstrated a full-colour change from red to blue with high uniformity over a cm2 scale, achieved by varying the mole fraction of monomers in the hydrogel. Owing to the low particle volume fraction, the elastomer film exhibited a colour change from red to blue upon stretching, and the maximum strain reached 120%. The film provided reversible and repeatable colour changes during extension without residual strain. The prepared films have the potential to be used in simple strain sensors to express invisible strains through colour change.
Introduction
Periodic arrays of monodisperse colloidal particles, that is, colloidal crystals, embedded in a soft polymer, act as tunable photonic crystals.1–21 Because the volume of the soft polymer can be altered by external stimuli, the lattice constant of the colloidal crystals varies, and hence, their optical properties can be tuned on demand. For example, colloidal crystals immobilized in a hydrogel can change the optical stop-band wavelength or Bragg reflection colour through changes in the gel volume corresponding to environmental changes such as temperature,1,2 pH,3,4 ionic strength,5–8 and solvent.9,10 These properties are also expected to be applicable in simple sensors and indicators that detect changes in environmental conditions and indicate those through changes in the reflection colour. One of the most intriguing applications of such tunable photonic crystals is a strain sensor.11,12 For example, when these sensors are attached to a constructed wall, such as a building, bridge, or tunnel, small amounts of strain which are normally invisible can be readily detected on sight through colour change without requiring expensive tools and prior training. However, hydrogels generally have low mechanical strength, especially against extension, and evaporation of swelling solvent into the ambient atmosphere,8 which hinders their practical use.
Solvent-free colloidal crystals immobilized in an elastomer are a possible solution.13–18 They can be prepared by filling interstices of opal-type close-packed colloidal crystals with elastomers.13–16 Elastomer-immobilized colloidal crystals without solvent are stable in the ambient atmosphere and have sufficient mechanical strength to allow stretching. However, maximum strain has been limited to approximately 50% because the extension caused a significant rearrangement of particles in the opal-based colloidal crystals with a very high particle volume fraction of more than 0.4.13–16 Recently, elastomer-immobilized colloidal crystals with a low particle volume fraction have been developed. They were prepared by polymerization of elastomer precursor solutions containing charge-stabilized colloidal crystals.17,18 Owing to the electrorepulsive interaction among particles, particles were separated from each other as they crystalized in the elastomer precursor solutions and were immobilized in the elastomer by polymerization at a low particle volume fraction (e.g. 0.23417 and 0.3318). Because the large interparticle separation suppressed particle rearrangement during the extension, the maximum strain was improved to approximately 70%.17,18 If elastomer-immobilized colloidal crystals with even lower particle volume fractions are prepared, they may exhibit a higher maximum strain.22 However, this preparation is a challenge because strong electrorepulsive interactions are not generally generated in elastomer precursor solutions. In addition, although optical qualities, especially the uniformity of Bragg reflection colour over a large area, are essential for practical applications, those of colloidal crystals prepared using the common methods are not high because they provide polycrystalline structures.
We have found that charge-stabilized colloidal crystals in water23–25 with a lower particle volume fraction of approximately 0.1 can be single-crystallized over a large area of a cm2 scale by a shear flow effect in a flat capillary cell.26–29 Moreover, loosely packed colloidal crystals can be immobilized in a hydrogel film without deterioration of the high crystalline qualities by the subsequent photopolymerization of gelation reagents previously dissolved in water.30,31 If water contained in the hydrogel film is replaced by an elastomer precursor solution that is then polymerized intact, elastomer-immobilized colloidal crystal films with high optical qualities and a lower particle volume fraction can be obtained. However, in general, when a hydrogel film is immersed in an elastomer precursor solution for replacement, the film shrinks significantly, destroying the crystal structure.
In this report, we demonstrate that elastomer-immobilized colloidal crystal films with a very low particle volume fraction exhibiting high optical qualities and high maximum strain can be achieved using gel-immobilized single-crystalline colloidal crystal films by the proper combination of the hydrogel and the elastomer. We used hydrogels composed of comonomers of N-isopropylacrylamide (NIPAM) and N-methylolacrylamide (NMAM) with different NIPAM mole fractions to adjust the affinity between the hydrogel and the elastomer precursor solution. Hydrophilic 4-hydroxybutyl acrylate (HBA) was used as the elastomer precursor. We examined the dependence of the particle volume fraction and Bragg reflection wavelength of elastomer-immobilized colloidal crystal films on the NIPAM mole fraction. In addition, the tunable properties of the elastomer-immobilized colloidal crystal films at extension were examined.
