
A novel yet simple strategy to fabricate visible light responsive C,N-TiO2/g-C3N4 heterostructures with significantly enhanced photocatalytic hydrogen generation†
Wei Chena,
Tian-Yu Liua,
Ting Huanga,
Xiao-Heng Liu*a,
Guo-Rong Duana,
Xu-Jie Yanga and
Shen-Ming Chen*b
aKey Laboratory of Education Ministry for Soft Chemistry and Functional Materials, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: xhliu@mail.njust.edu.cn
bElectroanalysis and Bioelectrochemistry Lab, Department of Chemical Engineering and Biotechnology, National Taipei University of Technology, No.1, Section 3, Chung-Hsiao East Road, Taipei 106, Taiwan, Republic of China. E-mail: smchen78@ms15.hinet.net
First published on 11th November 2015
Abstract
In this report, we first successfully designed and fabricated novel C,N co-doped titanium dioxide nanoparticles/graphite-like carbon nitrogen ultrathin nanosheets (C,N-TiO2 NPs/g-C3N4) heterostructures, wherein the C,N-TiO2 NPs were in situ grown on the porous g-C3N4 ultrathin nanosheets (NSs) by a simple one-pot solvothermal route with the assistance of concentrated nitric acid. The resulting C,N-TiO2 NPs/g-C3N4 nanocomposite photocatalysts were systematically characterized by powder X-ray diffraction (PXRD), Fourier transform infrared spectroscopy (FT-IR), diffuse reflectance spectroscopy (DRS), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), photoluminescence (PL) spectroscopy, transient photocurrent–time (I–t) curves and electrochemical impedance spectroscopy (EIS) Nyquist plots. The photocatalytic ability was evaluated by photocatalytic water splitting for hydrogen evolution. These studies indicate that C,N-TiO2 NPs/g-C3N4 composites exhibit superior ability for hydrogen generation compared to single C,N-TiO2 NPs and pure g-C3N4 NSs under visible light illumination. The optimal composites with 3 wt% C,N-TiO2 NPs/g-C3N4 showed the highest hydrogen evolution rate of 39.18 μmol g−1 h−1, which is about 10.9 and 21.3 times higher than those of C,N-TiO2 NPs and pure g-C3N4 NSs, respectively. The improved photocatalytic H2 evolution can be attributed to improved optical absorption and the lengthening lifetime of charge carrier pairs as a result of the C,N elemental codoping and the construction of intimate heterogeneous interfaces. This simple and feasible method for the fabrication of highly-efficient visible light responsive catalysts provides a great applied potential in energy generation.
1. Introduction
With the rapidly increasing population and high rate development of industrialization, recent years have witnessed a hugely serious energy crisis and environmental contamination resulting from the depletion of fossil fuels and the emission of poisonous and harmful substances. These challenges force us to develop new energies to replace the traditional energies. Hydrogen energy, widely popular as a clean energy, is a promising choice to resolve the above-mentioned bottlenecks since Fujishima and Honda reported the photoelectrochemical hydrogen evolution by using the TiO2 electrode in 1972.1 However, because of the relatively wide band gap with value of about 3.2 eV for the anatase phase and 3.0 eV for the rutile phase, the pure TiO2 just exhibits ultraviolet light response to generate holes and electrons pairs, which are important active species responsible for photocatalytic process. Unluckily, the ultraviolet light only makes up 4% of the total solar spectrum reaching the surface of the earth. The limited UV-driven activity largely inhibits the practical application of pure TiO2. Besides, the speedy recombination of photogenerated holes and electrons pairs is another fatal flaw, leading to low quantum efficiency of original TiO2. Therefore, huge efforts have been devoted to extending the spectrum absorption into visible light region and lengthening the lifespan of charge carrier pairs by means of noble metal loading,2–4 elemental doping5–7 or co-doping8,9 and heterogeneous coupling.10–12So now many practices with different fabrication routes have proven that elemental doping, such as C, N, S, F, Ti doping or co-doping, is a feasible strategy to modify the photocatalytic activity of TiO2 materials, which would not only narrow the band gap of TiO2 to increase the light absorption but also introduce more active sites to lengthen the lifetime of photogenerated holes and electrons pairs.13–15 To further optimize the photocatalytic activity, on the basis of original TiO2 or doped TiO2, researchers have constructed the heterogeneous composites by coupling with other materials.16 Graphitic carbon nitrogen (g-C3N4), with the features of appropriate band gap, abundant available, simple preparation and highly stability at different environments, were widely composited with TiO2 and modified TiO2. Recently, Wang et al.17 reported the fabrication of TiO2/g-C3N4 by calcination route and their highly-efficient photocatalytic activity for hydrogen generation from water splitting. Li et al.18 utilized the similar synthetic strategy to prepare the Ti3+ self-doped TiO2/g-C3N4 heterojunctions, which shows significantly enhanced photocatalytic performance under LED light irradiation. Also, Zhou et al.19 reported obvious photocatalytic enhancement of hybrid TiO2/g-C3N4 via ball milling method. Obviously, although the enhancement of photocatalytic activity has been realized as results of the construction of heterogeneous junctions by coupling between TiO2 (modified TiO2) and g-C3N4, the deficiency of these researches focuses on the composite with bulk g-C3N4, which often exhibits high recombination rate of photo-generated charge carriers due to the formation of a vast grain boundary defects during the thermal polycondensation and the low specific surface area.20 Han et al.21 in situ prepared the N-TiO2/g-C3N4 nanosheets heterostructures by a simple electrospinning process combined with a modified heat-etching method, which present highly efficient degradation activity and photocatalytic H2 production with the exposure of simulated solar light.
