
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Salt metathesis versus protonolysis routes for the synthesis of silylamide Hauser base (R2NMgX; X = halogen) and amido-Grignard (R2NMgR) complexes†
Conrad A. P.
Goodwin
,
Alex
Smith
,
Fabrizio
Ortu
,
Iñigo. J.
Vitorica-Yrezabal
and
David P.
Mills
*
School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.mills@manchester.ac.uk
First published on 5th August 2015
Abstract
The preparation of silylamide Hauser base (R2NMgX; X = halide) and amido-Grignard (R2NMgR) complexes from simple Grignard reagents using [K{N(SiMe2tBu)2}]n, [K{N(SiMe2tBu)(SiiPr3)}]n and [K{N(SiiPr3)2}]n, and their parent silylamines, was explored. Both salt metathesis and protonolysis routes proved ineffective with allylmagnesium chloride as a starting material due to complex Schlenk equilibria, with [Mg(NRR′)(μ-Cl)(THF)]2 (NRR′ = {N(SitBuMe2)2}−, 1; {N(SitBuMe2)(SiiPr3)}−, 2; {N(SiiPr3)2}−, 3) and [Mg{N(SiiPr3)2}(μ-C3H5)]∞ (4) identified as minor products. In contrast, salt metathesis protocols using potassium silylamides and methylmagnesium iodide gave [Mg(NRR′)(μ-CH3)]2 (NRR′ = {N(SitBuMe2)2}−, 7a; {N(SitBuMe2)(SiiPr3)}−, 8; {N(SiiPr3)2}−, 9) and [Mg{N(SitBuMe2)2}(CH3)(DME)] (7b), with [Mg{N(SitBuMe2)2}(μ-I)(THF)]2 (10) isolated as a side-product during the preparation of 7a. Unusually, methylmagnesium iodide, di-n-butylmagnesium and 7–9 did not react with HNRR′ under the conditions we employed. The synthesis of [Na{N(SitBuMe2)2}(THF)]2 (5a) and [Na{N(SitBuMe2)2}(DME)2] (5b) from benzyl sodium and HN(SitBuMe2)2, and a solvent-free structure of [K{N(SitBuMe2)2}] (6), are also reported. Complexes 1, 5b, 7a, 7b, 8, 9 and 10 are fully characterised by single crystal XRD, multinuclear NMR and IR spectroscopy and elemental analysis, whereas complexes 2–4, 5a and 6 were identified by XRD only.
Introduction
Grignard reagents are widely utilised due to their facile preparation and broad applicability in organic synthesis,1,2 yet in stark contrast the synthetic potential of Hauser bases, amido analogues with a N–Mg bond instead of a C–Mg bond, is only starting to be realised.3 This is remarkable as N-donor groups are harder than C-donors and a wider variety of synthetic routes are available to access homo- and heteroleptic N-donor alkaline earth (Ae) complexes4 than Ae organometallics.1,4,5 Furthermore, highly reactive heteroleptic magnesium complexes with N-donors, such as alkyls and hydrides, are desirable and useful reagents, as they can undergo σ-bond metathesis or protonolysis with a number of substrates, thereby providing access to various synthetic hetero-functionalisations.3f,6Sterically demanding N-donor ligands are commonly employed in Ae solution chemistry as they impede oligomerisation, complex Schlenk equilibria and other unwanted degradation pathways in ethereal solvents, particularly for the heavier Ae metals.4,5 Bochmann and co-workers have shown that cationic magnesium complexes with bulky amido ligands such as [Mg(N′′)(Et2O)3][BArF] (N′′ = {N(SiMe3)2}−, BArF = {B(C6F5)4}−) can act as potent ring opening polymerisation catalysts,7 an industrially significant process in which Chisholm and others have shown magnesium catalysts have great promise.8 In seminal work by Jones, bulky N-donor ligands have been used to stabilise the first examples of structurally characterised Mg(I) complexes,9 which have since proven their utility as selective one-electron reducing agents in a number of diverse transformations.10
Multidentate ligands such as guanidinates,9–11 amidinates12 and β-diketiminates6,9–11,13,14 dominate N-donor magnesium chemistry and there are relatively few examples of monodentate complexes.3b,e,9,15 In 1994 Power disclosed the first crystallographically authenticated Hauser base complex (R2NMgX; X = halide),16 [Mg(N′′)(μ-Cl)(Et2O)]2,17 which was originally prepared in 1972 by Wannagat et al. via a protonolysis reaction between HN′′ and propylmagnesium chloride in diethyl ether.18 To the best of our knowledge, the heavier congener [Mg(N′′)(μ-Br)(Et2O)]2,19 together with the multidentate N-heterocyclic carbene (NHC) supported complexes, [{Mg(N′′)}{Mg(Cl)}(μ-NHC3)(μ-Cl)] and [{Mg(N′′)}2(μ-NHC3)(μ-Cl)] (NHC3 = [N{CH2CH2[NCHCHN(Mes)C]}2], Mes = 2,4,6-trimethylphenyl),20 are the only other structurally characterised Hauser bases supported by silylated amides. Furthermore, expanding the search to halide Hauser bases of any monodentate amide yields only a handful more examples.3a,21
Amido-Grignard complexes, (R2NMgR),22 are better represented in the literature, but again there are few examples that contain silylamides.23 Recently, Mulvey reported the dimeric complex, [Mg(μ-N′′)(tBu)]2,24 and the NHC adduct, [Mg(N′′)(nBu)(IPr)] (IPr = 1,3-bis-(2,6-diisopropylphenyl)imidazol-2-ylidene);25 however, in general heteroleptic magnesium silylamide complexes remain relatively rare. Amido-Grignard complexes are key intermediates in the formation of monoalkyl-bis(amido) group 1/group 2 bimetallic systems, which are typically formed directly from secondary amines in the presence of alkali metal and organomagnesium reagents.26 Mulvey and co-workers have widely used such mixed metalation reactions with Hauser bases to prepare bimetallic systems with various degrees of complexity, ranging from simple heterobimetallic compounds to more elaborate architectures, e.g. inverse crown ethers.26 Such systems have been employed in synergic deprotonative metalation chemistry, providing a powerful tool for a number of transformations, and this pioneering work has recently been reviewed by Knochel and co-workers.3b,27 It follows that facile synthetic routes to a wider range of silylamide Hauser bases need to be developed in order to enable their future exploitation.
Recently, we have reported a series of bulky silylamines and we have utilised these starting materials to synthesise homo- and heteroleptic alkali metal28 and f-block28,29 complexes. These silylamides include the potassium salts [{K[μ-N(SiMe2But)2]}2(C7H8)]∞, [K{N(SiPri3)(SiMe2But)}]∞ and [K{N(SiPri3)2}]∞ (Fig. 1),28 which are used as starting materials herein. Jones and co-workers have also developed synthetic routes to bulky silylamide s-block complexes that are useful ligand transfer agents.30 In this paper, we report the reactions of bulky alkali metal silylamides and parent silylamines with commercially available Grignard reagents to afford a novel series of Hauser base and amido-Grignard complexes.