Results and discussion
Preparation of elastomer-immobilized colloidal crystal films
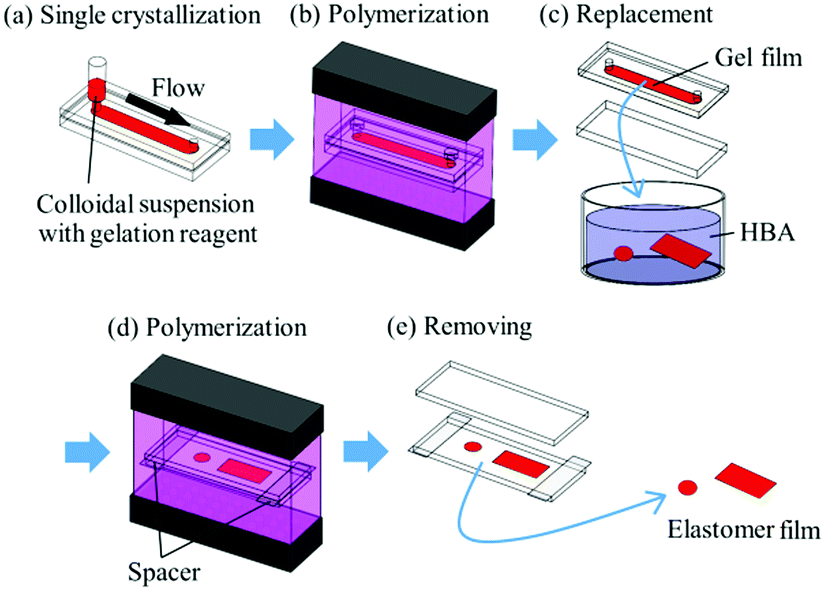
The method for preparing gel-immobilized single-crystalline colloidal crystal films has already been reported,30,31 and a brief description is given below. An aqueous suspension of monodisperse polystyrene particles (Thermo Scientific, particle diameter: 160 nm) was deionised by adding a mixed-bed ion-exchange resin (Bio-Rad, AG501-X8(D)) and stirring gently. Gelation reagent (NIPAM and NMAM as monomers, N,N′-methylenebisacrylamide (BIS) as a cross-linker, and 2,2′-azobis[2-methyl-N-(2-hydroxyethyl)propionamide] (VA) as a photo-initiator) and ultrapure water were added to the suspension to achieve monomer, cross-linker, photo-initiator, and particle concentrations of 800 mM, 40 mM, 0.35 mM, and approximately 10.7 vol%, respectively. The colloidal suspension with different NIPAM mole fractions x (x = [NIPAM]/([NIPAM] + [NMAM])) was shear-flowed into a flat capillary cell (channel height: 0.1 mm; width: 9 mm; length: 70 mm) to convert into a single-domain crystal (Fig. 1a).26–29 Ultraviolet (UV) light was irradiated uniformly from both sides of the cell to photopolymerize the gelation reagent (Fig. 1b).30,31 The obtained gel film containing single-crystalline colloidal crystals was removed from the cell and cut into circular fragments with a diameter of 4.5 mm (Fig. 1c). A rectangular fragment with a length of 20 mm and width of 9 mm was cut from the gel film with x = 0.50. The films were immersed in an elastomer precursor solution of HBA with 1 wt% of photo-initiator (BASF Japan Ltd, DAROCUR 1173) for approximately 24 h to replace the water contained in the gel film with the precursor solution. The films were placed between two glass slides using cover glasses as spacers, and the UV light was irradiated to photopolymerize the HBA (Fig. 1d).
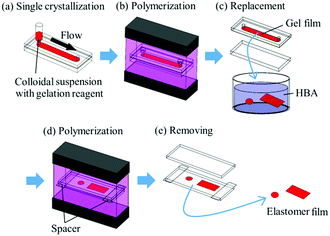 |
| | Fig. 1 Schematic representation of preparation process for elastomer-immobilized colloidal crystal films. | |
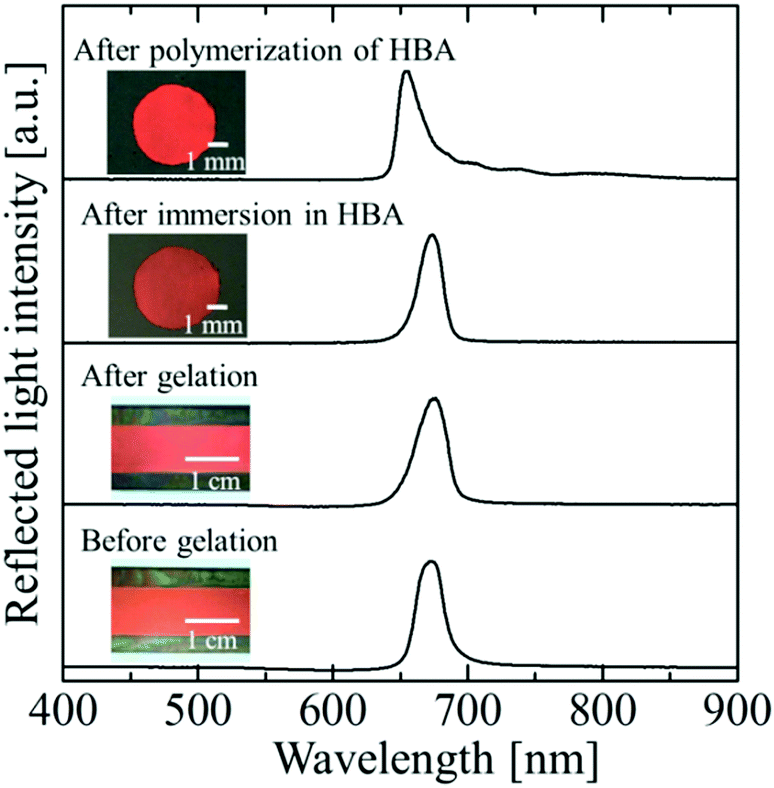
Fig. 2 shows photographs and reflection spectra of the colloidal crystal film with x = 0.50 at each preparation step. Before gelation, the colloidal crystal exhibited a uniform texture with a bright red colour over the entire sample area, which was achieved by the shear flow effect. The reflection spectrum showed a strong peak at 679 nm, which was derived from the Bragg reflection from the (111) lattice planes of the face-centred cubic (FCC) structure parallel to the sample surface.26–29 After gelation, uniform colour and an excellent spectral profile were preserved. When the gel-immobilized colloidal crystal film was cut into the circular fragments and immersed in the elastomer precursor solution, the uniform colour and the peak shape were retained. After the photopolymerization of the precursor, the film shrank, and the particle volume fraction increased to 0.155. As a result, the reflection peak was blue-shifted to 654 nm. However, excellent optical properties such as the uniform reflection colour and the strong reflection peak were almost fully retained.
