Azobenzene moiety variation directing self-assembly and photoresponsive behavior of azo-surfactants†
Received
18th February 2014
, Accepted 20th May 2014
First published on 21st May 2014
Abstract
The effect of varying the position of the azobenzene group within two comparable photoresponsive amphiphiles on their capability to form lyotropic liquid crystals (LLCs) was investigated in detail in this study. Two photoresponsive amphiphiles having comparable structures were designed and synthesized consisting of hydrophilic oligooxyethylene units, a hydrophobic alkyl chain and a light-sensitive azobenzene moiety. When the azobenzene group was located in the middle of the hydrophobic alkyl chain, multiple LLC phases were observed at various water contents in the azo-surfactant–water binary system. In contrast, when the azobenzene group was directly attached to the hydrophilic domain, the azo-surfactant–water binary system exhibited only lamellar phases. The temperature dependence of these self-organised nanostructures was also investigated by the combination of small angle X-ray scattering (SAXS), differential scanning calorimetry (DSC), and rheology. Under alternating UV and visible light irradiation, reversible trans–cis photoisomerization of the azobenzene group occurred efficiently in dilute solution for both azo-surfactants. However, only photoisomerization of the surfactant possessing the azobenzene group localized in the middle of the alkyl chain induced significant changes in the self-assembled structure and its bulk properties. This study demonstrates that self-assembly and photoresponsive behaviour of photosensitive amphiphiles is extremely sensitive to the position of the photoactive moiety within the surfactant molecular architecture.
Introduction
Self-assembly of amphiphilic molecules into a variety of well-defined molecular architectures with controlled size, orientation, shape and morphology have been drawing increasing attention in recent years due to their potential for application in the areas of drug delivery, imaging, sensing and catalysis.1–3 Driven by the reduction of surface energy, amphiphiles with different molecular structures can self-organise into various nanostructures according to their different chemical natures. For instance, highly ordered nanotubes with notable thermal and chemical stability have been prepared by self-assembly of amphiphilic peptides at different pHs.4,5 Apart from nanotubes, the self-assembly of amphiphilic molecules has also been used to develop morphologies such as nanofibres6,7 and spheres.8,9 Self-assembled lyotropic liquid crystals (LLCs) are another important class of materials, occupying an intermediate phase between solids and liquids. Unlike the more well-known single component thermotropic liquid crystal systems, LLC phase behaviour depends on both concentration in solution and temperature. LLC assemblies have immediate relevance in biology due to the prevalence of organized lipid structures in living systems.10 Considerable efforts have been devoted to the investigation of amphiphile self-assembly in water to generate LLCs.11–16 As a result, LLCs have been successfully applied to material synthesis,17–19 drug delivery,20,21 and protein crystallisation.22,23
Amphiphilic molecules functionalized with stimuli responsive groups which are able to be manipulated upon demand into desired structures, morphologies, and properties by external environmental stimuli have great potential for numerous practical applications such as drug delivery, catalysis, diagnostics, and biosensors.24 The stimuli may include temperature,25 ultrasound,3 an electric or magnetic field,26 light irradiation,27,28 and chemicals (pH, ionic strength),29 depending on the functionality of the stimuli responsive group employed. In particular, pH-sensitive amphiphiles are of interest in drug delivery due to the different pH conditions existing in normal tissue vs. tumours.30 Kataoka and co-workers reported that pH-responsive micelles formed by block copolymers containing citraconic amide as a pH-sensitive charge masking group selectively released active lysozyme in response to reduced pH.31 Light-sensitive amphiphiles are another attractive alternative since they possess a broad range of tunable parameters, e.g., wavelength, exposure duration, and intensity to manipulate their self-assembly behaviours. Several photosensitive units, such as azobenzene,32,33 stilbene,34,35 dithienylcyclopentene,36 naphthopyran,28,37 and merocyanine,38 show reversible conformational changes through external photo stimuli. Among those photosensitive groups, azobenzene has attracted much attention because polymers containing azobenzene have significant potential for use in holographic optical data-storage devices, surface relief graftings, sensors and artificial muscles.39
To date, the majority of reports concerning the self-assembly of azobenzene based amphiphiles describe simple colloidal aggregates such as micelles and vesicles while self-assembled LLC systems remain rarely reported. For instance, Zhao et al. found that azobenzene-containing amphiphilic diblock copolymers with poly(acrylic acid) as the hydrophilic block self-assembled into micelles in water and exhibited light-sensitive reversible morphological changes through alternating exposure to UV and visible light.32,40 Yu et al. prepared an amphiphilic diblock copolymer composed of a hydrophilic poly(ethylene oxide) block and a hydrophobic polymethacrylate block with photochromic azopyridine side groups. The resulting copolymer vesicles showed photoinduced fusion, damage and defect formation, disruption, disintegration and rearrangement under UV irradiation.41 More recently we reported a new class of photorheological materials with reversible rheological response of unprecedented magnitude based on the photo-induced conformational changes of an LLC forming azo-surfactant in solution.42 While Hatton et al.43,44 established that the change in shape and size of azobenzene based surfactants upon photoisomerization was the driving force behind their photoresponsiveness, the effect of surrounding molecular structure on their properties has not been systematically investigated for the corresponding LLC systems. Moreover, in order to develop and exploit new functional materials based on the photoresponsive behaviour of azobenzene-containing amphiphiles, a deeper understanding of the influence of molecular structure on azobenzene based photoreversible LLC is required.