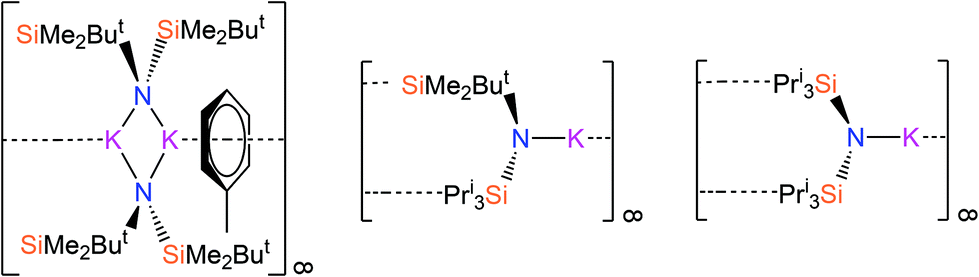 | ||
| Fig. 1 The potassium silylamide complexes [{K[μ-N(SiMe2But)2]}2(C7H8)]∞, [K{N(SiPri3)(SiMe2But)}]∞ and [K{N(SiPri3)2}]∞.28 | ||
Results and discussion
Reactions of allylmagnesium chloride with silylamines and potassium silylamides
We found that the reactions of both potassium silylamides, [K(NRR′)], and their parent silylamines with allylmagnesium chloride reproducibly gave mixtures of products and poor yields. Given that propylmagnesium chloride reacts with HN′′ in diethyl ether to give [Mg(N′′)(μ-Cl)(Et2O)]2,17,18 we added allylmagnesium chloride to THF solutions of HN(SitBuMe2)2, HN(SitBuMe2)(SiiPr3) and HN(SiiPr3)2 (Scheme 1). Work-up of the respective reaction mixtures after 8 hours reproducibly gave crystals of the chloride Hauser bases, [Mg(NRR′)(μ-Cl)(THF)]2 (NRR′ = {N(SitBuMe2)2}−, 1; {N(SitBuMe2)(SiiPr3)}−, 2; {N(SiiPr3)2}−, 3), which were minor components of intractable mixtures of products. Complexes 1–3 were structurally characterised (see below) but they could not be separated from the bulk material, which was shown to be a mixture of products by 1H NMR spectroscopy. By increasing the reaction time to 10 days, we were able to improve the crystalline yield of complex 1 to 19%. Improved yields of 2–3 were not achieved by this method so further analysis of these complexes could not be obtained. On the other hand, the attempted salt metathesis reactions between allylmagnesium chloride and the corresponding potassium amides, [K(NRR′)], did not give the expected amido-Grignard reagents, [Mg(NRR′)(C3H5)]n, despite several attempts at optimising the reaction conditions. However, in the attempted preparation of [Mg{N(SiiPr3)2}(C3H5)]n several crystals of complex 3 were identified together with a crystal of [Mg{N(SiiPr3)2}(μ-C3H5)]∞ (4) by single crystal XRD (see below). These observations suggested that complex Schlenk equilibria processes are taking place in these reaction mixtures.1,31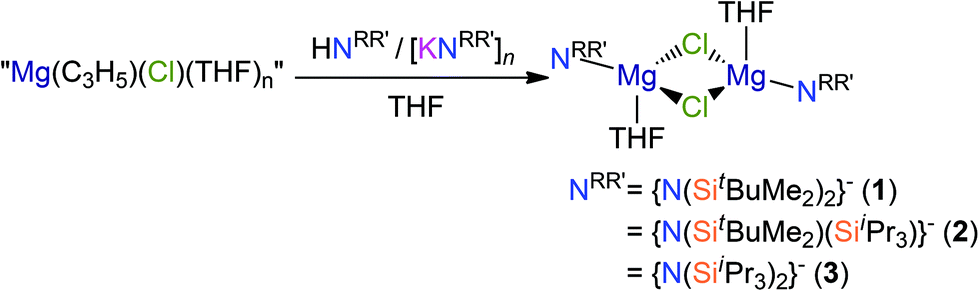 | ||
| Scheme 1 Synthesis of complexes 1–3. | ||
The 1H and 13C{1H} NMR spectra of 1 in d6-benzene are unremarkable and not shifted considerably from those reported for [K{N(SiMe2tBu)2}]2, although the 29Si{1H} NMR spectrum of 1 exhibits a single resonance (δ: −0.40 ppm) that is shielded in comparison to the related potassium complex (δ: −15.72 ppm).29a Microanalysis was performed multiple times on discrete crystalline samples of complex 1 and consistently low C values were obtained despite good agreement between measured and expected H and N content. This phenomenon was also encountered for complexes 5b, 7b and 9 (see below), even though the 1H, 13C{1H} and 29Si{1H} NMR spectra of these complexes exhibit only minor impurity signals (see ESI Fig. S1–S9†). Similar amounts of trace impurities were detected in the NMR spectra of other structurally characterised complexes reported herein, yet these have excellent agreement between predicted and observed microanalysis values. Therefore, we attribute these discrepancies to carbide formation, which has been cited previously as a recurring phenomenon in elemental analyses of silicon-rich complexes.29a,32
The identities of 1–4 were determined by single crystal XRD experiments (the molecular structure of 1 is depicted in Fig. 2; see ESI Fig. S10–S12† for the structures of 2–4. Selected bond lengths and angles are compiled in Table 1). The dataset obtained for 4 is of poor quality as the crystals were weakly diffracting. Therefore no discussion of the geometric parameters of 4 is given here, but as the connectivity is clear-cut the structure is included for completeness. The magnesium centres in 4 are 3-coordinate, which is rare for magnesium amide structures and even more so for monodentate ligands.21g,33 They are each bridged by two allyls, which generates an infinite 1D polymer comprised of two parallel-bridged chains. Complexes 1–3 exhibit similar motifs in the solid state, therefore only the structure of 1 is discussed for brevity. Complex 1 is dimeric with a central Mg2Cl2 rhomboid [Mg–Cl–Mg 92.15(6)°; Cl–Mg–Cl 87.85(6)°] and two 4-coordinate magnesium centres that are each bound by THF. The Mg–Cl [2.418(2) and 2.411(2) Å] and Mg–O [2.018(4) Å] distances are longer than those in [Mg(N′′)(μ-Cl)(Et2O)]2 [Mg–Cl 2.401(1) and 2.405(1) Å; Mg–O 2.000(3) Å],17 which we attribute to the increased steric demands of {N(SitBuMe2)2}−. Each magnesium coordination sphere of 1–3 is completed by two short Mg⋯C–H contacts [mean Mg⋯C 3.3485(8) Å (1); 3.192(4) Å (2); 3.267(6) Å (3)] and several short Mg⋯H distances [range Mg⋯H 2.71–2.75 Å (1); 2.63–3.02 Å (2); 2.56–2.67 Å (3)], where only one close Mg⋯C–H contact is present in [Mg(N′′)(μ-Cl)(Et2O)]2 [Mg⋯C 3.207(5) Å; mean Mg⋯H 3.02 Å].17
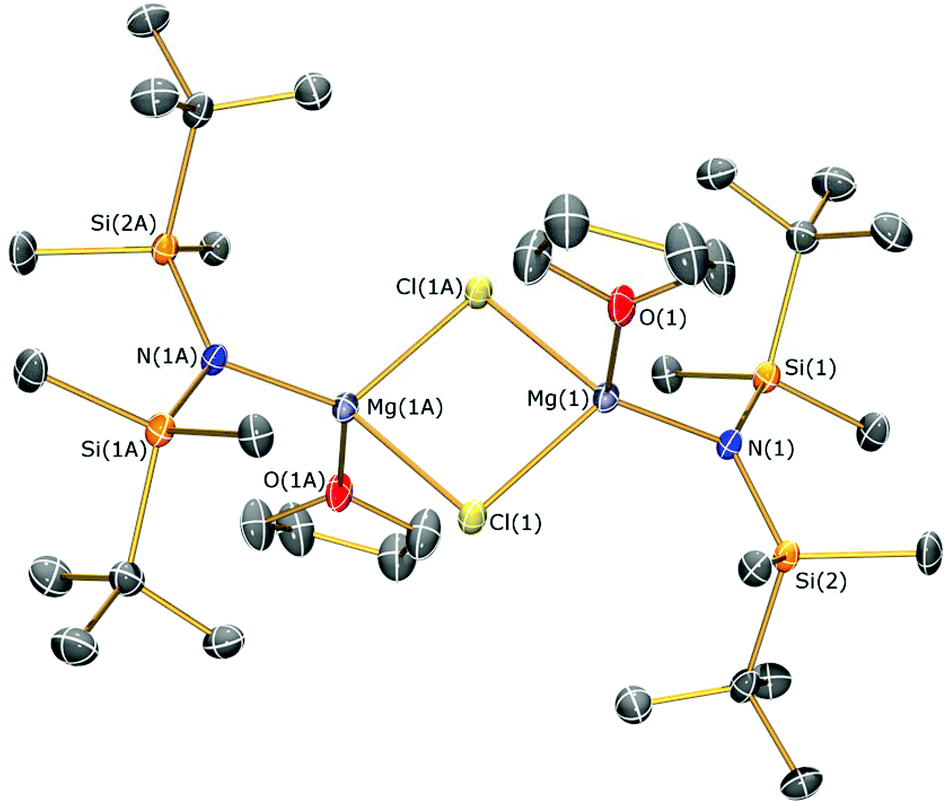 | ||
| Fig. 2 Molecular structure of 1 with selective atom labelling. Displacement ellipsoids set at 30% probability level and hydrogen atoms omitted for clarity. Symmetry operation to generate equivalent atoms: x − 1/2, −y − 1/2, z − 1/2. | ||
| 1 | |||
| N(1)–Mg(1) | 1.998(4) | Mg(1)–Cl(1) | 2.4178(17) |
| Mg(1)–Cl(1A) | 2.411(2) | N(1)–Si(1) | 1.722(4) |
| N(1)–Si(2) | 1.711(4) | Mg(1)–O(1) | 2.018(4) |
| Mg(1)⋯Mg(1A) | 3.478(4) | Cl(1)–Mg(1)–Cl(1A) | 87.85(6) |
| Mg(1)–Cl(1)–Mg(1A) | 92.15(6) | N(1)–Mg(1)–O(1) | 118.33(16) |
| N(1)–Mg(1)–Cl(1) | 123.38(13) | N(1)–Mg(1)–Cl(1A) | 125.18(14) |
| Si(1)–N(1)–Si(2) | 126.8(2) | ||
| 2 | |||
| N(1)–Mg(1) | 2.005(2) | Mg(1)–Cl(1) | 2.4098(11) |
| Mg(1)–Cl(1A) | 2.4073(11) | N(1)–Si(1) | 1.717(2) |
| N(1)–Si(2) | 1.707(2) | Mg(1)–O(1) | 2.031(2) |
| Mg(1)⋯Mg(1A) | 3.4324(16) | Cl(1)–Mg(1)–Cl(1A) | 89.11(4) |
| Mg(1)–Cl(1)–Mg(1A) | 90.89(4) | N(1)–Mg(1)–O(1) | 122.77(9) |
| N(1)–Mg(1)–Cl(1) | 125.72(7) | N(1)–Mg(1)–Cl(1A) | 117.93(7) |
| Si(1)–N(1)–Si(2) | 133.73(13) | ||
| 3 | |||
| N(1)–Mg(1) | 2.028(3) | Mg(1)–Cl(1) | 2.4169(14) |
| Mg(1)–Cl(1A) | 2.4376(15) | N(1)–Si(1) | 1.726(3) |
| N(1)–Si(2) | 1.724(3) | Mg(1)–O(1) | 2.046(3) |
