 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVertical profile of particle hygroscopicity and CCN effectiveness during winter in Beijing: insight into the hygroscopicity transition threshold of black carbon†
Dawei
Hu
 a,
Yu
Wang
a,
Chenjie
Yu
a,
Qiaorong
Xie
b,
Siyao
Yue
b,
Dongjie
Shang
c,
Xin
Fang
c,
Rutambhara
Joshi
a,
Dantong
Liu‡
a,
James
Allan
ad,
Zhijun
Wu
c,
Min
Hu
c,
Pingqing
Fu
b and
Gordon
McFiggans
*a
a,
Yu
Wang
a,
Chenjie
Yu
a,
Qiaorong
Xie
b,
Siyao
Yue
b,
Dongjie
Shang
c,
Xin
Fang
c,
Rutambhara
Joshi
a,
Dantong
Liu‡
a,
James
Allan
ad,
Zhijun
Wu
c,
Min
Hu
c,
Pingqing
Fu
b and
Gordon
McFiggans
*a
aSchool of Earth and Environmental Sciences, University of Manchester, UK. E-mail: g.mcfiggans@manchester.ac.uk
bState Key Laboratory of Atmospheric Boundary Layer Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing 100029, China
cState Key Joint Laboratory of Environmental Simulation and Pollution Control, International Joint Laboratory for Regional Pollution Control, Ministry of Education (IJRC), College of Environmental Sciences and Engineering, Beijing, 100871, China
dNational Centre for Atmospheric Science, University of Manchester, Manchester, UK
First published on 8th October 2020
Abstract
The hygroscopicity and ability of aerosol particles to act as cloud condensation nuclei (CCN) is important in determining their lifetime and role in aerosol–cloud interactions, thereby influencing cloud formation and climate. Previous studies have used the aerosol hygroscopic properties measured at the ground to evaluate the influence on cloud formation in the atmosphere, which may introduce uncertainty associated with aerosol hygroscopicity variability with altitude. In this study, the CCN behaviour and hygroscopic properties of daily filter collections of PM2.5 from three different heights (8, 120, 260 m) on a tower in Beijing were determined in the laboratory using water, water/methanol and methanol as the atomization solvents. Whilst there was substantial temporal variability in particle concentration and composition, there was little obvious difference in aerosol CCN and hygroscopic behaviour at different heights, although the planetary boundary layer height (PBLH) reduced to below the tower height during the nighttime, suggesting that use of surface hygroscopicity measurements is sufficient for the estimation of aerosol particle activation in clouds. Additionally, the critical coating thickness (in terms of mass ratio of coating/refractory BC, MRc) defining the BC transition between being hydrophobic to hydrophilic, was determined by combining hygroscopic tandem differential mobility analyser (H-TDMA), centrifugal particle mass analyzer (CPMA) and single particle soot photometer (SP2) measurements. The MRc of 250 nm BC-containing particles increased from a background value of between 0.8 and 1.6 to around 4.6 at the onset of the growth event of nanoparticles, decreasing monotonically back to the background level as the event progressed. This indicates that large particles do not act as an effective pre-existing condensation sink of the hygroscopic vapours during the nanoparticle growth events, leading to the 250 nm BC particles requiring more coating materials to transition between being hydrophobic and hydrophilic. These findings show that large particles may be less important in suppressing the new particle formation and subsequent growth in the atmosphere.
1 Introduction
According to the Intergovernmental Panel on Climate Change (IPCC), the impacts of aerosols on clouds are one of the largest uncertainties in estimates of global radiative forces. Aerosol hygroscopicity is one of the most important properties to determine if particles can act as a cloud condensation nucleus (CCN) and then form cloud droplets under atmospheric water saturation ratios.1–3 The hygroscopicity depends on the chemical composition of aerosols. Generally, inorganic salts such as sulfate, nitrate and chloride, are hydrophilic,4,5 organics are less hygroscopic, whilst black carbon and dust are more hydrophobic.6Over the last several decades, aerosol hygroscopicity has been intensively measured by hygroscopicity tandem differential mobility analyser (H-TDMA)7–12 and CCN counter (CCNc)8,13–16 under sub- and super-saturation, respectively.17 To associate aerosol hygroscopicity with its composition, the hygroscopicity parameter, kappa (κ), has been introduced.18 The kappa from online measurements has been used to compare with that predicted from chemical composition (κchem), which is obtained by simply volume weighting the kappa of each single compound in particles.19,20 Mismatch of kappa between measurement and prediction has been frequently observed but discrepancies remain difficult to interpret. Since the varying properties of organic compounds, such as solubility, surface activity and extent of dissociation in the atmosphere,21,22 the hygroscopicity of an organic material is difficult to quantify and most studies simply attribute discrepancies between the measurement and prediction to the organic fraction, and reconcile the discrepancy by tuning the kappa of organic (κorg).14,23–26 For example, Chang et al.14 parameterised κorg in terms of the oxidation degree (as O/C) based on measurements conducted in Canada during the spring of 2007, and presented κorg = (0.29 ± 0.05) × (O/C). Mei et al.24 found that κorg was better parameterized using f44 than O/C, as κorg = 2.10(±0.07) × f44 − 0.11(±0.01). Padró et al.26 provided a simple way to parameterize organic hygroscopicity in atmospheric models, i.e. κorg = 0.28 × εwsoc (where εwsoc is the volume fraction of water-soluble organic carbon (WSOC)). The influence of the organic fraction on a particle’s hygroscopicity and the appropriateness of reconciling the discrepancy between the measurement and prediction by simply tuning the κorg, remains unclear. Most previous field studies of aerosol hygroscopicity or ability to behave as CCN have been performed at or near ground level.17 These measurements have then typically been used to inform model descriptions of the aerosol hygroscopicity influence on cloud formation in the atmosphere. It is possible that such an approach may introduce uncertainty associated with aerosol hygroscopicity variability with altitude.
Black carbon (BC) is a widely investigated component of atmospheric aerosol because it can absorb solar radiation and heat the atmosphere.27 The hygroscopicity of BC aerosol significantly affects its lifetime and then influences its radiative forcing.28 Fresh BC particles are hydrophobic,29 but on acquisition of hygroscopic materials through atmospheric aging, they can transform from hydrophobic to hydrophilic to then more effectively act as a CCN.30,31 The hygroscopicity of BC particles has been explored extensively by coupling the BC identification instruments, such as the single particle soot photometer (SP2),32–34 downstream of the first35 or second differential mobility analyser (DMA)36–38 of H-TDMA systems. These studies found that the hygroscopicity of BC particles was mainly determined by the coating layer thickness and materials. However, the quantification of the critical coating thickness defining the transition between BC-containing particles being hydrophobic to hydrophilic has hitherto been reported. Such information can be used to simplify estimations of the lifetime of BC in the atmosphere and improve prediction of its radiative forcing.
In this study, we aim to address the two main questions identified above:
(i) To assess the vertical structure of composition affecting CCN behaviour, the CCN behaviour and hygroscopic properties of daily filter collections of PM2.5 from three different heights (8, 120, 260 m) on a tower in Beijing were determined in the laboratory using water, water/methanol and methanol as the atomization solvents, and compared with in situ measured hygroscopicity.
(ii) To determine the role of the BC mixing state in CCN in a dynamic urban environment, the critical coating thickness defining the BC transition between being hydrophobic to hydrophilic was determined by combining H-TDMA, scanning mobility particle sizer (SMPS), centrifugal particle mass analyzer (CPMA) and SP2 measurements.
