 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using highly time-resolved online mass spectrometry to examine biogenic and anthropogenic contributions to organic aerosol in Beijing†
Archit
Mehra
 a,
Manjula
Canagaratna
b,
Thomas J.
Bannan
a,
Stephen D.
Worrall
c,
Asan
Bacak
d,
Michael
Priestley
e,
Dantong
Liu
f,
Jian
Zhao
g,
Weiqi
Xu
g,
Yele
Sun
g,
Jacqueline F.
Hamilton
h,
Freya A.
Squires
h,
James
Lee
h,
Daniel J.
Bryant
h,
James R.
Hopkins
h,
Atallah
Elzein
h,
Sri Hapsari
Budisulistiorini
h,
Xi
Cheng
i,
Qi
Chen
i,
Yuwei
Wang
j,
Lin
Wang
j,
Harald
Stark
bkl,
Jordan E.
Krechmer
b,
James
Brean
m,
Eloise
Slater
n,
Lisa
Whalley
no,
Dwayne
Heard
n,
Bin
Ouyang
p,
W. Joe F.
Acton
p,
C. Nicholas
Hewitt
p,
Xinming
Wang
q,
Pingqing
Fu
r,
John
Jayne
b,
Douglas
Worsnop
b,
James
Allan
as,
Carl
Percival‡
a and
Hugh
Coe
*aj
a,
Manjula
Canagaratna
b,
Thomas J.
Bannan
a,
Stephen D.
Worrall
c,
Asan
Bacak
d,
Michael
Priestley
e,
Dantong
Liu
f,
Jian
Zhao
g,
Weiqi
Xu
g,
Yele
Sun
g,
Jacqueline F.
Hamilton
h,
Freya A.
Squires
h,
James
Lee
h,
Daniel J.
Bryant
h,
James R.
Hopkins
h,
Atallah
Elzein
h,
Sri Hapsari
Budisulistiorini
h,
Xi
Cheng
i,
Qi
Chen
i,
Yuwei
Wang
j,
Lin
Wang
j,
Harald
Stark
bkl,
Jordan E.
Krechmer
b,
James
Brean
m,
Eloise
Slater
n,
Lisa
Whalley
no,
Dwayne
Heard
n,
Bin
Ouyang
p,
W. Joe F.
Acton
p,
C. Nicholas
Hewitt
p,
Xinming
Wang
q,
Pingqing
Fu
r,
John
Jayne
b,
Douglas
Worsnop
b,
James
Allan
as,
Carl
Percival‡
a and
Hugh
Coe
*aj
aCentre for Atmospheric Science, School of Earth and Environmental Sciences, The University of Manchester, Manchester, M13 9PL, UK. E-mail: hugh.coe@manchester.ac.uk
bCenter for Aerosol and Cloud Chemistry, Aerodyne Research Inc, Billerica, Massachusetts, USA
cAston Institute of Materials Research, School of Engineering and Applied Science, Aston University, Birmingham, B4 7ET, UK
dTurkish Accelerator & Radiation Laboratory, Ankara University Institute of Accelerator Technologies, Gölbaşı Campus, 06830 Golbasi, Ankara, Turkey
eDepartment of Chemistry and Molecular Biology, University of Gothenburg, Gothenburg, Sweden
fDepartment of Atmospheric Sciences, School of Earth Sciences, Zhejiang University, Hangzhou, Zhejiang, China
gState Key Laboratory of Atmospheric Boundary Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing 100029, China
hWolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, UK
iState Key Joint Laboratory of Environmental Simulation and Pollution Control, College of Environmental Science and Engineering, Peking University, Beijing, China
jShanghai Key Laboratory of Atmospheric Particle Pollution and Prevention (LAP3), Department of Environmental Science & Engineering, Fudan University, Jiangwan Campus, Shanghai 200438, China
kCooperative Institute for Research in Environmental Sciences (CIRES), University of Colorado, Boulder, CO 80309, USA
lDepartment of Chemistry, University of Colorado, Boulder, CO 80309, USA
mDivision of Environmental Health and Risk Management, School of Geography, Earth and Environmental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
nSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
oNational Centre for Atmospheric Science, University of Leeds, Leeds, LS2 9JT, UK
pLancaster Environment Centre, Lancaster University, Lancaster, LA1 4YQ, UK
qGuangzhou Institute of Geochemistry, Guangzhou, China
rInstitute of Surface-Earth System Science, Tianjin University, Tianjin, China
sNational Centre for Atmospheric Science, University of Manchester, Manchester, UK
First published on 21st January 2021
Abstract
Organic aerosols, a major constituent of fine particulate mass in megacities, can be directly emitted or formed from secondary processing of biogenic and anthropogenic volatile organic compound emissions. The complexity of volatile organic compound emission sources, speciation and oxidation pathways leads to uncertainties in the key sources and chemistry leading to formation of organic aerosol in urban areas. Historically, online measurements of organic aerosol composition have been unable to resolve specific markers of volatile organic compound oxidation, while offline analysis of markers focus on a small proportion of organic aerosol and lack the time resolution to carry out detailed statistical analysis required to study the dynamic changes in aerosol sources and chemistry. Here we use data collected as part of the joint UK–China Air Pollution and Human Health (APHH-Beijing) collaboration during a field campaign in urban Beijing in the summer of 2017 alongside laboratory measurements of secondary organic aerosol from oxidation of key aromatic precursors (1,3,5-trimethyl benzene, 1,2,4-trimethyl benzene, propyl benzene, isopropyl benzene and 1-methyl naphthalene) to study the anthropogenic and biogenic contributions to organic aerosol. For the first time in Beijing, this study applies positive matrix factorisation to online measurements of organic aerosol composition from a time-of-flight iodide chemical ionisation mass spectrometer fitted with a filter inlet for gases and aerosols (FIGAERO-ToF-I-CIMS). This approach identifies the real-time variations in sources and oxidation processes influencing aerosol composition at a near-molecular level. We identify eight factors with distinct temporal variability, highlighting episodic differences in OA composition attributed to regional influences and in situ formation. These have average carbon numbers ranging from C5–C9 and can be associated with oxidation of anthropogenic aromatic hydrocarbons alongside biogenic emissions of isoprene, α-pinene and sesquiterpenes.
1. Introduction
Organic aerosols (OA) are a major constituent of fine particulate mass (PM)2 which impact upon climate, air quality and human health.3–5 Primary OA (POA) are directly emitted from natural and anthropogenic biomass burning alongside fossil fuel combustion, while secondary OA (SOA) are formed from the atmospheric oxidation of volatile organic compounds (VOC) emitted from both biogenic and anthropogenic sources.6 The relative contributions of biogenic and anthropogenic sources to OA remain poorly constrained owing to their diverse speciation7–9 and complex oxidation pathways.6,10,11Beijing is a megacity which continues to suffer from severe air pollution events despite recent reductions in primary emission of PM12–14 and nitrogen oxides (NOx).15 These have been associated with high emissions of VOC from diverse sources16,17 which lead to enhanced ozone production18,19 and SOA formation20 alongside regional transport which is an important source of semi-volatile organic compounds (SVOC), intermediate-volatility organic compounds (IVOC), POA and SOA in Beijing during the summertime.21,22
Historically, source apportionment of OA has been limited to low time-resolution high molecular precision offline measurements of molecular tracers, focusing on the behaviour of specific VOC systems including isoprene,23–26 α-pinene27 and toluene.28,29 More recently, highly time-resolved bulk composition measurements by aerosol mass spectrometry (AMS) and aerosol chemical speciation monitors (ACSM) have been used to apportion OA to broad source classifications including fossil fuels (FFOA), biomass burning (BBOA) and oxidised organic aerosol (OOA).18,21,30,31 These techniques alone are thus limited in their capability to constrain the dynamic sources and processes governing OA transformation at a molecular level. SOA typically contributes 52–64% of OA in Beijing,21 yet despite its diverse biogenic and anthropogenic sources, is classified solely on the basis of its bulk degree of oxidation into less-oxidised OOA (LO-OOA) and more-oxidised OOA (MO-OOA).31,32
Recently, highly-time resolved online chemical ionisation mass spectrometry (CIMS) techniques have been applied to in situ measurements of trace gas33–36 and aerosol composition37,38 providing sensitive and selective detection of molecules spanning an atmospherically relevant oxidation range.39–41 These have been employed for the study of VOC oxidation, driving novel insights into gas-phase and SOA chemistry,1,42–46 including detailed studies of highly oxygenated organic molecules (HOM),42,47,48 oligomers49 and new particle formation (NPF).50,51 Their application in ambient measurements have mostly focused on gas phase oxidised organic compounds,52–54 with fewer studies measuring OA composition directly.55,56
This study uses a Filter Inlet for Gases and Aerosols (FIGAERO)57 coupled to an online CIMS interface using an iodide reagent ion scheme for near-molecular identification of OA composition.58 This technique has been successfully applied in Beijing previously to study a small subset of OA components,59 while here we focus on characterising its bulk composition and sources. We couple laboratory measurements of SOA from oxidation of aromatic hydrocarbons1 with ambient measurements from summer 2017 and apply positive matrix factorisation (PMF)52,60–62 to integrated thermal desorption profiles from FIGAERO-I-CIMS for the first time in Beijing to identify anthropogenic and biogenic contributions to OA.
We make use of the abundance of chemical information, evaluated in terms of its uncertainty and carry out a single factor analysis for the whole dataset to show that the behaviour of ions can be broken down into distinct sub-groups. The different time periods are used to interpret the results of the single PMF analysis, both diurnally and across three time periods defined according to meteorological and chemical conditions. In order to evaluate these groups, we identify marker ions which can be related to previous work in the lab and field, where those ions are discrete and linked to particular sources and pathways. We identify these marker ions by correlation and use them throughout the paper to understand the sources.
Ions are selected for PMF using the approach outlined in ESI†. The temporal trends and bulk properties of the PMF factors are discussed in Section 3.1, following which the aromatic content is evaluated in Section 3.2 and the key marker ions are identified in Section 3.3. These together are used in the interpretation of the factors in Section 3.4. In Section 4, we compare the derived factors with air mass origins, and external markers.
2. Methodology
Measurements were carried out as part of the joint UK–China Air Pollution and Human Health (APHH) project, which involved an intensive summer measurement campaign during May–June 2017.63 The measurement site was located at the Institute of Atmospheric Physics, Chinese Academy of Sciences (IAP-CAS) in Beijing (39°58′28′′N, 116°22′16′′E), an urban site in the north of Beijing, between the third and fourth ring roads. The analysis in this paper will focus on a period near to the end of the summer measurement campaign from 2nd–18th June 2017, which was chosen owing to stable instrument parameters during this time period. Positive matrix factorisation (PMF) analysis was carried out on measurements from a time-of-flight chemical ionisation mass spectrometer (ToF-CIMS) using an iodide ionisation system58 coupled to a FIGAERO. This paper focuses on the results from the FIGAERO-CIMS, with other co-located measurements utilised to frame the discussion and for interpretation of factors. A detailed description of the methodology is provided in the ESI.†3. Results
3.1. Overview of FIGAERO-CIMS factor solution
FIGAERO-CIMS measurements during the period of 2nd–18th June 2017 are presented in this study alongside comparisons with various co-located measurements as part of the APHH-Beijing field campaign.63 Here, we briefly highlight the key features of relevance for description of the derived PMF factors oxidants (OH, O3 and NO3), ambient temperature and aerosol mass concentrations as measured by AMS.The measurements presented in this study have been split into three periods: period 1 (2017-06-02 13:00 to 2017-06-07 10:00), period 2 (2017-06-08 04:00 to 2017-06-13 18:00) and period 3 (2017-06-13 20:00 to 2017-06-18 10:00) based on differing meteorological and oxidant conditions. Specifically, the criteria used to separate these three periods were the relative differences in ambient temperature, NOx, OH and O3 mixing ratios.
