
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Routes of iron entry into, and exit from, the catalytic ferroxidase sites of the prokaryotic ferritin SynFtn†
Justin M.
Bradley
 a,
Jacob
Pullin
b,
Geoffrey R.
Moore
a,
Dimitri A.
Svistunenko
b,
Andrew M.
Hemmings
ac and
Nick E.
Le Brun
*a
a,
Jacob
Pullin
b,
Geoffrey R.
Moore
a,
Dimitri A.
Svistunenko
b,
Andrew M.
Hemmings
ac and
Nick E.
Le Brun
*a
aCentre for Molecular and Structural Biochemistry, School of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: n.le-brun@uea.ac.uk
bSchool of Biological Sciences, University of Essex, Wivenhoe Park, Colchester CO4 3SQ, UK
cSchool of Biological Sciences, University of East Anglia, Norwich, NR4 7TJ, UK
First published on 13th January 2020
Abstract
Ferritins are multimers comprised of 4 α-helical bundle monomers that co-assemble to form protein shells surrounding an approximately spherical internal cavity. The assembled multimers acquire Fe2+ from their surroundings by utilising channels that penetrate the protein for the transportation of iron to diiron catalytic centres buried within the monomeric units. Here oxidation of the substrate to Fe3+ is coupled to the reduction of O2 and/or peroxide to yield the precursor to a ferric oxy hydroxide mineral that is stored within the internal cavity. The rhombic dodecahedral quaternary structure results in channels of 4-fold and 3-fold symmetry, located at the vertices, which are common to all 24mer-ferritins. Ferritins isolated from higher eukaryotes have been demonstrated to take up Fe2+via the 3-fold channels. One of the defining features of ferritins isolated from prokaryotes is the presence of a further 24 channels, the B-channels, and these are thought to play an important role in Fe2+ uptake in this sub-family. SynFtn is an unusual ferritin isolated from the marine cyanobacterium Synechococcus CC9311. The reported structure of SynFtn derived from Fe2+ soaked crystals revealed the presence of a fully hydrated Fe2+ associated with three aspartate residues (Asp137 from each of the three symmetry related subunits) within each three-fold channel, suggesting that it might be the route for Fe2+ entry. Here, we present structural and spectro-kinetic data on two variants of SynFtn, D137A and E62A, designed to assess this possibility. Glu62 is equivalent to residues demonstrated to be important in the transfer of iron from the inner exit of the 3-fold channel to the catalytic centre in animal ferritins. As expected replacing Asp137 with a non-coordinating residue eliminated rapid iron oxidation by SynFtn. In contrast the rate of mineral core formation was severely impaired whilst the rate of iron transit into the catalytic centre was largely unaffected upon introducing a non-coordinating residue in place of Glu62 suggesting a role for this residue in release of the oxidised product. The identification of these two residues in SynFtn maps out major routes for Fe2+ entry to, and exit from, the catalytic ferroxidase centres.
Introduction
Iron is an essential micronutrient required for the correct functioning of proteins involved in such processes as electron transfer, catalysis and signalling. In many cases the biological function of iron is underpinned by the facile inter-conversion of the Fe2+ and Fe3+ oxidation states. However, in the presence of peroxide and superoxide, formed as by-products of aerobic respiration, iron redox chemistry can be detrimental as it catalyses the production of damaging hydroxyl radicals. Furthermore, Fe3+, the most stable oxidation state of iron at ambient dissolved oxygen concentrations, is poorly soluble at pH ≥ 3.1 Thus, living systems must sequester sufficient iron to meet requirements, and tightly regulate intracellular concentrations of the free metal ion. Members of the ferritin superfamily of proteins are critical to achieving this in many organisms ranging from prokaryotes to higher mammals.2–4Ferritins are typically composed of 24 four α-helical bundle subunits approximately 45 Å along their longest axis, which are of two types: H-chain and L-chain. Buried within H-chain type subunits is a catalytic diiron centre, called the ferroxidase centre, which is responsible for driving Fe2+ oxidation/mineralization. While animal ferritins are almost always heteropolymers of H- and L-chain subunits, plant and prokaryotic ferritins are composed of only H-chain type subunits. The 24 subunits co-assemble into hollow rhombic dodecahedra with external and internal diameters of 120 Å and 80 Å respectively. The protein cages are penetrated by channels 12–15 Å in length located at the 3-fold and 4-fold vertices of these dodecahedra (Fig. 1A and B). Following Fe2+ oxidation, up to several thousand equivalents of Fe3+ can be stored as a hydrated ferric-oxo mineral within the ferritin molecule's internal cavity.2–4 The diiron ferroxidase centres are located at the centre of H-chain type α-helical bundles. They are not accessible to Fe2+ directly through the subunit, and they are at least 20 Å away from any of the protein channels. Therefore, ferritin activity requires a route of Fe2+ transit from the exterior of the protein to the site of its oxidation.
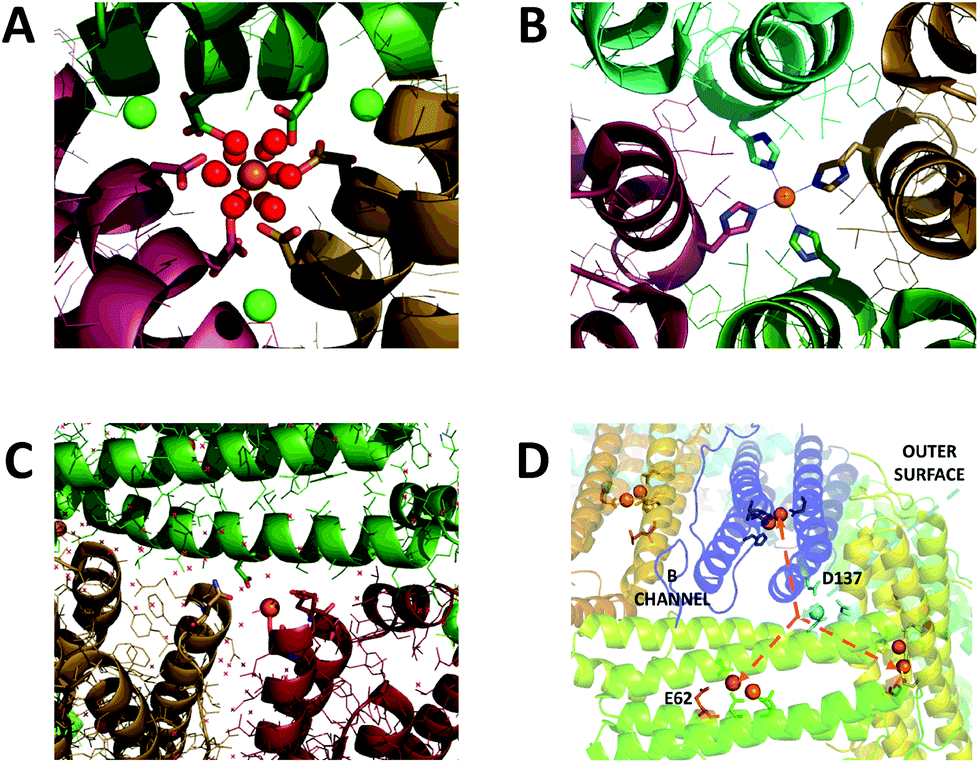 | ||
| Fig. 1 Iron binding sites in the channels of ferritin. (A) The 3-fold channel of Human H ferritin with two [Fe(H2O)6]2+ ions bound by conserved channel carboxylates. (B) The 4-fold channel of Human H ferritin showing a single non-hydrated iron ion coordinated by His173. (C) The B-channel of Pseudo-nitzschia multiseries Ftn showing a single iron ion bound to Asp30 and Glu35 of one of the monomeric units. (D) Cartoon representation of the structure of SynFtn showing the relative disposition of residues Asp137 (sidechains highlighted in pale blue), Glu62 (sidechains highlighted in orange) and the diiron catalytic centres (orange spheres). Iron is proposed to enter the protein via the 3-fold channel (blue dashed line) and from the inner exit is distributed to each of three symmetry equivalent catalytic sites (orange dashed lines). Images produced using PDB files 4YKH (HuHF), 4ZKH (PmFtn) and 6GKA (SynFtn). | ||
The route of iron transit has been elucidated for recombinantly expressed homopolymers of human H and frog M ferritin, largely due to the work of the groups of Theil, Turano and Mangani.5–10 The peptide sequences of the ferritins of higher eukaryotes are highly conserved with respect to residues located close to the 3-fold channel (Fig. S1†), and both crystallographic data and disruption of this channel by site directed mutagenesis demonstrate that this is the route of iron entry into these proteins.5,10,11 Specifically, two conserved carboxylate residues (Asp127 and Glu130, Frog M numbering) generate two symmetry-related binding sites for hexaquo-Fe2+ within each channel.