2 Experimental and methods
This study was conducted in the framework of the Air Pollution and Human Health-Beijing (APHH-Beijing) programme outlined in Shi et al.39 In this study, the measurements were conducted at the 325 m Institute of Atmospheric Physics (IAP) tower site (39°58′28′′N, 116°22′16′′E) in Beijing from 12th to 29th November, 2016. The site is located between the 3rd ring and 4th ring in the north of central Beijing.2.1 Filter sample collection and analysis
In this study, filter samples were collected daily for 9 days through 16th to 29th November, 2016. Aerosol samples (PM2.5) were collected on pre-combusted (450 °C for 6 h) quartz filters (PALLFLEX 2500QAT-UP, 20 × 25 cm) at 8, 120 and 260 m up the tower using a high-volume sampler (Kimoto AS-810B) with a flow rate of 0.6 m3 min−1. The interval of sample collection was 24 h, and the filters were replaced at 7:30 am (local time) each day. After sampling, the filters were kept separately in the pre-cleaned glass jar with a Teflon-lined screw cap and stored at −20 °C prior to analysis.A filter punch of 20 mm diameter from each filter was used to measure the OC and EC concentration using a carbon analyzer (Sunset Laboratory Inc., USA) with thermal/optical transmission method following the Interagency Monitoring of Protected Visual Environments (IMPROVE) method. The organic matter/OC (OM/OC) ratio of 1.4, the value commonly used for ambient aerosols in previous studies,41 was applied to derive the mass concentration of organics.
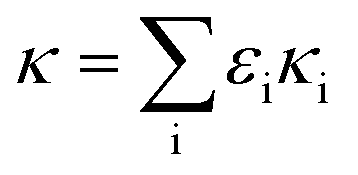 | (1) |
Since the chemical composition is reported in terms of the component inorganic ions, while the ZSR calculation requires the volume fraction and κ of each neutral salt, a simplified ion pairing scheme according to Gysel et al.23 is applied to convert ion mass to the mass concentrations of inorganic salts, expressed as:
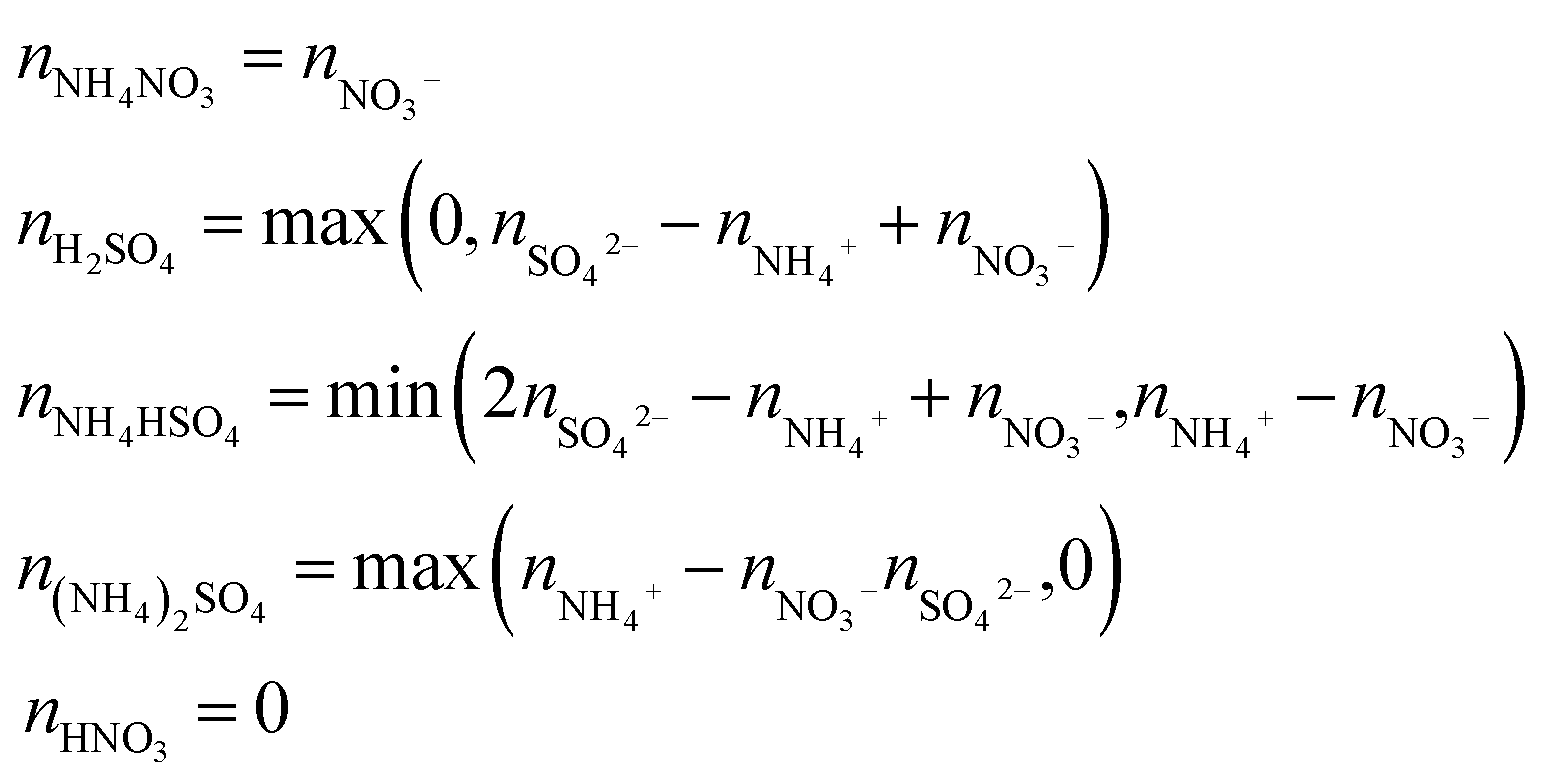 | (2) |
The generated aerosols were first passed through a Nafion drier to remove the water vapour, and subsequently passed through a diffusion drier with molecular sieve (type 5A, 3–4 mm dia.) to remove the methanol vapour in the sample stream. The dried particles were charged by a 90Sr diffusion charger and then measured by H-TDMA and a differential mobility particle sizer (DMPS) + CCNc system. The detailed description of the H-TDMA and DMPS + CCNc system can be found in Good et al.42 Briefly, for H-TDMA measurements, the charged polydisperse particles were introduced into the first DMA (DMA-1, Brechtel Manufacturing Inc., USA) to select the particle with a desired dry diameter (D0). Quasi-monodisperse particles exiting DMA-1 were subsequently exposed to a controlled humidity environment, i.e. a Nafion humidifier, where the RH was regulated to 90% in this study. The humidified particle size distribution was measured by the second DMA (DMA-2, Brechtel Manufacturing Inc., USA) combined with a condensation particle counter (CPC, Model 3786, TSI Inc., USA). The hygroscopic growth factor (GF), defined as the ratio of the humidified particle diameter (Dp) over D0, was obtained through the inversion of scanning DMA-2 data by the TDMAInv software.43 For DMPS + CCNc measurement, the dried and charged polydisperse particles were fed into a Vienna style DMA44 and then were split between the CCNc and CPC (Model 3775, TSI Inc., USA). By stepping through different dry sizes in DMA, the activation diameter (D50) of particles at a given water super-saturation (SS) was determined as the diameter at which 50% of the particle number concentration measured by the CPC were measured to be activated in the CCNc. The corresponding hygroscopicity parameter (κ) under sub- and super-saturated conditions was derived using the method developed by Petters and Kreidenweis.18
Before the experiment, DMAs were calibrated with certified polystyrene latex spheres (PSLs). Both the H-TDMA and DMPS + CCNc system were calibrated with (NH4)2SO4 particles.
2.2 Ambient aerosol measurement
In addition to the filter study, ambient measurement of aerosol hygroscopicity, size distribution and the properties of size-resolved BC particles were conducted during the measurement period at the ground site of the IAP at the foot of the tower on which the filter sampling was conducted, using H-TDMA, SMPS, CPMA and SP2, respectively.The interval of CPMA + SP2 measurement was 30 min. The CPMA was stepped from 0.3 to 15 fg for the first 20 min and paused for the following 10 min to allow the SP2 to measure poly-disperse ambient particles.
