
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of a Ti0.7W0.3O2/TiO2 nanocomposite interfacial photocatalyst and its photocatalytic degradation of phenol pollutants in wastewater†
Zemin
Dong
 *ab,
Rendan
Zhou
c,
Leyan
Xiong
*a,
Han
Li
d,
Qiang
Liu
a,
Longzhen
Zheng
a,
Zanru
Guo
a and
Zhaoxiang
Deng
e
*ab,
Rendan
Zhou
c,
Leyan
Xiong
*a,
Han
Li
d,
Qiang
Liu
a,
Longzhen
Zheng
a,
Zanru
Guo
a and
Zhaoxiang
Deng
e
aDepartment of Chemistry and Chemical Engineering, East China Jiao Tong University, Nanchang 330013, P. R. China. E-mail: xly12@ecjtu.edu.cn; zemin1987d@139.com
bJiangXi Institute for Veterinary Drug and Feedstuffs Control, Nanchang 330096, PR China
cAnalysis and Testing Center, Nan Chang University, Nanchang 330047, P. R. China
dCAS Key Laboratory of Crust-Mantle Materials and Environments, School of Earth and Space Sciences, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
eDepartment of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
First published on 12th December 2019
Abstract
A Ti0.7W0.3O2/TiO2 nanocomposite interfacial photocatalyst was designed and prepared for the photocatalytic degradation of phenol pollutants in wastewater. The detailed properties of the Ti0.7W0.3O2/TiO2 nanocomposite interface (NCI) were analyzed by XRD, SEM, EDX, DRS, UPS and XPS technologies, showing that anatase TiO2 nanospheres (NSs) were uniformly dispersed on the surface of rutile Ti0.7W0.3O2 nanoparticles (NPs) and formed the nanocomposite interface. The DRS and UPS results of 5 wt% Ti0.7W0.3O2/TiO2 NCI indicated a greatly broadened light response range with a wavelength shorter than 527 nm and a shorter band gap energy of 2.37 eV. The conduction band of TiO2 NSs, Ti0.7W0.3O2 NPs and 5 wt% Ti0.7W0.3O2/TiO2 NCI were measured based on the results of the valence band and band gap energy obtained via XPS and DRS, and then the energy level diagram of Ti0.7W0.3O2/TiO2 NCI was proposed. The photocatalytic degradation of phenol at Ti0.7W0.3O2/TiO2 NCI with different loading ratios of Ti0.7W0.3O2 NPs was investigated under optimum conditions (i.e., pH of 4.5, catalyst dosage of 0.45 g L−1 and phenol initial concentration of 95 ppm) under the illumination of ultraviolet visible light. Also, 5 wt% Ti0.7W0.3O2/TiO2 NCI exhibited the highest photocatalytic activity, with the initial rate constant (k) calculated as 0.09111 min−1. After recycling six times, Ti0.7W0.3O2/TiO2 NCI showed good stability and recyclability. The involvement of superoxide radicals in the initial reaction at Ti0.7W0.3O2/TiO2 NCI was evidenced by the use of a terephthalic acid (TA) fluorescent probe. Besides, UV-Vis spectroscopy, UHPLC-MS and GC-MS technologies were used to analyze the main intermediates in the photocatalytic degradation of phenol. The probable photocatalytic degradation mechanism of phenol at Ti0.7W0.3O2/TiO2 NCI was also proposed.
1. Introduction
Environment pollution has become a worldwide problem. Organic wastewater is one of the most typical pollution issues posing a serious threat to the health of humans and has consequently received extensive attention recently.1–3 Over the last several years, the utilization of direct solar light has become a much greener approach for energy generation as well as for environmental clean-up. Therefore, the development and design of a UV-Vis active photocatalyst for direct sunlight harvesting has drawn broad interdisciplinary attention and much research fascination. The photocatalytic decomposition of organic contaminants is one of the most promising techniques for wastewater treatment and purification.Among oxide semiconductor photocatalysts, TiO2 nanomaterials have been studied and applied widely in photocatalysis for their high photocatalytic activity, stability, nontoxicity and low cost. A variety of functional TiO2 nanomaterials have been synthetized, such as nanoparticles (NPs),4–9 nanotubes (NTs),10,11 nanowires (NWs),12–15 nanocrystal films16–20 and nanotube arrays,21–25 which have been widely used in solar energy storage and utilization, photodegradation of pollutants and noble metal recycling.
However, the total quantum efficiency of TiO2 is very low,26–28 which has limited the potential value of actual production and application of TiO2 nanomaterials. A lot of studies have been done to address the drawbacks mentioned above, with noble metal deposition considered as one of the most effective and promising solutions. Pt is one such representative noble metal, which has been widely used to improve the performance of TiO2 nanomaterials in wastewater treatment and air purification with a superior performance. Wang29 successfully synthesised a Pt/TiO2 NW photocatalyst. The recombination rate of electrons and holes was reduced greatly for Pt NPs, resulting in good conductivity. Pt NPs are superior electron acceptors on the photocatalyst surface and enable the timely transfer of electrons. Emilio et al.30 observed an increase in the lifetime of electrons by Pt modification on the TiO2 surface due to the better separation of charge carriers caused by the Schottky barrier between Pt and TiO2. As expected, this helped to enhance the photocatalytic efficiency of TiO2.5–9,11,14,15,19,20,22,25,31–33 However, noble metals are scarce and particularly expensive, which may limit their large-scale application. Thus, novel relatively economical photocatalysts are highly desirable.
In the present paper, a Ti0.7W0.3O2/TiO2 NCI was synthesized via a sol–gel and combustion technique, and was shown to possess several positive aspects, such as good stability, good visible light response range and effectively decreased recombination of charge carriers by a fast photogenerated electron transfer. The photocatalytic activity of the Ti0.7W0.3O2/TiO2 NCI was investigated for the degradation of phenol under simulated solar light illumination, and it showed higher photocatalytic activity. Furthermore, the main intermediates and mechanism for the photocatalytic degradation of phenol at the Ti0.7W0.3O2/TiO2 NCI were also analyzed and discussed. This type of a photocatalyst may find application in low concentration organic wastewater clean-up.
2 Experiment section
2.1 Materials and reagents
Titanium tetrachloride (99.9%), titanium trichloride (99.9%), WCl6 (99.9%), hexachloroplatinic acid (99.9%), carbolic acid (99.9%), HPLC-grade methanol, HPLC-grade acetonitrile and other reagents were all purchased from Shanghai Chemical Reagent Factory. Pure TiO2 samples were commercial Degussa P-25 (55 m2 g−1). Doubly distilled water was used throughout this study. All the chemicals were of analytical grade and were used without further purification.2.2 Synthesis of the TiO2NSs
The pure anatase TiO2 NSs was prepared via a hydrothermal method.34 First, 6 mM NaOH particles were added to 40 mL absolute ethyl alcohol and stirred for 10 min. Then, 2 mL titanium trichloride solution was added to the above NaOH solution drop-wise under vigorous stirring. After 10 min, the mixed solution was transferred to a 50 mL Teflon-lined autoclave and heated at 150 °C for 18 h. After cooling, the precipitate collected through centrifugation was rinsed with distilled water and pure ethanol several times until there were no residual ions left. Then, the products were calcined at 400 °C for 2 h after being dried at 80 °C.2.3 Synthesis of the Ti0.7W0.3O2 NPs
The Ti0.7W0.3O2 NPs were prepared by a modified sol–gel technique. First, 4 mM of WCl6 powder was added to 10 mL of absolute ethyl alcohol with stirring for 10 min. Then, 1 mL of titanium tetrachloride solution was added to 5 mL of distilled water drop-wise under vigorous stirring. The above solutions were mixed together under air-free conditions. Then, the above mixed solution was stirred by mechanical stirring under 40 °C constant temperature water for about 24 h to get a baby blue gel. The product was poured into a drying oven at 100 °C for 12 h. The dry power was added to a 50 mL Teflon-lined autoclave with a moderate amount of alcohol and heated at 180 °C for 8.5 h, and then the precipitate was dried and ground. Finally, the obtained powder was reduced at 1300 °C in a H2 atmosphere for 4 h to obtain a light tan powder. At this point, the W6+ was fully reduced to W4+.35,362.4 Synthesis of the Ti0.7W0.3O2/TiO2 NCI
The Ti0.7W0.3O2/TiO2 NCI was prepared via a simple method. First, 6 mM NaOH were added to 40 mL absolute ethyl alcohol and stirred for 10 min. Then, 2 mL titanium trichloride solution was added to the above NaOH solution drop-wise under vigorous stirring accompanied with a certain quality of pure rutile Ti0.7W0.3O2 NPs.34 After 30 min, the mixed solution was transferred to a 50 mL Teflon-lined autoclave and heated at 180 °C for 8.5 h, and then the precipitate obtained was dried and ground. Then, the obtained powders were calcined at 400 °C in a N2 atmosphere for 2 h. All referenced Ti0.7W0.3O2/TiO2 NCI samples were also prepared by the method described above and used for the photoactivity tests.2.5 Synthesis of the Pt/TiO2 nanocomposites
The synthesis of the Pt/TiO2 nanocomposites and the characterization results are described in the ESI (Section 1, Fig. S1–S4†).Meanwhile, the detailed information on the characterization, photocatalytic test and analysis of the intermediates in the photocatalytic degradation of phenol are shown in ESI (Section 2†).