| Mg(1)⋯Mg(1A) | 3.504(3) | Cl(1)–Mg(1)–Cl(1A) | 87.60(5) |
| Mg(1)–Cl(1)–Mg(1A) | 92.40(5) | N(1)–Mg(1)–O(1) | 121.27(13) |
| N(1)–Mg(1)–Cl(1) | 121.88(10) | N(1)–Mg(1)–Cl(1A) | 124.92(10) |
| Si(1)–N(1)–Si(2) | 134.07(19) | ||
| 5a | |||
| N(1)–Na(1) | 2.472(3) | N(2)–Na(2) | 2.499(3) |
| Na(1)–O(1) | 2.333(3) | Na(2)–O(2) | 2.361(3) |
| N(1)–Na(2) | 2.502(3) | N(2)–Na(1) | 2.485(3) |
| N(1)–Si(1) | 1.697(3) | N(1)–Si(2) | 1.692(3) |
| N(2)–Si(3) | 1.692(3) | N(2)–Si(4) | 1.698(3) |
| Na(1)⋯Na(2) | 2.998(2) | Na(1)⋯C(5) | 3.027(4) |
| Na(1)⋯C(8) | 3.373(5) | Na(1)⋯C(18) | 2.942(4) |
| Na(1)⋯C(22) | 3.484(5) | Na(2)⋯C(2) | 3.595(4) |
| Na(2)⋯C(12) | 2.828(4) | Na(2)⋯C(14) | 3.449(4) |
| Na(2)⋯C(24) | 2.921(4) | N(1)–Na(1)–N(2) | 106.61(10) |
| Na(1)–N(1)–Na(2) | 73.95(8) | Si(1)–N(1)–Si(2) | 126.85(16) |
| Si(3)–N(2)–Si(4) | 126.28(16) | N(1)–Na(1)–O(1) | 133.44(10) |
| N(2)–Na(2)–O(2) | 134.31(10) | ||
| 5b | |||
| N(1)–Na(1) | 2.350(3) | Na(1)–O(1) | 2.505(4) |
| Na(1)–O(2) | 2.428(3) | Na(1)–O(3) | 2.567(4) |
| Na(1)–O(4) | 2.425(3) | N(1)–Si(1) | 1.671(3) |
| N(1)–Si(2) | 1.665(3) | Na(1)⋯C(2) | 3.557(5) |
| Na(1)⋯C(5) | 3.713(5) | Na(1)⋯C(8) | 3.578(5) |
| Na(1)⋯C(12) | 3.621(5) | Si(1)–N(1)–Si(2) | 133.65(19) |
| 6 | |||
| N(1)–K(1) | 2.848(2) | N(1)–K(1A) | 2.864(3) |
| K(1)⋯K(1A) | 3.461(2) | N(1)–Si(1) | 1.677(3) |
| N(1)–Si(2) | 1.696(3) | K(1)⋯C(4A) | 3.343(3) |
| K(1)⋯C(5B) | 3.188(4) | K(1)⋯C(6) | 3.309(3) |
| K(1)⋯C(8A) | 3.482(4) | K(1)⋯C(12) | 3.289(4) |
| N(1)–K(1)–N(1A) | 105.41(6) | K(1)–N(1)–K(1A) | 74.59(6) |
| Si(1)–N(1)–Si(2) | 130.75(16) | ||
| 7a | |||
| N(1)–Mg(1) | 1.956(3) | Mg(1)–C(13) | |
| Mg(1)–C(13A) | 2.212(4) | Mg(1)⋯Mg(1A) | 2.692(2) |
| N(1)–Mg(1)–C(13) | 129.50(13) | N(1)–Mg(1)–C(13A) | 125.28(13) |
| Mg(1)–C(13)–Mg(1A) | 74.80(12) | C(13)–Mg(1)–C(13A) | 105.20(12) |
| Si(1)–N(1)–Si(2) | 132.02(15) | ||
| 7b | |||
| N(1)–Mg(1) | 2.0293(15) | Mg(1)–C(13) | 2.118(2) |
| Mg(1)–O(1) | 2.1213(14) | Mg(1)–O(2) | 2.1259(16) |
| N(1)–Mg(1)–C(13) | 133.97(8) | Si(1)–N(1)–Si(2) | 127.60(9) |
| 8 | |||
| N(1)–Mg(1) | 1.9658(18) | Mg(1)–C(16) | 2.241(3) |
| Mg(1)–C(16A) | 2.231(2) | Mg(1)⋯Mg(1A) | 2.7200(14) |
| N(1)–Mg(1)–C(16) | 130.90(9) | N(1)–Mg(1)–C(16A) | 123.91(9) |
| Mg(1)–C(16)–Mg(1A) | 74.93(8) | C(16)–Mg(1)–C(16A) | 105.06(9) |
| Si(1)–N(1)–Si(2) | 132.51(10) | ||
| 9 | |||
| N(1)–Mg(1) | 1.977(2) | Mg(1)–C(19) | 2.251(4) |
| Mg(1)–C(19A) | 2.212(3) | Mg(1)⋯Mg(1A) | 2.739(2) |
| N(1)–Mg(1)–C(19) | 128.97(11) | N(1)–Mg(1)–C(19A) | 126.47(14) |
| Mg(1)–C(19)–Mg(1A) | 75.72(11) | C(19)–Mg(1)–C(19A) | 104.28(13) |
| Si(1)–N(1)–Si(2) | 135.93(15) | ||
| 10 | |||
| N(1)–Mg(1) | 1.985(5) | Mg(1)–I(1) | 2.8280(18) |
| Mg(1)–I(2) | 2.8187(19) | Mg(1)–O(1) | 2.010(4) |
| Mg(1)⋯Mg(1A) | 4.068(4) | Mg(1)–I(1)–Mg(1A) | 91.99(7) |
| Mg(1)–I(2)–Mg(1A) | 92.38(7) | I(1)–Mg(1)–I(2) | 87.81(5) |
| N(1)–Mg(1)–O(1) | 111.85(19) | Si(1)–N(1)–Si(2) | 128.6(3) |
Reactions of allylmagnesium chloride with sodium silylamides
The reaction of sodium silylamides, [Na(NRR′)], with allylmagnesium chloride did not give improved yields of the expected amido-Grignard complexes. During the course of these studies, we considered that sodium silylamides could be employed as alternative ligand transfer agents that may be less susceptible to byproduct formation. HN(SitBuMe2)2 was treated with an excess of sodium hydride in THF, but this deprotonation strategy was laborious and only 30% conversion was observed by 1H NMR spectroscopy after 72 h reflux. In contrast, the deprotonation of HN(SitBuMe2)2 with benzyl sodium34 in THF proceeded at room temperature to give [Na{N(SitBuMe2)2}(THF)n] as a dark oil following work-up (Scheme 2). Attempted crystallisation from pentane gave only several crystals of [Na{μ-N(SitBuMe2)2}(THF)]2 (5a); therefore the residue was treated with DME and recrystallised from pentane to afford [Na{N(SitBuMe2)2}(DME)2] (5b) as a beige solid in fair yield (65%). The NMR spectra of 5b exhibit similar features to the potassium salt [K{N(SiMe2tBu)2}]n,29a although 5b contains two additional resonances for coordinated DME in both the 1H (δ = 2.80 and 2.88 ppm) and 13C (δ = 59.21 and 71.30 ppm) NMR spectra. 29Si{1H} NMR spectroscopy shows a single resonance for 5b (δ = −13.89 ppm; cf. [K{N(SiMe2tBu)2}]nδ = −15.73 ppm).29a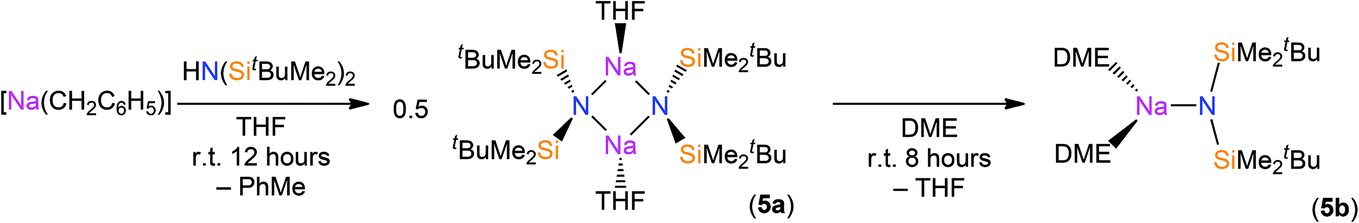 | ||
| Scheme 2 Synthesis of complexes 5a–b. | ||
The solid state structures of 5a and 5b are depicted in Fig. 3 and 4 respectively, and selected bond lengths and angles are shown in Table 1. Both complexes exhibit bond metrics typical of silylamide group 1 salts.28,35 Complex 5a is dimeric, with a central Na2N2 solvent-capped core and a mean Na–N distance of 2.4903(6) Å, which is longer than the mean Na–Nsilylamide bond lengths in [Na{μ-N(SiMe3)2}(THF)]2 [2.399(2) Å],35a [{Na[μ-N(SiMe3)2]}2(μ-TMEDA)]∞ (TMEDA = N,N,N′,N′-tetramethylethylenediamine) [2.4433(10) Å]35b and [Na{μ-N(SiMe2Ph)2}(THF)]2 [2.440(4) Å].35c This can be attributed to the increased steric demands of the silylamide ligands in 5a. The sodium coordination spheres in 5a are completed by multiple short Na⋯C–H/Na⋯H distances [Na⋯C range 2.827(4)–3.028(4) Å; Na⋯H range 2.48–2.88 Å], in common with the potassium salt [{K[μ-N(SiMe2But)2]}2(C7H8)]∞, which exhibits analogous K⋯C–H/K⋯H contacts.28 Complex 5b is monomeric in the solid state, with a 5-coordinate sodium centre and exhibits shorter Na–N distances [2.350(3) Å] than those in 5a, as would be expected for a terminally bound silylamide. The Na–O distances of 5b [range 2.425(3)–2.567(4) Å] are within the range of those for Na–DME interactions in the literature [2.402(9)–2.658(7) Å].36 To the best of our knowledge, [Sm{[N(Ar)C(CH2)]2CHC(NiPr)2Na(DME)2-κ3-N,N′,N′′}{N(SiMe3)2}] (Ar = C6H3iPr2-2,6)36d is the only other literature example containing a terminal, DME-capped N–Na bond. As with 5a, the coordination sphere of sodium in 5b is also completed by short Na⋯H contacts [range Na⋯H 2.84–3.06 Å].
