DOI:
10.1039/C6RA14555H
(Paper)
RSC Adv., 2016,
6, 69677-69684
Bisthienylethenes containing an imidazole bridge unit and their Ir(III) complexes: influence of substituent groups on photochromism and luminescence†
Received
5th June 2016
, Accepted 18th July 2016
First published on 18th July 2016
Abstract
Two new bisthienylethenes containing an imidazole bridge unit have been synthesized, namely tBuLH and tBuLMeH, which incorporate –C(CH3)3 and/or –CH3 substituent groups. Based on tBuLH and tBuLMeH, heteroleptic complexes [Ir(dfppy)2(tBuL)]·2CH3OH (1) and Ir(dfppy)2(tBuLMe)] (2) were prepared [dfppyH = 2-(2,4-difluorophenyl)-pyridine]. Crystal structures of tBuLH·HOAc and 1 were measured, indicating that two thiophene groups adopt antiparallel conformations in the former, while parallel conformations in the latter. In compound 1, a tBuL− ligand uses its phenol–imidazole moiety to coordinate with an {Ir(dfppy)2}+ unit, and neighboring [Ir(dfppy)2(tBuL)] molecules interact through van der Waals interactions. Upon irradiation with 326 nm/321 nm light, both tBuLH and tBuLMeH show photochromic behavior in CH2Cl2. However, no photochromism has been observed in 1 and 2. At room temperature, tBuLH and tBuLMeH in CH2Cl2 exhibit fluorescence emission at 464 nm and 479 nm, respectively. Compounds 1 and 2 in CH2Cl2 reveal phosphorescence emission at 514 nm and 507 nm, respectively, with a mixed 3MLCT/3LC character. This work discusses the influence of –C(CH3)3 and –CH3 substituent groups on the structures, photochromism and luminescence of tBuLH, tBuLMeH, 1 and 2.
Introduction
Bisthienylethenes and their metal complexes have attracted much attention due to their photochromism and additional functional properties,1 such as luminescence,2 nonlinear optical3 and magnetic properties.4 In these compounds, bisthienylethenes containing an imidazole bridge unit, i.e. BTE–imidazole compounds, form one class of promising organic ligand, mainly for the following two reasons. (1) BTE–imidazole compounds can not only be readily synthesized through a one-pot condensation reaction of an aldehyde, a diketone and ammonium acetate (Scheme 1), but also can be easily modified by attaching various substituent groups to the imidazole nitrogen atoms (NH and N).5 (2) In some BTE–imidazole compounds, coordinating R2 substituents on the imidazole rings that could efficiently coordinate to different metal ions, afforded metal–bisthienylethene complexes showing metal-perturbed photochromism from the BTE–imidazole ligand in addition to any metal-based functional properties, for example luminescence and magnetic behaviors.6–8 So far, some metal complexes have been prepared based on BTE–imidazole ligands. For example, Yam et al. reported a series of Re(I) and Pt(II) complexes incorporating BTE–imidazole ligands with 2-pyridyl moieties (denoted as BTE–imidazole–py, Scheme S1†).6 These complexes exhibit near-infrared photochromic behavior. In these complexes, the closed-form isomer of each complex shows a significantly red-shifted absorption band around 710 nm compared to that of BTE–imidazole–py (580 nm), which is due to metal coordination-assisted planarization of the BTE–imidazole–py ligand. Our group obtained three mononuclear Co(II) complexes,8 in which a Co(II) ion is chelated by two phenol–imidazole moieties from two nonequivalent BTE–imidazole–phenol ligands (i.e. L1H, BrLH and PLH in Schemes 2 and S2†). These Co(II) complexes reveal different field-induced slow magnetic relaxation behaviors and photochromic properties in CH2Cl2–CH3CN solution. Based on L1H and its derivatives L2H and hnbdtiH (Scheme S3†), four heteroleptic Ir(III) complexes have been prepared,7 namely [Ir(dfppy)2(L1)]·2CH3OH, [Ir(dfppy)2(L2)], [Ir(dfppy)2(hnbdti)]·2CH3OH and [Ir(ppy)2(hnbdti)]·CH3OH [dfppyH = 2-(2,4-difluoro-phenyl)-pyridine, ppyH = 2-phenyl-pyridine, seeing their molecular structures in Scheme S4†]. In these complexes, the N,O-donor of a BTE–imidazole ligand coordinates to an {Ir(dfppy)2}+/{Ir(ppy)2}+ unit. In CH2Cl2 solution, the former two complexes reveal phosphorescence emission with mixed 3MLCT/3LC character. The latter two complexes are non-luminescent in CH2Cl2, but [Ir(dfppy)2(hnbdti)]·2CH3OH exhibits aggregation-induced phosphorescence emission.7b In addition, these Ir(III) complexes have not shown photochromism. Up to now, the number of metal complexes incorporating BTE–imidazole ligands is quite limited, thus more examples of new BTE–imidazole and their metal complexes need to be explored to well understand the relationship between their structures and functional properties.
 |
| | Scheme 1 | |
 |
| | Scheme 2 | |
In this paper, we report the syntheses of two new BTE–imidazole compounds tBuLH and tBuLMeH, which contain the additional substituent groups –C(CH3)3 and/or –CH3 compared to L1H (Scheme 2). Based on tBuLH and tBuLMeH, heteroleptic complexes [Ir(dfppy)2(tBuL)]·2CH3OH (1) and [Ir(dfppy)2(tBuLMe)] (2) were obtained. Herein, we report the syntheses, crystal structures, photochromic and luminescent properties of tBuLH, tBuLMeH, 1 and 2, and discuss the influence of the substituent groups –C(CH3)3 and –CH3 on their structures and properties.
