
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe ligand influence in stereoselective carbene transfer reactions promoted by chiral metal porphyrin catalysts
Daniela
Intrieri
,
Daniela Maria
Carminati
and
Emma
Gallo
*
Chemistry Department of Milan University, Via Golgi 19, 20133 Milan, Italy. E-mail: emma.gallo@unimi.it; Fax: (+39) (0)2 50314405
First published on 17th June 2016
Abstract
The use of diazo reagents of the general formula N2C(R)(R1) as carbene sources to create new C–C bonds is of broad scientific interest due to the intrinsic sustainability of this class of reagents. In the presence of a suitable catalyst, diazo reagents react with several organic substrates with excellent stereo-control and form N2 as the only by-product. In the present report the catalytic efficiency of metal porphyrins in promoting carbene transfer reactions is reviewed with emphasis on the active role of the porphyrin skeleton in stereoselectively driving the carbene moiety to the target substrate. The catalytic performances of different metal porphyrins are discussed and have been related to the structural features of the ligand with the final aim of rationalizing the strict correlation between the three-dimensional structure of the porphyrin ligand and the stereoselectivity of carbene transfer reactions.
 Daniela Intrieri (Right), Daniela Maria Carminati (Left) and Emma Gallo (Centre) | Daniela Intrieri was born in Italy in 1984. In 2014 she received her Ph.D. degree in Chemical Science from the University of Milan (Italy) under the supervision of Prof. Emma Gallo. Her Ph.D. thesis focused on the synthesis of metal porphyrins to be employed as homogeneous catalysts for the functionalization of C–C and C–H bonds by carbene and nitrene transfer reactions as well as on the study of reaction mechanisms. After her Ph.D. she moved to the Trinity College of Dublin (Ireland) under the supervision of Prof. Mathias O. Senge to work in the field of organocatalysis and on the synthesis of highly substituted porphyrins. She is currently a post-doc researcher at Milan University in Prof. Emma Gallo's group. Daniela Maria Carminati was born in Italy in 1989. She received her master's degree in Industrial Chemistry and Management in 2014 with a thesis concerning the synthesis of a chiral iron(III) porphyrin complex to promote the enantioselective cyclopropanation of alkenes under the supervision of Prof. Emma Gallo in the Chemistry Department of Milan University. She is currently a PhD student at Milan University in Prof. Emma Gallo's group and her research activity is mainly devoted to the synthesis of metal porphyrin complexes and their use as catalysts in C–C or C–N bond formation reactions. Emma Gallo received her Ph.D. degree in Chemistry (1995) from the University of Lausanne (Switzerland) under the supervision of Prof. Carlo Floriani. Subsequently, she spent one year in Floriani's group as “Maitre assistant” before moving to Italy. After post-doctoral training with Prof. Sergio Cenini at Milan University (Italy) she became assistant professor and then associate professor at the same university. Research interests of her group include the synthesis of homogeneous catalysts for the synthesis of fine chemicals using sustainable catalytic processes. Particular attention is devoted to the study of reaction mechanisms to fine-tune catalytic performances. |
1. Introduction
Carbene transfer reactions mediated by metal transition complexes are largely employed in organic synthesis owing to the high versatility of this synthetic strategy. The metal carbene intermediate, formed by the reaction of a transition metal complex with a carbene precursor, can be involved in several organic transformations; among them, alkene cyclopropanation1–5 (Scheme 1, path a) and the functionalization of X–H (X = C, Si, N, S) bonds6–8 (Scheme 1, path c) represent efficient methodologies to synthesise high-added value chemicals starting from simple and cheap precursors.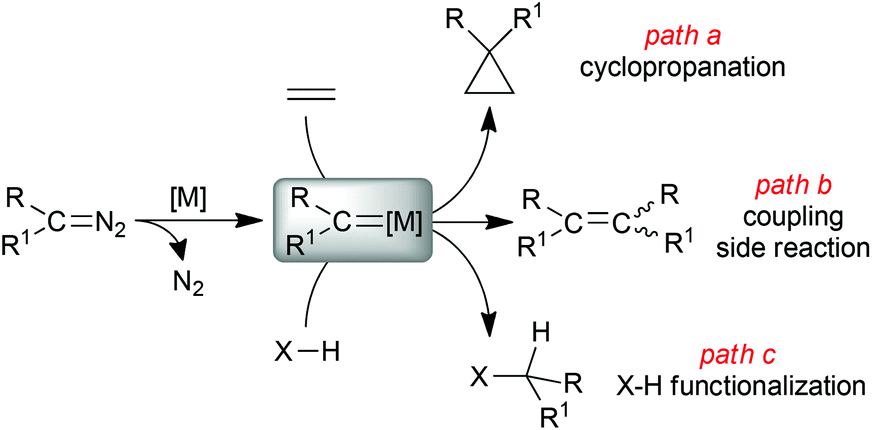 | ||
| Scheme 1 General formation of a metal carbene complex (R)(R1)C=[M] and its reactivity towards alkenes and X–H containing substrates. | ||
Diazo derivatives of the general formula N2C(R)(R1) are sustainable and atom-efficient starting materials of these reactions because the formation of the carbene [(R)(R1)C:] functionality occurs with the simultaneous extrusion of benign molecular nitrogen as the only by-product.9,10
Diazo derivatives can be efficiently synthesised by introducing R/R1 groups with different electronic behaviour (donor/donor, acceptor/donor or acceptor/acceptor) to fine-tune the reactivity of the carbene metal species with respect to the electrophilic/nucleophilic nature of the organic substrate. In addition, the steric nature of substituents on the carbene carbon atom can affect the reaction stereoselectivity by driving the approach of the metal carbene complex to the organic substrate. The electronic and steric features of the diazo reagent can also determine the occurrence of coupling side reactions (Scheme 1, path b), which must be minimized to guarantee productivity of the desired pathway. It is important to remember that the safe handling of diazo derivatives can be increased by managing them under continuous-flow technologies,11–14 which permit their synthesis and use on a large scale suitable for industrial applications.
Amongst the metal complexes which are effective in activating diazo derivatives in the alkene cyclopropanation and functionalization of X–H bonds, metal porphyrins are very efficient because they couple a high catalytic activity with an excellent chemical stability.15–22 Porphyrin ligands can be used to synthesise metal catalysts with very different electronic and steric characteristics thanks to the plethora of structural modifications that can be carried out on a porphyrin skeleton. In addition the presence of different axial ligand(s) on the active transition metal can further expand the chemical versatility of this class of catalysts (Fig. 1).
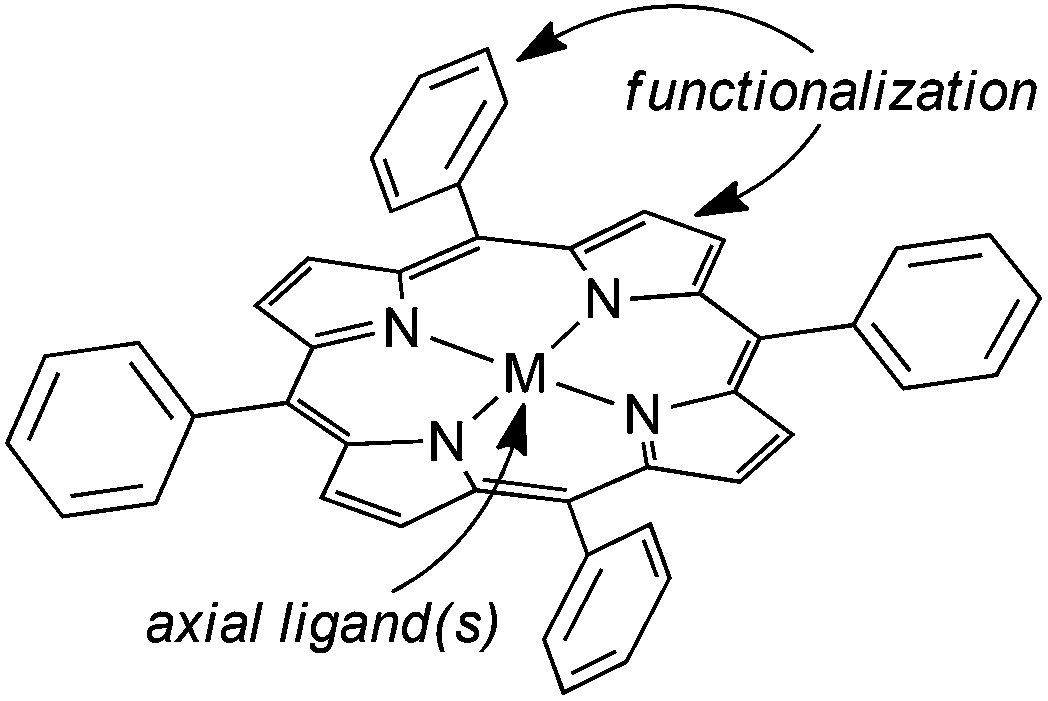 | ||
| Fig. 1 The parent structure of metal porphyrin complexes. | ||
Metal porphyrin complexes can also be obtained in a pure chiral form to promote enantioselective reactions which must be applied to the synthesis of pharmaceutical and/or biological compounds. Up to now many chiral metal porphyrins23 have been synthesised for different catalytic applications and in the present Perspective we would like to review their use in the enantioselective cyclopropanation of alkenes and the functionalization of X–H bonds. A selection of chiral porphyrin structures active in stereoselective carbene transfer reactions is shown in Fig. 2 and discussed below.
 | ||
| Fig. 2 General structures of the chiral porphyrins discussed herein. | ||
In this report, chiral porphyrins are grouped on the basis of the porphyrin skeleton symmetry due to the importance of the ligand structure in determining the catalytic performance.
We sincerely apologise in advance if some important contributions have been unintentionally omitted.
2. D 4-Symmetric catalysts
2.1 Chiral catalysts of group 8
The first enantioselective cyclopropanation promoted by a chiral ruthenium(II) porphyrin was independently published in 1997 by A. Berkessel24 and C.-M. Che.25 Both research groups employed the D4-symmetric chiral porphyrin (Fig. 2, type A) reported by L. Halterman and S. T. Jan26 to synthesise the ruthenium complexes (Halt*)Ru(CO) (1) and (Halt*)Ru(CO)(EtOH) (2) shown in Fig. 3.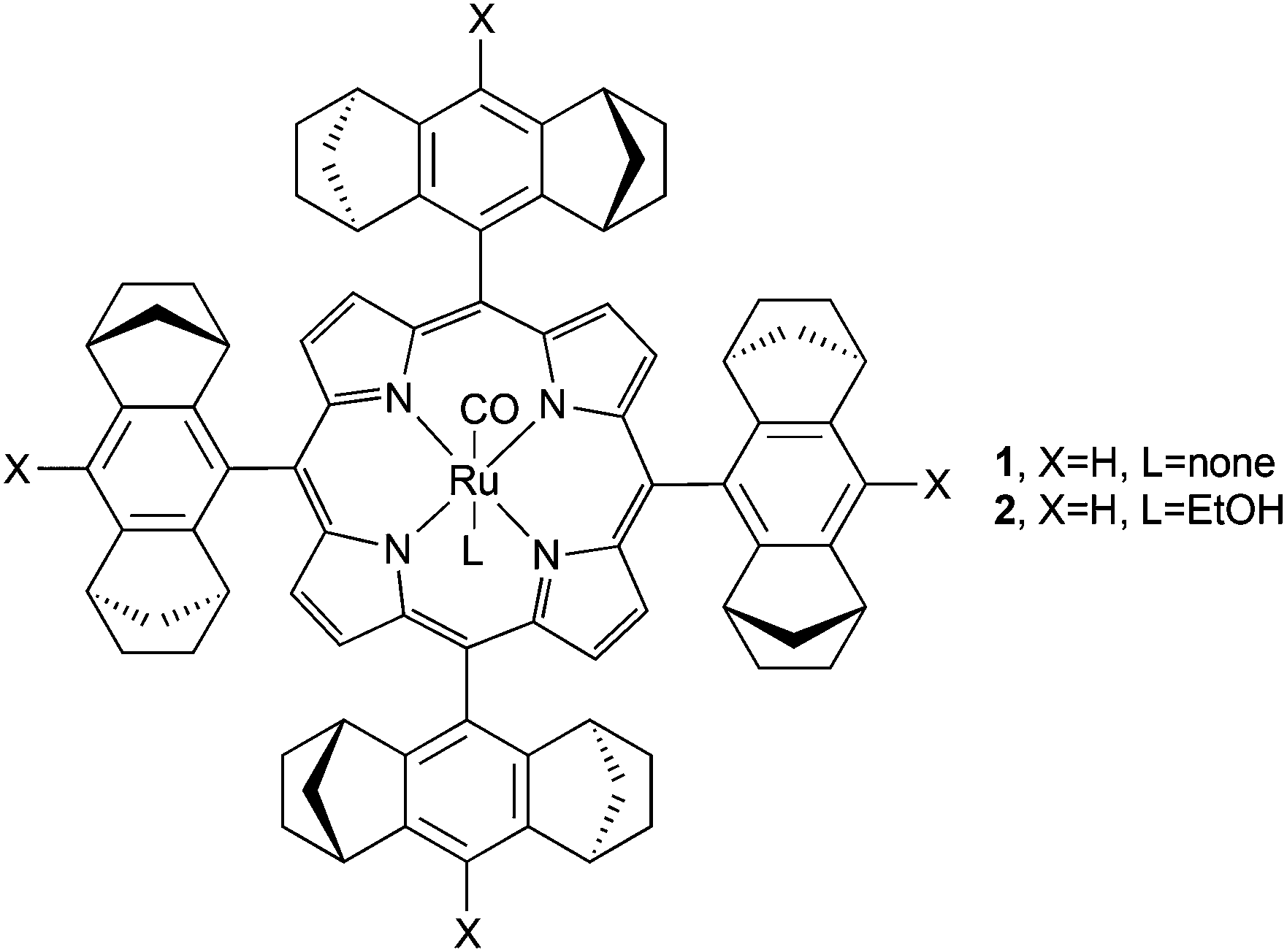
Complex 1 was obtained as a pentacoordinated complex and was active in the cyclopropanation of styrene by ethyl diazoacetate (EDA) at a low catalyst loading of 0.15 mol%. The cyclopropane was obtained in a quantitative yield with trans-diastereoselectivity ratios (d.r.trans) up to 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and trans-enantioselectivities (eetrans) up to 91%. Comparable results were obtained by C.-M. Che and co-authors by studying the cyclopropanation of styrene by EDA in the presence of 2 (0.2 mol%), which was also active in promoting the cyclopropanation of the other styrenes (Scheme 2) with differently substituted diazo reagents.27
:5 and trans-enantioselectivities (eetrans) up to 91%. Comparable results were obtained by C.-M. Che and co-authors by studying the cyclopropanation of styrene by EDA in the presence of 2 (0.2 mol%), which was also active in promoting the cyclopropanation of the other styrenes (Scheme 2) with differently substituted diazo reagents.27
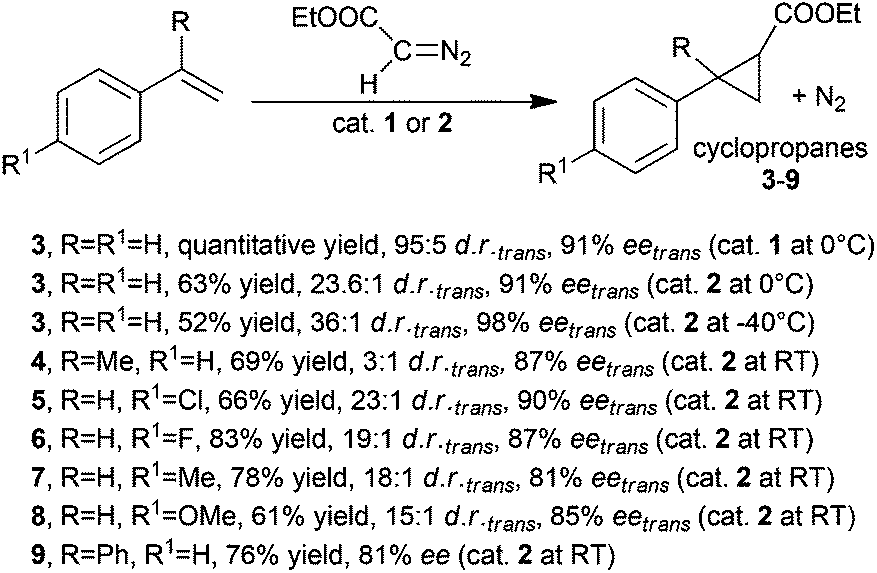
As shown in Scheme 2, the presence of the axial ligand EtOH on the ruthenium centre of 2 did not influence the catalytic efficiency and all the reactions occurred with good stereoselectivities. These first results evidenced the good stereo-control exhibited by porphyrin ligands in inducing the formation of the more thermodynamically stable trans-diastereomer almost independently of the electronic nature of the employed alkene reagents. Thus, catalyst 2 was tested in the cyclopropanation of styrene using diazo derivatives of the general formula N2CH(COOR) (10, R = Me; EDA, R = Et; 11, R = tBu; 12, R = L-menthyl; 13, R = D-menthyl), which showed a different steric hindrance of the R group. Achieved data indicated that the trans-diastereoselectivity was enhanced by increasing the R bulk from a methyl to a tert-butyl group. This trend was not confirmed when using diazoderivatives 12 and 13, where the stereoselectivity was worse than that observed in the reaction run with EDA as the carbene source.
Complex 2 was also active in promoting the intramolecular cyclopropanation of diazoacetate N2CH(COOCH2CH = CR1R2) which formed cyclopropyl lactones in ee up to 85%.27
In order to rationalise the observed catalytic efficiency of complex 2, C.-M. Che and co-authors investigated the reactivity of 2 towards diazo reagents. No products were isolated from the reaction of 2 with EDA, while when using diazo derivatives N2CPh2 (14) or N2C(Ph)(COOCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) (15) the two ruthenium carbene derivatives 16 and 17 were obtained and fully characterised including the X-ray molecular structure determination of complex 16 (Scheme 3).
CH2) (15) the two ruthenium carbene derivatives 16 and 17 were obtained and fully characterised including the X-ray molecular structure determination of complex 16 (Scheme 3).
