
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Field flow fractionation online with ICP-MS as novel approach for the quantification of fine particulate carbon in stream water samples and soil extracts†
Volker
Nischwitz
*a,
Nina
Gottselig
b,
Anna
Missong
b,
Thomas
Meyn
c and
Erwin
Klumpp
b
aCentral Institute for Engineering, Electronics and Analytics, Analytics (ZEA-3), Forschungszentrum Juelich, 52425 Juelich, Germany. E-mail: v.nischwitz@fz-juelich.de
bInstitute of Bio- and Geosciences, Agrosphere (IBG-3), Forschungszentrum Juelich, Juelich, Germany
cDepartment of Hydraulic and Environmental Engineering, Norwegian University of Science and Technology, Trondheim, Norway
First published on 3rd May 2016
Abstract
Reliable and efficient analytical techniques are required for quantitative size-resolved carbon determination of nanoparticles and colloids in complex sample matrices due to the key role of carbon in biological and environmental processes. Field flow fractionation (FFF) online with inductively coupled plasma mass spectrometry (ICP-MS) is a powerful technique for identification and quantification of particle bound metals, but has not been applied for quantitative determination of particulate carbon, yet, due to several challenges. Therefore, our study explores the potential of online particulate carbon detection by ICP-MS to overcome limitations of previously used UV detection or offline total organic carbon measurements. A novel organic carbon detector (OCD) was used as independent sensitive carbon detector to validate the ICP-MS results. Basic validation of organic carbon detection by offline quadrupole and sector-field ICP-MS was performed for fresh water samples using OCD as reference achieving recoveries of 107 ± 16% with Q-ICP-MS and 122 ± 22% with SF-ICP-MS. Limits of detection were 0.6 mg L−1 for Q-ICP-MS, 0.3 mg L−1 for SF-ICP-MS and 0.04 mg L−1 for OCD. The main focus was on comparison of FFF-ICP-MS and FFF-OCD for quantification of particulate carbon in fresh water samples, soil extracts as well as in bovine serum albumin (BSA) as candidate reference standard. Recoveries obtained by FFF-Q-ICP-MS with a flow-injection calibration approach were in a range from 90 to 113% for replicate analyses of fresh water samples compared to FFF-OCD and from 87 to 107% with an alternative post-channel calibration strategy.
Introduction
Particles in the nanometer and micrometer size range are ubiquitously present in environmental fresh water and soil systems. The far majority of these particles are generated by natural processes including metabolism and decay of organic matter as well as weathering of minerals and rocks. Therefore, these particles can be considered as natural nanoparticles and colloids representing a wide range of composition from mainly organic macromolecules (e.g. humic acids) to mainly inorganic particles (e.g. silicates, Fe–Mn-oxides, clay, Al-oxides) potentially with attached organic coatings. In addition synthetic (i.e. engineered) nanoparticles from consumer products are released into the environment in increasing amounts (e.g. TiO2 from sunscreen) and thus may also be present in water or soil samples. Regarding carbon, also soot particles from natural or anthropogenic origin, for example vehicle exhaust, are widely distributed in the environment. In order to understand the role of these complex mixtures of particles in environmental processes and to investigate changes due to anthropogenic influences and variation in climate detailed structural characterisation and quantification is required. Many studies have demonstrated that field flow fractionation (FFF) online with inductively coupled plasma mass spectrometry (ICP-MS) is a powerful analytical technique for the size-based elemental speciation or fractionation, respectively, both of natural and engineered nanoparticles.1–3Main focus of our recent studies was on the investigation of the role of fine colloids including natural nanoparticles in the transport of phosphorus as essential nutrient in environmental water and soil systems. According to the functional definition of Gustafsson and Gschwend the lower size range of colloids is 1–2 nm and the upper size range is around 1 μm.4 Fine colloids refer to the lower part of this range from 1 nm to about 500 nm. FFF has been optimised for size-fractionation of these naturally occurring particles and online detection with ICP-MS enabled monitoring and quantification of particle-bound phosphorus as well as the major matrix elements present in the particles (Al, Fe, Mn, Si).2 However, in addition carbon is a key element to distinguish between inorganic and organic particles as well as to investigate the extent of carbon fixation and storage having major influence on the cycling of climate relevant carbon dioxide.5,6 Consequently, there is a need for reliable carbon detection in parallel to the already existing elemental detection.
Initially, three options were considered for particulate carbon detection. First, fraction collection and carbon detection offline by classical total organic carbon (TOC) analyser was considered. Major drawbacks are the need for replicate particle separation (e.g. by FFF) to collect fractions and low peak resolution causing low efficiency. Second, previous studies have reported the use of UV detection for approximate carbon determination.7 This approach has been investigated in our former work,2 however the complex composition of the particulate carbon covering a large variation in size and chemical structure is likely to cause major changes in the UV response and thus correlation between carbon content and UV absorption is questionable and likely to suffer from significant systematic error. Third, an organic carbon detector (OCD) is commercially available since 1998 as online detector for liquid flow based separation systems but has found limited application up to now for FFF. The operation principle includes the acidification of the sample flow to remove inorganic carbon, followed by oxidation of the organic carbon in a thin film reactor to carbon dioxide which is subsequently quantified by infrared detection. Advantages are the high selectivity and low limits of detection (approximately 1–50 μg L−1).8 Therefore, the FFF online hyphenation with OCD is a promising technique for monitoring particle bound carbon.
In summary, having FFF-ICP-MS for monitoring of metals including phosphorus and FFF-OCD for monitoring of carbon, the challenge arises how to realise monitoring of all these parameters (quasi)-simultaneously for the same FFF run. The only feasible way seems to be flow splitting after the separation channel with appropriate flow rates going to ICP-MS and to OCD in parallel. This requires apart from the high instrumental cost careful optimisation of injection volume and split ratio resulting in quite a complex system. More specifically, particle bound phosphorus concentration in non-contaminated stream water samples are in the low μg L−1 range and therefore require high injection volume (5 mL) for adequate detection by FFF-ICP-MS. However particle bound carbon concentration is typically in the mg L−1 range and exceeding the calibration range (<5 mg L−1) of OCD when using the same high injection volume.
So why not doing this in a more simple way? In theory, ICP-MS has the potential to monitor also carbon in addition to metals and phosphorus, which would result in a much simpler and cheaper experimental setup for (quasi-) simultaneous monitoring of all relevant elements online with FFF. The use of ICP-MS for carbon monitoring has been reported by Vogl and Heumann for chromatographic fractions of humic substances using isotope dilution technique for quantification.9 This initial study demonstrated species-independent ionisation of carbon for three compounds with a molecular weight range from 198 Da to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da. Luong and Houk applied a modified dual detector ICP-MS for carbon isotope ratio measurements in aqueous solutions of amino acids, proteins and oligosaccharides.10 Application of ICP-MS for carbon isotope ratio measurement was reviewed by Santamaria-Fernandez.11 Smith et al. investigated the feasibility of using liquid chromatography online with ICP-MS for detection of low molecular weight organic compounds via the carbon signal.12 The results indicated that the carbon signal from ICP-MS is proportional to the carbon content of the investigated compounds and thus quantification is possible via external calibration. In spite of the promising results from these initial studies, organic carbon monitoring by ICP-MS has been rarely applied. Recently, ICP-tandem mass spectrometry was employed for carbon determination in plant digests, amino acids and peptides monitoring 12C+ and 12C16O+ achieving limits of detection of 0.42 mg L−1 and 0.17 mg L−1, respectively.13 Stolpe et al. monitored carbon during FFF-ICP-MS runs of natural water samples but results are given only as intensities without quantification.14 Quantitative determination of particulate carbon in natural or engineered nanoparticles and colloids by FFF-ICP-MS has not been reported.