Period 1 is characterised by lower-NOx conditions (26 ppb), lower temperatures (23 °C) and average ozone concentrations (42 ppb) alongside a 41% contribution of organics to total aerosol mass (Fig. S5†). In contrast, period 2 has elevated NOx concentrations (39 ppb) and low particle mass loadings (8 μg m−3), which were dominated by organics, making up 54% of aerosol mass (Fig. S5†). Period 3 is distinguished most by its high O3 (71 ppb, mean daytime 07:00–19:00 – 93 ppb), high overnight NO3 (16 ppt) and high ambient temperature (31 °C). In addition, period 3 showed high sulphate and organic contributions to aerosol mass, contributing 26% and 54%, respectively (Fig. S5†). It should be noted that at peak photochemistry, OH concentrations can peak at concentrations reaching as high as 2.8 × 107 molecules cm−3, over an order of magnitude higher than the global mean average concentration64 of 1.5 × 106 molecules cm−3.
Eight FIGAERO-CIMS organic aerosol factors were resolved during this study (Fig. 1 and 2). Here, we briefly introduce these factors, their bulk composition, temporal behaviour and trends across the measurement period. The bulk composition of each factor is shown in Table 1, and includes average formula and molecular weight alongside elemental ratios of oxygen-to-carbon (O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C), hydrogen-to-carbon (H:C) and nitrogen-to-carbon (N:C) which are used to calculate the carbon oxidation state (OSc) of each factor:65
:C), hydrogen-to-carbon (H:C) and nitrogen-to-carbon (N:C) which are used to calculate the carbon oxidation state (OSc) of each factor:65
| OSc = (2 × O:C) − H:C − (5 × N:C) |
 | ||
| Fig. 1 Diurnal trends (left) and time series (right) of PMF factors, AMS mass, temperature, O3, NOx, NO3 and OH. The PMF factors are ordered and named with a prefix according to diurnal trend that peak in the morning (AM), afternoon (PM), and night (NT). | ||
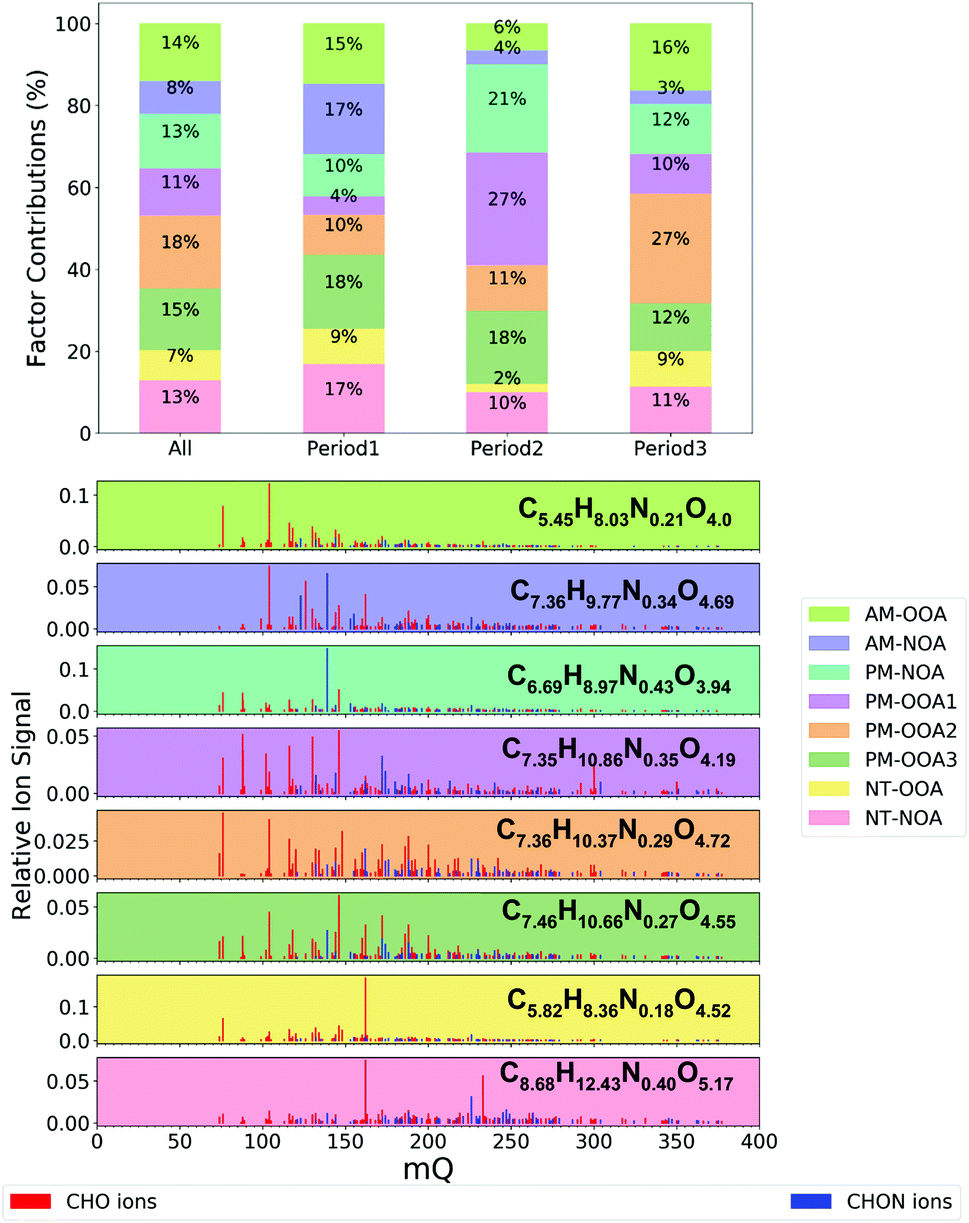 | ||
| Fig. 2 Contributions of factors to total observed signal during different time periods (top) and factor mass spectra (bottom) where mQ is the mass of the ions and the ion formulas shown are the signal weighted average ion formulas of each factor. | ||
| Factor | Signal weighted bulk properties | CHO (%) | CHON (%) | HOM (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| O:C |
H:C |
N:C |
OSc | Average formula | MW | ||||
| AM-NOA | 0.78 | 1.34 | 0.059 | −0.081 | C7.36H9.77N0.34O4.69 | 177.96 | 71 | 29 | 23 |
| AM-OOA | 0.89 | 1.51 | 0.038 | 0.092 | C5.45H8.03N0.21O4.0 | 140.58 | 84 | 16 | 10 |
| PM-NOA | 0.69 | 1.36 | 0.064 | −0.308 | C6.69H8.97N0.43O3.94 | 158.38 | 64 | 36 | 16 |
| PM-OOA3 | 0.72 | 1.43 | 0.034 | −0.162 | C7.46H10.66N0.27O4.55 | 176.92 | 79 | 21 | 20 |
| PM-OOA1 | 0.69 | 1.53 | 0.051 | −0.402 | C7.35H10.86N0.35O4.19 | 171.16 | 74 | 26 | 21 |
| PM-OOA2 | 0.79 | 1.42 | 0.042 | −0.051 | C7.36H10.37N0.29O4.72 | 178.48 | 78 | 22 | 27 |
| NT-NOA | 0.68 | 1.46 | 0.051 | −0.355 | C8.68H12.43N0.40O5.17 | 205.14 | 68 | 32 | 38 |
| NT-OOA | 0.89 | 1.48 | 0.028 | 0.156 | C5.82H8.36N0.18O4.52 | 153.06 | 87 | 13 | 14 |
This metric is widely used for comparison of oxidised components of organic aerosol. It makes the assumption that all the nitrogen atoms exist as nitrate groups and there may be associated uncertainties for nitro groups or amines and amides.65 Additionally, the contribution to factor mass spectra from ions containing carbon, hydrogen and oxygen only (CHO) and carbon, hydrogen, oxygen and nitrogen only (CHON) is included as well as the contribution of highly oxygenated organic molecules (HOM), defined here as ions which contain six or more oxygen atoms.48
The factors are distinguished by their diurnal profiles, splitting into two morning factors (AM), four afternoon factors (PM) and two night-time factors (NT). All factors have some nitrogen content, but to distinguish between them here we name those with higher nitrogen content (CHON% – Table 1) as NOA. The morning factors consist of nitrogen-containing organic aerosol (AM-NOA) and oxidised organic aerosol (AM-OOA). Of the afternoon factors, two are dominant diurnally during the higher-NOx conditions, and particularly abundant during period 2 (Fig. 2), one which is nitrogen-containing (PM-NOA) and one which is oxidised organic aerosol (PM-OOA2 and PM-OOA3). The remaining two afternoon factors show similar composition and are diurnally most prevalent during the lower-NOx afternoon hours (PM-OOA1 and PM-OOA2). The night-time factors consist of nitrogen-containing organic aerosol (NT-NOA) and oxidised organic aerosol (NT-OOA).
3.2. Comparison with aromatic SOA
VOC contributions to SOA are often estimated through application of yields derived from laboratory studies66–68 to ambient VOC concentrations.19,69 It has been suggested by such approaches that aromatic VOCs, abundant in Beijing during the summertime,70 are the dominant contributor to SOA,71 with contributions as high as 98.2%.19 The importance of aromatic VOCs was further highlighted during the Asia Pacific Economic Conference (APEC) emission controls, where a 50–80% reduction in aromatic VOCs from vehicle emissions and solvent evaporation72 was concurrent with a 37% reduction in SOA formation.73 However, these measurement approaches are indirect and thus limited by large variations in SOA yields,74 effects of VOC mixtures upon SOA chemistry10,75,76 and meteorology.77Recently, we studied the composition of SOA from several aromatic hydrocarbons using a FIGAERO-CIMS and identified a suite of ring-retaining products, including HOM, which contributed up to 43% of observed signal from single component aromatic experiments.1 Here we compare the ions observed in the ambient factors with those from this aromatic study to estimate the potential contribution that aromatics could have to measured SOA in Beijing. As discussed in Mehra et al. (2020),1 many ions observed from aromatics have ion formulas concurrent with products from oxidation of other precursors. Due to the potential for ring-scission products, in particular, to be attributed to many different sources, here we focus upon the ring-retaining products identified from aromatic oxidation in Mehra et al. (2020).1 Using these ions as markers provides a lower constraint on the contribution of aromatic SOA to the observed factor mass spectra, as many of the small oxidised products in the factors are likely to have an aromatic contribution from ring-scission.
Fig. 3 shows the proportion of mass spectral signal of each factor attributable to ion formulae measured during the aromatic oxidation experiments classified as ring-retaining (C6–C9 and double bound equivalent (DBE)≥ 4).1 This analysis indicates that the PM-NOA has the largest potential contribution from aromatic SOA products (26%), followed by AM-NOA (19%) and NT-NOA (16%). It should be noted that these contributions do not include nitro-aromatics which were not observed by Mehra et al. (2020)1 due to the lower HO2:NO ratios employed in these experiments. Though there is ambiguity in the potential sources of some specific ions, this comparison provides a useful basis for comparing SOA in different factors.