3 Results and discussion
3.1 Vertical profile of aerosol hygroscopicity and CCN ability
Fig. 1 shows the RH, temperature (T), wind speed and direction, and particle chemical composition measured at the 8 m height of the tower throughout the whole filter collection period. The period in between the two adjacent vertical grey dashed lines in Fig. 1(d) corresponds to the filter collection period each day. The corresponding information at 120 m and 260 m is presented in Fig. S1.† For all 3 heights, particle chemical composition and mass concentration changed significantly throughout the measurement. The mass concentration varied from 16 to 162 μg m−3. Lower particle mass concentrations were observed on the 19th, 23rd, and 27th November with the dominant wind direction from the north, so clean air from northern areas such as Inner Mongolia cleaned up the pollutants in Beijing. While, when the wind direction was dominantly from the south on the 18th, 24th, 25th, and 26th November, the pollutants were introduced into Beijing from southern areas, particles come from regional transport are more aged, as reflected by the large particle size (Fig. 1(d)) with a high fraction of organic materials (Fig. 1(c)).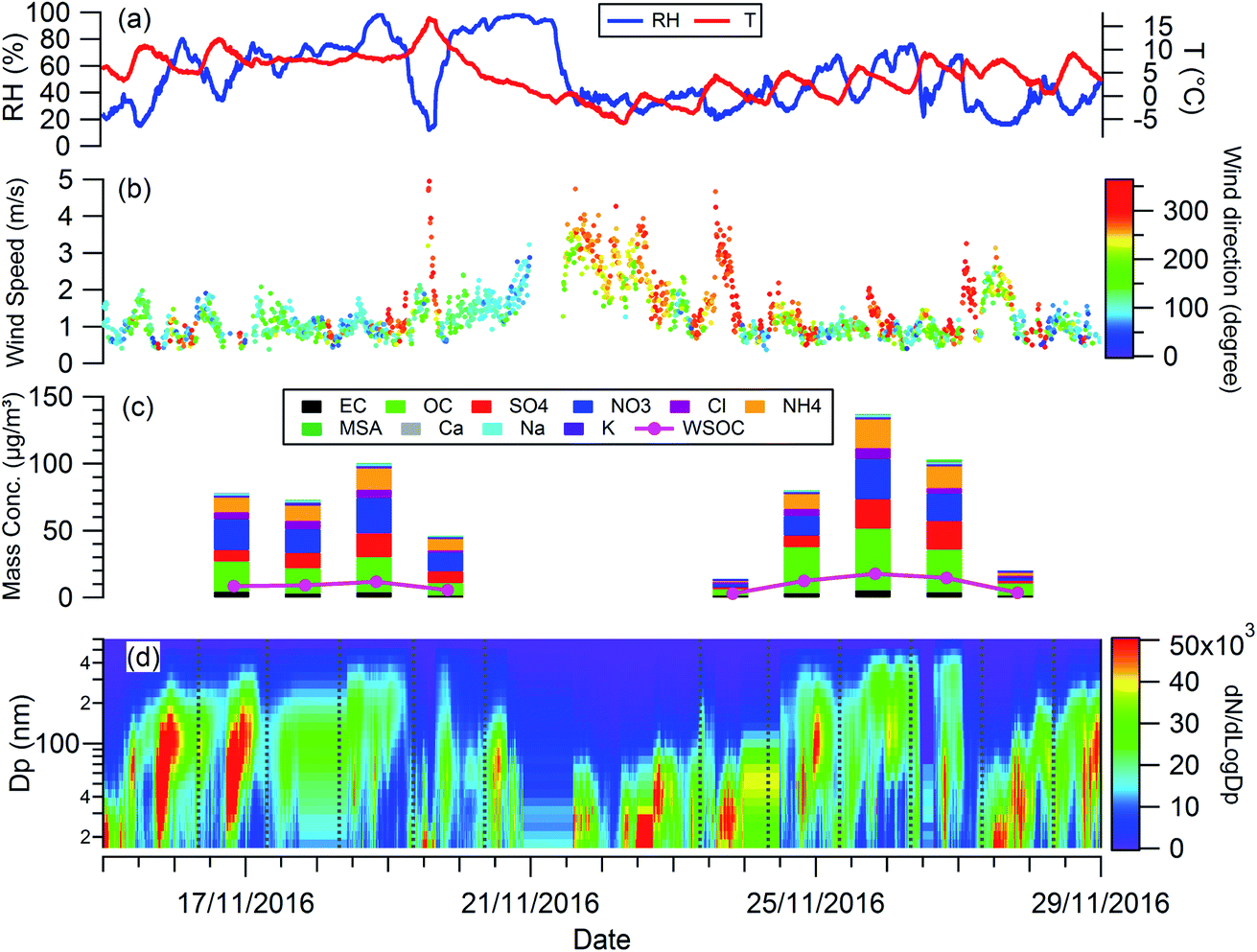 | ||
| Fig. 1 Temporal evolution of (a) RH, T; (b) wind speed and direction; (c) mass concentration of chemical compositions; (d) particle size distribution during the experiment. | ||
The planetary boundary layer height (PBLH) can vary dramatically over the day with responding changes in surface radiative forces from a few kilometres at noon to a few meters at night. Clearly such a change of PBLH will influence the vertical profile of pollutant concentrations and potentially particle properties. Fig. S2† presents the diurnal variation of PBLH during the measurement. The average PBLH reduced to below the top of the tower, and below our top measurement site at 260 m (red dashed line in Fig. S2†) during nighttime. This may lead to decoupling of the layers and possible differences in particle behaviour between that measured at 260 m and that at 8 and 120 m. The temporal and vertical profiles of kappa for 100 nm aerosols generated using water, water/methanol and methanol alone as the atomization solvents are presented in Fig. 2. It shows that, for the filters with the same solvent extraction, there is little obvious difference of the hygroscopicity and particle CCN effectiveness at 260 m compared with the other two heights. The average differences of the kappa with heights are 8 ± 10%, 14 ± 13% and 25 ± 22% for the 100 nm aerosols generated using water, water/methanol and methanol alone as the atomization solvents, respectively. This lack of obvious difference is consistent with the largely invariant aerosol composition with altitude presented in Fig. 1 and S1,† and suggests that the use of surface hygroscopicity measurements is sufficient for the estimation of aerosol particle activation in clouds.