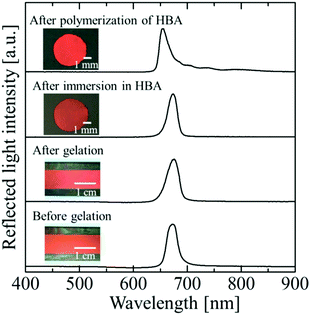 |
| | Fig. 2 Photographs and reflection spectra of the colloidal crystal film with x = 0.50 at each preparation step. | |
The change in the Bragg wavelength after the conversion from the gel film to the elastomer film can be quantitatively explained by the contraction of the FCC(111) lattice spacing and the change in the refractive index of the colloidal crystals using Bragg's law at normal incidence:
where
λhkl is the Bragg wavelength,
nc is the refractive index of the colloidal crystals, and
dhkl is the lattice spacing of the (
hkl) planes. The volume-weighted average of the refractive indices of the components of colloidal crystals is a good approximation of
nc:
8,32| | | nc = npϕp + nm(1 − ϕp), | (2) |
where
np and
nm are the refractive indices of the polystyrene particles (
np = 1.59) and the matrix of the elastomer film, respectively, and
ϕp is the volume fraction of polystyrene particles in the elastomer film. Because the matrix consists of poly(
N-isopropylacrylamide) (PNIPAM), poly(
N-methylolacrylamide) (PNMAM), and poly(4-hydroxybutyl acrylate) (PHBA),
nm can be approximated as
nm = (
nPNIPAMϕPNIPAM +
nPNMAMϕPNMAM +
nPHBA(1 −
ϕp −
ϕPNIPAM −
ϕPNMAM))/(1 −
ϕp), where
nPNIPAM,
nPNMAM, and
nPHBA are the refractive indices of the PNIPAM (
nPNIPAM = 1.45), PNMAM (
nPNMAM = 1.41), and PHBA (
nPHBA = 1.49), respectively, and
ϕPNIPAM and
ϕPNMAM are the volume fractions of the PNIPAM and PNMAM in the elastomer film, respectively. The
d111 value is determined using
ϕp and the diameter
d of colloidal particles:
| | 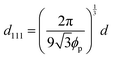 | (3) |
Because the volume change of the film is isotropic in three dimensions during solvent replacement and photopolymerization, the volume ratio
α of the elastomer film to the gel film is calculated using the measured diameters of the elastomer film
D and the gel film
D′ as follows:
| | 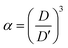 | (4) |
Assuming that the volumes of the colloidal particles, PNIPAM, and PNMAM are constant during the preparation process, the volume fractions of the particles ϕip, PNIPAM ϕiPNIPAM, and PNMAM ϕiPNMAM in the elastomer film can be estimated using α as
| | 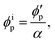 | (5) |
| | 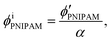 | (6) |
| | 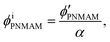 | (7) |
where
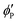
,
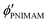
, and
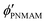
are the volume fractions of the colloidal particles, PNIPAM, and PNMAM in the gel film swollen in water, respectively. These values can be calculated from the Bragg wavelength and the masses of the gelation reagent added to the water.