Experimental
Materials and methods
1,2-Bis[2,5-dimethyl(3-thienyl)]ethane-1,2-dione and [Ir(dfppy)2Cl]2 dimer were prepared according to the literature.5a,9 All other reagents were commercially available and used without further purification. Elemental analyses were performed on a Perkin Elmer 240C elemental analyzer. IR spectra were obtained as KBr disks on a VECTOR 22 spectrometer. 1H NMR spectra were recorded at room temperature with a 300 or 500 MHz BRUKER spectrometer. Powder XRD patterns were recorded on a BRUKER D8 ADVANCE X-ray diffractometer. UV-vis absorption spectra were measured on a Cary 100 spectrophotometer. Luminescence spectra were measured using a Hitachi F-4600 fluorescence spectrometer. Photoirradiation was carried out using a 500 W Xe lamp and a monochromator.
Synthesis of 2-(3,5-di-tert-butyl-2-hydroxyphenyl)-4,5-bis[2,5-dimethyl(3-thienyl)]-1H-imidazole (tBuLH)
A mixture of 3,5-di-tert-butyl-2-hydroxybenzaldehyde (4 mmol, 0.9373 g), 1,2-bis[2,5-dimethyl(3-thienyl)]ethane-1,2-dione (3.5 mmol, 0.9748 g), ammonium acetate (21 mmol, 1.6180 g) and HOAc (20 mL) was refluxed for 24 hours. After evaporating HOAc under vacuum, the resultant solid was mixed with water (20 mL). The mixture was extracted with CHCl3 (20 mL × 3). The combined CHCl3 solution was dried with MgSO4, filtered, and then evaporated. The residue was refluxed in petroleum ether (25 mL) for one hour, and then the resultant white solid was filtered and dried in air. Yield: 954 mg (55% based on 1,2-bis[2,5-dimethyl(3-thienyl)]ethane-1,2-dione). Anal. found (calcd) for C29H36N2OS2: C, 70.57 (70.69); H, 7.51 (7.36), N, 5.82 (5.69). IR (KBr, cm−1): 3406(m), 3134(s), 2960(m), 1683(w), 1617(w), 1475(w), 1400(s), 1274(w), 1146(w), 1109(w), 1027(w), 824(w), 812(w), 772(w), 641(w), 601(w), 576(w), 495(w). 1H NMR (500 MHz, CDCl3), δ (ppm): 1.34 and 1.48 (18H from two tert-butyl groups), 2.06, 2.22 and 2.42 (12H from four –CH3 groups attached to two thiophene rings), 6.68 (2H from two thiophene rings), 7.22 and 7.34 (2H from phenyl group).
Synthesis of 2-(3,5-di-tert-butyl-2-hydroxyphenyl)-4,5-bis[2,5-dimethyl(3-thienyl)]-1-methyl-imidazole (tBuLMeH)
A mixture of tBuLH (1 mmol, 0.553 g) and K2CO3 (4 mmol, 0.552 g) in DMF (5 mL) was stirred at room temperature for 45 minutes. To this mixture was added the solution of CH3I (1 mmol, 62 μL) in DMF (4 mL) dropwise, and then the resultant solution was stirred for additional 20 hours. The reaction mixture was poured into water (150 mL), forming white solid. This solid was purified through flash column chromatography using petroleum ether–ethyl acetate (v/v = 100/0.2), obtaining white solid. Yield: 417 mg (82% based on tBuLH). Anal. found (calcd) for C30H38N2OS2: C, 71.21 (71.10); H, 7.75 (7.56), N, 5.65 (5.53). IR (KBr, cm−1): 3417(s), 3145(s), 2960(s), 2918(s), 2867(m), 1683–1617(br, w), 1429(m), 1399(s), 1360(s), 1297(m), 1254(w), 1243(m), 1225(w), 1204(w), 1182(w), 1144(w), 879(w), 869(w), 849(w), 837(w), 816(w), 776(w), 500(w). 1H NMR (300 MHz, CDCl3), δ (ppm): 1.35 and 1.49 (18H from two tert-butyl groups), 2.02, 2.25, 2.35 and 2.47 (12H from four –CH3 groups attached to two thiophene rings), 3.66 (3H from the –CH3 group attached to imidazole N atom), 6.50 and 6.60 (2H from two thiophene rings), 7.35 and 7.43 (2H from phenyl group).
Synthesis of [Ir(dfppy)2(tBuL)]·2CH3OH (1)
A mixture of tBuLH (0.33 mmol, 0.1626 g), [Ir(dfppy)2Cl]2 (0.15 mmol, 0.1827 g) and Na2CO3 (5 mmol, 0.5300 g) in 2-ethoxyethanol (30 mL) was heated in an oil bath (136 °C) under argon for 24 hours. After evaporation under vacuum, the resultant brown solid was mixed with water (15 mL). This mixture was extracted with CH2Cl2 (25 mL × 3). The combined CH2Cl2 solution was dried with MgSO4, filtrated, and then evaporated. The residue was purified through flash column chromatography using a petroleum ether–dichloromethane (v/v = 100/20) mixture, affording a yellow solid. A CH2Cl2–CH3OH solution of this solid was allowed to evaporate at room temperature for one week, resulting in yellow blocky crystals as a monophasic material based on the powder XRD pattern (Fig. S6†). Yield: 189 mg (56% based on [Ir(dfppy)2Cl]2). Anal. found (calcd) for C53H55F4IrN4O3S2: C, 56.51 (56.42); H, 5.03 (4.91), N, 5.11 (4.97). IR (KBr, cm−1): 3453(b, m), 3197(b, m), 2954(w), 2919(w), 2857(w), 1685(w), 1604(s), 1571(w), 1556(w), 1478(w), 1444(w), 1401(s), 1288(w), 1263(w), 1245(w), 1159(w), 1139(w), 1113(w), 843(w), 827(w), 809(w), 786(w), 756(w), 568(w), 526(w). 1H NMR (500 MHz, CDCl3), δ (ppm): 0.97 and 1.27 (18H from two tert-butyl groups), 1.55–2.27 (12H from four –CH3 groups attached to two thiophene rings), 4.95–7.16 and 7.58–9.28 (16H from two thiophene rings, one phenyl group and two dfppy− units).