We now present investigations of the effect of structural variation of azobenzene amphiphiles on their LLC self-assembly and photoresponsive behaviour, with a view to developing a fundamental understanding of relationship between molecular structure and the light-responsive properties of azo-surfactants. Two comparable photoresponsive amphiphiles containing an azobenzene moiety were designed and their ability to form LLC phases was evaluated. The photoresponsive amphiphiles consisted of oligooxyethylene units as the hydrophilic moiety, with a simple alkyl chain as the hydrophobic group. An azobenzene group was inserted as the photoresponsive site, systematically either in the middle of the hydrophobic part or between the hydrophilic and hydrophobic parts to produce two structurally comparable azo-surfactants. The study of the relationship between molecular structure and both morphological and photoresponsive properties of azo-surfactants may aid the design of appropriate photoresponsive amphiphiles for specific applications.
Experimental section
Chemicals and materials
Poly(ethylene glycol) methyl ether with number-average molecular weight of about 350 (Aldrich) was dried in vacuum oven at 80 °C overnight before use. Sodium nitrite, 4-butylaniline, phenol, ethyl 6-bromohexanoate, triethylamine, ethyl p-aminobenzoate, and 1-bromononane were purchased from Sigma-Aldrich and used without further purification. No purification was performed on the solvents except for the tetrahydrofuran (THF), which was dried using an Innovative Technologies, Inc., solvent purification system. Sodium hydrogen carbonate (NaHCO3), potassium hydroxide (KOH), hydrogen chloride (HCl, 37%), potassium carbonate (K2CO3), neutral alumina, and potassium iodide (KI) were from Merck Co. and used as received. Thionyl chloride was purchased from Scharlau and all the organic solvents were from Merck.
Nuclear magnetic resonance (1H and 13C NMR) spectra
1H and 13C NMR spectra were recorded on a Bruker NMR spectrometer at 400 MHz using deuterated chloroform as the solvent. 1H NMR chemical shifts (δ) in parts per millions (ppm) were referenced relative to chloroform (δ = 7.26 ppm) as an internal standard.
Mass spectra (MS)
Positive ion EI mass spectra were obtained on a ThermoQuest MAT95XL mass spectrometer using ionization energy of 70 eV. Perfluorokerosene (PFK) was used as the reference sample.
UV-vis spectroscopy
Photoisomerization of azo-surfactants in ethanol (5 × 10−6 mol L−1) was measured on a Cary 50 Bio UV-vis spectrophotometer (Varian) against a background of ethanol in a quartz cuvette. The isomerization degree at the photostationary state is estimated based on this equation:| |  | (1) |
where A0 is the absorbance at λmax before the light irradiation and Ai is the absorbance at the same wavelength measured at the photostationary state.45
UV-visible light exposures
Trans to cis isomerisation was induced by an EXFO Acticure 4000 light source via a liquid light-guide working at 365 nm wavelength. The UV light intensity was about 200 mW cm−2 for small angle X-ray scattering and photorheology measurements. Cis to trans isomerisation was induced by visible light at 38 mW cm−2. In order to slow down rate of the photoisomerization, a lower intensity UV and visible light was used for UV-vis spectroscopy measurements, 3.8 mW cm−2 and 12.1 mW cm−2, respectively. UV-vis data was collected every 30 s.
Polarized optical microscopy (POM)
Liquid crystal textures were observed with a Nikon Eclipse 80i cross-polarised optical microscope equipped with a Linkam hot stage and controller (LTS 120 with PE94 controller, Linkam UK). Images were captured with a Nikon Ds-Fi1 CCD camera equipped with DS-U2 controller (Nikon Australia Pty. Ltd.; Melbourne, Australia).
Differential scanning calorimetry (DSC)
DSC measurements were performed on a Mettler Toledo DSC 821 system. Neat samples weighing 5–15 mg were sealed in aluminium pans (40 μL) with pierced lids and heated or cooled at a scan rate 10 °C min−1. Thermograms were recorded in a nitrogen atmosphere, using empty aluminium pans as the reference.
Photo-rheometry
In situ monitoring of the phase transitions of binary water–surfactant systems was conducted using an ARES rheometer (TA Instruments, USA) as previously reported.42 The sample was loaded in the centre of two parallel plates of 20 mm in diameter. The gap between the two plates was set at 0.3 mm. The heating/cooling rate was 5 °C min−1. Storage shear modulus (G′), the loss shear modulus (G′′), and viscosity (η*) were measured as a function of temperature at a constant frequency of 10 rad s−1 and a strain of 1.0%.
