DOI:
10.1039/C8SC02126K
(Edge Article)
Chem. Sci., 2018,
9, 6411-6416
Ring-opening hydroarylation of monosubstituted cyclopropanes enabled by hexafluoroisopropanol†
Received
14th May 2018
, Accepted 28th June 2018
First published on 28th June 2018
Abstract
Ring-opening hydroarylation of cyclopropanes is typically limited to substrates bearing a donor–acceptor motif. Here, the transformation is achieved for monosubstituted cyclopropanes by using catalytic Brønsted acid in hexafluoroisopropanol (HFIP) solvent, constituting a rare example where such cyclopropanes engage in intermolecular C–C bond formation. Branched products are obtained when electron-rich arylcyclopropanes react with a broad scope of arene nucleophiles in accord with a simple SN1-type ring-opening mechanism. In contrast, linear products are obtained when cyclopropylketones react with electron-rich arene nucleophiles. In the latter case, mechanistic experiments and DFT-calculations support a homo-conjugate addition pathway.
Introduction
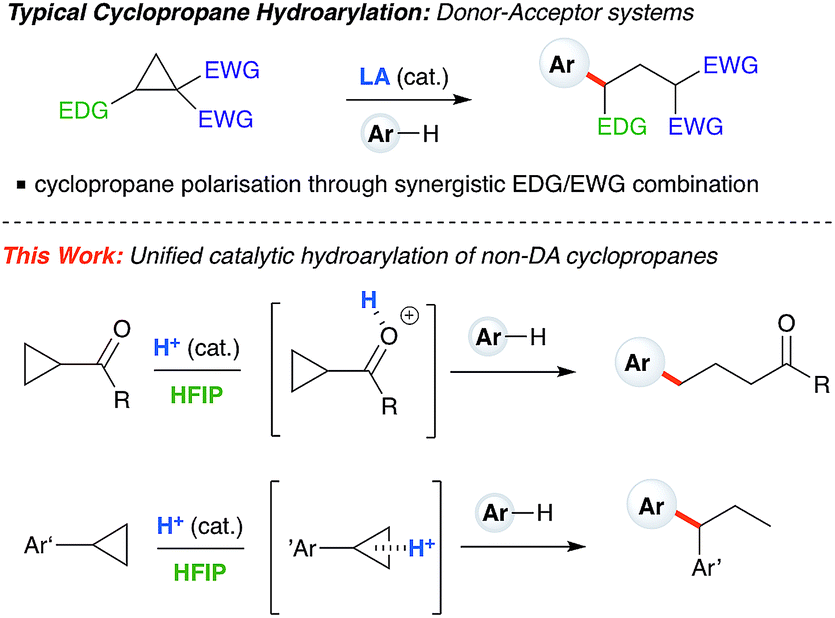
Cyclopropanes are readily accessed substrates whose strain-release ring opening gives rise to highly functionalized products.1 The ring-opening hydroarylation of cyclopropanes installs an aryl moiety and a hydrogen atom in a 1,3-relationship. Thus far, it has mostly been described for so-called donor–acceptor cyclopropanes, a class of cyclopropanes that are highly tractable synthetic precursors due to the presence of electron-donating and electron-withdrawing groups arranged in a 1,2-relationship.2,3 Intermolecular ring-opening hydroarylation can also occur in cyclopropanes bearing two geminal acceptor groups, but only for indoles under ultra-high pressure (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atm) conditions.4,5 Catalytic ring-opening hydroarylation of mono-substituted cyclopropanes has yet to be reported. Generally speaking, the reactivity of monosubstituted cyclopropanes is much less developed than for donor–acceptor cyclopropanes. Existing ring-opening reactions include oxidative addition into C–C bonds with transition metals,6–8 oxidative 1,3-difunctionalization,9 frustrated Lewis pairs,10,11 or addition of strong nucleophiles12–14 and mineral acids.15 One-pot iodination/alkylation procedures are also known.16 Our laboratory has begun to exploit the catalytically active aggregates formed between Brønsted acids and solvents such as hexafluoroisopropanol (HFIP) to enable challenging transformations.17,18 Herein, we report Brønsted acid catalyzed arylative ring-opening of mono-substituted cyclopropanes in HFIP (Scheme 1, this work). Mechanistic experiments and DFT calculations indicate that, depending on cyclopropane substitution, two differing mechanistic pathways appear to be operative. Cyclopropanes bearing an aryl group furnish branched products and proceed through a carbocation intermediate, whereas cyclopropanes bearing a carbonyl group lead to linear products and react via a homo-conjugate addition pathway.
000 atm) conditions.4,5 Catalytic ring-opening hydroarylation of mono-substituted cyclopropanes has yet to be reported. Generally speaking, the reactivity of monosubstituted cyclopropanes is much less developed than for donor–acceptor cyclopropanes. Existing ring-opening reactions include oxidative addition into C–C bonds with transition metals,6–8 oxidative 1,3-difunctionalization,9 frustrated Lewis pairs,10,11 or addition of strong nucleophiles12–14 and mineral acids.15 One-pot iodination/alkylation procedures are also known.16 Our laboratory has begun to exploit the catalytically active aggregates formed between Brønsted acids and solvents such as hexafluoroisopropanol (HFIP) to enable challenging transformations.17,18 Herein, we report Brønsted acid catalyzed arylative ring-opening of mono-substituted cyclopropanes in HFIP (Scheme 1, this work). Mechanistic experiments and DFT calculations indicate that, depending on cyclopropane substitution, two differing mechanistic pathways appear to be operative. Cyclopropanes bearing an aryl group furnish branched products and proceed through a carbocation intermediate, whereas cyclopropanes bearing a carbonyl group lead to linear products and react via a homo-conjugate addition pathway.