Synthesis of [Ir(dfppy)2(tBuLMe)] (2)
Compound 2 was synthesized by the same method as 1, using tBuLMeH instead of tBuLH. After the purification through flash column chromatography using a petroleum ether–dichloromethane (v/v = 100/20) mixture, and subsequent crystallization in CH2Cl2–CH3OH solution, yellow solid was obtained with a yield of 170 mg (53% based on [Ir(dfppy)2Cl]2). Anal. found (calcd) for C52H49F4IrN4OS2: C, 57.81 (57.92); H, 4.74 (4.58), N, 5.10 (5.20). IR (KBr, cm−1): 2957(w), 2916(w), 2864(w), 1602(s), 1570(w), 1555(w), 1476(s), 1443(w), 1423(w), 1401(m), 1304(w), 1288(w), 1268(w), 1244(w), 986(w), 844(w), 829(w), 814(w), 787(w), 755(w), 567(w), 526(w), 498(w). 1H NMR (500 MHz, CDCl3), δ (ppm): 0.94 and 1.22 (18H from two tert-butyl groups), 1.54–2.32 (12H from four –CH3 groups attached to two thiophene rings), 3.45–3.55 (3H from the –CH3 group attached to imidazole N atom), 4.92–7.22 and 7.48–9.40 (16H from two thiophene rings, one phenyl group and two dfppy− units).
X-ray crystallographic studies
Colorless blocky single crystals of tBuLH·HOAc were obtained by evaporation of a CHCl3 solution of tBuLH containing a drop of HOAc. We failed in obtaining the single crystals of tBuLMeH and 2 through evaporating a CHCl3 solution (containing HOAc or not) for the former, and a CH2Cl2–CH3OH solution for the latter. Single crystals of dimensions 0.10 × 0.08 × 0.06 mm3 for tBuLH·HOAc, and 0.20 × 0.18 × 0.13 mm3 for 1 were used for structural determinations on a Bruker SMART APEX CCD diffractometer using graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å) at room temperature (296 K). A hemisphere of data were collected in the θ range of 1.536–24.997° for tBuLH·HOAc, and 1.818–24.999° for 1 using a narrow-frame method with scan widths of 0.30° in ω and an exposure time of 10 s per frame. Numbers of observed and unique [I > 2σ(I)] reflections are 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 294 and 5471 (Rint = 0.0299) for tBuLH·HOAc, and 13793 and 8791 (Rint = 0.0490) for 1, respectively. The data were integrated using the Siemens SAINT program,10 with the intensities corrected for Lorentz factor, polarization, air absorption, and absorption due to variation in the path length through the detector faceplate. Multi-scan absorption corrections were applied. The structures were solved by direct methods and refined on F2 by full matrix least squares using SHELXTL.11 All non-hydrogen atoms were located from the Fourier maps, and were refined anisotropically. All H atoms were put in calculated positions using a riding model, and were refined isotropically, with the isotropic vibration parameters related to the non-H atom to which they are bonded. It should be noted that tBuLH·HOAc and 1 contain slightly/seriously disordered tert-butyl groups. Thus, ISOR, DELU, SADI and SIMU were used in their structural refinements (seeing the cif files containing the information of hkl and res files). In addition, some disagreeable reflections were omitted in the structural refinement of 1. The crystallographic data for tBuLH·HOAc and 1 are listed in Table 1, and selected bond lengths are given in Table 2. CCDC 1465203 and 1465204 contain the supplementary crystallographic data for this paper.
294 and 5471 (Rint = 0.0299) for tBuLH·HOAc, and 13793 and 8791 (Rint = 0.0490) for 1, respectively. The data were integrated using the Siemens SAINT program,10 with the intensities corrected for Lorentz factor, polarization, air absorption, and absorption due to variation in the path length through the detector faceplate. Multi-scan absorption corrections were applied. The structures were solved by direct methods and refined on F2 by full matrix least squares using SHELXTL.11 All non-hydrogen atoms were located from the Fourier maps, and were refined anisotropically. All H atoms were put in calculated positions using a riding model, and were refined isotropically, with the isotropic vibration parameters related to the non-H atom to which they are bonded. It should be noted that tBuLH·HOAc and 1 contain slightly/seriously disordered tert-butyl groups. Thus, ISOR, DELU, SADI and SIMU were used in their structural refinements (seeing the cif files containing the information of hkl and res files). In addition, some disagreeable reflections were omitted in the structural refinement of 1. The crystallographic data for tBuLH·HOAc and 1 are listed in Table 1, and selected bond lengths are given in Table 2. CCDC 1465203 and 1465204 contain the supplementary crystallographic data for this paper.