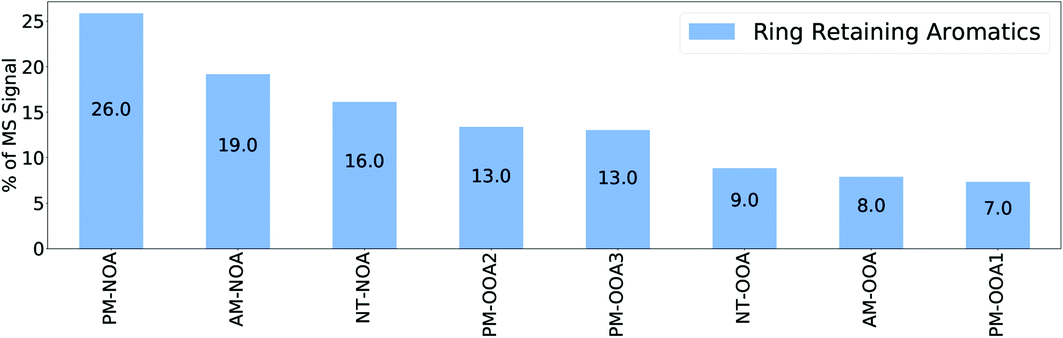 | ||
| Fig. 3 Contribution of ring-retaining aromatic SOA ions from Mehra et al. (2020)1 to PMF factor mass spectra. | ||
3.3. Identification of marker ions for factors
Many of the most abundant ions observed across the factor mass spectra (Fig. S5–S8†) are small oxidised ions which can be attributed to various sources including ring-scission pathways from aromatic oxidation1 and fragmentation pathways of biogenic oxidation.78 These ions are less likely to act as markers of a particular source and thus here we outline an approach taken to identify marker ions for the factors.We calculated the Pearson correlation coefficient (R) between the time series of each ion and the factors themselves, and those which have an R of greater than 0.5 are considered potential marker ions of the factor. These ions are ranked by decreasing correlation and the top twenty ions, where available, are classified as markers of a given factor. This threshold enables more ions to be included in this classification, as use of single ions as markers can lead to misinterpretation of factor profiles due to potential isomers from different sources.1 A similar approach has been applied previously with PMF analysis, whereby a cluster analysis of the ions and factors yielded unique or nearly unique markers for each factor.79
Marker ions span a broad range of chemical space, as shown in Fig. 4, and with the exception of AM-OOA and NT-OOA, which show similar composition, are more unique than the small oxidised ions abundant in the factor mass spectra (Fig. S5–S8†). Marker ions contribute differently to the mass spectra of the factors, with the highest contribution to the mass spectra from marker ions observed for NT-NOA (79%) followed by AM-OOA (61%), AM-NOA (57%), PM-OOA3 (53%), PM-OOA2 (25%) and PM-NOA (21%). In the case of PM-OOA1, no ions show a strong correlation with the time series of this factor, in some cases this can suggest factor splitting in the PMF solution.62 In order to establish if this was the case, 4–10 factor PMF solutions were evaluated, with this factor observed in all solutions. Due to its high contribution to the observed SOA signal in period 2 (27%), its correlation in this period alone was considered, and three potential marker ions were identified. This approach groups the marker ions whose details and likely origins will be discussed in the next section.
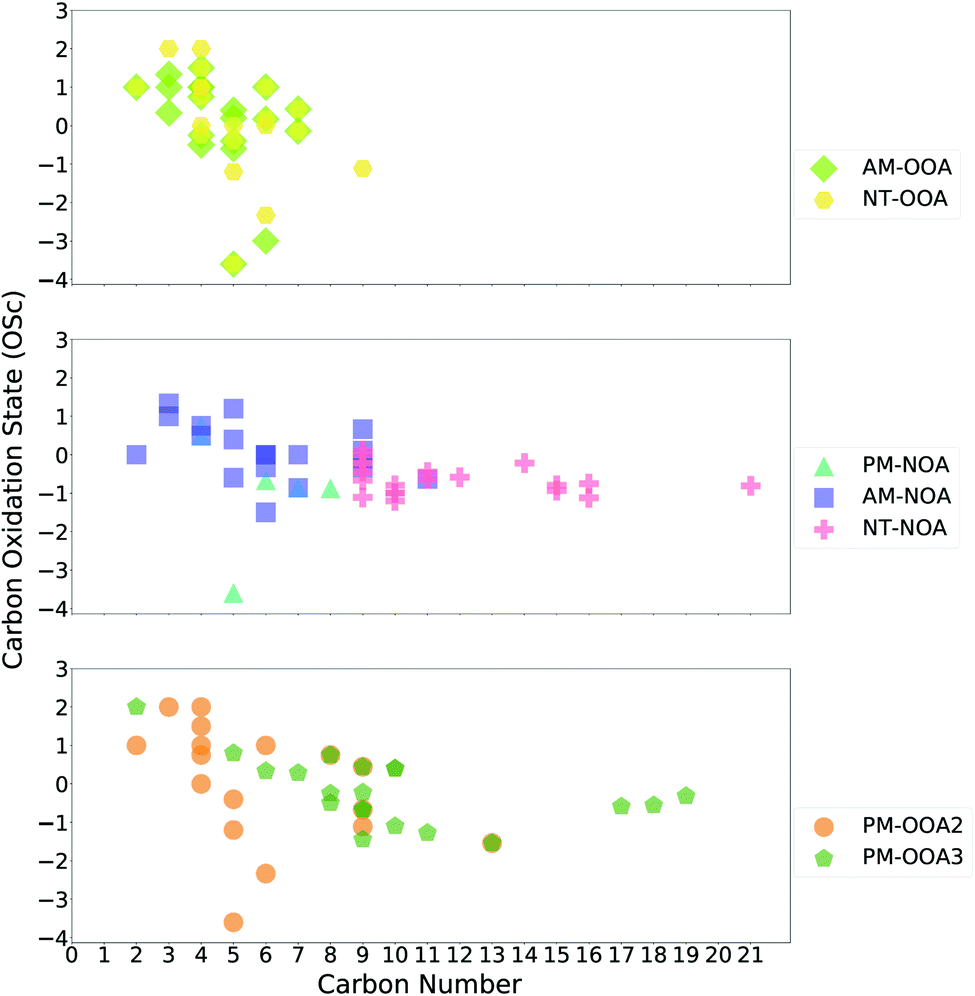 | ||
| Fig. 4 Oxidation state vs. carbon number plot of top 20 ions most correlated with factors (note: PM-OOA1 shows poor correlation with all ions across the whole time period and thus is not included; its few marker ions during period 2 are discussed in Section 3.4.2). | ||
3.4. Interpretation of factors
In this section, we bring together the bulk composition presented in Table 1, the comparisons with ring-retaining aromatic SOA products presented in Section 3.2 and the markers identified in Section 3.3, to discuss the likely sources of the factors. The marker ions discussed in this section are presented in Tables S9–S15† and the most abundant ions for each factor in Tables S1–S8,† with a summary of their potential sources from literature.AM-NOA consists of larger oxidised ions (C# 7.36, O# 4.69, MW 177.96), with a contribution of 29% from CHON ions and 23% from HOM ions suggesting less processed aerosol. Aromatic ring-retaining products contribute 19% to the mass spectrum, the second highest contribution of all factors. Of the 20 marker ions from this factor, 13 were observed in the aromatic SOA experiments, of which 2 have potential contributions from other sources. These are C3H4O4 and C5H6O4, which were also observed in AM-OOA, and their non-aromatic sources were highlighted as unlikely to be important given the temporal trends of these factors. Of the 7 marker ions not observed in the aromatic SOA experiments, the majority are nitroaromatics which have previously been observed from vehicle emissions86 and biomass burning84 alongside secondary formation from high-NOx oxidation of aromatics.43,87–89 Specifically, these included C6H5NO4 (4-nitrocatechol, Finewax et al., 2018),90 C7H5NO5 (perbenzoyl nitrate, Busilacchio et al., 2016),91 C7H7NO3 (methyl nitrophenol, Priestley et al., 2018)92 and C6H9NO7.43
PM-OOA1 consists of larger and more oxygenated ions (C# 7.35, O# 4.19, MW 171.16) alongside a higher HOM contribution (21%) and lower CHON contribution (26%). It also has the lowest contribution from aromatic ring-retaining SOA products (7%). Its marker ions can only be identified from period 2 and consist of tentatively identified C14H20O7, C17H18O8 and C15H16N2O5. C14H20O7 has been previously attributed to biomass burning,79 while C17H18O8 was observed during particle nucleation experiments from α-pinene93 and C15H16N2O5 has previously been reported as an oligomer formed during aqueous phase methylglyoxal oxidation.94 These observations suggest that this factor could have mixed sources, and more information is needed to establish its sources.
PM-OOA2 consists of large oxygenated ions (C# 7.36, O# 4.72, MW 178.48) and has the largest contribution (27%) from HOM ions of the afternoon factors and 13% contribution from aromatic ring-retaining SOA products. Of the 17 marker ions identified for this factor, 14 were observed in the aromatic SOA experiments, 4 of which have potential biogenic sources. Two of these ions are associated with oxidation of isoprene: C5H10O4 which is most abundant during low-NOx conditions45,82 and C4H8O4 which may correspond to 2-methyl glyceric acid, a major product of isoprene oxidation.95 The remaining two consist of C5H12O3 which is formed from oxidation of 2-methyl-3-buten-2-ol (MBO)96 under low-NOx conditions and C4H4O5 which has been previously attributed to the aqueous phase oxidation of α-pinene.80 The ions not observed in aromatic SOA include C9H12O, likely corresponding to trimethyl phenol and C13H26O3, which is potentially attributable to hydroxytridecanoic acid and has previously been reported in a cigarette smoke PMF factor.79 Of the aromatic marker ions, two have previously been attributed to 1,3,5-trimethyl benzene oxidation: C8H8O7 (ref. 97) and C9H16O5, which is a product attributed to a second OH attack42 and another to 1,2,4-trimethyl benzene oxidation C9H10O7.44 Together these observations suggest that this factor is highly related to low-NOx oxidation chemistry leading to HOM formation, with an additional strong influence from isoprene oxidation.
PM-OOA3 shows a similar bulk composition and contribution of aromatic ring-retaining ions to that of PM-OOA2, but a smaller contribution from HOM ions (20%). Of its marker ions, 13 are observed in the aromatic SOA experiments, of which 4 have other potential biogenic sources. These include C8H12O5 and C9H14O6 from limonene,98,99 C10H12O8 from pinanediol100 alongside C8H12O4 and C9H14O4 from α-pinene.101,102 Of the marker ions not observed from aromatic oxidation, C18H18O4, C19H18O6 were previously associated with degradation of lignin,103,104 potentially related to barbeque cooking emissions. Taken together these observations suggest that this factor is a mixture of biogenic and anthropogenic SOA, with monoterpene and cooking influences.
NT-NOA is the largest and most oxygenated of all the factors (C# 8.68, O# 5.17, MW 205.14) alongside containing the largest contribution from HOM (38%) and CHON (32%) ions. It also contains a 16% contribution from ring-retaining aromatic ions. Of its marker ions, only 2 were observed in aromatic SOA experiments. The remainder include terpene oxidation products such as C10H19NO7, C10H15NO6, C9H13NO8, C9H13NO7, C9H13NO9, C10H17NO5 and C9H15NO7 (ref. 60, 107 and 108) and sesquiterpene oxidation products including C15H25NO8, C15H25NO9 and C16H27NO7.109 Together these observations demonstrate that this factor corresponds to a terpene dominated night-time biogenic SOA factor likely associated with NO3 oxidation chemistry.