000 Da. Luong and Houk applied a modified dual detector ICP-MS for carbon isotope ratio measurements in aqueous solutions of amino acids, proteins and oligosaccharides.10 Application of ICP-MS for carbon isotope ratio measurement was reviewed by Santamaria-Fernandez.11 Smith et al. investigated the feasibility of using liquid chromatography online with ICP-MS for detection of low molecular weight organic compounds via the carbon signal.12 The results indicated that the carbon signal from ICP-MS is proportional to the carbon content of the investigated compounds and thus quantification is possible via external calibration. In spite of the promising results from these initial studies, organic carbon monitoring by ICP-MS has been rarely applied. Recently, ICP-tandem mass spectrometry was employed for carbon determination in plant digests, amino acids and peptides monitoring 12C+ and 12C16O+ achieving limits of detection of 0.42 mg L−1 and 0.17 mg L−1, respectively.13 Stolpe et al. monitored carbon during FFF-ICP-MS runs of natural water samples but results are given only as intensities without quantification.14 Quantitative determination of particulate carbon in natural or engineered nanoparticles and colloids by FFF-ICP-MS has not been reported.
The aim of this study in the framework of the TERENO project (Terrestrial Environmental Observatories) was to investigate the potential of our ICP-MS instrumentation for carbon monitoring using external calibration, avoiding the effort for isotope dilution, to establish a routine FFF-ICP-MS method for (quasi-) simultaneous quantification of particle-bound Al, Si, Fe, Mn, P and C. Multi-element detection using FFF-ICP-MS has been reported before and therefore this study focuses only on carbon detection using the same instrumental setup.2 The other elements were monitored in parallel to carbon with ICP-MS but results are not presented in detail in this manuscript. The performance both of quadrupole (Q) and sector-field (SF) ICP-MS was first compared for total organic carbon quantification in standard solutions and stream water samples using classical TOC and OCD as reference. Second, FFF-Q-ICP-MS was applied for particulate carbon quantification using two independent calibration strategies and results were compared to those obtained by FFF-OCD for the same samples.
Experimental
Chemicals and calibration standards
Citric acid was obtained from Merck, Germany, humic acid sodium salt and bovine serum albumin (>98%) from Sigma-Aldrich, Germany and sodium chloride, hydrochloric acid (Suprapur), nitric acid (Suprapur) from VWR, Germany. TOC standard (Certipur, potassium hydrogenphthalate in water, 1 g L−1 as carbon) and single element standards of Al, Si, Mn, Fe and P were purchased from VWR, Germany. Beryllium solution was prepared by dissolution of beryllium flakes (specpure, Johnson Matthey Chemicals Ltd., UK) in nitric acid (Suprapur, subboiled in house). Deionised water (18 MΩ cm−1) was prepared using a Millipore water purification system.Preparation of model solutions and samples
Three replicate citric acid solutions at a level of approximately 1 g L−1 as carbon were prepared by dissolution of the required amount of citric acid in deionised water. In analogy three replicate solutions of humic acid were prepared at an approximate concentration of 300 mg L−1 as carbon. Three replicate solutions of bovine serum albumin (BSA) were prepared in deionised water at an approximate concentration of 1 g L−1 as carbon. Stream water samples were obtained from sampling sites in the Eifel area (samples W4, W5 and B from TERENO research site Wüstebach; North Rhine Westphalia, Germany), in Sankt Englmar (samples W1, W2, W3, A and C from SPP1685 research site Mitterfels; Bavaria, Germany), next to Bad Brückenau (sample D from SPP1685 research site; Bavaria, Germany) and in Lettosuo, Finnland (sample 1, 2, 3). These environmental samples were filtered through 5 μm cellulose nitrate syringe filter prior to analysis. The syringe filters were first purged with an aliquot of the sample prior to filtration of the sample aliquot used for analysis in order to minimise contamination by potential release of carbon containing compounds from the filter membrane. Forest soil samples (BB8, BB9, BB10) were collected in Bad Brückenau (see above). Water dispersible colloids were extracted by suspension of the soil in deionised water (ratio 1:2, 6 h on horizontal shaker) followed by dilution with water, sedimentation and finally centrifugation at 4000g for 5 minutes to obtain the particle size fraction <500 nm in the supernatant.15
Determination of total organic carbon
FFF-ICP-MS analysis of bovine serum albumin solutions (at 10 fold dilution) used the same carrier and membrane as described above with an injection volume of 500 μL and 7 minutes focus time at a cross flow of 1 mL min−1. The cross flow was kept constant for 30 min and then reduced to 0 mL min−1 for channel purging. In addition BSA solutions were analysed without cross flow passing through the separation channel and also bypassing the separation channel in order to investigate the recovery of BSA.
Two alternative calibration procedures were applied for FFF-ICP-MS: (a) flow injection (FI) calibration using the FFF autosampler to inject standards containing the element(s) of interest in the carrier solution. The FFF channel was purged at constant tip flow without cross flow. Peak areas were determined by integration in excel and quantification of particle-bound elemental concentrations in the samples was performed via linear regression (b) post channel (PC) addition of calibration standards containing the element(s) of interest and Rh as an internal standard in 0.5% HCl via a Tee in the transfer line between FFF channel outlet and ICP-MS nebuliser. This calibration strategy has been previously applied for FFF-ICP-MS quantification of nanoparticles.3 During sample runs blank 0.5% HCl containing Rh as internal standard is added via the Tee; calibration is done in a separate run by replacing the post-channel diluting acid by calibration standards with increasing concentration of C and the target metals while purging the FFF channel with the carrier at the same detector flow without sample injection. Based on the obtained calibration the sample fractograms were converted to mass flow fractograms and the eluted absolute particle-bound metal content determined via peak integration followed by calculation of the metal concentration using the injection volume. Calibration for FFF-OCD was performed by the flow injection approach only.
Results and discussion
Challenges for carbon determination by ICP-MS
In spite of carbon being one of the key elements in living organisms and environmental processes including the contribution of carbon dioxide to climate change the potential of ICP-MS for carbon determination has hardly been investigated and utilised. This is due to several reasons: (i) carbon has quite a high ionisation potential of 11.26 eV and thus low ionisation efficiency leading to rather high limits of detection (LOD) in ICP-MS compared to metals.10 (ii) carbon background levels are rather high which makes LODs even worse. (iii) substantial amounts of organic compounds (for example using organic solvents in HPLC-ICP-MS) are known to affect plasma characteristics and stability and thus carbon quantification may suffer from matrix effects.16,17 (iv) finally, ICP-MS is predominantly applied by inorganic researchers focusing on the metallic elements and using organic mass spectrometry for complementary structural information.The key aim of our study was to develop FFF-ICP-MS for (quasi-) simultaneous monitoring and quantification of metals, phosphorus and carbon. More specifically, the focus was on organic carbon in order to compare results with TOC and OCD measurements. Considering that the levels of particle-bound phosphorus are rather low in uncontaminated natural stream water samples, the ICP-MS conditions were first optimised for phosphorus detection and then applied for carbon monitoring. Consequently, the decrease of carbon baseline levels, in particular due to inorganic carbon, was the only remaining option to improve LODs and stability of carbon monitoring. As described in the experimental section deionised water with various treatments was compared for the blank signal of carbon using SF-ICP-MS (MR). The results for 12C are shown in Fig. 1. Using glass vials instead of the routinely applied polystyrene vials led to approximately 14% lower baseline levels obviously due to lower (organic) carbon blank levels. When comparing various options for removal of inorganic carbon, in particular dissolved carbon dioxide and hydrogen carbonate, the combination of acidification and purging with argon was most efficient with about 30% reduction in blank levels. This pre-treatment was applied for all subsequent measurements of total organic carbon by ICP-MS. Additional sources of carbon blank levels, for example residual carbon in the plasma gas were not investigated at this stage.