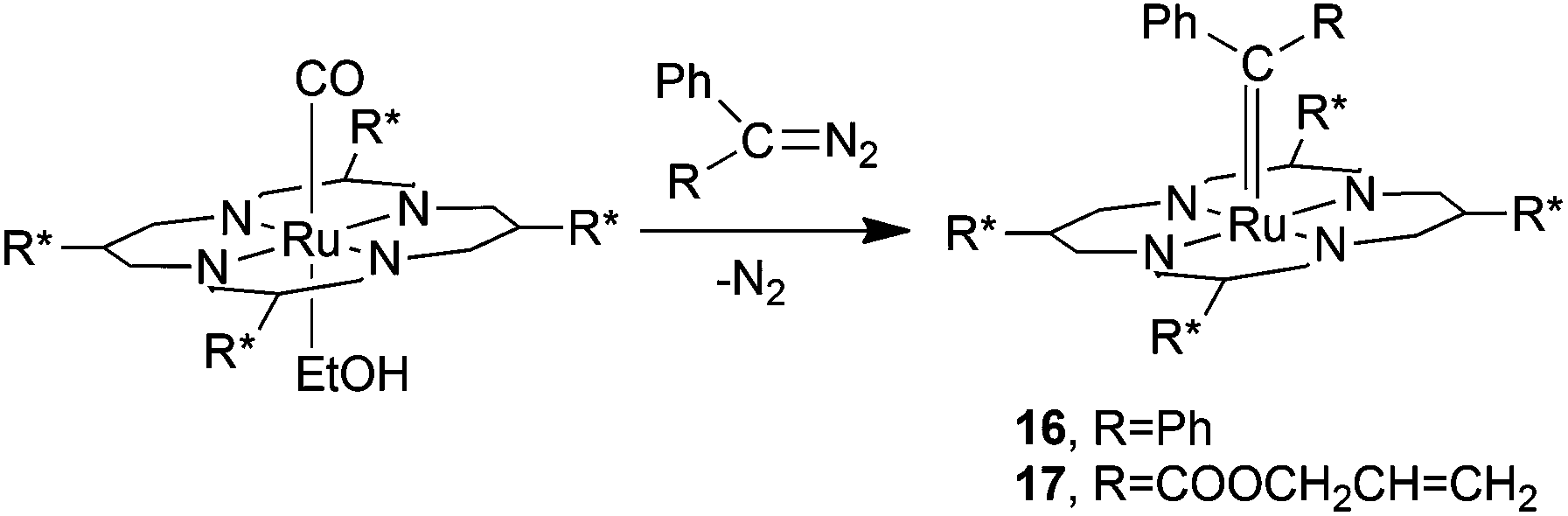 | ||
| Scheme 3 Synthesis of ruthenium carbene complexes 16 and 17.27 | ||
Both complexes 16 and 17 did not transfer the carbene moiety to styrene when treated with stoichiometric amounts of the alkene even at high temperatures. Conversely, complex 16 catalysed the cyclopropanation of styrenes by EDA with an efficiency comparable to that of its ruthenium precursor 2, indicating that pentacoordinated carbene complexes 16 and 17 are probably not the active intermediates of cyclopropanation reactions. In the 16-catalysed cyclopropanation of styrene by EDA, the carbene [Ph2C:] moiety could play the role of an activating L ligand with a strong trans effect to promote, on the other side of the porphyrin plane, the transfer of the [(COOEt)HC:] functionality deriving from EDA.
The observed diastereoselectivity was explained with the formation of the two transition states A and B described in Scheme 4, which were formed as a consequence of the end-on approach of the alkene to the ruthenium carbene species.
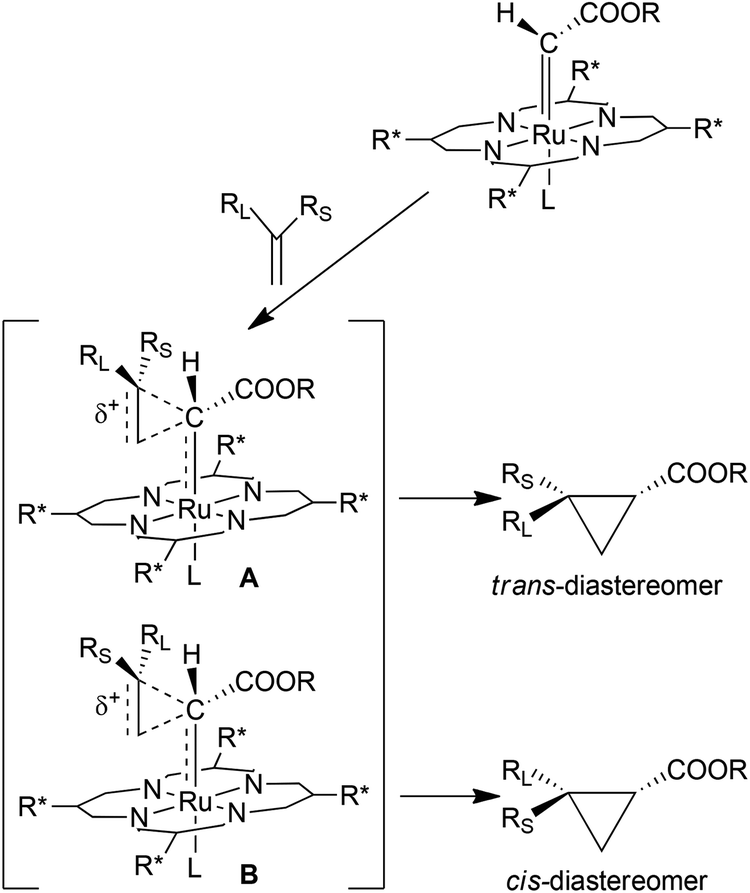 | ||
| Scheme 4 Proposed transition states A and B of the 2-catalysed alkene cyclopropanation.27 | ||
The observed trans-diastereoselectivity might be due to the higher stability of transition state A compared to B for the less extended steric interaction between the larger group RL and the ester group on the carbene moiety. The parallel approach of the alkene to the ruthenium carbene bond could be the reason why terminal alkenes were more reactive substrates than non-terminal ones in complex 2-mediated cyclopropanation reactions. The mechanism of Scheme 4 illustrated the dependence of the reaction stereoselectivity on the steric characteristics of substituents on carbene and alkene fragments.
The catalytic activity of pre-formed carbene complexes was also observed by G. Simonneaux and co-authors28 by testing the reactivity of (Halt*)Ru(CHCO2Ar)(THF) (Ar = 2,6tBu-4Me-C6H2) ruthenium complex 18 which was synthesised by a recrystallization of the product of the reaction of 1 with 2,6-di-tert-butyl-4-methylphenyl diazoacetate in the presence of THF. Complex 18 was catalytically active in the cyclopropanation of styrene by 2,6-di-tert-butyl-4-methylphenyl diazoacetate yielding the corresponding cyclopropane 19 in 62% yield, complete trans-diastereoselectivity and 60% of eetrans (Scheme 5).
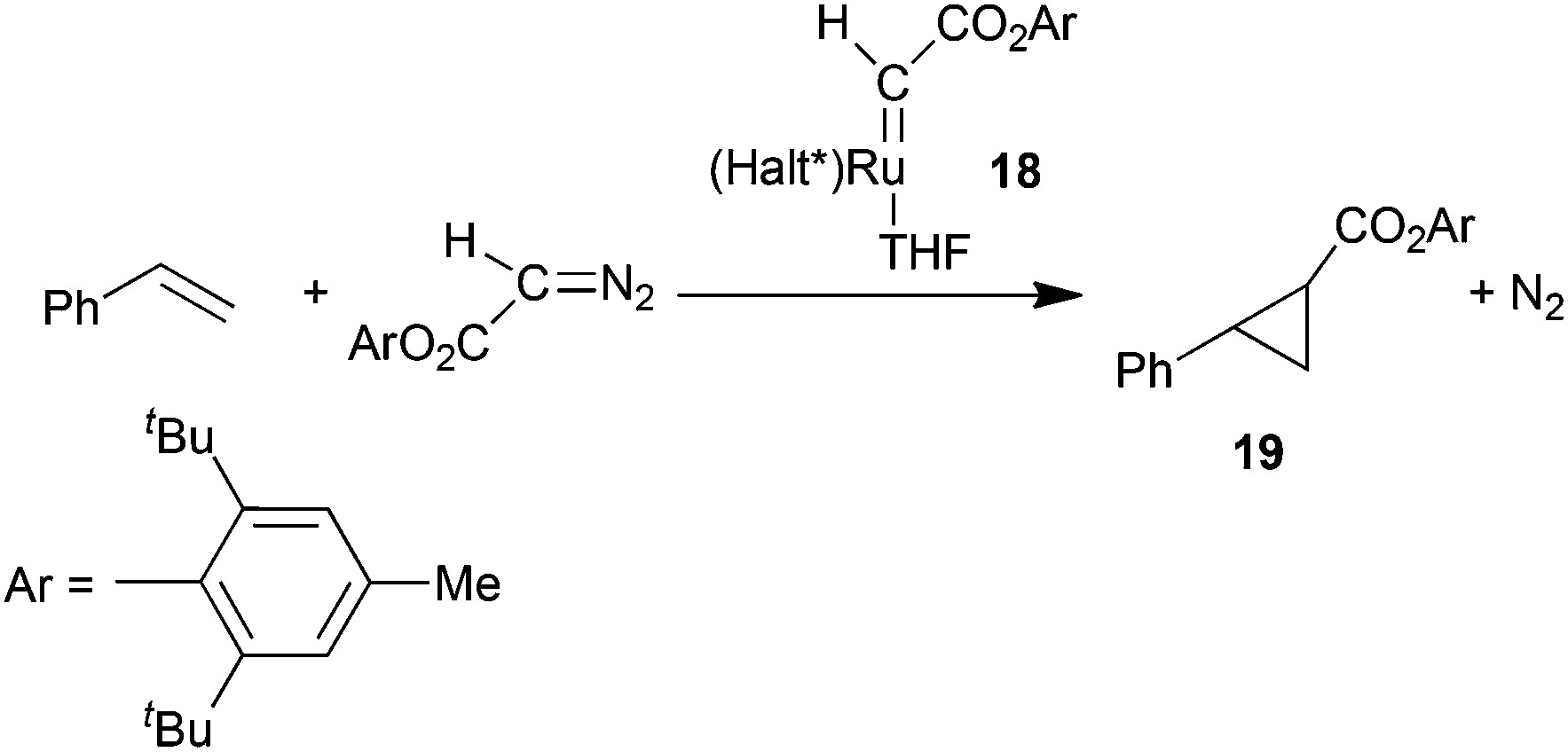 | ||
| Scheme 5 Structure of ruthenium carbene complexes 18 and its catalytic activity.28 | ||
In order to expand the study of the catalytic capacity of Halterman-type chiral porphyrins, A. Berkessel and co-authors synthesised ruthenium porphyrins similar to 1 (Fig. 3) which exhibit X = OMe (20), Me (21) and CF3 (22).29 In addition, also complex (Halt*)Ru(PF3) (23) was obtained to evaluate the effect of an axial ligand on the ruthenium centre. All of the complexes 20–23 showed a good catalytic efficiency in the cyclopropanation of styrenes, and achieved data showed that the tetrakis-CF3-substituted complex 22 was the most active and stable catalyst of all the tested catalysts. Considering that the steric hindrance of the Halterman ruthenium porphyrin 1 is not influenced by the introduction of a CF3 substituent on the X position (Fig. 3), the improvement of the catalytic performance was only due to electronic factors which can affect the catalyst stability more than the stereo-control of the reaction. In fact, a high TON (turnover number) of 7520 was observed in the 22-catalysed cyclopropanation of styrene by EDA.
The comparison of the catalytic activity of complexes 1 (Fig. 3, X = H, L = CO) with that of complex 23 (Fig. 3, X = H, L = PF3) revealed an increase in the reaction time with a contemporary decrease in the catalytic efficiency. The authors suggested that two different catalytic intermediates were formed from the two pre-catalysts 1 and 23. In the case of the 1-catalysed cyclopropanation, the CO axial ligand was lost to form a five-coordinated carbene active species, while when complex 23 was used as the catalyst, the PF3 axial ligand remained coordinated to the metal centre during the entire catalytic cycle and a pseudo-octahedral carbene intermediate was responsible for the cyclopropanation. It is important to note that complex 23 was active in the cyclopropanation of non-conjugated terminal olefins such as 1-octene where the corresponding cyclopropane was obtained with 99.5:0.5 of d.r.trans and 82% of eetrans. In addition, 23 promoted the cyclopropanation of styrene and α-methylstyrene by phenyl diazomethane (N2CH(Ph)) and the corresponding cyclopropanes were formed with a trans/cis ratio of 51:49 and 83:17 and eetrans of 72% and 96% respectively.29
The influence of axial ligands in determining the catalytic efficiency of ruthenium porphyrin complexes was also studied by C.-M. Che and co-authors, who reported the synthesis of (Halt*)Ru(IMe)2 (24) (IMe = 1,3-dimethyl-2,3-dihydro-1H-imidazole) which showed two N-heterocyclic carbene (NHC) axial ligands on the ruthenium centre.30 The cyclopropanation of styrene by EDA promoted by the low catalyst amount of 0.004 mol% occurred with 95% yield, 95:5 d.r.trans and 95% eetrans. Complex 24 was also active in catalysing the carbene insertion into a C–H bond of 1,4-cyclohexadiene using methyl phenyldiazoacetate (MPDA) (N2C(Ph)(COOMe)) as a carbene source.30 The insertion of the carbene functionality into the vinyl C–H bond afforded the desired compound in 80% yield with 92% ee (Scheme 6).
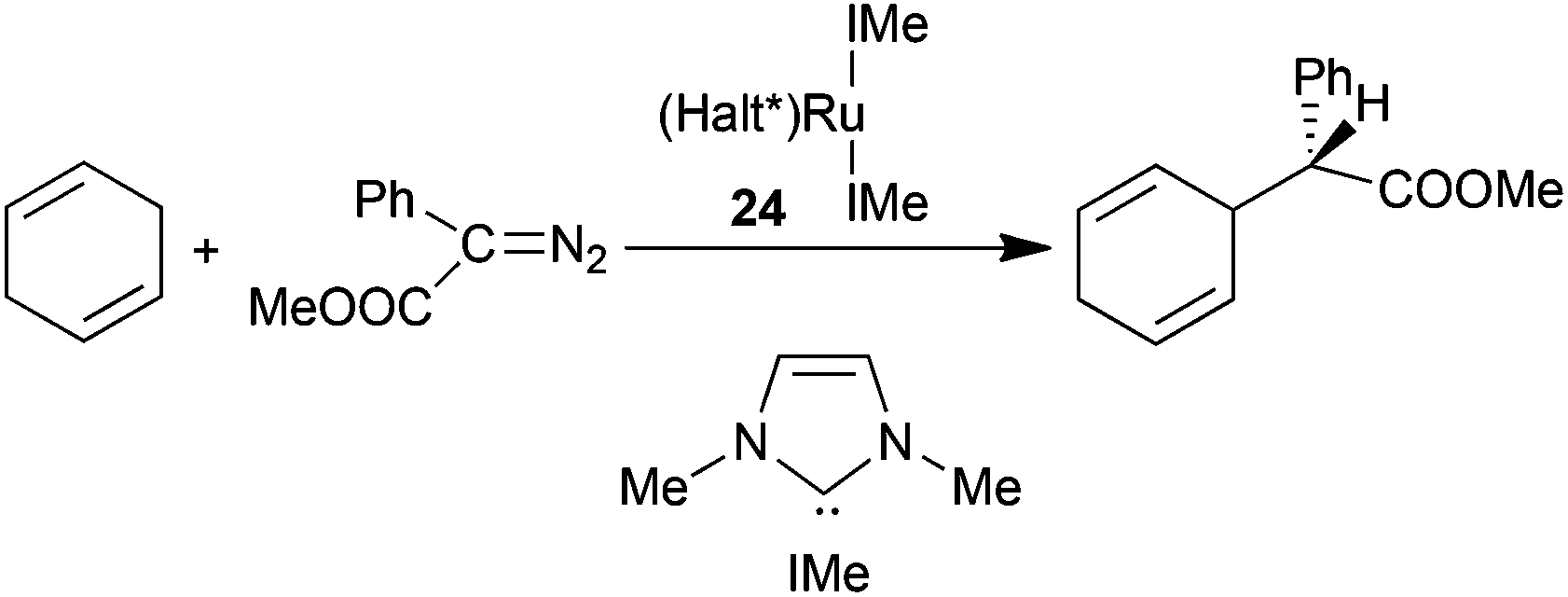 | ||
| Scheme 6 Complex 24-catalysed insertion of a carbene functionality into a vinyl C–H bond.30 | ||
As already discussed above,27 also in this case the presence of a carbene axial ligand had a positive effect on the efficiency of the cyclopropanation reaction. A DFT study indicated that the N-heterocyclic carbene ligands exhibited a strong σ donor strength which stabilises the ruthenium–carbene on the trans position with a consequential decrease of the activation barrier for the decomposition of diazo compounds.
In order to expand the reaction scope of enantioselective cyclopropanations catalysed by ruthenium porphyrin 1 (Fig. 3), G. Simonneaux and co-authors reported the use of diazo ketone N2CH(COPh), diisopropyl diazomethylphosphonate N2CH(OP(OiPr)2 (DAMP) and 2,2,2-trifluorodiazoethane N2CH(CF3) for the synthesis of optically active cyclopropylketones 25–29,31trans-cyclopropylphosphonates 30–3432 and trifluoromethylphenyl cyclopropanes 35–37,33 respectively (Scheme 7).
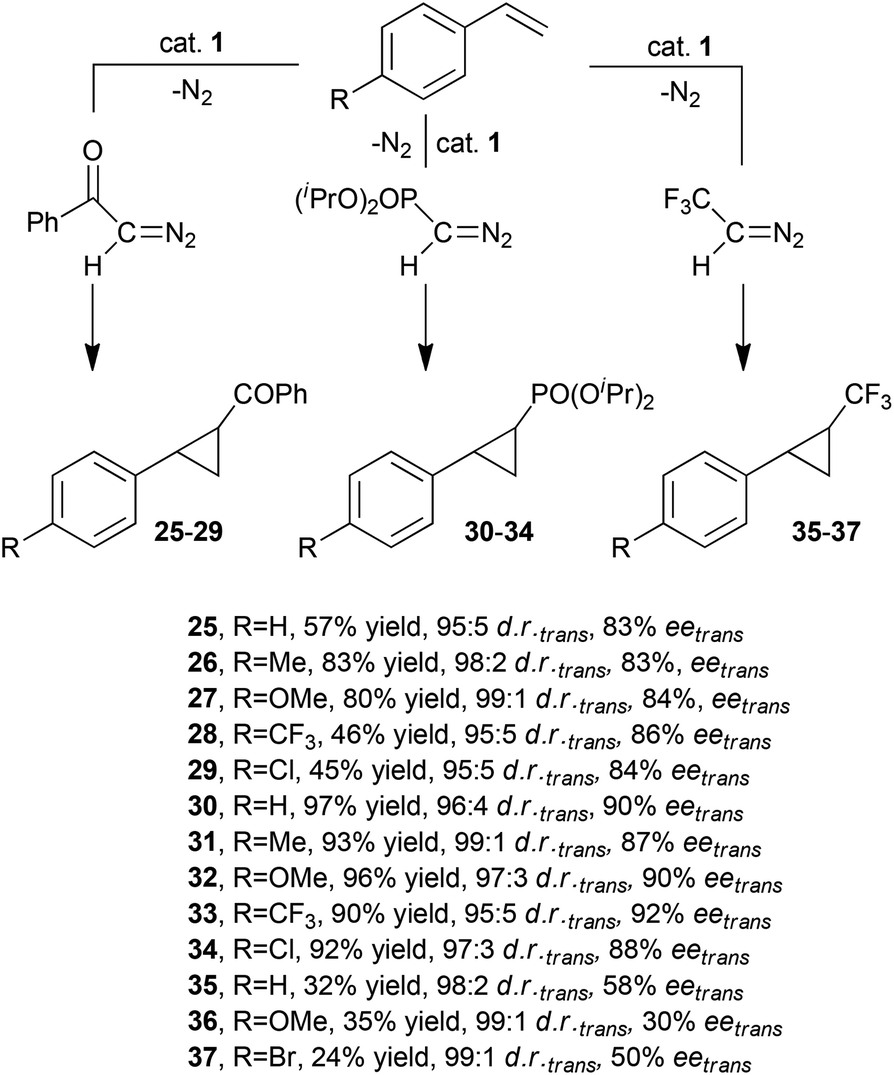 | ||
| Scheme 7 Complex 1-catalysed synthesis of cyclopropylketones 25–29,31trans-cyclopropylphosphonates 30–3432 and trifluoromethylphenyl cyclopropanes 35–37.33 | ||
As shown in Scheme 7, the synthesis of compounds 25–29 was more productive when using electron rich alkenes than electron poor ones, while the electronic features of the unsaturated substrate did not influence the reaction stereoselectivity. trans-Cyclopropylphosphonates 30–34 were obtained in comparable yields and stereoselectivities whereas the reaction efficiency of the synthesis of compounds 35–37 was modest.