3 Results and discussion
3.1 Characterization
PXRD analysis was carried out to investigate the impact of Ti0.7W0.3O2 modification on the phase structure and on the chemical composition of the TiO2 NSs, as these have a great influence on the photocatalytic activity. The PXRD patterns of the prepared samples are depicted in Fig. 1(A). According to previous studies, the photocatalytic property of anatase TiO2 is generally considered to be superior, attributed to a higher density of localized states, consequent surface-adsorbed hydroxyl radicals and slower charge-carrier recombination.37–39 All diffraction peaks of the samples (a and i) could be indexed to the International Centre for Diffraction data of pure anatase TiO2 (JCPDS no. 21-1272) and rutile TiO2 (JCPDS no. 21-1276), respectively. The crystallite sizes were calculated by Scherrer's formula given in eqn (1).D = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ | (1) |
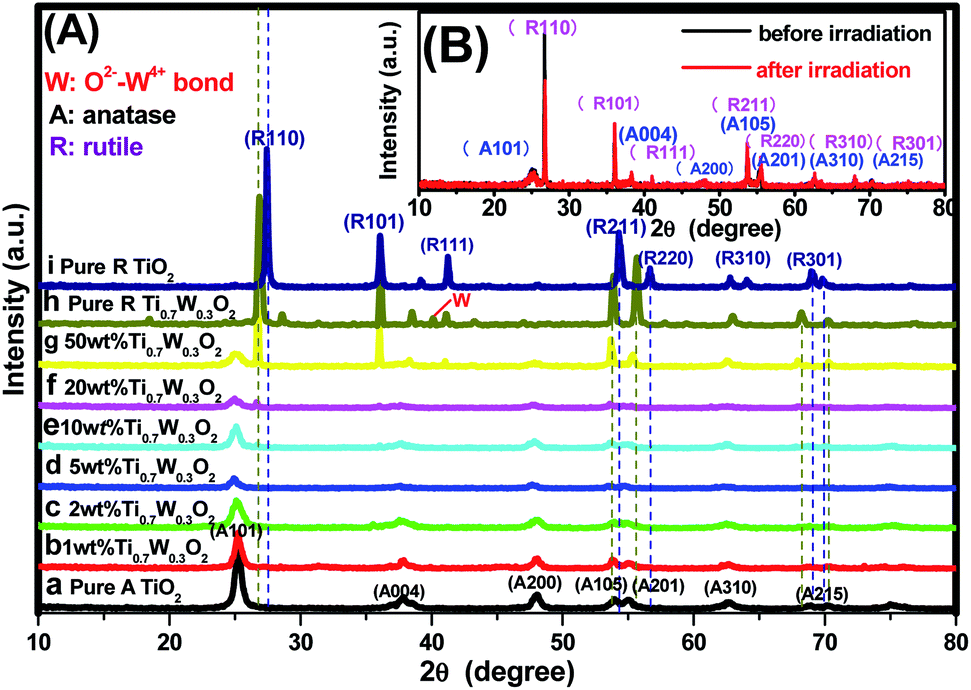 | ||
| Fig. 1 PXRD patterns of the tested samples: (A) (a) pure anatase TiO2, Ti0.7W0.3O2/TiO2 NCI of (b) 1 wt%, (c) 2 wt%, (d) 5 wt%, (e) 10 wt%, (f) 20 wt%, (g) 50 wt%, (h) pure rutile Ti0.7W0.3O2, (i) pure rutile TiO2; (B) 50 wt% Ti0.7W0.3O2/TiO2 NCI before and after six cycles of irradiation, respectively. | ||
The average particle size and distribution of the anatase TiO2 NSs and Ti0.7W0.3O2 NPs were obtained using a laser particle size analyzer and are shown in ESI (Section 3, Fig. S6†). The average particle size and distribution of anatase TiO2 NSs were determined to be about 287 nm, which was larger than the average crystalline size calculated from the major diffraction peak (101) in the XRD analysis. The average particle size and distribution of Ti0.7W0.3O2 NPs were determined to be about 1189 nm, which was consistent with the result calculated from the major diffraction peak (110) in the XRD analysis of the rutile Ti0.7W0.3O2 NPs. The possible reasons for the deviation were as follows. On the one hand, some TiO2 NSs reunite after high temperature calcination. On the other hand, the principles of the two kinds of detection methods were different, whereby the results of the XRD analysis were estimated using an empirical formula, whereas the laser particle size analyzer detection needed the samples to be dispersed in water, and the dispersion of the TiO2 NSs was not very good and they were prone to reunion. This might lead to an increase in the error of the result.
Obviously, some diffraction peaks of Ti0.7W0.3O2 NPs (h) were slightly shifted due to doping with W4+ compared with pure rutile TiO2 (i). This phenomenon indicated an expansion of the a-axis and a contraction of the c-axis due to W–W pairing in the doped compound, which has also been observed in WO2/TiO2 nanocomposites.40–45 Meanwhile, the diffraction peaks for O2−–W4+ were very weak, demonstrating the low levels of W4+.
The PXRD patterns of Ti0.7W0.3O2/TiO2 NCI loaded with 1 wt%, 2 wt%, 5 wt%, 10 wt%, 20 wt% and 50 wt% Ti0.7W0.3O2 NPs are shown in Fig. 1(A)(b–g). All the samples were identical with the pure anatase and rutile phase after calcination at 400 °C, respectively. None of the diffraction peaks were changed significantly after deposition, which indicated that the Ti0.7W0.3O2 NPs did not affect the phase structure and chemical composition of the TiO2 NSs. However, further observation showed that the diffraction peaks corresponding to TiO2 NSs exhibited relatively weaker peak intensities and broader diffraction peak widths. It could be inferred from this that the average crystallite size was slightly decreased by Ti0.7W0.3O2 NPs modification, indicating that the Ti0.7W0.3O2 NPs have a negative effect on the grain growth of TiO2 NSs. This is because the Ti0.7W0.3O2 NPs restrained the crystal growth in the solids by providing dissimilar boundaries and hindered the mass transportation, thus resulting in smaller crystallite sizes.46 Meanwhile, no diffraction peaks of Ti0.7W0.3O2 NPs were observed up to 10 wt% (e), indicating that the TiO2 NSs were uniformly dispersed on the surface of the Ti0.7W0.3O2 NPs.
The superimposed PXRD patterns for the Ti0.7W0.3O2/TiO2 NCI before and after six cycles of irradiation are shown in Fig. 1(B). It is obvious that the two PXRD patterns almost overlap, which indicates that the stability of the Ti0.7W0.3O2/TiO2 NCI was encouraging, with less decomposition, thus accounting for the higher photocatalytic activity. A feeble and relatively weaker peak intensity was also revealed for the loss of a certain amount of photocatalyst during the experiment.
The detailed morphological features of the Ti0.7W0.3O2/TiO2 NCI were characterized by SEM technology and are shown in Fig. 2(A–D). The pure anatase TiO2 was present as nanospheres, with a uniform size distribution as shown in Fig. 2(A). Fig. 2(B) shows the morphology of the pure rutile Ti0.7W0.3O2 NPs, which were irregular and schistose particles with a smooth surface and highly dense quality. Fig. 2(C) and (D) show that the TiO2 NSs were uniformly dispersed on the surface of the Ti0.7W0.3O2 NPs and formed the Ti0.7W0.3O2/TiO2 NCI.
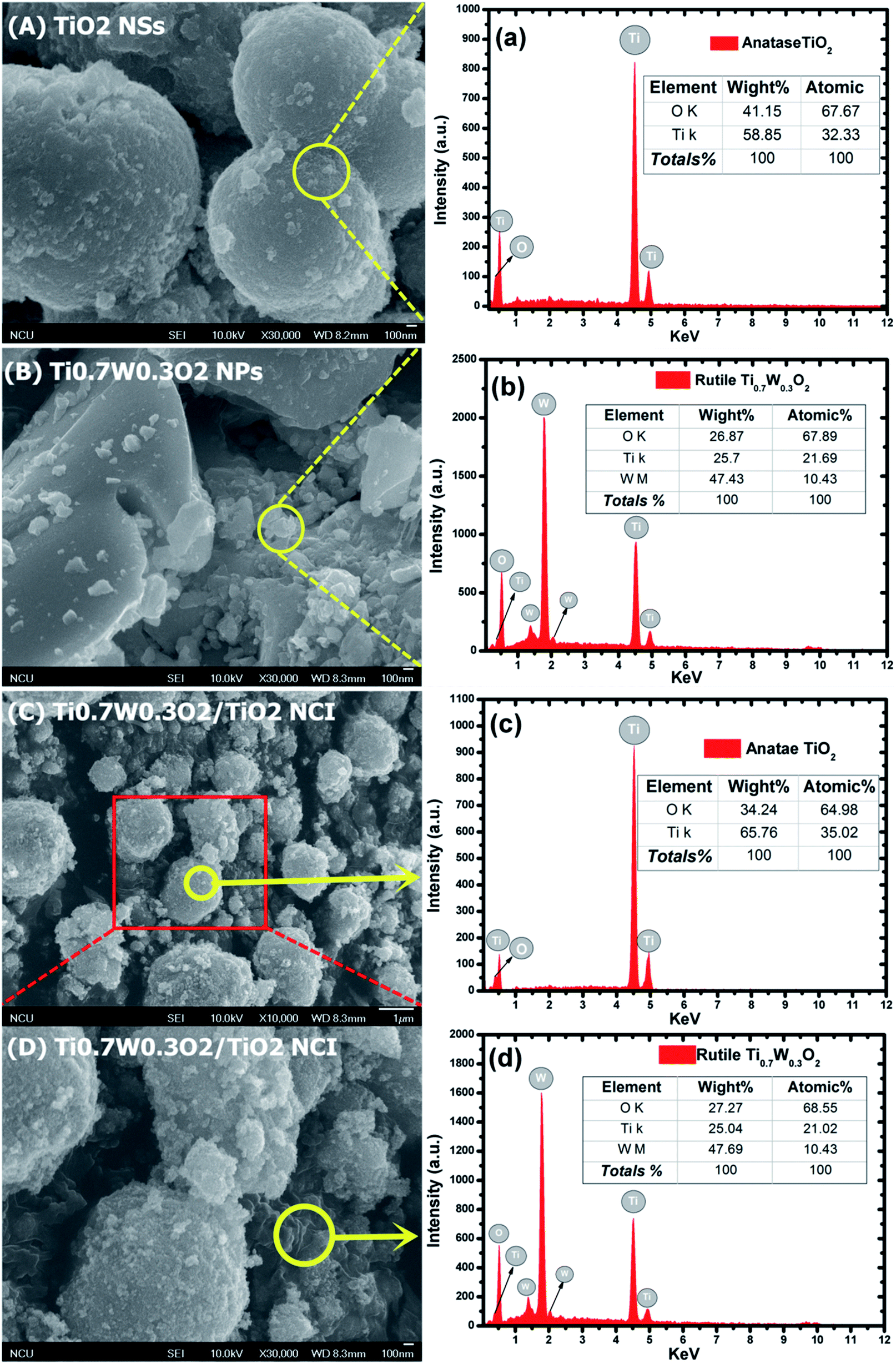 | ||
| Fig. 2 SEM images of: (A) anatase TiO2, (B) Ti0.7W0.3O2 and (C)/(D) 5 wt% Ti0.7W0.3O2/TiO2 NCI. The corresponding EDX test results of: (a) anatase TiO2, (b) Ti0.7W0.3O2 and (c) and (d) 5 wt% Ti0.7W0.3O2/TiO2 NCI, respectively. | ||
The elemental composition of the synthesis of the samples was confirmed by EDX spectra. In Fig. 2(a and b), the anatase TiO2 revealed major peaks of Ti and O, and the rutile Ti0.7W0.3O2 NPs revealed major peaks of Ti, W and O. Fig. 2(c and d) evidence the presence of Ti, W and O elements for the Ti0.7W0.3O2/TiO2 NCI, indicating the high purity, and we can easily distinguish between the two components in the composite photocatalyst.47–49
Furthermore, the atomic% of Ti/the atomic% of O of anatase TiO2 was measured as 0.48, which was close to the mol ratio of Ti/O (0.50) in TiO2. The atomic% of Ti/the atomic% of W of rutile Ti0.7W0.3O2 NPs was measured as 2.08, which was close to the mol ratio of Ti/W (2.33) in Ti0.7W0.3O2. The EDX results for each synthesized sample were a little different with the theoretical mol ratio of elements as EDX merely involved a local analysis of the entire surface of samples, and so these represented acceptable errors.50–52
However, the atomic% of Ti/the atomic% of O of anatase TiO2 and the atomic% of Ti/the atomic% of W of rutile Ti0.7W0.3O2 NPs in Ti0.7W0.3O2/TiO2 NCI were measured to be 0.54 and 2.02, respectively. The error of the results had thus increased, which might be due to the interaction between the two nanomaterials.