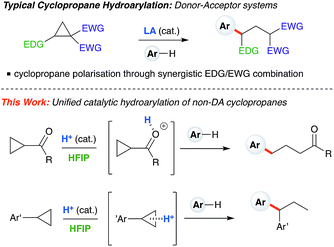 |
| | Scheme 1 Typical hydroarylation reactivity of cyclopropanes under Lewis acid catalysis using donor–acceptor substrates. This work: unified, HFIP-assisted, Brønsted acid-catalyzed cyclopropane hydroarylation. | |
Results and discussion
Synthetic studies
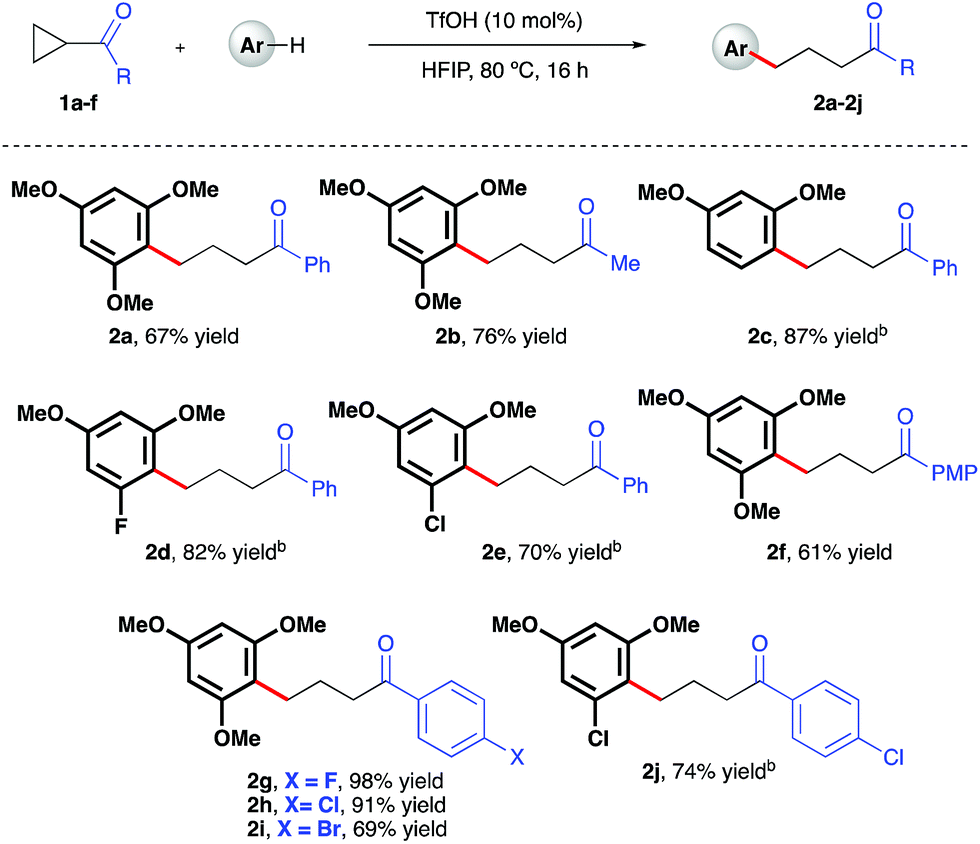
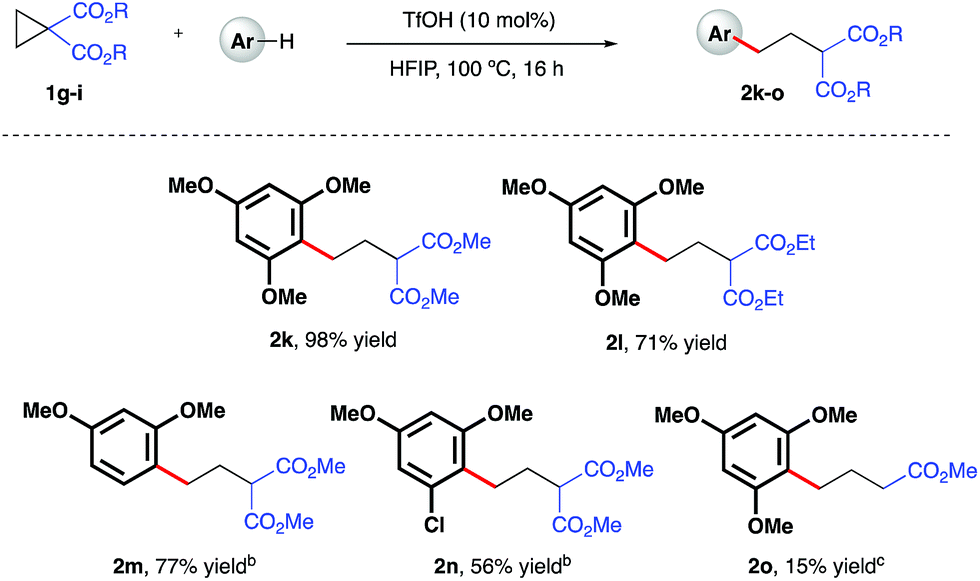
Initial investigations began with the optimization of the reaction between commercial cyclopropyl phenyl ketone 1a and 1,3,5-trimethoxybenzene. A screen of temperatures, acids, and solvents rapidly revealed that the combination of triflic acid (TfOH) catalyst in HFIP promoted smooth reactivity to the desired ring-opened adduct in near quantitative conversion and 67% isolated yield (Scheme 2, 2a). Application of these optimized reaction conditions to a series of ketone-bearing cyclopropanes19 gave access to a range of γ-arylated ketone products in good to excellent yields. The aryl ketone was replaced with a methyl ketone with almost no impact on reactivity (2b), and a series of cyclopropyl aryl ketones bearing a variety of substituents (2d–j) was arylatively ring-opened in 67–98% yield. In this series, variation of the nucleophile revealed a range of electron-rich arenes was well tolerated by the reaction system (entries 2c–e and 2j), provided that a 1,3-methoxy motif was present in all cases. Nucleophiles lacking this 1,3-pattern, even 1,2,3- and 1,2,4-trimethoxybenzene, were found to be ineffective in this reaction, as were 1,4-dimethoxybenzene, indole derivatives and cyclopropanes bearing nitrile groups.20 The optimal reaction conditions also proved applicable to the arylative ring-opening reaction of cyclopropanes bearing two ester groups (Scheme 3, entries 2k–n), with almost no impact on the reaction efficiency. However, a substrate bearing a single ester group proved recalcitrant, providing only poor yields of the γ-arylation product (2o). With conditions established for the preparation of linear, γ-arylated carbonyl derivatives, we next elected to probe simple aryl-cyclopropanes as reaction partners in this catalytic protocol. To the best of our knowledge, such substrates have yet to be engaged in catalytic ring-opening reactions with nucleophiles other than thiols21 and a single example of a Friedel–Crafts reaction.22
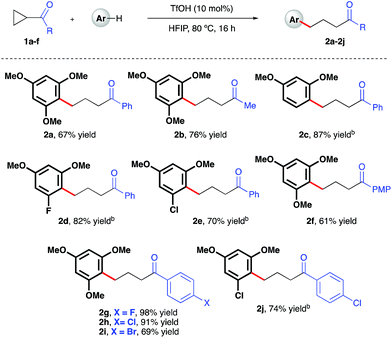 |