To our knowledge, there is no report involving the synthesis of C,N-TiO2 NPs/g-C3N4 ultrathin nanosheets heterostructures until now. Herein, we demonstrate the simple and novel strategy to prepare the C,N-TiO2 NPs/g-C3N4 NSs nanocomposites with highly efficient catalytic performance for hydrogen generation. The fabrication strategy of C,N-TiO2 NPs/g-C3N4 is simple and exclusive of calcination treatment at high temperature condition. Different from the previous reports of C-TiO2 and N-TiO2,5,7 here the anion group (–C4H9O) in tetrabutyl titanate (TBT) serve as carbon doping agent and the addition of concentrated HNO3 is used as inhibitor to control the rate of hydrolysis of TBT and employed as the source of N doping reagent.
2. Experimental
2.1 Preparation
Melamine (C6H6N6), tetrabutyl titanate (C16H36O4Ti), absolute ethanol (C2H6O) and concentrated nitric acid (HNO3) are provided by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). All reagents were used without further purification. Porous g-C3N4 ultrathin nanosheets were prepared according to our reported procedure.22 The specific surface area of g-C3N4 nanosheets was calculated to be 198.5 m2 g−1, with a nearly 9-fold enhancement in that of bulk g-C3N4 (22.3 m2 g−1). C,N-TiO2 NPs/g-C3N4 samples were fabricated by a simple solvothermal route with the presence of concentrated nitric acid. During the preparation process, a desired amount of TBT was dissolved in 30 mL of absolute ethanol containing 1 mL of concentrated HNO3 and 200 mg of porous g-C3N4 NSs. After 6 h stirring, the mixed solution was transferred to a 40 mL Teflon-lined stainless steel autoclave with up to 75% of the total volume. The auto-clave was sealed and kept at 453 K for 24 h. For comparison, the C,N-TiO2 NPs was also synthesized at same condition without addition of porous g-C3N4 NSs.2.2 Characterization
Powder X-ray diffraction (PXRD) patterns were recorded on a Bruker Advanced D8 diffractometer (Germany) at 40 kV and 40 mA with monochromatic high intensity Cu Kα radiation (λ = 0.15418 nm). Sample morphology and microstructure were observed by a transmission electron microscope (TEM) (JEOL JEM-2100) and high resolution transmission electron microscope (HRTEM) at 200 kV accelerating voltage. Fourier transform infrared (FT-IR) spectra were measured by a Nicolet IS10 infrared spectrometer. The optical properties of the samples were measured using UV-vis diffuse reflectance spectroscopy (Shimadzu UV-2500) with BaSO4 as the reflectance standard. The surface composition analysis and electronic binding energy of samples were acquired by a Thermo ESCALAB 250Xi X-ray photoelectron spectrometer under an Al Kα radiation (hν = 1486.6 eV). Solid-state fluorescence measurements (λex = 325 nm) were recorded from 350 to 700 nm on a JASCO FP-6500 type fluorescence spectrophotometer using 1 nm slit width. Raman spectra (RS) were recorded by a Renishaw Invia Raman Microscope using argon ion laser from 100 to 2000 cm−1. The photoelectrochemical properties were measured on a CHI-660E electrochemical workstation (Shanghai Chenhua Instrument Co., Ltd, China) in a standard three-electrode system using the prepared samples with an active area of ca. 0.5 cm × 0.5 cm as the working electrodes, a platinum wire as the counter electrode, and Ag/AgCl as the reference electrode and Na2SO4 (0.1 M) aqueous solution was used as the electrolyte. Electrochemical impedance spectra (EIS) were recorded at the open circuit potential by applying an AC voltage with 5 mV amplitude in the frequency range from 0.01 Hz to 100 kHz.2.3 Photocatalytic tests
