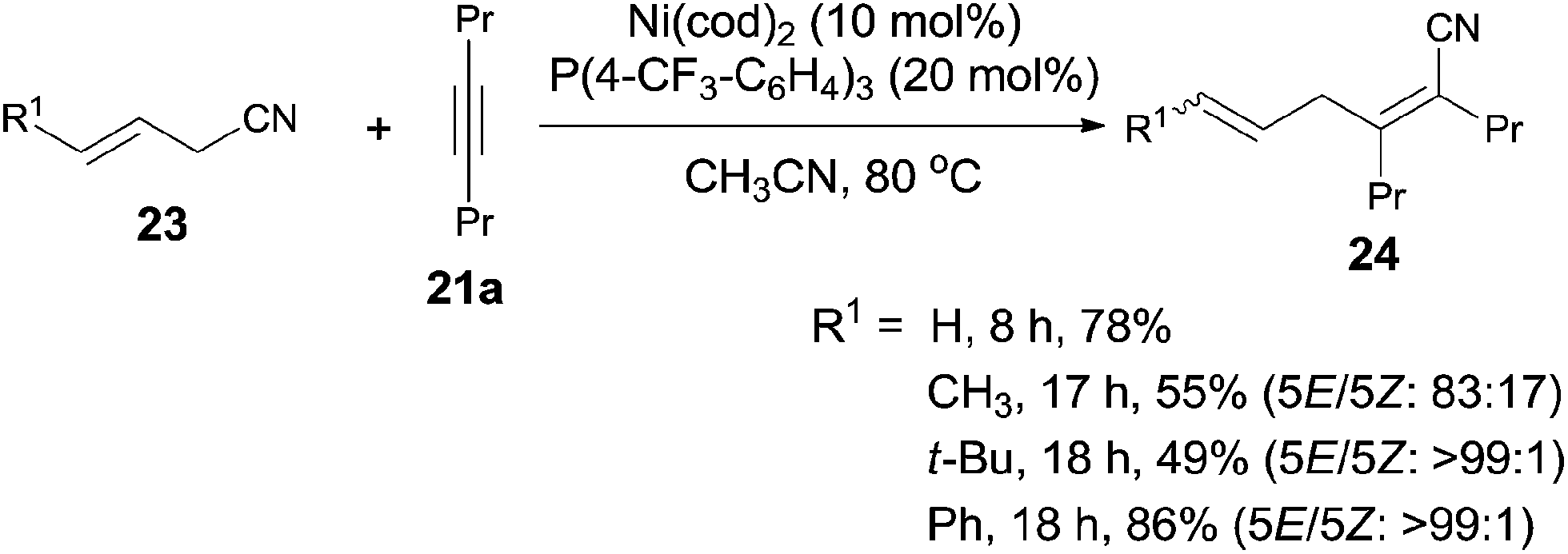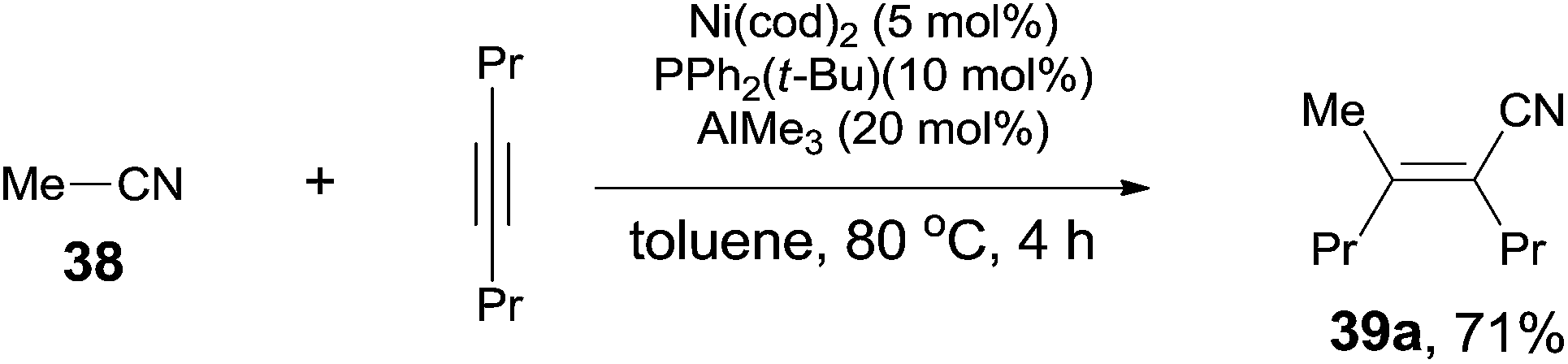