| | Scheme 2 TfOH-catalyzed, arylative ring-opening reactions of keto-bearing cyclopropanes. aYields of isolated products after column chromatography over silica. bCombined yield of regioisomeric products – see ESI† for further details. | |
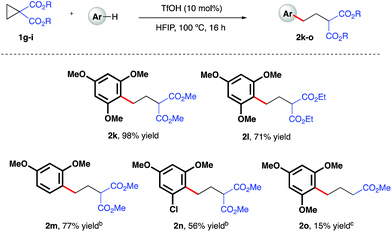 |
| | Scheme 3 TfOH-catalyzed, arylative ring-opening reactions of ester-bearing cyclopropanes. aYields of isolated products after column chromatography over silica. bCombined yield of regioisomeric products – see ESI† for further details. cReaction heated at 100 °C in 1,2-dichloroethane. | |
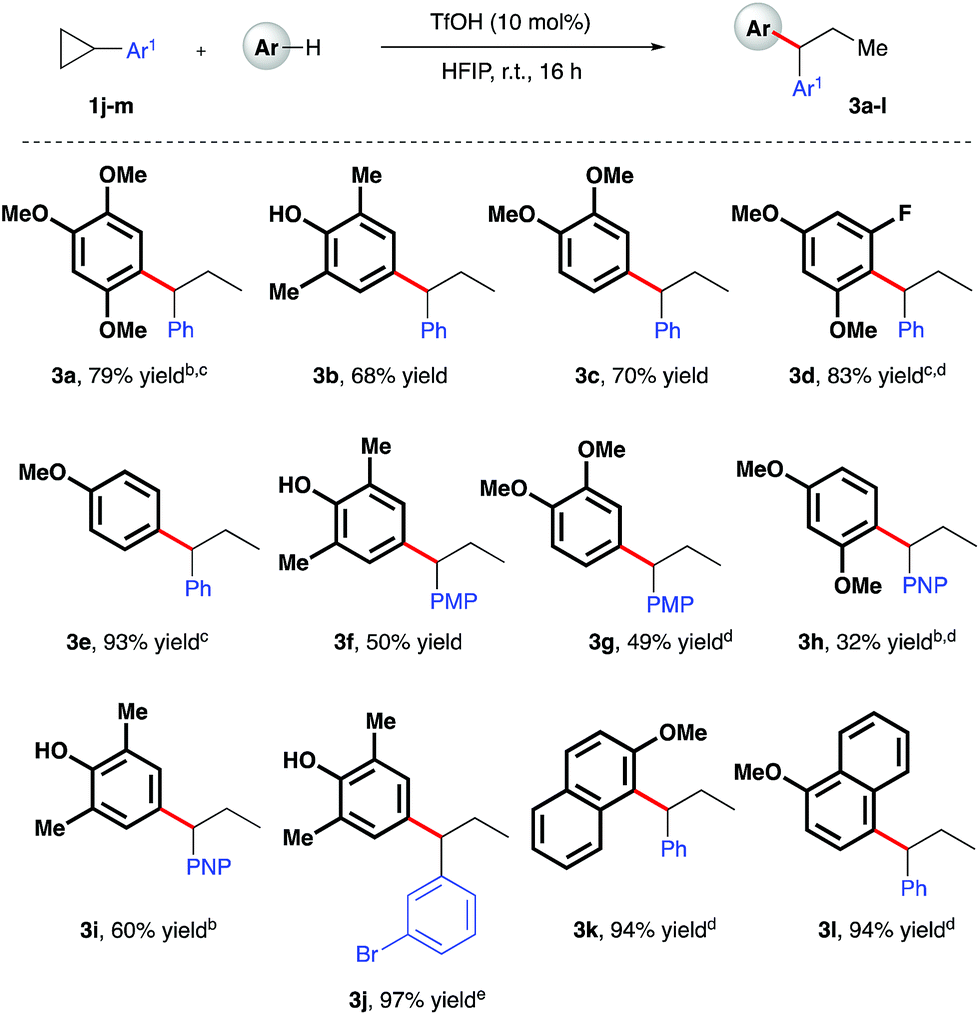
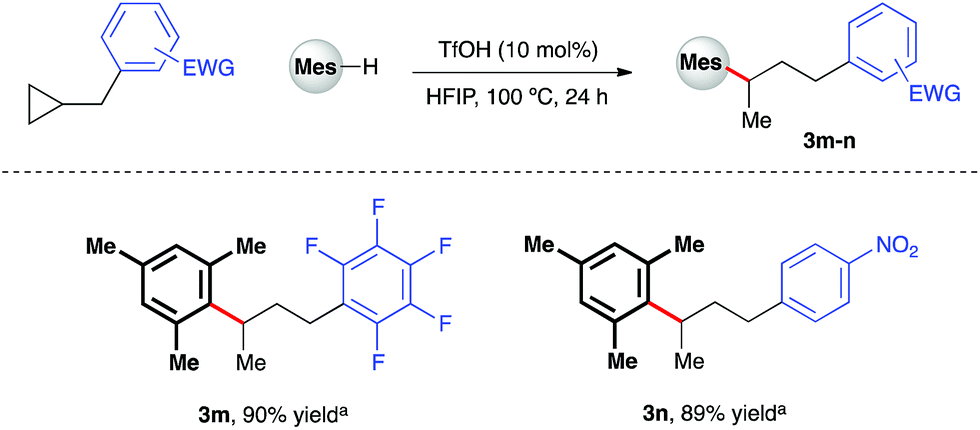
Given the polarity reversal of such substrates compared to their carbonyl-bearing counterparts, opposing reactivity to yield the branched ring-opening product was expected. Indeed, reaction of cyclopropylbenzene with 1,2,4-trimethoxybenzene yielded the desired, branched product in 79% yield (Scheme 4, 3a). A survey of the reaction scope with cyclopropylbenzene revealed a wider range of nucleophiles was tolerated in this reaction system (entries 3b–e, k, l), including less electron-rich arenes such as anisole (3e). Additionally, it was found that almost all of these reactions proceeded efficiently at room temperature. Variation of the aryl moiety of the cyclopropane proved unproblematic with electron-rich (3f and 3g), electron-deficient (3h and i) and halogenated ring systems (3j) all well tolerated. The catalytic reaction system was also successfully extended to the activation of benzylic cyclopropanes, in particular those bearing electron-deficient aryl rings (Scheme 5). In reaction with mesitylene, both pentafluorophenyl- and 4-nitrophenyl derivatives delivered methyl-substituted bis-aryls 3m and 3n respectively, presumably via Brønsted acid-mediated ring opening, followed by a 1,2-hydride shift to give the least inductively destabilized carbocation prior to nucleophilic capture by mesitylene.23
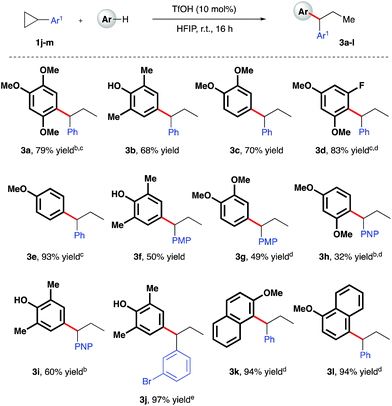 |
| | Scheme 4 TfOH-catalyzed, arylative ring-opening reactions of arylcyclopropanes. aYields of isolated products after column chromatography over silica. bReaction heated at 80 °C. cNMR yield. dCombined yield of regioisomeric products – see ESI† for further details. e1.1 equiv. of nucleophile was used. PMP = 4-methoxyphenyl-. PNP = 4-nitrophenyl-. | |
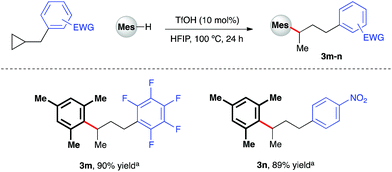 |