A UV-Vis spectrometer was used to record diffuse reflectance spectra in the range 200–800 nm. Fig. 3 (A) shows the DRS of pure TiO2 NSs, pure rutile Ti0.7W0.3O2 and 5 wt% Ti0.7W0.3O2/TiO2 NCI. The band gap values of the synthesized photocatalysts were calculated by plotting (F(R∞)hv)1/2versus the photo energy and the plot is shown in Fig. 3 (B). The pure TiO2 NSs and the pure rutile Ti0.7W0.3O2 demonstrated a photoabsorption modification ability for the UV light region with wavelength shorter than 396 and 598 nm, corresponding to band gap energies of 3.21 and 2.05 eV, respectively. The pure rutile Ti0.7W0.3O2 had a shorter band gap energy due to W4+ doped into the lattice of TiO2. When the pure rutile Ti0.7W0.3O2 was irradiated, conduction band electrons (ecb−) were generated and quickly spread to the valence band due to the shorter band gap energy, which might make the semiconductor have higher conductivity.53 The volume resistivity and conductivity of pure anatase TiO2, 0.3 wt% Pt/TiO2 and 5 wt% Ti0.7W0.3O2/TiO2 are shown in Fig. 3(F). The volume resistivity of pure anatase TiO2 was over 10 times that of 5 wt% Ti0.7W0.3O2/TiO2. The volume resistivity of 0.3 wt% Pt/TiO2 was over 2 times that of 5 wt% Ti0.7W0.3O2/TiO2. It was thus indicated that the conductivity of pure anatase TiO2 could be improved greatly by modifying the Ti0.7W0.3O2 NPs, and that the performance of Ti0.7W0.3O2 NPs was superior to that of Pt NPs, which could fully prove the above conjecture.
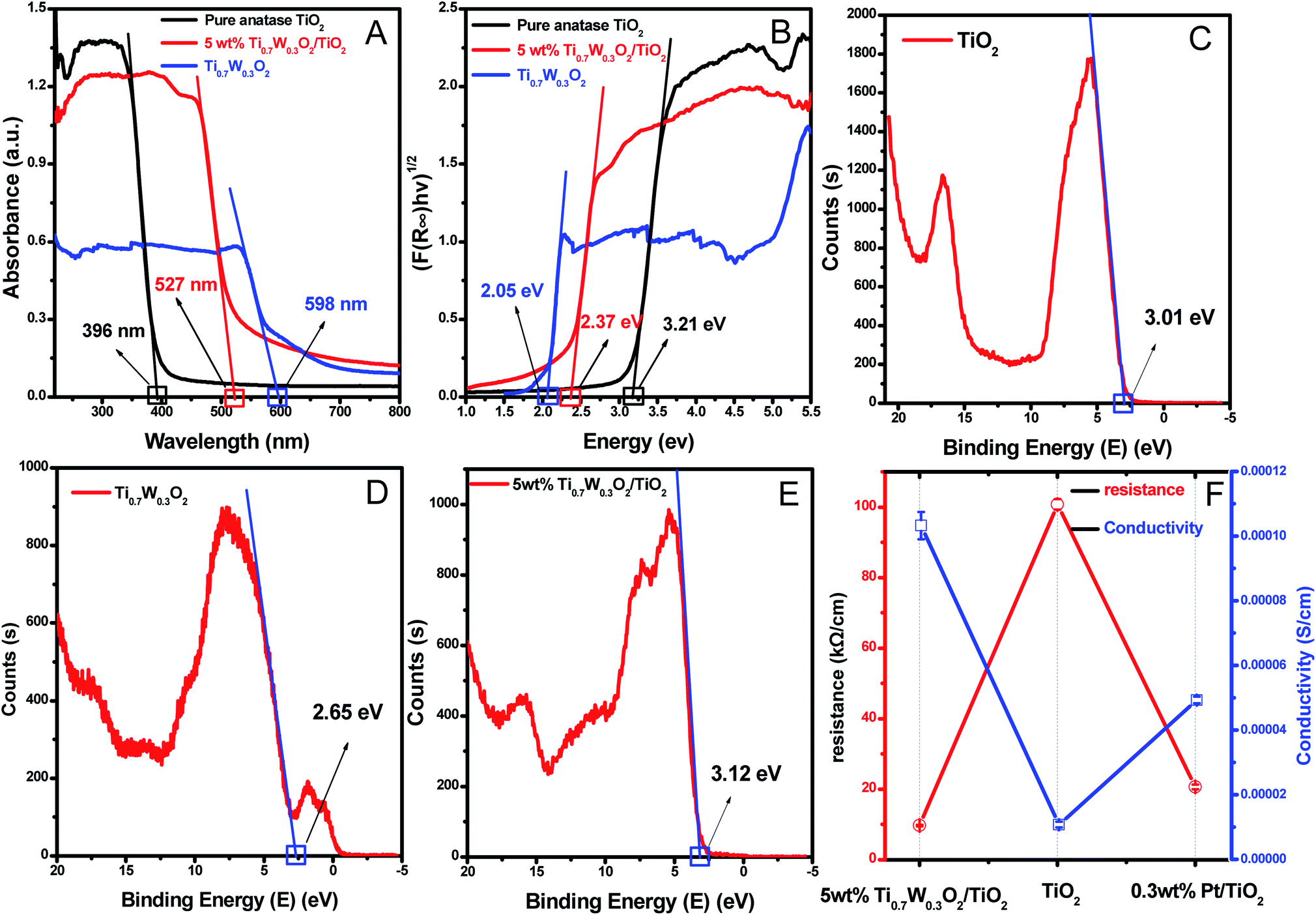 | ||
| Fig. 3 (A) DRS and (B) plots of the transformed Kubelka–Munk function versus the energy of absorbed light for the pure anatase TiO2, Ti0.7W0.3O2 and 5 wt% Ti0.7W0.3O2/TiO2 NCI. The XPS valence band scan spectra of: (C) pure anatase TiO2 NSs, (D) rutile Ti0.7W0.3O2 and (E) 5 wt% Ti0.7W0.3O2/TiO2 NCI. (F) The volume resistivity and conductivity of pure anatase TiO2, 0.3 wt% Pt/TiO2 and 5 wt% Ti0.7W0.3O2/TiO2 NCI. | ||
Meanwhile, the 5 wt% Ti0.7W0.3O2/TiO2 NCI was extended to the visible absorbance region with a wavelength shorter than 527 nm and had a shorter band gap energy of 2.37 eV. The above results were fully proved by ultraviolet photoemission spectroscopy (UPS) and the results are shown in ESI (Section 4, Fig. S7†). The band gap energies of the pure TiO2 NSs and 5 wt% Ti0.7W0.3O2/TiO2 NCI were 3.38 and 2.43 eV, respectively, which were slightly larger than that from the DRS. The reasons for the deviation may be due, on the one hand, to the detection depth of UPS technology, which was 10 atoms, while on the other hand, the carbon pollution signal would be higher for the solid powder sample.
In short, Ti0.7W0.3O2 NPs with higher conductivity can cause fast electron transfer and effectively restrain the recombination of ecb−–hvb+ pairs in Ti0.7W0.3O2/TiO2 NCI, which can diffuse to the surface and react with pollutants and produce more superoxide radical anions (˙O2−)/˙OH−. Furthermore, the Ti0.7W0.3O2/TiO2 NCI could also improve the utilization of visible light, which might account for the higher photocatalytic activity.
The XPS valence band scan spectra of pure anatase TiO2 NSs, rutile Ti0.7W0.3O2 and 5 wt% Ti0.7W0.3O2/TiO2 NCI are shown in Fig. 3(C–E). The valence bands of TiO2 NSs, rutile Ti0.7W0.3O2 and 5 wt% Ti0.7W0.3O2/TiO2 NCI were 3.01, 2.65 and 2.83 eV, respectively. According to the band gap energy result from DRS, the conduction bands were measured as −0.20, 0.60 and 0.46 eV, and the energy level diagram of Ti0.7W0.3O2/TiO2 NCI could be proposed and is shown in ESI (Section 4, Fig. S8†).
The rutile Ti0.7W0.3O2 was further characterized by XPS, as seen in Fig. 4(A–D), to illustrate the structural features and composition. The XPS survey scan spectrum of rutile Ti0.7W0.3O2 is shown in Fig. 4(A). The relative concentrations of Ti and W of Ti0.7W0.3O2 were determined by the respective XPS peak areas and atomic sensitivity factors and nTi/nW was measured as 2.13, which is close to the mol ratio of Ti/W (2.33) in Ti0.7W0.3O254 and consistent with the EDX result.