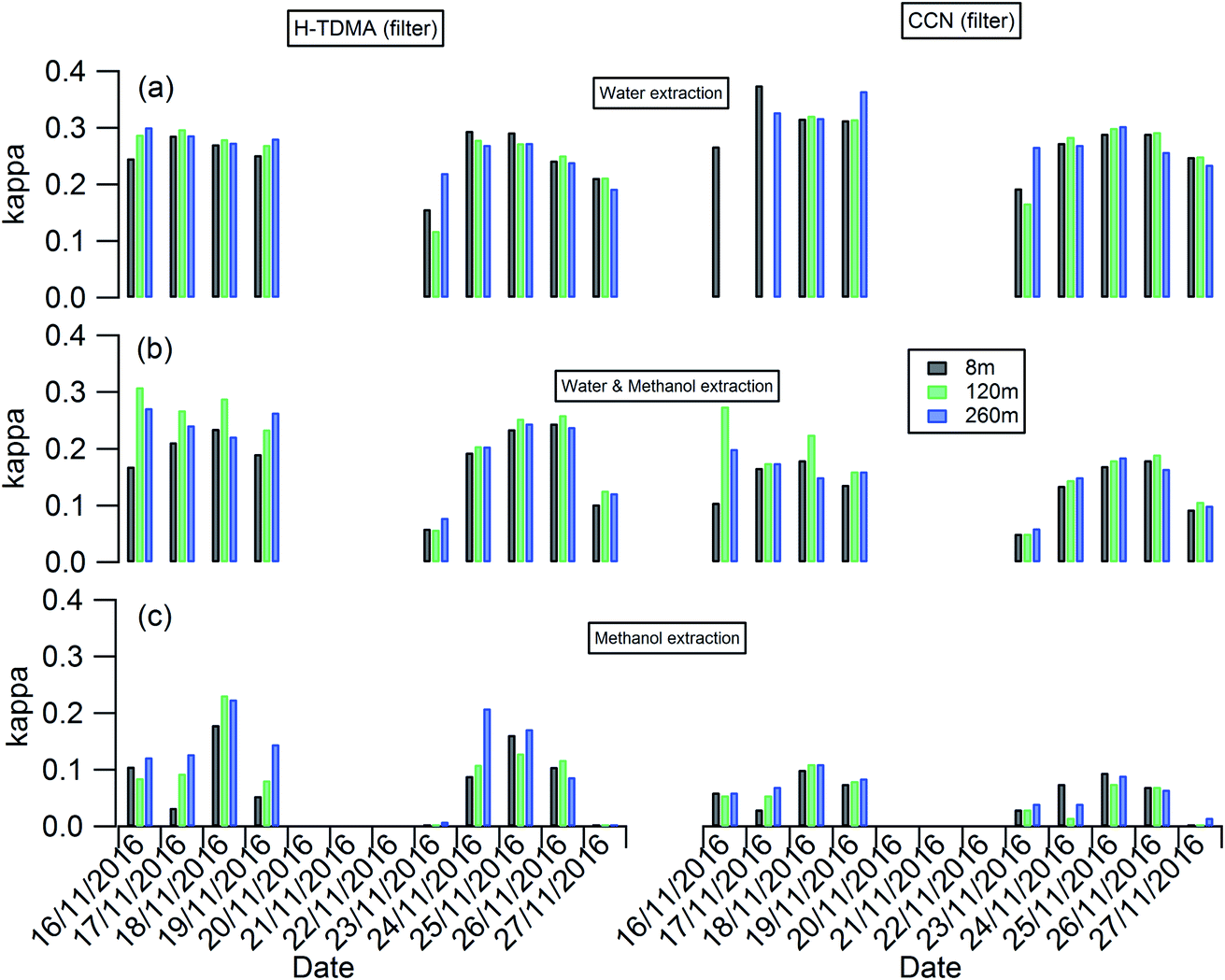 | ||
| Fig. 2 Temporal and vertical profile of kappa of 100 nm aerosols generated using (a) water; (b) water/methanol; and (c) methanol as the atomization solvents. | ||
The temporal evolution of kappa for 100 nm aerosols generated using water, water/methanol and methanol alone as the atomization solvents at 8 m height is presented alone in Fig. 3, to compare the kappa predicted from chemical composition and the daily averaged kappa from the H-TDMA ambient measurements. It is clear that the kappa of aerosols generated using water as the atomization solvent shows the highest kappa value, followed by that using water/methanol mixture, and methanol, as the atomization solvents. This is reasonable since the inorganic compounds (sulfate, nitrate) which are readily dissolved in water and are substantially less soluble in methanol and are more hygroscopic than the organics extracted in methanol. In summary: the mixed solvent (water/methanol) can dissolve both inorganics and organics, the presence of organics (inorganics) will decrease the fraction of inorganics (organics) in the atomized particles compared with that using water (methanol) as the only atomization solvent, and then it exhibits lower (higher) hygroscopic values than the latter.
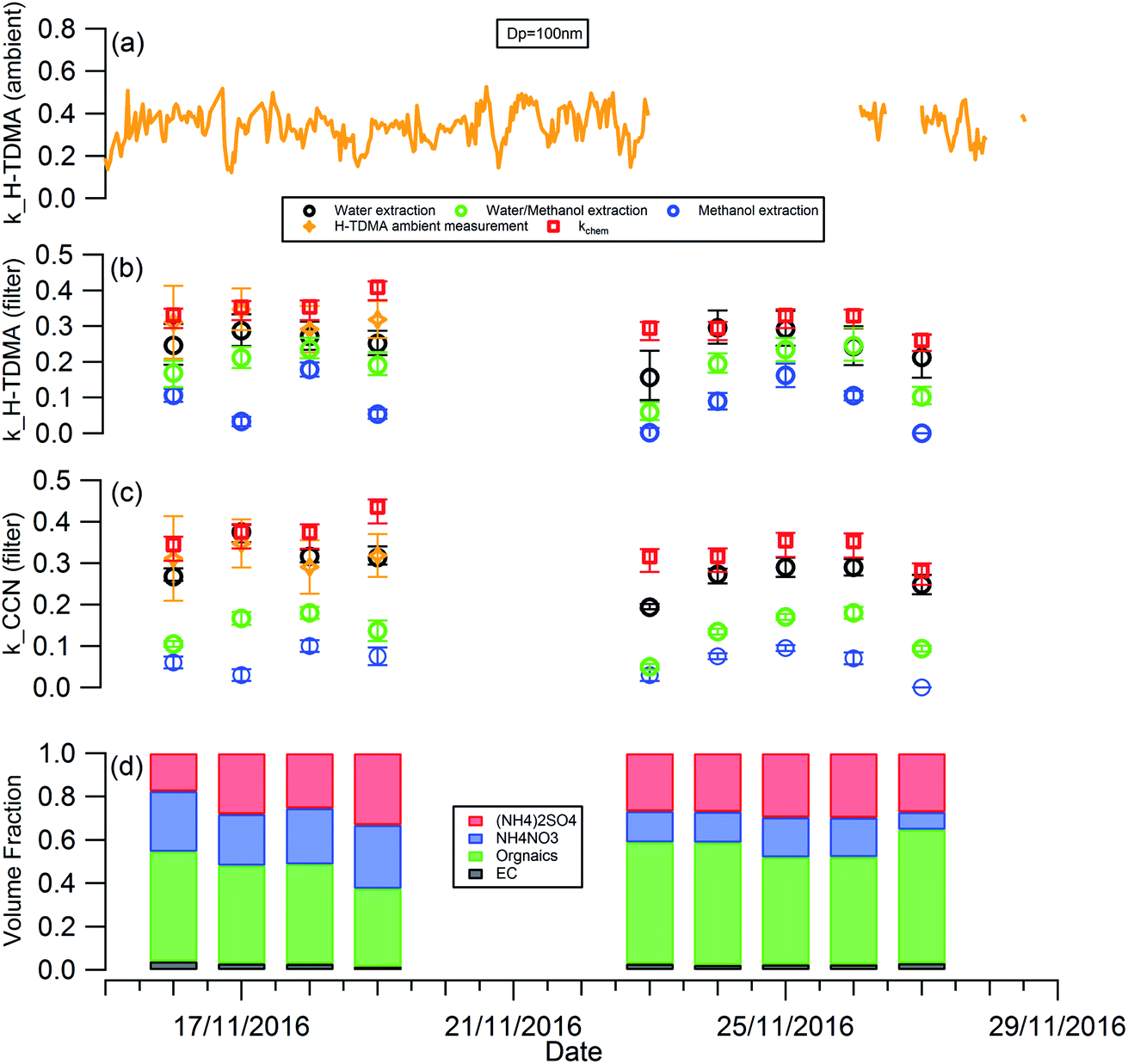 | ||
| Fig. 3 Temporal evolution of: (a) kappa from H-TDMA ambient measurements; (b) kappa from H-TDMA measurements of filter samples; (c) kappa from CCNc measurements of filter samples; and (d) volume fraction of aerosol composition. The daily averaged kappa from (a) and the predicted kappa (κchem) based on the chemical volume fraction in (d) are presented in (b) and (c) for comparison. | ||
As shown in Fig. 3(b), the aerosol hygroscopicity predicted using the chemical composition of filters slightly deviated from the H-TDMA ambient measurements. The discrepancy of both methods in characterizing aerosol hygroscopicity likely arises from: (1) the filter measurement being sensitive to larger particles which contribute the majority of the total mass, thus the bulk composition tends to mainly reflect the composition information for larger particles, but ambiguously hides the information for smaller particles. In contrast, the ambient H-TDMA measurements are number-based, and are thus able to provide more realistic information on smaller particles; (2) filter collection and analysis artifacts; and (3) gas-particle partition during filter collection, storage and analysis. The gas-particle partition issue may also occur in the H-TDMA system, but it is minimized in this study since the close loop of sheath air was used during the measurement, which will keep the surrounding environment of particles inside the DMA as close as that in the ambient.