The stoichiometric reaction of DAMP with complex 1 gave a ruthenium carbene complex, (Halt*)Ru=CHPO(OiPr)2 (38), which displayed a catalytic activity in the styrene cyclopropanation by DAMP similar to that reported for complex 1. These results support a possible involvement of 38 as a catalytic intermediate. However, the stoichiometric transfer reaction from 38 to styrene derivatives was not reported.
Catalyst 1 was also efficient in promoting the cyclopropanation of styrene by N- and O-protected 6-diazo-5-oxo-L-norleucine (DON), which is a biological compound with antibiotic features. The corresponding cyclopropane was obtained in 75% yield, with excellent trans-diastereoselectivity (trans/cis = 99:1) and 80% of eetrans. This study was undertaken to better understand some in vivo processes where enzymes containing metal porphyrins could be involved.34
In order to mimic biological processes where metal porphyrin complexes could play important roles, it was important to test the reactivity of porphyrin catalysts in water. In 2008 G. Simonneaux and co-authors synthesised the para-tetrasulfonated water soluble Halterman-type porphyrin 39 (Fig. 3, X = SO3Na, L = none).35 The 39-catalysed cyclopropanation of styrene occurred in 52% yield, with 96:4 of d.r.trans and 83% eetrans. The catalyst recycle was also investigated and a progressive decrease in the enantiomeric excess was observed during the second (eetrans = 62%) and the third (eetrans = 40%) cycles.
Ruthenium Halterman type porphyrins were also supported in heterogeneous systems to permit a better catalyst recyclability and reuse. The first example of heterogenization of chiral ruthenium porphyrin catalysts on ordered molecular sieves for cyclopropanation reactions was reported in 2002 by C.-M. Che.36 Catalyst 1 was supported on the ordered mesoporous silica MCM-48, which contains a three-dimensional porous structure to assure an efficient accessibility of reagents to catalytically active sites. Heterogeneous catalyst 40 was employed to promote the intramolecular cyclopropanation of trans-cinnamyl diazoacetate to yield the desired compound with a high enantioselectivity of 85%. The excellent stability of heterogeneous catalyst 40 endorsed recycling for four consecutive times at the high TON of 1.5 × 103 (Scheme 8).
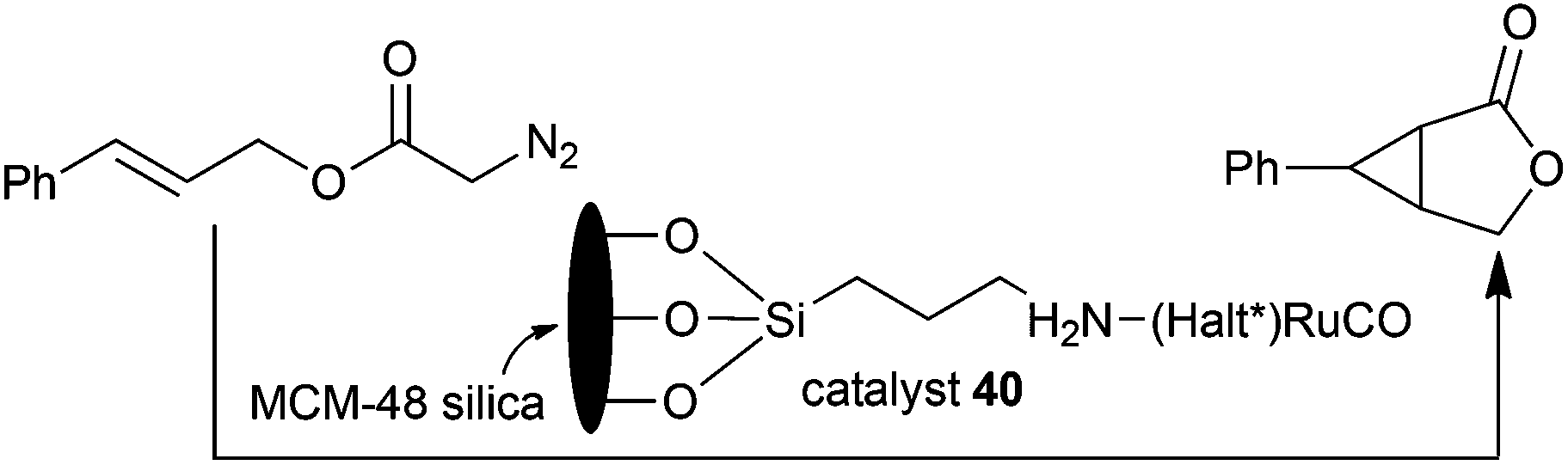 | ||
| Scheme 8 Heterogeneous intramolecular cyclopropanation of trans-cinnamyl diazoacetate catalysed by 40.36 | ||
Then, Halterman type ruthenium porphyrins 41 and 42 (Fig. 4), showing spirobifluorenyl groups on meso positions, were embedded in a polymeric support by an anodic oxidation in the presence of 9,9′-spirobifluorene (SBF) which provoked the coating of the electrode by an optically active film.37 The removal of the electrode produced a chiral polymer which was tested in the enantioselective cyclopropanation of styrene by EDA that occurred with good yields but only modest enantioselectivities (up to 53% at −40 °C). The good stability of catalysts poly-41 and poly-42 permitted their reuse for seven consecutive times without observing a considerable decrease in the catalytic efficiency in terms of both activity and enantioselectivity. The comparison of the catalytic activities of complexes 41 and poly-41 revealed a decrease in the reaction enantioselectivity from 76% to 53% after the anodic oxidation of the monomeric complex 41. It was envisaged that the cross-linked polymeric structure can hinder the interaction of substrates with the active sites and that a partial oxidation of ruthenium(II) to ruthenium(III) or (IV) occurred during the electropolymerization and it was responsible for a decrease of catalytically active ruthenium(II) centres.
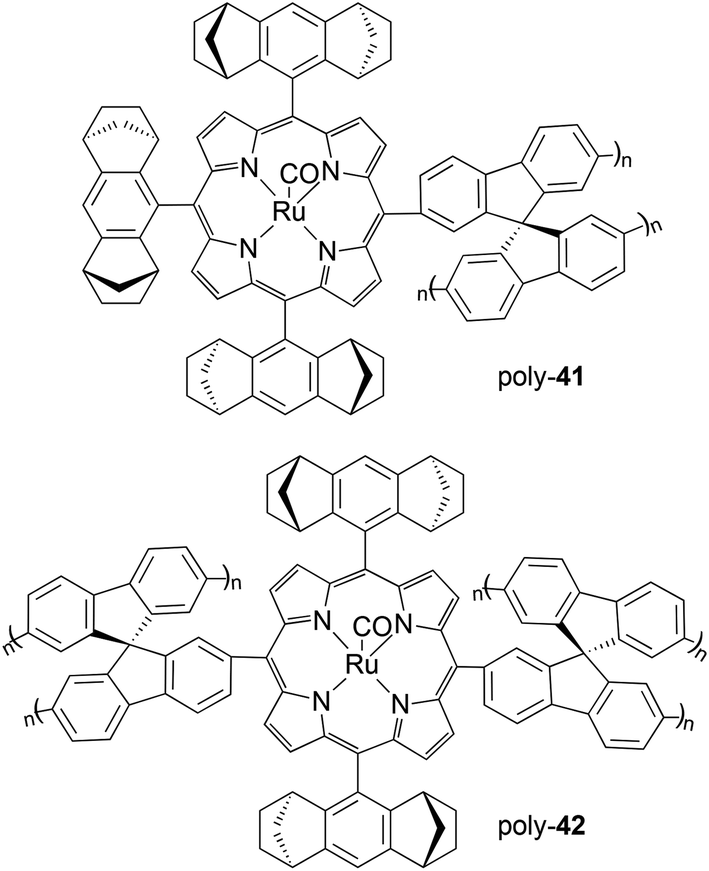 | ||
| Fig. 4 Structures of chiral ruthenium polymeric complexes poly-41 and poly-42.37 | ||
In order to improve the stereo-control accomplished by polymeric heterogeneous catalysts, G. Simonneaux and co-authors explored the polymerization of Halterman type ruthenium porphyrins functionalised with four vinyl groups.38 Complex 43 (Fig. 3, X = –CH2CH2, L = none) was polymerised with styrene and divinylbenzene forming the supported poly-43 ruthenium complex, which was tested in the cyclopropanation of styrenes by EDA,38 diazoacetonitrile38 (N2CH(CN)) and 2,2,2-trifluorodiazoethane33 (N2CH(CF3)) (Scheme 9). Desired compounds 3, 8 (Scheme 2) 35–37 (Scheme 7) and 44–47 (Scheme 9) were obtained with good enantiomeric excesses (up to 90%), which, in some cases, were comparable to those obtained using homogeneous chiral ruthenium porphyrins (compare data in Schemes 2 and 7 with those of Scheme 9).
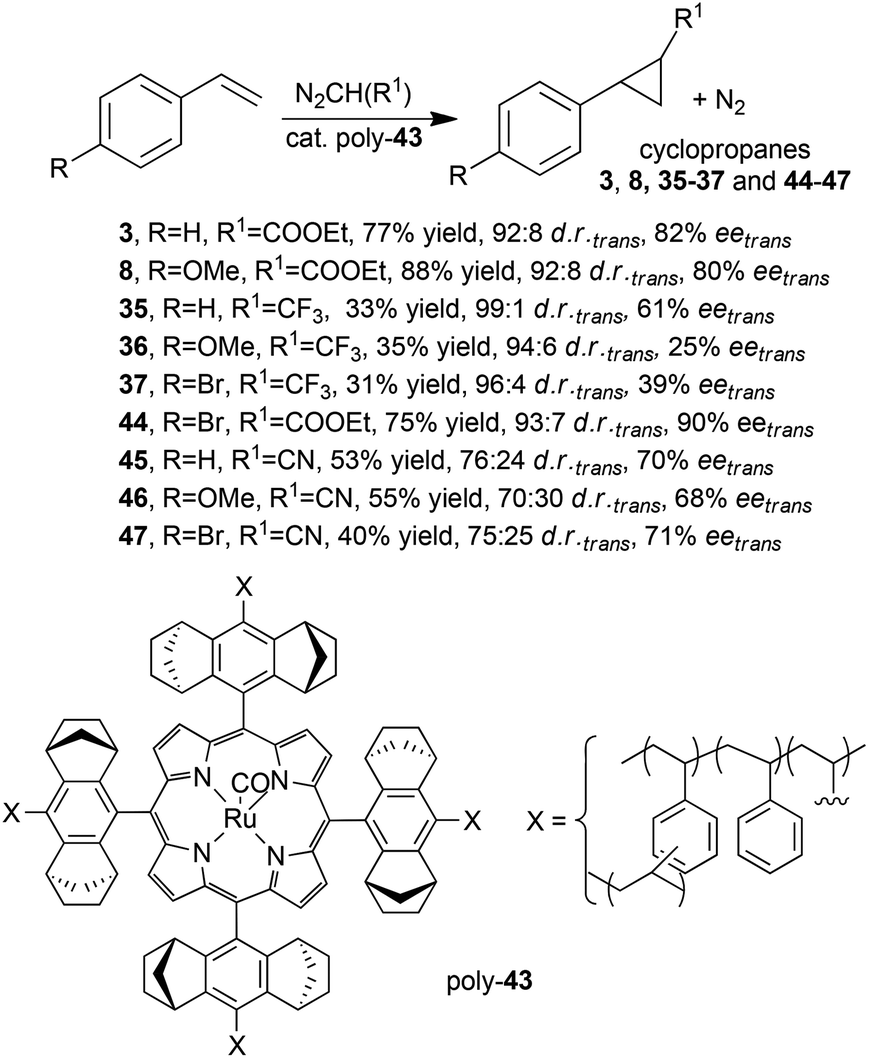 | ||
| Scheme 9 Catalytic activity of poly-43 in the cyclopropanation of styrenes by EDA,38 diazoacetonitrile38 or 2,2,2-trifluorodiazoethane.33 | ||
It important to point out that the asymmetric cyclopropanation with diazoacetonitrile afforded optically active trans-cyanocyclopropanes 45–47 which are precursors of biologically active compounds. The modest diastereoselectivities observed when diazoacetonitrile was used as the carbene precursor were probably due to the small size of the CN group which disfavours the trans-diastereoselectivity. The authors attributed the observed low yields of compounds 35–37 to the high volatility of the starting diazo 2,2,2-trifluorodiazoethane compound.
Chiral D4 symmetric porphyrins are also good ligands to synthesise active iron derivatives. The interest of the scientific community in developing iron porphyrin-based catalytic protocols for carbene transfer reactions is mainly due to the sustainability of the active, low toxic and cheap iron catalytic centre.39 C.-M. Che and co-authors reported in 200640 the first use of (Halt*)Fe(III)Cl complex 48 to catalyse carbene transfer reactions. The catalyst was slightly less active than the counterpart ruthenium derivative 1 or 2 (Fig. 3) to promote the formation of compounds 3–5 and 7–9 (Scheme 2). These cyclopropanes were obtained with comparable trans-diastereoselectivities but minor enantioselectivities of the trans isomer (Scheme 10).
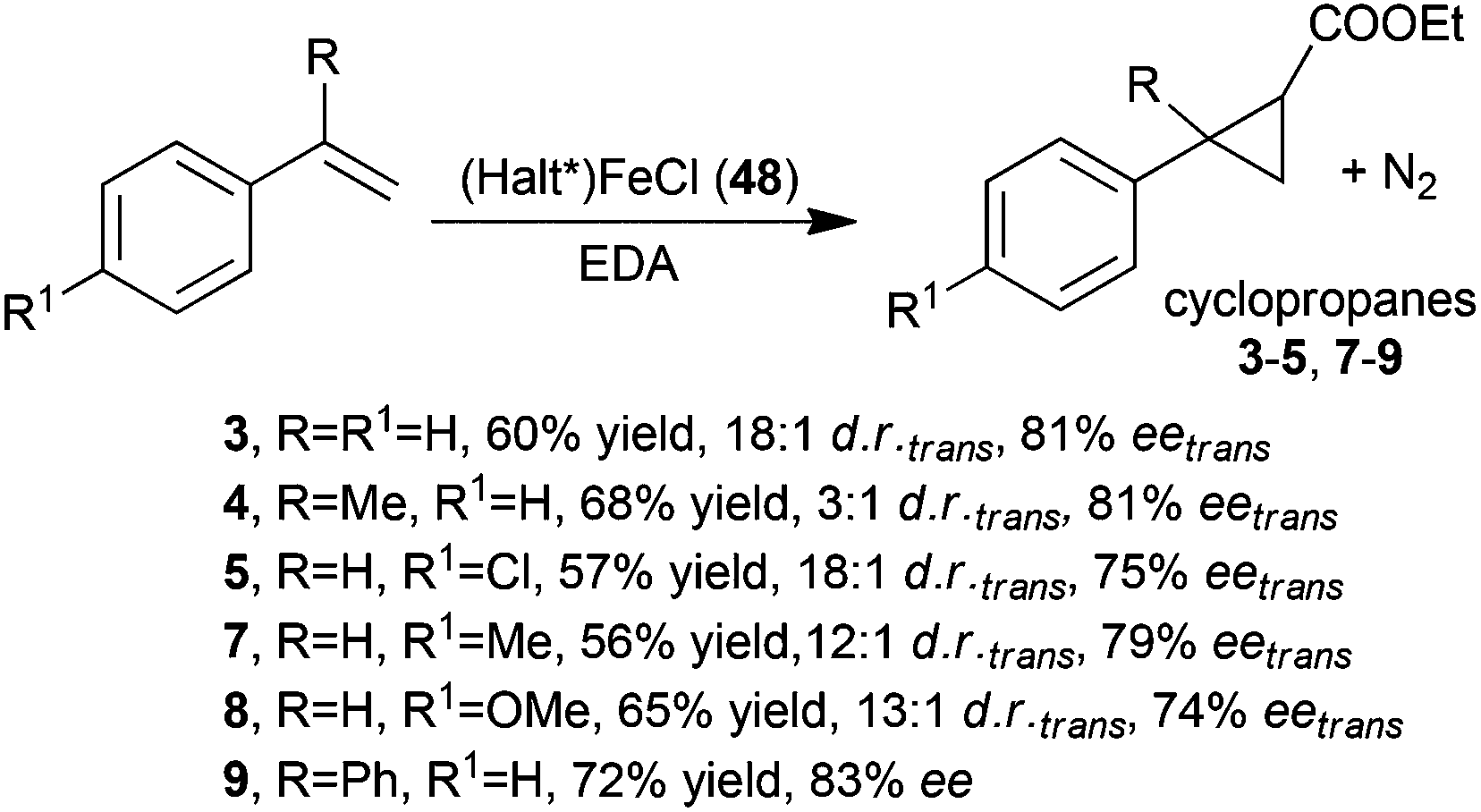 | ||
| Scheme 10 Iron catalyst 48-mediated cyclopropanation of styrenes by EDA.40 | ||
The cyclopropanation reactions occurred without the addition of a reductive species, which is supposed to reduce the iron(III) pre-catalyst into an iron(II) species able to react with the diazo reagent to form the iron(IV) carbene active intermediate.41 The authors proposed that EDA played a double role of a carbene source and a reductive agent as already proposed for copper(II) catalysed cyclopropanations.42 This last hypothesis was supported by the pronounced decrease of the catalytic efficiency observed when the reaction was run in air where a supposed iron(II) intermediate can be easily decomposed. The trans-selectivity of the reaction was enhanced by adding an axial ligand to the catalytic mixture to indicate the possible formation of an axially ligated monocarbene active intermediate. The existence of these species was supported by the electrospray mass spectrometry (ESMS) analysis of the reaction of 48 with EDA, run in the presence of pyridine (py) or 1-methylimidazole (MeIm), which detected the formation of complexes (Halt*)Fe(CHCOOEt)(py) (49) and (Halt*)Fe(CHCOOEt)(MeIm) (50), respectively. However the latter complexes were never isolated or detected in the presence of alkene substrates.