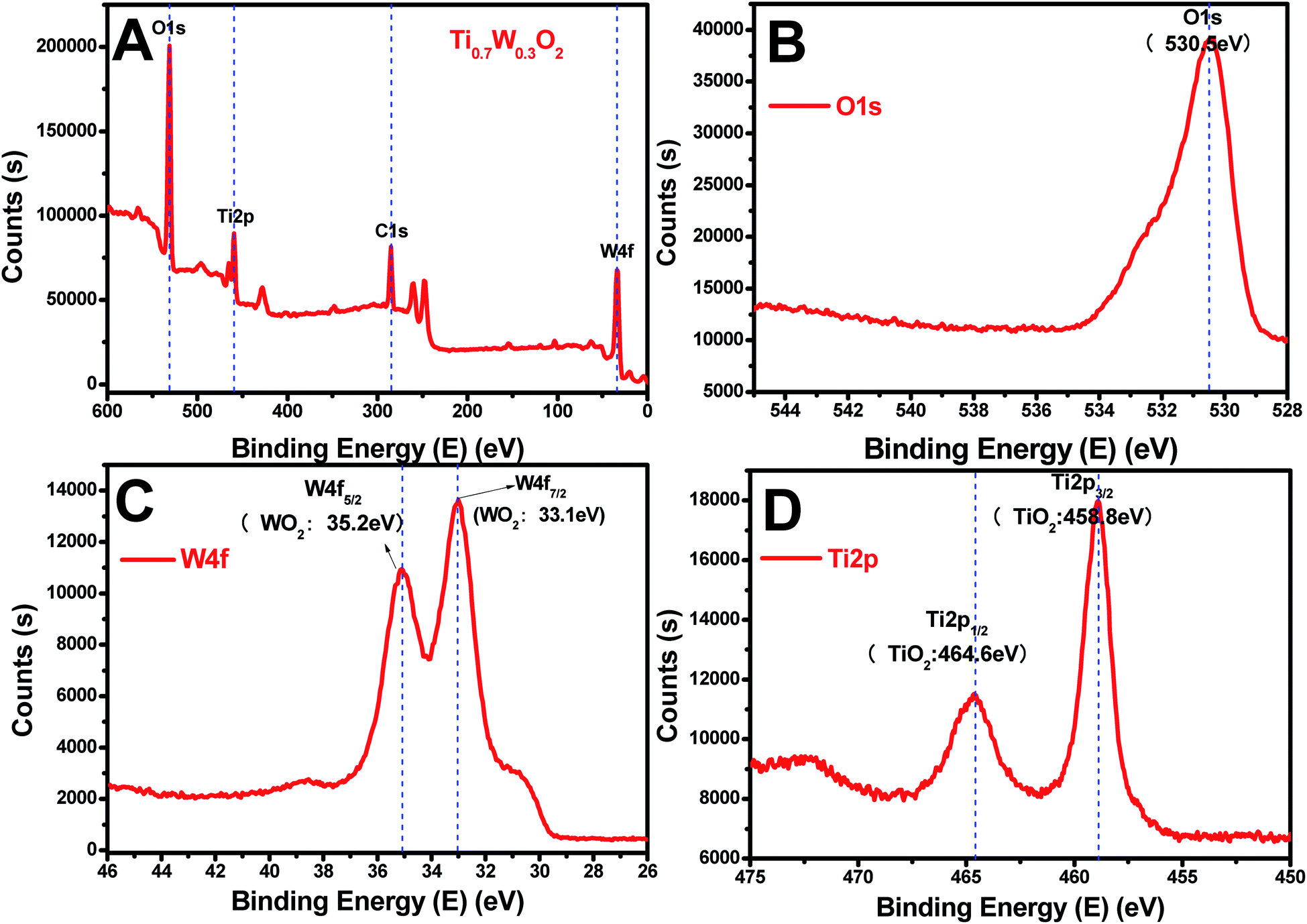 | ||
| Fig. 4 XPS survey scan spectra of: (A) rutile Ti0.7W0.3O2, and XPS narrow scans of: (B) O 1s, (C) W 4f and (D) Ti 2p of the rutile Ti0.7W0.3O2. | ||
The XPS narrow scan spectra of Ti 2p, W 4f and O 1s of the rutile Ti0.7W0.3O2 are shown in Fig. 4(B–D). The Ti 2p1/2 and Ti 2p3/2 peaks of Ti0.7W0.3O2 were located at 464.6 and 458.8 eV and assigned to TiO2, respectively. The W 4f5/2 and W 4f7/2 peaks were located at 34.2 and 33.1 eV, and assigned to WO2, proving that the W6+ was fully reduced to W4+ in Ti0.7W0.3O2.
3.2 Photocatalysis
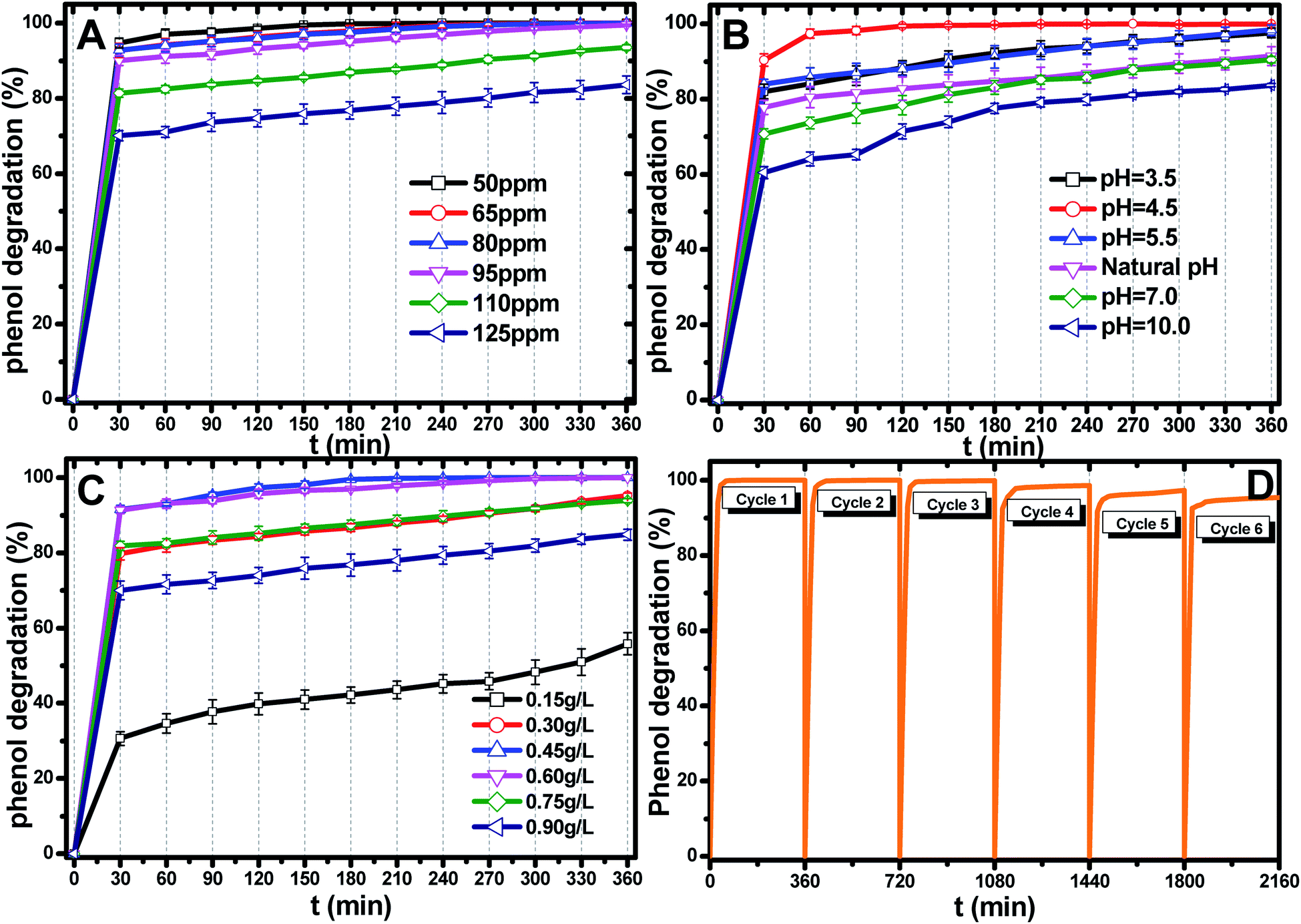 | ||
| Fig. 5 (A) Effect of the initial pH value on phenol photocatalytic degradation; catalyst dosage: 0.45 g L−1, phenol concentration: 95 ppm. (B) Effect of the initial catalyst dosage on phenol photocatalytic degradation; pH: 4.5, phenol concentration: 95 ppm. (C) Effect of the initial phenol concentration on phenol photocatalytic degradation; catalyst dosage: 0.45 g L−1, pH: 4.5. (D) Recyclability of the Ti0.7W0.3O2/TiO2 NCI in the phenol photodegradation process under visible light illumination through six cycles at initial concentration: 95 ppm, pH: 4.5, catalyst dosage: 0.45 g L−1. | ||
The most appropriate initial phenol concentration was investigated with the initial concentration ranging from 50–125 ppm and the results are shown in Fig. 5(B). Obviously, the complete photodegradation time of phenol increased with the increase in the initial concentration from 50–95 ppm, and the photodegradation of phenol at 95 ppm could be just finished with 360 min irradiation. The photocatalytic efficiencies of 110 ppm and 125 ppm phenol were 94.3% and 86.3% after 360 min irradiation, respectively. Further increases decreased the photocatalytic efficiency, indicating that there was an optimum value. The reasonable explanations for this are as follows: first, too many phenol molecules and its intermediates would also absorb a part of the irradiation and limit the light absorption capability of the photocatalysts. Second, excessive amounts of phenol molecules and its intermediates also deactivate more active sites and reduce the light penetration to active sites situated on the surfaces of Ti0.7W0.3O2/TiO2 NCI. The above two disadvantages also result in a lower production of superoxide radical anions (˙O2−)/˙OH− and ultimately a lower oxidation ability.57
The effect of the Ti0.7W0.3O2/TiO2 NCI dosage was investigated by varying the dosage from 0.15 g L−1 to 0.90 g L−1 and the results are shown in Fig. 5(C). When raising the Ti0.7W0.3O2/TiO2 NCI dosage from 0.15 g L−1 to 0.60 g L−1, the phenol photocatalytic efficiency increased from 58.7% to 100% as more active sites were available, increasing the response surface area and leading to a greater production of ˙O2−/˙OH−. However, further increasing, the dosage to 0.90 g L−1 decreased the photocatalytic efficiency. According to the literature,58–62 the reasons for this might be due to the following aspects: on the one hand, an excessive dosage of photocatalysts would result in lower solution transparency, light scattering and interception and the prevention of the light induction of some catalysts particles. On the other hand, too many photocatalysts particles would prevent the effective collisions between phenol molecules and a variety of free radicals. Moreover, the pore volume and available surface area of the photocatalysts would also be diminished with excessive dosage, resulting in a lower photocatalytic activity.