The photocatalytic hydrogen evolution from water splitting was detected using the PES-10A evaluation system. Typically, 100 mg of photocatalyst was dispersed in 100 mL of aqueous solution containing 10 vol% triethanolamine (TEOA) acted as sacrificial electron donors. Before the photocatalytic reaction, the system was evacuated and refilled with argon gas several times to remove the air inside the reaction system. This process was conducted until the residual air content checked by an online gas chromatography (GC-1690, Hangzhou JieDao Tech Co., Ltd, China) was negligible. Then, the reactor was irradiated from the side using a 300 W xenon lamp equipped with an optical cut-off filter to remove ultraviolet illumination (λ > 400 nm). The reaction was carried out for 8 h, and the produced hydrogen gas was analyzed by GC-1690 using an online sampling loop (3.5 mL) at intervals of 1 h. The GC 1690 system was equipped with a thermal conductivity detector. Reference experiments were performed without photocatalyst in the presence of light and with photocatalyst in the dark. In both cases, no production of hydrogen was observed.3. Results and discussion
The crystallographic structure and phase purity of C,N-TiO2 NPs/g-C3N4 nanocomposites were determined by X-ray diffraction technique as displayed in Fig. 1a In the case of pure g-C3N4 NSs, two obvious diffraction peaks with 2θ value of 13.2° and 27.8° belong to the (100) and (002) diffraction planes of g-C3N4 NSs. The strong (002) peak represents inter-layer stacking of conjugated aromatic system, whereas weak one of (100) plane arises from the in-plane structural packing motif of tri-s-triazine units, respectively.23,24 The XRD pattern of C,N-TiO2 NPs are in well agreement with anatase structure (JCPDS No. 21-1272). The characteristic diffraction peaks of (101) plane of C,N-TiO2 NPs and all diffraction peaks ascribed to g-C3N4 NSs are presented in C,N-TiO2 NPs/g-C3N4 composites, suggesting the coexistence of C,N-TiO2 NPs and g-C3N4 NSs in C,N-TiO2 NPs/g-C3N4 composites. No impurity phases are detected, indicative of high purity of as-prepared catalysts.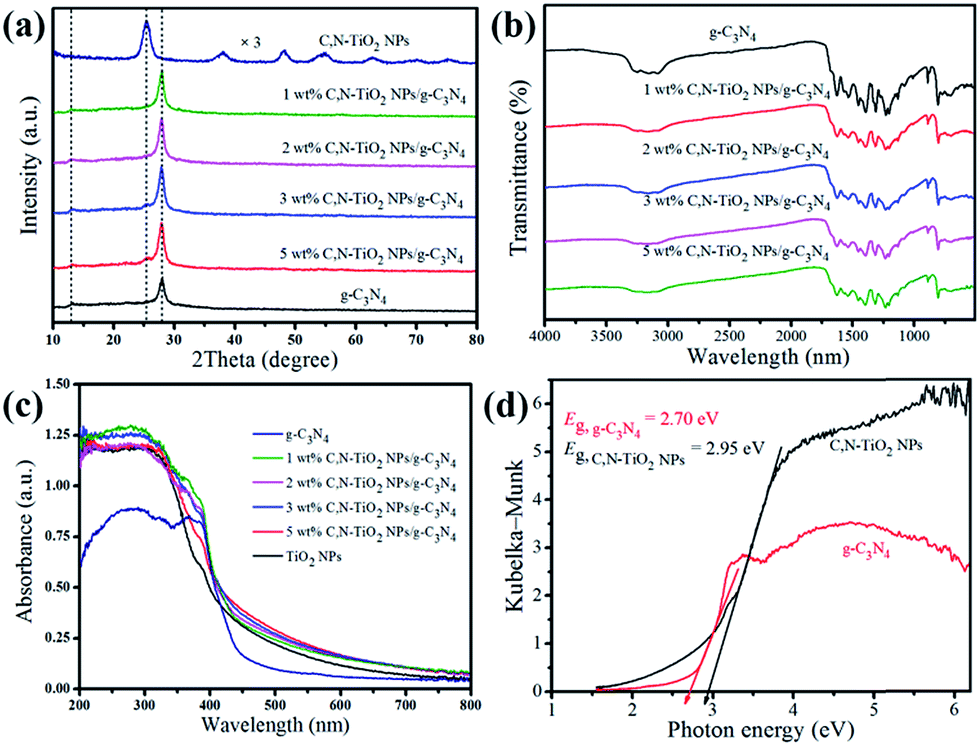 | ||
| Fig. 1 (a) Typical XRD pattern, (b) FT-IR spectra, (c) UV-vis diffuse reflectance spectra and (d) corresponding to Kubelka–Munk transformed function of different photocatalysts. | ||
The FT-IR spectra of pure g-C3N4 NSs and a series of C,N-TiO2 NPs/g-C3N4 composites are displayed in Fig. 1b. For pristine g-C3N4 NSs, a wide absorption band between 3000 cm−1 and 3600 cm−1 corresponds to stretching vibration of O–H of the absorbed water molecule and N–H stretching vibration modes of residual NH2 attached to the sp2 hybridized carbon or NH groups at the defect sites of the aromatic ring.25 While several strong characteristic peaks located between 1200 cm−1 and 1700 cm−1 are explained as typical stretching vibration of CN heterocycles.26 Additionally, a sharp peak at 808 cm−1 is assigned to breathing mode of triazine units.27 The FT-IR spectra of C,N-TiO2 NPs/g-C3N4 composites exhibit the overlap of the spectra of both C,N-TiO2 NPs (Fig. S2†) and g-C3N4 NSs, which suggests the coexistence of C,N-TiO2 NPs and g-C3N4 NSs in composite samples. These results are in well accordance with XRD analysis.