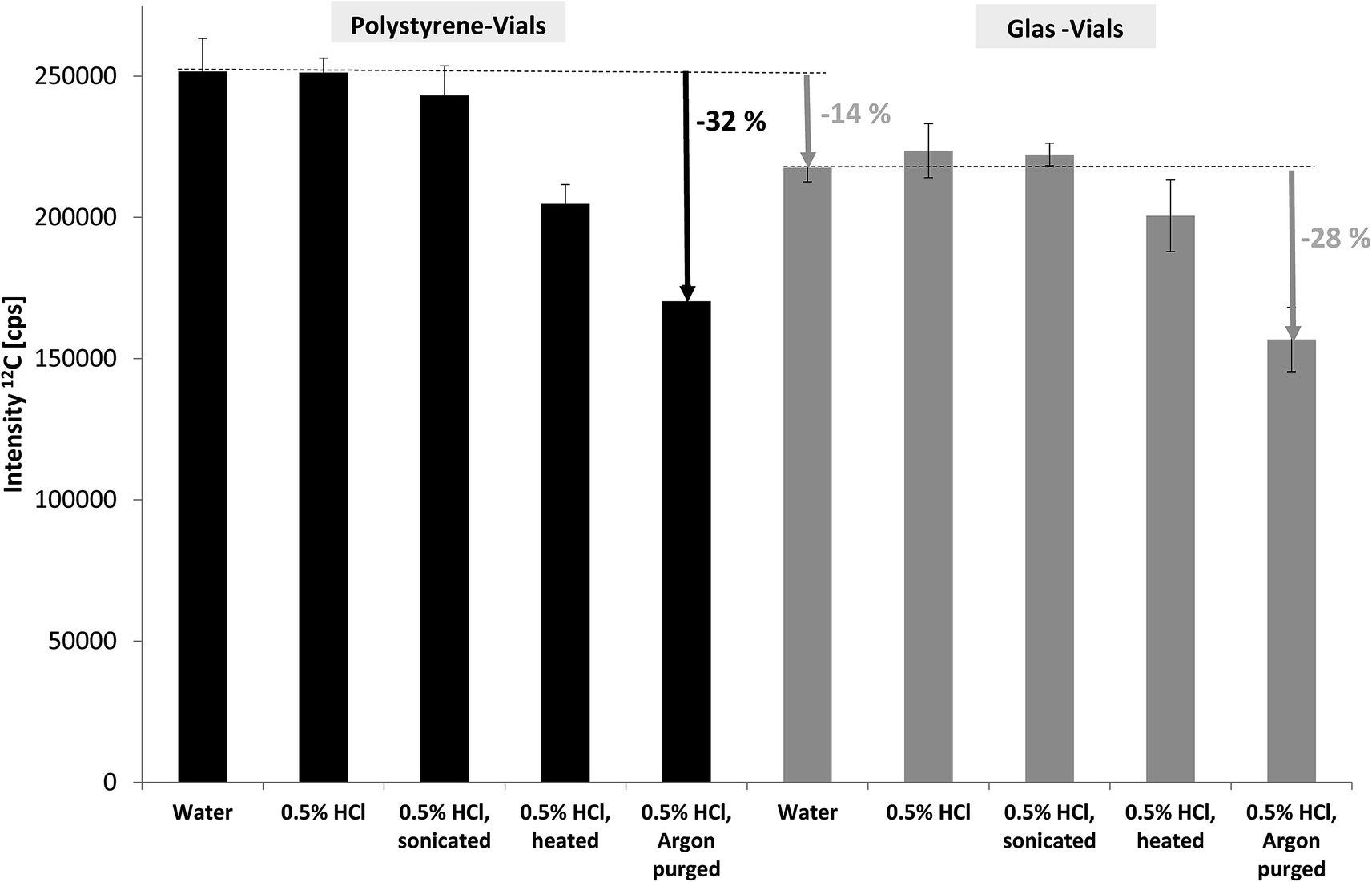 | ||
| Fig. 1 Effect of sample vials and sample pre-treatment on the carbon blank levels in deionised water monitored by SF-ICP-MS. | ||
Application of ICP-MS for the quantification of total organic carbon
Initially, the performance of ICP-MS was investigated offline for determination of total organic carbon in low molecular weight model solutions (citric acid) and high molecular weight model solutions (humic acid). Both quadrupole ICP-MS in He collision cell mode and sector-field ICP-MS in medium resolution were applied to evaluate potential differences in sample introduction, torch box and interface design on carbon quantification. Inorganic carbon in the diluted sample solutions was removed by acidification and purging with argon to achieve results for organic carbon, which could be compared to reference values obtained from TOC and OCD measurements of the same solutions. Therefore, all ICP-MS results in this section refer to organic carbon. Fig. 2 indicates that results from carbon measurements by ICP-MS are in good agreement with those obtained from OCD and TOC. This becomes even clearer when calculating the percentage of organic carbon in the solid citric acid (mean and standard deviation from replicate solutions calculated by propagation of error, n = 3): 38.9 ± 3.6% determined by Q-ICP-MS, 39.3 ± 2.7% by SF-ICP-MS, 35.8 ± 2.4% by TOC and 38.9 ± 1.9% by OCD. The results from the four techniques are matching well and also agree with the theoretical carbon concentration in citric acid of 37.4% (C6H8O7) under consideration of the obtained standard deviations, while the range of relative standard deviations from 5% to 9% is fit for purpose. For the high molecular weight model solution (humic acid) the following organic carbon results were obtained (mean and standard deviation from replicate solutions calculated by propagation of error, n = 3): 35.0 ± 1.8% by Q-ICP-MS, 30.3 ± 3.7% by SF-ICP-MS, 32.5 ± 1.5% by TOC and 29.4 ± 1.4% by OCD. Results are in good agreement, apart from Q-ICP-MS with slightly higher organic carbon concentration due to the low carbon concentration in the diluted humic acid solutions just about 12-fold above the LOD and potential contribution from residual inorganic carbon. Relative standard deviations are in an appropriate range from 5% to 12%.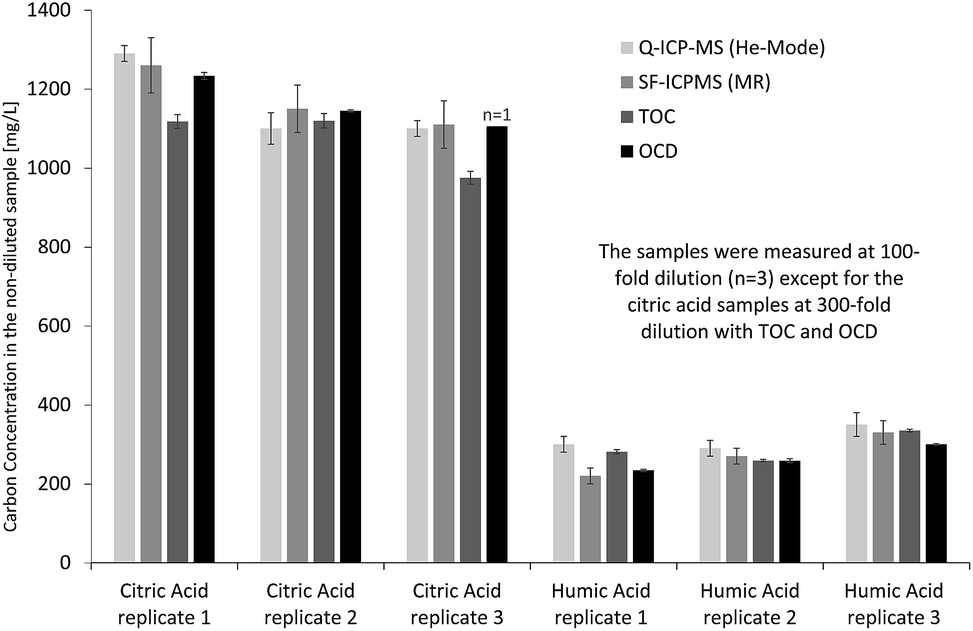 | ||
| Fig. 2 Determination of total organic carbon in citric acid and humic acid model solutions by Q-ICP-MS, SF-ICP-MS, TOC and OCD. | ||
In addition the same four techniques were applied for organic carbon quantification in real stream water samples (Fig. 3). For samples W1 and W5 results from all techniques are matching well, while for other samples the ICP-MS results, in particular SF-ICP-MS, are slightly higher compared to OCD and TOC. Average recoveries (n = 5) based on OCD results are 107 ± 16% for Q-ICP-MS, 122 ± 22% for SF-ICP-MS and 96 ± 7% for TOC. Instrumental limits of detection for organic carbon were calculated from linear regression based on the confidence interval: 0.6 mg L−1 for Q-ICP-MS, 0.3 mg L−1 for SF-ICP-MS, 0.2 mg L−1 for TOC and 0.04 mg L−1 for OCD. Sensitivity of ICP-MS and TOC is in same range and about 10-fold higher for OCD.