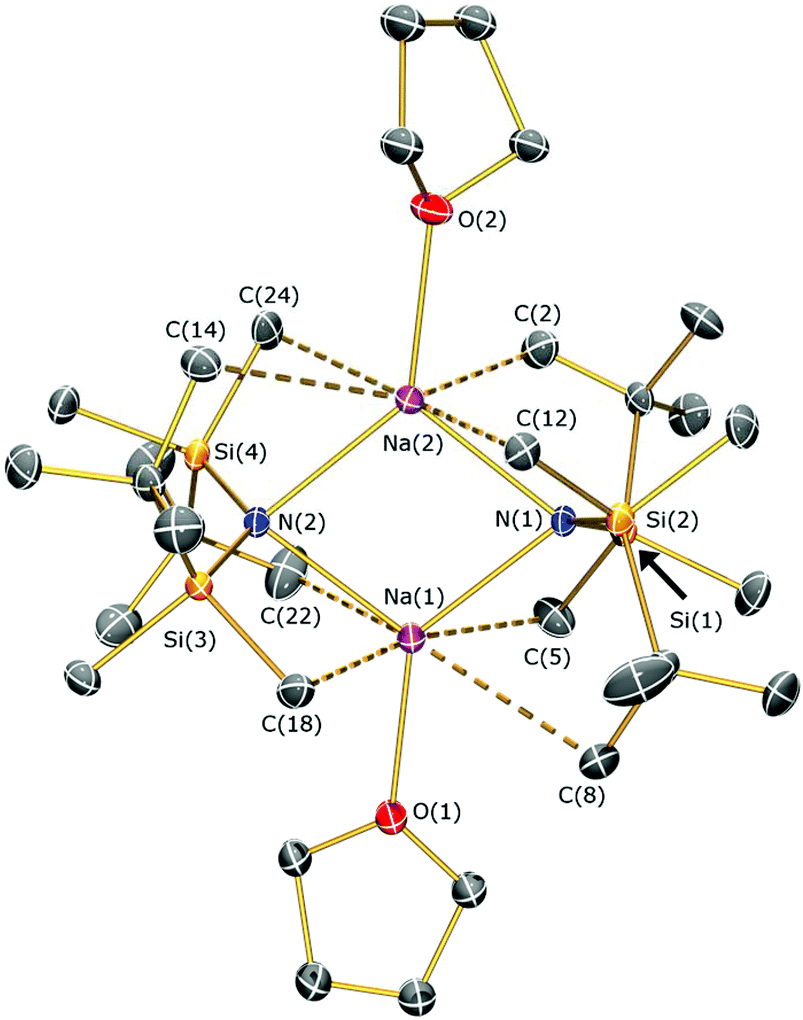 | ||
| Fig. 3 Molecular structure of 5a with selective atom labelling. Displacement ellipsoids set at 30% probability level and hydrogen atoms omitted for clarity. | ||
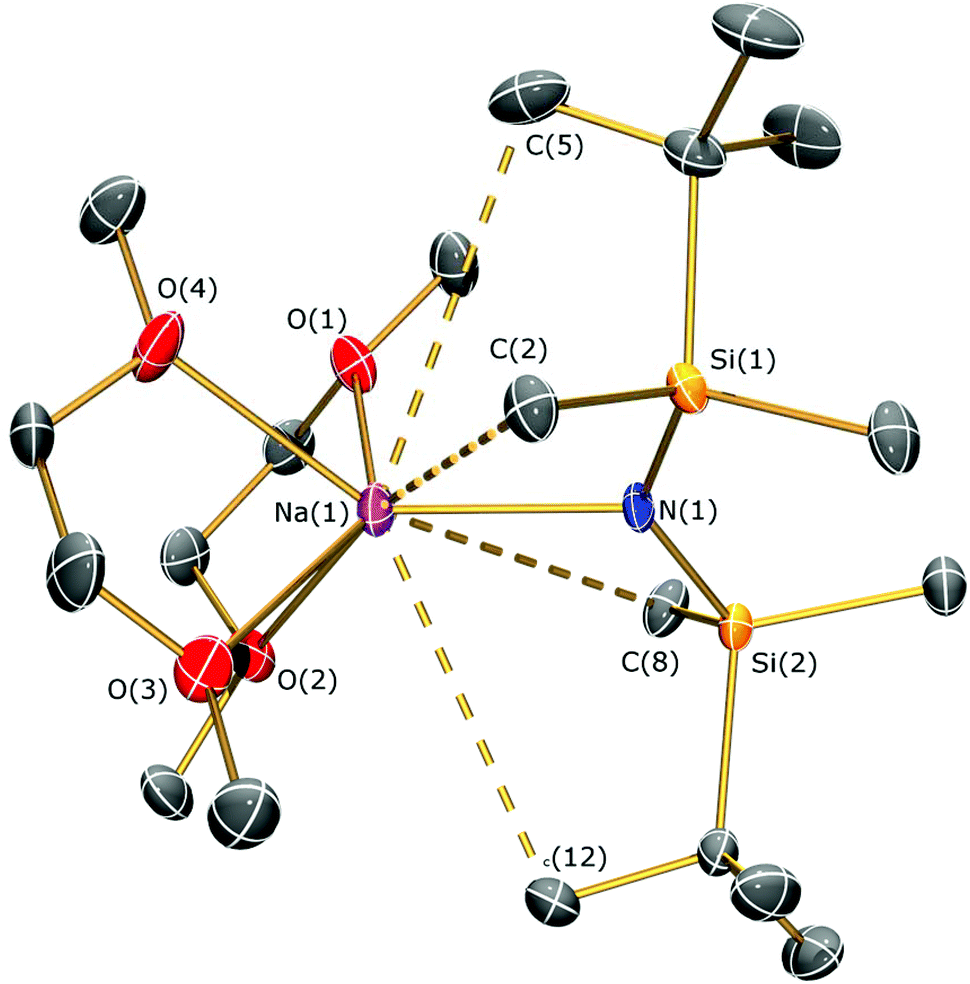 | ||
| Fig. 4 Molecular structure of 5b with selective atom labelling. Displacement ellipsoids set at 30% probability level and hydrogen atoms omitted for clarity. | ||
The reaction of 5b with allylmagnesium chloride in THF was performed in an attempt to synthesise [Mg{N(SiMe2But)2}(C3H5)]n. However, we were not able to isolate the amido-Grignard reagent from this reaction mixture, even after 10 days of stirring, and 1H NMR spectroscopy revealed a complex mixture of products. We concluded from these observations that allylmagnesium chloride is unsuitable for facile and reproducible synthetic routes to NRR′-containing Hauser bases, at least in the reaction conditions employed, and therefore we switched our attention to a different organomagnesium starting material (see below).