The optical properties of as-prepared C,N-TiO2 NPs, pure g-C3N4 NSs as well as C,N-TiO2 NPs/g-C3N4 composites were detected by UV-vis diffuse reflectance spectra (DRS) and the corresponding band gap energies were estimated by Kubelka–Munk equation. Fig. 1c shows the DRS spectra of the hybrid C,N-TiO2 NPs/g-C3N4 photocatalysts, from which, we can clearly found that g-C3N4 exhibits typical light absorption in the region ranging from 200 nm to 450 nm, which is ascribed to the charge transfer response of g-C3N4 NSs from the valence band formed with N 2p orbits to the conduction band populated by C 2p orbits.28 Interestingly, the C,N-TiO2 NPs is able to absorb visible light with the maximum wavelength up to 460 nm as a result of the doping level generated by C and N atom co-doping. Compared with that of single component, all the C,N-TiO2 NPs/g-C3N4 samples exhibit significantly improved light-harvesting capability. Such red shifts of light absorption might be attributed to the beneficial interaction between C,N-TiO2 NPs and g-C3N4 NSs resulted from the construction of close heterojunction interface. The band gap energies (Fig. 1d) of C,N-TiO2 NPs and pure g-C3N4 NSs calculated by Kubelka–Munk formula are 2.95 eV and 2.70 eV, respectively.
The sample morphology and microstructure information were investigated by TEM and HRTEM, as shown in Fig. 2. As expected, the C,N-TiO2 NPs with the diameter of several nanometers were homogeneously immobilized on the surface of ultrathin g-C3N4 NSs by one-pot solvothermal route, which is favorable to the formation of intimate heterogeneous interface. The size of C,N-TiO2 NPs in C,N-TiO2 NPs/g-C3N4 heterojunction photocatalysts is similar to that of pristine C,N-TiO2 NPs and no agglomeration structures of C,N-TiO2 NPs located on the surface of g-C3N4 NSs have be observed. This good dispersion is benefit to the separation of photo-induced holes and electrons pairs and thus is responsible for the enhancement of photocatalytic performance. From the high-resolution image of C,N-TiO2 NPs/g-C3N4 composites, the sharp-edged lattice images ascribed to C,N-TiO2 NPs are presented with the lattice spacing of 0.352 nm, corresponding to (101) crystallographic planes of anatase TiO2.
 | ||
| Fig. 2 (a) TEM image and (b) HR-TEM image of 3 wt% C,N-TiO2 NPs/g-C3N4 nanocomposites. | ||
To verify the elemental categories and chemical status in C,N-TiO2 NPs/g-C3N4 composites, the X-ray photoelectron spectra (XPS) of C,N-TiO2 NPs, g-C3N4 NSs as well as C,N-TiO2 NPs/g-C3N4 composites were measured. Fig. 3a displays the XPS survey of these three samples. It is obvious that C,N-TiO2 NPs/g-C3N4 composites is composed of C, N, Ti and O elements, which well confirms the coexistence of C,N-TiO2 NPs and g-C3N4 NSs in C,N-TiO2 NPs/g-C3N4 hybrid samples. The typical high resolution XPS spectra of C 1s, N 1s and Ti 2p in different samples are also shown in Fig. 3b–d. The C 1s high-resolution spectrum displays two peaks centering at 284.8, and 288.0 eV for pure g-C3N4 NSs, 284.8 and 287.9 eV for composite sample, which can be attributed to the contaminated carbon and the sp2-bonded carbon (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N) in graphitic carbon nitride, respectively.29 The C 1s peak, originating from the sp2 carbon, shifts to a lower binding energy direction after coupling with C,N-TiO2 NPs, indicating the variation of chemical environment of sp2 carbon via chemical bonds. While the N 1s spectrum of g-C3N4 NSs can be deconvoluted into four different Gaussian–Lorentzian peaks centered at the binding energies of 398.3, 398.9, 400.5 and 404.2 eV. The main peak at 398.3 eV could be ascribed to nitrogen atoms sp2-hybridized to carbon atom (CN–C).30 The characteristic peak at 398.9 eV is related to either N–(C)3 groups linking structural motif (C6N7) or amino groups ((C)2–N–H) connecting with structural defects and incomplete condensation.25,31 The weak peak with the binding energy of 400.5 eV is resulted from N bonded to three carbon atoms to develop the N–(C)3 group in the aromatic cycles.32 However, due to the coupling with C,N-TiO2 NPs, the peaks belonging to N 1s at 398.9 and 400.5 eV separately shift to 399.0 and 400.7 eV, which means the presence of interaction between C,N-TiO2 NPs and g-C3N4 NSs. We attribute this interaction to the generation of C–N–Ti bonds as evidence provided by high-resolution XPS spectra of Ti 2p region. The binding energies of Ti 2p3/2 and Ti 2p1/2 are located at 458.5 and 464.2 eV, which are very close with values of Ti4+ in N-TiO2.33 The obvious shifts of Ti 2p peaks in hybrid sample with respect to that of Ti4+ in C,N-TiO2 NPs further confirm the existence of C–N–Ti bonds at the interface of C,N-TiO2 NPs and g-C3N4 NSs.