Since water was used to extract the filter for aerosol chemical composition (inorganics and WSOC) analysis in this study, the kappa predicted by chemical composition was expected to be similar to that of the atomized aerosol with water extraction. As shown in Fig. 3, the predicted kappa (red square in Fig. 3(b) and (c)) is slightly higher than the measured value (black circle in Fig. 3(b) and (c)). There are three possible reasons: (1) the organic mass concentration used for kappa prediction was calculated from the OC concentration by applying the OM/OC ratio of 1.4, although this value could vary widely (1.17–2.67) based on previous studies.48,49 If the actual OM/OC ratio in our study is higher than the value of 1.4, the organic fraction will be under-estimated and then lead an over-estimated predicted kappa. (2) Some of the ammonium nitrate evaporates from the particles during the nebulization process, and losing some of the compounds with high hygroscopicity will decrease the hygroscopicity of the generated particles. (3) It should be noted that the aggregated composition volume, and hence mass fractions, were derived from a combination of instruments; the inorganic compounds by IC and organics were measured with an OC/EC analyser. There will be uncertainty associated with each technique which will propagate into the combined input into the κchem calculation. For example, the BC and organic mass measured by the OC/EC analyser using the thermal-optical analysis method could be biased by charring of organic carbon, which will lead to overestimating the BC mass concentration and underestimating the mass concentration of organics. To quantify the uncertainty of the κchem, the OC mass concentration was assumed to be underestimated by a maximum of 20% due to charring within the OC/EC analyser,50 and the conversion factor from OC to organic mass concentration was assumed to be in the range 1.2–1.6 with a mean value of 1.4. The analysis results are presented as the error bar for the red squares in Fig. 3(b) and (c).
Due to the low time resolution (1 day) and limited number of filter samples in our study, we are unable to establish a reliably robust relationship between κorg with the parameters of O/C, f44 or εwsoc as that presented by Chang et al.,14 Mei et al.24 and Padró et al.26 However, the validity and robustness of the methods mentioned above can be tested by our data. As shown in Fig. S3,† the κorg calculated using κorg = 0.28 × εwsoc which was presented by Padró et al.,26 was more appropriable for our study than the other two methods.
3.2 Hygroscopicity transition threshold of BC
The evolution of the hygroscopicity of BC was investigated by combining the H-TDMA, SMPS and CPMA + SP2 measurements, and the critical coating thickness defining the BC transition between hydrophobic and hydrophilic was determined by introducing a novel data analysis method. Since the H-TDMA and CPMA did not operate during the 22nd to 29th November 2016 due to an instrument issue, the subset of the measurement period from 12th to 22nd November, 2016 was investigated in this section. As shown in Fig. S4,† ambient BC-containing particles had the highest concentration around 200 nm. The SP2 can only detect the number concentration of BC-free particles with particle diameters larger than 200 nm, so 250 nm (240–260 nm) BC-containing particles were chosen for investigation.The particle number concentration detected by the different instruments was validated by comparison of the integrated number concentration of 250 nm (240–260 nm) particles from SMPS size distribution with that (BC-containing + BC-free particles) detected by the SP2. As shown in Fig. 4(c), the particle number concentration measured by the SMPS (orange line) robustly agrees well with that measured by the SP2 only (sum of black and grey bar). BC particles measured using the CPMA + SP2 combination (purple dashed line) were about 47% lower in number than those detected by the SP2 only (black bar), owing to the reduced transmission through the CPMA. In the discussion below, the SP2 particle number concentration was used, with the CPMA + SP2 measurement providing the coating thickness (MR) distribution of BC-containing particles, which remained uninfluenced by the CPMA particle loss.
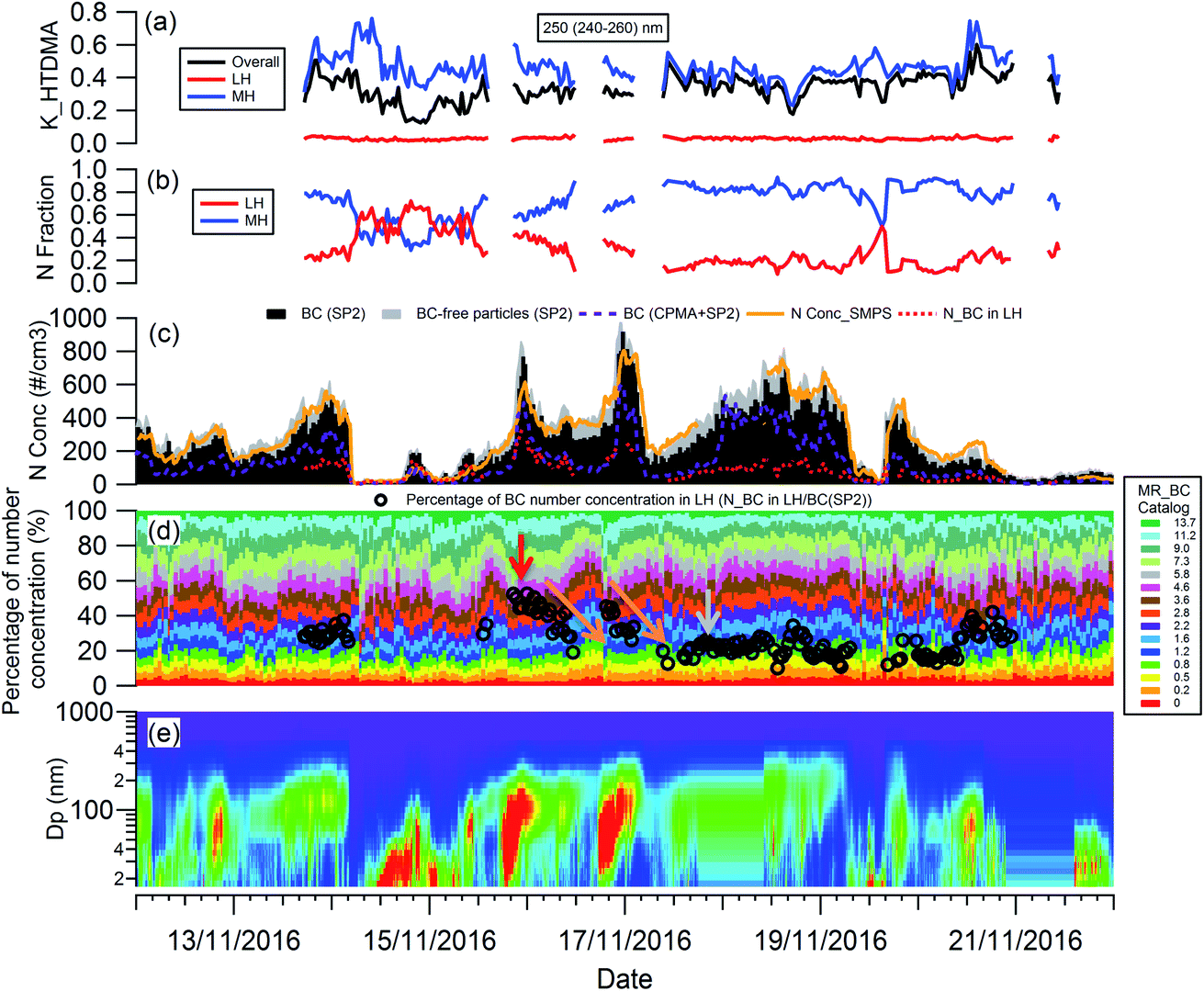 | ||