2,8 By substituting
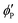
,
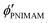
,
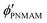
,
D, and
D′ into
eqn (4)–(7);
ϕip,
ϕiPNIPAM, and
ϕiPNMAM were determined. The values for
nc and
d111 of the elastomer film were calculated by substituting
ϕip,
ϕiPNIPAM, and
ϕiPNMAM into
eqn (2) and (3). Thus, the Bragg wavelength of the elastomer film was estimated to be 657 nm by substituting
nc and
d111 into
eqn (1). This value is in good agreement with the measured Bragg wavelength of 654 nm.
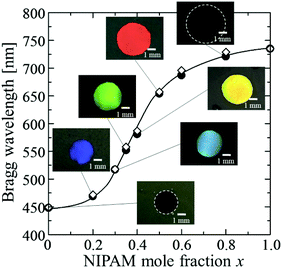
We found that the affinity of the gel film to HBA was greatly changed by x. Thus, the particle volume fraction and the Bragg reflection wavelength of the resultant elastomer-immobilized colloidal crystal film could be greatly altered by x (Fig. 3 and Fig. S1, ESI†). When x decreased, the gel film shrank significantly in the precursor solution. High uniformity was preserved, and the resultant elastomer film exhibited a full-colour change: red (x = 0.50), yellow (x = 0.40), green (x = 0.35), light blue (x = 0.30), and blue (x = 0.20) (Fig. 3). The particle volume fraction at x = 0 was calculated to be 0.503. In contrast, when x was increased, the gel film swelled well in the precursor solution. In particular, the swelling volume of the gel film with x = 1.0, in the precursor solution was larger than that of the gel film in water. As a result, surprisingly, the particle volume fraction of the elastomer film with x = 1.0 was 0.111. This signifies that the solvent-free elastomer film can realize a lower particle volume fraction than the gel-immobilized colloidal crystal film swollen in water (i.e. 0.114). The observed Bragg wavelengths of the elastomer films with various x values are plotted as filled circles in Fig. 3. At x values exceeding 0.80, the Bragg wavelength attained a near-IR range. These plots are in good agreement with the plots calculated using eqn (1)–(7), which are shown as open diamonds.
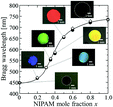 |
| | Fig. 3 Plots of the Bragg wavelength and photographs of the elastomer-immobilized colloidal crystal films as a function of x (●: experimentally measured Bragg wavelength, ⋄: calculated Bragg wavelength). | |
Tunable properties of the elastomer-immobilized colloidal crystal film at extension
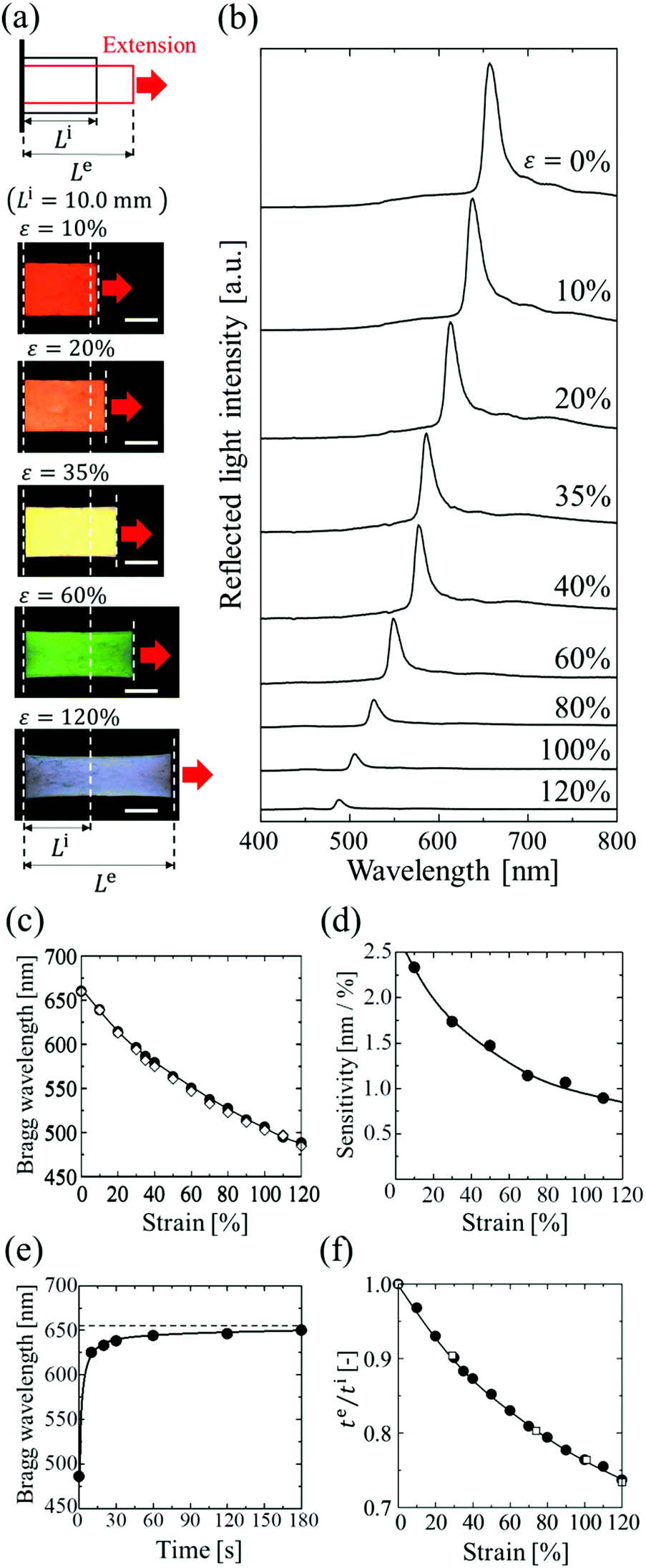
The elastomer-immobilized colloidal crystal film showed a full-colour change at extension and exhibited a maximum strain of 120%, which is, to date, the highest value reported for elastomer-immobilized colloidal crystals. As shown in Fig. 4a, when the film with x = 0.50 was stretched, various reflection colours (red, orange, yellow, green, and blue) were observed, corresponding to the strain (ε = ((Le − Li)/Li) × 100, Le: film length at extension, and Li: film length without extension), while preserving uniformity over a cm2 scale. In the reflection spectrum, the Bragg peak was gradually blue-shifted from 660 to 488 nm as the strain was increased to 120% (Fig. 4b and c). The intensity of the peak decreased upon stretching. This is explained by the decrease in the refractive index contrast between a slice containing the (111) plane at the middle and an intermediate slice.18Fig. 4d shows the sensitivity, that is, the amount of change in the Bragg wavelength responding to strain, estimated by the slope of the graph of the Bragg wavelength as a function of strain (Fig. 4c). The sensitivity decreased with increasing strain and became a very low value of less than 1 nm/% at a strain of 120%.