A further two conserved Glu residues (Glu57 and Glu136, Frog M numbering) were identified as important for transferring the Fe2+ substrate from the interior exits of the three-fold channels to the ferroxidase centres.6,8,9 Whilst crystallographic studies have not yet identified similar transient binding sites for oxidized iron en route from the ferroxidase centres to the interior cavity, electron density arising from iron has been observed close to the interior exit of the 4-fold channels.12 An NMR study of Frog M ferritin exploiting the paramagnetic nature of the oxo bridged Fe3+ clusters produced by ferroxidase centre activity has identified a route13 from the ferroxidase centre along the interior of the α-helical bundle to the interior four-fold channel exit.
The residues identified as important for Fe2+ uptake in animal ferritins are conserved in their counterparts isolated from plants (Fig. S1†), suggesting a similar route for substrate transport, but this is not the case for the prokaryotic ferritins (Ftns). Whilst there is a great deal of similarity in the peptide sequences at the 3-fold axes within the Ftns, these are distinct from those of the animal-and phyto-ferritins (Fig. S1†), such that the 3-fold pores would be expected to create a hydrophobic environment unsuitable for the transport of Fe2+ or other ionic species. Intriguingly however the ‘transfer carboxylates’ Glu57 and Glu136 of Frog M ferritin are conserved in the Ftn proteins suggesting that they may undertake varying or multiple roles.
Residues lining the 4-fold channels of ferritins are, in general, less well conserved than those at the 3-fold channels, and this remains the case within the Ftn subfamily. However, despite the variability in peptide sequence, the 4-fold channels of Ftns are again mostly comprised of residues with hydrophobic sidechains (Fig. S1†), and, as such, are unlikely to transport Fe2+ from bulk solution to the interior of the protein.
A third ferritin channel, named the B-channel, is found almost exclusively in proteins from prokaryotes.14–16 The B-channels are located at the intersection of three monomeric units (Fig. 1C) and are lined with two carboxylate side chains providing a favourable electrostatic environment for Fe2+ transport (Fig. S1†). Metal ions have been observed bound to these B-channels17 and their disruption by mutagenesis has been shown to impair Fe2+ oxidation and mineralisation16 suggesting that in at least a subset of prokaryotic ferritins these B-channels constitute the major route of iron uptake. However it is by no means clear that this is the case for the entire subfamily of Ftns.
A bacterial ferritin, called SynFtn, isolated from the coastal marine cyanobacterium Synechococcus strain CC9311 was recently shown to be unusual in that it more closely resembles eukaryotic H-chain ferritins than previously studied Ftn proteins.18 This study demonstrated that SynFtn oxidises iron via a mechanism that is unique among diiron proteins characterised to date. The protein as isolated was free from metal ions. Addition of Fe2+ led to rapid binding at the ferroxidase centre and, in the presence of O2, oxidation to Fe3+ in a multiphasic reaction. Quantification of the absorbance change associated with the most rapid of these phases revealed that this corresponded to the oxidation of only one equivalent of Fe2+ per protein monomer despite the product of O2 reduction being peroxide (a 2 electron reduction). Time resolved EPR showed this rapid burst of iron oxidation to be due to formation of a Fe2+/Fe3+ mixed valent ferroxidase centre (MVFC) from the di-Fe2+ site. A transient radical signal from a conserved tyrosine residue (Y40) also formed during the initial rapid iron oxidation event, but not in sufficient yield to provide the second reducing equivalent to oxygen. Rather, this residue served to initiate an electron transport chain enabling each of two di-Fe2+ sites to deliver a single electron to O2 bound at only one, generating two MVFC per O2 reduced. The MVFC was metastable in the presence of O2, decaying to an EPR silent species with a half-life of approximately 10 s. Quantification of the total absorbance change demonstrated that this was due to further oxidation of the MVFC to a di-Fe3+ state, akin to that reported for other ferritins, which is unstable leading to release of iron from the di-Fe3+ centres into the internal cavity, regenerating apo sites capable of reacting again with Fe2+ and O2 to support mineralisation of up to 2000 Fe per Ftn within the core of SynFtn. Thus in the overall reaction, 4 Fe2+ ions bind at 2 ferroxidase centres and react sequentially with 2 equivalents of O2 generating initially 2 MVFCs and one equivalent of H2O2, then 2 di-Fe3+ centres and a further equivalent of H2O2. Regeneration of the rapid activity associated with apo centres suggested that, in the absence of further incoming Fe2+, release of the mineral precursors into the interior cavity begins almost immediately but requires 60 min to reach completion.
The high resolution structure of SynFtn derived from crystals soaked in a Fe2+ containing solution for 20 min (PDB entry 6GKA) revealed a Fe2+ ion within the 3-fold channel.18 The original report focussed on the mechanism of iron oxidation and therefore the significance of this observation was not discussed. However, the presence of iron in the 3-fold channel of this prokaryotic ferritn provided our motivation to probe the route of Fe2+ entry into SynFtn.
The relative disposition of the interior exits of the 3-fold channels of ferritins and their ferroxidase centres (Fig. 1D) means that rapid, direct transfer of metal ions between the two is highly unlikely. In animal ferritins two transient iron binding sites on the inner surface of the protein, termed Fe3 and Fe4, are thought to rapidly guide incoming Fe2+ from the 3-fold channel to the site of its oxidation.8,9 SynFtn lacks coordinating residues at the equivalent position to site Fe4. However a glutamic acid residue (E62) is structurally equivalent to the Fe3 ligands H54 (Frog M) and Q58 (human H).
Here, we report structural and spectro-kinetic data acquired for two SynFtn variants and compare these to data previously reported for the wild type protein. SynFtn D137A was designed to investigate whether the 3-fold channel constitutes the major route of Fe2+ entry. SynFtn E62A was designed to investigate whether a common pathway linking the 3-fold channel to the ferroxidase centres via the inner surface of the protein is shared by SynFtn and the animal ferritins. The data, which only partially support a common transit route, reveal fundamental aspects of Fe2+ entry and Fe3+ exit routes to and from the ferroxidase centre of this prokaryotic ferritin.
Results and discussion
The protein coat channels of SynFtn
Comparison of the sequence of SynFtn to other selected Ftns revealed that the B channel contains an aliphatic residue in place of one of the carboxylate residues conserved amongst the other proteins (Fig. S1†) meaning the environment in the SynFtn B channel is more hydrophobic than that of other Ftns. Furthermore, the peptide sequence of SynFtn contains an N-terminal extension relative to other prokaryotic ferritins and inspection of the previously reported structure of the wild type protein18 revealed that this forms a cap over the exterior entrance to the B channel (Fig. S2†) meaning this is unlikely to be the route of Fe2+ entry to SynFtn.The 3-fold channel of SynFtn has little sequence similarity to those of the animal proteins for which Fe2+ entry via this route has been demonstrated (Fig. S1†), and the peptide sequence also lacks the conserved carboxylates that transfer the Fe2+ from the internal channel exits to the site of its oxidation. Nonetheless, the 3-fold channel of SynFtn is lined by carboxylates from three symmetry equivalent Asp137 residues, and the crystallographically observed Fe2+, present as [Fe(H2O)6]2+, was located only 4.0 Å away from oxygen atoms of these side chains (Fig. 2A). This suggests that the 3-fold channel may constitute the major route of Fe2+ uptake.