| Fig. 4 Temporal evolution of: (a) kappa of 250 nm ambient aerosols, and (b) number fraction of 250 nm ambient particles in a less-hygroscopic mode (LH) and more-hygroscopic mode (MH), from H-TDMA field measurements; (c) number concentration of 250 nm BC-containing and BC-free particles, and the calculated number concentration of 250 nm BC-containing particles in LH; (d) percentage of number concentration of 250 nm BC-containing particles with different coating thickness (MR_BC), the black circles represent the percentage number concentration of 250 nm BC-containing particles in LH; (e) size distribution of ambient aerosols. | ||
To analyze the hygroscopicity transition threshold of BC, we assume that the hygroscopicity of BC-containing particles increases with its coating thickness, and BC-free particles are more hygroscopic than BC-containing particles. This assumption is reasonable since fresh BC is hydrophobic29 and any increase in hygroscopicity of coated BC particles should be attributed to the coatings.30,31 As shown in Fig. 4(a), there are two hygroscopicity modes of 250 nm ambient particles detected by the H-TDMA system throughout the measurement: one was at the less hygroscopic mode (LH) with kappa around 0.1, and the other one was a more hygroscopic mode (MH) with kappa around 0.5. The number fraction of particles in each mode is presented in Fig. 4(b). By multiplying the particle number fraction in the LH mode with the total number concentration of particles at 250 nm measured by SP2, the particle number concentration in the LH mode (red dotted line in Fig. 4(c)) can be derived. According to the above assumption, BC particles with low coating thickness will first account for the LH mode. Since the particle number concentration in the LH mode was less than the BC particle concentration detected by the SP2 (black bar in Fig. 4(c)) in this study, there were evidently some BC particles with high coating thickness in the MH mode. The ratio of particle number concentration in the LH mode to total BC number concentration shows the proportion of BC particles in the LH mode (black circle in Fig. 4(d)). Combining the MR distribution of BC particles measured by the CPMA + SP2 (Fig. 4(d)), and through accumulating the proportion of BC particles from MR = 0 to match the proportion of BC calculated in the LH mode, the critical coating thickness (MRc of coating/rBC) defining the transition threshold between BC being hydrophobic to hydrophilic, can be determined as the coating thickness value indicated by the black circles in Fig. 4(d). Two examples of the derivation of MRc for BC are presented in Fig. 5, one is for the high MRc condition (Fig. 5(a)), indicated by the red arrow in Fig. 4(d) and the other is for the low MRc condition (Fig. 5(b)) which is indicated by the grey arrow in Fig. 4(d).
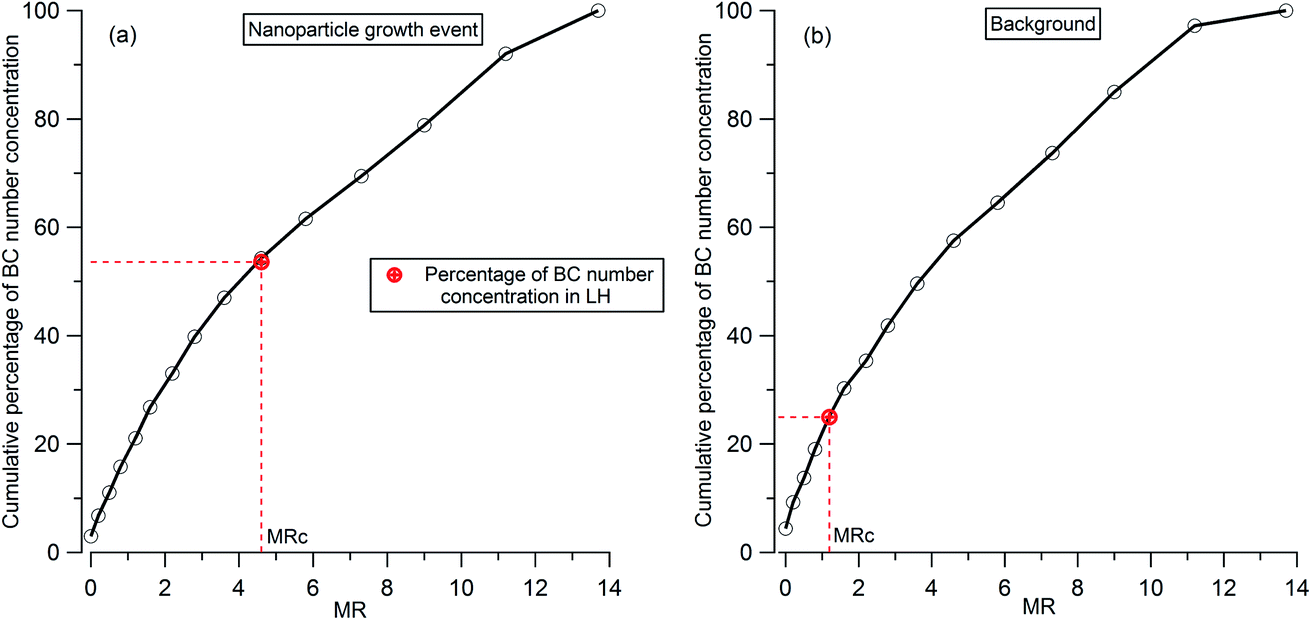 | ||
| Fig. 5 Examples of the derivation of critical coating thickness (MRc) for 250 nm BC-containing particles: (a) high MRc condition as indicated by the red arrow in Fig. 4(d); (b) low MRc condition as indicated by the grey arrow in Fig. 4(d). The red circle represents the percentage number concentration of 250 nm BC-containing particles in LH. | ||
Throughout the investigation period, the MRc of coating/rBC was stable and ranged from 0.8 to 1.6 with the exception of the afternoons of the 15th and 16th November. In the afternoons of the 15th and 16th November, nanoparticle growth events were observed and the MRc of coating/rBC increased from a background value to around 4.6 at the onset of the growth event, decreasing monotonically back to the background level as the growth event progressed (the trend is marked by the orange arrows in Fig. 4(d)). The high MRc of coating/rBC of BC-containing particles at the beginning of the growth event of nanoparticles indicates that the coating material is less hygroscopic compared with that during the background period, suggesting either that any condensing material with high hygroscopicity was captured by other sinks instead of the surface of BC particles during the growth event of nanoparticles, or that only non-hygroscopic material was available for condensation. This result suggests that large particles cannot act as an effective pre-existing condensation sink of the hygroscopic vapours during the growth event of nanoparticles.