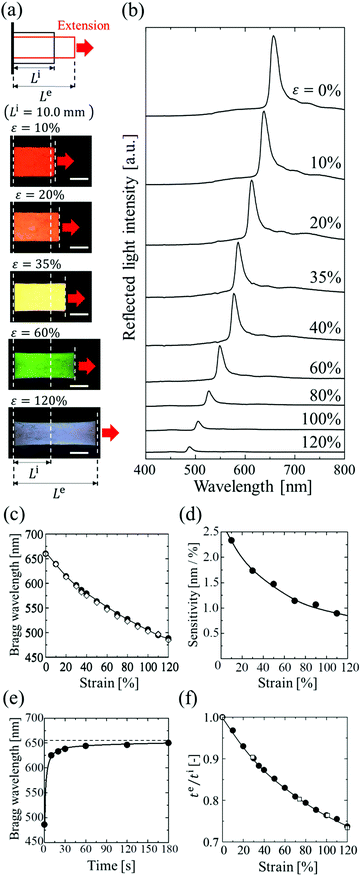 |
| | Fig. 4 (a) Photographs of elastomer-immobilized colloidal crystal film with x = 0.50 at various strains. The length of the scale bars is 5.0 mm. (b) Reflection spectra of the film at various strains. (c) Bragg wavelength of the film as a function of strain (●: experimentally measured Bragg wavelength, ⋄: calculated Bragg wavelength). (d) Sensitivity of the film estimated from c. (e) Time-course change in the Bragg wavelength after the release. (f) Thickness ratio of te to ti as a function of strain (●: elastomer film with x = 0.50, □: elastomer film without particles). | |
The blue shift of the Bragg peak is caused by the reduction of the lattice spacing perpendicular to the thickness direction of the film; hence, the Bragg wavelength during extension can be estimated from the film thickness. The lattice spacing dehkl at extension is given by the film thickness te at extension, film thickness ti without extension, and lattice spacing di111 without extension as follows:
| | 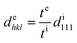 | (8) |
Because the particle volume is constant during the extension, the particle volume fraction ϕep at extension is given by the volume of the elastomer film Vi without extension and the volume of the elastomer film Ve at extension, which are calculated by the area and the thickness, as follows:
| | 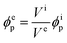 | (9) |
Assuming that
nm does not change by stretching,
nm is the refractive index of the matrix without extension.
nc under strain is determined by substituting
nm and
ϕep into
eqn (2). Thus, the Bragg wavelengths at various strains were estimated by substituting
dehkl and
nc into
eqn (1), and plotted as open diamonds in
Fig. 4c. These plots were in good agreement with the observed Bragg wavelengths.
Fig. 4e shows the time-course change in the Bragg wavelength of the elastomer film after releasing the extension with a maximum strain of 120%. At 10 s after release, the Bragg wavelength showed a 5% difference in the initial wavelength and became almost identical to the value after 1 min. Fig. 4f shows the thickness ratios of te to ti for the elastomer film with x = 0.50, and for the elastomer film without particles. The two films showed the same dependence of thickness on strain. In addition, the Poisson's ratios of both films had the same value of 0.39, which was estimated by the gradient of the plots of −ln(te/ti) vs. ln(Le/Li) up to a maximum strain of 120% (Fig. S2, ESI†).33 These data indicate that the colloidal crystals in the elastomer film do not affect the deformation of the elastomer even at a maximum strain of 120% because of the very low particle volume fraction. In addition, the relatively low Poisson's ratio of the elastomer film contributed to the high maximum strain.