Small angle X-ray scattering (SAXS)
SAXS experiments were performed at the Australian Synchrotron on the small/wide angle X-ray scattering beamline working at 12.0 KeV with the q-range of 0.2–9 nm−1. The magnitude of the scattering vector (q) is usually defined by this expansion46| | 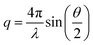 | (2) |
where λ is the wavelength of the X-ray and θ is the scattering angle. Samples were inserted into 1.0 mm borosilicate glass capillaries which were then sealed. The background correction was performed by measuring the scattering of an empty capillary and correcting for sample absorption. Sliver behenate was used to calibrate the sample to detector distance. The relative peak positions for a hexagonal phase are 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :√3:√4:√7 and for a lamellar phase are 1:2:3:4. Data analysis (calibration and integration) of data collected using a 2D detector was achieved using AXcess, a custom-written SAXS analysis program written by Dr Andrew Heron from Imperial College, London.47
:√3:√4:√7 and for a lamellar phase are 1:2:3:4. Data analysis (calibration and integration) of data collected using a 2D detector was achieved using AXcess, a custom-written SAXS analysis program written by Dr Andrew Heron from Imperial College, London.47
Synthesis of azo-surfactants
The chemical structures of the two comparable photoresponsive amphiphiles are shown in Scheme 1. Details about the synthesis and characterization of azo-surfactant A can be found in our previous work.42 Azo-surfactant B was prepared in a similar manner and details are included in the ESI.†
 |
| | Scheme 1 Chemical structures and molecular schematic of azo-surfactants A (a) and B (b). | |
Results and discussion
Concentration dependence of self-assembly
As shown in Scheme 1(a), azo-surfactant A has four main units: (1) oligooxyethylene units as the hydrophilic part; (2) an alkyl chain (C5H10) between hydrophilic part and the azobenzene unit; (3) an azobenzene unit as a photoresponsive component; (4) an alkyl tail group (C4H9) as a hydrophobic part. For azo-surfactant B, the azobenzene unit is directly attached to its hydrophilic oligooxyethylene block and it has a longer alkyl tail group (C9H19) as the hydrophobic part. The overall length of the hydrophobic alkyl chain has been kept constant in both azo-surfactants at C9. The effect of the chemical structure variation of the azo-surfactants on their self-assembly behaviour was first investigated as a function of concentration in water at 25 °C. POM and SAXS measurements were performed to evaluate their ability to form LLC phases. As for the POM images shown in Fig. 1(a1), no brightness was observed for the neat azo-surfactant A, suggesting it is in the isotropic state. After incorporating 20 wt% water, a streaked texture suggesting a lamellar structure was observed for azo-surfactant A as shown in Fig. 1(a2).48 At 50 wt% water, a smoke-like texture consistent with a hexagonal phase was observed as shown in Fig. 1(a3). In contrast, neat azo-surfactant B exhibits strong birefringence under polarised light (Fig. 1(b1)). Similar optical textures were also observed for the azo-surfactant B–water system with 20 wt% (Fig. 1(b2)) and 50 wt% H2O (Fig. 1(b3)), respectively. The observed anisotropic phases from azo-surfactant–water binary system were further investigated by SAXS in detail.
 |
| | Fig. 1 POM images for the azo-surfactant–water binary system at room temperature: (a1) neat azo-surfactant A; (a2) azo-surfactant A with 20 wt% H2O; (a3) azo-surfactant A with 50 wt% H2O; (b1) neat azo-surfactant B; (b2) azo-surfactant B with 20 wt% H2O; (b3) azo-surfactant B with 50 wt% H2O. (Magnification × 100). | |
As shown in Fig. 2(a), for water concentrations 0–10 wt% azo-surfactant A, no peaks were observed in the SAXS spectrum with q-range of 0.5–4 nm−1, indicating an absence of a long-range ordered structure. With increasing water content to 20 and 30 wt%, the SAXS spectrum with three peaks at q-spacing ratios of 1, 2 and 3 were observed, indicating the presence of a lamellar phase. In addition, it was found that the d-spacing of lamellar phase increased from 6.44 nm to 7.04 nm when the water content was increased from 20 to 30 wt%. Larger d-spacing may be attributed to swelling of the water containing interlayer of the lamellar phase. The formation of hexagonal phases at water contents of 40 to 60 wt% was confirmed by the observation of peaks in the SAXS spectrum at the q-spacing ratios of 1, √3, √4 and √7. The d-spacing for the hexagonal phase increased from 7.35 to 7.95 nm, and further to 8.72 nm for water contents of 40, 50 and 60 wt% respectively. The binary system evolved to isotropic phase when the water content was increased above 65 wt% (data not shown). The variety of LLCs observed in the azo-surfactant A–water binary system is similar to the results from commercial surfactants like Brij 97.49
 |
| | Fig. 2 SAXS patterns for azo-surfactant and water binary systems at various concentrations of water at 25 °C: (a) azo-surfactant A–water; (b) azo-surfactant B–water. Water contents are indicated and intensities offset for clarity. | |
Compared with the azo-surfactant A–water binary system, azo-surfactant B exhibits completely different self-assembly behavior in water at 25 °C. As shown in Fig. 2(b), for the neat azo-surfactant B, three scattering peaks with the ratios of 1:2:3 in the q-range of 0.5–4 nm−1 were observed, demonstrating the presence of a long-range ordered lamellar structure.50 This is probably because the hydrophobic domains phase separate from the hydrophilic oligooxyethylene units and this nanophase separation process promotes the bilayer packing of the molecules. Similar results have been reported for liquid-crystalline polymers.50,51 The d-spacing calculated using ‘AXcess’ for the neat azo-surfactant B is about 4.87 nm. By incorporating water into azo-surfactant B, the SAXS patterns in Fig. 2(b) show that the azo-surfactant B–water binary system retained lamellar structure upon water dilution, with SAXS peaks shifting toward a lower q range as water content increased. Quantitatively analysis of these SAXS patterns reveals that the d-spacing gradually increased from 5.03 to 7.38 nm when the water content in the binary system was increased from 10 to 60 wt%. The increase of d-spacing is likely due to the swelling of the hydrophilic layer formed by oligooxyethylene units with increasing water content. Comparing azo-surfactants A and B, these results clearly indicate that the different position of the azobenzene group brought about significantly different self-assembly behaviors.