Reactions of methylmagnesium iodide with silylamines and potassium silylamides
Methylmagnesium iodide reacts with potassium silylamides, [K(NRR′)], to give amido-Grignard complexes in fair yields, but protonolysis reactions with the parent silylamines were unsuccessful. As the reaction of [K{N(SitBuMe2)2}]n or 5b with allylmagnesium chloride is slow and gives poor yields of 1 (see above), we selected methylmagnesium iodide as an alternative starting material, which has been extensively used in the literature as a reagent for the preparation of L–Mg–X and L–Mg–R complexes.6,9–14,36 In the course of synthesising potassium silylamide precursors for this study, crystals of [{K[μ-N(SitBuMe2)2]}2]∞ (6) were obtained and identified by single crystal XRD (Fig. 5, selected bond lengths and angles are shown in Table 1). We previously reported that desolvation of [{K[μ-N(SiMe2But)2]}2(C7H8)]∞ under vacuum yielded 6, but this was not structurally characterised.28 In the solid state, complex 6 is dimeric with a K2N2 core. K⋯C–H agostic interactions [K⋯C 3.188(4) Å] between the dimers form a 1D chain and numerous other K⋯C–H/K⋯H agostic and anagostic interactions are also present. Each potassium centre in 6 is close to two nitrogen atoms, with N–K distances [2.856(3) Å mean] and K–N–K [74.59(6)°] and N–K–N [105.41(6)°] angles that are comparable to those in [{K[μ-N(SiMe2But)2]}2(C7H8)]∞ [K–N 2.874(2) Å mean; K–N–K 78.37(5)°; N–K–N 101.63(5)°]28 and [K(μ-N′′)]2 [K–N 2.787(3) Å mean; K–N–K 85.53(9)°; N–K–N 94.47(9)°].37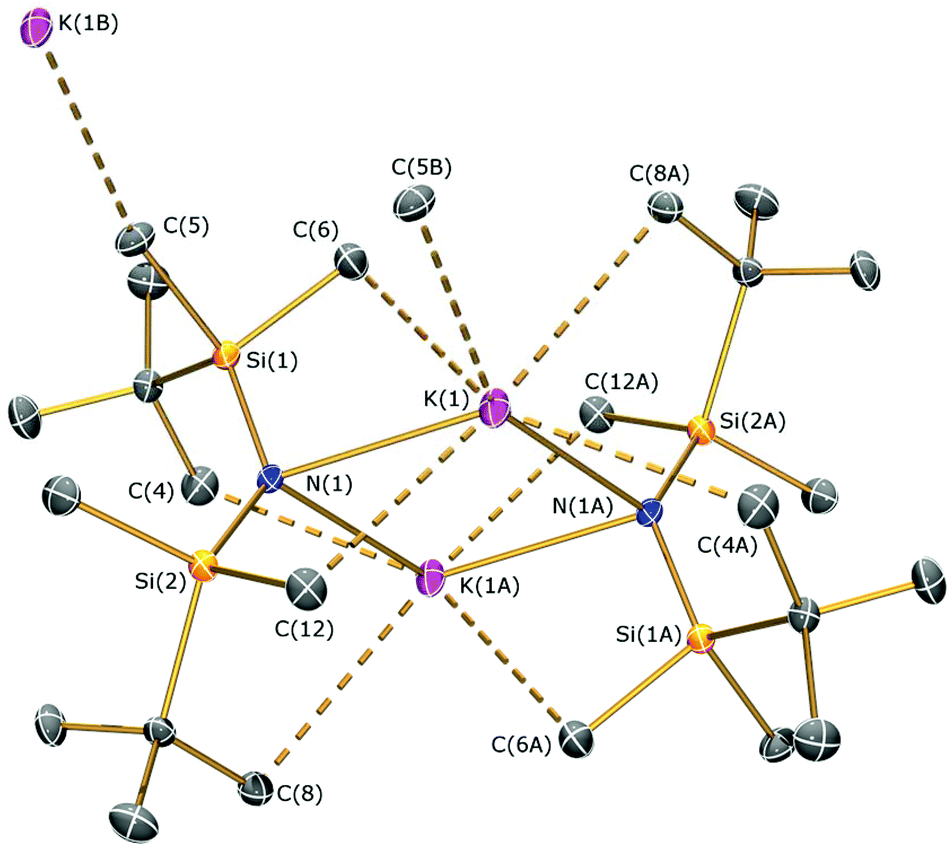 | ||
| Fig. 5 Molecular structure of 6 with selective atom labelling. Displacement ellipsoids set at 30% probability level and hydrogen atoms omitted for clarity. Symmetry operation to generate equivalent atoms: −x, −y, −z. | ||
The amido-Grignard complexes [Mg(NRR′)(μ-CH3)]2 (NRR′ = {N(SitBuMe2)2}−, 7a; {N(SitBuMe2)(SiiPr3)}−, 8; {N(SiiPr3)2}−, 9) were obtained by salt-metathesis reactions using the appropriate potassium amides and methylmagnesium iodide in diethyl ether, and treatment of 7a with DME gave [Mg{N(SitBuMe2)2}(CH3)(DME)] (7b) (Scheme 3). Workup and crystallisation from hot toluene afforded colourless crystals in poor (7a and 7b), fair (9) and excellent (8) yields, respectively. On one occasion a small crop of the Hauser base [Mg{N(SitBuMe2)2}(μ-I)(THF)]2 (10) was obtained (4%) when attempting to isolate 7a, which has presumably formed via complex Schlenk equilibria.1 Treatment of methylmagnesium iodide with HNRR′ in diethyl ether gave no reaction even after extended reaction times (>3 days at room temperature) or heating the reaction mixture under reflux for three hours. Furthermore, we found that 7a does not react with HN(SitBuMe2)2 under similar forcing conditions in hexanes. Germane to this, no reaction was observed between di-n-butylmagnesium and HN(SitBuMe2)2 in a mixture of heptane and hexanes. Refluxing this reaction mixture for extended periods gave an intractable mixture of products. Together, these experiments illustrate the sluggishness of protonolysis reactions between alkylmagnesium complexes and HNRR′. Complexes 7–10 were characterised by single crystal XRD studies (see below), elemental analysis and NMR and IR spectroscopies. To the best of our knowledge, 7a, 8 and 9 are the first examples of structurally characterised dimeric CH3-bridged Hauser bases with monodentate N-donor ligands. Complex 7b is unusual as it is monomeric with a terminal Mg–CH3 group, and there are few examples of this motif in the literature.38
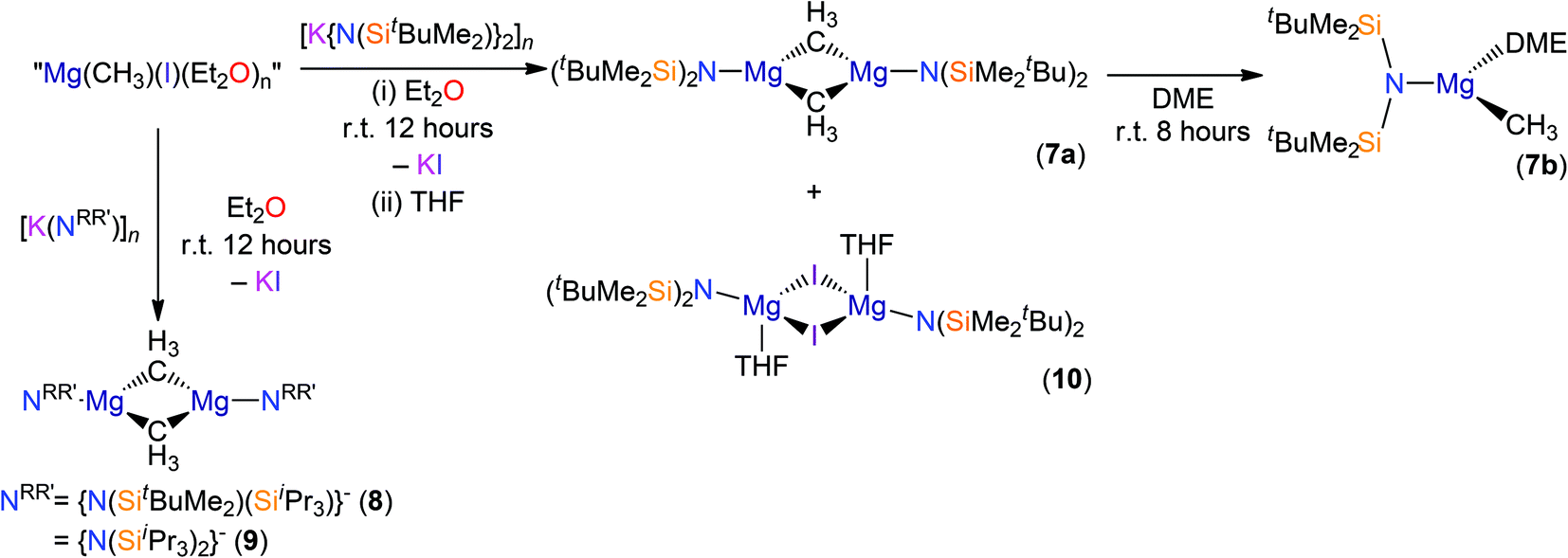 | ||
| Scheme 3 Synthesis of complexes 7–10. | ||
The 1H and 13C{1H} NMR spectra of 7–10 contain unremarkable silylamide ligand resonances, but the shielded methyl resonances in 7–9 [δH: −0.71 ppm (7a); −0.99 ppm (7b); −0.81 ppm (8); −0.80 ppm (9); δC: −10.04 ppm (7a), −13.65 ppm (7b), −13.02 ppm (8), −12.19 ppm (9)] are in agreement with those of structurally characterised examples in the literature (δH range −2.00 ppm to −0.67 ppm).38 The similarity of the methyl group chemical shifts in 7–9 with multidentate N-donor complexes in the literature suggests that the spectator ligand does not greatly influence these values.6,39,40 One resonance is observed in the 29Si{1H} NMR spectra of 7a, 7b, 9 and 10 and two resonances were found for 8 [δSi: −1.93 ppm (7a); −2.43 ppm (7b); −2.90 and −1.76 ppm (8); −3.15 ppm (9); −1.19 ppm (10)], correlating with the number of unique silicon environments.