Fig. 5a shows the spectral change of the elastomer film by repeating the stretch and release between strains of 10% and 35%. The Bragg peak blue-shifted from 637 to 586 nm during the stretch and returned to the initial wavelength after the release, preserving the excellent spectral profile (Fig. 5a). The uniform reflection colour was also intact after the repetitions (Fig. 5b). The film was fully reversible, and the colour change was repeatable with elastic deformation, which was achieved by the large interparticle separation in the colloidal crystals with a very low particle volume fraction. The variations in the Bragg wavelength and the intensity of the reflection did not exceed a few percent after repeating the extension with the maximum strain of 120% and the release 100 times, and no damage was observed on the film.
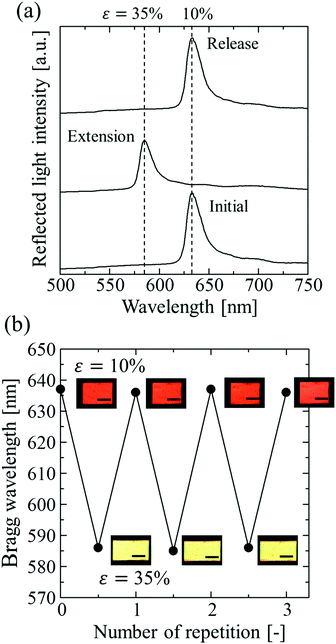 |
| | Fig. 5 (a) Spectral change of the elastomer-immobilized colloidal crystal film with x = 0.50 by repeating the stretch and release. (b) Photographs and Bragg wavelength of the film by repeating the stretch and release 3 times. The length of the scale bars is 5.0 mm. | |
Conclusions
In summary, we succeeded in preparing elastomer-immobilized colloidal crystal films with high optical qualities and a low particle volume fraction, realizing the benefits of hydrogel-immobilized single-crystalline colloidal crystal films processed by shear flow. By the proper combination of the copolymer hydrogel consisting of NIPAM and NMAM monomers and the hydrophilic HBA elastomer precursor, the shrinkage of the gel film during solvent replacement was suppressed; thus, the high-quality colloidal crystals could be immobilized in solvent-free elastomer films intact. Furthermore, we found that the gel film composed of the NIPAM monomer swelled to a greater degree in the HBA than in water, and therefore, the particle volume fraction in the resultant elastomer film could be greatly altered between 0.111 and 0.503 by varying the mole fraction of monomers. This leads to elastomer films exhibiting a full-colour change from red to blue with high uniformity over a cm2 scale. Owing to the very low particle volume fraction, the elastomer film exhibited a full-colour change from red to blue when stretched, without rearrangement of particles, and the maximum strain reached 120%. This is the highest value reported to date for elastomer-immobilized colloidal crystals. In addition, because the particles remain untouched during stretching, no residual strain remains. Thus, the film exhibited reversible and repeatable colour changes during extension. The films have the potential to be used in simple strain sensors to detect invisible strains through colour change.
Experimental
Preparation of gel-immobilized colloidal crystal films
An ion-exchange resin (Bio-Rad, AG501-X8(D)) was added to an aqueous suspension of monodisperse polystyrene particles (Thermo Scientific, particle diameter: 160 nm), and the mixture was gently stirred for deionization. The formed charge-stabilized colloidal crystals were centrifuged to increase the particle concentration. Gelation reagents (NIPAM (FUJIFILM Wako Pure Chemical Corp.) and NMAM (FUJIFILM Wako Pure Chemical Corp.) as monomers, BIS (FUJIFILM Wako Pure Chemical Corp.) as a cross-linker, and VA (FUJIFILM Wako Pure Chemical Corp.) as a photo-initiator) and ultrapure water (Millipore, Milli-Q system) were added to the colloidal crystals so that the concentrations of the monomers, cross-linker, photo-initiator, and particle were 800 mM, 40 mM, and 0.35 mM, and approximately 10.7 vol%, respectively. Colloidal crystals with different NIPAM mole fractions x (x = [NIPAM]/([NIPAM] + [NMAM])) were prepared and bubbled with Ar gas for 5 min to remove dissolved O2 and CO2. The colloidal crystals were shear-flowed into a flat capillary cell (channel height: 0.1 mm; width: 9 mm; length: 70 mm) to convert into a single-domain crystal. The obtained particle array of the single crystal was immobilized in the hydrogel network by UV light irradiation using a UV light device (MORITEX SCHOTT, MBRL-CUV7530). Photographs of the samples before and after UV light irradiation were taken using a CCD camera (Sony, XCD-V60CR) through a macro zoom lens (Edmund Optics, MLH-10X), and the reflection spectra were measured using a multichannel spectrometer (Soma Optics, Fastevert S-2630).