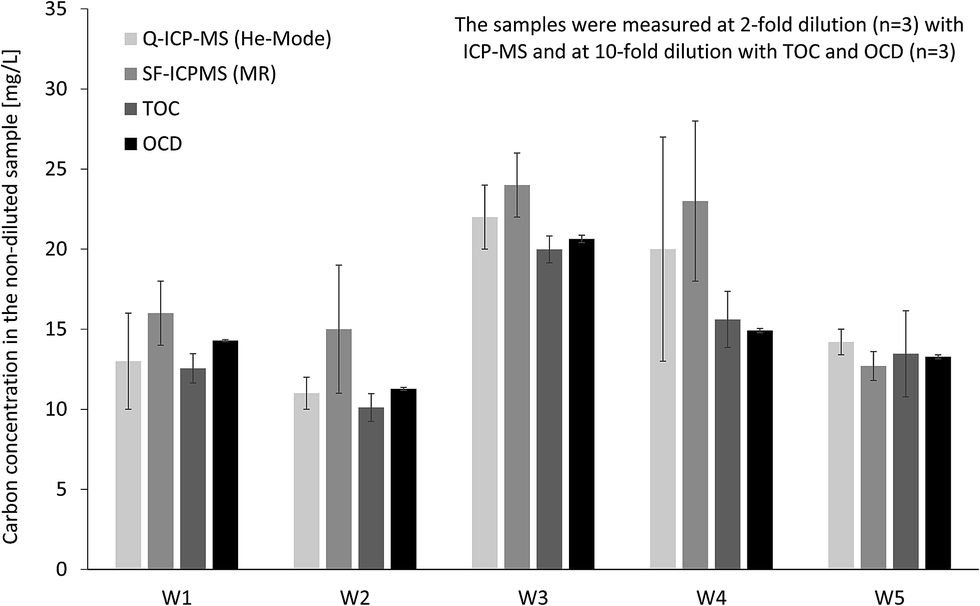 | ||
| Fig. 3 Determination of total organic carbon in fresh water samples by Q-ICP-MS, SF-ICP-MS, TOC and OCD. | ||
In summary, these data determined for model solutions and real fresh water samples indicate that both Q- and SF-ICP-MS provide quantitative organic carbon results matching those from TOC and OCD within average recoveries fit for purpose. Slightly elevated organic carbon results from ICP-MS may be due to potentially incomplete manual removal of inorganic carbon which is automated in TOC and OCD instruments. In addition organic carbon results in the diluted model solutions and samples used for measurement were in a range of approximately 3 to 12 mg L−1 and thus only 5 to 20 fold above the LOD of Q-ICP-MS. The presented results were obtained by evaluation of the 12C signal; the results obtained from the 13C signal were agreeing well within the calculated standard deviations, but the limit of detection was higher in particular for SF-ICP-MS due to the 92-fold lower natural abundance of 13C compared to 12C. The use of medium resolution in SF-ICP-MS and He collision cell mode in Q-ICP-MS decreased transmission of 12C+ ions and thus the 12C signal was well within the detection range and used in the following experiments. The aim of this comparison is not to establish ICP-MS as novel technique to determine TOC in solution, which is routinely performed by TOC-analysers with much lower effort and cost, but to demonstrate the feasibility of ICP-MS to provide meaningful and sufficiently accurate carbon quantification. This is the prerequisite for exploring the performance of ICP-MS as online carbon detector in the next section.
Comparison of FFF-Q-ICP-MS and FFF-OCD for particulate carbon monitoring
The main goal of this study is the use of ICP-MS for monitoring and quantification of transient carbon signals as obtained from the fractionation of carbon containing particles using FFF. Due to the lack of well characterised reference samples with known concentration of particulate carbon, validation of the FFF-ICP-MS results was performed via analysis of representative samples both with FFF-UV-ICP-MS and FFF-UV-OCD. Two relevant environmental sample matrices were chosen due to their high importance in studying the function and life cycle of particulate elemental contents: stream water samples and soil extracts. Stream water samples typically contain particulate carbon concentrations in the low mg L−1 range and can be analysed by FFF without pre-treatment apart from filtration. Soil samples require aqueous extraction of the nanoparticulate and colloidal fraction prior to FFF separation, but extracted particulate carbon concentrations are about 10-fold higher compared to stream water samples. Therefore, the different properties of these sample matrices are well suited to compare the performance of both detection systems. Regarding comparison of particulate carbon results from FFF-ICP-MS and FFF-OCD it is important to keep in mind that the OCD includes automatic online removal of inorganic carbon while in case of ICP-MS the effluent from FFF was analysed without pre-treatment. Therefore, in the following sections particulate carbon from FFF-OCD refers to particulate organic carbon while particulate carbon from FFF-ICP-MS refers to particulate total carbon. However, the fraction of inorganic carbon binding to particles in these environmental samples is considered negligible and thus results from FFF-OCD and FFF-ICP-MS are essentially both referring to particulate organic carbon and are thus comparable.The same FFF parameters were applied with exception of the injection volume for water samples which was increased for ICP-MS detection due to the low concentration of phosphorus. Of course, the use of a carbon-free eluent is required; low concentration of sodium chloride in deionised water was applied based on previous optimisation.2 Quantification was in both cases based on the same TOC standard solution (Certipur). On some occasions elevated carbon baseline levels were observed after installation of a new membrane which slowly decreased after purging of the separation channel. This is supposed to be due to membrane preservation by the manufacturer with glycerol to prevent drying of the membrane.