C–N) in graphitic carbon nitride, respectively.29 The C 1s peak, originating from the sp2 carbon, shifts to a lower binding energy direction after coupling with C,N-TiO2 NPs, indicating the variation of chemical environment of sp2 carbon via chemical bonds. While the N 1s spectrum of g-C3N4 NSs can be deconvoluted into four different Gaussian–Lorentzian peaks centered at the binding energies of 398.3, 398.9, 400.5 and 404.2 eV. The main peak at 398.3 eV could be ascribed to nitrogen atoms sp2-hybridized to carbon atom (CN–C).30 The characteristic peak at 398.9 eV is related to either N–(C)3 groups linking structural motif (C6N7) or amino groups ((C)2–N–H) connecting with structural defects and incomplete condensation.25,31 The weak peak with the binding energy of 400.5 eV is resulted from N bonded to three carbon atoms to develop the N–(C)3 group in the aromatic cycles.32 However, due to the coupling with C,N-TiO2 NPs, the peaks belonging to N 1s at 398.9 and 400.5 eV separately shift to 399.0 and 400.7 eV, which means the presence of interaction between C,N-TiO2 NPs and g-C3N4 NSs. We attribute this interaction to the generation of C–N–Ti bonds as evidence provided by high-resolution XPS spectra of Ti 2p region. The binding energies of Ti 2p3/2 and Ti 2p1/2 are located at 458.5 and 464.2 eV, which are very close with values of Ti4+ in N-TiO2.33 The obvious shifts of Ti 2p peaks in hybrid sample with respect to that of Ti4+ in C,N-TiO2 NPs further confirm the existence of C–N–Ti bonds at the interface of C,N-TiO2 NPs and g-C3N4 NSs.
 | ||
| Fig. 3 XPS spectra of original C,N-TiO2 NPs, pure g-C3N4 and 3 wt% C,N-TiO2 NPs/g-C3N4: (a) survey spectra, (b) C 1s, (c) N 1s and (d) Ti 2p region. | ||
The photocatalytic activities of those C,N-TiO2 NPs/g-C3N4 composites were investigated by hydrogen production from water splitting with the addition of 10 vol% TEOA as sacrificial reagent under the visible light irradiation. Controllable experiments suggested that no appreciable H2 produced in the absence of either photocatalyst or light irradiation. Fig. 4 displays the H2 generation curves (Fig. 4a) and the average rate of H2 evolution (Fig. 4b) over C,N-TiO2 NPs/g-C3N4 hybrid samples with different concentration of C,N-TiO2 NPs loadings. As shown in Fig. 4a, the total amount of H2 evolution during the 8 h photoredox reaction increases upon enhancing the C,N-TiO2 NPs contents from 0 to 3 wt% and then decreases with further enhancement to 5 wt%. Obviously, the pure C,N-TiO2 NPs and g-C3N4 NSs alone show negligible photocatalytic performance for H2 production with average value of 3.59 μmol g−1 h−1 and 1.84 μmol g−1 h−1, respectively. The extremely low photocatalytic activity of g-C3N4 NSs possibly originates from some vast grain boundary defects formed during the high temperature preparation process, which hugely promote the faster recombination of photo-induced holes and electrons pairs.19 However, after assembly of C,N-TiO2 NPs onto the g-C3N4 NSs, the obtained C,N-TiO2 NPs/g-C3N4 nanocomposites can response broader visible light region and form more photo-induced holes and electrons, thus benefiting the photocatalytic activities for H2 evolution. In this hybrid system, the optimal content of C,N-TiO2 NPs immobilized on the g-C3N4 NSs to achieve the highest photocatalytic water splitting is determined to 3 wt%. The average H2 evolved rate over 3 wt% C,N-TiO2 NPs/g-C3N4 is 39.18 μmol g−1 h−1, which is ca. 10.9 times higher than that of sole C,N-TiO2 NPs and 21.3 times higher than that of pure g-C3N4 NSs.