Preparation of elastomer-immobilized colloidal crystal films
The gel film containing single-crystalline colloidal crystals was removed from the cell and cut into circular fragments of 4.5 mm diameter. For the tensile testing, the gel film with x = 0.50 was cut into a rectangular fragment 20 mm long and 9 mm wide. The gel films were immersed in HBA (Tokyo Chemical Industry Co., Ltd) with 1 wt% of a photo-initiator (BASF Japan Ltd, DAROCUR 1173) for approximately 24 h to replace the water in the gel with the elastomer precursor solution. Slide glasses were coated with toluene (FUJIFILM Wako Pure Chemical Corp.) with 5 wt% of silicone oil (Shin-Etsu Chemical Co., Ltd, KF-96A-50CS) as a release reagent and heated at 350 °C for 1 h in an electric furnace (Tokyo Rikakikai Co., Ltd, TMF-4100). The gel films, swollen in the elastomer precursor solution, were placed between the two glass slides using cover glasses (thickness: approximately 0.15 mm) as spacers. The films were irradiated with UV light (MORITEX SCHOTT, MBRL-CUV7530), both above and below. Photographs of the films before and after UV light irradiation were taken using a CCD camera (Sony, XCD-V60CR) through a macro zoom lens (Edmund Optics, MLH-10X), and reflection spectra were measured using a multichannel spectrometer (Soma Optics, Fastevert S-2630).
Tensile testing of elastomer-immobilized colloidal crystal film
The film was pinched 5 mm from both edges and stretched at 1.0 mm intervals. At each step, photographs of the film were taken from above using a CCD camera (Sony, XCD-V60CR) through a macro zoom lens (Edmund Optics, MLH-10X). The side view of the film was observed using a CCD camera through a video lens (Edmund Optics, VIDEO LENS-VZM 600) to measure the film thickness. The reflection spectra of the stretched films were measured using a multichannel spectrometer (Soma Optics, Fastevert S-2630).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge Dr Tsutomu Sawada (NIMS) for helpful discussions. This work was supported by JSPS KAKENHI (grant number 25289237).
Notes and references
- J. M. Weissman, H. B. Sunkara, A. S. Tse and S. A. Asher, Science, 1996, 274, 959 CrossRef CAS PubMed.
- T. Kanai, H. Yano, N. Kobayashi and T. Sawada, ACS Macro Lett., 2017, 6, 1196 CrossRef CAS.
- K. Lee and S. A. Asher, J. Am. Chem. Soc., 2000, 122, 9534 CrossRef CAS.
- T. Sakiyama, H. Takata, T. Toga and K. Nakanishi, J. Appl. Polym. Sci., 2001, 81, 667 CrossRef CAS.
- J. Holtz and S. A. Asher, Nature, 1997, 389, 829 CrossRef CAS PubMed.
- H. Saito, Y. Takeoka and M. Watanabe, Chem. Commun., 2003, 2126 RSC.
- C. Fenzl, S. Wilhelm, T. Hirsch and O. S. Wolfbeis, ACS Appl. Mater. Interfaces, 2013, 5, 173 CrossRef CAS PubMed.
- T. Kanai, S. Yamamoto and T. Sawada, Macromolecules, 2011, 44, 5865 CrossRef CAS.
- A. Toyotama, T. Kanai, T. Sawada, J. Yamanaka, K. Ito and K. Kitamura, Langmuir, 2005, 21, 10268 CrossRef CAS PubMed.
- C. Katsura, S. Nobukawa, H. Sugimoto, E. Nakanishi and K. Inomata, Colloid Polym. Sci., 2017, 295, 1709 CrossRef CAS.
- S. H. Foulger, P. Jiang, A. C. Lattam, D. W. Smith Jr and J. Ballato, Langmuir, 2001, 17, 6023 CrossRef CAS.
- K. Yoshino, Y. Kawagishi, M. Ozaki and A. Kose, Jpn. J. Appl. Phys., 1999, 38, L786 CrossRef CAS.
- H. Fudouzi and Y. N. Xia, Adv. Mater., 2003, 15, 892 CrossRef CAS.
- H. Fudouzi and T. Sawada, Langmuir, 2006, 22, 1365 CrossRef CAS PubMed.
- X. Sun, J. Zhang, X. Lu, X. Fang and H. Peng, Angew. Chem., Int. Ed., 2015, 54, 3630 CrossRef CAS PubMed.
- J. Zhang, S. He, L. Liu, G. Guan, X. Lu, X. Sun and H. Peng, J. Mater. Chem. C, 2016, 4, 2127 RSC.
- E. Miwa, K. Watanabe, F. Asai, T. Seki, K. Urayama, J. Odent, J. M. Raquez and Y. Takeoka, ACS Appl. Polym. Mater., 2020, 2, 4078 CrossRef CAS.