4. Discussion
4.1. Regional influences
Regional transport is an important source of organic aerosol in Beijing and thus here we compare the factors with co-located measurements and air mass fractions derived from HYSPLIT back trajectories110 to better understand the regional influences. A map showing the regional boundaries defined in Liu et al.110 is included in the ESI (Fig. S9†). Fig. 5 shows that the air masses change frequently in period 1, with a cycling between the northern plateau and the western and eastern north China plain (NCP) regions on timescales of <1 day, while periods 2 and 3 have a prolonged contribution from eastern and western NCP air masses, respectively. | ||
| Fig. 5 Comparison of fractional factor contributions with HYSPLIT fractional air mass contributions. | ||
Upon inspection of the factors shown in Fig. 5 alongside the air mass fractions, five of the factors show relevant patterns with regional influences and are thus included in Fig. 6. The factors most likely associated with regional transport include AM-OOA which relates to the NCP (Eastern and Western), PM-OOA1 which relates to air masses from the eastern NCP and NT-OOA which relates to air masses from the western NCP. AM-OOA and NT-OOA are marked by a strong influence from small oxidised ions indicative of highly processed or aged aerosol whose sources are numerous while PM-OOA1 shows few distinctive markers with mixed sources as discussed in Section 3.4. The remaining three factors do not show a distinctive regional influence and are more influenced by biogenic oxidation chemistry, these are shown in Fig. 7 and discussed further in Section 4.3.
 | ||
| Fig. 6 Comparison of factors with regional influence with external tracers and air mass origins. Events A–F are defined in this figure for illustrative purposes. | ||
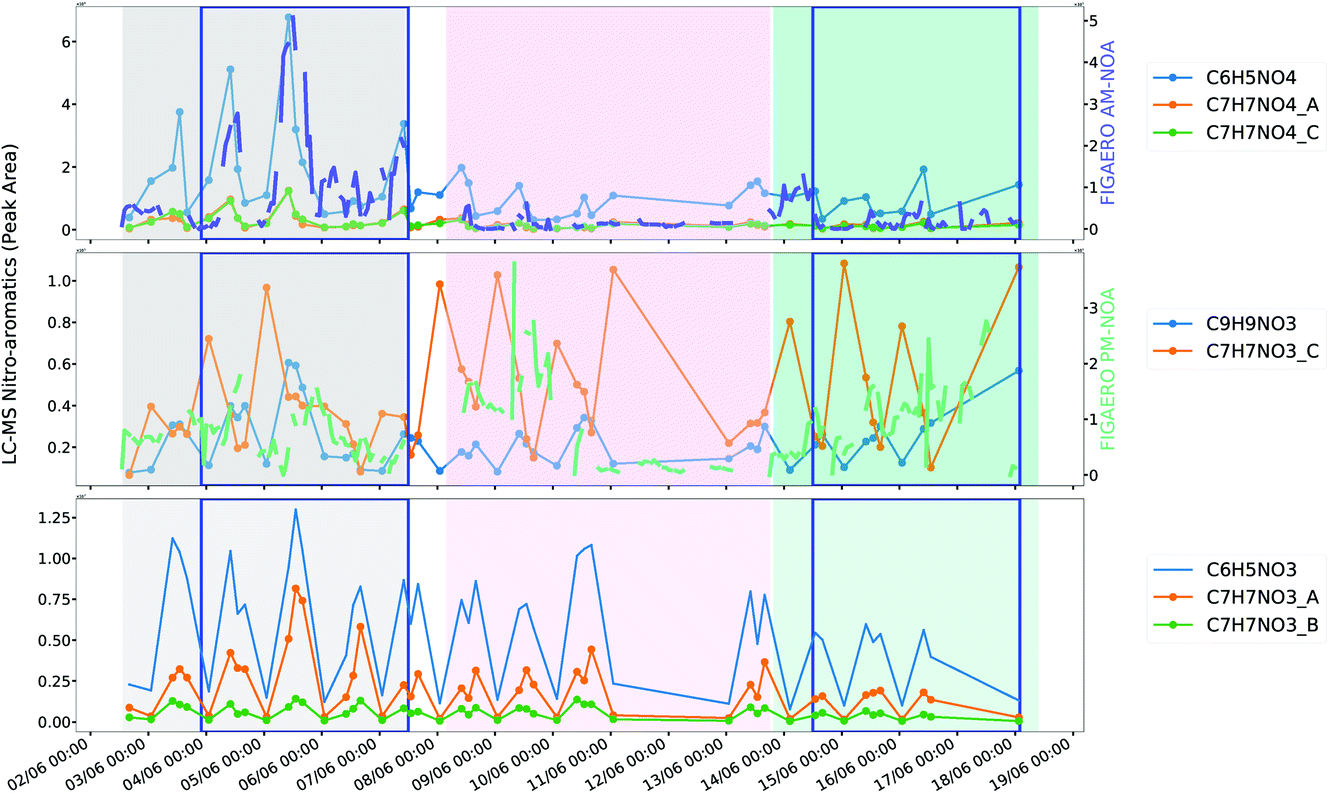 | ||
| Fig. 7 Comparison of AM-NOA and PM-NOA with nitroaromatic ions measured by UPLC-MS. Sample midpoint has been used to plot the offline filter data. | ||
Fig. 6 shows six events (A–F) which are defined for illustrative purposes. AM-OOA is classified as a morning factor owing to its sharp rises typically occurring during the early morning during events A, B, D and E. Its composition is highly processed and includes ions associated with aqueous phase oxidation of α-pinene and methyl glyoxal alongside potential influence from guaiacol oxidation as outlined in Section 3.4.1. Its sharp rise during event B is concurrent with a shift in air mass origin to that from the eastern NCP which is associated with a spike in SO2, HCN, aromatic hydrocarbons and chloride aerosol indicative of industrial sources. There is also a slight enhancement in sulphate aerosol and C6H10O5, typically attributed to levoglucosan, a marker for biomass burning. This spike occurs at around 6 am and thus is likely to be associated with a breakdown and mixing of the residual layer into the boundary layer during the early hours of the day. The behaviour of AM-OOA during period 3 is different owing to a shift in the dominant air mass origin to that from the western NCP. Enhancement of the factor during this period is concurrent with only C6H10O5 and aromatics during event D, while during event E and F its enhancements are associated with slight increases in aromatics, sulphate and SO2. Events E and F in particular show a stronger association of this factor with C6H10O5, indicating that the air masses from the western NCP are more related to biomass burning than industrial sources.
NT-OOA appears to represent air masses from the western NCP throughout the measurement period, showing greatest enhancement during event F in period 3 shown in Fig. 6. The polar plot of NT-OOA (Fig. S12†) shows that this factor is highest at high wind speeds from the west which correspond to the strongest enhancement in C6H10O5 during event F. This particular event is not associated with enhancements in any of the other markers and thus suggests it is most likely associated with long range transport of biomass burning aerosol from the western NCP. This is further supported by levoglucosan being both a marker of this factor and its most abundant ion (Tables S2 and S10†).
PM-OOA1 shows a correspondence with the prolonged air mass contribution from the Eastern NCP during period 2. Its key markers, as discussed in Section 3.4.2, are few and provide limited insight into its sources. During event C in Fig. 6, when this factor is most abundant, the wind direction is strongly from the southeast, where the industrial sources lie and this period contains two spikes in SO2 which suggest an industrial plume. It should be noted that this factor is the most abundant under high-NOx conditions when aerosol concentrations were also relatively low.
4.2. Sources and formation of nitro-aromatics
Nitro-aromatics can be attributed to various sources, including biomass burning111 and vehicle emissions,112 alongside secondary formation by gas, particle and aqueous phase oxidation processes113,114 and are an important contributor to brown carbon in the polluted atmosphere (BrC).115,116 AM-NOA and PM-NOA are characterised by nitro-aromatic marker ions and show different temporal trends as well as composition, most likely representing different sources and formation pathways.AM-NOA, which has the second highest aromatic content of the factors is abundant during the same time periods as the highly-aged AM-OOA (Fig. 6) which suggests that its source may be associated with regional transport and early morning mixing from the residual layer as the boundary layer develops. One potential source of nitro-aromatics in this factor can be related to biomass burning, however its temporal trends are dissimilar to that of C6H10O5 (levoglucosan) suggesting that its sources may be different. Its early morning peak suggests a potential influence from traffic, further supported by its polar plot (Fig. S12†) which shows it is elevated at low wind speeds and those from the north/north-east, which may suggest a traffic source from the Jingzang Highway to the east and fourth ring road to the north as has been reported previously for NOx emission flux.70 This source could be further supported by the increase in HCN, which, though widely thought to be a marker of biomass burning,117 has been shown to be an important emission from traffic in urban areas.118 However, unlike NOx, which has a reasonably consistent source across the measurement period,70 this factor is most abundant during period 1 and appears more episodic in nature than direct source emissions which would typically exhibit more regular diurnal behaviour. In addition, its key nitro-aromatic marker ion is C6H5NO4 (4-nitrocatechol), which is typically not observed from direct vehicle emissions and is more related to biomass burning or secondary formation.88,90 Several other ions measured by UPLC-MS including isomers of C7H7NO4 are strongly correlated with AM-NOA as shown in Fig. 7.
C6H5NO4 (4-nitrocatechol) is expected to form in the aqueous phase under polluted environmental conditions119 and night-time sources such as nitrate radical reactions have been previously suggested to be attributable to their formation.90 These factors may suggest, that as with the transported AM-OOA which appears to be introduced into the daytime boundary layer through mixing of the residual layer in the morning, this is related to nitro-aromatics formed in this residual layer overnight. The formation of nitro-aromatics in the residual layer can occur due to a decoupling from the nocturnal boundary layer in which high O3 and low-NO conditions lead to rapid conversion of NO2 to NO3. The potential for aqueous phase pathways to be important are further supported by AM-OOA whose markers include ions associated with aqueous phase oxidation of methyl glyoxal, as discussed in Section 3.3.
The reduction in AM-NOA, which is larger and less oxidised, is concurrent with an increase in AM-OOA (Fig. 6), which is smaller and more highly processed, potentially corresponding to its processing in Beijing while its reduction in period 3 could also be attributed to the elevated temperature which is known to impact upon gas–particle partitioning of nitro-aromatics120 alongside gas-phase photolysis121 and oxidation due to the higher OH concentrations measured during period 3. Together, these observations suggest that this factor is strongly influenced by aromatics with a potential influence of transported biomass burning sources or secondary formation in a decoupled residual layer, which are introduced from its mixing as the daytime boundary layer develops.
PM-NOA, which has the highest contribution from ring-retaining aromatic SOA products of all the factors, shows a different temporal profile and a more consistent source throughout the measurement period. In addition, it peaks around peak photochemistry suggesting that it is related to direct emission, potentially from traffic sources or from secondary formation. The markers of this factor include C6H5NO3, which can be related to 4-nitrophenol speciated by the UPLC-MS during this measurement period and is typically emitted from vehicle emissions112,122 and C7H7NO3 which is also measured by the UPLC-MS and show more regular diurnal profiles than that of the nitro-aromatic species associated with AM-NOA (Fig. 7).