Table 1 Crystallographic data and refinement for tBuLH·HOAc and 1
| |
tBuLH·HOAc |
1 |
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Formula |
C31H40N2O3S2 |
C53H55F4IrN4O3S2 |
| M |
552.77 |
1128.33 |
| Crystal system |
Monoclinic |
Triclinic |
| Space group |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| T/K |
296(2) |
296(2) |
| a/Å |
11.1823(6) |
12.9987(16) |
| b/Å |
26.5152(14) |
13.2108(16) |
| c/Å |
11.5015(6) |
17.001(2) |
| α/° |
|
69.213(2) |
| β/° |
114.4590(10) |
69.523(2) |
| γ/° |
|
73.873(2) |
| V/Å−3 |
3104.2(3) |
2517.4(5) |
| Z |
4 |
2 |
| Dc/g cm−3 |
1.183 |
1.489 |
| F(000) |
1184 |
1140 |
| GooF on F2 |
1.027 |
1.025 |
| R1, wR2 [I > 2σ(I)]a |
0.0597, 0.2039 |
0.0461, 0.0832 |
| R1, wR2 (all data)a |
0.0780, 0.2209 |
0.0747, 0.0894 |
| (Δρ)max, (Δρ)min (e Å−3) |
0.450, −0.477 |
1.265, −1.311 |
Table 2 Selected bond lengths (Å) for tBuLH·HOAc and 1
| tBuLH·HOAc |
| N1–C15 |
1.328(3) |
O1–C1 |
1.360(3) |
| N2–C15 |
1.347(3) |
C30–O2 |
1.219(4) |
| C30–O3 |
1.350(5) |
|
|
|
| Compound 1 |
| Ir1–C22 |
1.986(7) |
Ir1–O1 |
2.144(4) |
| Ir1–C11 |
1.996(6) |
Ir1–N1 |
2.167(5) |
| Ir1–N4 |
2.006(5) |
N1–C37 |
1.329(7) |
| Ir1–N3 |
2.046(5) |
N2–C37 |
1.353(7) |
| C23–O1 |
1.320(7) |
|
|
Results and discussion
Crystal structures of tBuLH·HOAc and 1
Compound tBuLH·HOAc crystallizes in the monoclinic space group P21/n. Its asymmetric unit contains one tBuLH molecule and one HOAc molecule (Fig. 1), and they connect each other through hydrogen bond N2–H⋯O2 = 2.847(1) Å. In the tBuLH molecule, an intra-molecular hydrogen bond O1–H⋯N1 = 2.563(1) Å is formed between phenol and imidazole moieties. The dihedral angle between the imidazolyl and phenyl rings is 7.1(1)°. Two thiophene groups of tBuLH adopt photoactive antiparallel conformation. Neighboring tBuLH molecules interact only through van der Waals interactions probably because of the bulky –C(CH3)3 groups in tBuLH (Fig. S7†).
 |
| | Fig. 1 Molecular structure of tBuLH·HOAcH. All H atoms attached to carbon atoms are omitted for clarity. | |
Compound 1 crystallizes in the triclinic space group P. In its molecular structure (Fig. 2), a tBuL− ligand uses its phenolate oxygen O1 and imidazole nitrogen N1 to chelate an {Ir(dfppy)2}+ unit. Around Ir(III) ion, two cyclometallated dfppy− ligands adopt the C,C-cis and N,N-trans configurations as those in [Ir(dfppy)2Cl]2.12 The O1 and N1 atoms in the tBuL− ligand are respectively trans to corresponding σ-bound C22 and C11 atoms in the dfppy− ligands. Therefore, the Ir–N(O)tBuL bond distances [2.167(5) and 2.144(4) Å, respectively] are significantly longer than the Ir–C(N)dfppy distances [1.986(7)–2.046(5) Å].13 In compound 1, the tBuL− ligand adopts a non-photoactive parallel conformation, which is different from the photoactive antiparallel conformation in tBuLH·HOAc. The dihedral angle between the imidazolyl and phenyl rings in 1 [35.3(1)°] is significantly larger than that in tBuLH·HOAc [7.1(1)°]. This could be assigned to the coordination of the phenol–imidazole moiety to Ir(III) ion, with this twist necessary to form a six-membered chelate ring with sensible bond distances.
 |
| | Fig. 2 Molecular structure of 1. Two lattice CH3OH molecules and all H atoms attached to carbon atoms are omitted for clarity. | |
Compound 1 shows an intra-molecular aromatic stacking interaction between a thiophene ring of tBuL− ligand and a phenyl ring from a dfppy− ligand [distance between C17 and the center of thiophene ring: 3.388(1) Å] (Fig. 2).14 Each [Ir(dfppy)2(tBuL)] molecule connects to two lattice CH3OH molecules through hydrogen bonds N2–H⋯O2 = 2.785(1) Å and O2–H⋯O3 = 2.723(1) Å (Fig. 3). Neighboring [Ir(dfppy)2(tBuL)] molecules are held together by van der Waals interactions. Clearly, the packing structure of 1 is greatly different from that of [Ir(dfppy)2(L1)]·2CH3OH (Fig. S8†).7a In the latter, neighboring molecules are linked through two CH3OH molecules into right- or left-handed 21 helical chains. The different packing structure between 1 and [Ir(dfppy)2(L1)]·2CH3OH should be due to their different auxiliary ligand (tBuL− and L1−, respectively). In other words, the presence of two –C(CH3)3 groups in tBuL− ligand could significantly influence the molecular stacking mode of 1.