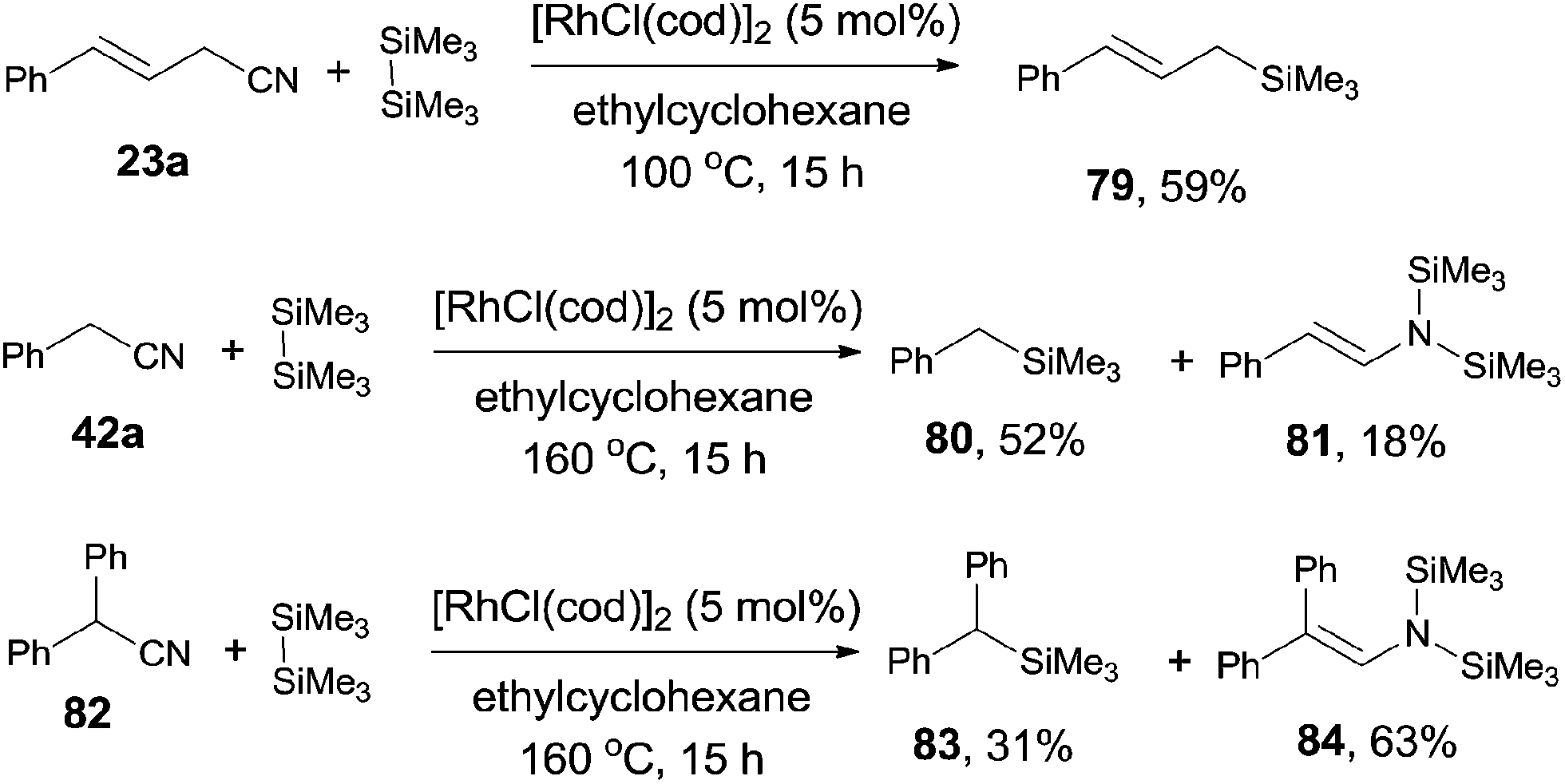