| | Scheme 5 TfOH-catalyzed arylative ring-opening of benzylic cyclopropanes. aYields of isolated products after column chromatography over silica. Mes = mesityl. | |
Mechanistic & computational studies
In situ
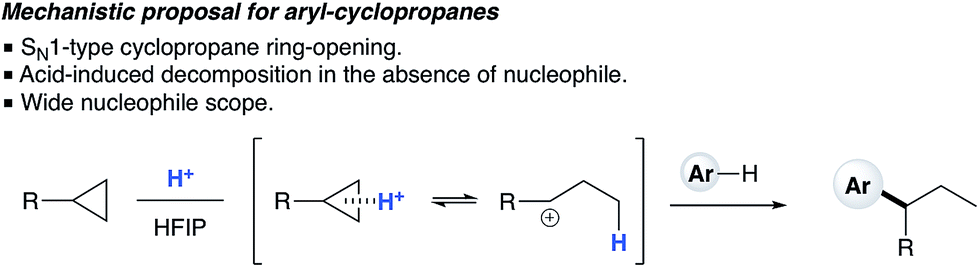
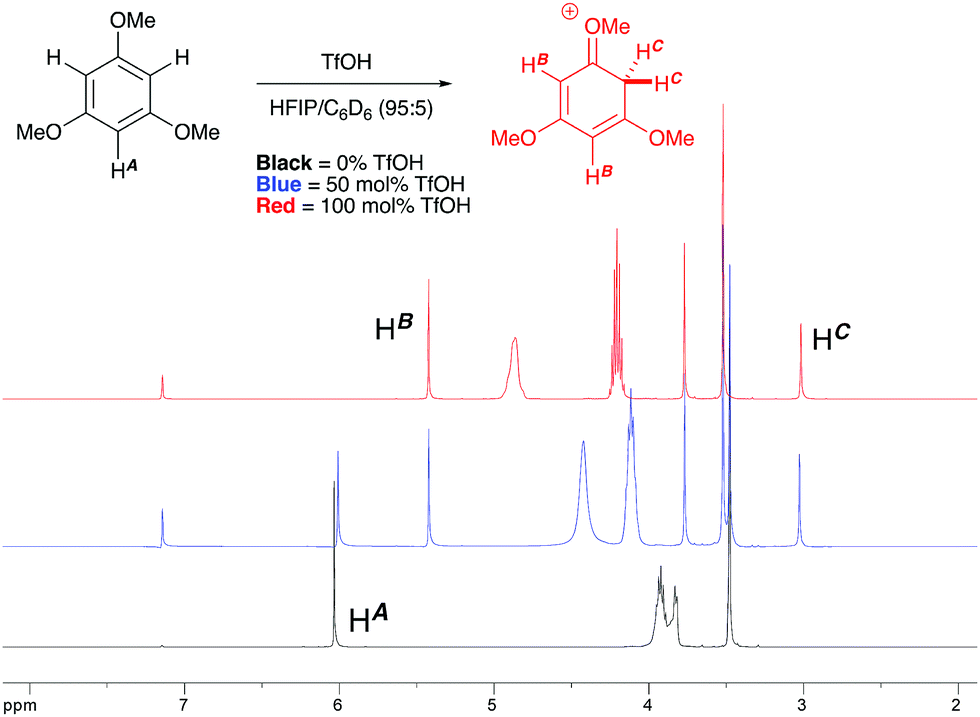
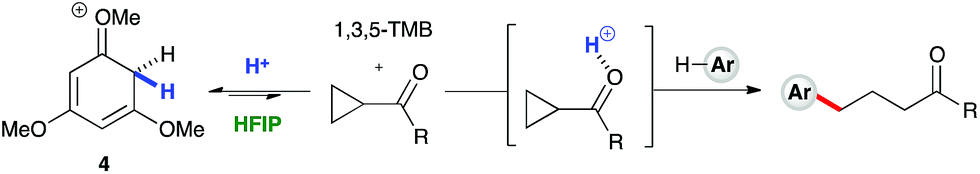
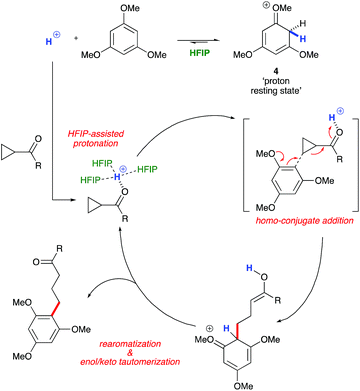
1H NMR titration studies of various reaction components in HFIP with TfOH were carried out in order to better rationalize the observed reactivity trends. Addition of 10 mol% TfOH to cyclopropylbenzene in HFIP in the absence of any nucleophile gave a highly exothermic reaction and an intense orange colour, with 1H NMR analysis suggesting decomposition of starting material. This is in accord with prior literature on the reaction of similar cyclopropanes with mineral acids, in which a direct protonation yields an intermediate carbocation followed by collapse of the resulting ion pair. These observations, in combination with the wide nucleophile scope tolerated by such substrates, suggest that aryl cyclopropanes ring-open in the presence of TfOH and react via an SN1-type mechanistic pathway (Scheme 6). In contrast, initial in situ1H NMR experiments with cyclopropyl ketones suggest no spontaneous reactivity with TfOH in the absence of nucleophile. Rather, addition of TfOH to a 1:1 mixture of cyclopropyl phenyl ketone and 1,3,5-trimethoxybenzene revealed a complete preference for protonation of the latter. Titration of a solution of the 1,3,5-trimethoxybenzene (1,3,5-TMB) nucleophile in HFIP with TfOH revealed that 1,3,5-TMB is rapidly and completely protonated to yield a dearomatized cationic diene (Fig. 1).24 At room temperature, a 1:1 mixture of cyclopropyl phenyl ketone and 1,3,5-TMB shows complete protonation of the latter upon titration of TfOH.25 While an intriguing observation, thus far it has not been possible to establish whether this protonated nucleophile plays a more intimate role than simply being a ‘resting state’ for the catalyst. Additionally, this dearomatizing protonation is not observed in other solvents, suggesting that HFIP plays a more intimate role in the reaction beyond that of a bulk solvent.18d Thus, a tentative mechanistic scenario can be proposed where TfOH in HFIP initially enables protonation of 1,3,5-TMB (Scheme 7). Protonated TMB 4 can subsequently act as a reversible proton-reservoir26 to promote a homo-conjugate addition typical of donor–acceptor cyclopropanes.