First, four fresh water samples were analysed with both techniques (n = 1) to compare elution profiles for carbon and feasibility of quantification. The UV signal was monitored for both techniques and used as qualitative reference to overlay the fractograms from the two FFF systems. The example in Fig. 4 shows one main carbon fraction at an elution time of about 5 minutes. The corresponding fractograms of Al, Si, Fe, Mn and P are shown in Fig. S1 of the ESI.† UV signals from both FFF-systems are suffering from detector overflow (Fig. 4), but are matching well with the ICP-MS signal for 12C regarding retention time and peak shape. The carbon signal from OCD is detected significantly later at about 11 minutes. In addition, the peak width in half height is 1.5 min for the ICP-MS C-signal and 3.1 min for the OCD C-signal. The observed shift in elution time and significant peak broadening is due to the high internal volume of the thin film reactor of the OCD. However, the carbon baseline from OCD is much more stable than the carbon baseline from ICP-MS. Calibration was performed in both systems by injection of carbon standard solutions prepared in the carrier matrix (25 μmol L−1 aqueous NaCl) via the FFF autosampler in analogy to the samples but keeping the tip flow at the detector flow of 0.5 mL min−1 and the cross flow at 0 mL min−1 (FI calibration approach). Standard solutions for OCD were prepared by dilution of the Certipur TOC standard. Standard solutions for ICP-MS were prepared from citric acid stock solution previously quantified for TOC against the Certipur standard and purged with argon prior to injection. Direct use of the Certipur standard was found not suitable for FFF with multi-element detection by ICP-MS due to very high concentration of phosphorus in the TOC standard, which was not compatible with parallel monitoring of low particulate phosphorus concentrations. Limits of detection for carbon calculated from the linear regression were 1.4 mg L−1 for FFF-Q-ICP-MS and 0.08 mg L−1 for FFF-OCD. Particulate carbon concentrations in the first FFF-fraction (main peak) were in good agreement for fresh water samples C and D with 6.7 mg L−1 and 15.7 mg L−1 obtained by FFF-ICP-MS compared to 7.1 mg L−1 and 15.4 mg L−1 obtained by FFF-OCD (Table 1). Particulate carbon concentrations in samples A and B were much lower (2.1 mg L−1 and 5.1 mg L−1 as determined by FFF-OCD) and thus too close to the LOD from FFF-ICP-MS (resulting in <1.4 mg L−1 and 3.1 mg L−1). These initial results were promising to continue the comparison with another type of environmental sample.
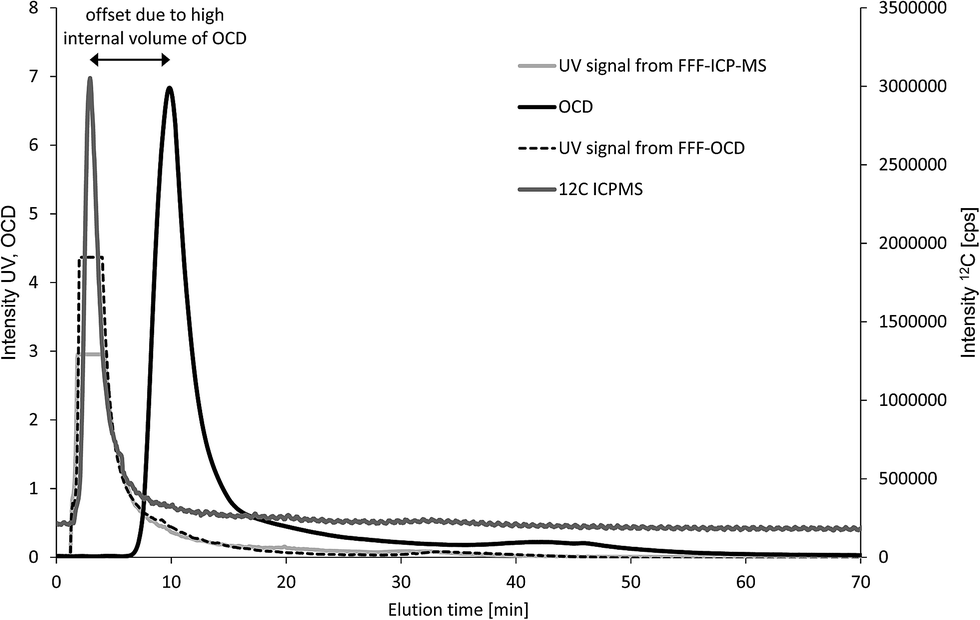 | ||
| Fig. 4 Overlay of fractograms of a fresh water sample recorded by FFF-UV-Q-ICP-MS and FFF-UV-OCD. The UV signals suffer from detector overload and were only used for qualitative comparison of the elution profiles from the two FFF systems. | ||
| Water samples | Sample A | Sample B | Sample C | Sample D |
|---|---|---|---|---|
| FFF-OCD particulate C [mg L−1] | 2.1 | 5.1 | 7.1 | 15.4 |
| FFF-ICP-MS particulate C [mg L−1] | <1.4 | 3.1 (61%) | 6.7 (94%) | 15.7 (102%) |
| Soil extracts | BB8 | BB9 | BB10 |
|---|---|---|---|
| FFF-OCD particulate C [mg L−1] | 75 | 101 | 97 |
| FFF-ICP-MS particulate C [mg L−1] | 57 (76%) | 73 (72%) | 79 (81%) |
| FFF-ICP-MS particulate C [mg L−1] (post-channel calibration) | 77 (103%) | 91 (90%) | 98 (101%) |
Second, three soil extracts were analysed with FFF-UV-ICP-MS and FFF-UV-OCD (n = 1). Particle bound elemental concentrations in these extracts are much higher compared to the fresh water samples. Therefore, 25-fold lower injection volume was applied. The same shift in elution time and peak broadening as in Fig. 4 was observed for FFF-OCD. An example overlay of carbon fractograms from OCD and ICP-MS detection is shown in Fig. 5 (retention time offset was corrected as explained in the figure caption). The corresponding fractograms of Al, Si, Fe, Mn, P and Ca are shown in Fig. S2 of the ESI.† The peak obtained for 12C is noisier due to 40-fold lower signal intensity but still well above the LOD in spite of the low injection volume. In addition the larger particle size in soil extracts compared to water samples may contribute to increased signal noise, which is typically observed for metallic nanoparticles with increasing size.3 Compared to the water sample in Fig. 4 the particle size distribution of the predominant fraction for carbon is much broader which is partly due to slight differences in the FFF separation conditions, but mainly due to higher concentration of larger carbon containing particles in the soil extracts. Quantification was performed via the same flow injection calibration approach as used before for the water samples. LOD for ICP-MS detection was calculated from the linear regression as 1.9 mg L−1, which is only slightly higher compared to the use of 5 mL injection volume for water samples due to the much smaller peak width with lower injection volume. The results from FFF-ICP-MS detection of particulate carbon concentrations for the three soil extracts were 57, 73 and 79 mg L−1 and thus about 70–80% recovery compared to the results from OCD detection at 75, 101 and 97 mg L−1 (Table 1). The low recovery may be due to potential effects of the soil matrix on the measurement of the carbon calibration standards, which was performed after the samples.