4.3. Biogenic influenced chemistry
While the other factors show a strong relationship from air mass trends, PM-OOA2, PM-OOA3 and NT-NOA show a stronger relationship with that of oxidant, radical and VOC concentrations as shown in Fig. 8. As discussed in Section 3.3, these factors are compositionally most influenced by species with biogenic origin, or those formed from low-NOx oxidation pathways of aromatics such as HOM. These factors are most dynamic and abundant during period 3, which is least influenced by regional transport. | ||
| Fig. 8 Comparison of SOA factors with external markers of biogenic sources and chemistry. | ||
PM-OOA2 shows a regular diurnal pattern which is consistent throughout the measurement period, with the exception of the end of period 2, when its levels drop down significantly under higher NO conditions shown in Fig. 8. It shows an enhancement in period 3 which corresponds with an increase in measured isoprene and ozone mixing ratios, which along with its composition further suggest this factor is isoprene related. SOA from terpenoid emissions has previously been reported to have a strong exponential relationship with temperature, and is also observed for PM-OOA2 as can be seen in Fig. 9.123–125 Though other factors show an elevated contribution under the higher temperatures to a small extent, this is by virtue of them being afternoon factors and the lack of an exponential relationship along with composition less influenced by biogenic SOA products confirms that they are less related to terpenoid emissions. This factor also shows a similar temporal trend to the sum of HOM ions, measured in the gas phase by I-CIMS, consistent with it being the afternoon factor which contains the largest contribution from particle phase HOM ions. This shows that it is not only related to biogenics but more generically to low-NO oxidation pathways, which have recently been identified as an important pathway for isoprene oxidation chemistry in Beijing.24
 | ||
| Fig. 9 Temperature dependence of afternoon factors with signal binned hourly by ambient temperature (night is 7 pm to 7 am). | ||
PM-OOA3 shows little correlation with radicals and oxidants during periods 1 and 2, but a strong correspondence with measured RO2 (described in ESI 1.2.1†) and ozone during period 3, suggesting it is related to local formation during this time period. It is likely that during period 1, the sources of these ions are less related to secondary formation pathways and more related to cooking emissions which contribute to its factor profile and to regional transport given that the measured RO2 radical concentrations are much lower during this time (Fig. 7).
NT-NOA, which is compositionally strongly related to monoterpene and sesquiterpene oxidation and contains the largest contribution from HOM ions, shows a poor correlation with NO3 during periods 1 and 2, again suggesting that during this time period the regional influence to organic aerosol is stronger and the formation cannot be resolved by the PMF and potentially the formation of these products is occurring in the residual layer. In contrast, this factor shows a strong diurnal pattern similar to that of NO3 as well as monoterpene and sesquiterpene concentrations in period 3, as well as a polar plot which shows highest levels under low wind speeds which suggest local formation (Fig. S12†).
4.4. Comparison with AMS
A factor analysis of AMS measurements during the same time period identified factors associated with cooking organic aerosol (COA), fossil fuel organic aerosol (FFOA) and three oxidised factors referred to as oxidised primary organic aerosol (OPOA), less-oxidised oxidised organic aerosol (LO-OOA) and more-oxidised oxidised organic aerosol (MO-OOA). Fig. 10 shows the potential correspondences of the FIGAERO factor components with those derived from the AMS, where OPOA, FFOA and COA are related to the factors most influenced by vehicle emissions and regional sources (AM-OOA, NT-OOA, AM-NOA and PM-NOA) and LO-OOA and MO-OOA are related to those formed in the afternoon during peak photochemistry and related to biogenic sources (PM-OOA1, PM-OOA2, PM-OOA3 and NT-NOA). It should be noted that the iodide reagent ion scheme would typically not be expected to detect all of the AMS OA, as it is particularly sensitive to multi-functional oxidised species, and thus would be expected to detect the most oxidised portion of what is observed by the AMS. Therefore, the differences in primary–secondary split of the FIGAERO and AMS factors is likely attributable to an increased proportion of less oxidised POA detected by the AMS which the CIMS does not observe, which is likely to be the case during period 2 when the AMS POA fraction is dominant. | ||
| Fig. 10 Comparison in relative contribution of CIMS and AMS PMF factors during different time periods. | ||
Looking at the relative ratios of these factors during different time periods, it is clear that the CIMS factors show additional variation in OA chemical composition that is not captured by the AMS factors. Correlations are observed between increases in AMS factors and CIMS factors between periods suggesting some broad correspondence between them. The most noticeable relationship between these factors is that the AMS shows an enhanced contribution during period 3 from LO-OOA which corresponds to the elevated contribution from PM-OOA2 in the CIMS measurements. This is the factor associated with enhanced SOA formation relating to increased temperature and strong isoprene influence. In contrast, MO-OOA is more abundant during periods 1 and 2, and this likely corresponds best with an increase in relative contributions of PM-OOA3.
5. Conclusions and implications
This study presents the first PMF factor analysis of FIGAERO-CIMS measurements from Beijing, identifying eight factors representing anthropogenic and biogenic contributions to OA alongside regional influences.Regional contributors to OA in Beijing include air masses from the eastern NCP (AM-OOA and PM-OOA1) and those from the western NCP (NT-OOA) which contribute on average 14%, 11% and 7%, respectively to FIGAERO-CIMS OA factors. Anthropogenic contributions to OA include those from PM-NOA which is most influenced by ring-retaining aromatic SOA products and AM-NOA, which is potentially associated with nocturnal processing of aromatic hydrocarbons in the residual layer, which contribute 13% and 8%. Biogenic contributions, specifically related to isoprene, monoterpenes and sesquiterpenes exist in mixed factors associated with their chemical oxidation regimes in PM-OOA2, PM-OOA3 and NT-NOA, and contribute on average 13%, 18% and 15% to OA respectively.
Together these observations show that aromatic hydrocarbons are important for SOA in Beijing, both from daytime oxidation and potential regional transport and nocturnal processing. In addition, the oxidation of biogenic emissions is dominated by isoprene during the afternoon, with monoterpene and sesquiterpenes becoming important for NO3 oxidation pathways leading to formation of large oxidised aerosol products overnight. Regional transport is most significant from the eastern NCP, which brings highly polluted air masses which participate in nocturnal processing and are introduced into the urban atmosphere in the morning.
Author contributions
AM operated the FIGAERO-CIMS, carried out the analysis and wrote the manuscript working closely with HC and MRC. JA, HC, CP, AM, TB, SDW and AB deployed the FIGAERO-CIMS in the field. MRC and HS assisted with the FIGAERO-CIMS and PMF analysis. ES, LW and DH provided the RO2 and OH data. JFH, AE, SHB and DB provided the LC-MS analysis. JRH provided the GC-FID data. JB provided the SMPS data. BO provided the NO3 data. WJFA, CNH and XW provided the PTR VOC data. DL carried out the hysplit air mass back trajectory analysis. YS, JZ and WX provided the AMS data and PMF analysis. YS and PF provided the meteorological data. LW, YW and JEK contributed to the aromatic SOA experiments. FS and JL provided the NO, NO2, SO2 and O3 measurements. MP, XC, QC, JJ, DW assisted with the interpretation and development of the results in the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Archit Mehra is fully funded by the Natural Environment Research Council (NERC) and acknowledges his funding through the NERC EAO Doctoral Training Partnership (NE/L002469/1) and CASE partnership support from Aerodyne Research Inc. Funding for this work was provided by the UK Natural Environment Research Council as part of AIRPRO (NE/N00695X/1) and AIRPOLL (NE/N007123/1) alongside UK Natural Environment Research Council and UK Medical Research Council funding under the framework of the Newton Innovation Fund (grant NE/N006976/1).References
- A. Mehra, Y. Wang, J. E. Krechmer, A. Lambe, F. Majluf, M. A. Morris, M. Priestley, T. J. Bannan, D. J. Bryant, K. L. Pereira, J. F. Hamilton, A. R. Rickard, M. J. Newland, H. Stark, P. Croteau, J. T. Jayne, R. Worsnop, M. R. Canagaratna, L. Wang and H. Coe, Atmos. Chem. Phys. Discuss., 2020, 161, 1–28 Search PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS.
- J. Haywood and O. Boucher, Rev. Geophys., 2000, 38, 513–543 CrossRef CAS.
- U. Pöschl, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef.
- S. Fuzzi, U. Baltensperger, K. Carslaw, S. Decesari, H. Denier Van Der Gon, M. C. Facchini, D. Fowler, I. Koren, B. Langford, U. Lohmann, E. Nemitz, S. Pandis, I. Riipinen, Y. Rudich, M. Schaap, J. G. Slowik, D. V. Spracklen, E. Vignati, M. Wild, M. Williams and S. Gilardoni, Atmos. Chem. Phys., 2015, 15, 8217–8299 CrossRef CAS.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. Mcfiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- J. K. Pearson, Atmos. Environ., 2019, 204, 118–124 CrossRef CAS.
- K. Sindelarova, C. Granier, I. Bouarar, A. Guenther, S. Tilmes, T. Stavrakou, J.-F. Müller, U. Kuhn, P. Stefani and W. Knorr, Atmos. Chem. Phys., 2014, 14, 9317–9341 CrossRef.
- G. McFiggans, T. F. Mentel, J. Wildt, I. Pullinen, S. Kang, E. Kleist, S. Schmitt, M. Springer, R. Tillmann, C. Wu, D. Zhao, M. Hallquist, C. Faxon, M. Le Breton, Å. M. Hallquist, D. Simpson, R. Bergström, M. E. Jenkin, M. Ehn, J. A. Thornton, M. R. Alfarra, T. J. Bannan, C. J. Percival, M. Priestley, D. Topping and A. Kiendler-Scharr, Nature, 2019, 565, 587–593 CrossRef CAS.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- Z. Chen, D. Chen, M.-P. Kwan, B. Chen, B. Gao, Y. Zhuang, R. Li and B. Xu, Atmos. Chem. Phys., 2019, 19, 13519–13533 CrossRef CAS.
- Q. Zhang, Y. Zheng, D. Tong, M. Shao, S. Wang, Y. Zhang, X. Xu, J. Wang, H. He, W. Liu, Y. Ding, Y. Lei, J. Li, Z. Wang, X. Zhang, Y. Wang, J. Cheng, Y. Liu, Q. Shi, L. Yan, G. Geng, C. Hong, M. Li, F. Liu, B. Zheng, J. Cao, A. Ding, J. Gao, Q. Fu, J. Huo, B. Liu, Z. Liu, F. Yang, K. He and J. Hao, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24463–24469 CrossRef CAS.
- J. Cheng, J. Su, T. Cui, X. Li, X. Dong, F. Sun, Y. Yang, D. Tong, Y. Zheng, Y. Li, J. Li, Q. Zhang and K. He, Atmos. Chem. Phys., 2019, 19, 6125–6146 CrossRef CAS.
- B. Zheng, D. Tong, M. Li, F. Liu, C. Hong, G. Geng, H. Li, X. Li, L. Peng, J. Qi, L. Yan, Y. Zhang, H. Zhao, Y. Zheng, K. He and Q. Zhang, Atmos. Chem. Phys., 2018, 18, 14095–14111 CrossRef CAS.
- A. Guenther, ISRN Atmos. Sci., 2013, 2013 DOI:10.1155/2013/786290.
- G. Huang, R. Brook, M. Crippa, G. Janssens-Maenhout, C. Schieberle, C. Dore, D. Guizzardi, M. Muntean, E. Schaaf and R. Friedrich, Atmos. Chem. Phys., 2017, 17, 7683–7701 CrossRef CAS.
- Y. Sun, Z. Wang, H. Dong, T. Yang, J. Li, X. Pan, P. Chen and J. T. Jayne, Atmos. Environ., 2012, 51, 250–259 CrossRef CAS.
- Q. Li, G. Su, C. Li, P. Liu, X. Zhao, C. Zhang, X. Sun, Y. Mu, M. Wu, Q. Wang and B. Sun, Sci. Total Environ., 2020, 720, 137536 CrossRef CAS.
- Z. An, R.-J. Huang, R. Zhang, X. Tie, G. Li, J. Cao, W. Zhou, Z. Shi, Y. Han, Z. Gu and Y. Ji, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8657–8666 CrossRef CAS.
- Y. Sun, W. Xu, Q. Zhang, Q. Jiang, F. Canonaco, A. S. H. Prévôt, P. Fu, J. Li, J. Jayne, D. R. Worsnop and Z. Wang, Atmos. Chem. Phys., 2018, 18, 8469–8489 CrossRef CAS.