3.3 Atmospheric implications
Global modeling simulations show that new particle formation (NPF) and the following growth of nanoparticles can contribute around half of the CCN population in the atmosphere.51,52 NPF and the following growth of the nanoparticles has been observed in various environments around the world including urban, forested areas and marine/coastal regions.53–55 A clean environment was acknowledged as favorable for NPF and its following growth, because of less pre-existing particles competing with the newly formed nanoparticles to capture the condensing vapours.53 Our results show that, even though large particles pre-exist in the atmosphere, they may not act as an effective pre-existing condensation sink of the hygroscopic vapours during the growth event of nanoparticles; the BC particles here are shown only to scavenge the condensing vapours with the lower hygroscopicity, either because there are no hygroscopic vapours available to scavenge or because they preferentially leave the condensing vapors with higher hygroscopicity for the newly formed particles. These finding show that large particles may be less important in suppressing the new particle formation and subsequent growth in the atmosphere.4 Conclusions
In this study, the vertical profile of aerosol hygroscopicity and CCN ability during winter in Beijing is examined using laboratory measurements of the water uptake of particles nebulised from extractions of daily filter collections of PM2.5 from three different heights (8, 120, 260 m) at the IAP tower. Our results provide evidence that using surface hygroscopicity measurements is sufficient for the estimation of aerosol particle activation in clouds, because there was no significant difference in aerosol CCN and hygroscopic behaviour at different heights throughout the measurement period, even though substantial temporal variability in particle concentration and composition was observed. In addition, the critical coating thickness (in terms of mass ratio of coating/refractory BC, MRc) defining the BC transition between being hydrophobic to hydrophilic was determined by applying a novel analysis method to the data from the H-TDMA, CPMA and SP2 ambient measurements. The high MRc observed during the growth events of nanoparticles indicates that the coating material of BC-containing particles is less hygroscopic during the growth event of nanoparticles compared with that during the background period, and then it requires more coating materials for transition between being hydrophobic and hydrophilic. This suggests either that any condensing material with high hygroscopicity was captured by other sinks instead of the surface of BC particles during the growth event of nanoparticles, or that only non-hygroscopic material was available for condensation. Our results indicate that large BC-containing particles do not act as an effective pre-existing condensation sink of the hygroscopic vapours during these nanoparticle growth events.Author contributions
G. M., D. H. and P. F. designed the research; D. H. and Y. W. performed the H-TDMA lab experiments; Q. X. and S. Y. performed filter analysis; C. Y., R. J., D. L. and J. A. performed CMPA + SP2 ambient measurements; Y. W. D. S., X. F., Z. W. and M. H. performed SMPS and H-TDMA ambient measurements; D. H. carried out data analysis; D. H. and G. M. co-wrote the paper.Data availability
Processed data are available through the APHH project archive at the Centre for Environmental Data Analysis (http://data.ceda.ac.uk/badc/aphh/data/beijing/). Raw data is archived at the University of Manchester and is available on request.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported through the UK Natural Environment Research Council (NERC) in the Air Pollution and Human Health-Beijing (APHH-Beijing) programme (grant ref: NE/N00695X/1). Thanks to Prof. Yele Sun's group for providing the AMS data.References
- G. McFiggans, P. Artaxo, U. Baltensperger, H. Coe, M. C. Facchini, G. Feingold, S. Fuzzi, M. Gysel, A. Laaksonen, U. Lohmann, T. F. Mentel, D. M. Murphy, C. D. O’Dowd, J. R. Snider and E. Weingartner, Atmos. Chem. Phys., 2006, 6, 2593–2649 CrossRef CAS.
- U. Dusek, G. P. Frank, L. Hildebrandt, J. Curtius, J. Schneider, S. Walter, D. Chand, F. Drewnick, S. Hings, D. Jung, S. Borrmann and M. O. Andreae, Science, 2006, 312, 1375–1378 CrossRef CAS.
- D. O. Topping and G. McFiggans, Atmos. Chem. Phys., 2012, 12, 3253–3260 CrossRef CAS.
- D. Hu, J. Chen, X. Ye, L. Li and X. Yang, Atmos. Environ., 2011, 45, 2349–2355 CrossRef CAS.
- D. Hu, L. Qiao, J. Chen, X. Ye, X. Yang, T. Cheng and W. Fang, Aerosol Air Qual. Res., 2010, 10, 255–264 CrossRef CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, New York, 2006 Search PubMed.
- M. C. Yeung, B. P. Lee, Y. J. Li and C. K. Chan, J. Geophys. Res.: Atmos., 2014, 119, 9864–9883 Search PubMed.
- Y. Y. Wang, Z. Q. Li, Y. J. Zhang, W. Du, F. Zhang, H. B. Tan, H. B. Xu, T. Y. Fan, X. A. Jin, X. X. Fan, Z. P. Dong, Q. Y. Wang and Y. L. Sun, Atmos. Chem. Phys., 2018, 18, 11739–11752 CrossRef CAS.
- H. Wex, G. McFiggans, S. Henning and F. Stratmann, Geophys. Res. Lett., 2010, 37, L10805 CrossRef.
- W. Xu, J. Ovadnevaite, K. N. Fossum, C. Lin, R. J. Huang, C. O’Dowd and D. Ceburnis, Atmos. Chem. Phys., 2020, 20, 3777–3791 CrossRef CAS.
- Y. Yan, P. Q. Fu, B. Jing, C. Peng, S. K. R. Boreddy, F. Yang, L. F. Wei, Y. L. Sun, Z. F. Wang and M. F. Ge, Sci. Total Environ., 2017, 578, 307–316 CrossRef CAS.
- E. Swietlicki, H. C. Hansson, K. Hämeri, B. Svenningsson, A. Massling, G. McFiggans, P. H. McMurry, T. Petäjä, P. Tunved, M. Gysel, D. Topping, E. Weingartner, U. Baltensperger, J. Rissler, A. Wiedensohler and M. Kulmala, Tellus B, 2008, 60, 432–469 CrossRef.
- S. S. Gunthe, D. Rose, H. Su, R. M. Garland, P. Achtert, A. Nowak, A. Wiedensohler, M. Kuwata, N. Takegawa, Y. Kondo, M. Hu, M. Shao, T. Zhu, M. O. Andreae and U. Poschl, Atmos. Chem. Phys., 2011, 11, 11023–11039 CrossRef CAS.
- R. Y. W. Chang, J. G. Slowik, N. C. Shantz, A. Vlasenko, J. Liggio, S. J. Sjostedt, W. R. Leaitch and J. P. D. Abbatt, Atmos. Chem. Phys., 2010, 10, 5047–5064 CrossRef CAS.
- Y. Li, F. Zhang, Z. Q. Li, L. Sun, Z. Z. Wang, P. Li, Y. Sun, J. Y. Ren, Y. Y. Wang, M. Cribb and C. Yuan, Atmos. Res., 2017, 188, 80–89 CrossRef CAS.
- G. C. Roberts and A. Nenes, Aerosol Sci. Technol., 2005, 39, 206–221 CrossRef CAS.
- H. C. Che, X. Y. Zhang, Y. Q. Wang, L. Zhang, X. J. Shen, Y. M. Zhang, Q. L. Ma, J. Y. Sun, Y. W. Zhang and T. T. Wang, Sci. Rep., 2016, 6, 14 CrossRef.
- M. D. Petters and S. M. Kreidenweis, Atmos. Chem. Phys., 2007, 7, 1961–1971 CrossRef CAS.
- R. H. Stokes and R. A. Robinson, J. Phys. Chem., 1966, 70(7), 2126–2131 CrossRef CAS.
- A. Zdanovskii, Zh. Fiz. Khim., 1948, 22, 1475–1485 CAS.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS.
- M. Gysel, J. Crosier, D. O. Topping, J. D. Whitehead, K. N. Bower, M. J. Cubison, P. I. Williams, M. J. Flynn, G. B. McFiggans and H. Coe, Atmos. Chem. Phys., 2007, 7, 6131–6144 CrossRef CAS.
- F. Mei, A. Setyan, Q. Zhang and J. Wang, Atmos. Chem. Phys., 2013, 13, 12155–12169 CrossRef.
- S. K. R. Boreddy, K. Kawamura, S. Mkoma and P. Q. Fu, J. Geophys. Res.: Atmos., 2014, 119, 12233–12245 CAS.
- L. T. Padró, D. Tkacik, T. Lathem, C. J. Hennigan, A. P. Sullivan, R. J. Weber, L. G. Huey and A. Nenes, J. Geophys. Res.: Atmos., 2010, 115, D09204 CrossRef.
- T. C. Bond and R. W. Bergstrom, Aerosol Sci. Technol., 2006, 40, 27–67 CrossRef CAS.
- T. C. Bond, S. J. Doherty, D. W. Fahey, P. M. Forster, T. Berntsen, B. J. DeAngelo, M. G. Flanner, S. Ghan, B. Karcher, D. Koch, S. Kinne, Y. Kondo, P. K. Quinn, M. C. Sarofim, M. G. Schultz, M. Schulz, C. Venkataraman, H. Zhang, S. Zhang, N. Bellouin, S. K. Guttikunda, P. K. Hopke, M. Z. Jacobson, J. W. Kaiser, Z. Klimont, U. Lohmann, J. P. Schwarz, D. Shindell, T. Storelvmo, S. G. Warren and C. S. Zender, J. Geophys. Res.: Atmos., 2013, 118, 5380–5552 CAS.