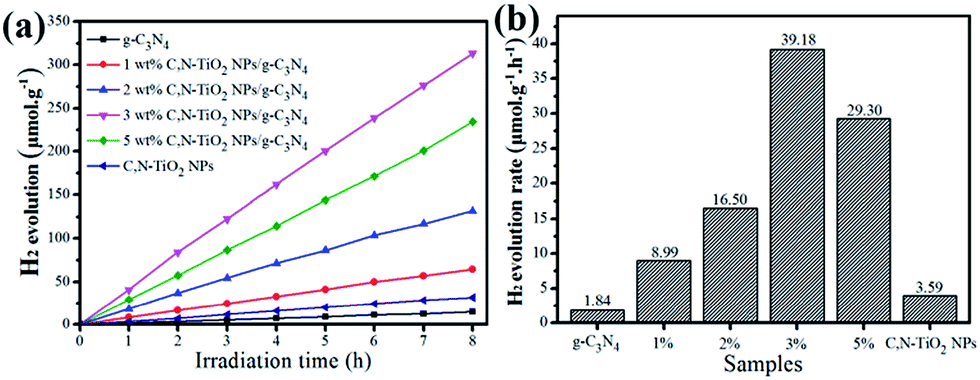 | ||
| Fig. 4 (a) Hydrogen evolution and (b) corresponding to hydrogen production rate from water splitting by different samples under visible light irradiation. | ||
The important factors for 3 wt% C,N-TiO2 NPs/g-C3N4 nanocomposites with the highest photocatalytic activity lie in the synergistic effect of element doping of C and N into TiO2 and the construction of intimate heterojunction structure between C,N-TiO2 NPs and g-C3N4 NSs, namely, the enhancement of light absorption and the lengthen of charge carrier lifetime. The C,N-TiO2 NPs in hybrid system could remarkably strengthen the light absorption to extend into visible light region, from which, the doped TiO2 NPs here was similar with the role cocatalyst played. Moreover, due to the construction of intimate heterojunction interface and well-matched band edge, the C,N-TiO2 NPs also could act as electron acceptors and transfer channels, thus improving the charge separation efficiency. As can be seen in Fig. 5, upon photo-irradiation, C,N-TiO2 NPs and g-C3N4 could easily absorb visible light to generate hole–electron pairs. According to previous reports, the conduction band (CB) and valence band (VB) potential could be determined to be −1.12 and +1.58 eV for g-C3N4, −0.29 and 2.91 eV for pristine TiO2.18,22 Therefore, the photo-generated electrons in conduction band (CB) of the g-C3N4 would directly inject into that of C,N-TiO2 NPs, meanwhile the holes could migrate from C,N-TiO2 NPs to VB of g-C3N4. However, the electrons accumulated in the CB of C,N-TiO2 NPs would be extracted by oxygen vacancy originated from elemental doping.34 Such electron transfer paths effectively suppress their recombination thus benefiting the photocatalytic activity.
 | ||
| Fig. 5 Schematic illustration of charge transfer and separation in C,N-TiO2 NPs/g-C3N4 photocatalysts for hydrogen production under visible light irradiation. | ||
Besides photocatalytic activity, the photo-stability is also an important parameter to estimate the catalysts for practical application. Therefore, recycling experiments of water splitting for H2 generation were carried out at same photocatalytic condition. Fig. 6 shows the H2 evolution curve of 3 wt% C,N-TiO2 NPs/g-C3N4 photocatalyst in four consecutive days cycling photocatalytic runs. The results indicate that the photocatalytic performance of the composite presents no apparent deactivation with the exposure of irradiation for 32 h, indicating the excellent stability of C,N-TiO2 NPs/g-C3N4 photocatalysts.
 | ||
| Fig. 6 Recycling tests of hydrogen evolution over 3 wt% C,N-TiO2 NPs/g-C3N4 nanocomposites under visible light irradiation. | ||
It was well known that many factors, including the crystallinity, morphology, phase structure, light absorption, separation rate of photogenerated hole–electron pairs, play a key role in affecting the photocatalytic activity.35,36 In the current work, the similarity of crystallinity, phase structure and microstructure should not be responsible for the remarkable enhancement of photocatalytic performance for H2 production. The most important point deciding the photocatalytic activity may be the construction of close heterojunction interface, which dramatically faster the separation rate of charge carrier pairs. Conceivably, the importance of the heterogeneous interface in prompt charge migration was also illustrated in several previous reports.37–41 To further prove this hypothesis, the faster separation rate of charge carrier pairs of C,N-TiO2 NPs/g-C3N4 composites were characterized by photocurrent responses, EIS measurements and PL spectra. Fig. 7a exhibits the photocurrent–time (I–t) curves of C,N-TiO2 NPs, pure g-C3N4 as well as C,N-TiO2 NPs/g-C3N4 hybrid material electrodes with several on–off cycles of intermittent irradiation. Obviously, the photocurrent obtained using 3 wt% C,N-TiO2 NPs/g-C3N4 is significantly enhanced as comparison with the unitary component. As we known, the photocurrent is formed mainly by the diffusion of the photogenerated electrons to the back contact and meanwhile the photoinduced holes are taken by the hole acceptor in the electrolyte.42 As a result, the enhanced photocurrent responses over the 3 wt% C,N-TiO2 NPs/g-C3N4 hybrid composites reveals better charge separation and efficient electron transfer within the hybrid structure compared with those of bare C,N-TiO2 NPs and pure g-C3N4. The more efficient transfer of charge carriers could be presented by the result of electrochemical impedance spectroscopy (EIS). The smaller frequency semicircle of the arc in an EIS Nyquist plot indicates the smaller resistance at the interface and the smaller charge-transfer resistance on the electrode surface.43 The results of description in Fig. 7b present the sequence of the frequency semicircles is: g-C3N4 > C,N-TiO2 NPs > 3 wt% C,N-TiO2 NPs/g-C3N4, which suggests the highest separation rate of charge carrier pairs over 3 wt% C,N-TiO2 NPs/g-C3N4 photoanode, lowest over g-C3N4 photoanode. This also confirms that the construction of heterogeneous interface induces a more efficient separation of the photo-excited electron–hole pairs and lengthens the lifespan of the photo-generated charge carriers.