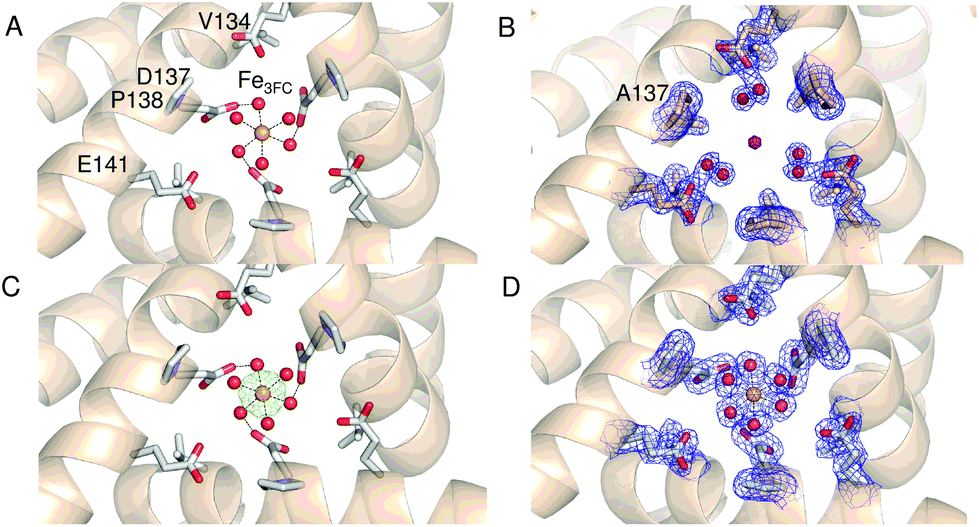 | ||
| Fig. 2 The 3-fold channel of SynFtn. The channels of wild type (PDB entry 6GKA) (A), variant D137A (B) and variant E62A (C, D) SynFtn viewed along the symmetry axis located at the center of the channel. In each case crystals from which the diffraction data were collected were soaked in a 5 mM Fe2+-containing solution for 20 min prior to freezing. Carbon is shown in white, nitrogen in blue, oxygen in red and iron as bright orange spheres with associated water molecules as red spheres. In panels (B) and (D) the blue mesh shows the double difference Fourier (2mFo-DFc) map contoured at 1.1 σ, while in panel (C) the light green mesh shows the anomalous difference Fourier map calculated from data collected at the iron K-edge and contoured at 10 σ. | ||
Crystal structures of D137A SynFtn reveal iron at the ferroxidase sites but not in the three-fold channels
The 2.13 Å crystal structure of the D137A variant of SynFtn (PDB entry 6SOM) revealed no significant structural perturbation other than the sidechain of the substituted residue (Fig. S3, Tables S1 and S3†). However, structures obtained from crystals aerobically soaked in a 5 mM Fe2+ ion solution for 20 min (PDB entry 6SOO) contained no electron density associated with iron within the 3-fold channel (Fig. 2B). The iron-soaked structures did contain additional areas of electron density at the ferroxidase centre sites and data sets collected at the iron absorption edge were consistent with these arising from the binding of iron (Fig. S4†). However, significant differences in metal binding were apparent between the iron-soaked structures of D137A SynFtn and those reported for the wild type protein.18 Crystals soaked in Fe2+ for 2 min (PDB entry 6SON) contained iron bound at both the FeA and FeB sites of the ferroxidase centre (Fig. 3), but with low occupancy (Table S3†). The distance between the two metals was 3.54 Å, shorter than the 3.90 Å observed for the equivalent 2 min soak of wild type protein crystals. The ferroxidase centre reaction requires the simultaneous occupancy of both iron binding sites together with the availability of O2. Hydrolysis of the resulting oxidised ferroxidase centre species leads to incorporation of a bridging (hydr)oxo ion between the metal centres.18 Therefore the lack of any bridging density between the metals, together with the low occupancy indicates that this structure represents a superposition of unreacted Fe2+ ions in singly occupied catalytic centres.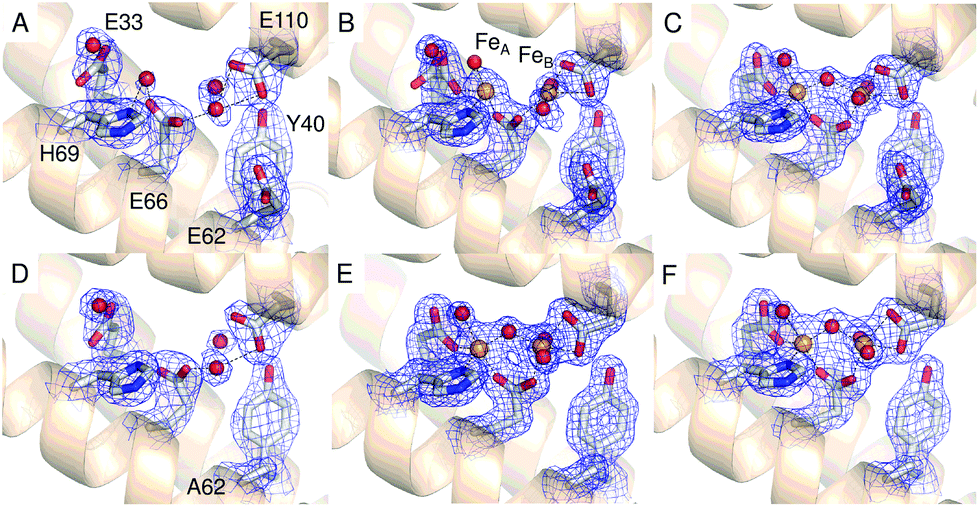 | ||
| Fig. 3 The ferroxidase center of SynFtn variants. The di-iron site of apo D137A SynFtn (A) and following the soaking of crystals in a 5 mM Fe2+ solution for either 2 min (B) or 20 min (C). (D–F) as in (A–C), but for crystals of E62A SynFtn. Carbon is shown in white, nitrogen in blue, oxygen in red and iron as bright orange spheres with associated water molecules or (hydr)oxide ions as red spheres. The blue mesh in each of panels A to F shows the double difference Fourier (2mFo-DFc) map contoured at 1.1 σ. | ||
Increasing the soaking time to 20 min led to an increase in the occupancy of both the FeA and FeB sites (Table S3†), and the appearance of bridging density between the metals that was modelled as water/(hydr)oxo. These observations, together with an inter-metal distance of 3.62 Å indicates that this structure represents the di-ferric form of the active site.12 This interpretation of the structural data was supported by the solution phase kinetics of iron oxidation observed for D137A.
Rapid Fe2+ oxidation, but not mineralization, is abolished in D137A SynFtn
In common with most ferritins, SynFtn exhibits kinetically distinct phases of iron oxidation following addition of Fe2+ to the metal free protein. The apo protein contains 48 vacant metal binding sites, two at each of 24 ferroxidase centres. For catalytically active proteins, aerobic addition of up to 48 equivalents of Fe2+ results in rapid transport of the metal to the ferroxidase centre where it binds and is oxidised to the Fe3+ state in 10–20 s. The mineralisation activity of ferritins is only observed spectroscopically when Fe2+ is added in excess of 48 equivalents. Here 48 equivalents are again rapidly oxidised at the ferroxidase sites but further reaction requires that the Fe3+ product be cleared from the catalytic centres, forming the mineral core, to allow further Fe2+ binding. This process typically occurs on the timescale of minutes. Thus, whilst Fe3+ contained within ferritins absorbs light at 340 nm regardless of whether it is bound at ferroxidase centres or located in the core, the process monitored by spectrophotometric assays is readily interpreted by considering the rate of oxidation in conjunction with the excess of Fe2+ over protein added.Stopped flow absorbance measurements conducted under identical conditions to those reported for wild type SynFtn18 revealed that the rapid phase of iron oxidation was abolished by the D137A substitution (Fig. 4A and B). In contrast, assays of iron mineralisation activity (conducted with 400 Fe2+ per Ftn) showed that the variant protein was capable of laying down a mineral core at 75% of the initial rate reported for the wild type protein. Pre-treatment of the protein with Zn2+, a potent inhibitor of ferroxidase centres, resulted in a reduction in the rate of mineral formation (Fig. S5†) demonstrating that the catalytic centre is required for the activity observed for D137A. Thus, the data indicate that the ferroxidase centre remains competent in variant D137A and that the loss of rapid iron oxidation, for which Fe2+-binding is rate limiting even in wild type protein, is the result of disruption of the route of iron transit.