Temperature dependence of self-assembly
Although LLCs are characterized by the fact that concentration is the determining factor in their phase transitions, temperature also plays an important role. Temperature dependence of the self-assembly behavior of azo-surfactants A and B in water was also investigated. Fig. 3(a) presents the SAXS spectra for azo-surfactant A–water binary system with 50 wt% H2O at the temperature ranging from 25 to 60 °C. From the SAXS spectrum at 25 and 30 °C, it can be clearly seen that there exists hexagonal phase in this system due to the presence of SAXS peaks situated at the positions 1:√3:√4:√7. Comparable lattice spacing for the hexagonal phases, 7.92 nm at 25 °C and 8.01 nm at 30 °C, were obtained. With further increasing temperature to 35 °C, SAXS peaks assignable to hexagonal phase faded away and characteristic peaks with relative positions 1:2:3 for a lamellar phase emerged when the temperature reached 40 °C. Similar results concerning the temperature induced phase transition from hexagonal to lamellar have been reported for poly(styrene-b-isoprene) (SI) diblock copolymers in selective solvents.52 This order–order transition (OOT) is believed to be related to the fact that the selectivity of water for the hydrophilic oxyethylene units and the hydrophobic domains decreases as the temperature increases. The phase transition from hexagonal to lamellar was also observed for the azo-surfactant A–water binary system with 45 wt% H2O (ESI Fig. S3†). Meanwhile, this OOT from hexagonal to lamellar was also verified by the rheology measurements. Fig. 4 shows the temperature dependence of viscosity for the azo-surfactant A–water binary system with 50 wt% H2O. At initial 25 °C, it exhibits very high viscosity around 2000 Pa s, which can be attributed to the presence of a highly ordered hexagonal phase. A sharp decrease of viscosity was observed as the temperature increased from 25 to 33 °C. This may be due to the melting of hexagonal phase and the initiation of highly ordered packing of the azo-surfactant A molecules in hexagonal phase was destroyed, resulting in the decrease of viscosity. After that, the viscosity levelled off at around 10 Pa s in the temperature range of 33–50 °C. These steady state viscosities were associated with the lamellar phases according to the SAXS patterns in Fig. 3(a). Compared to hexagonal phase, the units in lamellar phase were organized in sheets and that may slide over each other. The relatively increased mobility of the chains in the lamellar phase sheet gives rise to lower viscosity.53 With further increase of temperature to 60 °C, the azo-surfactant A–water binary system eventually transformed to the isotropic phase with extremely low viscosity. It is worth noting that these phase transitions were reversible. As shown in Fig. 4, the viscosity nearly returns to the initial values for each state as the temperature decreases, which indicates its phase transition as the function of temperature was reversible. The partial phase diagram derived from the SAXS measurements for azo-surfactant A with a concentration accuracy of 5 wt% water content and a temperature accuracy of 5 °C is plotted in Fig. 5. It can be clearly observed that the azo-surfactant A and water binary systems exhibit rich liquid crystal phases and an interesting order–order transition (OOT) from hexagonal to lamellar can be induced by increasing temperature at high water contents (35–50 wt%).
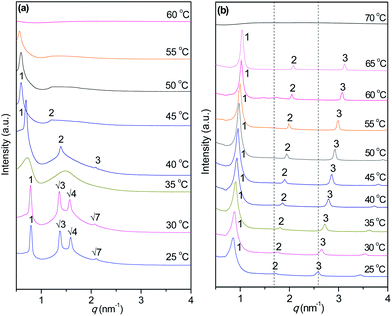 |
| | Fig. 3 SAXS patterns for azo-surfactant and water binary systems at various temperatures with 50 wt% water: (a) azo-surfactant A–water; (b) azo-surfactant B–water. Intensity offset for clarity. | |
 |
| | Fig. 4 Plot of the viscosity of azo-surfactant A–water system with 50 wt% water as a function of temperature. | |
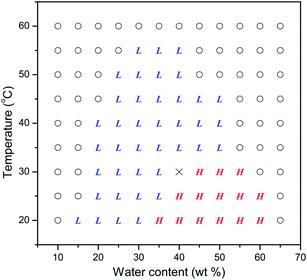 |
| | Fig. 5 Data points for the partial phase diagram derived from SAXS patterns of the azo-surfactant A–water binary system. L: lamellar phase; H: hexagonal phase; O: isotropic phase; ×: lamellar and hexagonal. | |
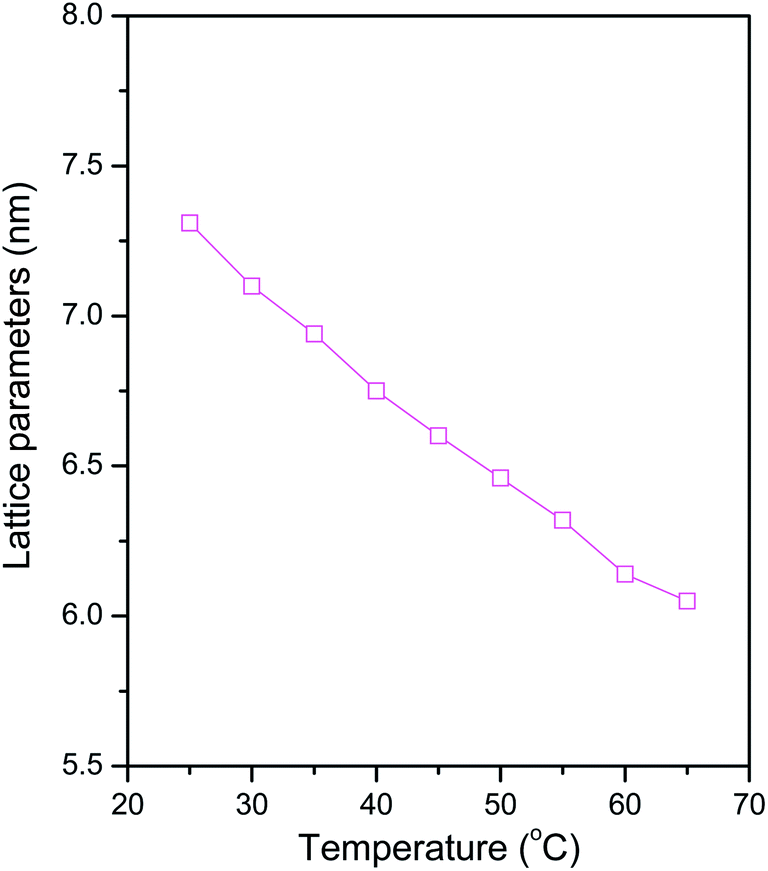