G. Simonneaux and co-authors reported that iron complex 48 was also active in the cyclopropanation of styrenes by diazoacetophenone43 and 2,2,2-trifluorodiazoethane33 that were efficiently promoted by ruthenium complex 1.31,33
Data shown in Scheme 11 revealed that 48 was less efficient than 1 in terms of cyclopropane yields and trans-enantioselectivities; however similar trans-diastereoselectivities were observed.
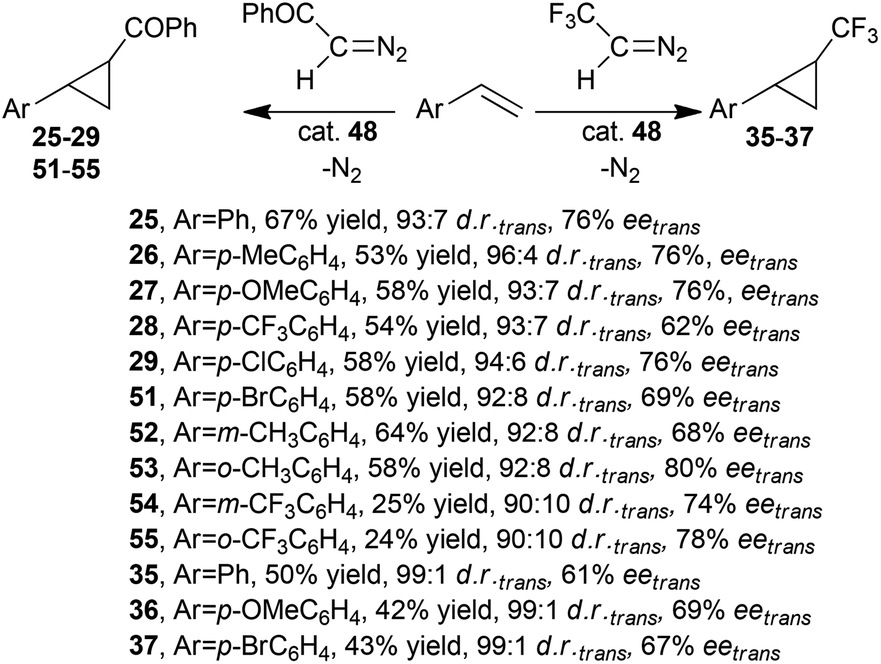 | ||
| Scheme 11 Catalytic activity of (Halt*)FeCl (48) in the cyclopropanation of styrenes by diazoacetophenone43 and 2,2,2-trifluorodiazoethane.33 | ||
G. Simonneaux and co-authors also reported a heterogeneous version of the synthesis of compounds 35–37 using an iron compound which displays a structure analogous to that shown in Scheme 9 for the ruthenium derivative (Ru(II)CO of poly-43 was replaced by Fe(III)Cl in the porphyrin core to yield poly-48).33 Complex poly-48 showed a catalytic efficiency comparable to the homogeneous counterpart 48 for yields and diastereoselectivities, whereas a decrease in the enantioselectivities was observed in all the tested reactions (35, 52% yield, 97:3 d.r.trans and 56% eetrans; 36, 51% yield, 96:4 d.r.trans and 17% eetrans; 37, 42% yield, 97:3 d.r.trans and 37% eetrans).
In order to envisage the application of iron chiral porphyrins for the synthesis of biologically active compounds, the reactivity of pharmaceutically active diazo derivatives as well as the use of water as the medium of catalytic reactions were studied. First, catalyst 48 was active to promote the reactivity of the antibiotic species N- and O-protected 6-diazo-5-oxo-L-norleucine (DON) towards styrene yielding the corresponding cyclopropane in 95% yield, 95:5 trans-diastereoselectivity ratio and 80% eetrans.34 Then, the introduction of sulfonate groups of the porphyrin skeleton of 48 yielded complex 56, which catalysed the reaction between styrene and EDA in water to yield cyclopropane 3 in 85% yield, 92:8 d.r.trans and 83% d.r.trans (Scheme 12).35
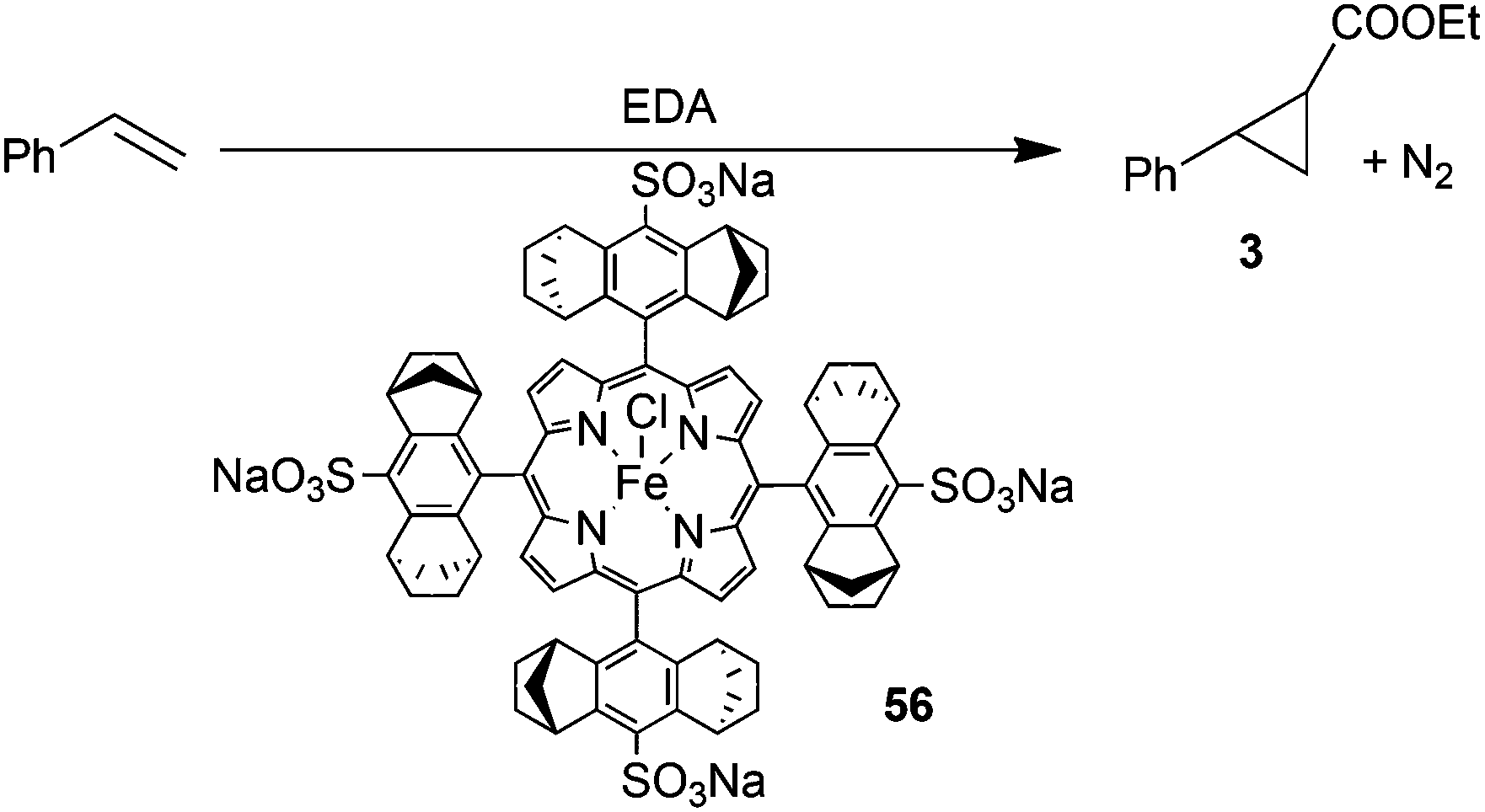 | ||
| Scheme 12 Iron catalyst 56-mediated cyclopropanation of styrene by EDA in water.35 | ||
It should be noted that the reaction of Scheme 12 occurred in the presence of cobaltocene which seems necessary to reduce the starting iron(III) pre-catalyst into an iron(II) species active towards the diazo compounds.
2.2 Chiral catalysts of group 9
Considering the fact that all the Halterman-type porphyrins discussed up to now do not present substantial structural dissimilarities in their ligand skeleton, the differences which were observed in the reaction stereoselectivities should be due to the nature of the catalytically active metal centre.Therefore the synthesis of rhodium(III)44,45 and iridium(III)46,47 complexes of Halterman chiral porphyrin (Fig. 5) was of great interest in comparing their catalytic performances in carbene transfer reactions with those already reported for iron and ruthenium derivatives.
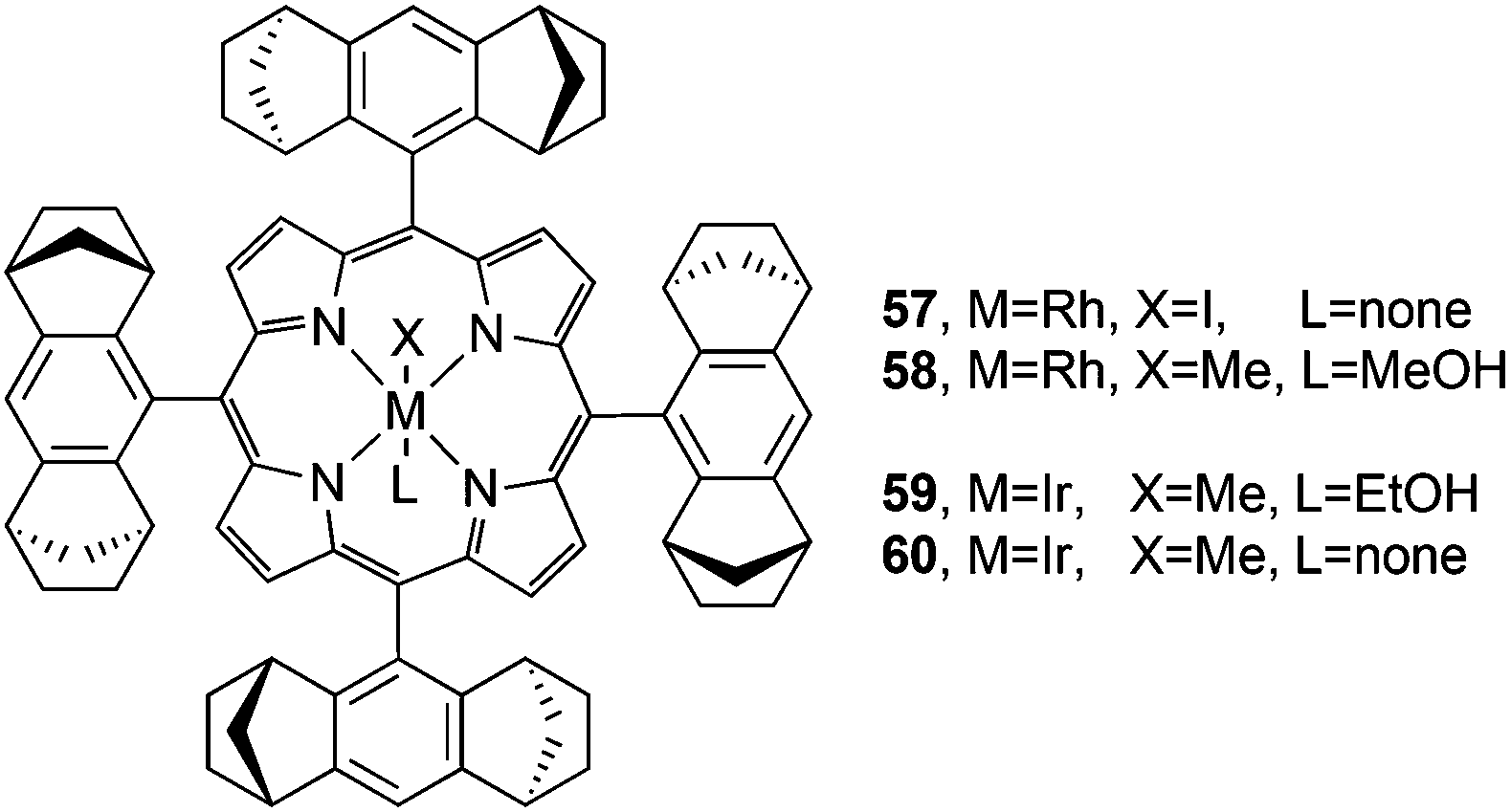 | ||
| Fig. 5 Structures of rhodium and iridium 57,4458,455946 and 6047 catalysts used to promote carbene transfer reactions. | ||
The synthesis of compounds 3–9 was performed in the presence of catalyst 57 and achieved data shown in Scheme 13 revealed minor performance of the rhodium catalyst with respect to the ruthenium counterpart (compare Schemes 13 and 2).
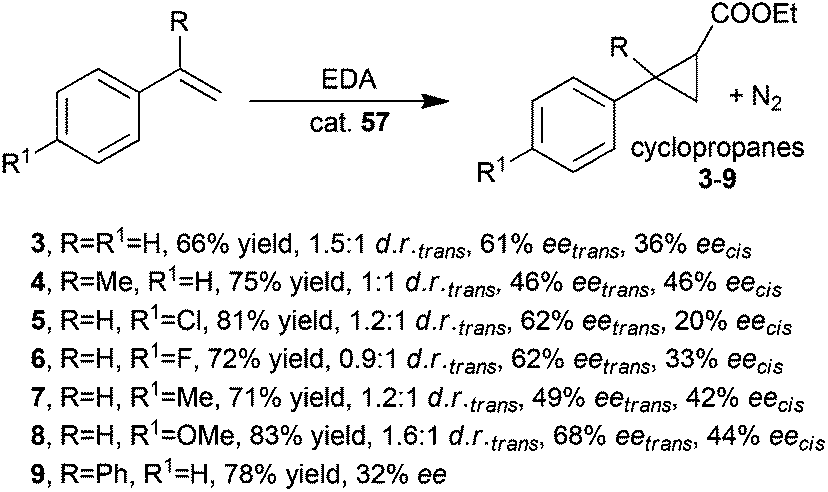 | ||
| Scheme 13 Cyclopropanation of styrenes promoted by complex 57.44 | ||
Complex 57 was also active in catalysing the intramolecular cyclopropanation of alkenes yielding the corresponding lactones 61–70 in moderate to good yields but with modest enantioselectivities (Scheme 14).44
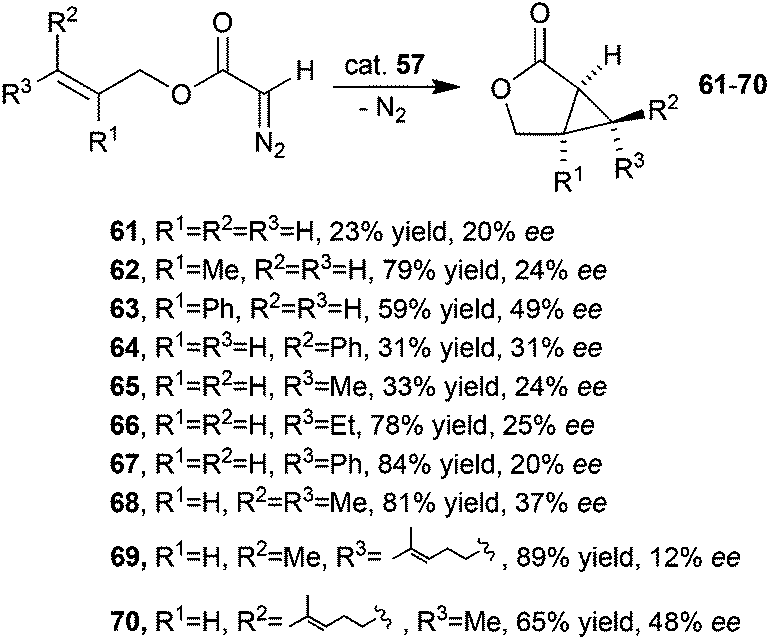 | ||
| Scheme 14 Synthesis of lactones 61–70 by 57-catalysed intramolecular cyclopropanation.44 | ||
Rhodium(III) complex 58 (Fig. 5) displayed a very good catalytic activity in the intermolecular carbenoid insertion into C–H bonds using N2C(Ph)(COOMe) (MPDA) as the carbene source.45 Compounds 71–77 shown in Scheme 15 were formed in up to 80% yield and up to 93% ee. It should be noted that the methodology was more efficient for primary than for secondary C–H bonds and 71a and 72a were formed in a better yield than the other isomers 71b/71c and 72b, respectively.