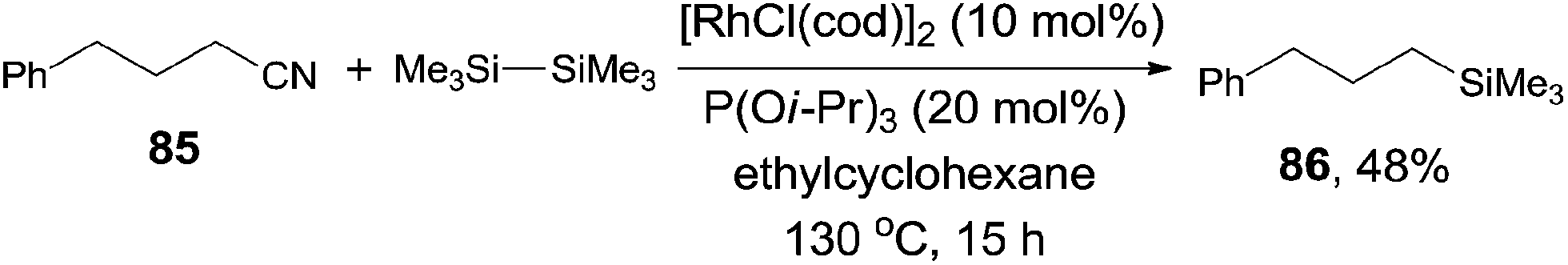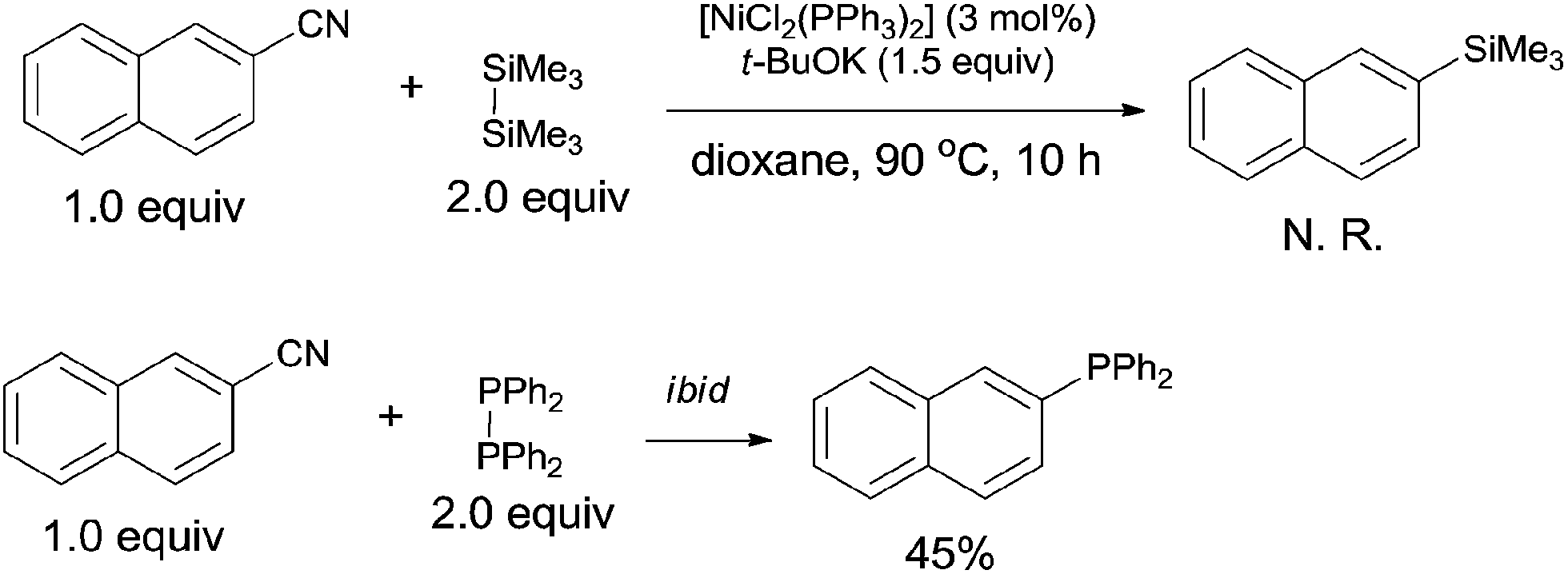