 |
| | Fig. 3 Packing structure of 1. Big red balls are O atoms from CH3OH molecules. | |
Electronic absorption spectra
The UV-vis absorption spectra of tBuLH, tBuLMeH, 1 and 2 were measured in CH2Cl2 at room temperature (Fig. 4, Table S1†). Compounds tBuLH and tBuLMeH show similar absorption bands, including one strong absorption peak (228 nm and 230 nm, respectively) and the other two moderate broad bands (around 291 nm and 326 nm for each case). Compounds 1 and 2 reveal a broad high-energy absorption band with maxima at 248 nm and 245 nm, respectively, which can be assigned to spin-allowed ligand-centered (1LC) transitions (dfppy−, tBuL− and tBuLMe− ligands). The lower-energy absorption bands around 379 nm in 1 and 346 nm in 2 most likely have spin-allowed metal-to-ligand charge transfer (1MLCT) and possibly ligand-centered (1LC) character, because of the high extinction coefficient (ε = 0.59 × 104 M−1 cm−1 for 1 and ε = 0.82 × 104 M−1 cm−1 for 2).7a,13,15 The weaker bands around 450 nm for 1 and 2 are mainly attributed to 3MLCT and possible 3LC absorptions.16
 |
| | Fig. 4 UV-vis absorption spectra of tBuLH, tBuLMeH, 1 and 2 in CH2Cl2. | |
Photochromism
Compound tBuLH shows photochromic behavior in CH2Cl2 solution at room temperature, which is confirmed by its UV-vis absorption spectra and 1H NMR spectra. Under irradiation with 326 nm light, two original strong absorption bands at 227 and 326 nm gradually became weak, and two other new broad absorption bands were formed, around 473 nm and 700 nm (Fig. 5). After this irradiation for 2.5 minutes, the CH2Cl2 solution of tBuLH reached a photostationary state (PSS), i.e. a structure–transition equilibrium between the open form and the closed form (Scheme S5† top).17a,b These new absorption bands at 473 and 700 nm should be from the closed-form tBuLH with a greater degree of π-electron conjugation compared with its open form (Scheme S5† top). The electrospray mass spectrometry (ES-MS) measurement excludes the possible decomposition of tBuLH after 2.5 minute irradiation (Fig. S9 and S10†). In addition, these new absorption bands could gradually disappear when the solution was subsequently irradiated with 700 nm light or allowed to stay in the dark at room temperature (Fig. S11†), indicating that the photochromic behavior of tBuLH in CH2Cl2 is reversible. Before and after irradiation with 326 nm light, the 1H NMR spectra of tBuLH show clear differences in the signals of the four methyl groups attached to two thiophene groups and two H atoms belonging to the phenyl group (Fig. 6, S1 and S12†). The signals of the four methyl groups appear as three peaks at 2.06, 2.22 and 2.42 ppm before UV irradiation, but are converted to two peaks at 2.09 and 2.39 ppm after UV irradiation. Two H atoms from phenyl group appear as separate signals at 7.22 and 7.34 ppm before UV irradiation, but appear as a broad peak at 7.39 ppm after UV irradiation. Similar photoisomerisation-induced 1H NMR changes were also observed in the other BTE compounds.7a,8b,17c In addition, the CDCl3 solution of tBuLH changed its color from colorless to yellowish-brown after UV irradiation (Fig. 6, inset).
 |
| | Fig. 5 UV-vis absorption-spectra changes of tBuLH in CH2Cl2 (c = 1.0 × 10−5 M) upon UV irradiation (λ = 326 nm) for 0–2.5 minutes. | |
 |
| | Fig. 6 1H NMR spectra of tBuLH before and after UV irradiation (λ = 326 nm), in which the peaks with * indicate partial conversion from the open form to the closed form. | |
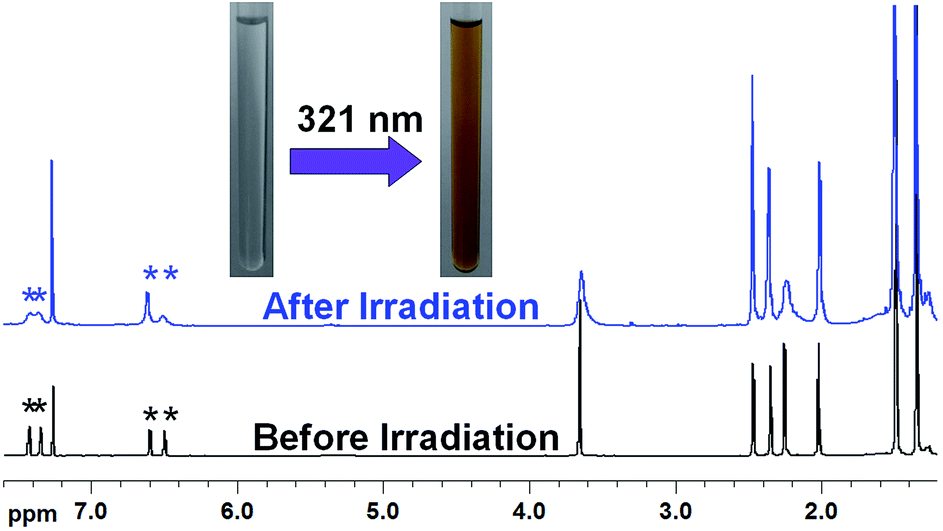
Compared to tBuLH, compound tBuLMeH shows weaker photochromism ability in CH2Cl2, which could be assigned to the influence of the –CH3 group attached to the imidazole nitrogen atom in tBuLMeH (Scheme 2). As shown in Fig. 7, a new weak broad around 570 nm was gradually formed upon irradiation with 321 nm light, and a photostationary state (PSS) was observed after two-minute irradiation. This new absorption band gradually disappeared when the solution was subsequently irradiated with 570 nm light or allowed to stay in the dark at room temperature (Fig. S13†). These changes in UV-vis absorption spectra indicate that tBuLMeH can occur reversible structural transition between the open form and the closed form (Scheme S5† bottom). The photochromic behavior of tBuLMeH was further confirmed by its 1H NMR spectra before and after irradiation with 321 nm light (Fig. 8, S2 and S14†). The peaks of H atoms from two thiophene rings (at 6.50 and 6.60 ppm, respectively) show a peak-area ratio 1.03:1.04 before irradiation, while 0.77:1.33 after irradiation. The signals of two H atoms from the phenyl group in tBuLMeH appear as two well separated peaks at 7.35 and 7.41 ppm before irradiation, but are converted to two overlapping peaks at 7.35 and 7.43 ppm after irradiation. In addition, the CDCl3 solution of tBuLMeH revealed a color change from colorless to yellowish-brown after UV irradiation (Fig. 8, inset). This color change is similar to that of tBuLH upon UV irradiation (Fig. 6, inset).