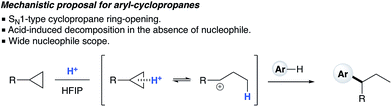 |
| | Scheme 6 Proposed SN1-type nucleophilic ring opening of arylcyclopropanes. | |
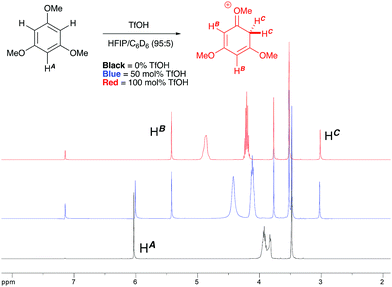 |
| | Fig. 1
1H NMR titration of 1,3,5-TMB with TfOH in HFIP leading to the observation of a dearomatized diene intermediate. | |
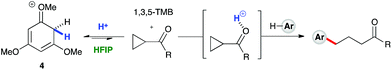 |
| | Scheme 7 A plausible mechanistic scenario: Brønsted acid catalyzed homo-conjugate addition. | |
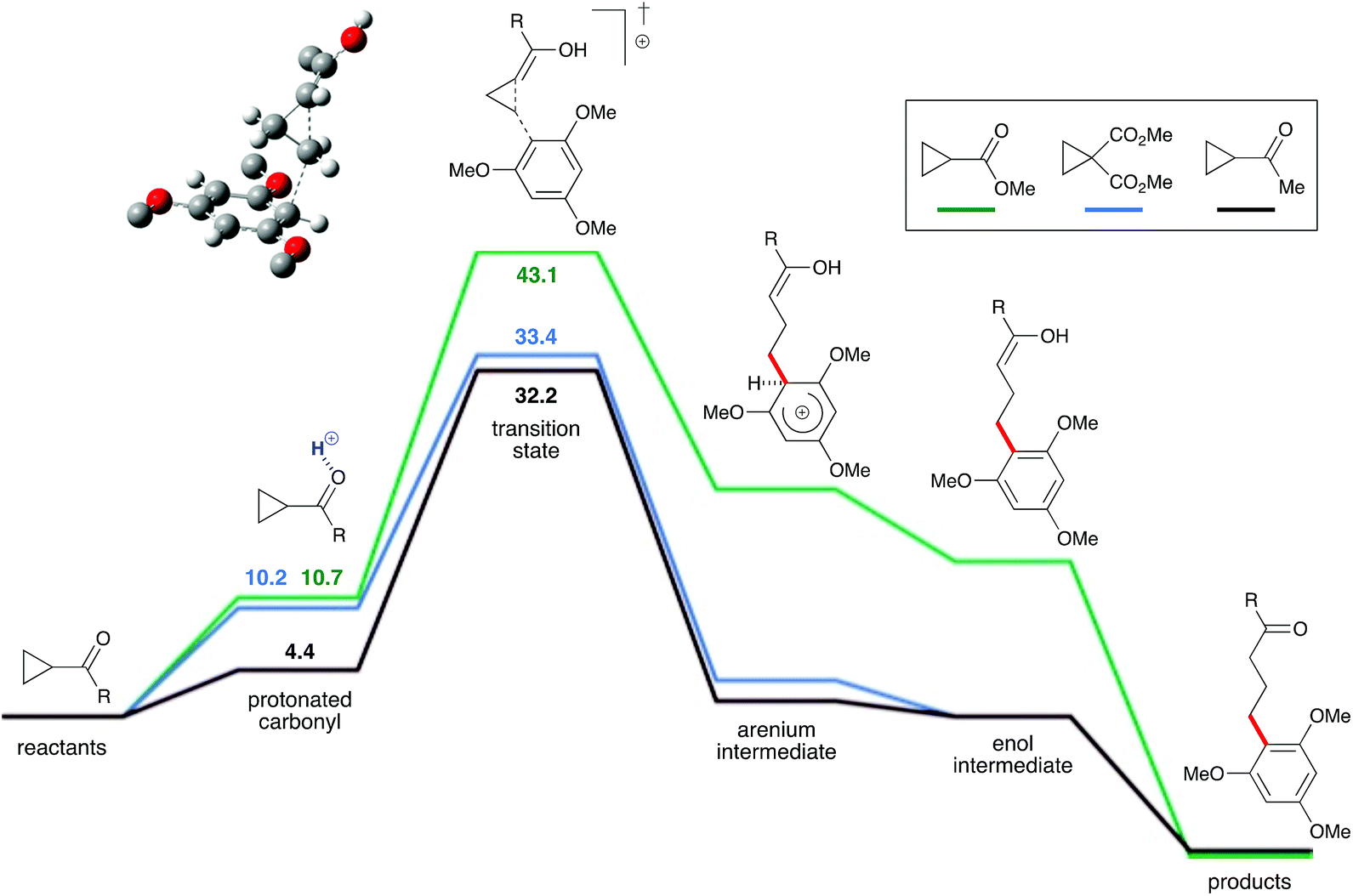
To evaluate this hypothesis, this reaction system was probed via density functional theory calculations (ωB97X-D/def2-TZVP). A plausible mechanism was identified for the proposed Brønsted-acid-catalyzed homo-conjugate (Fig. 2). The ketone oxygen was predicted to be moderately basic for most cyclopropyl ketones, allowing it to be protonated by strong acids like TfOH. The protonated cyclopropyl ketone is activated for reaction with the arene, due to its partial enol character. This protonated cyclopropyl is then intercepted by the arene to form an arenium intermediate, which is the rate-limiting step. Deprotonation of the arenium intermediate yields an enol, which then tautomerizes to the ketone product. The calculated free energy profiles are generally consistent with the observed reactivity. Cyclopropyl methyl ester 1i (green line) did not react with 1,3,5-trimethoxybenzene, which is consistent with the calculations that show the barrier to this reaction is 9.7 kcal mol−1 higher than for the cyclopropyl dimethyl ester 1g (blue line). The calculated barrier for cyclopropyl dimethyl ester addition is only 1.2 kcal mol−1 higher than for the cyclopropyl methyl ketone 1b (black line), consistent with the high yields for these reactions. Likewise, the calculated activation energies for addition of 1,2-, 1,2,3- and 1,2,4- methoxy-substituted arenes are significantly higher than for the reaction with 1,3,5-trimethoxybenzene.25
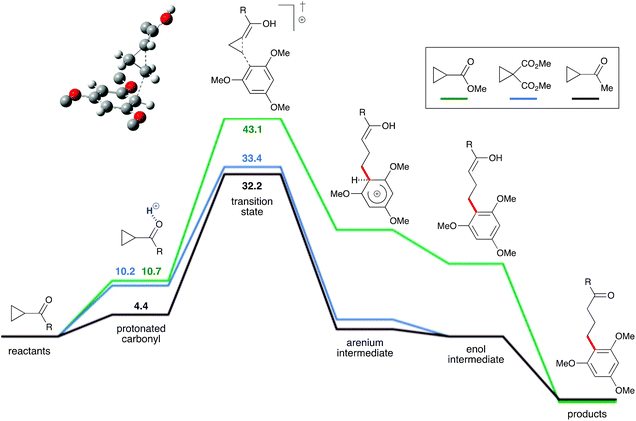 |
| | Fig. 2 DFT-calculated reaction profiles for the addition of cyclopropyl ketones or esters to arenes. Energy values in kcal mol−1. | |
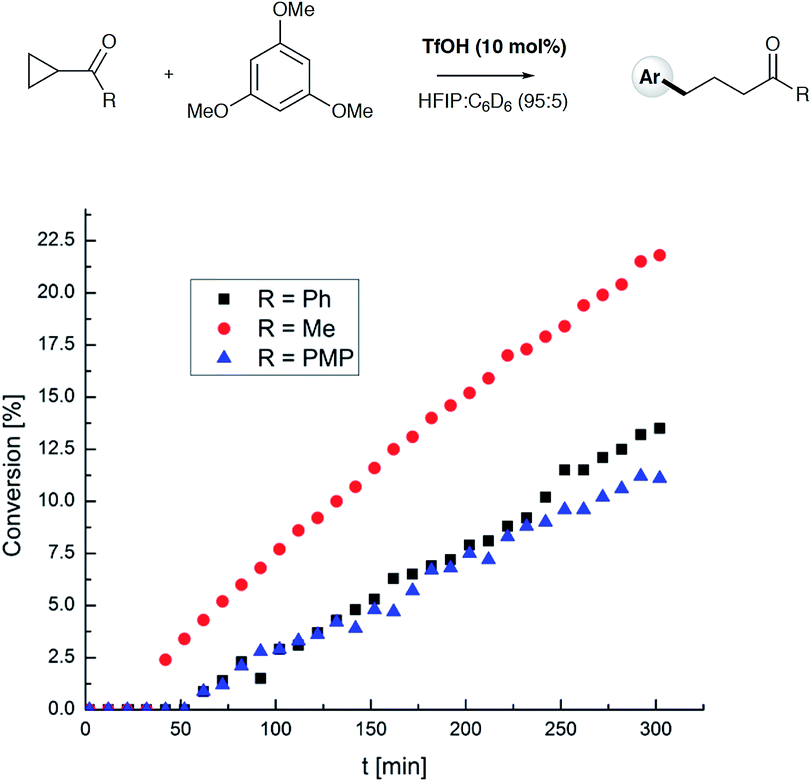