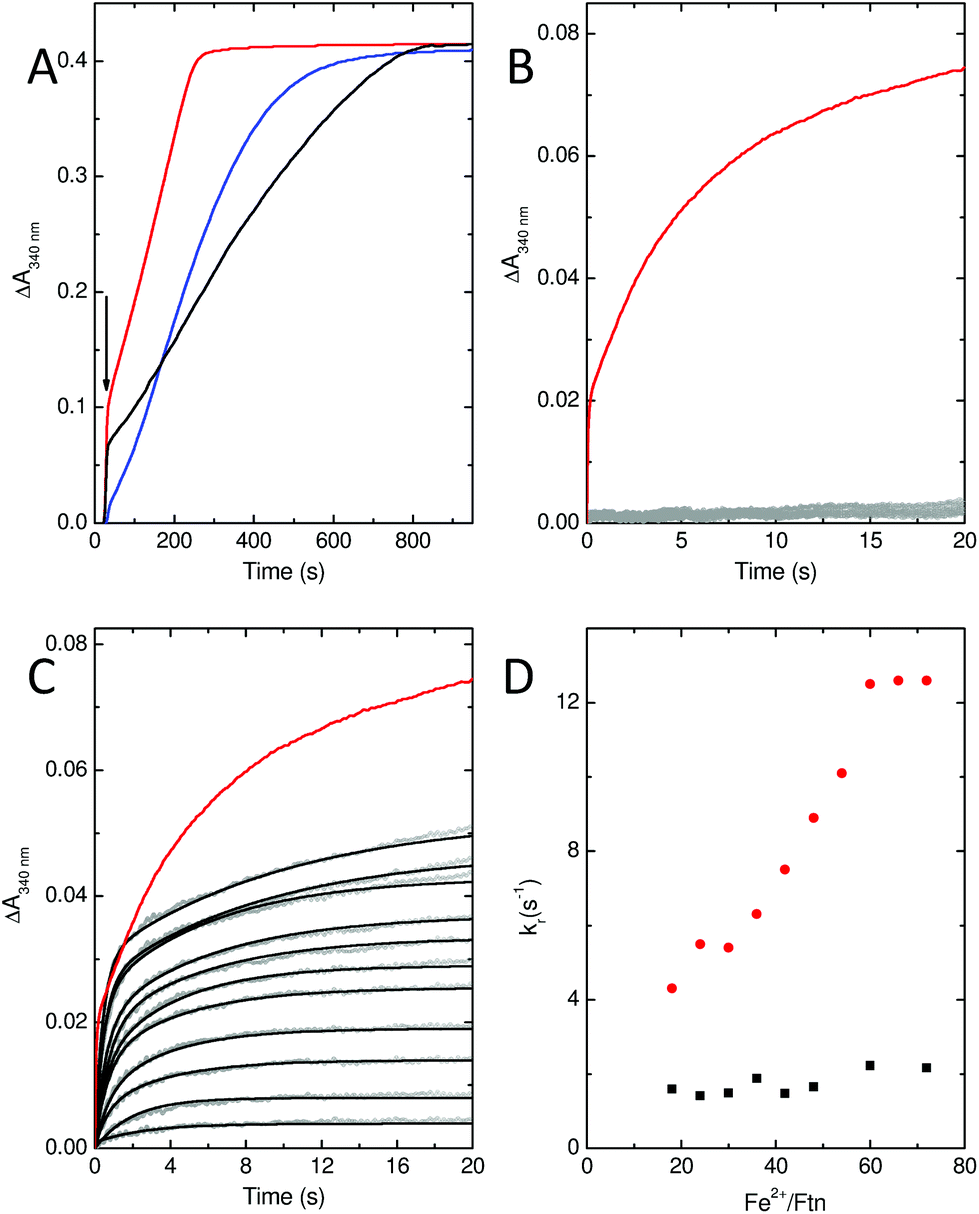 | ||
| Fig. 4 Iron oxidation kinetics of SynFtn variants D137A and E62A. (A) Mineral core formation following the addition of 200 μM Fe2+ to 0.5 μM SynFtn variant E62A (black trace) or D137A (blue trace). Red trace shows the response of the wild type protein for comparison. (B) Iron oxidation following rapid mixing of 1 μM D137A SynFtn with an equal volume of 6–96 μM Fe2+. (C) As (B) but for SynFtn variant E62A. Black traces in panel C represent bi-exponential fits to the data. In panels B and C, data points are shown as gray circles, red traces represent the response of wild type protein upon mixing with 96 μM Fe2+ for comparison. (D) Apparent rate constant of the rapid phase extracted from exponential fits shown in panel (C) (black squares) together with the equivalent data reported for the wild type protein (red circles) for comparison. All measurements were conducted at pH 6.5 and 25 °C. | ||
MVFC and Tyr radical formation are observed in D137A SynFtn
As a consequence of the reduced rate of iron flux through variant D137A SynFtn, occupancy of the FeA and FeB sites in D137A crystals was lower than that observed for the wild type protein crystals treated equivalently, and so the structure of the MVFC form of the catalytic centre was not observed. Mechanistically unperturbed functioning of the ferroxidase centre in this variant, with a reaction cycle that proceeds through formation of a MVFC, was, however, confirmed using the time dependence of the EPR spectrum (Fig. 5). Apo SynFtn is essentially EPR silent whilst samples frozen at varying time points after the aerobic mixing of D137A apo protein with 72 equivalents of Fe2+ contained the same three paramagnetic species identified in the wild type protein: mono-nuclear high spin Fe3+, a tyrosyl radical and the MVFC18 (Fig. S6†). Importantly, though, the concentration of these species reached maximum intensity approximately 20 s after mixing at ambient temperature (Fig. 5 and Fig. S6†), compared to 1 s at 4 °C for the wild type protein.18 The MVFC signal then decayed over the following 40 s, the concomitant increase in 340 nm absorbance in equivalent samples showing this to be due to formation of the EPR silent di-Fe3+ state. Variant D137A forms exactly the same paramagnetic species as wild type SynFtn suggesting the same unusual mechanism of iron oxidation is utilised by this protein. However the kinetics of the ferroxidase reaction are reduced by at least an order of magnitude. The data are, therefore, fully consistent with variant D137A containing a competent catalytic centre within a protein cage that has an impaired ability to acquire Fe2+ from solution.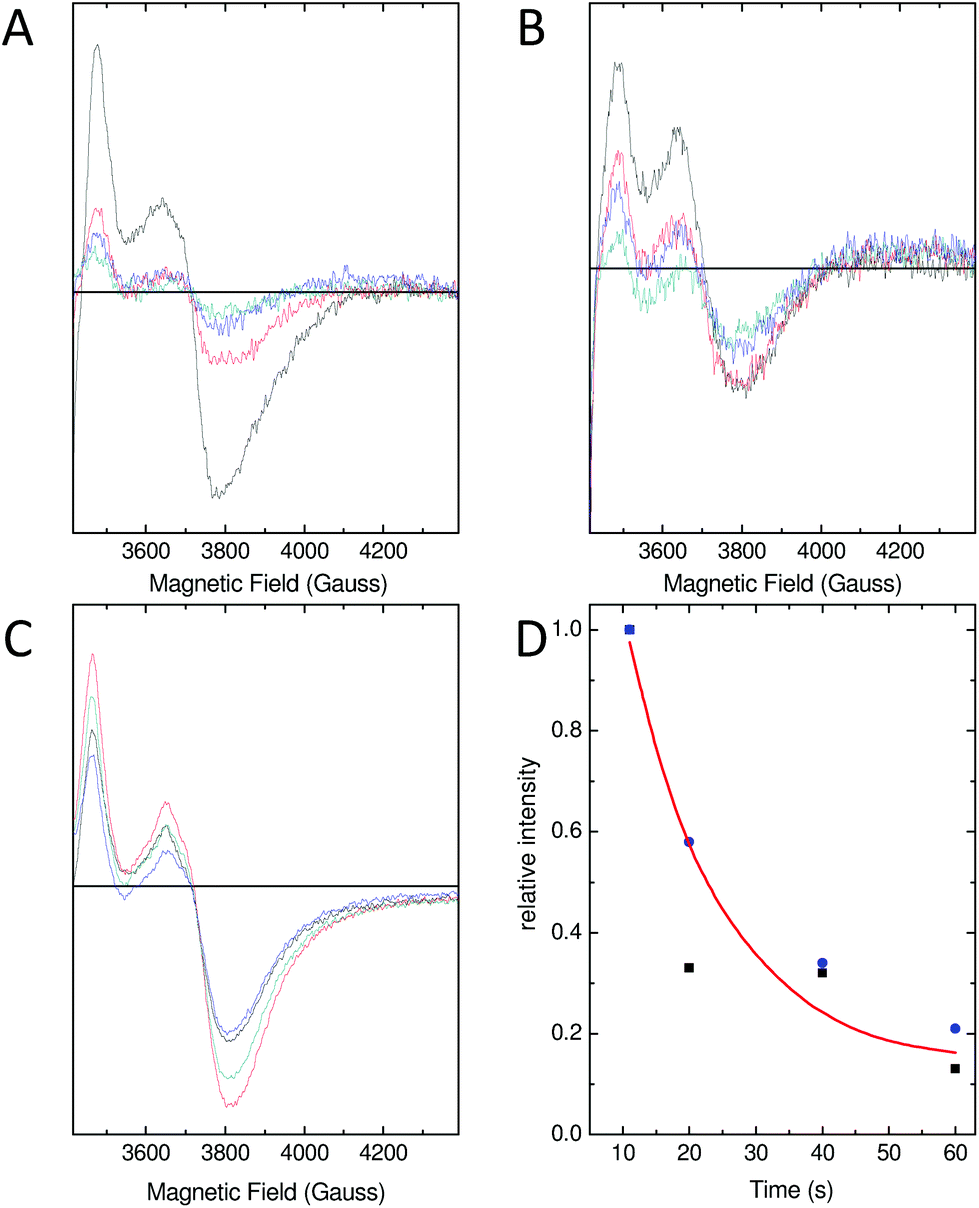 | ||
| Fig. 5 Characterization of the MVFC in E62A and D137A SynFtn and comparison with wild wild type. The EPR signal due to the Fe2+/Fe3+ MVFC of (A) wild type and (B) variant E62A and (C) variant D137A. For wild type and E62A SynFtn, samples were frozen 10 s (black trace), 20 s (red trace), 40 s (blue trace) and 60 s (cyan trace) after the addition of 200 μM Fe2+ to a 4.17 μM (100 μM monomer) protein solution in 100 mM MES pH 6.5. For D137A, samples were frozen 10 s (black trace), 20 s (red trace), 30 s (blue trace) or 40 s (gray trace) following aerobic mixing with Fe2+ at a final concentration of 300 μM with protein of 100 μM (monomer concentration) in 100 mM MES pH 6.5. Panel D shows the relative intensity associated with the MVFC observed as a function of freezing time for wild type (black squares) and variant E62A (blue circles) SynFtn. | ||
Residue Glu62 does not form part of a transit site for Fe2+ entry