 |
| | Fig. 7 UV-vis absorption-spectra changes of tBuLMeH in CH2Cl2 (c = 1.0 × 10−5 M) upon UV irradiation (λ = 321 nm) for 0–2 minutes. | |
 |
| | Fig. 8 1H NMR spectra of tBuLH before and after UV irradiation (λ = 321 nm), in which the peaks with * indicate partial conversion from the open form to the closed form. | |
The fatigue resistance properties of both tBuLH and tBuLMeH were measured in CH2Cl2 at room temperature, alternately using irradiation lights with 326 and 700 nm for the former, and 321 and 570 nm for the latter. Fig. S15 and S16† show the UV-vis absorbance changes at 700 nm for tBuLH, and at 570 nm for tBuLMeH in five cycles. After 1–2 cycles, the absorbance of two compounds (at 700 and 570 nm, respectively) gradually declined, and the similar behaviors were also observed in several BTE compounds.18 This indicates that both tBuLH and tBuLMeH are poor in their fatigue resistance.
The photochromic behaviors of both tBuLH and tBuLMeH are clearly different from that of the previously-reported L1H,8a due to their different molecular structures (Scheme 2). Upon irradiation with 321 nm light, the latter shows a color change from colorless to pink (Fig. S17†). The photochromism comparisons among tBuLH, tBuLMeH and L1H indicate that the variation of substituent groups [H, –C(CH3)3, –CH3] attached to the phenyl group and/or the imidazole unit in three compounds can modify their photochromic properties. Moreover, no photochromic behavior was observed for the CH2Cl2 solutions of 1 and 2 upon UV irradiation with 320–380 nm light. This is in agreement with our reported result that [Ir(dfppy)2(L1)]·2CH3OH has no photochromic behavior although both L1H and its complex Co(L1)2·3CH3OH show photochromism.7a,8a The absence of photochromism in 1 and 2 could be mainly due to the following two factors. The first is that the UV excitation energy is transferred to the {Ir(dfppy)2}+ unit, preventing any photocyclization process of tBuL−/tBuLMe− ligand.7a The second is that the coordination of ligand tBuL−/tBuLMe− to {Ir(dfppy)2}+ unit results in a crowded space which hinders tBuL−/tBuLMe− ligand from occurring photocyclization reaction.17c
Luminescence properties
The luminescence spectra of tBuLH, tBuLMeH, 1 and 2 were measured in CH2Cl2 solution at room temperature, using excitation light at 340 nm for tBuLH and tBuLMeH, and 370 nm for 1 and 2 (Fig. 9, Table S2†). Luminescence from tBuLH and tBuLMeH occur at 464 nm and 479 nm, respectively, at longer wavelength than that of L1H (442 nm) due to the electron-donating nature of the substituent groups –C(CH3)3 and –CH3 in tBuLH and tBuLMeH. Compared to tBuLH and tBuLMeH, compounds 1 and 2 exhibit significantly red-shifted emission, with a broad band at 514 nm and 507 nm, respectively. Therefore, compounds tBuLH, tBuLMeH, 1 and 2 reveal different emission colors upon irradiation with 365 nm light, cyan for tBuLH, cornflower blue for tBuLMeH, lime green for 1, and medium spring green for 2 (Fig. 10).19 We further measured the luminescence spectra of 1 and 2 at 77 K (Fig. S18†). Both cases show the emissions that is blue-shifted with respect to their emissions at room temperature, occurring at 498 nm for 1 and 492 nm for 2. This slight rigidchromism indicates a mixing of 3MLCT and 3LC character for the luminescence of 1 and 2.20 It should be noted that the blue-shifted Δλ = 16 nm for 1 and 15 nm for 2 are clearly smaller than that of [Ir(dfppy)2(L1)]·2CH3OH (blue-shift Δλ = 37 nm) (these Δλ values were calculated based the data in Table S2†). This suggests that the luminescence of 1 and 2 has relatively less 3MLCT character than that of [Ir(dfppy)2(L1)]·2CH3OH. Both the different fluorescence emissions among tBuLH, tBuLMeH and L1H, and the different phosphorescence emissions among 1, 2 and [Ir(dfppy)2(L1)]·2CH3OH, indicate that the substituent groups –C(CH3)3 and –CH3 in tBuLH, tBuLMeH, 1 and 2 could significantly influence their luminescence behaviors.