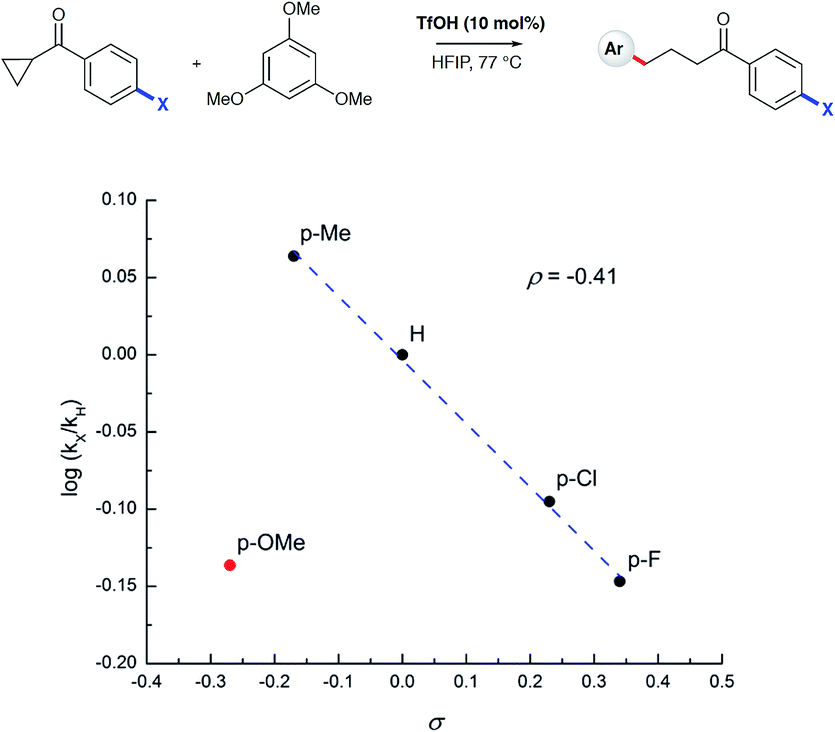
To test the findings of the DFT-investigations experimentally, a series of reactions between 1,3,5-trimethoxybenzene and various cyclopropyl aryl ketones was performed and monitored by 1H NMR spectroscopy over time. In an initial survey of cyclopropyl methyl- 1b, cyclopropyl phenyl- 1a, and cyclopropyl 4-methoxyphenyl-1c ketones, relative rates in agreement with the computed transition state energies were observed with cyclopropyl methyl ketone exhibiting a significantly faster rate of arylation (Fig. 3). A further comparison of the initial reaction rates of para- substituted aryl cyclopropanes (4-Me, 4-MeO, 4-F and 4-Cl), relative to that of the reaction of the parent cyclopropyl phenyl ketone, allowed a Hammett plot to be constructed for this reaction by plotting relative reaction rates vs. Hammett substituent parameter (sigma) (Fig. 4). Examination of the resulting plot reveals a strong linear relationship for most data, with a negative slope; an observation in agreement with the generation of positive charge on the substrate in the transition state. The magnitude of this slope is also strongly suggestive of an SN2-like mechanism, i.e. homo-conjugate addition rather than a discrete carbocationic intermediate. Whilst most inductively-stabilising substituents follow the expected trend and fall nicely on the observed line-of-best-fit, the data-point obtained for the reaction of the 4-methoxyphenyl cyclopropane ketone is a notable outlier. This observation can be rationalized from the computational data, which predicts the protonated form of this ketone to be exceptionally stable, so its rate of reaction is slowest if the activation energies are calculated relative to a protonated-ketone resting state. Taken together, this combined experimental and computational insight provide strong support for the proposed homo-conjugate addition mechanism, whereby initial HFIP-assisted protonation of the cyclopropyl carbonyl group induces nucleophilic ring-opening via a homo-conjugate addition pathway, leading to the enol form of the observed linear product (Scheme 8).