The identities of 7a, 7b, 8, 9 and 10 were determined by single crystal XRD and are depicted herein (7a: Fig. 6; 7b: Fig. 7; 10: Fig. 8; see ESI Fig. S13 and S14† for the structures of 8 and 9), with selected bond lengths and angles compiled in Table 1. The structures of 7a, 8 and 9 are broadly similar, with 3-coordinate Mg centres and bridging CH3 moieties to form central Mg2C2 rhomboids with centres of inversion. The Mg–N distances [7a: 1.956(3) Å, 8: 1.9658(18) Å; 9: 1.977(2) Å] are slightly shorter than in 1, 2, and 3 respectively, which is a reflection of the absence of coordinated solvent molecules in 7–9. The mean Mg–C bond lengths in 7a, 8 and 9 are comparable [7a: 2.217(6) Å; 8: 2.236(3) Å; 9: 2.232(5) Å] and are typical of methylmagnesium amides [previously reported range Mg–C 1.977(3)–2.434(13) Å].33 The MgNSi2 fragments of all three ligands are roughly planar in 7a, 8 and 9; these planes are twisted relative to the central Mg2C2 plane by differing amounts [7a: 74.38(11)°; 8: 63.95(8)°; 9: 67.77(10)°] with no clear trend. In the dimeric unit, the two ligand Si–N–Si fragments are also co-planar with each other (to within 0.1°) for all three structures. The coordination spheres of the magnesium centres in 7a, 8 and 9 are completed by a number of short Mg⋯C–H/Mg⋯H distances. Complex 7b is monomeric due to the coordinated DME molecule and the Mg–N [2.0293(15) Å] and Mg–C [2.118(2) Å] bonds are correspondingly shorter than those in 7a. However, the coordination sphere of the magnesium centre is again completed by short Mg⋯C–H/Mg⋯H distances. Complex 10 exhibits a near-square Mg2I2 central motif, with the sum of the four internal angles calculated at 359.99(12)°. The two Mg–I bond lengths in 10 [2.8280(18) and 2.8187(19) Å] are similar to those observed for literature examples that contain N–Mg–I moieties and bridging iodide ligands [range 2.7766(12)–2.901(3) Å].6,19,41 Finally, in common with 1–3, the coordination sphere of Mg is completed by a THF molecule [Mg–O 2.010(4) Å] and multiple short Mg⋯C–H/Mg⋯H distances.
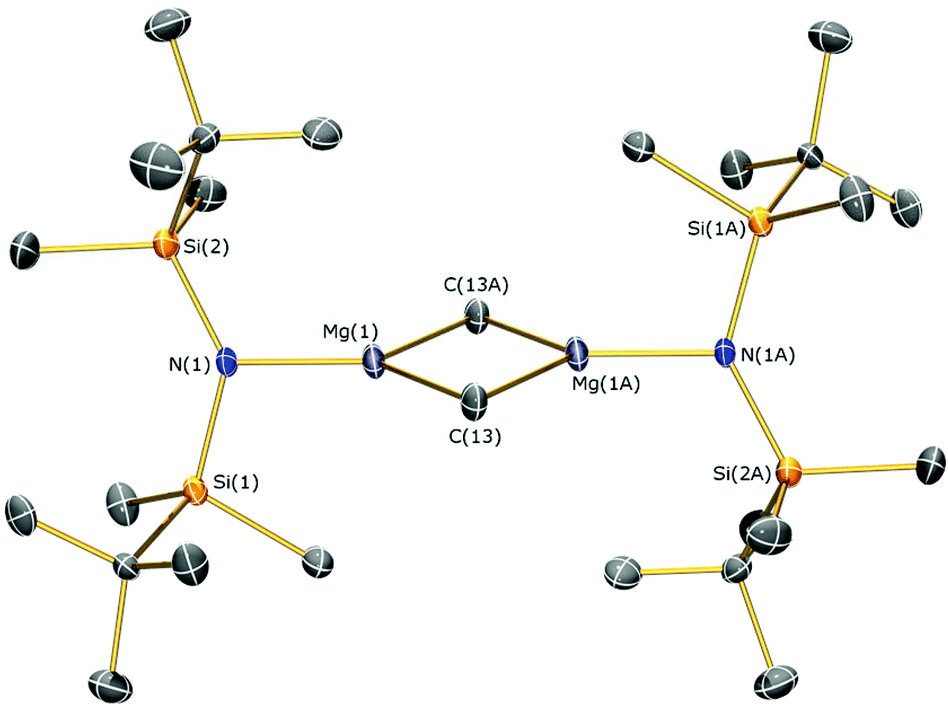 | ||
| Fig. 6 Molecular structure of 7a with selective atom labelling. Displacement ellipsoids set at 30% probability level and hydrogen atoms omitted for clarity. Symmetry operation to generate equivalent atoms: −x, −y, −z. | ||
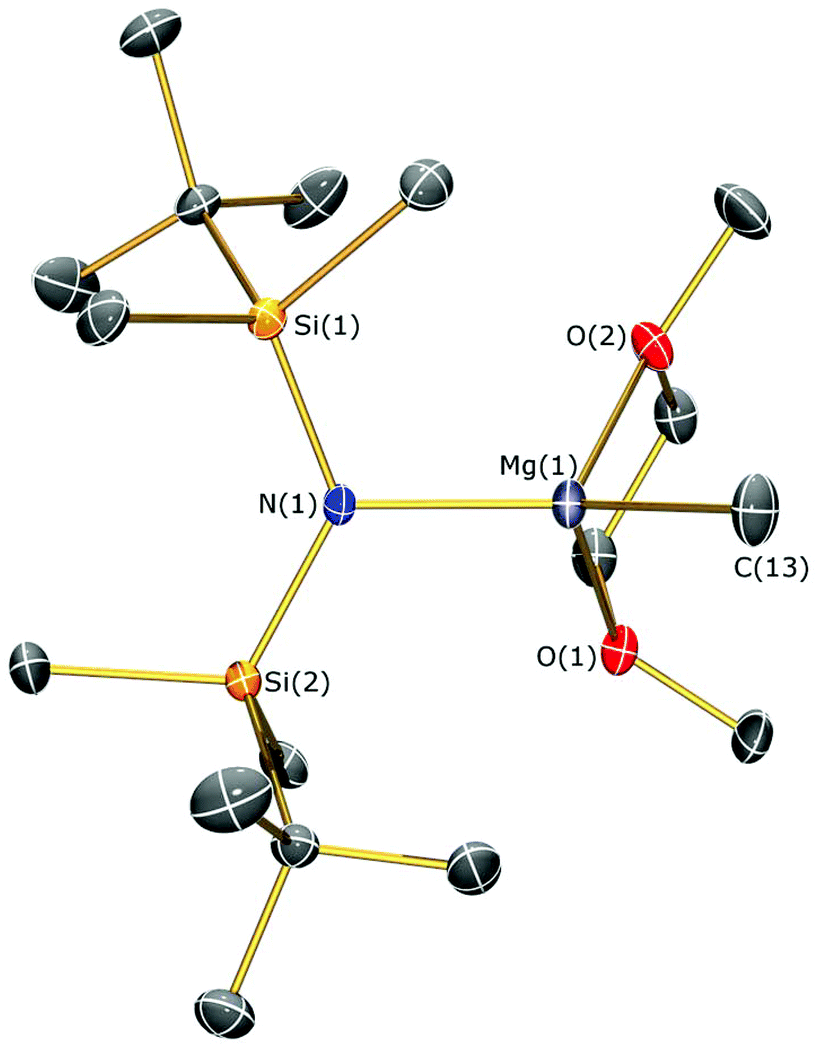 | ||
| Fig. 7 Molecular structure of 7b with selective atom labelling. Displacement ellipsoids set at 30% probability level and hydrogen atoms omitted for clarity. | ||
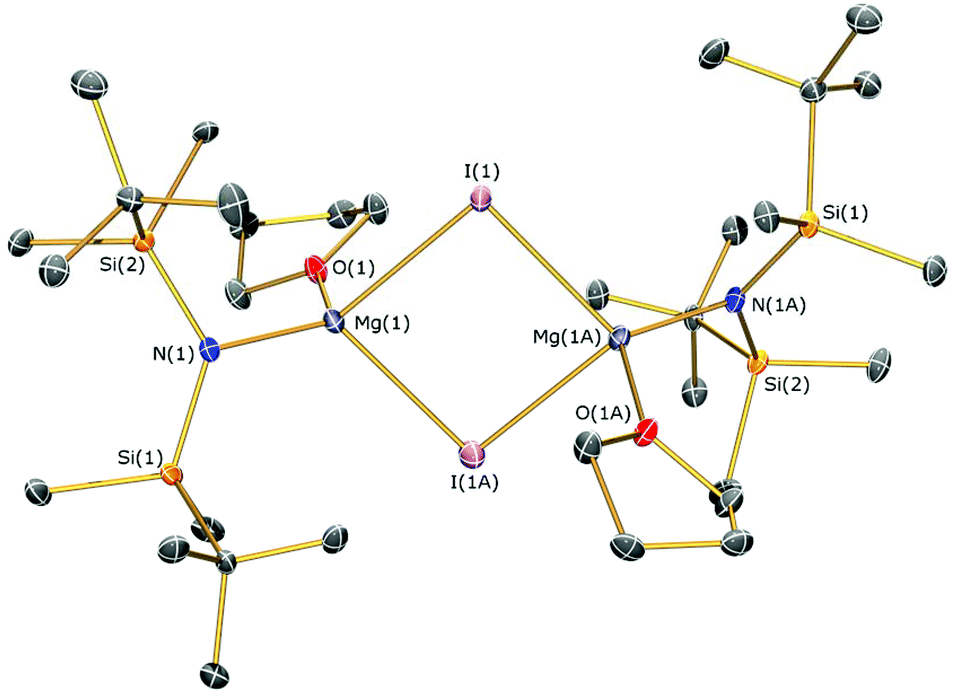 | ||
| Fig. 8 Molecular structure of 10 with selective atom labelling. Displacement ellipsoids set at 30% probability level and hydrogen atoms omitted for clarity. Symmetry operation to generate equivalent atoms: x + 1/2, −y + 1/2, z − 1/2. | ||
Conclusions
We have structurally characterised a series of silylamide Hauser bases during our exploration of straightforward synthetic routes to these complexes. Protonolysis routes to Hauser base complexes by treating allylmagnesium chloride, di-n-butylmagnesium or methylmagnesium iodide with a series of silylamines were found to be slow and prone to complex Schlenk equilibria. A fully characterised isolated product, 1, was achieved on only one occasion by using extended reaction times. The salt metathesis reactions of allylmagnesium chloride with potassium silylamides, and the novel sodium silylamide, 5b, gave intractable mixtures. We concluded from these studies that, using these ligand systems and methodologies, allylmagnesium chloride is unsuitable as a starting material for reproducible syntheses of silylamide Hauser base and amido-Grignard complexes.Salt metathesis reactions of potassium silylamides with methylmagnesium iodide were found to be a far more successful strategy, giving modest to excellent yields of the amido-Grignard complexes 7–9. The isolation of a small amount of the iodide Hauser base complex 10 indicated that complex Schlenk equilibria also operate in these reaction mixtures, slightly reducing the yields of the target complexes. Despite these side-reactions, facile synthetic routes to amido-Grignard complexes of three different silylamide ligands have been achieved. We envisage that such amido-Grignard complexes could be useful reagents for heterofunctionalisation reactions. Additionally, such compounds could pave the way for the preparation of novel bimetallic systems with alkali metals, which could potentially be employed in synergic metalation reactions.