A transient site (referred to as Fe3 by Pozzi et al.8,9) close to the ferroxidase centre is thought to form part of a conserved route of Fe2+ transport from three-fold channels to ferroxidase centres in ferritins of the higher eukaryotes. Fe3 is ligated by four residues in frog M ferritin but only three in human H-chain ferritin.8,9SynFtn contains potential Fe2+ ligands at equivalent positions to the three residues that form part of the Fe3 site in both of the eukaryotic proteins. Of these, Glu110 acts as a ferroxidase centre ligand whilst Asp65 of SynFtn is structurally equivalent to glutamic acid residues that act as a ligand to both sites Fe3 and Fe4 in the animal ferritins. Therefore, the remaining residue, Glu62, was selected for substitution in an attempt to disrupt any potential site in SynFtn analogous to Fe3.The 1.93 Å structure of E62A SynFtn (PDB entry 6SOP) revealed no significant changes beyond the substituted sidechain (Fig. S7 and Tables S2 and S3†); in particular, the disposition of the amino acid sidechains making up the ferroxidase centre were unaffected (Fig. 3D and Fig. S7†). Crystals soaked for 2 min in an aerobic Fe2+ solution (PDB entry 6SOQ) revealed two areas of electron density at the ferroxidase centre. Datasets collected at the iron K absorption edge (1.7389 Å) showed strong anomalous scattering associated with these two areas of electron density, which were modelled as iron accordingly (Table S3†). However, in contrast to variant D137A, the metal ions were at a separation of 3.34 Å, the shortest Fe–Fe distance observed in crystals of SynFtn to date, with a clear area of bridging electron density between them (Fig. 3E). Therefore, the structural data suggest that following a 2 min Fe2+ soak the majority of iron containing ferroxidase centres in crystals of E62A SynFtn have bound and reacted with O2. However these centres are not in the MVFC state. Increasing the soaking time to 20 min (PDB entry 6SOR) resulted in no change to the metal–metal distance (3.34 Å) suggesting that in both metal soaked structures the form of the ferroxidase centre is most likely oxo-bridged di-Fe3+ that is only observed in 20 min Fe2+ soaks for wild type and D137A SynFtn.
As with variant D137A, formation of a MVFC was confirmed using EPR spectroscopy (Fig. 5B and Fig. S6†), suggesting that the failure to observe this state in crystal soaking experiments was the result of altered iron transport and oxidation kinetics. The expected consequence of disruption of an Fe3-like site by the E62A substitution was impaired transit of Fe2+ to the catalytic centre, similar to that observed in variant D137A. However, the observation at both long and short soaking times of oxo-bridged iron clusters at the ferroxidase centre with structure very similar to that of the 20 min Fe2+ soak of wild type protein was not consistent with this. Therefore the kinetics of Fe2+ oxidation by E62A SynFtn in solution were investigated to further probe the effect of this substitution.
Stopped-flow absorbance measurements, fitted using eqn (1) (see Experimental section), revealed the presence of a rapid phase of Fe2+ oxidation (Fig. 4). The apparent rate constant for this, kr, was almost independent of iron, being approximately 2 s−1 in all cases. Therefore, in variant E62A, the binding of iron at ferroxidase centre sites is not rate limiting for Fe2+ oxidation. Consequently the rate of Fe2+ oxidation cannot be used to infer the rate of its transport to the ferroxidase centre other than to say that the apparent rate constant of this process must be greater than 2 s−1 in all cases. As such these data demonstrate that Glu62 is not required for rapid binding of Fe2+ at the ferroxidase centre in SynFtn, suggesting that this protein does not contain an Fe3-like transit site. Nevertheless absorbance monitored activity assays conducted at the higher iron loading of 400 Fe2+ per Ftn indicated that Glu62 is important for SynFtn activity, since variant E62A was able to lay down a mineral core at a rate only ∼38% of the wild type protein and ∼50% that of D137A (Fig. 4A). Impaired core formation in variant E62A cannot, therefore, be attributed to disruption of the transport of substrate into the catalytic centre, given that variant D137A, in which all rapid oxidation activity is abolished, is able to support a greater rate of mineralization. Therefore, it appears that the principal role of residue Glu62 is not in delivery of substrate to the catalytic centre, but, rather, occurs after the initial Fe2+ oxidation event.