The corresponding temperature dependence of phase behavior for the azo-surfactant B–water binary system with 50 wt% H2O is shown in Fig. 3(b). The SAXS patterns with relative peak positions 1:2:3 were observed for this binary system at 25 °C, indicating the presence of a lamellar phase. Moreover, the lamellar phase remains unchanged with increasing the temperature up to 65 °C. Nevertheless, it was clearly observed that these SAXS peaks shift towards higher q as the temperature increases. The d-spacing plotted as a function of temperature is shown in Fig. 6. It shows that d-spacing decreased gradually from 7.31 nm to 6.05 nm with increasing temperature from 25 to 65 °C. Compared with azo-surfactant A, as shown in Scheme 1, azo-surfactant B has a longer hydrophobic alkyl chain (C9H19) adjacent to the rigid azobenzene group. As the temperature increases, the flexibility of the long hydrophobic alkyl chain of azo-surfactant B is increased, which results in the shrinking of the structure. This decreasing tendency of lattice parameter as a function of temperature is commonly observed in LLCs systems with long hydrocarbon amphiphile molecules and lipids.49,54–56 With further increasing temperature to 70 °C, no long range ordered structures could be observed, as evidenced by the absence of the periodic peaks in the SAXS spectrum. This was presumably due to the melting of the lamellar phases, which was further confirmed by the DSC results discussed in the following text.
 |
| | Fig. 6 Variation of d-spacing for the azo-surfactant B–water system with 50 wt% H2O as a function of temperature. | |
The melting points of these lamellar phases were determined by DSC measurements. Fig. 7 shows the DSC curves from 25 to 90 °C of neat azo-surfactant B and azo-surfactant B–water binary system with different water contents. For the neat azo-surfactant B, one endothermic peak at 58 °C was recorded due to the melting of the surfactant crystals. Upon incorporation of 10 wt% water into azo-surfactant B, the melting point increased to 63 °C. The melting points further increased to 66 °C and 68 °C for the binary system with 20 and 30 wt% H2O respectively. After that, an identical melting point 69 °C was obtained for the binary system with 40, 50 and 60 wt% H2O respectively. The increase of the melting point is most likely caused by the enhanced segregation of hydrophilic and hydrophobic domains after adding water into the azo-surfactant B. The highly separated bilayer structure results in the improved thermal stability. The DSC measurements for the azo-surfactant B–water binary system with 50 wt% H2O were in good agreement with those of SAXS spectra shown in Fig. 3(b).
 |
| | Fig. 7 DSC curves for the azo-surfactant B–water binary system with different water content. | |
Photoresponse of azo-surfactants
As both azo-surfactants A and B have a potentially photosensitive azobenzene unit within them, the photoresponsive behaviour of azo-surfactant in dilute solution as well as in LLCs was investigated. The UV-vis spectra for both azo-surfactants are shown in Fig. 8. As shown in Fig. 8(a), before UV light irradiation, the trans-form of azo-surfactant A exhibits a maximum absorption band at around 350 nm. This absorbance peak decreased after UV-light irradiation due to trans–cis isomerization and a photoequilibrium was established within 10 min (Fig. 8(a), spectrum 2). Overlapping spectra recorded at different time points confirmed that the photostationary state was established. A more detailed kinetic study of azo-surfactant A can be found in our previous publication.42 The trans–cis transition was confirmed by the appearance of a weak absorption band around 440 nm based on n–π* transition of cis-isomer.32,57 Isomerization degrees calculated based on eqn (1) for azo-surfactant A and B were 89% and 86% respectively. After UV irradiation, the sample was irradiated with visible light for 10 min and the UV-vis spectrum was recorded. The maximum absorption band at round 350 nm fully recovered and the spectrum essentially overlapped the original UV-vis spectrum, indicating a completely reversible trans–cis isomerization was achieved under alternating UV and visible light irradiation. Meanwhile, Fig. 8(b) shows that a similar reversible behaviour was observed for azo-surfactant B. A comparison of the kinetic plots of UV spectra of azo-surfactants A and B under UV and visible light exposure indicated that they had very similar rates of photoisomerisations (data not shown). Based upon the efficient reversible photoisomerization of these azo-surfactants in dilute solution, a similar approach was used to assess material properties of their self-assembled LLCs aggregations as outline in the following section.