- J. Li, Z. Han, J. Li, R. Liu, Y. Wu, L. Liang and R. Zhang, Sci. Total Environ., 2020, 703, 134937 CrossRef CAS.
- Y. Wang, M. Hu, S. Guo, Y. Wang, J. Zheng, Y. Yang, W. Zhu, R. Tang, X. Li, Y. Liu, M. Le Breton, Z. Du, D. Shang, Y. Wu, Z. Wu, Y. Song, S. Lou, M. Hallquist and J. Yu, Atmos. Chem. Phys., 2018, 18, 10693–10713 CrossRef CAS.
- M. J. Newland, D. J. Bryant, R. E. Dunmore, T. J. Bannan, W. J. F. Acton, B. Langford, J. R. Hopkins, F. A. Squires, W. Dixon, W. S. Drysdale, P. D. Ivatt, M. J. Evans, P. M. Edwards, L. K. Whalley, D. E. Heard, E. J. Slater, R. Woodward-Massey, C. Ye, A. Mehra, S. D. Worrall, A. Bacak, H. Coe, C. J. Percival, C. N. Hewitt, J. D. Lee, T. Cui, J. D. Surratt, X. Wang, A. C. Lewis, A. R. Rickard and J. F. Hamilton, Atmos. Chem. Phys. Discuss. DOI:10.5194/acp-2020-35.
- J. Li, G. Wang, C. Wu, C. Cao, Y. Ren, J. Wang, J. Li, J. Cao, L. Zeng and T. Zhu, Sci. Rep., 2018, 8, 535 CrossRef.
- D. Bryant, W. Dixon, J. Hopkins, R. Dunmore, K. Pereira, M. Shaw, F. Squires, T. Bannan, A. Mehra, S. Worrall, A. Bacak, H. Coe, C. Percival, L. Whalley, D. Heard, E. Slater, B. Ouyang, T. Cui, J. Surratt, D. Liu, Z. Shi, R. Harrison, Y. Sun, W. Xu, A. Lewis, J. Lee, A. Rickard and J. Hamilton, Atmos. Chem. Phys. Discuss., 2019, 1–30 Search PubMed.
- R. Tang, Z. Z. Wu, X. Li, Y. Wang, D. Shang, Y. Xiao, M. Li, L. Zeng, Z. Z. Wu, M. Hallquist, M. Hu and S. Guo, Atmos. Chem. Phys., 2018, 18, 4055–4068 CrossRef CAS.
- Y. Gao, H. Wang, X. Zhang, S. Jing, Y. Peng, L. Qiao, M. Zhou, D. D. Huang, Q. Wang, X. Li, L. Li, J. Feng, Y. Ma and Y. Li, Environ. Sci. Technol., 2019, 53, 8664–8671 CrossRef CAS.
- F. Yang, K. Kawamura, J. Chen, K. Ho, S. Lee, Y. Gao, L. Cui, T. Wang and P. Fu, Atmos. Environ., 2016, 124, 166–175 CrossRef CAS.
- Q. Zhang, J. L. Jimenez, M. R. Canagaratna, I. M. Ulbrich, N. L. Ng, D. R. Worsnop, Y. Sun, Q. Zhang, Y. Sun, J. L. Jimenez, I. M. Ulbrich, M. R. Canagaratna, N. L. Ng and D. R. Worsnop, Anal. Bioanal. Chem., 2011, 401, 3045–3067 CrossRef CAS.
- J. Li, Z. Liu, W. Gao, G. Tang, B. Hu, Z. Ma and Y. Wang, Atmos. Environ., 2020, 220, 117070 CrossRef CAS.
- W. Hu, M. Hu, W. Hu, J. L. Jimenez, B. Yuan, W. Chen, M. Wang, Y. Wu, C. Chen, Z. Wang, J. Peng, L. Zeng and M. Shao, J. Geophys. Res.: Atmos., 2016, 121, 1955–1977 CAS.
- T. J. Bannan, A. Bacak, J. B. A. Muller, A. M. Booth, B. Jones, M. Le Breton, K. E. Leather, M. Ghalaieny, P. Xiao, D. E. Shallcross and C. J. Percival, Atmos. Environ., 2014, 83, 301–310 CrossRef CAS.
- L. G. Huey, Mass Spectrom. Rev., 2007, 26, 166–184 CrossRef CAS.
- P. Veres, J. M. Roberts, C. Warneke, D. Welsh-Bon, M. Zahniser, S. Herndon, R. Fall and J. De Gouw, Int. J. Mass Spectrom., 2008, 274, 48–55 CrossRef CAS.
- R. Woodward-Massey, Y. M. Taha, S. G. Moussa and H. D. Osthoff, Atmos. Environ., 2014, 98, 693–703 CrossRef CAS.
- B. H. Lee, F. D. Lopez-Hilfiker, E. L. D’ambro, P. Zhou, M. Boy, T. Petäjä, L. Hao, A. Virtanen and J. A. Thornton, Atmos. Chem. Phys., 2018, 18, 11547–11562 CrossRef CAS.
- F. D. Lopez-Hilfiker, C. Mohr, E. L. D’ambro, A. Lutz, T. P. Riedel, C. J. Gaston, S. Iyer, Z. Zhang, A. Gold, J. D. Surratt, B. H. Lee, T. Kurten, W. W. Hu, J. Jimenez, M. Hallquist and J. A. Thornton, Environ. Sci. Technol., 2016, 50, 2200–2209 CrossRef CAS.
- G. Isaacman-VanWertz, P. Massoli, R. E. O’Brien, J. B. Nowak, M. R. Canagaratna, J. T. Jayne, D. R. Worsnop, L. Su, D. A. Knopf, P. K. Misztal, C. Arata, A. H. Goldstein and J. H. Kroll, Faraday Discuss., 2017, 200, 579–598 RSC.
- G. Isaacman-VanWertz, P. Massoli, R. O’Brien, C. Lim, J. P. Franklin, J. A. Moss, J. F. Hunter, J. B. Nowak, M. R. Canagaratna, P. K. Misztal, C. Arata, J. R. Roscioli, S. T. Herndon, T. B. Onasch, A. T. Lambe, J. T. Jayne, L. Su, D. A. Knopf, A. H. Goldstein, D. R. Worsnop and J. H. Kroll, Nat. Chem., 2018, 10, 462–468 CrossRef CAS.
- M. Riva, P. Rantala, J. E. Krechmer, O. Peräkylä, Y. Zhang, L. Heikkinen, O. Garmash, C. Yan, M. Kulmala, D. Worsnop and M. Ehn, Atmos. Meas. Tech., 2019, 12, 2403–2421 CrossRef CAS.
- U. Molteni, F. Bianchi, F. Klein, I. El Haddad, C. Frege, M. J. Rossi, J. Dommen and U. Baltensperger, Atmos. Chem. Phys., 2018, 18, 1909–1921 CrossRef CAS.
- E. Tsiligiannis, J. Hammes, C. M. Salvador, T. F. Mentel and M. Hallquist, Atmos. Chem. Phys., 2019, 19, 15073–15086 CrossRef CAS.
- A. Zaytsev, A. R. Koss, M. Breitenlechner, J. E. Krechmer, K. J. Nihill, C. Y. Lim, J. C. Rowe, J. L. Cox, J. Moss, J. R. Roscioli, M. R. Canagaratna, D. R. Worsnop, J. H. Kroll and F. N. Keutsch, Atmos. Chem. Phys., 2019, 19, 15117–15129 CrossRef CAS.
- E. L. D’ambro, B. H. Lee, J. Liu, J. E. Shilling, C. J. Gaston, F. D. Lopez-Hilfiker, S. Schobesberger, R. A. Zaveri, C. Mohr, A. Lutz, Z. Zhang, A. Gold, J. D. Surratt, J. C. Rivera-Rios, F. N. Keutsch and J. A. Thornton, Atmos. Chem. Phys., 2017, 17, 159–174 CrossRef.
- D. Aljawhary, A. K. Y. Lee and J. P. D. Abbatt, Atmos. Meas. Tech., 2013, 6, 3211–3224 CrossRef CAS.
- O. Garmash, M. P. Rissanen, I. Pullinen, S. Schmitt, O. Kausiala, R. Tillmann, C. Percival, T. J. Bannan, M. Priestley, Å. M. Hallquist, E. Kleist, A. Kiendler-Scharr, M. Hallquist, T. Berndt, G. McFiggans, J. Wildt, T. F. Mentel and M. Ehn, Atmos. Chem. Phys., 2020, 20, 515–537 CrossRef CAS.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS.
- W. Huang, H. Saathoff, A. Pajunoja, X. Shen, K.-H. Naumann, R. Wagner, A. Virtanen, T. Leisner and C. Mohr, Atmos. Chem. Phys., 2018, 18, 2883–2898 CrossRef CAS.
- C. Mohr, F. D. Lopez-Hilfiker, T. Yli-Juuti, A. Heitto, A. Lutz, M. Hallquist, E. L. D’Ambro, M. P. Rissanen, L. Hao, S. Schobesberger, M. Kulmala, R. L. Mauldin, U. Makkonen, M. Sipilä, T. Petäjä and J. A. Thornton, Geophys. Res. Lett., 2017, 44, 2958–2966 CrossRef CAS.
- L. Yao, O. Garmash, F. Bianchi, J. Zheng, C. Yan, J. Kontkanen, H. Junninen, S. B. Mazon, M. Ehn, P. Paasonen, M. Sipilä, M. Wang, X. Wang, S. Xiao, H. Chen, Y. Lu, B. Zhang, D. Wang, Q. Fu, F. Geng, L. Li, H. Wang, L. Qiao, X. Yang, J. Chen, V. M. Kerminen, T. Petäjä, D. R. Worsnop, M. Kulmala and L. Wang, Science, 2018, 361, 278–281 CrossRef CAS.
- C. Yan, W. Nie, M. Äijälä, M. P. Rissanen, M. R. Canagaratna, P. Massoli, H. Junninen, T. Jokinen, N. Sarnela, S. Häme, S. Schobesberger, F. Canonaco, A. S. H. Prevot, T. Petäjä, M. Kulmala, M. Sipilä, D. R. Worsnop and M. Ehn, Atmos. Chem. Phys., 2016, 16, 12715–12731 CrossRef CAS.
- Y. Zhang, O. Peräkylä, C. Yan, L. Heikkinen, M. Äijälä, K. R. Daellenbach, Q. Zha, M. Riva, O. Garmash, H. Junninen, P. Paatero, D. Worsnop and M. Ehn, Atmos. Chem. Phys., 2020, 20, 5945–5961 CrossRef CAS.
- J. Brean, R. M. Harrison, Z. Shi, D. C. S. Beddows, W. J. F. Acton, C. N. Hewitt, F. A. Squires and J. Lee, Atmos. Chem. Phys., 2019, 19, 14933–14947 CrossRef CAS.
- C. Mohr, J. A. Thornton, A. Heitto, F. D. Lopez-Hilfiker, A. Lutz, I. Riipinen, J. Hong, N. M. Donahue, M. Hallquist, T. Petäjä, M. Kulmala and T. Yli-Juuti, Nat. Commun., 2019, 10, 4442 CrossRef CAS.
- V. Pospisilova, F. D. Lopez-Hilfiker, D. M. Bell, I. El Haddad, C. Mohr, W. Huang, L. Heikkinen, M. Xiao, J. Dommen, A. S. H. Prevot, U. Baltensperger and J. G. Slowik, Sci. Adv., 2020, 6(11), eaax8922 CrossRef CAS.