Residue Glu62 facilitates transfer of Fe3+ from the ferroxidase centre to the cavity
Glu62 could play an important role after the initial oxidation of Fe2+ at the ferroxidase centre by affecting the oxidation of the MVFC, such that the E62A mutation leads to an increase in the activation energy for oxidation of the MVFC and a consequent reduction of the rate of formation of di-Fe3+ centres. Alternatively, Glu62 might be important for the transport of Fe3+ to the internal cavity, such that its substitution prolongs the lifetime of the di-Fe3+ form. A combination of time-resolved EPR and stopped-flow absorbance measurements was employed in order to distinguish these possibilities.As with the wild type protein,18 the observed increase in absorbance at 340 nm in the first several seconds following the aerobic mixing of apo protein and Fe2+ could not be fitted to a single phase when the iron loading was ≥18 Fe2+ per Ftn, but was well described by a bi-exponential function (Fig. 4). This biphasic response is due to formation of the MVFC in the first several hundred milliseconds followed by further oxidation to the di-Fe3+ state at a much lower rate over the following several seconds.18 The rate of the slower of these two processes, ks, is almost invariant with Fe2+ concentration proceeding with an apparent rate constant of approximately 0.1 s−1 in all cases, as observed for the wild type protein. This phase is due to the reaction between MVFCs and a second equivalent of O2 that generates the di-Fe3+ centre. Furthermore decay of the MVFC EPR signal intensity, caused by its oxidation to a diamagnetic di-Fe3+ cluster, also occurs at a similar rate in both proteins (Fig. 5). The data therefore suggest that the rate of the second oxidative step, that of conversion of the MVFC to a di-Fe3+ centre, is unaffected by mutation of Glu62 consistent with the failure to identify this form of the catalytic centre crystallographically. However, we note that the amplitude of the slower phase following addition of 96 Fe2+ per protein is significantly lower in E62A SynFtn compared to the wild type protein. In fact, the amplitude observed here corresponds to that expected for the oxidation of 48 Fe2+ ions per protein for wild type SynFtn, suggesting that Fe2+ oxidation subsequent to the oxidation of the initial 48 Fe2+ at ferroxidase centres, i.e. the mineralisation process, is severely affected.
To explore this possibility further, the rate at which iron was released from fully oxidised centres was assessed using stopped-flow absorbance. Protein was incubated with 400 Fe2+ per Ftn and the mineralization reaction allowed to proceed to completion. The rate of iron oxidation was then monitored following mixing with a further 72 Fe2+ per Ftn. In the case of the wild type protein, the maximum recovery of the amplitude of the rapid phase of oxidation associated with vacant ferroxidase centres (corresponding to 80% of the value observed for apo protein) was observed at 60 min after the addition of 400 equivalents of Fe2+.18 The rate of recovery of rapid oxidation was much slower for variant E62A (Fig. 6) with only 60% of the amplitude observed for apo protein recovered following incubation with 400 Fe per Ftn for 16 h. Therefore, the lower rate of mineralization in this protein is most likely due to impaired release of the oxidized ferric-oxo product to regenerate empty catalytic centres capable of binding further equivalents of Fe2+.
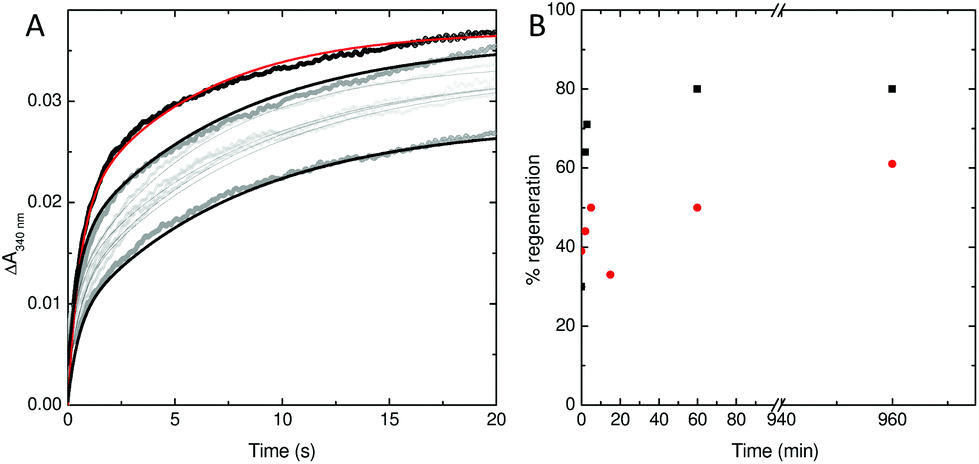 | ||
| Fig. 6 Stability of the di-Fe3+ ferroxidase center in wild type and E62A SynFtn. (A) regeneration of rapid Fe2+ oxidation activity in E62A SynFtn following incubation of 1 μM protein with 400 μM Fe2+. Gray circles represent the absorbance at 340 nm as a function of time upon mixing with an equal volume of 72 μM Fe2+ at increasing times up to 16 h following complete oxidation of the initial Fe2+ addition and black lines the fits of the experimental data to bi-exponential functions. The equivalent response of apo protein is shown as black circles and a red trace for comparison. (B) The percentage of rapid activity recovered as a function of time following incubation with 400 equivalents of Fe2+ for wild type (black squares) and variant E62A (red circles) SynFtn. All measurements were performed at 25 °C in 100 mM MES pH 6.5. | ||
Experimental
Protein overexpression and purification
Plasmids for the expression of SynFtn variants E62A and D137A based on pET21a (Novagen) were purchased from Genscript (New Jersey, USA). The encoded proteins were overexpressed in E. coli strain BL21(DE3) (Promega). Cultures were grown in LB containing 100 μg mL−1 ampicillin at 37 °C, 200 rpm shaking until they reached an optical density at 600 nm of 0.6–0.8. Overexpression was induced by addition of isopropyl β-D-1 thiogalactopyranoside (IPTG) to a final concentration of 50 μM and cultures grown for a further 3 h at 37 °C, 200 rpm shaking prior to harvesting by centrifugation. Cells were re-suspended in 20 mM HEPES pH 7.8 containing 100 mM KCl, 0.1 mM EDTA (buffer A), disrupted by sonication and debris removed by centrifugation at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 45 min. Thermally unstable proteins were precipitated from the supernatant by heating to 65 °C for 15 min and removed by a further round of centrifugation (40000g for 45 min). Protein was precipitated from the supernatant via the addition of ammonium sulfate to a concentration of 0.55 g mL−1. The precipitate was solubilized in the minimum volume of buffer A and dialyzed against 1 L of identical buffer for 12 h. Contaminating proteins were removed by size exclusion chromatography (HiPrep 26/60 Sephacryl S-300HR, GE Healthcare) and contaminating DNA by anion exchange chromatography (HiTrap Q FF, GE Healthcare). For the latter, protein solutions were loaded in buffer A and eluted by stepping to 50% buffer B (20 mM HEPES pH 7.8 containing 100 mM KCl, 1.0 M NaCl, 0.1 mM EDTA). Protein as isolated contained small quantities of iron that was removed according to the method of Bauminger et al.19 Following iron removal, protein was exchanged into 100 mM MES pH 6.5 by centrifugation over a 10 kDa molecular weight cut off cellulose membrane (Millipore). Sample purity was assessed using SDS-PAGE and proteins judged to be free of DNA contamination once the ratio of absorbance at 277 nm and 260 nm reached 1.5.
000g for 45 min. Thermally unstable proteins were precipitated from the supernatant by heating to 65 °C for 15 min and removed by a further round of centrifugation (40000g for 45 min). Protein was precipitated from the supernatant via the addition of ammonium sulfate to a concentration of 0.55 g mL−1. The precipitate was solubilized in the minimum volume of buffer A and dialyzed against 1 L of identical buffer for 12 h. Contaminating proteins were removed by size exclusion chromatography (HiPrep 26/60 Sephacryl S-300HR, GE Healthcare) and contaminating DNA by anion exchange chromatography (HiTrap Q FF, GE Healthcare). For the latter, protein solutions were loaded in buffer A and eluted by stepping to 50% buffer B (20 mM HEPES pH 7.8 containing 100 mM KCl, 1.0 M NaCl, 0.1 mM EDTA). Protein as isolated contained small quantities of iron that was removed according to the method of Bauminger et al.19 Following iron removal, protein was exchanged into 100 mM MES pH 6.5 by centrifugation over a 10 kDa molecular weight cut off cellulose membrane (Millipore). Sample purity was assessed using SDS-PAGE and proteins judged to be free of DNA contamination once the ratio of absorbance at 277 nm and 260 nm reached 1.5.