 | ||
| Fig. 7 (a) Transient photocurrent responses and (b) electrochemical impedance spectroscopy (EIS) Nyquist plots of C,N-TiO2 NPs, g-C3N4 and C,N-TiO2 NPs/g-C3N4 nanocomposites in 0.1 M Na2SO4 solution. | ||
Photoluminescence (PL) spectroscopy originated from the recombination of photo-induced holes and electrons, the intensity of which could reflect the recombination rate of those free charge carrier pairs.44,45 Fig. 8 shows the fluorescence emission spectroscopy of pure g-C3N4 sample and 3 wt% C,N-TiO2 NPs/g-C3N4 hybrid sample. It is clear that the fluorescence intensities of 3 wt% C,N-TiO2 NPs/g-C3N4 is smaller than that of g-C3N4, signifying that the hybrid sample has lower recombination rate of the photoinduced electron–hole pairs than pure g-C3N4 sample. These results are in well agreement with the photocurrent test and EIS result, which further confirm it is the heterojunction interface that promotes the enhancement of separation rate of photo-generated hole–electron pairs, thereby significantly improving photocatalytic efficiency.
 | ||
| Fig. 8 Room temperature PL spectra of g-C3N4 sample and 3 wt% C,N-TiO2 NPs/g-C3N4 nanocomposites. | ||
4. Conclusions
In conclusion, we present the design, fabrication and the related characterizations of C,N-TiO2 NPs/g-C3N4 hybrid heterostructures for the photocatalytic H2 evolution from water splitting. Compared to the bare C,N-TiO2 NPs and pure g-C3N4 NSs, the sample of C,N-TiO2 NPs/g-C3N4 composites exhibits much higher catalytic activity, especially 3 wt% C,N-TiO2 NPs/g-C3N4 composite. Its H2 generation rate is 10.9 folds and 21.3 folds than those of C,N-TiO2 NPs and pure g-C3N4 NSs, respectively. The highly-efficient photocatalytic performance of the C,N-TiO2 NPs/g-C3N4 composite is attributed to improved light absorption and the close interfacial contact between the two components inducing the fast interfacial charge transfer from C,N-TiO2 NPs to g-C3N4 nanosheets. Overall, this work provides a simple fabrication strategy yet promising prospect for the utilization of C,N-TiO2 NPs/g-C3N4 as highly-efficient visible-light-driven photocatalyst for H2 production without the presence of any noble metal co-catalyst.Acknowledgements
This investigation was supported by the National Natural Science Foundation of China (Grant no. 51272107, 51372118 and 51572126) and the Fundamental Research Funds for the Central Universities (Grant no. 30920140132038).Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–39 CrossRef CAS PubMed
.
- A. G. Dosado, W. T. Chen, A. Chan, D. Sun-Waterhouse and G. I. N. Waterhouse, J. Catal., 2015, 330, 238–254 CrossRef CAS
- D. Zheng, X. Pang, M. Wang, Y. He, C. Lin and Z. Lin, Chem. Mater., 2015, 27, 5271–5278 CrossRef CAS
- Z. H. N. Al-Azri, W. T. Chen, A. Chan, V. Jovic, T. Ina, H. Idriss and G. I. N. Waterhouse, J. Catal., 2015, 329, 355–367 CrossRef CAS
- L. L. Presti, M. Ceotto, F. Spadavecchia, G. Cappelletti, D. Meroni, R. G. Acres and S. Ardizzone, J. Phys. Chem. C, 2014, 118, 4797–4807 Search PubMed
- Q. Li, J. Bian, L. Zhang, R. Zhang, G. Wang and D. H. L. Ng, ChemPlusChem, 2014, 79, 454–461 CrossRef CAS
- B. Sambandam, A. Surenjan, L. Philip and T. Pradeep, ACS Sustainable Chem. Eng., 2015, 3, 1321–1329 CrossRef CAS
- J. W. J. Hamilton, J. A. Byrne, P. S. M. Dunlop, D. D. Dionysiou, M. Pelaez, K. O'Shea, D. Synnott and S. C. Pillai, J. Phys. Chem. C, 2014, 118, 12206–12215 CAS
- H. Bai, K. S. Y. Kwan, Z. Liu, X. Song, S. S. Lee and D. D. Sun, Appl. Catal., B, 2013, 129, 294–300 CrossRef CAS
- Y. Li, J. Wang, Y. Yang, Y. Zhang, D. He, Q. An and G. Cao, J. Hazard. Mater., 2015, 292, 79–89 CrossRef CAS PubMed
- J. Liu, H. Bai, Y. Wang, Z. Liu, X. Zhang and D. D. Sun, Adv. Funct. Mater., 2010, 20, 4175–4181 CrossRef CAS