- F. D. Lopez-Hilfiker, C. Mohr, M. Ehn, F. Rubach, E. Kleist, J. Wildt, T. F. Mentel, A. Lutz, M. Hallquist, D. Worsnop and J. A. Thornton, Atmos. Meas. Tech., 2014, 7, 983–1001 CrossRef.
- B. H. Lee, F. D. Lopez-Hilfiker, C. Mohr, T. Kurtén, D. R. Worsnop, J. A. Thornton, T. Kurte, D. R. Worsnop, J. A. Thornton, T. Kurtén, D. R. Worsnop and J. A. Thornton, Environ. Sci. Technol., 2014, 48, 6309–6317 CrossRef CAS.
- M. Le Breton, Y. Wang, Å. M. Hallquist, R. K. Pathak, J. Zheng, Y. Yang, D. Shang, M. Glasius, T. J. Bannan, Q. Liu, C. K. Chan, C. J. Percival, W. Zhu, S. Lou, D. Topping, Y. Wang, J. Yu, K. Lu, S. Guo, M. Hu and M. Hallquist, Atmos. Chem. Phys., 2018, 18, 10355–10371 CrossRef CAS.
- P. Massoli, H. Stark, M. R. Canagaratna, J. E. Krechmer, L. Xu, N. L. Ng, R. L. Mauldin, C. Yan, J. Kimmel, P. K. Misztal, J. L. Jimenez, J. T. Jayne and D. R. Worsnop, ACS Earth Space Chem., 2018, 2, 653–672 CrossRef CAS.
- Y. Chen, M. Takeuchi, T. Nah, L. Xu, M. R. Canagaratna, H. Stark, K. Baumann, F. Canonaco, A. S. H. Prévôt, L. G. Huey, R. J. Weber and N. L. Ng, Atmos. Chem. Phys. Discuss., 2020, 20, 8421–8440 CrossRef CAS.
- I. M. Ulbrich, M. R. Canagaratna, Q. Zhang, D. R. Worsnop and J. L. Jimenez, Atmos. Chem. Phys., 2009, 9, 2891–2918 CrossRef CAS.
- Z. Shi, T. Vu, S. Kotthaus, R. M. Harrison, S. Grimmond, S. Yue, T. Zhu, J. Lee, Y. Han, M. Demuzere, R. E. Dunmore, L. Ren, D. Liu, Y. Wang, O. Wild, J. Allan, W. J. Acton, J. Barlow, B. Barratt, D. Beddows, W. J. Bloss, G. Calzolai, D. Carruthers, D. C. Carslaw, Q. Chan, L. Chatzidiakou, Y. Chen, L. Crilley, H. Coe, T. Dai, R. Doherty, F. Duan, P. Fu, B. Ge, M. Ge, D. Guan, J. F. Hamilton, K. He, M. Heal, D. Heard, C. N. Hewitt, M. Hollaway, M. Hu, D. Ji, X. Jiang, R. Jones, M. Kalberer, F. J. Kelly, L. Kramer, B. Langford, C. Lin, A. C. Lewis, J. Li, W. Li, H. Liu, J. Liu, M. Loh, K. Lu, F. Lucarelli, G. Mann, G. McFiggans, M. R. Miller, G. Mills, P. Monk, E. Nemitz, F. O’Connor, B. Ouyang, P. I. Palmer, C. Percival, O. Popoola, C. Reeves, A. R. Rickard, L. Shao, G. Shi, D. Spracklen, D. Stevenson, Y. Sun, Z. Sun, S. Tao, S. Tong, Q. Wang, W. Wang, X. Wang, X. Wang, Z. Wang, L. Wei, L. Whalley, X. Wu, Z. Wu, P. Xie, F. Yang, Q. Zhang, Y. Zhang, Y. Zhang and M. Zheng, Atmos. Chem. Phys., 2019, 19, 7519–7546 CrossRef CAS.
- J. Lelieveld, S. Gromov, A. Pozzer and D. Taraborrelli, Atmos. Chem. Phys., 2016, 16, 12477–12493 CrossRef CAS.
- J. H. Kroll, N. M. Donahue, J. L. Jimenez, S. H. Kessler, M. R. Canagaratna, K. R. Wilson, K. E. Altieri, L. R. Mazzoleni, A. S. Wozniak, H. Bluhm, E. R. Mysak, J. D. Smith, C. E. Kolb and D. R. Worsnop, Nat. Chem., 2011, 3, 133–139 CrossRef CAS.
- C. Xavier, A. Rusanen, P. Zhou, C. Dean, L. Pichelstorfer, P. Roldin and M. Boy, Atmos. Chem. Phys., 2019, 19, 13741–13758 CrossRef CAS.
- N. L. Ng, J. H. Kroll, A. W. H. Chan, P. S. Chhabra, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2007, 7, 3909–3922 CrossRef CAS.
- J. Odum, T. Hoffmann, F. Bowman, D. Collins, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 1996, 30, 2580–2585 CrossRef CAS.
- W. Wu, B. Zhao, S. Wang and J. Hao, J. Environ. Sci., 2017, 53, 224–237 CrossRef CAS.
- F. A. Squires, E. Nemitz, B. Langford, O. Wild, W. S. Drysdale, W. J. F. Acton, P. Fu, C. B. Sue Grimmond, J. F. Hamilton, C. N. Hewitt, M. Hollaway, S. Kotthaus, J. Lee, S. Metzger, N. Pingintha-Durden, M. Shaw, A. R. Vaughan, X. Wang, R. Wu, Q. Zhang and Y. Zhang, Atmos. Chem. Phys. Discuss., 2020, 20, 8737–8761 CrossRef CAS.
- J. Sun, F. Wu, B. Hu, G. Tang, J. Zhang and Y. Wang, Atmos. Environ., 2016, 141, 560–570 CrossRef CAS.
- W. Yang, Y. Zhang, X. Wang, S. Li, M. Zhu, Q. Yu, G. Li, Z. Huang, H. Zhang, Z. Wu, W. Song, J. Tan and M. Shao, Atmos. Chem. Phys., 2018, 18, 12663–12682 CrossRef CAS.
- Y. Sun, Z. Wang, O. Wild, W. Xu, C. Chen, P. Fu, W. Du, L. Zhou, Q. Zhang, T. Han, Q. Wang, X. Pan, H. Zheng, J. Li, X. Guo, J. Liu and D. R. Worsnop, Sci. Rep., 2016, 6, 20668 CrossRef CAS.
- N. M. Donahue, K. E. Huff Hartz, B. Chuong, A. A. Presto, C. O. Stanier, T. Rosenhørn, A. L. Robinson and S. N. Pandis, Faraday Discuss., 2005, 130, 295–309 RSC.
- C. L. Faiola, A. Buchholz, E. Kari, P. Yli-Pirilä, J. K. Holopainen, M. Kivimäenpää, P. Miettinen, D. R. Worsnop, K. E. J. Lehtinen, A. B. Guenther and A. Virtanen, Sci. Rep., 2018, 8, 3053 CrossRef CAS.
- E. Ahlberg, J. Falk, A. Eriksson, T. Holst, W. H. Brune, A. Kristensson, P. Roldin and B. Svenningsson, Atmos. Environ., 2017, 161, 210–220 CrossRef CAS.
- T. U. Ansari, O. Wild, J. Li, T. Yang, W. Xu, Y. Sun and Z. Wang, Atmos. Chem. Phys., 2019, 19, 8651–8668 CrossRef CAS.
- N. M. Donahue, K. M. Henry, T. F. Mentel, A. Kiendler-Scharr, C. Spindler, B. Bohn, T. Brauers, H. P. Dorn, H. Fuchs, R. Tillmann, A. Wahner, H. Saathoff, K.-H. Naumann, O. Möhler, T. Leisner, L. Müller, M.-C. Reinnig, T. Hoffmann, K. Salo, M. Hallquist, M. Frosch, M. Bilde, T. Tritscher, P. Barmet, A. P. Praplan, P. F. Decarlo, J. Dommen, A. S. H. Prévôt and U. Baltensperger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13503–13508 CrossRef CAS.
- L. Qi, M. Chen, G. Stefenelli, V. Pospisilova, Y. Tong, A. Bertrand, C. Hueglin, X. Ge, U. Baltensperger, A. S. H. Prévôt and J. G. Slowik, Atmos. Chem. Phys., 2019, 19, 8037–8062 CrossRef CAS.
- M. Takeuchi and N. L. Ng, Atmos. Chem. Phys., 2019, 19, 12749–12766 CrossRef CAS.
- C. H. Clark, S. Nakao, A. Asa-Awuku, K. Sato and D. R. Cocker, Aerosol Sci. Technol., 2013, 47, 1374–1382 CrossRef CAS.
- T. Berndt, H. Herrmann, M. Sipilä and M. Kulmala, J. Phys. Chem. A, 2016, 120(51), 10150–10159 CrossRef CAS.
- K. H. Bates, T. B. Nguyen, A. P. Teng, J. D. Crounse, H. G. Kjaergaard, B. M. Stoltz, J. H. Seinfeld and P. O. Wennberg, J. Phys. Chem. A, 2016, 120, 106–117 CrossRef CAS.
- L. D. Yee, K. E. Kautzman, C. L. Loza, K. A. Schilling, M. M. Coggon, P. S. Chhabra, M. N. Chan, A. W. H. Chan, S. P. Hersey, J. D. Crounse, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2013, 13, 8019–8043 CrossRef.
- D. O. De Haan, E. Tapavicza, M. Riva, T. Cui, J. D. Surratt, A. C. Smith, M.-C. Jordan, S. Nilakantan, M. Almodovar, T. N. Stewart, A. de Loera, A. C. De Haan, M. Cazaunau, A. Gratien, E. Pangui and O. Doussin, Environ. Sci. Technol., 2018, 52, 4061–4071 CrossRef CAS.
- M. A. J. Harrison, S. Barra, D. Borghesi, D. Vione, C. Arsene and R. Iulian Olariu, Atmos. Environ., 2005, 39, 231–248 CrossRef CAS.
- L. Wang, X. Wang, R. Gu, H. Wang, L. Yao, W. Liang, F. Zhu, W. Wang, L. Xue, L. Yang, K. Lu, J. Chen, T. Wang, Y. Zhang and W. Wang, Atmos. Chem. Phys., 2018, 18, 4349–4359 CrossRef CAS.
- M. Li, X. Wang, C. Lu, R. Li, J. Zhang, S. Dong, L. Yang, L. Xue, J. Chen and W. Wang, Sci. Total Environ., 2020, 714, 136760 CrossRef CAS.
- K. L. Pereira, J. F. Hamilton, A. R. Rickard, W. J. Bloss, M. S. Alam, M. Camredon, M. W. Ward, K. P. Wyche, A. Mun, T. Vera, N. Va, E. Borra and M. Ro, Environ. Sci. Technol., 2015, 49, 13168–13178 CrossRef CAS.
- Z. Finewax, J. A. De Gouw and P. J. Ziemann, Environ. Sci. Technol., 2018, 52, 1981–1989 CrossRef CAS.
- M. Busilacchio, P. Di Carlo, E. Aruffo, F. Biancofiore, C. D. Salisburgo, F. Giammaria, S. Bauguitte, J. Lee, S. Moller, J. Hopkins, S. Punjabi, S. Andrews, A. C. Lewis, M. Parrington, P. I. Palmer, E. Hyer and G. M. Wolfe, Atmos. Chem. Phys., 2016, 16, 3485–3497 CAS.
- M. Priestley, M. Le Breton, T. J. Bannan, K. E. Leather, A. Bacak, E. Reyes-Villegas, F. De Vocht, B. M. A. Shallcross, T. Brazier, M. Anwar Khan, J. Allan, D. E. Shallcross, H. Coe and C. J. Percival, J. Geophys. Res.: Atmos., 2018, 123, 7687–7704 CAS.