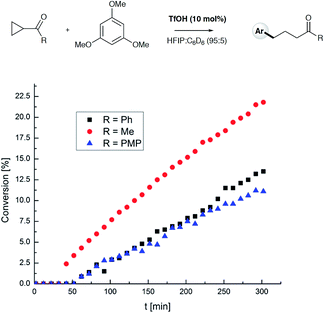 |
| | Fig. 3 Comparison of relative reaction rates for cyclopropyl phenyl ketone 1a (black squares), cyclopropyl methyl ketone 1b (red circles), and cyclopropyl 4-methoxyphenyl ketone 1c (blue triangles) in reaction with 1,3,5-TMB. Reactions were monitored by 1H NMR spectroscopy at 65 °C and conversion (%) is plotted vs. time (min). | |
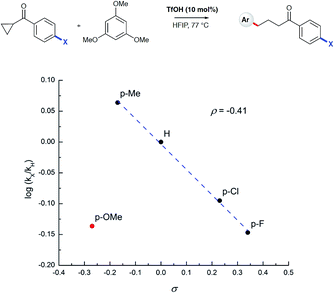 |
| | Fig. 4 Hammett plot of log(kX/kH) vs. sigma value for the depicted reaction. Initial rates were determined by plotting [product] vs. time as determined by 1H NMR spectroscopy. | |
 |
| | Scheme 8 A proposed homo-conjugate addition mechanistic pathway consistent with combined experimental and computational data. | |
Conclusions
In conclusion, a general catalytic system for the arylative ring opening of mono-substituted cyclopropanes has been described. Triflic acid in combination with hexafluoroisopropanol provides a superior Brønsted acid catalyst system, engaging weakly-polarized substrates in Friedel–Crafts-type reactivity. The regioselectivity and operative mechanism varies depending on the cyclopropane substitution, with carbonyl-bearing cyclopropanes reacting via a homo-conjugate addition pathway. Computational and mechanistic investigations are congruent with these findings.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 639170) and by grants from LabEx “Chemistry of Complex Systems”. V. D. V. thanks the French government for an MRT fellowship. We thank Dr Helmut Muenster of MasCom for his assistance with HRMS analysis. F. S. and C. N. R. thank NSERC of Canada for funding through the Discovery Grant program (Application 418505-2012). Computational resources were provided by Compute Canada (RAPI: djk-615-ab). F. S. thanks Dr Liqin Chen for a graduate scholarship.
Notes and references
- H. N. C. Wong, M.-Y. Hon, C. W. Tse, Y.-C. Yip, J. Tanko and T. Hudlickym, Chem. Rev., 1989, 89, 165 CrossRef.
- For comprehensive overviews of the broad chemistry of donor–acceptor cyclopropanes, see:
(a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef PubMed;
(b) M. Yu and B. L. Pagenkopf, Tetrahedron, 2005, 61, 321 CrossRef;
(c) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef PubMed.
- For examples of catalytic arylative ring-opening of donor–acceptor cyclopropanes, see:
(a) S. J. Gharpure, M. K. Shukla and U. Vijayasree, Org. Lett., 2009, 11, 5466 CrossRef PubMed;
(b) S. J. Gharpure, U. Vijayasree and S. Raja Bhushan Reddy, Org. Biomol. Chem., 2012, 10, 1735 RSC;
(c) X. Jiang, Z. Lim and Y.-Y. Yeung, Tetrahedron Lett., 2013, 54, 1798 CrossRef;
(d) R. Talukdar, A. Saha, D. P. Tiwari and M. K. Ghorai, Tetrahedron, 2016, 72, 613 CrossRef;
(e) T. N. Nguyen, T. S. Nguyen and J. A. May, Org. Lett., 2016, 18, 3786 CrossRef PubMed;
(f) T. Kaicharla, T. Roy, M. Thangaraj, R. G. Gonnade and A. T. Biju, Angew. Chem., Int. Ed., 2016, 55, 10061 CrossRef PubMed.
- P. Harrington and M. A. Kerr, Tetrahedron Lett., 1997, 38, 5949 CrossRef.
- An annulative cyclopropane arylation using superstoichiometric TfOH has been reported: G.-Q. Chen, X.-Y. Tang and M. Shi, Chem. Commun., 2012, 48, 2340 RSC.
- For reviews, see:
(a) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef PubMed;
(b) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 17, 9404 CrossRef PubMed. For a leading reference, see:
(c) S. C. Bart and P. J. Chirik, J. Am. Chem. Soc., 2003, 125, 886 CrossRef PubMed.
-
(a) D. M. Roundhill, D. N. Lawson and G. Wilkinson, J. Chem. Soc. A, 1968, 845 RSC;
(b) Y. Koga and K. Narasaka, Chem. Lett., 1999, 28, 705 CrossRef;
(c) M. H. Shaw and J. F. Bower, Chem. Commun., 2016, 52, 10817 RSC.