 |
| | Fig. 8 Influence of photo-irradiation upon absorption spectra of azo-surfactant A (a) and azo-surfactant B (b): (1) before UV irradiation; (2) after UV irradiation (10 min); (3) after UV irradiation followed by visible light exposure (10 min). | |
To determine the effect of photoisomerization of azobenzene group within azo-surfactants on their LLC phase behaviour, SAXS studies were carried out for the above two azo-surfactants in binary water systems before UV irradiation, after UV irradiation, and followed by visible light exposure respectively. The obtained SAXS spectra are given in Fig. 9. As shown in Fig. 9(a), the azo-surfactant A–water system with 50 wt% H2O before UV light irradiation shows an ordered hexagonal phase. After UV light irradiation, these periodic SAXS peaks for a hexagonal phase faded away and a SAXS pattern with two broad peaks was recorded. This may be correlated to an isotropic state because no brightness was observed when the sample was visualised between two polarized films under UV irradiation (data not shown). According to the previous UV-vis spectroscopy measurements, this order-disorder transition (ODT) is most likely attributed to the local isomerization of azobenzene units in azo-surfactant A. Under UV irradiation, the rodlike trans conformation azobenzene units in azo-surfactant A transformed to their cis ‘bent’ conformation. The disruption of hexagonal phase may be due either to molecular-geometrical changes or to the difference in hydrophobicity of the two isomers of azo-surfactant A. Azobenzene based surfactants in the cis form usually have greater hydrophilicity.58 After switching off UV light and turning on visible light, the well-defined periodic SAXS peaks for a hexagonal phase returned. Referring to the UV-vis spectroscopic results in Fig. 8(a), under visible light exposure, the bent azobenzene units in cis form relaxed back to its rod-like trans form and hence azo-surfactant A assembled back to hexagonal phase. The detailed photoresponsive behaviour of azo-surfactant A has been reported in our previous work.42
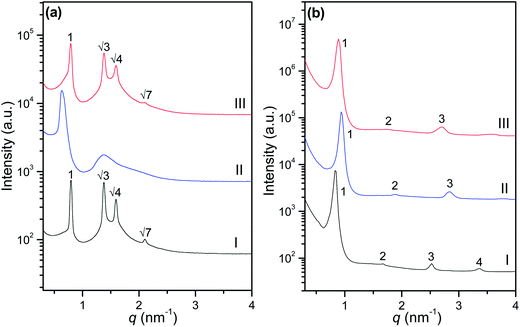 |
| | Fig. 9 SAXS patterns for the azo-surfactant–water systems with 50 wt% H2O before UV irradiation (I), after UV irradiation (5 min) (II), and followed by visible exposure (5 min) (III) at 25 °C: (a) azo-surfactant A–water; (b) azo-surfactant B–water. | |
A corresponding photoresponsive study for the azo-surfactant B and water binary system with 50 wt% H2O was also conducted and the SAXS profiles are shown in Fig. 9(b). Before UV irradiation, it shows periodic SAXS peaks with relative positions 1:2:3 for lamellar phase. Surprisingly, the SAXS pattern still exhibits characteristic peaks for a lamellar phase after UV irradiation, which is in contrast to order-disorder transition for azo-surfactant A and water binary system. It should be noted that these periodic peaks shifted towards higher q and the resulting d-spacing decreased from 7.31 to 6.66 nm after UV irradiation. This presumably results from the trans–cis isomerization of azobenzene units in azo-surfactant B. The effective chain length of azo-surfactant B in the bent cis form is decreased under UV irradiation, resulting in the decrease of d-spacing. Although azo-surfactant B showed similar UV-vis spectra as azo-surfactant A in dilute solution during tran–cis isomerization (Fig. 8), the isomerization of azobenzene group did not disrupt the bilayer packing of azo-surfactant B molecules in water. This may be due to that the azobenzene group of surfactant B was directly attached to a flexible PEG chain, the changes in azobenzene conformation under UV irradiation were dissipated when it was attached to the flexible PEG chain. After switching off UV light and turning on visible light, the bent cis form azo-surfactant B molecules relaxed back to rod-like trans form and therefore, it was observed that the periodic SAXS peaks for lamellar phase shifted back to low q range. It should be noted, however, that the d-spacing (7.01 nm) did not fully recover after a long (more than 10 min) visible light exposure. Unlike the mixture of azo-surfactant A–water (50 wt%) which exhibited a large rapid reversible change in modulus upon exposure to UV and visible light,42 no significant rheological change was observed for the azo-surfactant B with 50 wt% water under alternating UV and visible light irradiation due to their highly stable ordered lamellar phase. A schematic of the different phase behaviours and stimulus-response properties of azo-surfactants A and B at 50 wt% H2O content are schematically shown in Fig. 10.
 |
| | Fig. 10 Schematic illustration of different phase behaviours and stimulus-response properties of azo-surfactants A and B at 50 wt% H2O content respectively. | |
Conclusions
In summary, two structurally similar and directly comparable photoresponsive azo-surfactants were successfully synthesized and their capability to form LLCs and the photoresponsive behaviour of these LLCs were investigated. The results show that the self-organised structures of azo-surfactants A and B in water were significantly different depending on the position of azobenzene unit. Neat azo-surfactant A with azobenzene group localized in the centre of its hydrophobic alkyl chain did not show liquid crystal structures at room temperature. The presence of water, however, increases the segregation between the hydrophilic and hydrophobic blocks of azo-surfactant A and induced ordering in the amphiphilic system. This was evident in the formation of lamellar LLCs in the azo-surfactant A–water system with 20 wt% water at room temperature, which evolved to hexagonal phase when the water content reached to 40 wt%. In contrast, the neat azo-surfactant B with azobenzene unit directly attached to the hydrophilic block exhibited lamellar phase which remains the bilayer structures upon water dilution. Quantitative analysis of SAXS patterns reveals that the lamellar phase for azo-surfactant B–water system tended to swell with increasing water content. In addition, an order-to-order transition from hexagonal to lamellar was observed for the azo-surfactant A–water binary system with 50 wt% water when the temperature was increased from 25 to 80 °C. However, the phase behaviour of the azo-surfactant B–water binary systems only showed slight changes with temperature, i.e., the slight decrease in lattice spacing. Under alternating UV and visible light irradiation, reversible trans–cis photoisomerization of azobenzene group was observed in the dilute ethanol solution for both azo-surfactants. As a result, a dynamically switchable phase transition between lyotropic hexagonal phase and isotropic phase was observed for the azo-surfactant A–water binary system with 50 wt% water. Localized alkyl chain rigidity may amplify the effect of the azobenzene conformational changes in azo-surfactant A, resulting in much more significant changes to the self-assembled structure. In contrast, the azobenzene group of surfactant B is directly attached to a flexible PEG chain, the changes in azobenzene conformation are dissipated in the flexible PEG chain. This paper sheds new light on the critical importance of the position of the azo-benzene group within azo-surfactants in order to maximise photoresponsive LLC behaviour which should aid the future design of improved stimuli responsive self-assembly systems.