The stability and recyclability of all heterogeneous photocatalysts are critically important for application in wastewater treatment plants. The stability and recyclability of Ti0.7W0.3O2/TiO2 NCI were investigated in a batch reactor under pH 4.5, a catalyst dosage of 0.45 g L−1 and phenol concentration of 95 ppm. After each experiment, the used photocatalyst was collected from the suspension turbid solution and washed with 50% ethanol solution to remove residue phenol and other photodegradation products on the photocatalysts surface. Then, the wet photocatalyst was dried at 105 °C for 4 h. This sequence was repeated six times and the phenol photodegradation efficiency of each cycle recorded and the results are shown in Fig. 5(D). After six recycles, the photocatalytic degradation efficiency of Ti0.7W0.3O2/TiO2 NCI was reduced from 100% to 94.5%, indicating that the Ti0.7W0.3O2/TiO2 NCI showed high photocatalytic activity with good stability and recyclability. The reduction could be explained by a loss of photocatalyst during the washing process, which was consistent with the PXRD results.
Fig. 6(A and B) present the phenol photocatalytic degradation by P-25, Pt/TiO2 and Ti0.7W0.3O2/TiO2 NCI, showing the differences in the phenol degradation activity with the varying loading rates of Ti0.7W0.3O2/TiO2 and Pt/TiO2. The phenol degradation rate of Ti0.7W0.3O2/TiO2 NCI increased with the loading value of Ti0.7W0.3O2 up to 5 wt%; however, a further increase would decrease the photocatalytic activity, indicating that there was an optimum loading value. The optimum value had a close relationship with the dispersion and particle sizes of Ti0.7W0.3O2 NPs. Meanwhile, one could easily find that the phenol degradation rate of 5 wt% Ti0.7W0.3O2/TiO2 NCI was always higher than that of 0.3 wt% Pt/TiO2 at any synchronous irradiation time, revealing its higher photocatalytic activity.
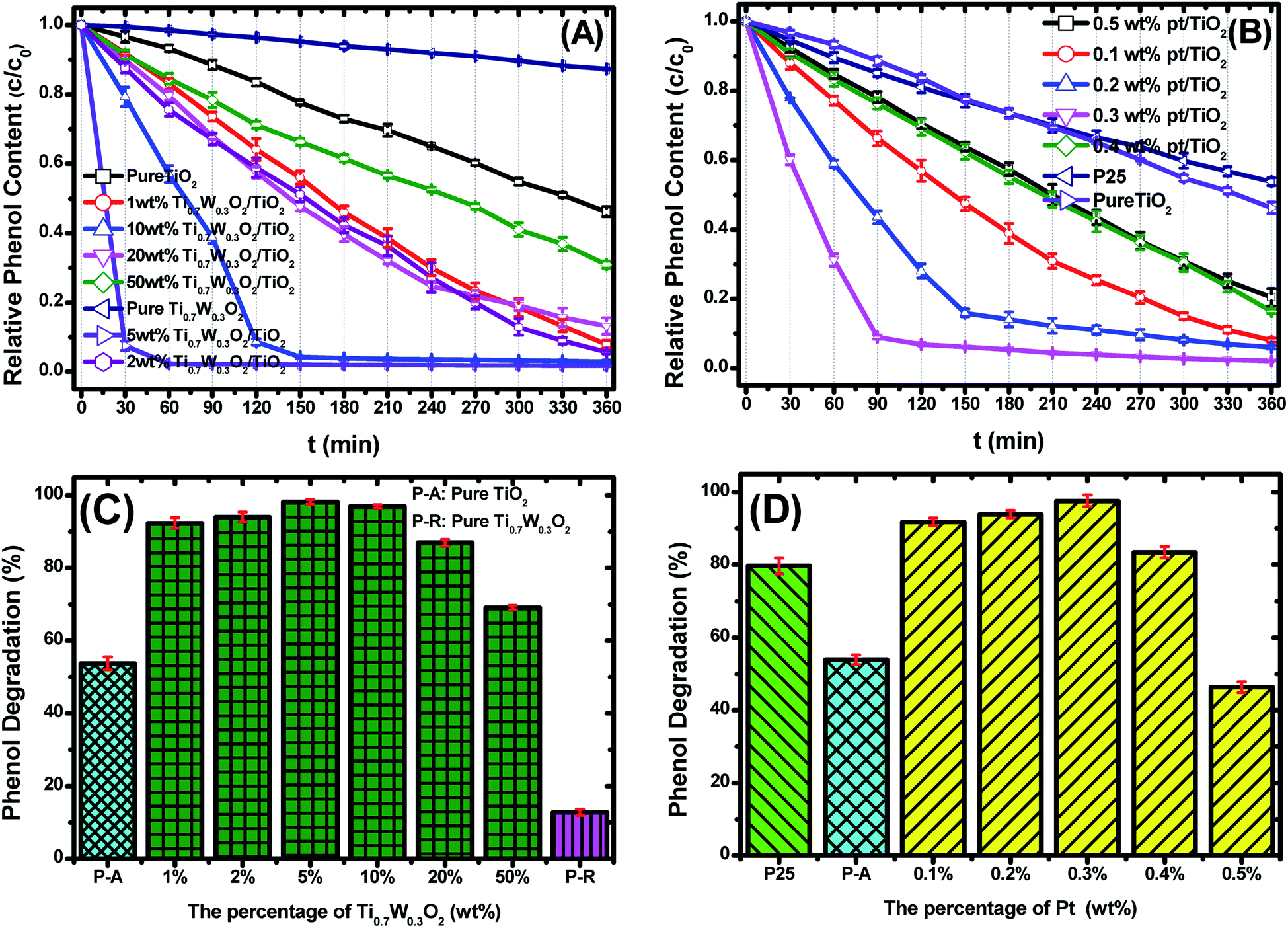 | ||
| Fig. 6 Photocatalytic degradation of phenol over: (A) Ti0.7W0.3O2/TiO2 NCI, (B) P-25 and Pt/TiO2, c = concentration, c0 = initial concentration. The phenol photocatalytic degradation rates of: (C) Ti0.7W0.3O2/TiO2 NCI, (D) P-25 and Pt/TiO2 after 360 min irradiation. | ||
Fig. 6(C and D) compare the phenol photocatalytic degradation rates of P-25, Pt/TiO2 and Ti0.7W0.3O2/TiO2 NCI after 360 min irradiation. The phenol photocatalytic degradation rate after various intervals of time was estimated using the following eqn (2).
| Phenol photocatalytic degradation rate (%) = (ct=0 − ct)/ct=0 × 100% | (2) |
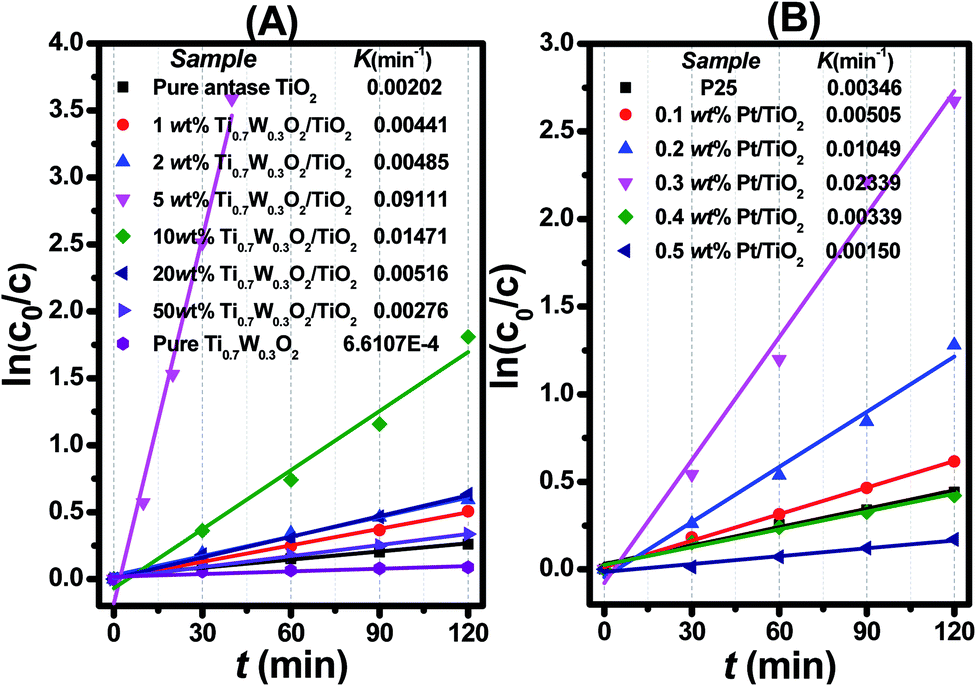 | ||
| Fig. 7 Dependence of In (c0/c) on the irradiation time (t) for: (A) Ti0.7W0.3O2/TiO2 NCI, (B) P25 and Pt/TiO2. | ||
The initial rate constant (k) for phenol photocatalytic degradation in P-25 was calculated as 0.00346 min−1, while the initial rate constant (k) for phenol photocatalytic degradation in 5 wt% Ti0.7W0.3O2/TiO2 and 0.3 wt% Pt/TiO2 was about 26 and 6.8 times that of P-25, respectively. This indicated that the photocatalytic activity of TiO2 was enormously improved with the proper amount of loading Ti0.7W0.3O2 and Pt NPs. Furthermore, the initial rate constant (k) for 5 wt% Ti0.7W0.3O2/TiO2 was over 3.9 times that in 0.3 wt% Pt/TiO2, illustrating that the as-prepared Ti0.7W0.3O2 NPs may be much superior to Pt NPs in embellishing the photocatalytic properties of TiO2 nanomaterials and could even replace them.