Experimental
Materials and methods
All manipulations were carried out using standard Schlenk and glove box techniques under an atmosphere of dry argon. Solvents were dried by refluxing over potassium and degassed before use. All solvents were stored over potassium mirrors (with the exception of THF and DME which were stored over activated 4 Å molecular sieves). Deuterated solvents were distilled from potassium, degassed by three freeze–pump–thaw cycles and stored under argon. [K{N(SitBuMe2)2}]n,29a [K{N(SitBuMe2)(SiiPr3)2}]n,28 [K{N(SiiPri3)2}]n,29b were prepared according to published procedures. [Na(CH2C6H5)] was prepared via a modification of published procedures.34 All other chemicals were used as purchased and stored appropriately. Most solid reagents were dried under vacuum for four hours and most liquid reagents were dried over 4 Å molecular sieves and distilled before use. 1H, 13C{1H} and 29Si{1H} NMR spectra were recorded on a spectrometer operating at 400.2, 100.6 and 79.5 MHz, respectively; chemical shifts are quoted in ppm and are relative to TMS. FTIR spectra were recorded as Nujol mulls in KBr discs on a Perkin Elmer Spectrum RX1 spectrometer. Elemental microanalyses were carried out by Mr Stephen Boyer at the Microanalysis Service, London Metropolitan University, UK.Synthetic procedures
Acknowledgements
We thank the EPSRC and The University of Manchester for generously supporting this work. This work was funded by the Engineering and Physical Sciences Research Council (grant numbers EP/K039547/1 and EP/L014416/1).References
- G. S. Silverman and P. E. Rakita, Handbook of Grignard Reagents, Marcel Dekker, Inc., New York, NY, 1996 Search PubMed.
- (a) J. K. Stille and W. J. Scott, J. Am. Chem. Soc., 1986, 108, 3033 CrossRef; (b) E. C. Ashby and A. B. J. Goel, Inorg. Chem., 1978, 17, 1862 CrossRef CAS; (c) K. Kobayashi, M. Kawakita, S. Irisawa, H. Akamatsu, K. Sakashita, O. Morikawa and H. Konishi, Tetrahedron, 1998, 54, 2691 CrossRef CAS; (d) D. Bonafoux, M. Bordeau, C. Brian and P. Cazeau, J. Org. Chem., 1996, 61, 5532 CrossRef CAS; (e) N. A. Van Draanen, S. Arseniyadis, M. T. Crimmins and C. H. Heathcock, J. Org. Chem., 1991, 56, 2499 CrossRef CAS; (f) Y. Kondo, A. Yoshida and T. Sakamoto, J. Chem. Soc., Perkin Trans. 1, 1996, 2331 RSC; (g) K. A. Swiss, C. Woo-Baeg, D. C. Liotta and A. F. Abdel-Maryanoff, J. Org. Chem., 1991, 56, 5978 CrossRef CAS; (h) J. F. Allan, K. W. Henderson and A. R. Kennedy, J. Chem. Soc., Chem. Commun., 1997, 1149 Search PubMed.
- (a) P. García-Álvarez, D. V. Graham, E. Hevia, A. R. Kennedy, J. Klett, R. E. Mulvey, C. T. O'Hara and S. Weatherstone, Angew. Chem., Int. Ed., 2008, 47, 8079 CrossRef PubMed; (b) R. E. Mulvey and S. D. Robertson, Top. Organomet. Chem., 2013, 45, 103 CrossRef CAS; (c) A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. London, Ser. A, 2010, 466, 927 CrossRef CAS; (d) S. Harder, Chem. Rev., 2010, 110, 3852 CrossRef CAS PubMed; (e) J.-F. Carpentier and Y. Sarazin, Top. Organomet. Chem., 2013, 45, 141 CrossRef CAS; (f) M. R. Crimmin and M. S. Hill, Top. Organomet. Chem., 2013, 45, 191 CrossRef CAS; (g) M. F. Lappert, A. Protchenko, P. P. Power and A. Seeber, Metal Amide Chemistry, John Wiley & Sons Ltd, Wiltshire, UK, 2009 Search PubMed.
- (a) K. Ruhlandt-Senge, A. Torvisco and A. Y. O'Brien, Coord. Chem. Rev., 2011, 255, 1268 CrossRef PubMed; (b) W. D. Buchanan, D. G. Allis and K. Ruhlandt-Senge, Chem. Commun., 2010, 46, 4449 RSC.
- M. Westerhausen, Coord. Chem. Rev., 2008, 252, 1516 CrossRef CAS PubMed.
- (a) M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826 RSC; (b) D. J. Liptrot, M. S. Hill and M. F. Mahon, Angew. Chem., Int. Ed., 2014, 53, 6224 CrossRef CAS PubMed.
- Y. Sarazin, M. Schormann and M. Bochmann, Organometallics, 2004, 23, 3296 CrossRef CAS.
- (a) M. H. Chisholm and N. W. Eilerts, Chem. Commun., 1996, 853 RSC; (b) M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iver, V. Pacol and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845 CrossRef CAS; (c) M. H. Chisholm, J. Galluci and K. Phomphrai, Chem. Commun., 2003, 48 RSC.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754 CrossRef CAS PubMed.
- (a) A. Stasch and C. Jones, Dalton Trans., 2011, 40, 5659 RSC; (b) A. Stasch, Angew. Chem., Int. Ed., 2014, 53, 10200 CrossRef CAS PubMed; (c) C. Jones and A. Stasch, Top. Organomet. Chem., 2013, 45, 73 CrossRef CAS.
- (a) S. Krieck, L. Yu, M. Reiher and M. Westerhausen, Eur. J. Inorg. Chem., 2010, 197 CrossRef CAS PubMed; (b) S. Krieck and M. Westerhausen, Chem. Unserer Zeit, 2009, 43, 384 CrossRef CAS PubMed; (c) M. Westerhausen, Angew. Chem., Int. Ed., 2008, 47, 2185 CrossRef CAS PubMed.
- G. J. Moxey, F. Ortu, L. G. Sidley, H. N. Strandberg, A. J. Blake, W. Lewis and D. L. Kays, Dalton Trans., 2014, 43, 4838 RSC , and references therein.
- L. Bourget-Merle, M. F. Lappert and J. S. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS PubMed.
- For reviews of the closely related bis(iminophospharano)methanide ligand see: (a) T. K. Panda and P. W. Roesky, Chem. Soc. Rev., 2009, 38, 2782 RSC; (b) S. T. Liddle, D. P. Mills and A. J. Wooles, Top. Organomet. Chem., 2010, 36, 29 CAS; (c) S. T. Liddle, D. P. Mills and A. J. Wooles, Chem. Soc. Rev., 2011, 40, 2164 RSC.
- See: M. Westerhausen, Coord. Chem. Rev., 1998, 176, 157 CrossRef CAS ; for a review on silylamide Ae chemistry.
- (a) L. Meunier, C. R. Hebd. Seances Acad. Sci., 1903, 136, 758 CAS; (b) C. R. Hauser and H. G. Walker, J. Am. Chem. Soc., 1947, 69, 295 CrossRef CAS; (c) C. R. Hauser and F. C. Frostick, J. Am. Chem. Soc., 1949, 71, 1350 CrossRef.
- R. A. Bartlett, M. M. Olmstead and P. P. Power, Inorg. Chem., 1994, 33, 4800 CrossRef CAS.
- U. Wannagat, H. Autzen, H. Kuckertz and H.-J. Wismar, Z. Anorg. Allg. Chem., 1972, 394, 254 CrossRef CAS PubMed.
- K.-C. Yang, C.-C. Chang, J.-Y. Huang, C.-C. Lin, G.-H. Lee, Y. Wang and M. Y. Chiang, J. Organomet. Chem., 2002, 648, 176 CrossRef CAS.
- P. L. Arnold, I. S. Edworthy, C. D. Carmichael, A. J. Blake and C. Wilson, Dalton Trans., 2008, 3739 RSC.