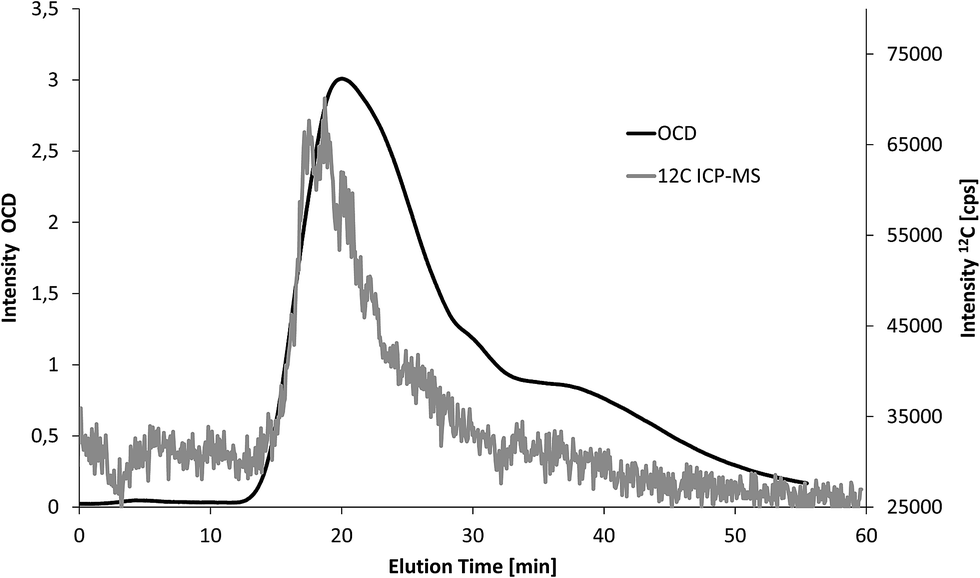 | ||
| Fig. 5 Overlay of fractograms of a soil extract recorded by FFF-Q-ICP-MS and FFF-OCD (the offset of 6 minutes between the fraction elution times from OCD and ICP-MS detection due to the larger internal volume of the OCD was determined by flow injection without FFF separation and subtracted from the elution time). | ||
These initial results for fresh water samples and soil extracts demonstrated the feasibility of using ICP-MS for online carbon monitoring. However, the quantification required further improvement in order to be a competitive alternative to OCD detection.
Two independent calibration strategies for FFF-ICP-MS
The initially applied flow injection (FI) calibration for ICP-MS detection was compared to a post-channel (PC) calibration approach, which has been previously applied for quantification of transient signals when species-specific calibration standards were not available.3 The Rh signal from post-channel addition was used as internal standard for both calibration approaches. PC calibration was first applied for the three soil extracts reported in the previous section. The resulting particulate carbon concentrations of 77, 91 and 98 mg L−1 were in much better agreement with the results from OCD detection (recoveries 91–102%) (Table 1). The experimental setup and calibration data including calibration curves for both calibration strategies are summarized in Fig. 6 for the example of the soil extracts.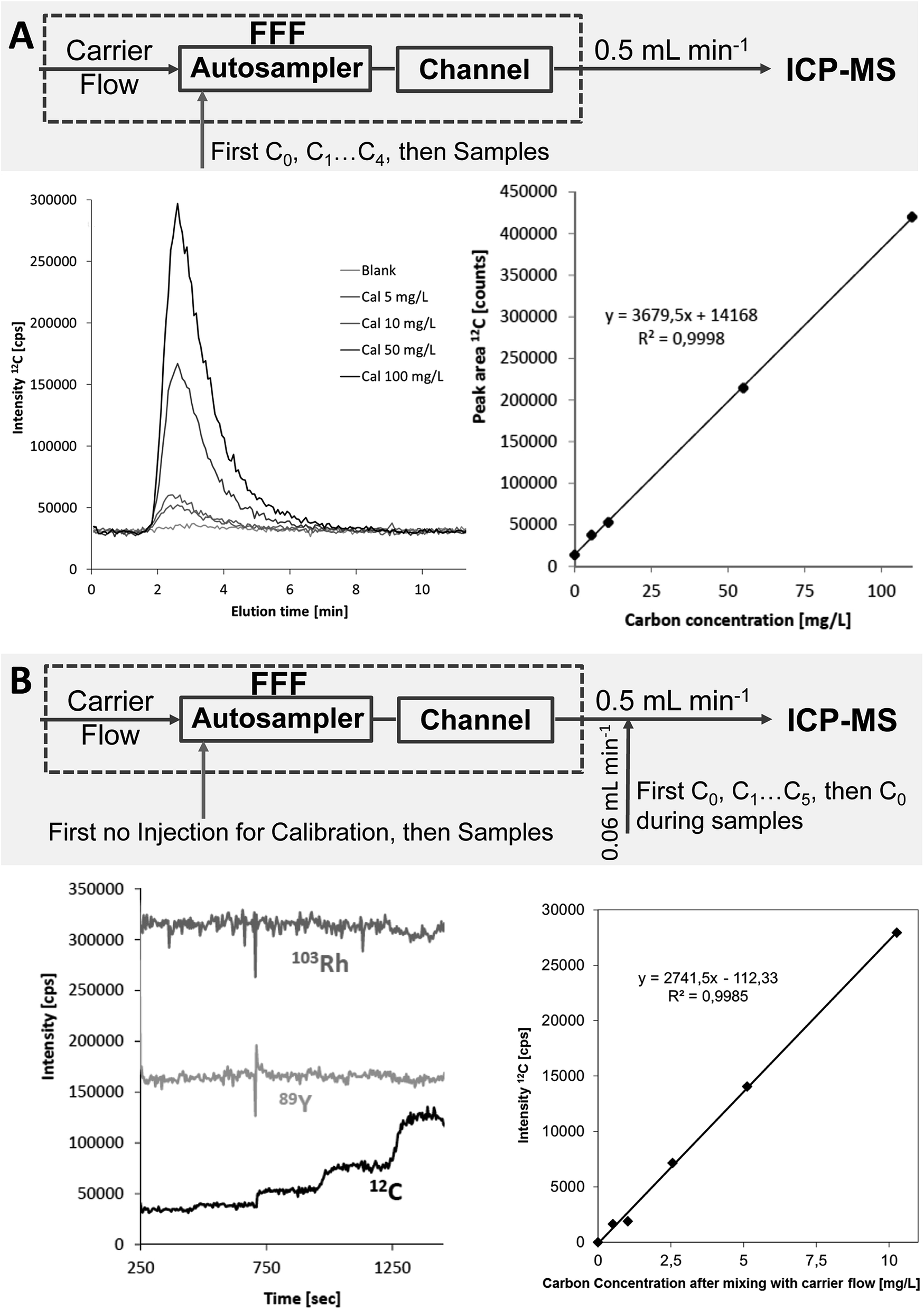 | ||
| Fig. 6 Experimental setup, calibration raw data and calibration curves both for the flow injection calibration (A) and for the post-channel calibration (B) approach using the example of the soil extracts. | ||
Both quantification strategies were studied in more detail by analysing three stream water samples in triplicate with FFF-ICP-MS and FFF-OCD. The sequence of the FFF-ICP-MS measurements started with PC calibration followed by FI calibration, first measurement of each sample, quality control (QC) standard (FI, 5 mg L−1 carbon), second measurement of each sample, QC standard, third measurement of each sample, QC standard and finally another PC calibration. Mean concentrations with standard deviation are summarised in Table 2. Results from linear regression of the FI calibration achieved recoveries in the range of 101–113% compared to OCD results. Relative standard deviations ranged from 4% to 12%. When using the QC standards analysed before and after each sample block for drift correction the recoveries were in the range of 90–100% and RSDs improved (range of 1–9%). Results from PC calibration showed recoveries from 95% to 107% compared to OCD results; RSDs were in the range of 4–11%. There was no significant difference in the results from the PC calibration at the beginning of the sequence and the PC calibration performed at the end of the sequence. When using the QC standards for drift correction the recoveries shifted from 87% to 97% and RSDs ranged from 1% to 8%.