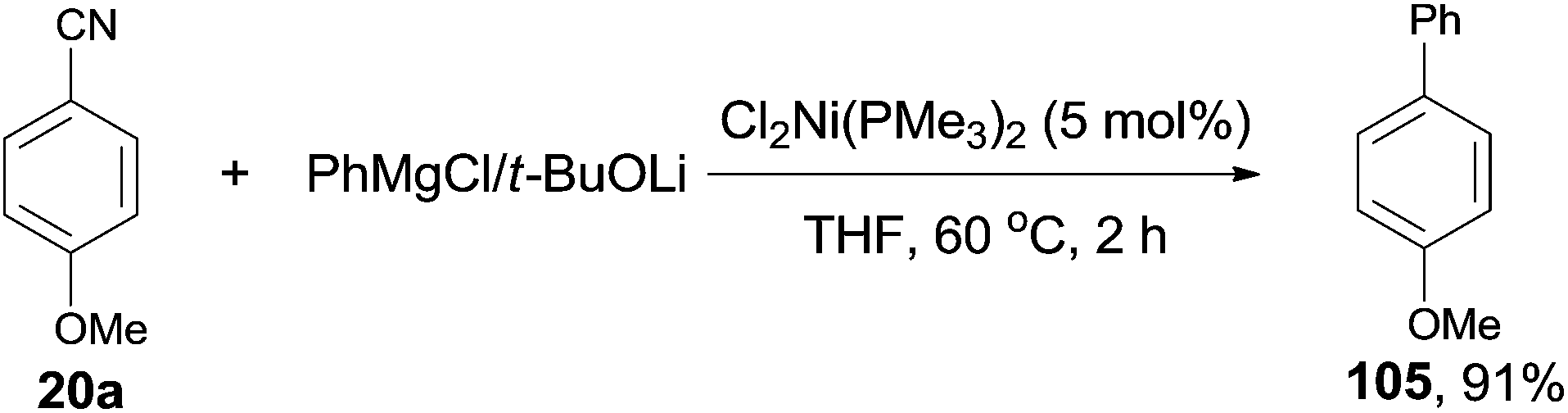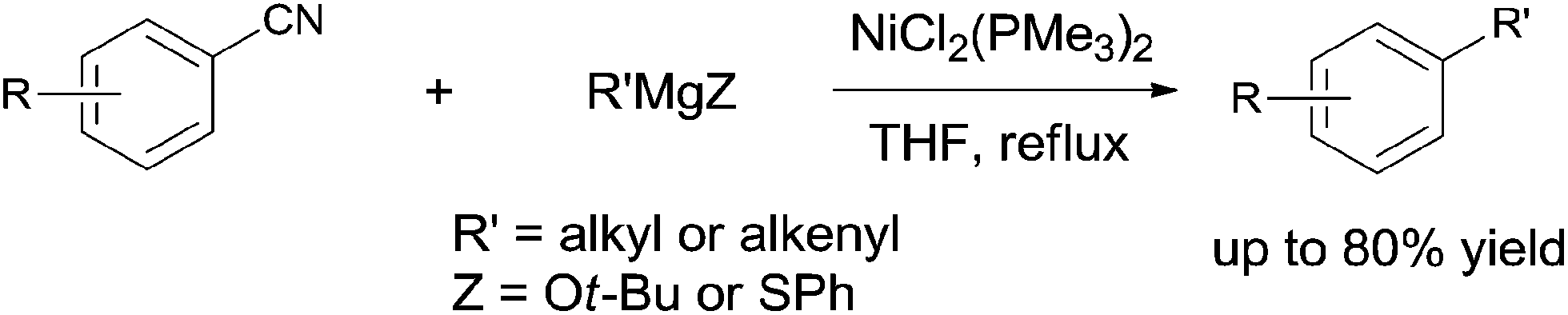