 |
| | Fig. 9 Luminescence spectra of tBuLH, tBuLMeH, 1 and 2 in CH2Cl2 (c = 4 × 10−5 M). | |
 |
| | Fig. 10 Under room light and 365 nm light, the photographs of the CH2Cl2 solutions of tBuLH (a and a′), tBuLMeH (b and b′), 1 (c and c′), and 2 (d and d′). | |
Considering the photochromic behaviors of both tBuLH and tBuLMeH in solution, we further studied the influence of UV irradiation on their luminescence at room temperature. As shown in Fig. S20,† their emission intensities significantly decreased after three-minute UV irradiation with 326 nm light for tBuLH and 321 nm for tBuLMeH. This could be due to the UV-irradiation-induced structural transition of tBuLH and tBuLMeH from the open form to the closed form.21 As shown in Fig. 5 and 7, after UV irradiation, the closed-form species of tBuLH and tBuLMeH exhibit broad absorption bands at 350–510 nm, and 400–650 nm, respectively. These absorption bands completely overlap with the emission band at 464 nm for tBuLH and 479 nm for tBuLMeH, respectively, resulting in the decrease in emission intensity. In additionally, the emission-intensity decrease of tBuLH (about 44%) is more significant than that of tBuLMeH (25%). This is in agreement with the fact that tBuLMeH shows weaker photochromism ability than tBuLH (seeing the part of photochromism).
Conclusions
In this paper, we have reported the bisthienylethenes tBuLH and tBuLMeH which contain the substituent groups –C(CH3)3 and/or –CH3, and their complexes [Ir(dfppy)2(tBuL)]·2CH3OH (1) and Ir(dfppy)2(tBuLMe)] (2). The crystal structures of tBuLH·HOAc and 1 were determined. Compared to the packing structure of [Ir(dfppy)2(L1)]·2CH3OH,7a neighboring [Ir(dfppy)2(tBuL)] molecules in 1 have not been connected by lattice CH3OH molecules through hydrogen bonds, which could be due to the two bulky –C(CH3)3 groups on the tBuL− ligand. Both tBuLH and tBuLMeH reveal photochromic behaviors in CH2Cl2, with a color change from colorless to yellowish-brown upon UV irradiation with 326 nm/321 nm light. However, the photochromism ability of the latter is relatively weaker than that of the former due to the different molecular structures between tBuLH and tBuLMeH. The absence of photochromism in both 1 and 2 indicates that the coordination of tBuL−/tBuLMe− ligand to {Ir(dfppy)2}+ unit could suppress its photochromic behavior, mainly due to the energy transfer of the UV excitation energy to the {Ir-(dfppy)2}+ unit, and/or because of insufficient space to allow photocyclization of the tBuL−/tBuLMe− ligand to take place. At room temperature, the fluorescence emissions of tBuLH (464 nm) and tBuLMeH (479 nm) in CH2Cl2 occur at longer wavelengths than that of L1H (442 nm). After photochromism reaction, both tBuLH and tBuLMeH show the decrease in emission intensity. The phosphorescence emissions of 1 (514 nm) and 2 (507 nm) have mixed 3MLCT and 3LC character. It should be noted that the luminescence of 1 and 2 have relatively less 3MLCT character than that of [Ir(dfppy)2(L1)]·2CH3OH, which was confirmed by their reduced rigidchromism (the blue-shifted Δλ = 16, 15, and 37 nm, respectively). Our experimental results demonstrate that the substituent groups –C(CH3)3 and/or –CH3 in tBuLH, tBuLMeH, 1 and 2 could significantly modify the structures, photochromism and luminescence of the complexes.
Acknowledgements
We acknowledge financial support from the NSF of Jiangsu Province, China (BK 20141314), and the NSF of China–Royal Society of UK International Exchanges Scheme–Cost Share Program (no. 21411130119).
References
-
(a) V. Guerchais, L. Ordronneau and H. Le Bozec, Coord. Chem. Rev., 2010, 254, 2533 CrossRef CAS;
(b) H. L. Bozec and V. Guerchais, C. R. Chim., 2013, 16, 1172 CrossRef.
-
(a) I. Lee, Y. You, S. J. Lim and S. Y. Park, Chem. Lett., 2007, 36, 888 CrossRef CAS;
(b) V. Aubert, L. Ordronneau, M. Escadeillas, J. A. G. Williams, A. Boucekkine, E. Coulaud, C. Dragonetti, S. Righetto, D. Roberto, R. Ugo, A. Valore, A. Singh, J. Zyss, I. Ledoux-Rak, H. L. Bozec and V. Guerchais, Inorg. Chem., 2011, 50, 5027 CrossRef CAS PubMed;
(c) S. Monaco, M. Semeraro, W. Tan, H. Tian, P. Ceroni and A. Credi, Chem. Commun., 2012, 48, 8652 RSC.
- J. Boixel, V. Guerchais, H. L. Bozec, D. Jacquemin, A. Amar, A. Boucekkine, A. Colombo, C. Dragonetti, D. Marinotto, D. Roberto, S. Righetto and R. D. Angelis, J. Am. Chem. Soc., 2014, 136, 5367 CrossRef CAS PubMed.
-
(a) M. Morimoto, H. Miyasaka, M. Yamashita and M. Irie, J. Am. Chem. Soc., 2009, 131, 9823 CrossRef CAS PubMed;
(b) T. Shiga, H. Miyasaka, M. Yamashita, M. Morimotob and M. Irie, Dalton Trans., 2011, 40, 2275 RSC;
(c) M. Milek, F. W. Heinemann and M. M. Khusniyarov, Inorg. Chem., 2013, 52, 11585 CrossRef CAS PubMed.