| Sample 1 [mg L−1] | Sample 2 [mg L−1] | Sample 3 [mg L−1] | |
|---|---|---|---|
| a Recoveries for QC sample for drift correction: QC1 106%; QC2 118%; QC3 125%. b Recoveries for QC sample for drift correction: QC1 108%; QC2 121%; QC3 129%. c Total run time of the sequence was 25 hours. | |||
| FFF-OCD | 9.59 ± 0.56 | 11.02 ± 0.30 | 11.11 ± 0.15 |
| FFF-ICP-MS | 10.24 ± 1.15 | 11.55 ± 0.54 | 10.60 ± 0.45 |
| Post channel | (106.8%) | (104.8%) | (95.4%) |
| FFF-ICP-MS | 9.31 ± 0.09 | 10.56 ± 0.76 | 9.70 ± 0.76 |
| Post channel (drift corrected)a | (97.1%) | (95.8%) | (87.3%) |
| FFF-ICP-MS | 10.81 ± 1.27 | 12.26 ± 0.60 | 11.21 ± 0.50 |
| Flow-injection | (112.8%) | (111.2%) | (100.9%) |
| FFF-ICP-MS | 9.62 ± 0.12 | 10.97 ± 0.88 | 10.03 ± 0.86 |
| Flow-injection (drift corrected)b | (100.3%) | (99.5%) | (90.3%) |
The flow injection calibration approach has the principal challenge that the calibration standards need to pass through the FFF channel and thus may interact with the membrane and potentially with residual matrix from previously analysed samples remaining in the channel. In case of particulate carbon in environmental water and soil samples well characterised species-specific standards are not available. Therefore, well characterised low molecular weight carbon species were selected as calibration standards assuming that these standards are fully recovered from the channel when using flow injection with zero cross flow. The problem of high phosphorus concentration in the Certipur standard was overcome by using citric acid standards previously quantified against Certipur TOC standard. Separate calibration solutions are required for other metals monitored in parallel (P, Mn, Fe, Al, and Si) because the citric acid contains elevated concentration of some of these metals and also causes significant wash-out of metals from the FFF system. Purging the standard solutions with argon prior to injection significantly improved the performance by minimising interferences from dissolved carbon dioxide. Analysis time for the measurement of each calibration standard is about 20–40 minutes depending on injection volume.
The post channel calibration approach simplifies the introduction of calibration standards by addition via a Tee between FFF and ICP-MS. Acidified standard solutions can be used and high phosphorus concentrations in Certipur TOC standard are not critical. Calibration of carbon and other elements (e.g. P, Mn, Fe, Al) anyway needs to be performed in separate calibrations because elemental standards typically contain elevated carbon blank levels. Time requirement is much lower with about 5 minutes per calibration standard. Slight disadvantage is that the PC standards are currently introduced manually, however this might be done via a liquid autosampler in the future. Further requirements of the PC approach are the necessity to measure the flow rate of the post channel added standard (or diluting acid respectively) to convert the intensity data into a mass flow fractogram and also the need to know (determine) the exact injection volume for calculation of the carbon concentration from the absolute mass of carbon obtained by peak integration of the mass flow converted fractogram. Both parameters add to the overall uncertainty of this calibration approach but the results shown above indicate that these contributions are not critical.
Application of FFF-ICP-MS for quantification of carbon in bovine serum albumin
As initially discussed there is a lack of well characterised particulate carbon reference materials and thus method validation was mainly based on comparison of FFF-ICP-MS and FFF-OCD results for the same samples. A potential candidate for such a reference material could be bovine serum albumin, which is available in sufficient purity to calculate the carbon concentration of 53.2% from the known stoichiometry (C3071H4826N816O927S40, molecular weight 69293.4 Da).18,19 BSA is routinely applied for performance check of FFF-UV systems using 10 kDa molecular weight cut off regenerated cellulose membrane with 1 mol L−1 aqueous NaCl carrier.20 Moreover, BSA was employed for size calibration of colloid separation by FFF.21 Modified separation conditions were reported for hyphenation with ICP-MS using 100 mM ammonium acetate as carrier for detection of sulfur in human serum albumin.22 Both carriers are obviously not suitable for monitoring of carbon in BSA using FFF-ICP-MS. Therefore, the same membrane (1 kDa polyethersulfon) and the same carrier (25 μmol L−1 NaCl in deionised water) as used for the environmental samples in this study were applied and elution was performed with constant cross flow of 1 mL min−1. This resulted in a predominant BSA peak at 13.5 min detected in the carbon and sulfur fractograms (Fig. 7). A minor carbon peak at 8 min coeluted with copper indicating separation of apo–BSA and Cu–BSA under the applied conditions with very low ionic strength carrier. Commercially available apo–BSA usually contains small amounts of residual bound copper which is useful for detection of BSA by hyphenated ICP-MS techniques with higher sensitivity compared to sulfur and carbon. Finally, another minor carbon containing fraction was detected at 43 min when the cross flow was set to zero for cleaning of the channel. Duplicate analysis of the three replicate BSA solutions using the flow injection calibration approach resulted in a mean carbon concentration of 36.2% with absolute standard deviation of 4.3%, which equals a mean recovery of 68.0% with standard deviation 8.2% based on the theoretical carbon concentration. Only the main fraction at 13.5 min was used for quantification. Recovery of BSA from the membrane is likely to be incomplete under the applied low ionic strength. Therefore, the same BSA solutions were analysed with zero cross flow in analogy to the calibration standards. This improved recovery of BSA carbon concentration to 83.4% with standard deviation of 4.7%. Finally, the analysis was repeated with zero cross flow bypassing the separation channel to exclude interaction of BSA with the membrane. This increased the mean recovery to 94.8% with a non-expanded standard deviation of 2.8% showing essentially complete recovery considering a purity of >98% and the fact, that there was no correction for moisture applied. For the purpose of quality control of the carbon quantification in a macromolecule of low nm size range using the flow injection calibration approach with citric acid calibration standards it is of minor relevance whether the BSA is passing the separation channel or not. Therefore, the low recovery due to interaction with the membrane can be overcome by bypassing the FFF separation channel to achieve a well-shaped transient BSA signal in the FFF carrier for online quantification by ICP-MS.