Crystallization and structure determination
Protein (10 mg mL−1) exchanged into 20 mM MES pH 6.5 in 2 μL drops was mixed with an equal volume of well solution (0.1 M sodium acetate, 2.0 M sodium chloride pH 4.6) and equilibrated in sitting drops by vapour diffusion against 200 μL of the same well solution. Crystals of bi-pyramidal morphology appeared within 24 h and grew to optimum size (100–150 μm) in approximately 1 week. Apo crystals were transferred to cryo-protectant comprising the well solution with pH adjusted to 6.5 containing 30% (v/v) glycerol prior to flash freezing in liquid nitrogen. Iron containing crystals were prepared by soaking for either 2 or 20 min in well solution containing Fe2+ ions at 5 mM concentration and pH adjusted to 6.5. Crystals were then cryo-protected and frozen as above but using a solution containing 5 mM Fe2+ in addition to 30% (v/v) glycerol. Diffraction data was collected at the Diamond Light Source using beamline i03 (variant E62A) and i04 (variant D137A). The wavelength used was 0.9795 Å in all cases. Additional, highly redundant anomalous scattering data were collected either from the same or identically treated iron-containing crystals of E62A and D137A SynFtn at wavelengths corresponding to the peak of the iron K-edge (around 1.7389 Å). All data was indexed and processed using XDS and Aimless as part of the automatic xia2 pipeline.20 Reprocessing was carried out as necessary using Aimless as part of the CCP4 programme suite.21 Statistics for X-ray data collection, structure solution and refinement are summarized in Tables S1 and S2.†Structure solution was performed by molecular replacement using phenix.phaser MR22 with the 2.05 Å resolution structure of E. coli FtnA, PDB entry 1EUM, as the search model. In all cases the asymmetric unit contained a single copy of the monomer subunit. Placement of metal ions was confirmed by reference to Bijvoet-difference Fourier maps calculated from anomalous scattering data. Model refinement employed iterative cycles using phenix.refine and manual correction using COOT.23 No metal coordination restraints were applied to metal sites during refinement of iron-containing structures. Anisotropic temperature factor refinement was employed for all metal ions and their occupancies were manually adjusted to ensure that the average B factor of the metal fell within ±14% of the B factors of atoms of their environment. The coordination geometry of metal binding sites was analysed after refinement using the CheckMyMetal web server.24 Statistics relating to the metal binding sites in the refined structures can be found in Table S3.†
Kinetic analysis of iron oxidation and mineralization by SynFtn
Rates of Fe2+ oxidation were deduced from the rate of increase in absorbance at 340 nm due to the resulting ferric-oxo species, be they iron bound at the ferroxidase centre or in the mineral core. Assays employed 0.5 μM SynFtn in 100 mM MES pH 6.5 at 25 °C. Aerobic oxidation of ferroxidase centre bound Fe2+ following addition to apo wild type SynFtn was complete in ∼20 s. Accordingly the ability of variants E62A and D137A to support similar chemistry was monitored using stopped-flow absorbance spectroscopy. 1.0 μM protein in 100 mM MES pH 6.5 was mixed with an equal volume of Fe2+ of the appropriate concentration in 1 mM HCl using an Applied Photophysics Bio-Sequential DX.17MV spectrophotometer with a 1 cm path length observation cell. In the case of variant E62A the time dependences of absorbance increase at 340 nm were fitted to the sum of two exponential processes, encompassing rapid (r) and slow (s) components, using OriginPro 8 (OriginLab):| ΔA340(t) = ΔA(tot)340 − ΔAr340e−krt − ΔAs340e−kst. | (1) |
The extent to which oxidized Fe3+ vacates the ferroxidase centres of E62A SynFtn was investigated by monitoring the regeneration of the rapid phase of Fe2+ oxidation associated with the apo protein. 1 μM protein was incubated with 400 μM Fe2+ at 25 °C until the absorbance at 340 nm became invariant with time. Equivalent samples were then mixed with an equal volume of 72 μM Fe2+ in 1 mM HCl either immediately or following a further period of incubation of 2, 5, 15 or 60 min. An additional sample incubated at 25 °C for 60 min was subsequently incubated for further 15 h at 4 °C. After re-equilibration at 25 °C the protein was mixed with an equal volume of 72 μM Fe2+ in 1 mM HCl as above.
Assays to assess the rate at which the proteins were able to mineralize iron within the internal cavity employed a higher iron to protein stoichiometry of 400 Fe2+ per SynFtn. Dependences of absorbance on time were recorded on a Hitachi U-2900 spectrometer following manual mixing of 6.4 μL of a 50 mM Fe2+ solution in 50 mM HCl to a 1.6 mL sample of 0.5 μM protein in 100 mM MES pH 6.5. The increased ratio of Fe2+:protein resulted in dependences that were approximately linear for the initial 180 s of the reaction. Initial rates of iron mineralization were deduced from the gradient of this linear region and an extinction coefficient for the mineral core calculated from the net absorbance change upon complete oxidation of the 200 μM Fe2+ added.
Electron paramagnetic resonance (EPR) spectroscopy
EPR spectra were recorded at 10 K on a Bruker EMX (X-band) EPR spectrometer equipped with an Oxford Instruments liquid helium system and a spherical high-quality ER 4122 SP 9703 Bruker resonator. Protein samples in EPR tubes were mixed with the appropriate volume of a 25 mM stock Fe2+ solution and frozen at least 10 s thereafter by plunging the tubes into methanol cooled with solid CO2. Final protein concentration was 4.17 μM (100 μM in monomer) in all cases. Proteins were in 100 mM MES pH 6.5 and Fe2+ solutions in 50 mM HCl. Instrument parameters for EPR measurements were as follows: microwave frequency υMW = 9.4657 GHz, modulation frequency υM = 100 kHz, time constant τ = 82 ms, microwave power = 3.19 mW, modulation amplitude AM = 5 G, scan rate = 22.6 G s−1.Conclusions
The channels that penetrate ferritin protein cages have attracted considerable study, not only with the aim of understanding how Fe2+ substrate and Fe3+ product are transported to and from an active site buried within an α-helical bundle, but also due to their potential as mimics for membrane ion channels that are embedded within a water soluble matrix.5,6,10,16,25 As such, it is desirable to determine which of the channels and residues lining the interior of the channels are required for efficient transport of the substrate. The 3-fold channel of animal ferritins has been identified as the route of iron entry in Frog M and Human H chain ferritin, and the peptide sequences of other animal ferritins are strictly conserved in the vicinity of this channel. Together with the identification of conserved glutamate residues at positions intermediate between the channel exits and the ferroxidase centre (Glu57 and Glu136 in Frog M numbering) this is suggestive of a common route of iron entry across all animal (and possibly plant) ferritins. In contrast, the B-channel of prokaryotic ferritins have been proposed to be a major route of Fe2+ entry and this has been demonstrated for E. coli bacterioferritin.16The data presented here identifies the 3-fold channel of the prokaryotic ferritin SynFtn as the major route of iron entry into this protein, despite significant variation in its composition compared to those in animal ferritins. Three-fold channels of the animal proteins contain three symmetry related copies of each of two carboxylates, an Asp and a Glu (Asp127 and Glu130, Frog M numbering). Disruption of either by site directed mutagenesis significantly affected the rate of Fe2+ oxidation supported by the ferroxidase centre, even in the case of conservative substitutions such as E130D. Replacement of either carboxylate by Ala resulted in almost undetectable rapid oxidation of Fe2+ by the catalytic centres at Fe2+ loadings ≥96 Fe per cage.25 However, the three-fold channel of SynFtn contains only one symmetry-related carboxylate, Asp137, which is at a position equivalent to Glu130, and is shown here to be essential for rapid oxidation of Fe2+ at the ferroxidase centre. At the equivalent position to Asp127 of animal ferritins, it contains a Val (V134). Thus, the SynFtn three-fold channel has a significantly lower concentration of negatively charged carboxylates. Despite this, it is capable of supporting Fe2+ transport at a rate comparable to those of the animal ferritins.