Acknowledgements
SAXS/WAXS research was undertaken at the Australian Synchrotron, Victoria, Australia and the authors thank Dr Nigel Kirby for his assistance. S. P. gratefully acknowledges the Deakin University and CSIRO for provision of a collaborative PhD scholarship and thanks Dr Deng Hong (Zhejiang University, China & CSIRO, Australia) for assistance with chemical synthesis and purification.
References
- Z. Ge and S. Liu, Macromol. Rapid Commun., 2009, 30, 1523–1532 CrossRef CAS PubMed.
- N. Mizoshita and T. Seki, Soft Matter, 2006, 2, 157–165 RSC.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- H. Xu, Y. M. Wang, X. Ge, S. Y. Han, S. J. Wang, P. Zhou, H. H. Shan, X. B. Zhao and J. A. R. Lu, Chem. Mater., 2010, 22, 5165–5173 CrossRef CAS.
- J. D. Hartgerink, T. D. Clark and M. R. Ghadiri, Chem. – Eur. J., 1998, 4, 1367–1372 CrossRef CAS.
- J. W. Sadownik, J. Leckie and R. V. Ulijn, Chem. Commun., 2011, 47, 728–730 RSC.
- K. J. C. van Bommel, A. Friggeri and S. Shinkai, Angew. Chem., Int. Ed., 2003, 42, 980–999 CrossRef CAS PubMed.
- S. Q. Zhou, C. Burger, B. Chu, M. Sawamura, N. Nagahama, M. Toganoh, U. E. Hackler, H. Isobe and E. Nakamura, Science, 2001, 291, 1944–1947 CrossRef CAS PubMed.
- M. Han and M. Hara, J. Am. Chem. Soc., 2005, 127, 10951–10955 CrossRef CAS PubMed.
- D. L. Gin, W. Q. Gu, B. A. Pindzola and W. J. Zhou, Acc. Chem. Res., 2001, 34, 973–980 CrossRef CAS PubMed.
- C. H. Li, J. H. He, J. H. Liu, Z. Q. Yu, Q. L. Zhang, C. X. He and W. L. Hong, J. Colloid Interface Sci., 2010, 342, 354–360 CrossRef CAS PubMed.
- A. Firouzi, F. Atef, A. G. Oertli, G. D. Stucky and B. F. Chmelka, J. Am. Chem. Soc., 1997, 119, 3596–3610 CrossRef CAS.
- M. U. Araos and G. G. Warr, J. Phys. Chem. B, 2005, 109, 14275–14277 CrossRef CAS PubMed.
- P. Alexandridis, U. Olsson and B. Lindman, Langmuir, 1998, 14, 2627–2638 CrossRef CAS.
- P. Alexandridis, D. L. Zhou and A. Khan, Langmuir, 1996, 12, 2690–2700 CrossRef CAS.
- E. Z. Radlinska, T. Gulik-Krzywicki, D. Langevin and F. Lafuma, Langmuir, 1998, 14, 5070–5076 CrossRef CAS.
- Z. F. Chen, T. L. Greaves, R. A. Caruso and C. J. Drummond, J. Mater. Chem., 2012, 22, 10069–10076 RSC.
- G. S. Attard, J. C. Glyde and C. G. Goltner, Nature, 1995, 378, 366–368 CrossRef CAS.
- C. G. Goltner and M. Antonietti, Adv. Mater., 1997, 9, 431–436 CrossRef CAS PubMed.
- C. J. Drummond and C. Fong, Curr. Opin. Colloid Interface Sci., 1999, 4, 449–456 CrossRef CAS.
- C. Y. Guo, J. Wang, F. L. Cao, R. J. Lee and G. X. Zhai, Drug Discovery Today, 2010, 15, 1032–1040 CrossRef CAS PubMed.
- M. Caffrey, Curr. Opin. Struct. Biol., 2000, 10, 486–497 CrossRef CAS.
- M. Caffrey, Biochem. Soc. Trans., 2011, 39, 725–732 CAS.
- X. Q. Xue, J. Zhu, Z. B. Zhang, N. C. Zhou, Y. F. Tu and X. L. Zhu, Macromolecules, 2010, 43, 2704–2712 CrossRef CAS.
- Y. Yin, L. Wang, H. Jin, C. Lv, S. Yu, X. Huang, Q. Luo, J. Xu and J. Liu, Soft Matter, 2011, 7, 2521–2529 RSC.
- T. Hao, Adv. Mater., 2001, 13, 1847–1857 CrossRef CAS.