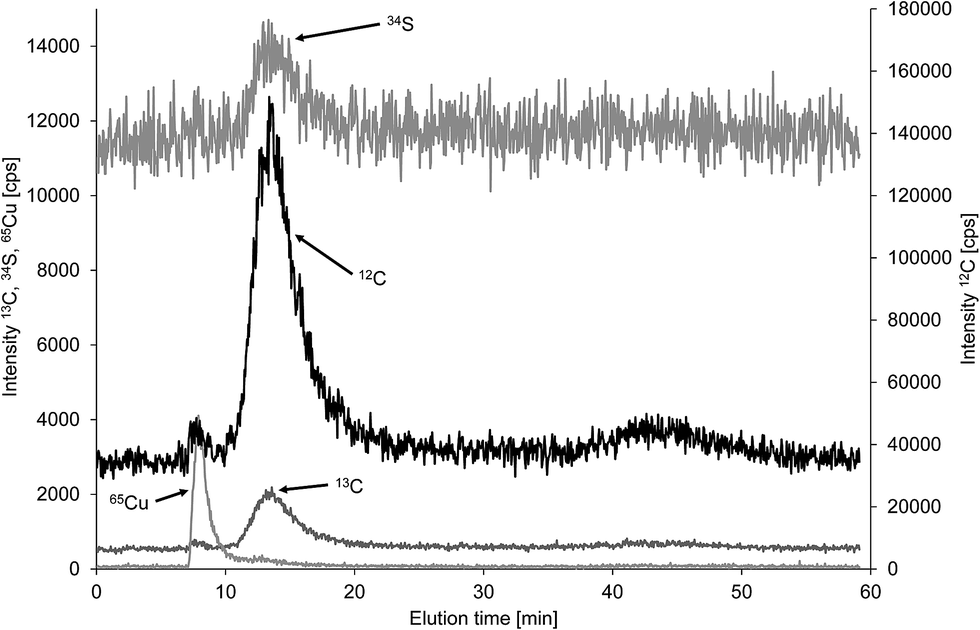 | ||
| Fig. 7 FFF-Q-ICP-MS fractogram of bovine serum albumin monitoring carbon, sulfur and copper. | ||
Conclusion
The comparison of four techniques for total organic carbon determination demonstrated the feasibility of using ICP-MS for (organic) carbon monitoring and quantification in standard solutions and fresh water samples containing dissolved and particulate organic carbon species. The results obtained in our study indicate that external calibration provides sufficiently accurate and precise total organic carbon quantification without the need for isotope dilution techniques. There was no systematic difference observed between organic carbon recoveries for citric acid and humic acid model solutions showing that quantification is species-independent as already reported by Vogl and Heumann.9Qualitative and quantitative detection of particulate carbon using FFF-ICP-MS was successfully demonstrated by comparison with results from FFF-OCD for the same set of fresh water samples and soil extracts. Both, flow injection calibration and post-channel calibration approaches were applied for FFF-ICP-MS showing good agreement with FFF-OCD. This indicates that the contribution of inorganic particulate carbon to the ICP-MS results of the investigated samples was negligible. Limits of detection for particulate carbon are at least 10-fold higher using FFF-ICP-MS compared to FFF-OCD, but sufficient for monitoring of natural carbon containing nanoparticles and colloids in real environmental samples. Soot particles or carbon included in mineral associates could be critical for OCD detection because these particles may not be efficiently oxidised in the thin film reactor. Complete atomisation of those particles is expected for ICP-MS at least in the nanometer size range. However, this needs more detailed investigation which is currently in progress. Determination of the carbon concentration of BSA using the same membrane and carrier suffered from incomplete recovery of BSA from the separation channel. However, flow injection without channel proved complete recovery and thus accurate quantification of the carbon concentration by ICP-MS. The advanced development of FFF-ICP-MS for parallel monitoring of carbon and metals substantially increases the application range of this technique and reduces the instrumental effort for monitoring of interactions between carbon and other elements in a wide range of samples containing natural and engineered (nano)particles with carbon-based core or organic coatings. In addition, there is potential for further optimisation and application to other liquid flow based separation systems online with ICP-MS.
Acknowledgements
Funding of this work and international cooperation between Forschungszentrum Juelich and University of Trondheim by DAAD PPP exchange is gratefully acknowledged. The authors also would like to gratefully acknowledge the support of Annalea Lohila and Tuomas Laurila (Finnish Meteorological Institute) for sampling at Lettosuo and logistical support, which was conducted in the framework of another study in cooperation with Nina Gottselig.References
- S. Dubascoux, I. Le Hecho, M. Hassellöv, F. Von Der Kammer, M. Potin Gautier and G. Lespes, J. Anal. At. Spectrom., 2010, 25, 613–623 RSC.
- N. Gottselig, R. Bol, V. Nischwitz, H. Vereecken, W. Amelung and E. Klumpp, Vadose Zone J., 2014, 13(7), 1–11 Search PubMed.
- V. Nischwitz and H. Goenaga-Infante, J. Anal. At. Spectrom., 2012, 27, 1084–1092 RSC.
- Ö. Gustafsson and P. M. Gschwend, Limnol. Oceanogr., 1997, 42, 519–528 CrossRef.
- N. T. Chaopricha and E. Marin-Spiotta, Soil Biol. Biochem., 2014, 69, 251–264 CrossRef CAS.
- A. Bar-Even, E. Noor and R. Milo, J. Exp. Bot., 2012, 63, 2325–2342 CrossRef CAS PubMed.
- E. Neubauer, F. Van Der Kammer and T. Hofmann, J. Chromatogr. A, 2011, 1218, 6763–6773 CrossRef CAS PubMed.
- http://www.doc-labor.de/Specs.html .
- J. Vogl and K. G. Heumann, Anal. Chem., 1998, 70, 2038–2043 CrossRef CAS.
- E. T. Luong and R. S. Houk, J. Am. Soc. Mass Spectrom., 2003, 14, 295–301 CrossRef CAS PubMed.
- R. Santamaria-Fernandez, Anal. Bioanal. Chem., 2010, 397, 973–978 CrossRef CAS PubMed.
- C. Smith, B. Packert Jensen, I. D. Wilson, F. Abou-Shakra and D. Crowther, Rapid Commun. Mass Spectrom., 2004, 18, 1487–1492 CrossRef CAS PubMed.
- C. D. B. Amaral, R. S. Amais, L. L. Fialho, D. Schiavo, A. R. A. Nogueira and J. A. Nobrega, Microchem. J., 2015, 122, 29–32 CrossRef CAS.
- B. Stolpe, M. Hasellöv, K. Andersson and D. R. Turner, Anal. Chim. Acta, 2005, 535, 109–121 CrossRef CAS.
- J. M. Sequaris and H. Lewandowski, Colloids Surf., 2003, 217, 93–99 CrossRef CAS.
- G. Grindlay, J. Mora, M. De Loos-Vollebregt and F. Vanhaecke, Spectrochim. Acta, Part B, 2013, 86, 42–49 CrossRef CAS.
- A. L. Molinero, J. R. Castillo, P. Chamorro and J. M. Muniozguren, Spectrochim. Acta, Part B, 1997, 52, 103–112 CrossRef.
- SIB ExPASy, Bioinformatics Resources Portal, http://www.expasy.org.
- P. Artimo, M. Jonnalagedda, K. Arnold, D. Baratin, G. Csardi, E. de Castro, S. Duvaud, V. Flegel, A. Fortier, E. Gasteiger, A. Grosdidier, C. Hernandez, V. Ioannidis, D. Kuznetsov, R. Liechti, S. Moretti, K. Mostaguir, N. Redaschi, G. Rossier, I. Xenarios and H. Stockinger, Nucleic Acids Res., 2012, 40, W597–W603 CrossRef CAS PubMed.
- Postnova FFF installation test.
- B. Stolpe, L. Guo, A. M. Shiller and M. Hassellöv, Mar. Chem., 2010, 118, 119–128 CrossRef CAS.
- J. Heroult, T. Harvard and H. Goenaga-Infante, Column, 2012, 8, 11–15 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ja00027d |
| This journal is © The Royal Society of Chemistry 2016 |