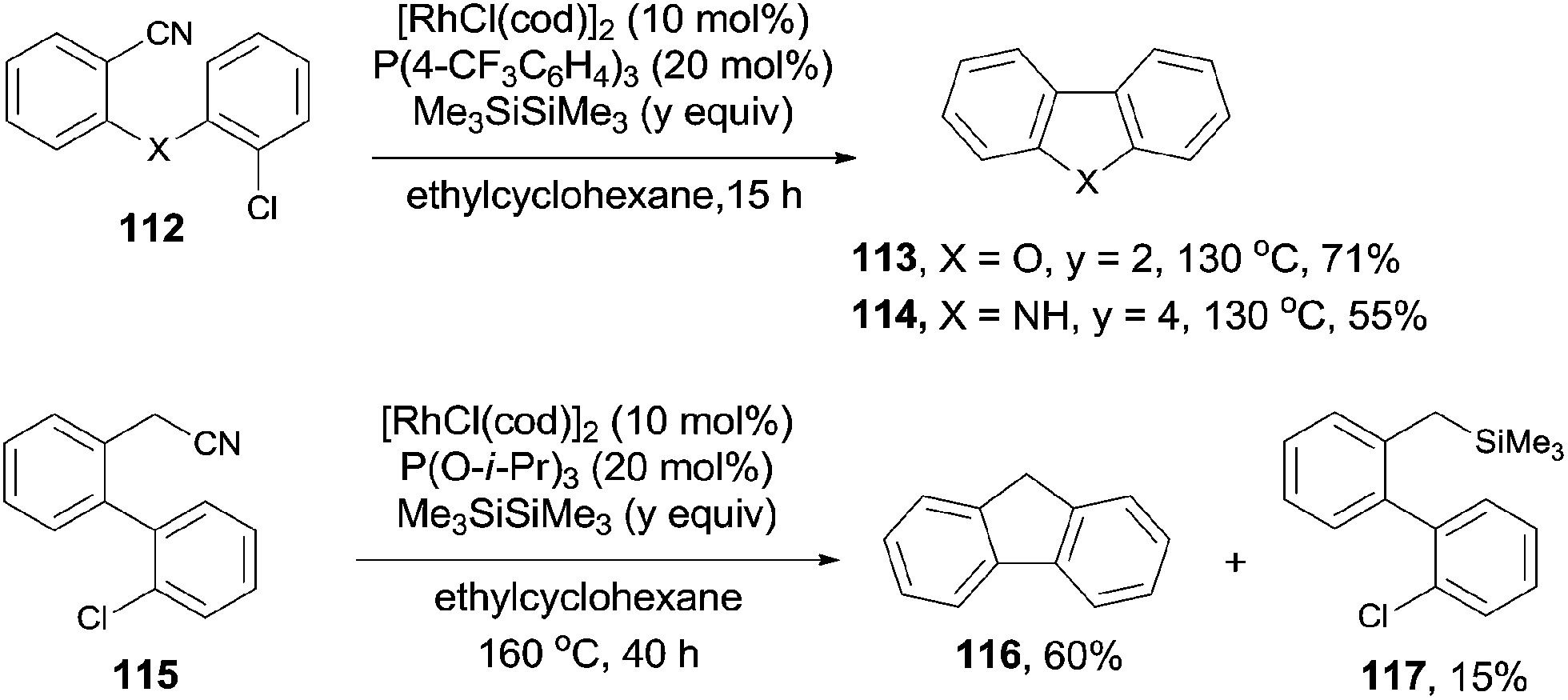