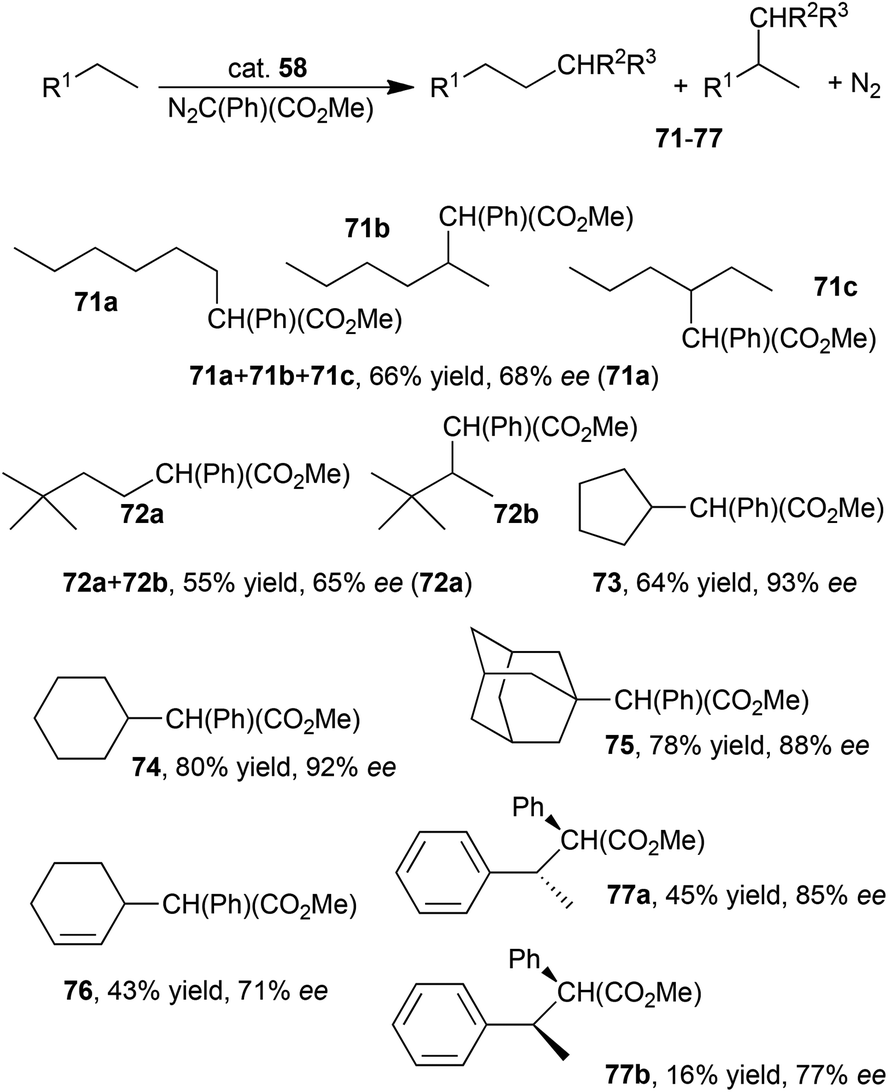 | ||
| Scheme 15 Complex 58-catalysed intermolecular C–H bond insertion.45 | ||
Complex 58 showed a very good chemical stability which allowed the catalyst to be reused for at least five consecutive times. In addition, the synthesis of compound 74 was also performed on a large scale affording 2.88 g of the desired reaction products with 73% yield and 91% ee.
Iridium complex 59 was employed by C.-M. Che and co-authors for catalysing the stereoselective intermolecular carbene insertion into both C–H and Si–H bonds.46 The intermolecular alkylation of 1,4-cyclohexadiene with methyl phenyl diazoacetate (MPDA) was firstly studied and then the reaction scope was investigated by testing the reactivity of other methyl aryl diazoacetates. The obtained data indicated that substituents at positions 3 and 4 of the aryl moiety did not influence the reaction performance as well as α-thienyl and naphthyl substituents. The obtained alkylated compounds 78–85 are shown in Scheme 16.
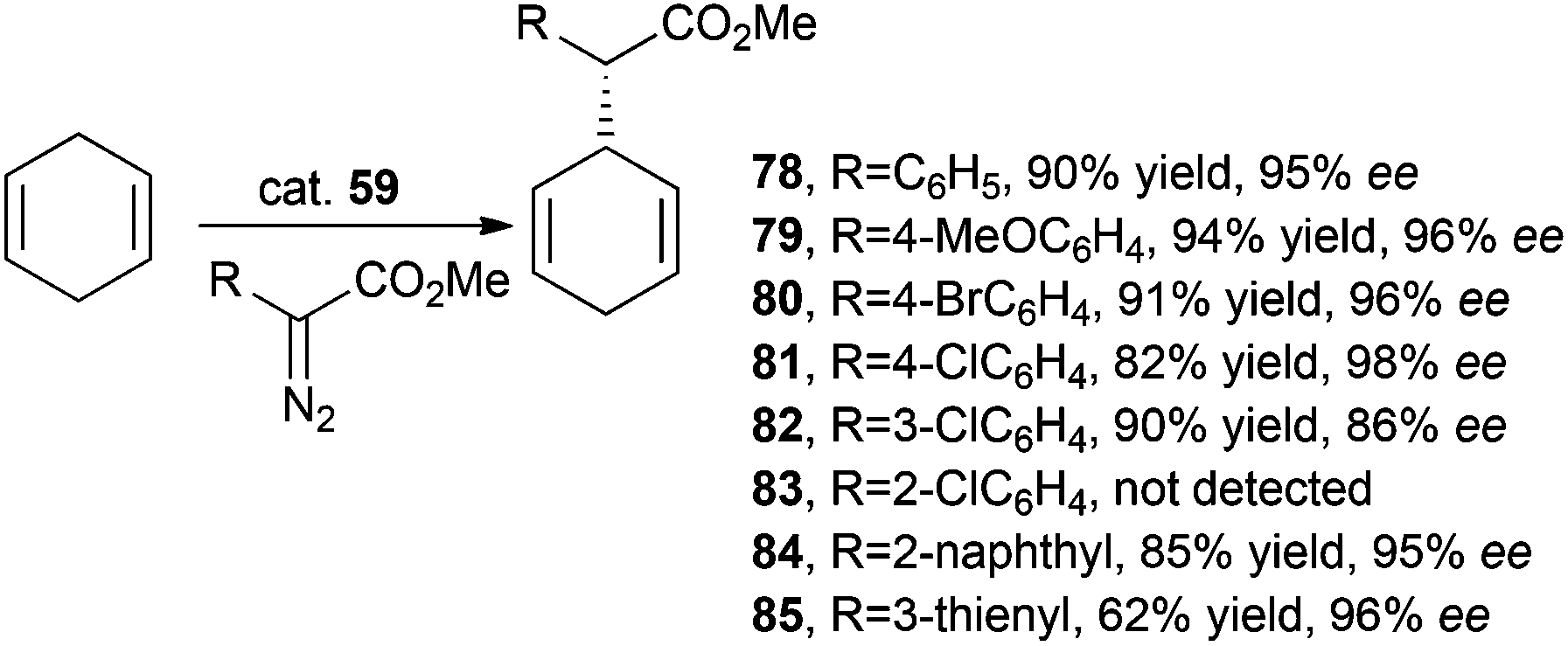 | ||
| Scheme 16 Complex 59-catalysed synthesis of compounds 78–85.46 | ||
Iridium complex 59 was efficient in catalysing the reaction between tetrahydrofurane (THF) and methyl aryl diazoacetate derivatives. Carbene transfer reactions occurred in good yields, with trans/cis diastereoselectivities up to 20:1 and trans-enantioselectivities up to 97%. Only the synthesis of compounds 90 and 93 occurred in low yields, but with very good enantioselectivities (Scheme 17).
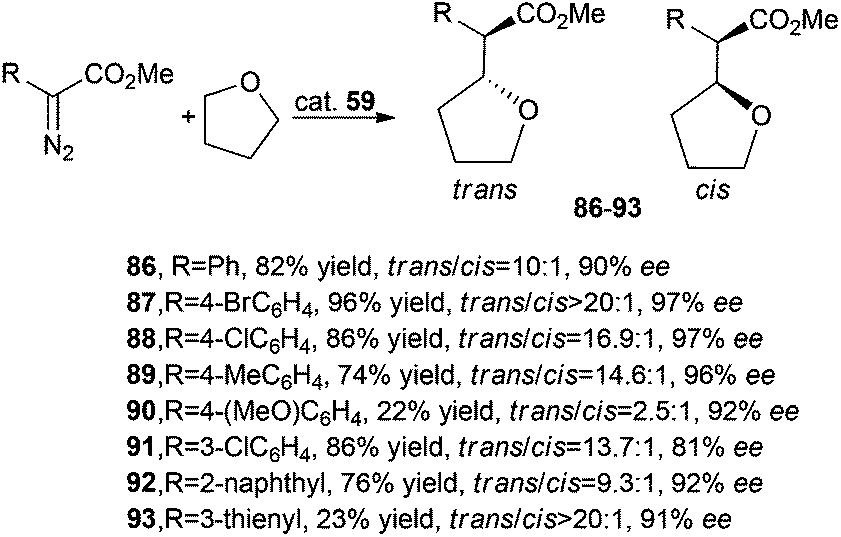 | ||
| Scheme 17 Complex 59-catalysed synthesis of compounds 86–93.46 | ||
Complex 59 was also active in promoting the insertion of a carbene moiety into Si–H bonds.46 Products 94–100 shown in Scheme 18 were obtained in high yields and enantioselectivities.
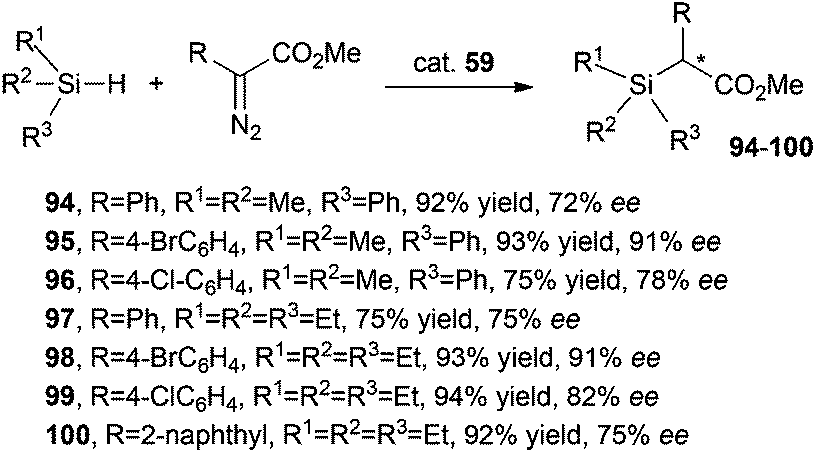 | ||
| Scheme 18 Complex 59-catalysed synthesis of compounds 94–100.46 | ||
The activity of iridium complex 60 was investigated by C.-M. Che and co-authors in catalysing the intramolecular C–H carbene insertion using several α-diazoesters as starting materials to selectively synthesise cis-β-lactones.47 Achieved data indicated that electron-donating R1 groups, such as a methyl group, were responsible for lower yields while the presence of an encumbered R2 substituent had a negative effect on the reaction efficiency due to steric reasons (Scheme 19).
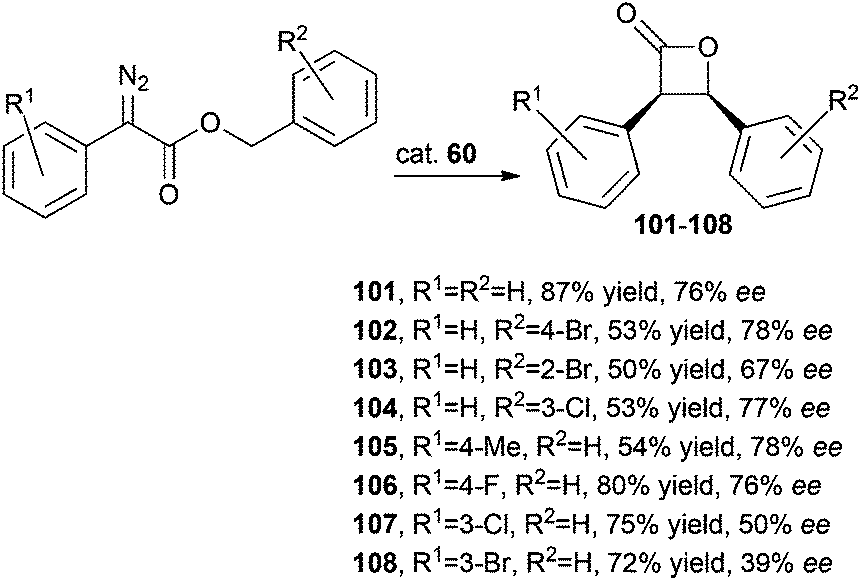 | ||
| Scheme 19 Complex 60-catalysed synthesis of enantiopure cis-β-lactones 101–108.47 | ||
As described up to now, Halterman-type porphyrins have been extensively used for synthesising metal complexes active in carbene transfer reactions. The observed stereo-control, due to the chiral ligand skeleton, was only in part modified by the nature of the porphyrin core, which was more responsible for the reaction chemoselectivity. In fact, when other D4 symmetrical chiral porphyrins were used in promoting carbene transfers, worse results were achieved, indicating the active role of the Halterman porphyrin skeleton in determining the stereo-control of the reaction. Metal porphyrins 109,4811049 and 11149 were tested in the cyclopropanation of styrene and α-methylstyrene by EDA affording compounds 3 and 4, respectively (Scheme 2).
It should be noted that, even if the reaction always occurred with modest to poor stereoselectivities (Scheme 20), cobalt(II) catalyst 109 promoted a cis-diastereoselectivity conversely to that which was reported for the Halterman-type-catalysed cyclopropanations discussed so far.
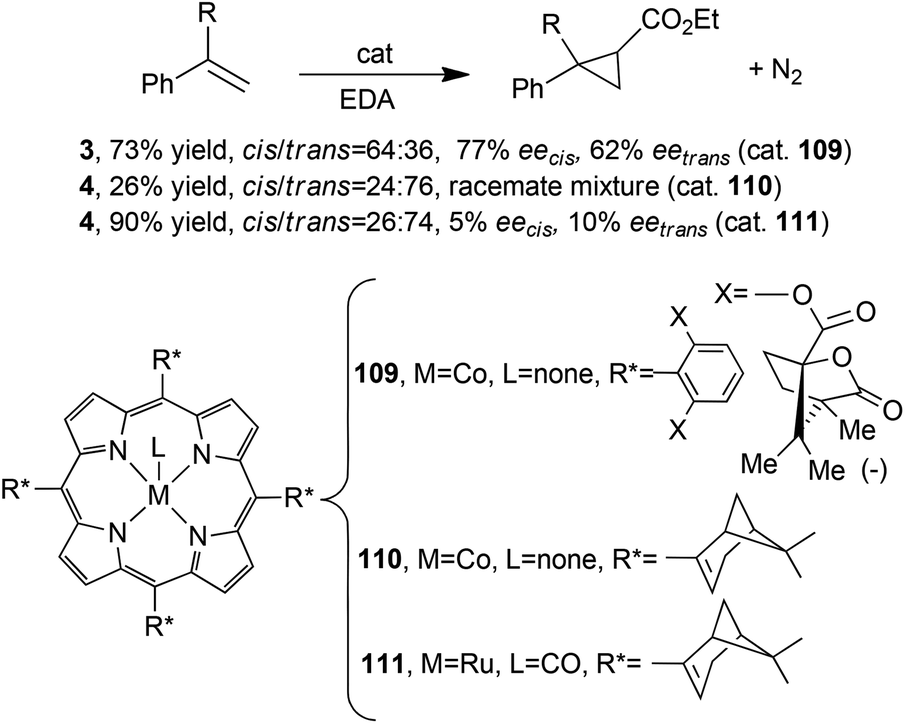
3. D 2-Symmetric catalysts
Considering that the stereo-control of carbene transfer reactions is strictly related to the porphyrin skeleton, many efforts have been made up to now to structurally modify the ligand to create a befitting ‘active pocket’ around the active metal where the catalytic reaction takes place.In 1992 T. Kodadek and co-authors reported the synthesis of two rhodium complexes 112 and 113 deriving from αβαβ porphyrin precursors (Fig. 2, structure C), namely ‘Chiral Wall’50 and ‘Chiral Fortress’,51 respectively (Fig. 6).
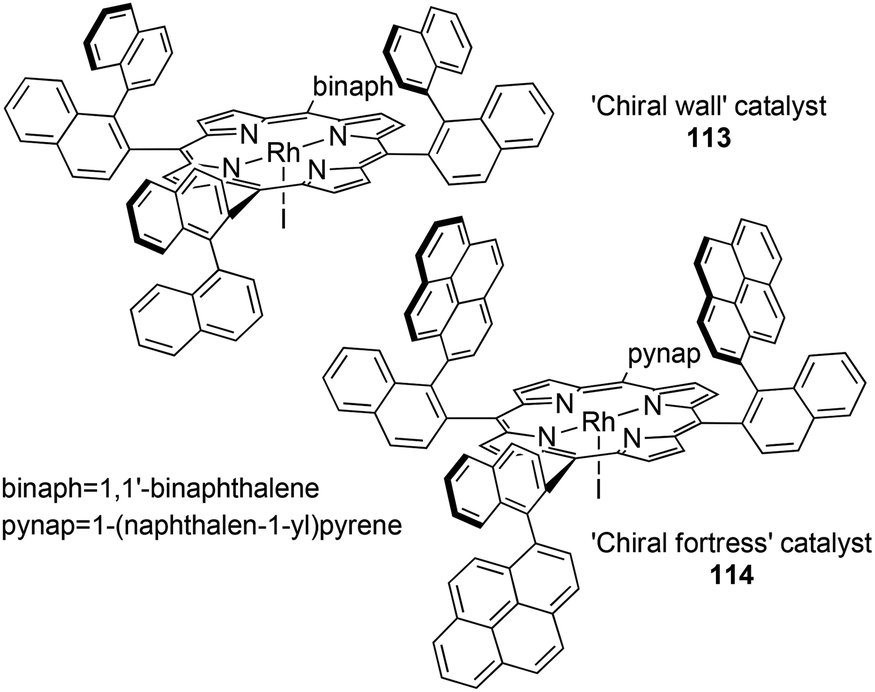
Complexes 112 and 113 were tested in the cyclopropanation of styrenes by EDA but unfortunately the stereo-control was poor with both catalysts but, in every tested case, a cis-diastereoselectivity (up to cis/trans = 14.2:1) was observed. This result indicated a different orientation of the olefin approach with respect to Halterman-type-catalysed cyclopropanations where a trans-diastereoselectivity was always observed.
The authors ascribed the modest observed enantioselectivity (up to 60%) to a large number of non-equivalent orientations of the carbene moiety, which can be responsible for the lack of stereo-control.
Better results in terms of enantioselectivity were achieved using the chiral ruthenium, rhodium and cobalt porphyrins 114–11748,52,53 (Fig. 2, type C) shown in Scheme 21 together with the synthesised cyclopropanes 3, 5–8 (Scheme 2) and 44 (Scheme 9).