- R. Kaplan, B. Erjavec, G. Dražić, J. Grdadolnik and A. Pintar, Appl. Catal., B, 2016, 181, 465–474 CrossRef CAS
- Z. Hua, Z. Dai, X. Bai, Z. Ye, H. Gu and X. Huang, J. Hazard. Mater., 2015, 293, 112–121 CrossRef CAS PubMed
- K. Sasan, F. Zuo, Y. Wang and P. Feng, Nanoscale, 2015, 7, 13369–13372 RSC
- S. W. Shin, J. Y. Lee, K. S. Ahn, S. H. Kang and J. H. Kim, J. Phys. Chem. C, 2015, 119, 13375–13383 CAS
- S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS PubMed
- J. Wang, J. Huang, H. Xie and A. Qu, Int. J. Hydrogen Energy, 2014, 39, 6354–6366 CrossRef CAS
- K. Li, S. Gao, Q. Wang, H. Xu, Z. Wang, B. Huang, Y. Dai and J. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 9023–9030 CAS
- J. Zhou, M. Zhang and Y. Zhu, Phys. Chem. Chem. Phys., 2015, 17, 3647–3652 RSC
- Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and L. Wu, Appl. Catal.,
B, 2015, 163, 135–142 CrossRef CAS
- C. Han, Y. Wang, Y. Lei, B. Wang, N. Wu, Q. Shi and Q. Li, Nano Res., 2015, 8, 1199–1209 CrossRef CAS
- W. Chen, T. Y. Liu, T. Huang, X. H. Liu, J. W. Zhu, G. R. Duan and X. J. Yang, Appl. Surf. Sci., 2015, 355, 379–387 CrossRef CAS
- H. J. Li, B. W. Sun, L. Sui, D. J. Qing and M. Chen, Phys. Chem. Chem. Phys., 2015, 17, 3309–3315 RSC
- W. Chen, T. Y. Liu, T. Huang, X. H. Liu, J. W. Zhu, G. R. Duan and X. J. Yang, J. Mater. Sci., 2015, 50, 8142–8152 CrossRef CAS
- K. Li, Z. Zeng, L. Yan, S. Luo, X. Luo, M. Huo and Y. Guo, Appl. Catal., B, 2015, 165, 428–437 CrossRef CAS
- M. Xiong, L. Chen, Q. Yuan, J. He, S. L. Luo, C. T. Au and S. F. Yin, Carbon, 2015, 86, 217–224 CrossRef CAS
- Y. He, L. Zhang, B. Teng and M. Fan, Environ. Sci. Technol., 2015, 49, 649–656 CrossRef CAS PubMed
- Y. Yao, F. Lu, Y. Zhu, F. Wei, X. Liu, C. Lian and S. Wang, J. Hazard. Mater., 2015, 297, 224–233 CrossRef CAS PubMed
- J. Sun, Y. Fu, G. He, X. Sun and X. Wang, Appl. Catal., B, 2015, 165, 661–667 CrossRef CAS
- W. Chen, G. R. Duan, T. Y. Liu, S. M. Chen and X. H. Liu, Mater. Sci. Semicond. Process., 2015, 35, 45–54 CrossRef CAS
- K. Li, L. Yan, Z. Zeng, S. Luo, X. Luo, X. Liu, H. Guo and Y. Guo, Appl. Catal., B, 2014, 156–157, 141–152 CrossRef CAS
- H. Li, L. Zhou, L. Wang, Y. Liu, J. Lei and J. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 17406–17412 RSC
- X. Li, P. Liu, Y. Mao, M. Xing and J. Zhang, Appl. Catal., B, 2015, 164, 352–359 CrossRef CAS
- Y. Cong, J. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2007, 111, 6976–6982 CAS
- X. Bai, L. Wang and Y. Zhu, ACS Catal., 2012, 2, 2769–2778 CrossRef CAS
- Z. Luo, A. S. Poyraz, C. H. Kuo, R. Miao, Y. Meng, S. Y. Chen, T. Jiang, C. Wenos and S. L. Suib, Chem. Mater., 2015, 27, 6–17 CrossRef CAS
- S. Meng, X. Ning, T. Zhang, S. F. Chen and X. Fu, Phys. Chem. Chem. Phys., 2015, 17, 11577–11585 RSC
- S. Chen, Y. Hu, S. Meng and X. Fu, Appl. Catal., B, 2014, 150–151, 564–573 CrossRef CAS
- J. Zhang, Y. Hu, X. Jiang, S. Chen, S. Meng and X. Fu, J. Hazard. Mater., 2014, 280, 713–722 CrossRef CAS PubMed
- J. Di, J. Xia, S. Yin, H. Xu, L. Xu, Y. Xu, M. He and H. Li, J. Mater. Chem. A, 2014, 2, 5340–5351 CAS
- M. Lu, Z. Pei, S. Weng, W. Feng, Z. Fang, Z. Zheng, M. Huang and P. Liu, Phys. Chem. Chem. Phys., 2014, 16, 21280–21288 RSC
- L. Yuan, M. Q. Yang and Y. J. Xu, J. Mater. Chem. A, 2014, 2, 14401–14412 CAS
- J. Tong, L. Zhang, F. Li, M. Li and S. Cao, Phys. Chem. Chem. Phys., 2015, 17, 23532–23537 RSC
- W. Chen, Z. Chen, T. Liu, Z. Jia and X. Liu, J. Environ. Chem. Eng., 2014, 2, 1889–1897 CrossRef CAS
- S. Pany and K. M. Parida, Phys. Chem. Chem. Phys., 2015, 17, 8070–8077 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18302b |
| This journal is © The Royal Society of Chemistry 2015 |