Herein, to better assess the photocatalytic activity of the synthesized Ti0.7W0.3O2/TiO2 NCI, we compared our results with the photodegradation of phenol reported in previous studies, as shown in Table 1. The Ti0.7W0.3O2/TiO2 NCI showed several advantages in the photocatalytic performance, photocatalytically degrading the most amount of phenol with the least irradiation time and catalyst dosage. The initial rate constant (k) of the Ti0.7W0.3O2/TiO2 NCI was over 2.6 times that of SnS2/TiO2 nanocomposite catalyst, which showed a higher photocatalytic activity than the other catalysts. Therefore, it was concluded that the Ti0.7W0.3O2/TiO2 NCI was one of the most efficient catalysts for the photocatalytic degradation of phenol under the selected experimental parameters.
| Catalyst | Concentration (mg L−1)/volume (mL) of phenol | Catalyst amount (g L−1) | Degradation (%) | Irradiation time (min) | Initial rate constant (k) (min−1) | Reference |
|---|---|---|---|---|---|---|
| Pt–ZnO | 15 | — | >95 | 540 | — | 63 |
| ZnO | 50/200 | 1.0 | 69.75 | 480 | 0.0150 | 64 |
| GO/TiO2 | 14/100 | 1.48 | 100 | 180 | — | 65 |
| RGO/TiO2 | 50/1700 | — | 96 | 180 | 0.0154 | 66 |
| MWCNT/TiO2 | 50/800 | 1.0 | 96 | 300 | 0.0074 | 67 |
| Fe/S/TiO2 | 20/60 | 1.0 | 99.4 | 600 | — | 68 |
| CNT/Ce–TiO2 | 50/500 | 0.4 | 95 | 180 | 0.0012 | 69 |
| BiPO4 | 50/100 | 0.5 | 100 | 240 | 0.0370 | 70 |
| Co/Pd/BiVO4 | 18.4/100 | 0.8 | 90 | 180 | 0.0130 | 71 |
| ZnO/TiO2 | 60/250 | 0.6 | 100 | 160 | 0.0124 | 72 |
| TiO2−xBx | 94/50 | 6 | 97 | 240 | 0.0084 | 73 |
| BiMnO4 | 20/100 | 1.0 | 90 | 480 | 0.0049 | 74 |
| Fe(III)–TiO2 | 100/1500 | 0.5 | 93.8 | 210 | 0.0190 | 75 |
| TiO2/Ag/C | 20/100 | 1.0 | 95 | 60 | — | 76 |
| N–TiO2@CS | 9.4/40 | 2.5 | 90 | 180 | — | 77 |
| V2O5/N,S–TiO2 | 100/20 | 1.0 | 88 | 240 | — | 78 |
| Pt/TiO2 | — | — | 87.7 | 180 | — | 79 |
| TiO2–Fe2O3–graphene | 5/100 | 1.5 | — | 150 | 0.01415 | 80 |
| SnS2/TiO2 | 10/100 | 0.5 | — | 150 | 0.03595 | 81 |
| Ti 0.7 W 0.3 O 2 /TiO 2 NCI | 95/1000 | 0.45 | 98.7 | 50 | 0.09111 | This study |
The UV-Vis absorption spectra from the photodegradation of phenol over P-25, Pt/TiO2 and Ti0.7W0.3O2/TiO2 are compared in ESI (Section 6, Fig. S10†). At P-25, besides the characteristic absorption bands at 270 nm of phenol, a new absorption band at 289 nm appeared, which might be attributed to the ring-retaining compounds.29,59,86,87 However, besides the two absorption bands at 270 and 289 nm, there were two new absorption bands at 247 and 257 nm for Pt/TiO2, and another two new absorption bands at 333 and 363 nm for Ti0.7W0.3O2/TiO2, which might be attributed to ring-opened produciits.87,88 It can be concluded that the phenol photodegradation pathway over Ti0.7W0.3O2/TiO2 NCI was partially different from that over P-25 and Pt/TiO2.
The aqueous solutions of phenol degradation over P25, Pt/TiO2 and Ti0.7W0.3O2/TiO2 were detected by UV-Vis spectrometry, UHPLC-MS and GC-MS. Then, the main intermediates were analyzed and inferred by the molecular ions and mass fragment peaks present and from library data. The LC chromatograms, UV-Vis spectrograms and mass spectra from HPLC-MS are shown in ESI (Section 7, Fig. S11a–k†). The GC chromatograms and mass spectra from GC-MS of the intermediates are shown in ESI (Section 8, Fig. S12 and S13†). The analytical results and possible structures of each intermediate are shown in ESI (Section 9, Table S1 and Fig. S14†).
In short, five and six kinds of intermediates were identified in the aqueous suspension of P-25 and Pt/TiO2, respectively. Six kinds of intermediates were found in the aqueous suspension of Ti0.7W0.3O2/TiO2. The further degradation of all the intermediates might include oxidative hydroxylation and oxidative decarboxylation products, etc. from several reaction pathways operating simultaneously.
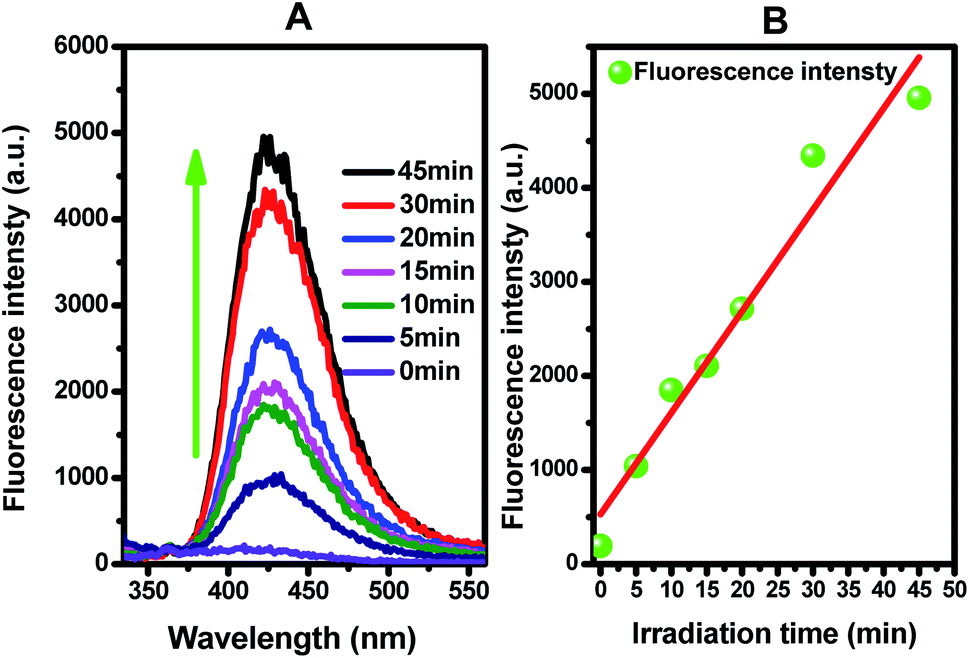 | ||
| Fig. 8 (A) Fluorescence spectral changes observed during the illumination of Ti0.7W0.3O2/TiO2 NCI in 0.01 M NaOH and 3 mM terephthalic acid solution (under 315 nm excitation); (B) plots showing the induced fluorescence intensities (425 nm) against light illumination time for terephthalic acid. | ||
Figure 8(B) presents the fluorescence intensity as a function of the duration of irradiation. The fluorescence intensity increased linearly with the irradiation time, showing that the formation superoxide radical follows the quasi-first-order-type kinetics, as evidenced by the linear relationship between the concentration of superoxide radical and irradiation time within a certain range.
Therefore, based on the present experimental data and the referenced studies,29,59,91,94–97 the different intermediates of P-25, Pt/TiO2 and Ti0.7W0.3O2/TiO2 indicate the different phenol degradation processes, as clearly illustrated in Fig. 9.
 | ||
| Fig. 9 Proposed photocatalytic degradation process of phenol over: (A) P-25, (B) Pt/TiO2 and (C) Ti0.7W0.3O2/TiO2 NCI. | ||
In addition, we believe that the photocatalytic degradation of phenol over P-25 follows a radical reaction mechanism. The photocatalytic degradation of phenol over Pt/TiO2 and Ti0.7W0.3O2/TiO2 follows both a radical reaction mechanism and holes reaction mechanism, which proceed in parallel.
The phenol photocatalytic degradation mechanism involves initial reactions at the Ti0.7W0.3O2/TiO2 NCI, as shown in Scheme 1. It was well established that conduction band electrons (ecb−) and valence band holes (hvb+) were generated when the suspension was irradiated. A Schottky barrier might be formed at the NCI between TiO2 NSs and Ti0.7W0.3O2 NPs, leading to a greater formation of hvb+ and ecb− and an enhanced photocatalytic activity.59 Ti0.7W0.3O2 NPs with high conductivity might cause fast electron transfer and effectively restrain the recombination of ecb−–hvb+ pairs in the bulk catalyst, which can then diffuse to the surface and react with pollutants.54 Hydroxyl radical (˙OH−) formation occurred at the Ti0.7W0.3O2/TiO2 NCI by hvb+ trapping absorbed hydroxyl and hydration molecules (OH−/H2O). Meanwhile, ecb− could react with molecular oxygen (O2) adsorbed at the NCI and produce superoxide radical anion (˙O2−). Acidic conditions could generate a higher affinity towards unpaired ecb− of NCI, leading to the formation of more hydroxyl radicals (˙HO2−).98,99 The hvb+ could also oxidize pollutants directly. These might account for the extraordinary photocatalytic activity of Ti0.7W0.3O2/TiO2 NCI.