- C. Zuth, PhD thesis, Johannes Gutenberg-Universitat Mainz, 2018.
- P. K. Aiona, H. J. Lee, R. Leslie, P. Lin, A. Laskin, J. Laskin and S. A. Nizkorodov, ACS Earth Space Chem., 2017, 1, 522–532 CAS.
- C. Giorio, A. Monod, L. Brégonzio-Rozier, H. L. Dewitt, M. Cazaunau, B. Temime-Roussel, A. Gratien, V. Michoud, E. Pangui, S. Ravier, A. T. Zielinski, A. Tapparo, R. Vermeylen, M. Claeys, D. Voisin, M. Kalberer and J.-F. Doussin, J. Phys. Chem. A, 2017, 121, 7641–7654 CrossRef CAS.
- H. Zhang, Z. Zhang, T. Cui, Y.-H. Lin, N. A. Bhathela, J. Ortega, D. R. Worton, A. H. Goldstein, A. Guenther, J. L. Jimenez, A. Gold and J. D. Surratt, Environ. Sci. Technol. Lett., 2014, 1, 242–247 CrossRef CAS.
- A. P. Praplan, K. Hegyi-Gaeggeler, P. Barmet, L. Pfaffenberger, J. Dommen and U. Baltensperger, Atmos. Chem. Phys., 2014, 14, 8665–8677 CrossRef.
- J. F. Hamilton, M. R. Alfarra, K. P. Wyche, M. W. Ward, A. C. Lewis, G. B. Mcfiggans, N. Good, P. S. Monks, T. Carr, I. R. White and R. M. Purvis, Atmos. Chem. Phys., 2011, 11, 5917–5929 CrossRef CAS.
- S. Kundu, R. Fisseha, A. L. Putman, T. A. Rahn and L. R. Mazzoleni, Atmos. Chem. Phys., 2012, 12, 5523–5536 CrossRef CAS.
- S. Schobesberger, H. Junninen, F. Bianchi, G. Lönn, M. Ehn, K. Lehtipalo, J. Dommen, S. Ehrhart, I. K. Ortega, A. Franchin, T. Nieminen, F. Riccobono, M. Hutterli, J. Duplissy, J. Almeida, A. Amorim, M. Breitenlechner, A. J. Downard, E. M. Dunne, R. C. Flagan, M. Kajos, H. Keskinen, J. Kirkby, A. Kupc, A. Kürten, T. Kurtén, A. Laaksonen, S. Mathot, A. Onnela, A. P. Praplan, L. Rondo, F. D. Santos, S. Schallhart, R. Schnitzhofer, M. Sipilä, A. Tomé, G. Tsagkogeorgas, H. Vehkamäki, D. Wimmer, U. Baltensperger, K. S. Carslaw, J. Curtius, A. Hansel, T. Petäjä, M. Kulmala, N. M. Donahue and D. R. Worsnop, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17223–17228 CrossRef CAS.
- X. Zhang, R. C. Mcvay, D. D. Huang, N. F. Dalleska, B. Aumont, R. C. Flagan and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14168–14173 CrossRef CAS.
- H. Zhang, L. D. Yee, B. H. Lee, M. P. Curtis, D. R. Worton, G. Isaacman-VanWertz, J. H. Offenberg, M. Lewandowski, T. E. Kleindienst, M. R. Beaver, A. L. Holder, W. A. Lonneman, K. S. Docherty, M. Jaoui, H. O. T. Pye, W. Hu, D. A. Day, P. Campuzano-Jost, J. L. Jimenez, H. Guo, R. J. Weber, J. De Gouw, A. R. Koss, E. S. Edgerton, W. Brune, C. Mohr, F. D. Lopez-Hilfiker, A. Lutz, N. M. Kreisberg, S. R. Spielman, S. V. Hering, K. R. Wilson, J. A. Thornton and A. H. Goldstein, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2038–2043 CrossRef CAS.
- E. A. Dalluge, PhD thesis, Iowa State University, 2013.
- L. Khachatryan, M. Barekati-Goudarzi, D. Kekejian, G. Aguilar, R. Asatryan, G. G. Stanley and D. Boldor, Energy Fuels, 2018, 32, 12597–12606 CrossRef CAS.
- B. Wang and A. Laskin, J. Geophys. Res.: Atmos., 2014, 119, 3335–3351 CAS.
- W. W. Hu, P. Campuzano-Jost, B. B. Palm, D. A. Day, A. M. Ortega, P. L. Hayes, J. E. Krechmer, Q. Chen, M. Kuwata, Y. J. Liu, S. S. de Sá, K. McKinney, S. T. Martin, M. Hu, S. H. Budisulistiorini, M. Riva, J. D. Surratt, J. M. St Clair, G. Isaacman-Van Wertz, L. D. Yee, A. H. Goldstein, S. Carbone, J. Brito, P. Artaxo, J. A. de Gouw, A. Koss, A. Wisthaler, T. Mikoviny, T. Karl, L. Kaser, W. Jud, A. Hansel, K. S. Docherty, M. L. Alexander, N. H. Robinson, H. Coe, J. D. Allan, M. R. Canagaratna, F. Paulot and J. L. Jimenez, Atmos. Chem. Phys., 2015, 15, 11807–11833 CrossRef CAS.
- B. H. Lee, C. Mohr, F. D. Lopez-Hilfiker, A. Lutz, M. Hallquist, L. Lee, P. Romer, R. C. Cohen, S. Iyer, T. Kurten, W. Hu, D. A. Day, P. Campuzano-Jost, J. L. Jimenez, L. Xu, N. L. Ng, H. Guo, R. J. Weber, R. J. Wild, S. S. Brown, A. Koss, J. de Gouw, K. Olson, A. H. Goldstein, R. Seco, S. Kim, K. McAvey, P. B. Shepson, T. Starn, K. Baumann, E. S. Edgerton, J. Liu, J. E. Shilling, D. O. Miller, W. Brune, S. Schobesberger, E. L. D’Ambro and J. A. Thornton, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1516–1521 CrossRef CAS.
- C. M. Boyd, J. Sanchez, L. Xu, A. J. Eugene, T. Nah, W. Y. Tuet, M. I. Guzman and N. L. Ng, Atmos. Chem. Phys., 2015, 15, 7497–7522 CrossRef CAS.
- M. N. Chan, J. D. Surratt, A. W. H. Chan, K. Schilling, J. H. Offenberg, M. Lewandowski, E. O. Edney, T. E. Kleindienst, M. Jaoui, E. S. Edgerton, R. L. Tanner, S. L. Shaw, M. Zheng, E. M. Knipping and J. H. Seinfeld, Atmos. Chem. Phys., 2011, 11, 1735–1751 CrossRef CAS.
- D. Liu, R. Joshi, J. Wang, C. Yu, J. D. Allan, H. Coe, M. J. Flynn, C. Xie, J. Lee, F. Squires, S. Kotthaus, S. Grimmond, X. Ge, Y. Sun and P. Fu, Atmos. Chem. Phys., 2019, 19, 6749–6769 CrossRef CAS.
- A. Laskin, J. S. Smith and J. Laskin, Environ. Sci. Technol., 2009, 43, 3764–3771 CrossRef CAS.
- S. Inomata, A. Fushimi, K. Sato, Y. Fujitani and H. Yamada, Atmos. Environ., 2015, 110, 93–102 CrossRef CAS.
- Y. Wang, M. Hu, Y. Wang, J. Zheng, D. Shang, Y. Yang, Y. Liu, X. Li, R. Tang, W. Zhu, Z. Du, Y. Wu, S. Guo, Z. Wu, S. Lou, M. Hallquist, J. Zhen Yu and M. Hu, Atmos. Chem. Phys., 2019, 19, 7649–7665 CrossRef CAS.
- K. Vidović, D. Lašič Jurković, M. Šala, A. Kroflič and I. Grgić, Environ. Sci. Technol., 2018, 52, 9722–9730 CrossRef.
- C. Wu, G. Wang, J. Li, J. Li, C. Cao, S. Ge, Y. Xie, J. Chen, X. Li, G. Xue, X. Wang, Z. Zhao and F. Cao, Atmos. Chem. Phys., 2020, 20, 2017–2030 CrossRef CAS.
- S. H. Budisulistiorini, M. Riva, M. Williams, J. Chen, M. Itoh, J. D. Surratt and M. Kuwata, Environ. Sci. Technol., 2017, 51, 4415–4423 CrossRef CAS.
- M. Le Breton, A. Bacak, J. B. A. Muller, S. J. O’Shea, P. Xiao, M. N. R. Ashfold, M. C. Cooke, R. Batt, D. E. Shallcross, D. E. Oram, G. Forster, S. J. B. Bauguitte and C. J. Percival, Atmos. Chem. Phys., 2013, 13, 9217–9232 CrossRef.
- S. G. Moussa, A. Leithead, S.-M. Li, T. W. Chan, J. J. B. Wentzell, C. Stroud, J. Zhang, P. Lee, G. Lu, J. R. Brook, K. Hayden, J. Narayan and J. Liggio, Atmos. Environ., 2016, 131, 185–195 CrossRef CAS.
- E. H. Hoffmann, A. Tilgner, R. Wolke, O. Bö, A. Walter and H. Herrmann, Phys. Chem. Chem. Phys., 2018, 20, 10960–10977 RSC.
- B. Yuan, J. Liggio, J. Wentzell, S.-M. Li, H. Stark, J. M. Roberts, J. Gilman, B. Lerner, C. Warneke, R. Li, A. Leithead, H. D. Osthoff, R. Wild, S. S. Brown and J. A. De Gouw, Atmos. Chem. Phys., 2016, 16, 2139–2153 CrossRef CAS.
- I. Bejan, I. Barnes, R. Olariu, S. Zhou, P. Wiesen and T. Benter, Phys. Chem. Chem. Phys., 2007, 9, 5686–5692 RSC.
- C. Lu, X. Wang, S. Dong, J. Zhang, J. Li, Y. Zhao, Y. Liang, L. Xue, H. Xie, Q. Zhang and W. Wang, Environ. Res., 2019, 179, 108709 CrossRef CAS.
- W. R. Leaitch, A. M. Macdonald, P. C. Brickell, J. Liggio, S. J. Sjostedt, A. Vlasenko, J. W. Bottenheim, L. Huang, S. M. Li, P. S. K. Liu, D. Toom-Sauntry, K. A. Hayden, S. Sharma, N. C. Shantz, H. A. Wiebe, W. Zhang, J. P. D. Abbatt, J. G. Slowik, R. Y. W. Chang, L. M. Russell, R. E. Schwartz, S. Takahama, J. T. Jayne and N. L. Ng, Atmos. Environ., 2011, 45, 6696–6704 CrossRef CAS.
- A. Vlachou, K. R. Daellenbach, C. Bozzetti, B. Chazeau, G. A. Salazar, S. Szidat, J.-L. Jaffrezo, C. Hueglin, U. Baltensperger, I. El Haddad and A. S. H. Prévôt, Atmos. Chem. Phys., 2018, 18, 6187–6206 CrossRef CAS.
- G. Stefenelli, V. Pospisilova, F. D. Lopez-Hilfiker, K. R. Daellenbach, C. Hüglin, Y. Tong, U. Baltensperger, A. S. H. Prévôt and J. G. Slowik, Atmos. Chem. Phys., 2019, 19, 14825–14848 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0fd00080a |
| ‡ Current address: NASA Jet Propulsion Laboratory, California Institute of Technology, Pasadena, CA, USA. |
| This journal is © The Royal Society of Chemistry 2021 |