In further contrast to animal ferritins, disruption of a potential Fe3 transient binding site in SynFtn through substitution of Glu62 impaired the rate of release of the oxidised ferric-oxo product from catalytic centres, rather than the uptake of Fe2+ substrate. Therefore, it seems likely that the only requirement for rapid transport of Fe2+ through the 3-fold channels of ferritins is a carboxylate residue with a sidechain directed toward the mid-channel position (D137 in SynFtn, E130 in Frog M ferritin). The stringent requirements on animal ferritins for a Glu at the mid-channel position and an Asp at the interior channel exit most likely arise from the need to efficiently deliver the substrate into the Fe4 transit site rather than for rapid transport through the channel itself.
The route utilised to transfer Fe2+ from the interior exit of the 3-fold channel of SynFtn to the ferroxidase centre is as yet undefined, but must either require a different trajectory of incoming substrate, or be more tolerant of variation in it, allowing the channel to function efficiently with only the mid-channel carboxylate.
Although the D137A substitution abolished rapid Fe2+ oxidation at the ferroxidase centres of SynFtn, it is noteworthy that overall mineralization activity was ∼75% that of the wild type protein. In the E62A variant, initial oxidation was rapid, but mineralization was only ∼38% of wild type SynFtn. These data indicate that, for SynFtn, while the rate-limiting step of the initial ferroxidase centre Fe2+ oxidation reaction is the binding of Fe2+, this is not the case for the overall mineralization process. Instead, the rate-limiting step of mineralization is associated with Fe3+ egress from the ferroxidase centre.
Finally, whilst iron uptake has now been demonstrated to occur through both the 3-fold and B-channels in different prokaryotic ferritins, and Fe2+ binding in a B-channel has been observed in another Ftn-like example,16,17 data on iron transit in the Ftn proteins remains, on the whole, scarce. Whilst the prokaryotic ferritins appear to exhibit greater variability in their mechanism of iron uptake than their animal counterparts, the examples characterized to date are drawn from distinct subfamilies. E. coli bacterioferritin, in which the B-channel is the major route in for Fe2+, is a heme-containing ferritin with a distinct ferroxidase centre architecture and mechanism of Fe2+ oxidation,26,27 whilst SynFtn represents the first characterized example of a subset of ferritins, found almost exclusively within marine picocyanobacteria, which combine structural features previously considered to define ferritins as either eukaryotic or prokaryotic.18 Therefore, the field would benefit from further work towards elucidating the route for Fe2+ entry into the most widely distributed class of prokaryotic Ftns, of which EcFtnA remains the best characterized example.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Dr Nick Watmough (University of East Anglia) for access to the stopped-flow instrument. This work was supported by the UK's Biotechnology and Biological Sciences Research Council through grant BB/R002363/1.Notes and references
- K. H. Gayer and L. Wootner, J. Am. Chem. Soc., 1956, 78, 3944–3946 CrossRef CAS.
- P. Arosio and S. Levi, Free Radical Biol. Med., 2002, 33, 457–463 CrossRef CAS PubMed.
- N. E. Le Brun, A. Crow, M. E. P. Murphy, A. G. Mauk and G. R. Moore, Biochim. Biophys. Acta, 2010, 1800, 732–744 CrossRef CAS PubMed.
- E. C. Theil, R. K. Behera and T. Tosha, Coord. Chem. Rev., 2013, 257, 579–586 CrossRef CAS PubMed.
- T. Tosha, H. L. Ng, O. Bhattasali, T. Alber and E. C. Theil, J. Am. Chem. Soc., 2010, 132, 14562–14569 CrossRef CAS PubMed.
- R. K. Behera and E. C. Theil, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7925–7930 CrossRef CAS PubMed.
- C. Bernacchioni, S. Ciambellotti, E. C. Theil and P. Turano, Biochim. Biophys. Acta, 2015, 1854, 1118–1122 CrossRef CAS PubMed.
- C. Pozzi, F. Di Pisa, C. Bernacchioni, S. Ciambellotti, P. Turano and S. Mangani, Acta Crystallogr., Sect. D: Struct. Biol., 2015, 71, 1909–1920 CrossRef CAS PubMed.
- C. Pozzi, F. Di Pisa, D. Lalli, C. Rosa, E. Theil, P. Turano and S. Mangani, Acta Crystallogr., Sect. D: Struct. Biol., 2015, 71, 941–953 CrossRef CAS PubMed.
- B. Chandramouli, C. Bernacchioni, D. Di Maio, P. Turano and G. Brancato, J. Biol. Chem., 2016, 291, 25617–25628 CrossRef CAS PubMed.
- R. K. Behera, R. Torres, T. Tosha, J. M. Bradley, C. W. Goulding and E. C. Theil, J. Biol. Inorg. Chem., 2015, 20, 957–969 CrossRef CAS PubMed.
- I. Bertini, D. Lalli, S. Mangani, C. Pozzi, C. Rosa, E. C. Theil and P. Turano, J. Am. Chem. Soc., 2012, 134, 6169–6176 CrossRef CAS PubMed.
- P. Turano, D. Lalli, I. C. Felli, E. C. Theil and I. Bertini, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 545–550 CrossRef CAS PubMed.
- S. Macedo, C. V. Romao, E. Mitchell, P. M. Matias, M. Y. Liu, A. V. Xavier, J. LeGall, M. Teixeira, P. Lindley and M. A. Carrondo, Nat. Struct. Biol., 2003, 10, 285–290 CrossRef CAS PubMed.
- A. Marchetti, M. S. Parker, L. P. Moccia, E. O. Lin, A. L. Arrieta, F. Ribalet, M. E. P. Murphy, M. T. Maldonado and E. V. Armbrust, Nature, 2009, 457, 467–470 CrossRef CAS PubMed.
- S. G. Wong, J. C. Grigg, N. E. Le Brun, G. R. Moore, M. E. Murphy and A. G. Mauk, J. Biol. Chem., 2015, 290, 3732–3739 CrossRef CAS PubMed.
- S. Pfaffen, J. M. Bradley, R. Abdulqadir, M. R. Firme, G. R. Moore, N. E. Le Brun and M. E. Murphy, J. Biol. Chem., 2015, 290, 28416–28427 CrossRef CAS PubMed.
- J. M. Bradley, D. A. Svistunenko, J. Pullin, N. Hill, R. K. Stuart, B. Palenik, M. T. Wilson, A. M. Hemmings, G. R. Moore and N. E. Le Brun, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2058–2067 CrossRef CAS PubMed.
- E. R. Bauminger, P. M. Harrison, D. Hechel, I. Nowik and A. Treffry, Biochim. Biophys. Acta, 1991, 1118, 48–58 CrossRef CAS.
- G. Winter, J. Appl. Crystallogr., 2010, 43, 186–190 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352–367 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- H. P. Zheng, M. D. Chordia, D. R. Cooper, M. Chruszcz, P. Muller, G. M. Sheldrick and W. Minor, Nat. Protoc., 2014, 9, 156–170 CrossRef CAS PubMed.
- S. Haldar, L. E. Bevers, T. Tosha and E. C. Theil, J. Biol. Chem., 2011, 286, 25620–25627 CrossRef CAS PubMed.
- J. M. Bradley, D. A. Svistunenko, T. L. Lawson, A. M. Hemmings, G. R. Moore and N. E. Le Brun, Angew. Chem., Int. Ed., 2015, 54, 14763–14767 CrossRef CAS PubMed.
- A. Crow, T. L. Lawson, A. Lewin, G. R. Moore and N. E. Le Brun, J. Am. Chem. Soc., 2009, 131, 6808–6813 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt03570b |
| This journal is © The Royal Society of Chemistry 2020 |