- E. Verploegen, J. Soulages, M. Kozberg, T. Zhang, G. McKinley and P. Hammond, Angew. Chem., Int. Ed., 2009, 48, 3494–3498 CrossRef CAS PubMed.
- T. Kosa, L. Sukhomlinova, L. Su, B. Taheri, T. J. White and T. J. Bunning, Nature, 2012, 485, 347–349 CrossRef CAS PubMed.
- Z. Ge, J. Xu, J. Hu, Y. Zhang and S. Liu, Soft Matter, 2009, 5, 3932–3939 RSC.
-
Superamolecular Soft Matter: Applications in Materials and Organic Electronics, ed. T. Nakanishi, John Wiley & Sons, Inc, 2011 Search PubMed.
- Y. Lee, S. Fukushima, Y. Bae, S. Hiki, T. Ishii and K. Kataoka, J. Am. Chem. Soc., 2007, 129, 5362–5363 CrossRef CAS PubMed.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911–8917 CrossRef CAS.
- Y. Y. Wang, S. L. Lin, M. H. Zang, Y. H. Xing, X. H. He, J. P. Lin and T. Chen, Soft Matter, 2012, 8, 3131–3138 RSC.
- S. Miljanic, L. Frkanec, Z. Meic and M. Zinic, Langmuir, 2005, 21, 2754–2760 CrossRef CAS PubMed.
- J. Eastoe, M. Sanchez-Dominguez, P. Wyatt and R. K. Heenan, Chem. Commun., 2004, 2608–2609 RSC.
- J. J. D. de Jong, P. R. Hania, A. Pugžlys, L. N. Lucas, M. de Loos, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. van Esch, Angew. Chem., Int. Ed., 2005, 44, 2373–2376 CrossRef CAS PubMed.
- S. A. Ahmed, X. Sallenave, F. Fages, G. Mieden-Gundert, W. M. Muller, U. Muller, F. Vogtle and J. L. Pozzo, Langmuir, 2002, 18, 7096–7101 CrossRef CAS.
- H.-Y. Lee, K. K. Diehn, K. Sun, T. Chen and S. R. Raghavan, J. Am. Chem. Soc., 2011, 133, 8461–8463 CrossRef CAS PubMed.
- J. M. Schumers, C. A. Fustin and J. F. Gohy, Macromol. Rapid Commun., 2010, 31, 1588–1607 CrossRef CAS PubMed.
- Y. Zhao, Macromolecules, 2012, 45, 3647–3657 CrossRef CAS.
- L. Lin, Z. Yan, J. S. Gu, Y. Y. Zhang, Z. Feng and Y. L. Yu, Macromol. Rapid Commun., 2009, 30, 1089–1093 CrossRef CAS PubMed.
- S. H. Peng, Q. P. Guo, T. C. Hughes and P. G. Hartley, Langmuir, 2014, 30, 866–872 CrossRef CAS PubMed.
- T. G. Shang, K. A. Smith and T. A. Hatton, Langmuir, 2003, 19, 10764–10773 CrossRef CAS.
- C. T. Lee, K. A. Smith and T. A. Hatton, Macromolecules, 2004, 37, 5397–5405 CrossRef CAS.
- Y. H. Deng, Y. B. Li and X. G. Wang, Macromolecules, 2006, 39, 6590–6598 CrossRef CAS.
-
P. Linder and T. Zemb, Neutrons, X-Rays and Light Scattering Methods Applied to Soft Condensed Matter, Elsevier, 2002 Search PubMed.
- J. M. Seddon, A. M. Squires, C. E. Conn, O. Ces, A. J. Heron, X. Mulet, G. C. Shearman and R. H. Templer, Philos. Trans. R. Soc., A, 2006, 364, 2635–2655 CrossRef CAS PubMed.
- B. Soberats, E. Uchida, M. Yoshio, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2014, 136, 9552–9555 CrossRef CAS PubMed.
- Z. F. Chen, T. L. Greaves, C. Fong, R. A. Caruso and C. J. Drummond, Phys. Chem. Chem. Phys., 2012, 14, 3825–3836 RSC.
- X. Li, L. Fang, L. Hou, L. Zhu, Y. Zhang, B. Zhang and H. Zhang, Soft Matter, 2012, 8, 5532–5542 RSC.
- K. Okano, Y. Mikami and T. Yamashita, Adv. Funct. Mater., 2009, 19, 3804–3808 CrossRef CAS PubMed.
- T. P. Lodge, B. Pudil and K. J. Hanley, Macromolecules, 2002, 35, 4707–4717 CrossRef CAS.
- G. T. Dimitrova, T. F. Tadros and P. F. Luckham, Langmuir, 1995, 11, 1101–1111 CrossRef CAS.
-
Handbook of Applied Surface and Colloid Chemistry, ed. K. Holmberg, John Wiley & Sons, Ltd, 2001 Search PubMed.
- S. M. Sagnella, C. E. Conn, I. Krodkiewska and C. J. Drummond, Soft Matter, 2009, 5, 4823–4834 RSC.
- R. Mezzenga, C. Meyer, C. Servais, A. I. Romoscanu, L. Sagalowicz and R. C. Hayward, Langmuir, 2005, 21, 3322–3333 CrossRef CAS PubMed.
- T. Hayashita, T. Kurosawa, T. Miyata, K. Tanaka and M. Igawa, Colloid Polym. Sci., 1994, 272, 1611–1619 CAS.
- J. Eastoe and A. Vesperinas, Soft Matter, 2005, 1, 338–347 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tc00321g |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a