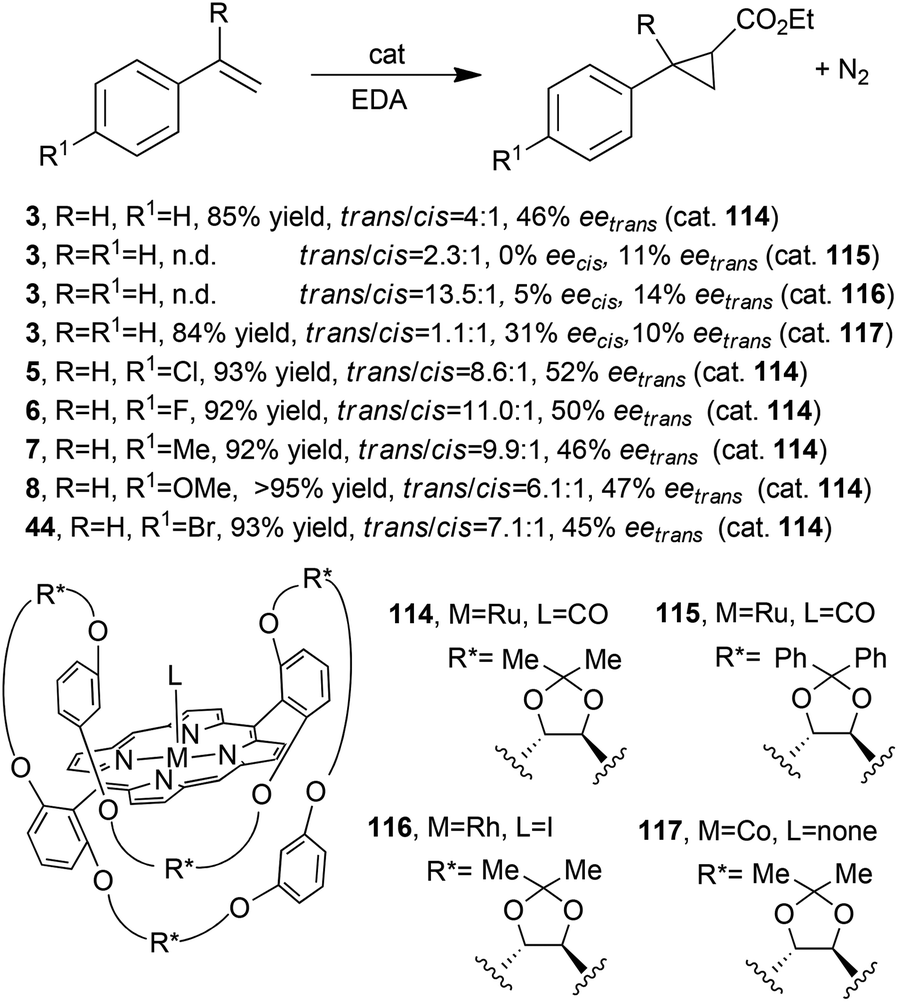
Catalyst 114 was also used to promote the cyclopropanation of styrene by DAMP; the desired compound 30 (Scheme 7) was obtained in 92% yield, 97:3 d.r.trans and 33% eetrans.54
Even if better stereoselectivities were observed using catalysts 114–117, it is evident that those structural modifications of the porphyrin skeleton were not effective to achieve high reaction stereo-control.
The D2 symmetrical dioxo-ruthenium(IV) picket-fence complex 118 (Fig. 2, type C) bearing α-methoxy-α-(trifluoromethyl)phenylacetyl chiral groups (Fig. 7) was also used to catalyse the styrene cyclopropanation by EDA. Compound 3 (Scheme 2) was formed with a trans/cis ratio of 9:1, eecis = 34% and eetrans = 14%.55
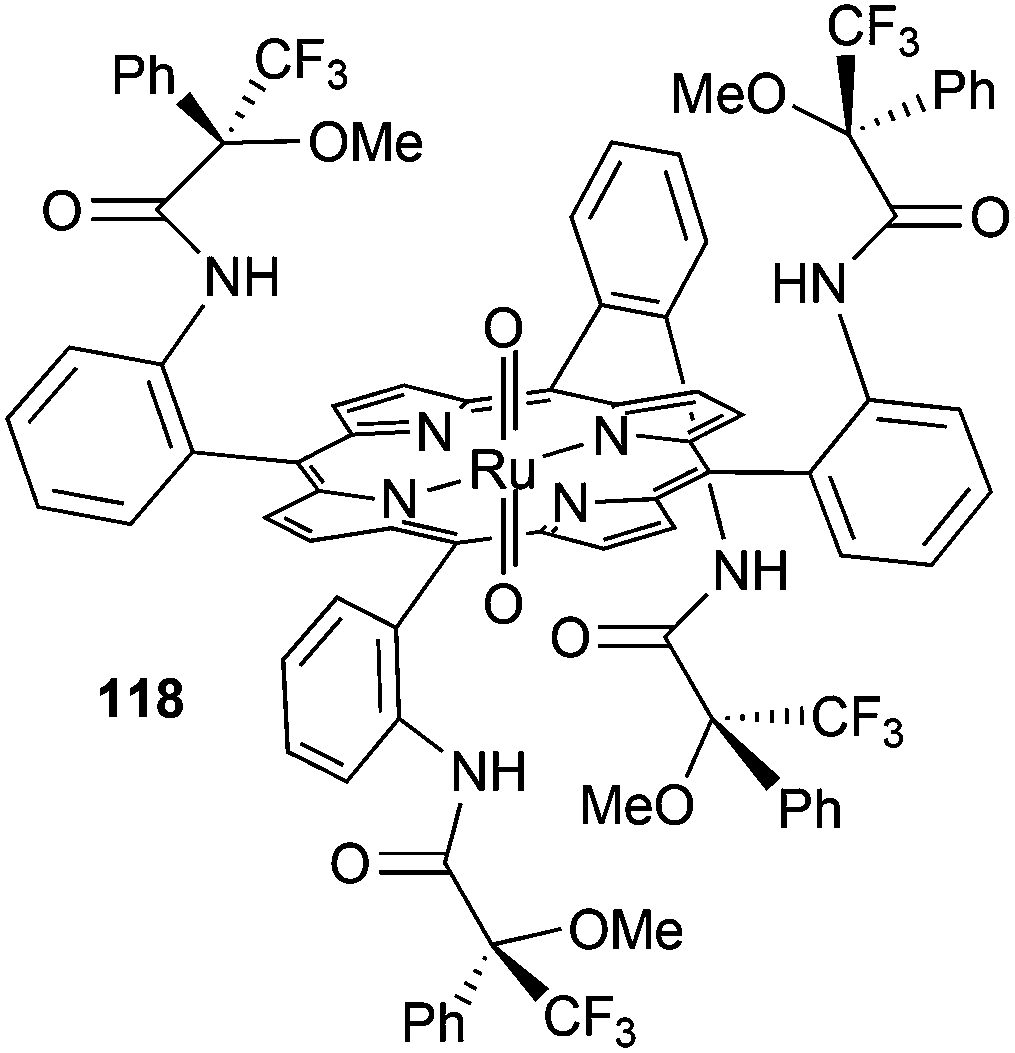 | ||
| Fig. 7 Structure of the D2 symmetrical dioxo-ruthenium(IV) picket-fence complex 118. | ||
The authors suggested that, independently of the oxidation state of the ruthenium pre-catalyst, the active carbene intermediate of the cyclopropanation is the same that proposed for the reaction catalysed by ruthenium(II) porphyrin complexes.
Considering the importance of driving the carbene transfer reaction into a confined chiral space to achieve a high stereo-control, X. P. Zhang and co-authors developed a pool of chiral cobalt porphyrins showing a skeleton which has an active role in controlling the reaction outcome (Fig. 2, type B).
The D2 symmetrical structure of catalysts 119–13156–60 (Fig. 8) presents, on two of the four meso positions, amido functionalities which are fundamental in determining the reaction stereoselectivity by establishing hydrogen bonding interactions with the carbene functionality.
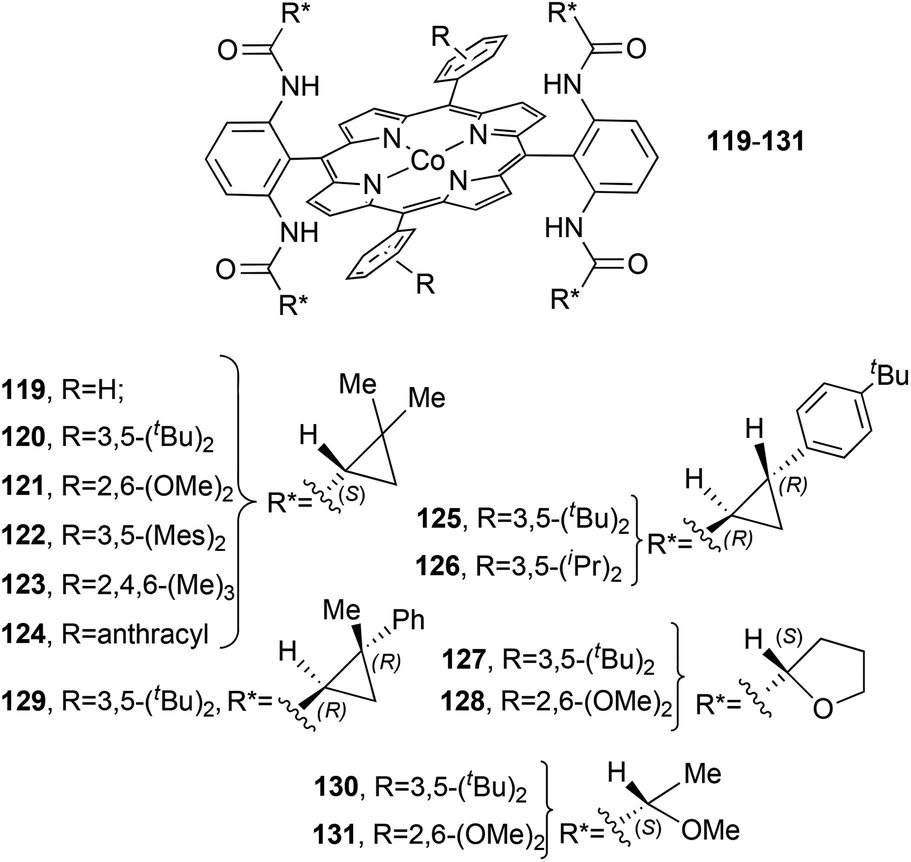 | ||
| Fig. 8 Structures of the D2 symmetrical cobalt porphyrins 119–131.56–60 | ||
Among all the catalysts shown in Fig. 8, complex 120 was largely employed to catalyse the cyclopropanation of substituted alkenes by diazo reagents displaying a variety of electronic features (Chart 1).
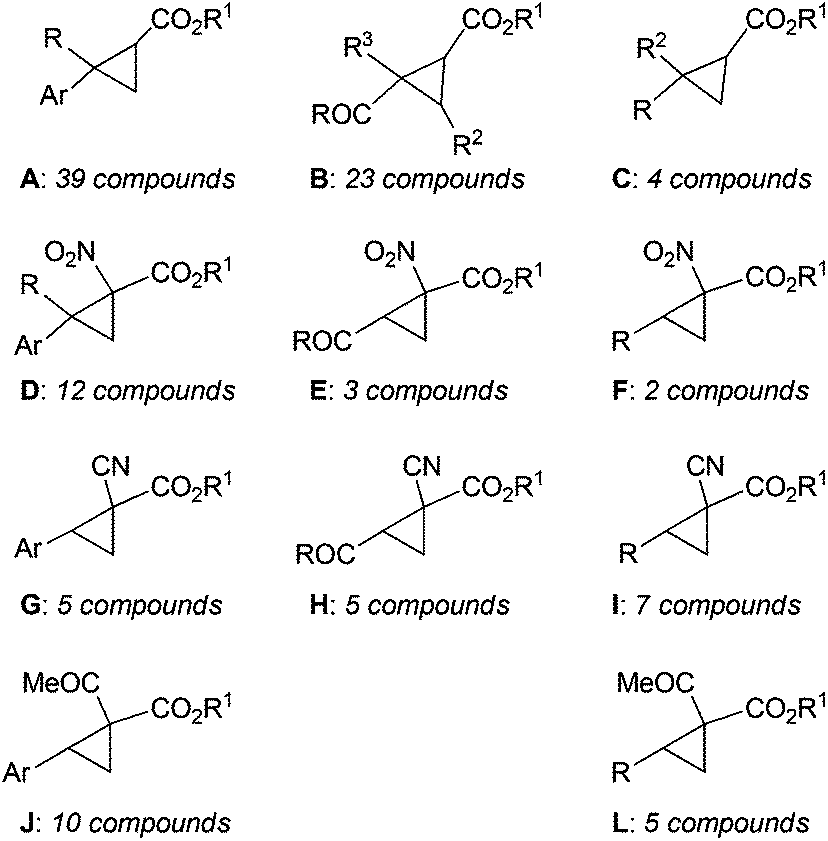 | ||
| Chart 1 General structures of cyclopropanes synthesised in the presence of cobalt(II) catalyst 120.61–66 | ||
A large number of cyclopropane derivatives were synthesised starting from aromatic alkenes61–65 (Chart 1, type A, D, G and J), electron deficient non-aromatic alkenes62–66 (Chart 1, type B, E and H) and aliphatic alkenes62,63,65 (Chart 1, type C, F, I and L) using both mono-substituted61,64,66 (Chart 1, type A–C) and acceptor/acceptor bis-substituted62,63,65 (Chart 1, type D–L) diazo derivatives.
Type A cyclopropanes (Chart 1), in the presence of catalyst 120, were formed with excellent trans-diastereoselectivities (up to 99:1) and enantioselectivities (up to 98% eetrans) using both EDA and tert-butyl diazoacetate (tBDA) as the diazo reagents and 4-dimethylaminopyridine (DMAP) as the co-catalyst.61
It was proposed that a coordinating ligand such as DMAP, with a relevant trans effect, can favour the asymmetric induction by forming a more catalytically active five-coordinated cobalt(II) intermediate from the starting four-coordinated cobalt(II) porphyrin complex.67
The catalytic performance was unaffected by the electronic nature of the starting alkene and the presence of several functional substituents on the alkene was well tolerated in the catalytic reaction. The reaction was also effective when using succinimidyl diazoacetate which reacted with aromatic alkenes forming cyclopropane succinimidyl esters, which are useful synthons of the synthesis of optically active cyclopropyl carboxamides.64
Furthermore, EDA, tBDA and succinimidyl diazoacetate reacted very well in the presence of 120 with electron deficient alkenes forming electrophilic cyclopropane derivatives in high yields and high stereoselectivities (Chart 1, type B and C).64,66
Complex 120 catalysed the cyclopropanation of aromatic alkenes using acceptor/acceptor bis-substituted diazo derivatives such as α-nitrodiazoacetates (e.g. N2C(NO2)(COOEt))62 (Chart 1, type D), α-cyanodiazoacetates (e.g. N2C(CN)(COOEt))63 (Chart 1, type G) and α-ketodiazoacetates (e.g. N2C(COMe)(COOtBu))65 (Chart 1, type J). Excellent results in terms of diastereo- and enantioselectivity were achieved albeit these diazo reagents usually display a low reactivity in carbene transfer reactions and perform with a poor enantioselectivity. It should be noted that the above cited acceptor/acceptor bis-substituted diazo derivatives also efficiently reacted with electron deficient non-aromatic alkenes (Chart 1, type E and H) and aliphatic alkenes (Chart 1, type F, I and L).
As reported above, the catalytic activity of D2 symmetric cobalt(II) complex 120 was independent of the electronic characteristics of the employed reagents. This catalytic versatility was ascribed to the formation of hydrogen-bonds between amide NH groups, located on the porphyrin ligand, and the oxygen atom of the carbonyl groups on the carbene moiety in the suggested cobalt(III) carbene radical intermediate (Scheme 22). The authors proposed that the formation of hydrogen interactions was fundamental in correctly driving the carbene addition to the alkene double bond and could be responsible for the high stereo-control observed.
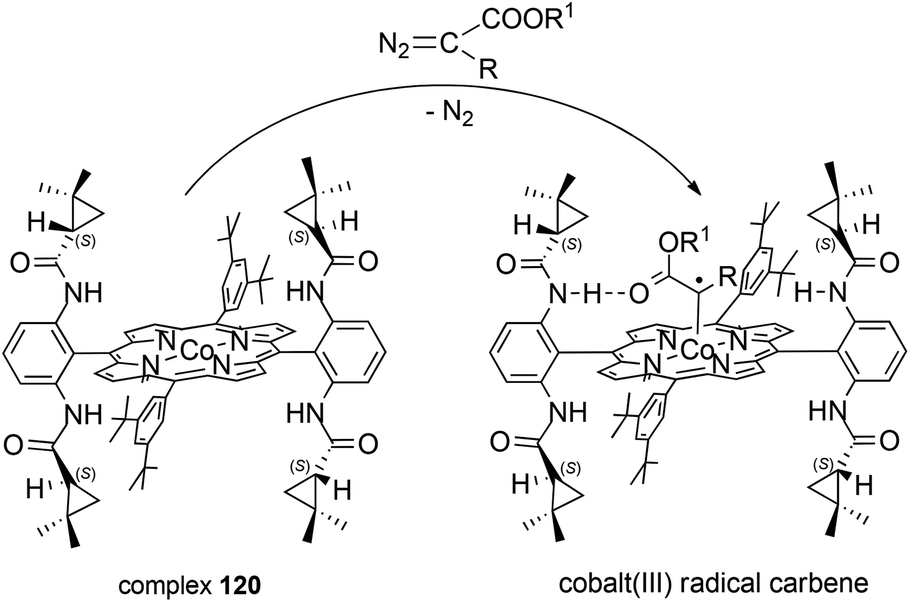 | ||
| Scheme 22 Reaction of complex 120 with a diazo reagent forming cobalt(III) radical carbene.74–77 | ||
Until now several studies have been performed to shed some light on the mechanism of cobalt(II) porphyrin-catalysed cyclopropanations68–73 and over the last few years X. P. Zhang, B. de Bruin and co-authors have published several papers to support the radical nature of the alkene cyclopropanation and the active role of D2 symmetrical porphyrin catalysts in influencing the reaction stereoselectivity.74–77
A combination of experimental EPR/ESI-MS and theoretical DFT studies of the cyclopropanation reaction was reported to support the formation of a Fisher-type radical species which interacts with the parallel alkene approaching (Scheme 23).
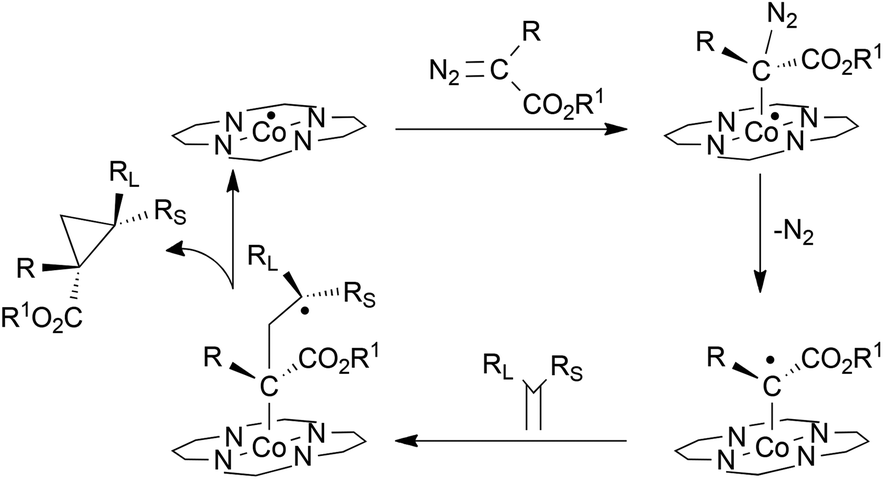 | ||
| Scheme 23 Proposed mechanism of the D2 symmetric cobalt porphyrin-catalysed alkene cyclopropanation.74–77 | ||
Considering the high influence of the porphyrin skeleton in inducing an optimised stereo-control, the catalytic activity of other cobalt D2 symmetric chiral porphyrins was investigated by X. P. Zhang and co-authors. The use of the sterically hindered catalyst 128 (Fig. 8) thus achieved a very good stereoselectivity in the cyclopropanation of several alkenes with different diazosulfones N2CH(SO2Ar).57,58 Better results in terms of both diastereo- and enantioselectivities were observed in the presence of 128 with respect to that observed when the reaction was performed in the presence of catalyst 120. These results could be due to the presence of a more rigid and polar chiral environment, which in turn is the result of intramolecular hydrogen bonding interactions and the presence of cyclic structures.