- U. Dusek, G. P. Reischl and R. Hitzenberger, Environ. Sci. Technol., 2006, 40, 1223–1230 CrossRef CAS.
- T. Tritscher, Z. Jurányi, M. Martin, R. Chirico, M. Gysel, M. F. Heringa, P. F. DeCarlo, B. Sierau, A. S. H. Prevot, E. Weingartner and U. Baltensperger, Environ. Res. Lett., 2011, 6, 034026 CrossRef.
- B. Zuberi, K. S. Johnson, G. K. Aleks, L. T. Molina, M. J. Molina and A. Laskin, Geophys. Res. Lett., 2005, 32, L01807 CrossRef.
- M. Shiraiwa, Y. Kondo, N. Moteki, N. Takegawa, Y. Miyazaki and D. R. Blake, Geophys. Res. Lett., 2007, 34, L16803 CrossRef.
- J. P. Schwarz, A. E. Perring, M. Z. Markovic, R. S. Gao, S. Ohata, J. Langridge, D. Law, R. McLaughlin and D. W. Fahey, J. Aerosol Sci., 2015, 81, 110–126 CrossRef CAS.
- S. Ding, D. T. Liu, D. L. Zhao, K. Hu, P. Tian, W. Zhou, M. Y. Huang, Y. Yang, F. Wang, J. J. Sheng, Q. Liu, S. F. Kong, P. Y. Cui, Y. D. Huang, H. He, H. Coe and D. P. Ding, Environ. Sci. Technol., 2019, 53, 11112–11121 CrossRef CAS.
- B. Sarangi, S. Ramachandran, T. A. Rajesh and V. K. Dhaker, Atmos. Environ., 2019, 200, 110–118 CrossRef CAS.
- D. Liu, J. Allan, J. Whitehead, D. Young, M. Flynn, H. Coe, G. McFiggans, Z. L. Fleming and B. Bandy, Atmos. Chem. Phys., 2013, 13, 2015–2029 CrossRef.
- G. R. McMeeking, N. Good, M. D. Petters, G. McFiggans and H. Coe, Atmos. Chem. Phys., 2011, 11, 5099–5112 CrossRef CAS.
- K. N. Li, X. N. Ye, H. W. Pang, X. H. Lu, H. Chen, X. F. Wang, X. Yang, J. M. Chen and Y. J. Chen, Atmos. Chem. Phys., 2018, 18, 15201–15218 CrossRef CAS.
- Z. Shi, T. Vu, S. Kotthaus, R. M. Harrison, S. Grimmond, S. Yue, T. Zhu, J. Lee, Y. Han, M. Demuzere, R. E. Dunmore, L. Ren, D. Liu, Y. Wang, O. Wild, J. Allan, W. J. Acton, J. Barlow, B. Barratt, D. Beddows, W. J. Bloss, G. Calzolai, D. Carruthers, D. C. Carslaw, Q. Chan, L. Chatzidiakou, Y. Chen, L. Crilley, H. Coe, T. Dai, R. Doherty, F. Duan, P. Fu, B. Ge, M. Ge, D. Guan, J. F. Hamilton, K. He, M. Heal, D. Heard, C. N. Hewitt, M. Hollaway, M. Hu, D. Ji, X. Jiang, R. Jones, M. Kalberer, F. J. Kelly, L. Kramer, B. Langford, C. Lin, A. C. Lewis, J. Li, W. Li, H. Liu, J. Liu, M. Loh, K. Lu, F. Lucarelli, G. Mann, G. McFiggans, M. R. Miller, G. Mills, P. Monk, E. Nemitz, F. O’Connor, B. Ouyang, P. I. Palmer, C. Percival, O. Popoola, C. Reeves, A. R. Rickard, L. Shao, G. Shi, D. Spracklen, D. Stevenson, Y. Sun, Z. Sun, S. Tao, S. Tong, Q. Wang, W. Wang, X. Wang, X. Wang, Z. Wang, L. Wei, L. Whalley, X. Wu, Z. Wu, P. Xie, F. Yang, Q. Zhang, Y. Zhang, Y. Zhang and M. Zheng, Atmos. Chem. Phys., 2019, 19, 7519–7546 CrossRef CAS.
- Y. Yan, P. Fu, B. Jing, C. Peng, S. K. R. Boreddy, F. Yang, L. Wei, Y. Sun, Z. Wang and M. Ge, Sci. Total Environ., 2017, 578, 307–316 CrossRef CAS.
- W. H. White and P. T. Roberts, Atmos. Environ., 1977, 11, 803–812 CrossRef CAS.
- N. Good, H. Coe and G. McFiggans, Atmos. Meas. Tech., 2010, 3, 1241–1254 CrossRef.
- M. Gysel, G. B. McFiggans and H. Coe, J. Aerosol Sci., 2009, 40, 134–151 CrossRef CAS.
- W. Winklmayr, G. P. Reischl, A. O. Lindner and A. Berner, J. Aerosol Sci., 1991, 22, 289–296 CrossRef CAS.
- Y. Wang, Z. Wu, N. Ma, Y. Wu, L. Zeng, C. Zhao and A. Wiedensohler, Atmos. Environ., 2018, 175, 184–191 CrossRef CAS.
- D. Liu, J. Whitehead, M. R. Alfarra, E. Reyes-Villegas, D. V. Spracklen, C. L. Reddington, S. Kong, P. I. Williams, Y.-C. Ting, S. Haslett, J. W. Taylor, M. J. Flynn, W. T. Morgan, G. McFiggans, H. Coe and J. D. Allan, Nat. Geosci., 2017, 10, 184–188 CrossRef CAS.
- C. Yu, D. Liu, K. Broda, R. Joshi, J. Olfert, Y. Sun, P. Fu, H. Coe and J. D. Allan, Atmos. Chem. Phys., 2020, 20, 3645–3661 CrossRef CAS.
- S. G. Brown, T. Lee, P. T. Roberts and J. L. Collett, J. Air Waste Manage. Assoc., 2013, 63, 1422–1433 CrossRef CAS.
- H. Simon, P. V. Bhave, J. L. Swall, N. H. Frank and W. C. Malm, Atmos. Chem. Phys., 2011, 11, 2933–2949 CrossRef CAS.
- H. Yang and J. Z. Yu, Environ. Sci. Technol., 2002, 36, 5199–5204 CrossRef CAS.
- H. Gordon, et al. , J. Geophys. Res.: Atmos., 2017, 122, 8739–8760 Search PubMed.
- F. Yu and G. Luo, Atmos. Chem. Phys., 2009, 9, 7691–7710 CrossRef CAS.
- V.-M. Kerminen, X. Chen, V. Vakkari, T. Petäjä, M. Kulmala and F. Bianchi, Environ. Res. Lett., 2018, 13, 103003 CrossRef.
- M. Kulmala, H. Vehkamäki, T. Petäjä, M. Dal Maso, A. Lauri, V. M. Kerminen, W. Birmili and P. H. McMurry, J. Aerosol Sci., 2004, 35, 143–176 CrossRef CAS.
- Z. Wang, Z. Wu, D. Yue, D. Shang, S. Guo, J. Sun, A. Ding, L. Wang, J. Jiang, H. Guo, J. Gao, H. C. Cheung, L. Morawska, M. Keywood and M. Hu, Sci. Total Environ., 2017, 577, 258–266 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0fd00077a |
| ‡ Currently at the Department of Atmospheric Sciences, School of Earth Sciences, Zhejiang University, Hangzhou, Zhejiang, China. |
| This journal is © The Royal Society of Chemistry 2021 |