- (a) P. C. Junk, C. L. Raston, B. W. Skelton and A. H. White, J. Chem. Soc., Chem. Commun., 1987, 1162 RSC; (b) S. Yuan, S. Bai, D. Liu and W.-H. Sun, Organometallics, 2010, 29, 2132 CrossRef CAS; (c) A. S. Batsanov, P. D. Bolton, R. C. B. Copley, M. G. Davidson, J. A. K. Howard, C. Lustig and R. D. Price, J. Organomet. Chem., 1998, 550, 445 CrossRef CAS; (d) D. R. Armstrong, P. García-Álvarez, A. R. Kennedy, R. E. Mulvey and J. A. Parkinson, Angew. Chem., Int. Ed., 2010, 49, 3185 CrossRef CAS PubMed; (e) T. Hascall, M. M. Olmstead and P. P. Power, Angew. Chem., Int. Ed., 1994, 33, 1000 CrossRef PubMed; (f) J. A. Rood, S. E. Hinman, B. C. Noll and K. W. Henderson, Eur. J. Inorg. Chem., 2008, 3935 CrossRef CAS PubMed; (g) F. Ortu, G. J. Moxey, A. J. Blake, W. Lewis and D. L. Kays, Chem. – Eur. J., 2015, 21, 6949 CrossRef CAS PubMed.
- P. E. Eaton, C.-H. Lee and Y. Xiong, J. Am. Chem. Soc., 1989, 111, 8016 CrossRef CAS.
- (a) L. M. Engelhardt, B. S. Jolly, P. C. Junk, C. L. Raston, B. W. Skelton and A. H. White, Aust. J. Chem., 1986, 39, 1337 CrossRef CAS; (b) H. Schumann, A. Steffens and M. Hummert, Z. Anorg. Allg. Chem., 2009, 635, 1041 CrossRef CAS PubMed; (c) T. Y. Her, C. C. Chang, G. H. Lee, S. M. Peng and Y. Wang, Inorg. Chem., 1994, 33, 99 CrossRef CAS; (d) B. Conway, E. Hevia, A. R. Kennedy, R. E. Mulvey and S. Weatherstone, Dalton Trans., 2005, 1532 RSC.
- A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Dalton Trans., 2010, 39, 9091 RSC.
- P. C. Andrikopoulos, D. R. Armstrong, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, R. B. Rowlings and S. Weatherstone, Inorg. Chim. Acta, 2007, 360, 1370 CrossRef CAS PubMed.
- (a) D. J. Gallagher, K. W. Henderson, A. R. Kennedy, C. T. O'Hara, R. B. Rowlings and R. E. Mulvey, Chem. Commun., 2002, 376 RSC; (b) E. Hevia, F. R. Kenley, A. R. Kennedy, R. E. Mulvey and R. B. Rowlings, Eur. J. Inorg. Chem., 2003, 3347 CrossRef CAS PubMed; (c) E. Hevia, D. J. Gallagher, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and C. Talmard, Chem. Commun., 2004, 2422 RSC; (d) P. C. Andrikopoulos, D. R. Armstrong, W. Clegg, C. J. Gilfillan, E. Hevia, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, J. A. Parkinson and D. M. Tooke, J. Am. Chem. Soc., 2004, 126, 11612 CrossRef CAS PubMed; (e) K. J. Drewette, K. W. Henderson, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and R. B. Rowlings, Chem. Commun., 2002, 1176 RSC.
- See, for example: (a) A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 2958 CrossRef CAS PubMed; (b) W. Lin, O. Baron and P. Knochel, Org. Lett., 2006, 8, 5673 CrossRef CAS PubMed; (c) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794 CrossRef CAS PubMed; (d) A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834 CrossRef PubMed.
- C. A. P. Goodwin, K. C. Joslin, S. J. Lockyer, A. Formanuik, G. A. Morris, F. Ortu, I. J. Vitorica-Yrezabal and D. P. Mills, Organometallics, 2015, 34, 2314 CrossRef CAS.
- (a) C. A. P. Goodwin, F. Tuna, E. J. L. McInnes, S. T. Liddle, J. McMaster, I. J. Vitorica-Yrezabal and D. P. Mills, Chem. – Eur. J., 2014, 20, 14579 CrossRef CAS PubMed; (b) N. F. Chilton, C. A. P. Goodwin, D. P. Mills and R. E. P. Winpenny, Chem. Commun., 2015, 51, 101 RSC.
- J. Li, A. Stasch, C. Schenk and C. Jones, Dalton Trans., 2011, 40, 10448 RSC.
- Z. Rappoport and I. Marek, The Chemistry of Organomagnesium Compounds, John Wiley & Sons Ltd, West Sussex, UK, 2008 Search PubMed.
- P. B. Hitchcock, M. F. Lappert, L. Maron and A. V. Protchenko, Angew. Chem., Int. Ed., 2008, 47, 1488 CrossRef CAS PubMed.
- CSD version 5.36, November 2014, update 3 (May 2015); F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- (a) M. G. Davidson, G. Garcia-Vico, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 3364 CrossRef CAS PubMed; (b) P. J. Bailey, R. A. Coxall, C. M. Dick, S. Fabre, L. C. Henderson, C. Herber, S. T. Liddle, D. Loroño-González, A. Parkin and S. Parsons, Chem. – Eur. J., 2003, 9, 4820 CrossRef CAS PubMed; (c) D. Hoffmann, W. Bauer, F. Hampe, N. J. R. van Eikema Hommes, P. v. R. Schleyer, P. Otto, U. Pieper, D. Stalke, D. S. Wright and R. J. Snaith, J. Am. Chem. Soc., 1994, 116, 528 CrossRef CAS; (d) M. Schlosser and J. Hartmann, Angew. Chem., Int. Ed. Engl., 1973, 12, 508 CrossRef PubMed.
- (a) M. Karl, G. Seybert, W. Massa, K. Harms, S. Agarwal, R. Maleika, W. Stelter, A. Greiner, W. Heitz, B. Neumuller and K. Dehnicke, Z. Anorg. Allg. Chem., 1999, 625, 1301 CrossRef CAS; (b) A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, S. D. Robertson and G. M. Robertson, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m1468 CAS; (c) W. J. Evans, D. B. Rego and J. W. Ziller, Inorg. Chem., 2006, 45, 3437 CrossRef CAS PubMed , and references therein.
- (a) M. L. Cole, A. J. Davies, C. Jones and P. C. Junk, J. Organomet. Chem., 2004, 689, 3093 CrossRef CAS PubMed; (b) C. Glock, H. Gorls and M. Westerhausen, Eur. J. Inorg. Chem., 2011, 5288 CrossRef CAS PubMed; (c) M. L. Cole and P. C. Junk, Dalton Trans., 2003, 2109 RSC; (d) P. Liu, H. Chen, Y. Zhang, M. Xue, Y. Yao and Q. Shen, Dalton Trans., 2014, 43, 5586 RSC.
- K. F. Tesh, T. P. Hanusa and J. C. Huffman, Inorg. Chem., 1990, 29, 1584 CrossRef CAS.
- (a) V. C. Gibson, J. A. Segal, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2000, 122, 7120 CrossRef CAS; (b) P. J. Bailey, R. A. Coxall, C. M. Dick, S. Fabre and S. Parsons, Organometallics, 2001, 20, 798 CrossRef CAS; (c) M. Arrowsmith, B. Maitland, G. Kociok-Köhn, A. Stasch, C. Jones and M. S. Hill, Inorg. Chem., 2014, 53, 10543 CrossRef CAS PubMed; (d) S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938 CrossRef CAS PubMed.
- M. Veith, W. Frank, F. Tollner and H. Lange, J. Organomet. Chem., 1987, 326, 315 CrossRef CAS.
- (a) P. J. Bailey, R. A. Coxall, C. M. E. Dick, S. Fabre, S. Parsons and L. J. Yellowlees, Chem. Commun., 2005, 4563 RSC; (b) T.-Y. Her, C.-C. Chang and L.-K. Liu, Inorg. Chem., 1992, 31, 2291 CrossRef CAS; (c) P. J. Bailey, C. M. E. Dick, S. Fabre and S. Parsons, J. Chem. Soc., Dalton Trans., 2000, 1665 Search PubMed.
- (a) M. Veith, A. Spaniol, J. Poehlmann, F. Gross and V. Huch, Chem. Ber., 1993, 126, 2625 CrossRef CAS PubMed; (b) B. Lian, M. Christophe, O. L. Casagrande Jr., T. Roisnel and J.-F. Carpentier, Polyhedron, 2007, 26, 3817 CrossRef CAS PubMed; (c) W. Zhang, J.-P. Hu, D. Xiao-Feng, Y.-J. Wu and Z.-W. Ye, Inorg. Chem. Commun., 2003, 6, 1185 CrossRef CAS; (d) M. L. H. Green, G. A. Moser, I. Packer, F. Petit, R. A. Forder and K. Prout, J. Chem. Soc., Chem. Commun., 1974, 839 RSC; (e) P. Haiss, A. Kuhn, N. Kuhn, C. Maichle-Mößmer, S. Laufer, M. Steimann and K.-P. Zeller, Eur. J. Inorg. Chem., 2011, 22, 3284 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1408436–1408447 for 1–3, 5a–b, 6, 7a–b, 8–10 and 4. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02535d |
| This journal is © The Royal Society of Chemistry 2016 |