Catalyst 128 was active in promoting the synthesis of compounds 132–143 shown in Scheme 24 with trans-diastereoselectivities up to 99:1 and eetrans up to 97%.
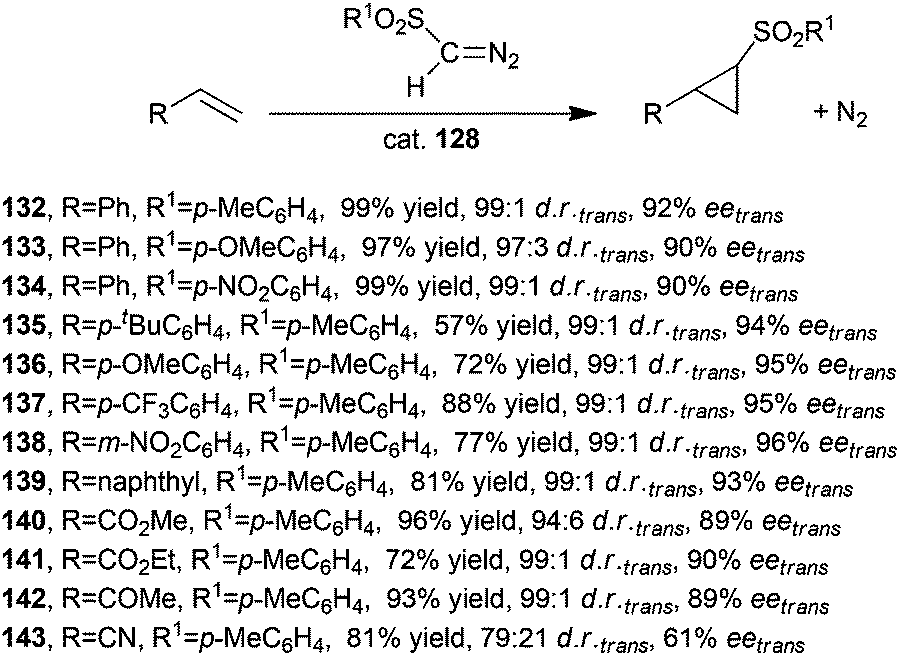 | ||
| Scheme 24 Synthesis of compounds 132–143 catalysed by cobalt catalyst 128.57,58 | ||
It should be noted that compounds shown in Scheme 24 were obtained using alkene as the limiting agent and without requiring the slow addition of the diazo reagent, which is generally applied to limit dimerization reactions of starting diazo molecules.
Considering the excellent results achieved in various intermolecular cyclopropanations, X. P. Zhang and co-authors tested the activity of D2-symmetric chiral porphyrins in intramolecular cyclopropanations. Catalyst 129 (Fig. 8) showed a very good catalytic efficiency in promoting the asymmetric intramolecular cyclopropanation of various allylic diazoacetates providing the differently functionalized 3-oxabicyclo[3.1.0]hexan-2-one derivatives 144–161 bearing three contiguous quaternary and tertiary stereocentres. It should be noted that compounds 160 and 161 were obtained starting from the corresponding cis and trans starting alkenes, respectively (Scheme 25).
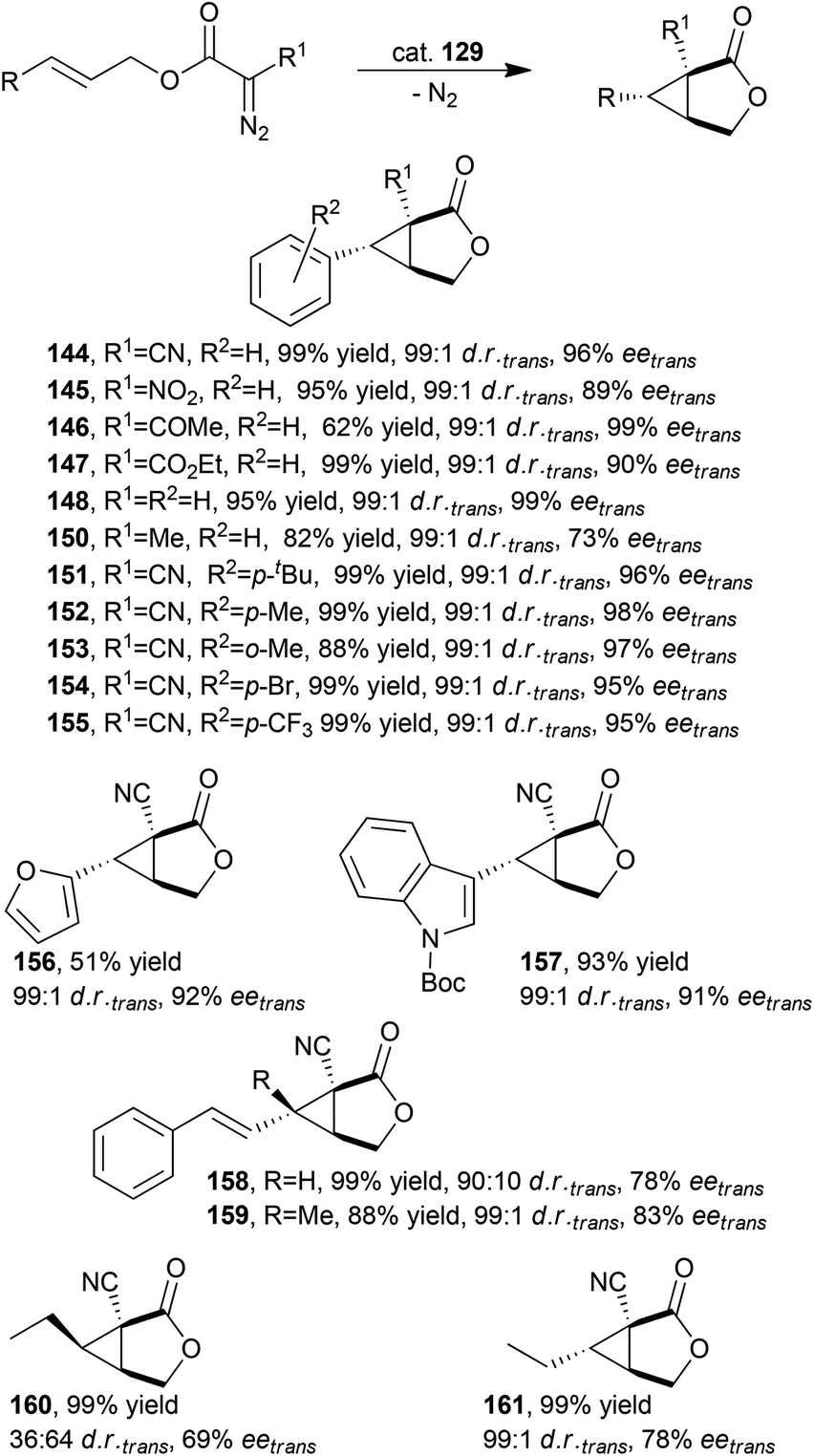 | ||
| Scheme 25 Synthesis of compounds 144–161 catalysed by cobalt catalyst 129.60 | ||
D
2 symmetric cobalt porphyrin 126 (Fig. 8) was used to promote the stereoselective intramolecular alkylation of α-methoxycarbonyl-α-diazosulfone compounds to form trans-sulfolane derivatives by a C–H bond activation.59 The synthesised compounds 162–173 were obtained in high yields, with trans-diastereoselectivities up to 97:3 and eetrans up to 94% (Scheme 26).
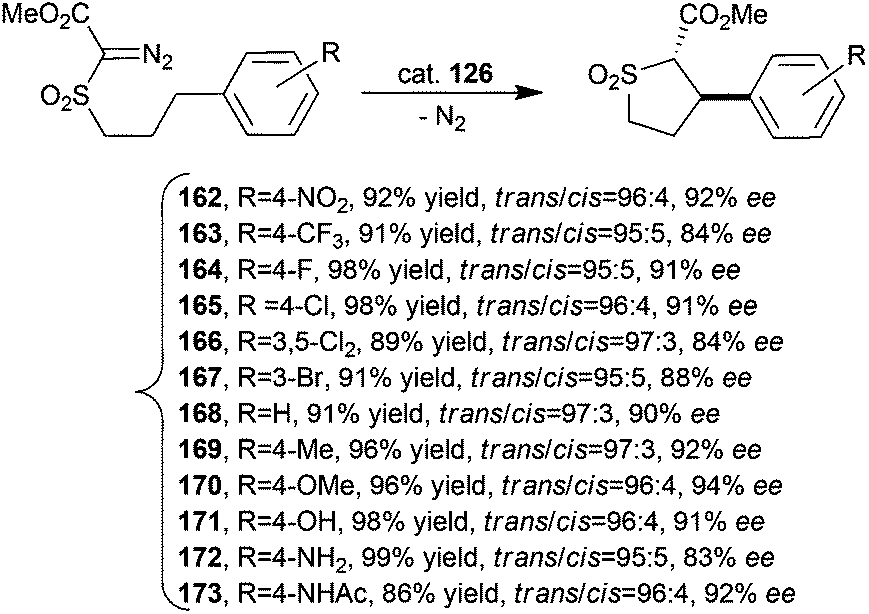 | ||
| Scheme 26 Synthesis of compounds 162–173 catalysed by cobalt catalyst 126.59 | ||
As already suggested for intermolecular reactions, the authors proposed that the very good catalytic activity could be ascribed to the formation of hydrogen bonds between the substrate and amide groups on the porphyrin skeleton. The study of the reaction scope revealed that the procedure was efficient for reacting substrates showing different electronic characteristics. The independence of the catalytic performance from the electronic nature of the involved starting materials was explained by the occurrence of a metal–radical mechanism.
D 2 symmetric cobalt porphyrins shown in Fig. 8 were also very efficient in catalysing the cyclopropenation of alkynes using acceptor/acceptor substituted diazo reagents. Complex 122 (Fig. 8) showed a very good catalytic competency and compounds shown in Scheme 27 were formed with an excellent stereo-control;78 the reactions occurred in high yields (up to 97%) and enantiomeric excesses up to 99%.
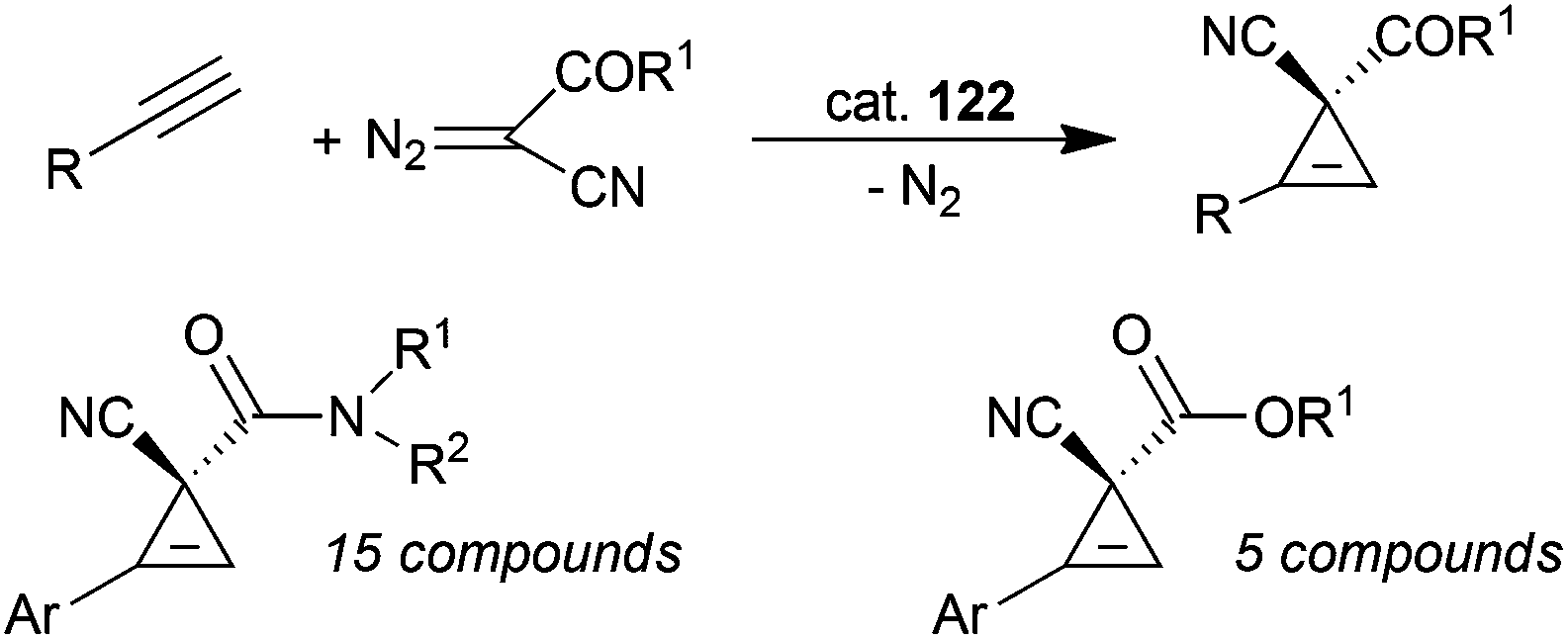 | ||
| Scheme 27 Complex 122-catalysed cyclopropenation of alkynes.78 | ||
4. C 2-Symmetric catalysts
Data reported up to now on the catalytic efficiency of D2 symmetric chiral cobalt catalysts clearly indicate that the catalytic performance in terms of stereo-control is due to a synergistic action of the active metal centre with the ligand periphery. Consequently, porphyrin ligands could be considered ‘non-innocent ligands’ of which the three-dimensional structure could be essential to opportunely drive the carbene moiety to the target substrate.The functionalization of ααββ porphyrin atropoisomers with chiral moieties affords C2 symmetric ligands (Fig. 2, type D) which can be used for the synthesis of metal chiral catalysts.
The chiral porphyrin 174, achieved by reacting the atropoisomer ααββ-tetrakis-(2-aminophenyl)porphyrin with diacid chlorides derived from binaphthyl, was used by E. Gallo, E. Rose and co-authors to synthesise cobalt(II) complex 175 (Fig. 9), active in the cyclopropanation of styrenes.79
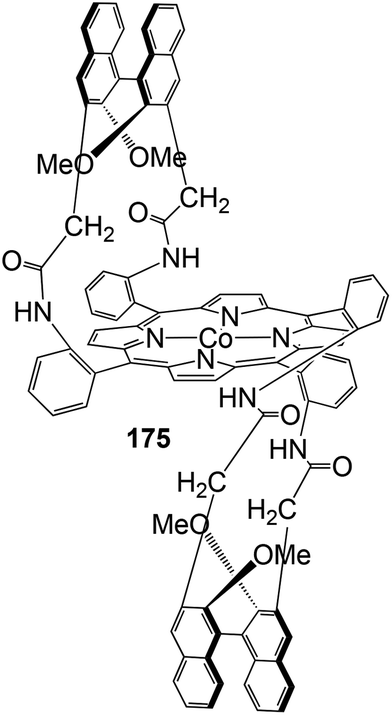 | ||
| Fig. 9 Structure of complex 175 used for the cyclopropanation of styrenes.79 | ||
Reactions occurred with modest stereo-control, albeit good yields (85–99%) of desired cyclopropanes were observed. The best result was observed in the cyclopropanation of α-methylstyrene by EDA in the presence of N-methyl imidazole (NMI) as a co-catalyst. Even if the cis/trans diastereoselectivity was only 34:66, the cis-diastereomer was obtained with 90% ee and a TON of 200.
The reasons for the low degree of stereo-control observed in cyclopropanations catalysed by complex 175 were considered from a theoretical point of view by L. Toma and co-authors, who studied the conformational properties of cobalt catalyst 175.80 E. Gallo, E. Rose and co-authors proposed that complex 175-catalysed cyclopropanation occurred through the formation of an active carbene radical intermediate in which the two axial positions on the cobalt atom were occupied by NMI and CHCO2Et carbene ligands, respectively. The modelling of the supposed carbene species revealed that the cavity originated by the binaphthyl moiety surmounting the porphyrin plane was not large enough to host the carbene ethyl group. The latter, placed outside the cavity, presented a high degree of freedom which did not permit adequate chiral discrimination.