-
(a) L. Liu and J. Montgomery, J. Am. Chem. Soc., 2006, 128, 5348 CrossRef PubMed;
(b) L. Liu and J. Montgomery, Org. Lett., 2007, 9, 3885 CrossRef PubMed;
(c) T. Tamaki, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2011, 50, 12607 CrossRef PubMed.
- For a recent, transition-metal free oxidative 1,3-difunctionalization of cyclopropanes, see: S. M. Banik, K. M. Mennie and E. N. Jacobsen, J. Am. Chem. Soc., 2017, 139, 9152 CrossRef PubMed.
- J. G. Morton, M. A. Dureen and D. W. Stephan, Chem. Commun., 2010, 46, 8947 RSC.
- Z.-Y. Zhang, Z.-Y. Liu, R.-T. Guo, Y.-Q. Zhao, X. Li and X.-C. Wang, Angew. Chem., Int. Ed., 2017, 56, 4028 CrossRef PubMed.
- For a short review, see: S. Danishefsky, Acc. Chem. Res., 1978, 12, 66 CrossRef.
- W. A. Bone and W. H. Perkin, J. Chem. Soc., Trans., 1895, 67, 108 RSC.
-
(a) W. E. Truce and L. B. Lindy, J. Org. Chem., 1961, 26, 1463 CrossRef;
(b) A. B. Smith and R. B. Scarborough Jr, Tetrahedron Lett., 1978, 19, 1649 CrossRef;
(c) R. K. Dieter and S. Pounds, J. Org. Chem., 1982, 47, 3174 CrossRef.
- For HX-mediated transformations, see:
(a) W. H. Perkin and T. R. Marshall, J. Chem. Soc., Trans., 1891, 59, 853 RSC;
(b) J. B. Lambert, J. J. Napoli, K. K. Johnson, K. N. Taba and B. S. Packard, J. Org. Chem., 1985, 50, 1291 CrossRef;
(c) W. Xu, W. R. Dolbier Jr and J. Salazar, J. Org. Chem., 2008, 73, 3535 CrossRef PubMed. Additionally, an example of intramolecular bromo-cyclization has also been reported:
(d) C. Roesner and U. Hennecke, Org. Lett., 2015, 17, 3226 CrossRef PubMed.
- A stoichiometric CeCl3·7H2O/LiI promoted reaction between indoles and cyclopropyl ketones has also been reported: J. S. Yadav, B. V. S. Reddy, D. Chandrakanth and G. Satheesh, Tetrahedron Lett., 2007, 48, 8040 CrossRef.
-
(a) M. Dryzhakov, M. Hellal, E. Wolf, F. C. Falk and J. Moran, J. Am. Chem. Soc., 2015, 137, 9555 CrossRef PubMed;
(b) M. Dryzhakov and J. Moran, ACS Catal., 2016, 6, 3670 CrossRef;
(c) M. Dryzhakov, E. Richmond, G. Li and J. Moran, J. Fluorine Chem., 2017, 193, 45 CrossRef;
(d) V. D. Vukovic, E. Richmond, E. Wolf and J. Moran, Angew. Chem., Int. Ed., 2017, 56, 3085 CrossRef PubMed;
(e) E. Richmond, V. Vuković and J. Moran, Org. Lett., 2018, 20, 574 CrossRef PubMed.
- For examples of other HFIP-enabled strong Brønsted acid catalyzed reactions, see:
(a) A. Saito, M. Takayama, A. Yamazaki, J. Numaguchi and Y. Hanzama, Tetrahedron, 2007, 63, 4039 CrossRef;
(b) H. F. Motiwala, M. Charaschanya, V. W. Day and J. Aubé, J. Org. Chem., 2016, 81, 1593 CrossRef PubMed;
(c) X. Zeng, S. Liu and B. Xu, Org. Lett., 2016, 18, 4770 CrossRef PubMed;
(d) W. Liu, H. Wang and C.-J. Li, Org. Lett., 2016, 18, 2184 CrossRef PubMed;
(e) T. Kamitanaka, K. Morimoto, K. Tsuboshima, D. Koseki, H. Takamuro, T. Dohi and Y. Kita, Angew. Chem., Int. Ed., 2016, 55, 15535 CrossRef PubMed. For a more generał overview of HFIP, see:
(f) O. Holloczki, A. Berkessel, J. Mars, M. Mezger, A. Wiebe, S. R. Waldvogel and B. Kirchner, ACS Catal., 2017, 7, 1846 CrossRef.
- See ESI† for structural details of all cyclopropane starting materials.
- For further details of ineffective substrate combinations, see the ESI file.†.
- Y. S. Shabarov, L. G. Saginova and S. V. Veselovskaya, Zh. Org. Khim., 1986, 22, 768 Search PubMed.
- A Friedel–Crafts product was also isolated in 12% yield as a side product in: T. Ohwada, M. Kasuga and K. Shudo, J. Org. Chem., 1990, 55, 2717 CrossRef.
- In support of this mechanistic hypothesis, benzylic substrates bearing electron-rich aryl rings were found to give mixtures of regioisomeric products. Further studies into the nucleophilic ring-opening of benzylic cyclopropanes are ongoing.
- Such behaviour has previously been observed with 1,3,5-TMB using superacids such as HF-SbF5 as solvent, see:
(a) G. A. Olah and Y. K. Mo, J. Am. Chem. Soc., 1972, 94, 5341 CrossRef. It has also been observed in HFIP using laser flash photolysis, see:
(b) S. Steenken and R. A. McClelland, J. Am. Chem. Soc., 1990, 112, 9648 CrossRef;
(c) N. Mathivanan, F. Cozens, R. A. McClelland and S. Steenken, J. Am. Chem. Soc., 1992, 114, 2198 CrossRef.
- See ESI† for further information and NMR spectra of these competitive titration studies. Additionally, 13C and 1H NMR titration studies of cyclopropyl methyl ketone and TfOH in HFIP in the absence of nucleophile revealed no spontaneous cyclopropane opening, but a strong downfield shift of both the keto-carbonyl and methyl 13C signals was observed.
- D. Hoefler, M. van Gemmeren, P. Wedemann, K. Kaupmees, I. Leito, M. Leutzsch, J. B. Lingnau and B. List, Angew. Chem., Int. Ed., 2017, 55, 1411 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization data, experimental procedures, characterization of new compounds, computational methods and spectral data (PDF). See DOI: 10.1039/c8sc02126k |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Christopher N.
Rowley
b,
Christopher N.
Rowley