-
(a) M. M. Krayushkin, S. N. Ivanov, A. Yu. Martynkin, B. V. Lichitsky, A. A. Dudinov and B. M. Uzhinov, Russ. Chem. Bull., 2001, 50, 116 CrossRef CAS;
(b) H.-H. Liu and Y. Chen, J. Phys. Chem. A, 2009, 113, 5550 CrossRef CAS PubMed;
(c) Z. Li, J. Xia, J. Liang, J. Yuan, G. Jin, J. Yin, G.-A. Yu and S. H. Liu, Dyes Pigm., 2011, 90, 290 CrossRef CAS;
(d) H.-H. Liu and Y. Chen, J. Phys. Org. Chem., 2012, 25, 142 CrossRef CAS;
(e) H.-H. Liu and Y. Chen, New J. Chem., 2012, 36, 2223 RSC;
(f) H.-H. Liu and Y. Chen, Tetrahedron, 2013, 69, 1872 CrossRef CAS;
(g) J.-Q. Zhang, Q.-C. Wang, L. Zou and C.-Y. Jia, Chin. Chem. Lett., 2014, 25, 762 CrossRef CAS.
-
(a) P. H.-M. Lee, C.-C. Ko, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 6058 CrossRef CAS PubMed;
(b) H.-L. Wong, N. Zhu and V. W.-W. Yam, J. Organomet. Chem., 2014, 751, 430 CrossRef CAS.
-
(a) D.-K. Cao, R.-H. Wei, X.-X. Li, J.-F. Chen and M. D. Ward, Dalton Trans., 2015, 44, 4289 RSC;
(b) R.-H. Wei, J.-F. Chen, J.-Q. Feng, J.-S. Hu and D.-K. Cao, RSC Adv., 2015, 5, 14359 RSC.
-
(a) D.-K. Cao, J.-Q. Feng, M. Ren, Y.-W. Gu, Y. Song and M. D. Ward, Chem. Commun., 2013, 49, 8863 RSC;
(b) D.-K. Cao, R.-H. Wei, X.-X. Li and Y.-W. Gu, Dalton Trans., 2015, 44, 5755 RSC.
- S. Kappaun, S. Eder, S. Sax, K. Mereiter, E. J. W. List and C. Slugovc, Eur. J. Inorg. Chem., 2007, 4207 CrossRef CAS.
- SAINT, Program for Data Extraction and Reduction, Siemens Analytical X-ray Instruments, Madison, WI, 1994–1996 Search PubMed.
-
(a) SHELXTL, Reference Manual, version 5.0, Siemens Indus-trial Automation, Analytical Instruments, Madison, WI, 1997 Search PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- E. S. Andreiadis, D. Imbert, J. Pecaut, A. Calborean, I. Ciofini, C. Adamo, R. Demadrille and M. Mazzanti, Inorg. Chem., 2011, 50, 8197 CrossRef CAS PubMed.
- E. Marchi, R. Sinisi, G. Bergamini, M. Tragni, M. Monari, M. Bandini and P. Ceroni, Chem.–Eur. J., 2012, 18, 8765 CrossRef CAS PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- T. Hofbeck and H. Yersin, Inorg. Chem., 2010, 49, 9290 CrossRef CAS PubMed.
-
(a) M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. D. Cola, Adv. Funct. Mater., 2010, 20, 1812 CrossRef CAS;
(b) Y. You, S. Lee, T. Kim, K. Ohkubo, W.-S. Chae, S. Fukuzumi, G.-J. Jhon, W. Nam and S. J. Lippard, J. Am. Chem. Soc., 2011, 133, 18328 CrossRef CAS PubMed.
-
(a) R. Wang, P. Ren, S. Pu, G. Liu and S. Cui, J. Photochem. Photobiol., A, 2014, 294, 44 CrossRef CAS;
(b) G.-T. Xu, B. Li, J.-Y. Wang, D.-B. Zhang and Z.-N. Chen, Chem.–Eur. J., 2015, 21, 3318 CrossRef CAS PubMed;
(c) D.-K. Cao, J.-S. Hu, M.-Q. Li, D.-P. Gong, X.-X. Li and M. D. Ward, Dalton Trans., 2015, 44, 21008 RSC.
- Q. Luo, M. Liu, R. Cheng and Y. Liu, Dyes Pigm., 2015, 113, 602 CrossRef CAS.
- The various colors and their names can be found via http://www.wenku.baidu.com/link?url=lsGFDZd2y14AMB6Bo9x2ZnOH4ZFtorsELAMXNNZtdFLcROu9wJvNB70klYe_EjEJKilV3bPm8MconP0wVBh84aK_cPzMZq7C6euZZX_O0nW.
-
(a) A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS PubMed;
(b) F. Kessler, R. D. Costa, D. D. Censo, R. Scopelliti, E. Orti, H. J. Bolink, S. Meier, W. Sarfert, M. Gratzel, M. K. Nazeeruddin and E. Baranoff, Dalton Trans., 2012, 41, 180 RSC.
- D.-P. Gong, J.-F. Chen, Y. Zhao and D.-K. Cao, Dalton Trans., 2016, 45, 3443 RSC , and references therein.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format, UV-vis and luminescence date, some schemes of compounds, figures including 1H NMR spectra, XRD patterns, structures, UV-vis spectra and luminescence spectra. CCDC 1465203 and 1465204. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra14555h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.