- G. H. Lee, T. M. Choi, B. Kim, S. H. Han, J. M. Lee and S. H. Kim, ACS Nano, 2017, 11, 11350 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, H. Wang, Y. Yu, Q. Zhong and Y. Zhao, Small, 2019, 15, 1902198 CrossRef PubMed.
- F. Fu, Z. Chen, H. Wang, C. Liu, Y. Liu and Y. Zhao, Nanoscale, 2019, 11, 10846 RSC.
- H. Wang, Y. Liu, Z. Chen, L. Sun and Y. Zhao, Sci. Adv., 2020, 6, eaay1438 CrossRef CAS PubMed.
- J. M. Clough, C. Weder and S. Schrettl, Macromol. Rapid Commun., 2021, 42, 2000528 CrossRef CAS PubMed.
- K. Ito, K. Sumaru and N. Ise, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 3105 CrossRef PubMed.
- A. P. Gast and W. B. Russel, Phys. Today, 1998, 51, 24 CrossRef CAS.
- T. Kanai, T. Sawada, J. Yamanaka and K. Kitamura, Langmuir, 2005, 21, 7633 CrossRef CAS PubMed.
- T. Kanai, T. Sawada and K. Kitamura, Langmuir, 2003, 19, 1984 CrossRef CAS.
- T. Kanai, T. Sawada, I. Maki and K. Kitamura, Jpn. J. Appl. Phys., 2003, 42, L655 CrossRef CAS.
- T. Kanai, T. Sawada and K. Kitamura, Chem. Lett., 2005, 34, 904 CrossRef CAS.
- T. Kanai, T. Sawada, A. Toyotama and K. Kitamura, Adv. Funct. Mater., 2005, 15, 25 CrossRef CAS.
- T. Kanai, T. Sawada and J. Yamanaka, J. Ceram. Soc. Jpn., 2010, 118, 370 CrossRef CAS.
- T. Kanai, J. Ceram. Soc. Jpn., 2012, 120, 87 CrossRef CAS.
- P. A. Hiltner and I. M. Krieger, J. Phys. Chem., 1969, 73, 2386 CrossRef CAS.
- K. Urayama, T. Takigawa and T. Masuda, Macromolecules, 1993, 26, 3092 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reflection spectra and plots of true strains in the thickness direction vs. those in the stretching direction. See DOI: 10.1039/d1ma00133g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*

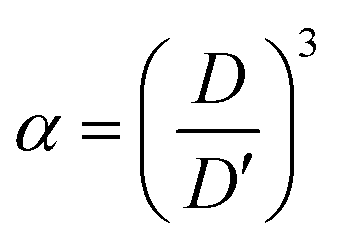
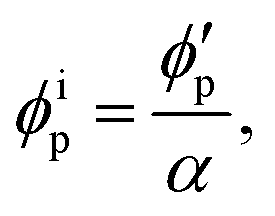
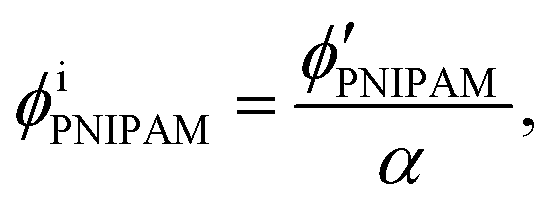

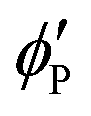 ,
,  , and
, and 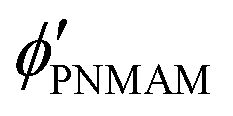 are the volume fractions of the colloidal particles, PNIPAM, and PNMAM in the gel film swollen in water, respectively. These values can be calculated from the Bragg wavelength and the masses of the gelation reagent added to the water.2,8 By substituting
are the volume fractions of the colloidal particles, PNIPAM, and PNMAM in the gel film swollen in water, respectively. These values can be calculated from the Bragg wavelength and the masses of the gelation reagent added to the water.2,8 By substituting  ,
,  ,
,  , D, and D′ into eqn (4)–(7); ϕip, ϕiPNIPAM, and ϕiPNMAM were determined. The values for nc and d111 of the elastomer film were calculated by substituting ϕip, ϕiPNIPAM, and ϕiPNMAM into eqn (2) and (3). Thus, the Bragg wavelength of the elastomer film was estimated to be 657 nm by substituting nc and d111 into eqn (1). This value is in good agreement with the measured Bragg wavelength of 654 nm.
, D, and D′ into eqn (4)–(7); ϕip, ϕiPNIPAM, and ϕiPNMAM were determined. The values for nc and d111 of the elastomer film were calculated by substituting ϕip, ϕiPNIPAM, and ϕiPNMAM into eqn (2) and (3). Thus, the Bragg wavelength of the elastomer film was estimated to be 657 nm by substituting nc and d111 into eqn (1). This value is in good agreement with the measured Bragg wavelength of 654 nm.