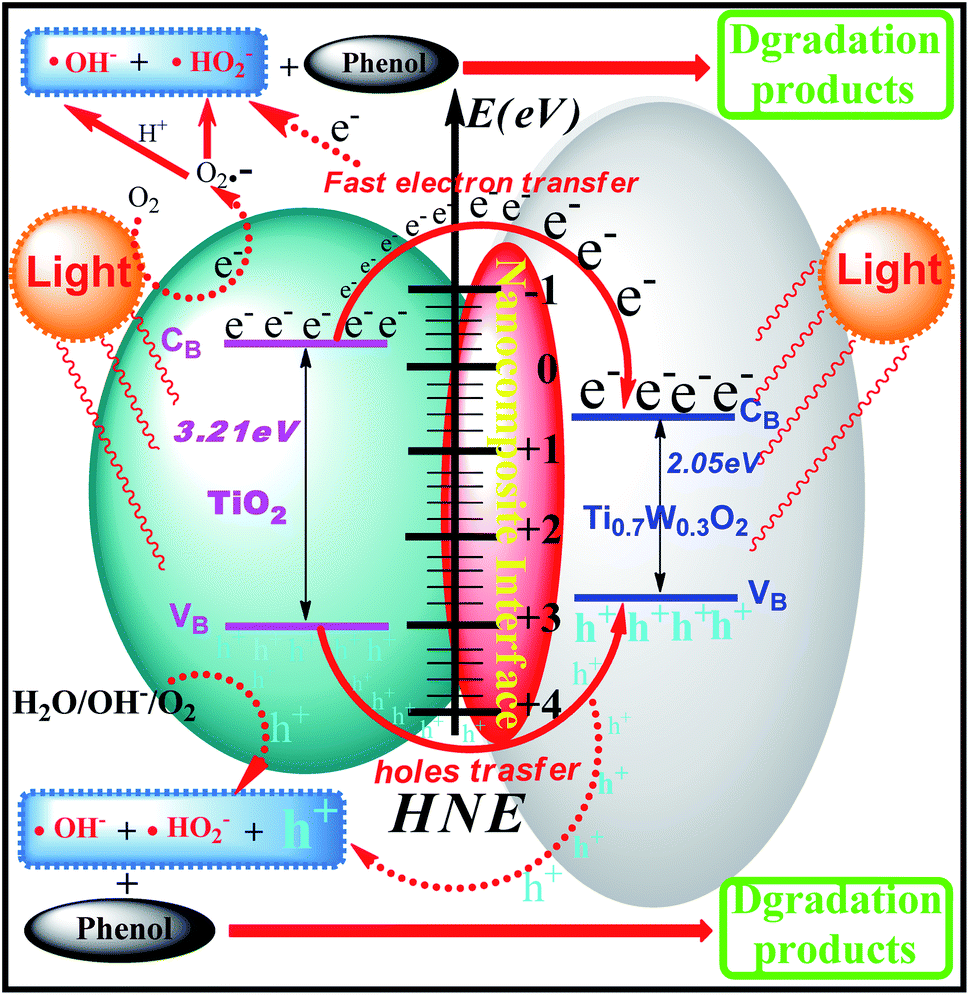 | ||
| Scheme 1 Proposed phenol photocatalytic degradation mechanism at the Ti0.7W0.3O2/TiO2 NCI. | ||
4. Conclusions
In summary, Ti0.7W0.3O2/TiO2 nanocomposite interfacial photocatalysts with loading of different weight ratios of Ti0.7W0.3O2NPs were designed and synthesized for the photocatalytic degradation of phenol in wastewater under the illumination of ultraviolet visible light. The optimum photocatalytic degradation of phenol conditions were pH 4.5, a catalyst dosage of 0.45 g L−1 and phenol initial concentration of 95 ppm. The 5 wt% Ti0.7W0.3O2 NPs was the best loading level, and the initial rate constant (k) for 5 wt% Ti0.7W0.3O2/TiO2 NCI was over 3.9 times that in 0.3 wt% Pt/TiO2. The valence bands of TiO2 NSs, Ti0.7W0.3O2 NPs and 5 wt% Ti0.7W0.3O2/TiO2 NCI were 3.01, 2.65 and 2.83 eV, the band gap energies were 3.21, 2.05 and 2.37 eV, respectively. Then the conduction bands of the above three materials were measured to be −0.20, 0.60 and 0.46 eV. Ti0.7W0.3O2/TiO2 NCI showed a greatly broadened light response range, shorter band gap energy and good stability and recyclability. A Schottky barrier was formed at the NCI between TiO2 NSs and Ti0.7W0.3O2 NPs, leading to a higher formation of hvb+ and ecb−, and the Ti0.7W0.3O2 NPs could quickly transfer ecb− and effectively restrain the recombination of ecb−–hvb+ pairs for high conductivity. A large number of superoxide radicals were generated in the suspension in the photocatalytic degradation of phenol by the Ti0.7W0.3O2/TiO2 NCI, which enhanced the photocatalytic activity. Besides, five and six kinds of intermediates were identified in the suspension of P-25 and Pt/TiO2, respectively, and six kinds of intermediates were found in the suspension of Ti0.7W0.3O2/TiO2. The photocatalytic degradation of phenol over P-25 followed a radical reaction mechanism. The photocatalytic degradation of phenol over Pt/TiO2 and Ti0.7W0.3O2/TiO2 followed both a radical reaction mechanism and hole reaction mechanism, which proceeded in parallel.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21802041; No. 21465011), the Project of JiangXi Academic and Technological leader (No. 20133BCB22007), the Project of Science and Technology of JiangXi Province education department (No. 180304) and the Natural Science Foundation of JiangXi Province (No. 20192BAB206014; No. 20132BAB203012).Notes and references
- D. F. Zhang, Monatsh. Chem., 2011, 706, 631–632 Search PubMed.
- C. Galidino, P. Jacques and T. Kalt, J. Photochem. Photobiol., A, 2001, 141, 47–56 CrossRef.
- X. Zhang, Y. Liu, K. Yan and H. Wu, J. Biosci. Bioeng., 2007, 104, 104–110 CrossRef CAS PubMed.
- P. Wang, S. M. Zakeeruddin, P. Comte, R. Charvet, R. Humphry-Baker and M. Gr, J. Phys. Chem. B, 2003, 107, 14336–14341 CrossRef CAS.
- L. Ma, G. Wang, C. Jiang, H. Bao and Q. Xu, Appl. Surf. Sci., 2017, 430, 263–272 CrossRef.
- G. Zhao, J. Zou, C. Li, J. Yu, X. Jiang, Y. Zheng, W. Hu and F. Jiao, J. Mater. Sci.: Mater. Electron., 2018, 29, 7002–7014 CrossRef CAS.
- T. H. Liou, L. W. Hung, H. S. Syu and L. Chu, J. Nanosci. Nanotechnol., 2018, 18, 20–29 CrossRef CAS PubMed.
- K. Alamelu, V. Raja, L. Shiamala and B. M. Jaffar, Appl. Surf. Sci., 2018, 430, 585–594 CrossRef.
- D. Hu, L. Rui, M. Li, J. Pei, F. Guo and S. Zhang, Mater. Res. Express, 2018, 5, 20–29 CrossRef.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Adv. Mater., 1999, 11, 1307–1311 CrossRef CAS.
- E. Han, F. Hu, S. Zhang and B. Luan, Energy Fuels, 2018, 32, 3627–3636 CrossRef.
- Z. Miao, D. Xu, S. Ou, J. H. yang, G. L. Guo, X. S. Zhao and Y. Q. Tang, Nano Lett., 2002, 2, 717–720 CrossRef CAS.
- J. Q. Huang, Y. G. Cao, Q. F. Huang, H. He, Y. Liu, W. Guo and M. C. Hong, Cryst. Growth Des., 2009, 9, 3632–3637 CrossRef CAS.
- F. Fu, F. Wang, T. Li, C. Jiao, Y. Zhang and Y. Chen, Materials, 2018, 11, 1022 CrossRef PubMed.
- W. Wang, Z. Wu, E. Eftekhari, Z. Huo, X. Li, M. Trad, Y. Chen, Z. Yan, C. Li, Q. Li and D. Zhao, Catal. Sci. Technol., 2018, 8, 1039–1050 RSC.
- S. D. Burnside, V. Shklover, C. Barbe, P. Comte, F. Arendse, K. Brooks and M. Gratzel, Chem. Mater., 1998, 10, 2419–2425 CrossRef CAS.
- H. Yang and J. Yang, RSC Adv., 2018, 8, 11921–11929 RSC.
- S. Mondal and D. Basak, Appl. Surf. Sci., 2018, 427, 147–155 CrossRef.
- J. Li, D. Ren, Z. Wu, J. Xu, Y. Bao, S. He and Y. Chen, J. Colloid Interface Sci., 2018, 530, 78–87 CrossRef CAS PubMed.
- X. Yang, Y. Min, S. Li and D. Wang, Catal. Sci. Technol., 2018, 8, 1357–1365 RSC.
- O. K. Varghese, D. W. Gong, M. Paulose, K. G. Ong, E. C. Dickey and C. A. Grimes, Adv. Mater., 2003, 15, 624–627 CrossRef CAS.
- M. Motola, L. Satrapinskyy, M. Čaplovicová, T. Roch, M. Gregor, B. Grancic, J. Gregus, L. Caplovic and G. Plesch, Appl. Surf. Sci., 2018, 434, 1257–1265 CrossRef CAS.
- Y. Yang, L. Kao, Y. Liu, K. Sun, H. Yu, J. Guo, S. Liou and M. Hoffmann, ACS Catal., 2018, 8, 4278–4287 CrossRef CAS PubMed.
- F. Feng, W. Yang, S. Gao, C. Sun and Q. Li, ACS Sustain. Chem. Eng., 2018, 6, 6166–6174 CrossRef CAS.
- J. Low, S. Qiu, D. Xu, C. Jiang and B. Cheng, Appl. Surf. Sci., 2018, 434, 423–432 CrossRef CAS.
- P. Fu, P. Zhang and J. Li, Appl. Catal., B, 2011, 105, 220–228 CrossRef CAS.
- M. Fox and M. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS.
- M. Hoffmann, S. Martin, W. Choi and D. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- C. Wang, L. Yin, L. Zhang, N. Liu, N. Lun and Y. Qi, ACS Appl. Mater. Interfaces, 2010, 2, 3373–3377 CrossRef CAS PubMed.
- C. A. Emilio, M. I. Litter, M. Kunst, M. Bouchard and C. Colbeau-Justin, Langmuir, 2006, 22, 3606–3613 CrossRef CAS PubMed.
- Z. Q. He, L. Xie, J. J. Tu, S. Song, W. P. Liu, Z. W. Liu, J. Q. Fan, Q. Liu and J. M. Chen, J. Phys. Chem. C, 2010, 114, 526–532 CrossRef CAS.
- D. Tsukamoto, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2012, 134, 6309–6315 CrossRef CAS PubMed.
- Y. Shiraishi, D. Tsukamoto, Y. Sugano, A. Shiro, S. Ichikawa, S. Tanaka and T. Hirai, ACS Catal., 2012, 2, 1984–1992 CrossRef CAS.
- C. S. Guo, M. Ge, L. Liu, G. D. Gao, Y. C. Feng and Y. Q. Wang, Environ. Sci. Technol., 2010, 44, 419–425 CrossRef CAS PubMed.
- C. V. Subban, Q. Zhou, A. Hu, T. E. Moylan, F. T. Wagner and F. DiSalvo, J. Am. Chem. Soc., 2010, 132, 17531–17536 CrossRef CAS PubMed.
- M. Marezio, P. Bordet, J. J. Capponi, R. J. Cava, C. Chaillout, J. Chenavas, A. W. Hewat, E. A. Hewat, J. L. Hodeau and P. Strobel, Physica C, 1989, 281, 281–284 CrossRef.
- A. Sclafani and J. M. Herrmann, J. Phys. Chem., 1996, 100, 13655–13661 CrossRef CAS.
- K. Hadjiivanov and D. G. Klissurski, Chem. Soc. Rev., 1996, 25, 61–69 RSC.
- E. Setiawati and K. Kawano, J. Alloys Compd., 2008, 451, 293–296 CrossRef CAS.
- D. J. Palmer and P. G. Dickens, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 2199–2201 CrossRef.