Then, to strengthen the binaphthyl arms and increase the dimension of the active space to favour the formation of the carbene intermediate in the chiral cavity, the CH2 linker of porphyrin 174 was replaced by a benzylic moiety by reacting porphyrin 17681 with (R)-(2,2′-dimethoxy-[1,1′-binaphthalene]-3,3′-diyl)diboronic acid in the presence of (Ph3P)4Pd. Chiral porphyrin ligand 177 was obtained as shown in Scheme 28.82
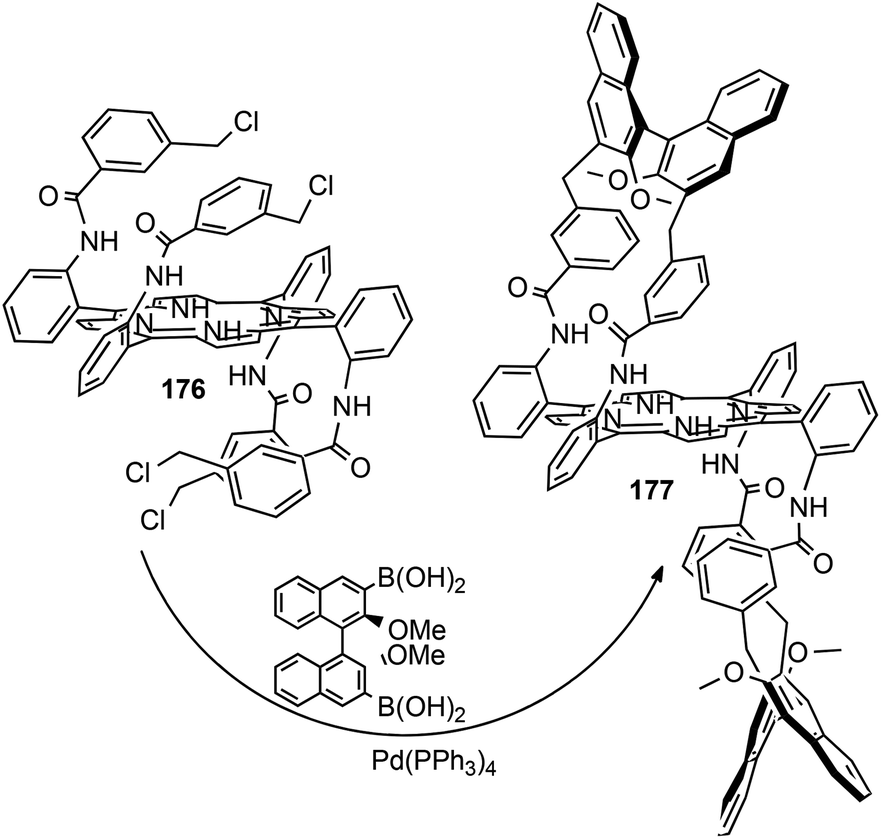 | ||
| Scheme 28 Synthesis of bis-strapped porphyrin 177.82 | ||
Porphyrin 177 was employed to synthesise the iron(III) methoxy derivative Fe(177)(OMe) which was able to promote the synthesis of cyclopropanes 4, 5, 7–9 (Scheme 2) and 178–186 listed in Scheme 29.82,83
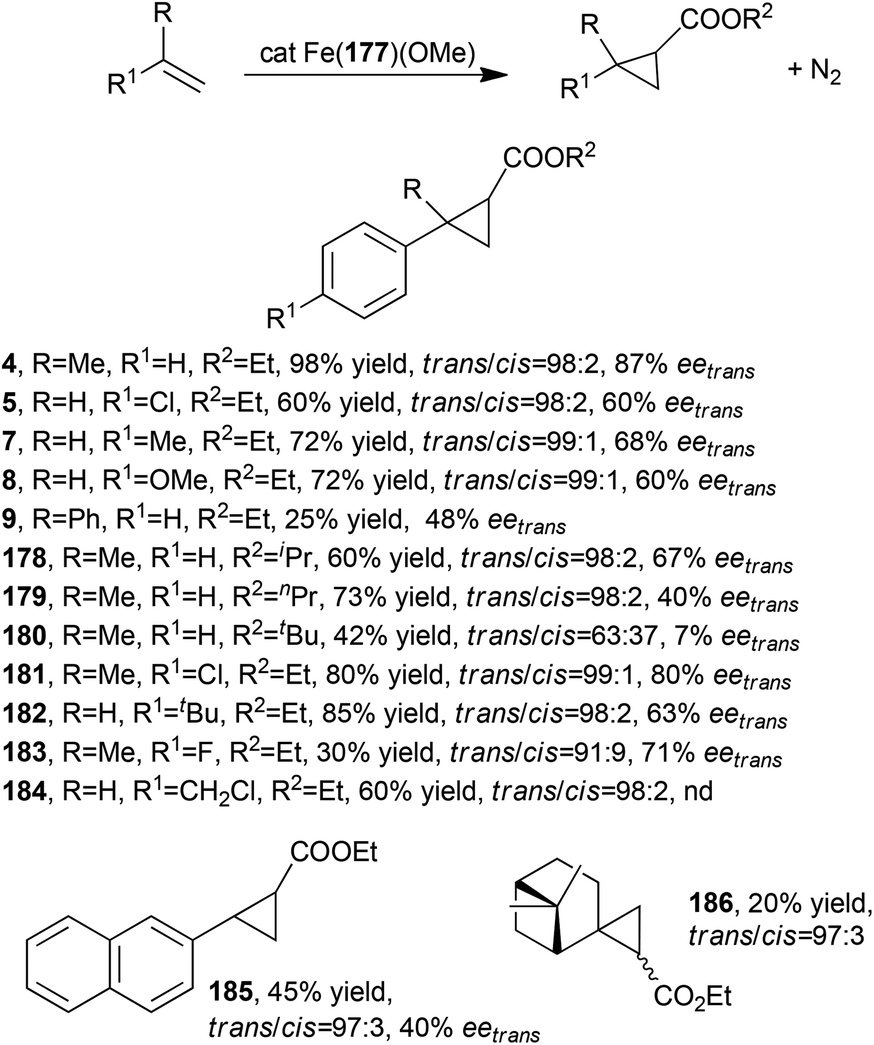 | ||
| Scheme 29 Complex Fe(177)(OMe)-catalysed synthesis of compounds 4, 5, 7–9, and 178–186.82,83 | ||
The synthesis of compounds shown in Scheme 29 was performed using alkene as the limiting reagent; a slight excess of the diazo compound was fundamental in achieving very good yields and stereoselectivities. In all tested reactions excellent trans-diastereoselectivities were observed and a general decrease of stereo-control was observed when using high sterically hindered starting compounds. The cyclopropanation of (1S)-β-pinene yielded only two of the four possible diastereomers of 186 and 1R,2R,2′R diastereomer was isolated as the major reaction product.
It should be noted that a catalyst loading of 0.01% (TON = 10000) was used for the synthesis of selected cyclopropanes where very short reaction times were observed resulting in excellent TOF values (up to 120000 h−1). A combination of experimental and DFT studies suggested the formation of an iron carbene intermediate showing a methoxy axial ligand which plays an important role in determining the catalytic performance of the iron complex.83 The theoretical modelling of involved carbene intermediates disclosed that the observed diastereoselectivities were due to the three-dimensional arrangements of the two arms surrounding the porphyrin plane, which are able to efficiently interact with the approaching alkene. Conversely, the reaction enantiocontrol was not always satisfactory for the long distance between the chiral portion of the ligand and the incoming unsaturated substrate.
5. Conclusions
The present Perspective gives an overview of the employment of chiral porphyrin metal catalysts to promote carbene transfer reactions. The influence of the porphyrin ligand structure in controlling the reaction stereoselectivity was stressed to underline the effect of the three-dimensional ligand arrangement in driving the carbene towards the target molecule. The most important class of chiral porphyrin catalysts used to activate diazo compounds have been discussed here with the final aim of summarising relevant synthetic strategies yielding porphyrin skeletons capable of high reaction stereo-control.The analysis of published data revealed that chiral porphyrin complexes are very effective enantioselective catalysts and generally promote the alkene cyclopropanation with a high trans-diastereoselectivity.
We can conclude that a very fascinating challenge for the near future could be the development of new porphyrin chiral catalysts able to couple a cis-diastereoselectivity with an excellent enantiocontrol.
Acknowledgements
We gratefully acknowledge the MIUR-Italy for the financial support over the years.References
- G. Maas, Chem. Soc. Rev., 2004, 33, 183–190 RSC
.
- G. Bartoli, G. Bencivenni and R. Dalpozzo, Synthesis, 2014, 979–1029 Search PubMed
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed
-
M. M. K. Boysen, in Carbohydrates – Tools for Stereoselective Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, pp. 313–318 Search PubMed
- A. Caballero, A. Prieto, M. M. Díaz-Requejo and P. J. Pérez, Eur. J. Inorg. Chem., 2009, 1137–1144 CrossRef CAS
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed
- M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379–3394 CrossRef PubMed
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed
- N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937–2981 CrossRef CAS PubMed
- N. M. Roda, D. N. Tran, C. Battilocchio, R. Labes, R. J. Ingham, J. M. Hawkins and S. V. Ley, Org. Biomol. Chem., 2015, 13, 2550–2554 Search PubMed
- S. T. R. Müller and T. Wirth, ChemSusChem, 2015, 8, 245–250 CrossRef PubMed
- B. J. Deadman, S. G. Collins and A. R. Maguire, Chem. – Eur. J., 2015, 21, 2298–2308 CrossRef CAS PubMed
- B. Castano, E. Gallo, D. J. Cole-Hamilton, V. Dal Santo, R. Psaro and A. Caselli, Green Chem., 2014, 16, 3202–3209 RSC
- C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC
- D. Intrieri, A. Caselli and E. Gallo, Eur. J. Inorg. Chem., 2011, 5071–5081 CrossRef CAS
-
C.-Y. Zhou, V. K.-Y. Lo and C.-M. Che, in Handbook of Porphyrin Science, ed. K. M. Kadish, R. Guilard and K. Smith, World Scientific Publishing Company, Hackensack, NJ, 2012, vol. 21, pp. 321–376 Search PubMed
-
B. J. Anding and L. K. Woo, in Handbook of Porphyrin Science, ed. K. M. Kadish, R. Guilard and K. Smith, World Scientific Publishing Company, Hackensack, NJ, 2012, vol. 21, pp. 145–319 Search PubMed
- H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899–1909 RSC
- C.-Y. Zhou, J.-S. Huang and C.-M. Che, Synlett, 2010, 2681–2700 CAS
-
J. V. Ruppel, K. B. Fields, N. L. Snyder and X. P. Zhang, in Handbook of Porphyrin Science, ed. K. M. Kadish, R. Guilard and K. Smith, World Scientific Publishing Company, Hackensack, NJ, 2010, vol. 10, pp. 1–84 Search PubMed
- C.-Y. Zhou, J.-S. Huang and C.-M. Che, Synlett, 2010, 2681–2700 CAS
- G. Simonneaux and P. Le Maux, Coord. Chem. Rev., 2002, 228, 43–60 CrossRef CAS
- M. Frauenkron and A. Berkessel, Tetrahedron Lett., 1997, 38, 7175–7176 CrossRef CAS
- W.-C. Lo, C.-M. Che, K.-F. Cheng and T. C. W. Mak, Chem. Commun., 1997, 1205–1206 RSC
- R. L. Halterman and S. T. Jan, J. Org. Chem., 1991, 56, 5253–5254 CrossRef CAS
- C.-M. Che, J.-S. Huang, F.-W. Lee, Y. Li, T.-S. Lai, H.-L. Kwong, P.-F. Teng, W.-S. Lee, W.-C. Lo, S.-M. Peng and Z.-Y. Zhou, J. Am. Chem. Soc., 2001, 123, 4119–4129 CrossRef CAS PubMed
- P. Le Maux, T. Roisnel, I. Nicolas and G. Simonneaux, Organometallics, 2008, 27, 3037–3042 CrossRef CAS
- A. Berkessel, P. Kaiser and J. Lex, Chem. – Eur. J., 2003, 9, 4746–4756 CrossRef CAS PubMed
- K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982–2987 CrossRef CAS PubMed
- I. Nicolas, P. L. Maux and G. Simonneaux, Tetrahedron Lett., 2008, 49, 2111–2113 CrossRef CAS
- Y. Ferrand, P. Le Maux and G. Simonneaux, Org. Lett., 2004, 6, 3211–3214 CrossRef CAS PubMed
- P. Le Maux, S. Juillard and G. Simonneaux, Synthesis, 2006, 1701–1704 CAS
- P. Le Maux, I. Nicolas, S. Chevance and G. Simonneaux, Tetrahedron, 2010, 66, 4462–4468 CrossRef CAS
- I. Nicolas, P. L. Maux and G. Simonneaux, Tetrahedron Lett., 2008, 49, 5793–5795 CrossRef CAS
- J.-L. Zhang, Y.-L. Liu and C.-M. Che, Chem. Commun., 2002, 2906–2907 RSC
- Y. Ferrand, C. Poriel, P. Le Maux, J. Rault-Berthelot and G. Simonneaux, Tetrahedron: Asymmetry, 2005, 16, 1463–1472 CrossRef CAS
- Y. Ferrand, P. Le Maux and G. Simonneaux, Tetrahedron: Asymmetry, 2005, 16, 3829–3836 CrossRef CAS
- I. Bauer and H.-J. Knolker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed
- T.-S. Lai, F.-Y. Chan, P.-K. So, D.-L. Ma, K.-Y. Wong and C.-M. Che, Dalton Trans., 2006, 4845–4851 RSC
- J. R. Wolf, C. G. Hamaker, J.-P. Djukic, T. Kodadek and L. K. Woo, J. Am. Chem. Soc., 1995, 117, 9194–9199 CrossRef CAS
- R. G. Salomon and J. K. Kochi, J. Am. Chem. Soc., 1973, 95, 3300–3310 CrossRef CAS
- I. Nicolas, T. Roisnel, P. L. Maux and G. Simonneaux, Tetrahedron Lett., 2009, 50, 5149–5151 CrossRef CAS
- P.-F. Teng, T.-S. Lai, H.-L. Kwong and C.-M. Che, Tetrahedron: Asymmetry, 2003, 14, 837–844 CrossRef CAS
- H.-Y. Thu, G. S.-M. Tong, J.-S. Huang, S. L.-F. Chan, Q.-H. Deng and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9747–9751 CrossRef CAS PubMed
- J.-C. Wang, Z.-J. Xu, Z. Guo, Q.-H. Deng, C.-Y. Zhou, X.-L. Wan and C.-M. Che, Chem. Commun., 2012, 48, 4299–4301 RSC
- J.-C. Wang, Y. Zhang, Z.-J. Xu, V. K.-Y. Lo and C.-M. Che, ACS Catal., 2013, 3, 1144–1148 CrossRef CAS
- L. Huang, Y. Chen, G.-Y. Gao and X. P. Zhang, J. Org. Chem., 2003, 68, 8179–8184 CrossRef CAS PubMed
- A. Caselli, E. Gallo, F. Ragaini, F. Ricatto, G. Abbiati and S. Cenini, Inorg. Chim. Acta, 2006, 359, 2924–2932 CrossRef CAS
- J. L. Maxwell, S. O'Malley, K. C. Brown and T. Kodadek, Organometallics, 1992, 11, 645–652 CrossRef CAS
- S. O'Malley and T. Kodadek, Organometallics, 1992, 11, 2299–2302 CrossRef
- E. Galardon, S. Roué, P. Le Maux and G. Simonneaux, Tetrahedron Lett., 1998, 39, 2333–2334 CrossRef CAS
- Z. Gross, N. Galili and L. Simkhovich, Tetrahedron Lett., 1999, 40, 1571–1574 CrossRef CAS
- C. Paul-Roth, F. De Montigny, G. Rethoré, G. Simonneaux, M. Gulea and S. Masson, J. Mol. Catal. A: Chem., 2003, 201, 79–91 CrossRef CAS
- E. Galardon, P. Le Maux and G. Simonneaux, Chem. Commun., 1997, 927–928 RSC
- Y. Chen, K. B. Fields and X. P. Zhang, J. Am. Chem. Soc., 2004, 126, 14718–14719 CrossRef CAS PubMed
- S. Zhu, J. V. Ruppel, H. Lu, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2008, 130, 5042–5043 CrossRef CAS PubMed
- S. Zhu, X. Cui and X. P. Zhang, Eur. J. Inorg. Chem., 2012, 2012, 430–434 CrossRef CAS
- X. Cui, X. Xu, L.-M. Jin, L. Wojtas and X. P. Zhang, Chem. Sci., 2015, 6, 1219–1224 RSC
- X. Xu, H. Lu, J. V. Ruppel, X. Cui, S. Lopez de Mesa, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 15292–15295 CrossRef CAS PubMed
- Y. Chen and X. P. Zhang, J. Org. Chem., 2007, 72, 5931–5934 CrossRef CAS PubMed
- S. Zhu, J. A. Perman and X. P. Zhang, Angew. Chem., Int. Ed., 2008, 47, 8460–8463 CrossRef CAS PubMed
- S. Zhu, X. Xu, J. A. Perman and X. P. Zhang, J. Am. Chem. Soc., 2010, 132, 12796–12799 CrossRef CAS PubMed
- J. V. Ruppel, T. J. Gauthier, N. L. Snyder, J. A. Perman and X. P. Zhang, Org. Lett., 2009, 11, 2273–2276 CrossRef CAS PubMed
- X. Xu, S. Zhu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2013, 52, 11857–11861 CrossRef CAS PubMed
- Y. Chen, J. V. Ruppel and X. P. Zhang, J. Am. Chem. Soc., 2007, 129, 12074–12075 CrossRef CAS PubMed
- Y. Chen and X. P. Zhang, Synthesis, 2006, 1697–1700 CAS
- A. W. Johnson, D. Ward, P. Batten, A. L. Hamilton, G. Shelton and C. M. Elson, J. Chem. Soc., Perkin Trans. 1, 1975, 2076–2085 RSC
- H. J. Callot and E. Schaeffer, J. Organomet. Chem., 1978, 145, 91–99 CrossRef CAS
- H. J. Callot and E. Schaeffer, Nouv. J. Chim., 1980, 4, 307–309 CAS
- P. Chattopadhyay, T. Matsuo, T. Tsuji, J. Ohbayashi and T. Hayashi, Organometallics, 2011, 30, 1869–1873 CrossRef CAS
- I. Iwakura, H. Tanaka, T. Ikeno and T. Yamada, Chem. Lett., 2004, 33, 140–141 CrossRef CAS
- A. Penoni, R. Wanke, S. Tollari, E. Gallo, D. Musella, F. Ragaini, F. Demartin and S. Cenini, Eur. J. Inorg. Chem., 2003, 1452–1460 CrossRef CAS
- W. I. Dzik, X. Xu, X. P. Zhang, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2010, 132, 10891–10902 CrossRef CAS PubMed
- W. I. Dzik, X. P. Zhang and B. de Bruin, Inorg. Chem., 2011, 50, 9896–9903 CrossRef CAS PubMed
- H. Lu, W. I. Dzik, X. Xu, L. Wojtas, B. de Bruin and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8518–8521 CrossRef CAS PubMed
- J. L. Belof, C. R. Cioce, X. Xu, X. P. Zhang, B. Space and H. L. Woodcock, Organometallics, 2011, 30, 2739–2746 CrossRef CAS PubMed
- X. Cui, X. Xu, H. Lu, S. Zhu, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 3304–3307 CrossRef CAS PubMed
- S. Fantauzzi, E. Gallo, E. Rose, N. Raoul, A. Caselli, S. Issa, F. Ragaini and S. Cenini, Organometallics, 2008, 27, 6143–6151 CrossRef CAS
- E. Gallo, E. Rose, B. Boitrel, L. Legnani and L. Toma, Organometallics, 2014, 33, 6081–6088 CrossRef CAS
- A. Didier, L. Michaudet, D. Ricard, V. Baveux-Chambenoît, P. Richard and B. Boitrel, Eur. J. Org. Chem., 2001, 1927–1926 CrossRef
- D. Intrieri, S. Le Gac, A. Caselli, E. Rose, B. Boitrel and E. Gallo, Chem. Commun., 2014, 50, 1811–1813 RSC
- D. M. Carminati, D. Intrieri, A. Caselli, S. Le Gac, B. Boitrel, L. Toma, L. Legnani and E. Gallo, submitted.
| This journal is © The Royal Society of Chemistry 2016 |