- M. Aryanpour, R. Hoffmann and F. DiSalvo, Chem. Mater., 2009, 21, 1627–1635 CrossRef CAS.
- C. Subban, Q. Zhou, B. Leonard, C. Ranjan, H. M. Edvenson, S. Munie, J. Hunting and F. DiSalvo, Philos. Trans. R. Soc., A, 2010, 368, 3243–3253 CrossRef CAS PubMed.
- P. Chatterjee and S. P. Sen Gupta, J. Appl. Crystallogr., 1999, 32, 1060–1068 CrossRef CAS.
- U. Gesenhues, J. Appl. Crystallogr., 2005, 38, 749–756 CrossRef CAS.
- J. Pelleg, E. Elish and D. Mogilyanski, Metall. Mater. Trans. A, 2005, 36, 3187–3194 CrossRef.
- P. Chen and X. G. Zhang, Clean: Soil, Air, Water, 2010, 36, 507–511 Search PubMed.
- M. Maleki and M. Haghighi, J. Mol. Catal. A: Chem., 2016, 424, 283–296 CrossRef CAS.
- Y. Chen, X. Xie, X. Xin, Z. Tang and Y. Xu, ACS Nano, 2019, 13, 295–304 CrossRef CAS PubMed.
- T. Parandhaman, N. Pentela, B. Ramalingam, D. Samantaand and S. Das, ACS Sustainable Chem. Eng., 2017, 5, 489–501 CrossRef CAS.
- T. Parandhamanaand and S. Das, Biomater. Sci., 2018, 6, 3356–3372 RSC.
- S. Das, A. Dasand and A. Guha, Langmuir, 2009, 25, 8192–8199 CrossRef CAS PubMed.
- B. Weng, K. Lu, Z. Tang, H. Chen and Y. Xu, Nat. Commun., 2018, 9, 1543–1551 CrossRef PubMed.
- M. Mehrad, Mater. Sci. Semicond. Process., 2015, 30, 599–604 CrossRef CAS.
- L. Zheng, L. Xiong, Q. Liu, J. Xu, X. Kang, Y. Wang, S. Yang, J. Xia and Z. Deng, Electrochim. Acta, 2014, 150, 197–204 CrossRef CAS.
- A. Khataee, B. Kayan, P. Gholami, P. Gholami, D. Kalderis and S. Akay, Ultrason. Sonochem., 2017, 39, 540–549 CrossRef CAS.
- S. Kim and W. Choi, J. Phys. Chem. B, 2005, 109, 5143–5149 CrossRef CAS PubMed.
- A. Taufik, A. Albert and R. Saleh, J. Photochem. Photobiol. A Chem., 2017, 344, 149–162 CrossRef CAS.
- S. Jorfi, S. Pourfadakari and B. Kakavandi, Chem. Eng. J., 2018, 343, 95–107 CrossRef CAS.
- M. Ahmadi, B. Kakavandi, N. Jaafarzadeh and A. Babaei, Separ. Purif. Technol., 2017, 177, 293–303 CrossRef CAS.
- I. Tsuji, H. Kobayashi, A. Kudo and H. Kato, J. Am. Chem. Soc., 2004, 126, 13406–13413 CrossRef CAS PubMed.
- R. Ezzat, N. Elham, A. A. Zinatizadehc and Z. Hadis, RSC Adv., 2016, 6, 96554–96562 RSC.
- A. Sobczynski, L. Duczmal and W. Zmudzinski, J. Mol. Catal. A Chem., 2004, 213, 225–230 CrossRef CAS.
- N. Morales-Flores, U. Pal and E. Sánchez Mora, Appl. Catal., A, 2011, 394, 269–275 CrossRef CAS.
- H. Benhebal, M. Chaib, T. Salmon, J. Geens, A. Leonard, S. D. Lambert, M. Crine and B. Heinrichs, Alexandria Eng. J., 2013, 52, 517–523 CrossRef.
- R. Shahbazi, A. Payan and M. Fattahi, J. Photochem. Photobiol., A, 2018, 364, 564–576 CrossRef CAS.
- H. Adamu, P. Dubey and J. A. Anderson, Chem. Eng. J., 2016, 284, 380–388 CrossRef CAS.
- W. Wang, P. Serp, P. Kalck and J. L. Faria, J. Mol. Catal. A: Chem., 2005, 235, 194–199 CrossRef CAS.
- Y. Niu, M. Xing, J. Zhang and B. Tian, Catal. Today, 2013, 201, 159–166 CrossRef CAS.
- N. Shaari, S. H. Tan and A. R. Mohamed, J. Rare Earths, 2012, 30, 651–658 CrossRef CAS.
- Y. Liu, Y. Zhu, J. Xu, X. Bai, R. Zong and Y. Zhu, Appl. Catal., B, 2013, 142–143, 561–567 CrossRef CAS.
- K. Zhang, Y. Liu, J. Deng, S. Xie, X. Zhao, J. Yang, Z. Han and H. Dai, Appl. Catal., B, 2018, 224, 350–359 CrossRef CAS.
- F. Hayati, A. A. Isari, M. Fattahi, B. Anvaripour and S. Jorfibc, RSC Adv., 2018, 8, 40035–40053 RSC.
- L. Y. Xiong, L. Z. Zheng, J. P. Xu, D. Zheng, J. H. Li, X. J. Li, J. Sun, Q. Liu, L. N. Niu, S. M. Yang and J. Xia, Environ. Chem. Lett., 2010, 39, 1201–1208 Search PubMed.
- W. Yin, W. Wang and S. Sun, Catal. Commun., 2010, 11, 647–650 CrossRef CAS.
- S. H. Borji, S. Nasseri, A. H. Mahvi, R. Nabizadeh and A. H. Javadi, J. Environ. Health Sci. Eng., 2014, 12, 101–111 CrossRef PubMed.
- M. Shang, W. Wang, S. Sun, E. P. Gao, Z. J. Zhang, L. Zhang and R. P. O'Hayre, Nanoscale, 2013, 5, 5036–5042 RSC.
- P. Soumyashree, K. M. Parida and B. Naik, RSC Adv., 2013, 3, 4976–4984 RSC.
- S. Martha, D. P. Das, N. Biswal and K. M. Parida, J. Mater. Chem., 2012, 22, 10695–10703 RSC.
- M. A. Barakat, R. I. Al-Hutailah, E. Qayyumet, J. Rashid and J. N. Kuhn, Environ. Technol., 2014, 35, 137–144 CrossRef CAS.
- L. M. He, L. Q. Jing, Z. J. Li and W. T. Sun, RSC Adv., 2013, 3, 7438–7444 RSC.
- Z. Zhang, C. Shao, X. Li, Y. Y. Sun, M. Y. Zhang, J. B. Mu, P. Zhang, Z. C. Guo and Y. C. Liu, Nanoscale, 2013, 5, 606–618 RSC.
- B. Weng, M. Y. Qi, C. Han, Z. R. Tang and Y. J. Xu, ACS Catal., 2019, 9, 4642–4687 CrossRef CAS.
- N. Zhang, M. Q. Yang, S. Q. Liu, Y. G. Sun and Y. J. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS.
- K. Nagaveni, G. Sivalingam, M. S. Hegde and M. Giridhar, Environ. Sci. Technol., 2004, 38, 1600–1604 CrossRef CAS.
- I. Ilisz and A. Dombi, Appl. Catal., A, 1999, 180, 35–45 CrossRef CAS.
- S. X. Liu and X. Y. Chen, J. Hazard Mater., 2008, 152, 48–55 CrossRef CAS PubMed.
- B. Toyoda, A. W. Morawski, I. Michio and T. Masahiro, Appl. Catal., B, 2006, 65(1–2), 215–221 Search PubMed.
- J. B. Wang, W. P. Zhu, S. X. Yang, W. Wang and Y. R. Zhou, Appl. Catal., B, 2008, 78, 30–37 CrossRef CAS.
- M. Andersson, L. Sterlund, S. Ljungstro and A. Palmqvist, J. Phys. Chem. B, 2002, 106, 10674–10679 CrossRef CAS.
- D. Wodka, E. Bielanska, R. P. Socha, M. E. Wodka, J. Gurgul and P. Nowak, ACS Appl. Mater. Interfaces, 2010, 2, 1945–1953 CrossRef CAS PubMed.
- T. Hirakawa and Y. Nosaka, Langmuir, 2002, 18, 3247–3254 CrossRef CAS.
- B. Ramalingam, T. Parandhaman and S. K. Das, ACS Appl. Mater. Interfaces, 2016, 8, 4963–4976 CrossRef CAS PubMed.
- R. Li, N. D. Mansukhani, L. M. Guiney, Z. X. Ji, Y. C. Zhao, C. H. Chang, C. T. French, J. F. Miller, M. C. Hersam, A. E. Nel and T. Xia, ACS Nano, 2016, 10, 10966–10980 CrossRef CAS.
- L. Liu, H. J. Liu, Y. P. Zhao, Y. Q. Wang, Y. Q. Duan, G. D. Gao, M. Ge and W. Chen, Environ. Sci. Technol., 2008, 42, 2342–2348 CrossRef CAS.
- E. B. Azevedo, F. R. Neto and M. Dezotti, Appl. Catal., B, 2004, 54, 165–173 CrossRef CAS.
- V. Iliev, A. Mihaylova and L. Bilyarska, J. Mol. Catal. A: Chem., 2002, 184, 121–130 CrossRef CAS.
- V. Iliev, J. Photochem. Photobiol., A, 2002, 151, 195–199 CrossRef CAS.
- B. Naik, K. M. Parida, C. S. Gopinath and B. Naik, J. Phys. Chem. C, 2010, 114, 19473–19482 CrossRef CAS.
- I. K. Konstantinou, V. A. Sakkas and T. A. Albanis, Water Res., 2002, 36, 2733–2742 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis and characterization of Pt/TiO2 nanocomposite; the detail information of characterization, photocatalytic test and analysis of intermediates of photocatalytic degradation of phenol; particle size analysis; the Ultraviolet Photoemission Spectroscopy (UPS) analysis; the UV-Vis spectra of phenol degradation catalyzed by P-25, Pt/TiO2 and Ti0.7W0.3O2/TiO2 NCI; compared the characteristic UV-Vis spectra of phenol degradation intermediates; the LC chromatogram, UV-Vis spectrogram and mass spectrum of main intermediates; the UV-Vis absorption spectra, GC chromatogram and mass spectrum of GC-MS for intermediates; the analysis result and possible structure of each intermediate. See DOI: 10.1039/c9na00478e |
| This journal is © The Royal Society of Chemistry 2020 |