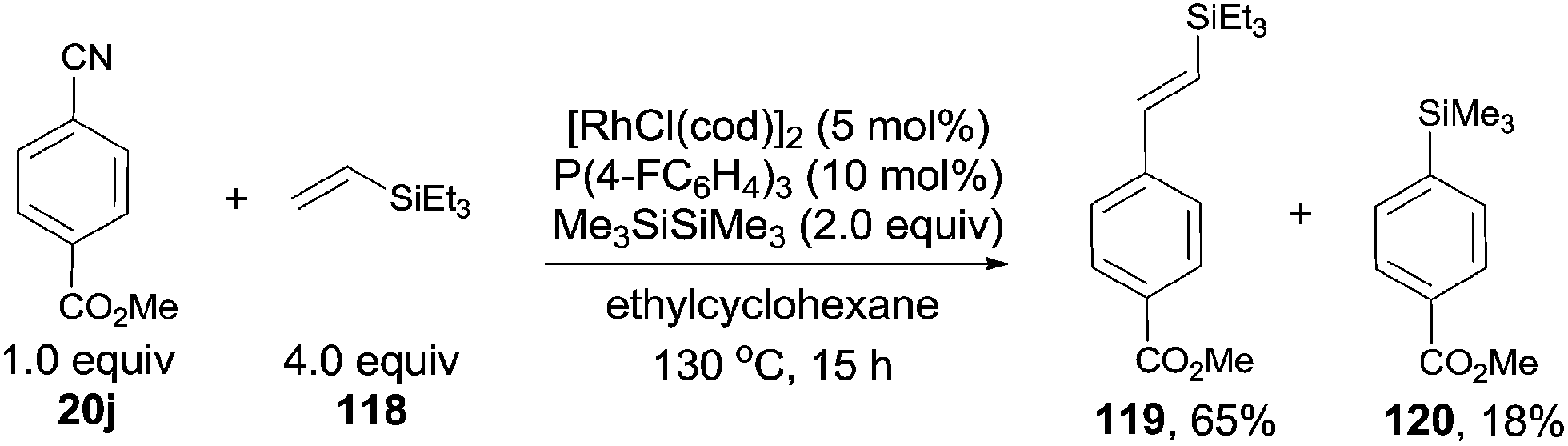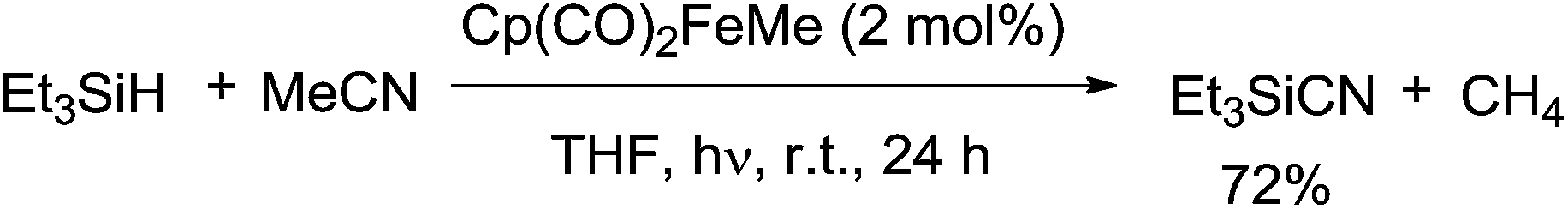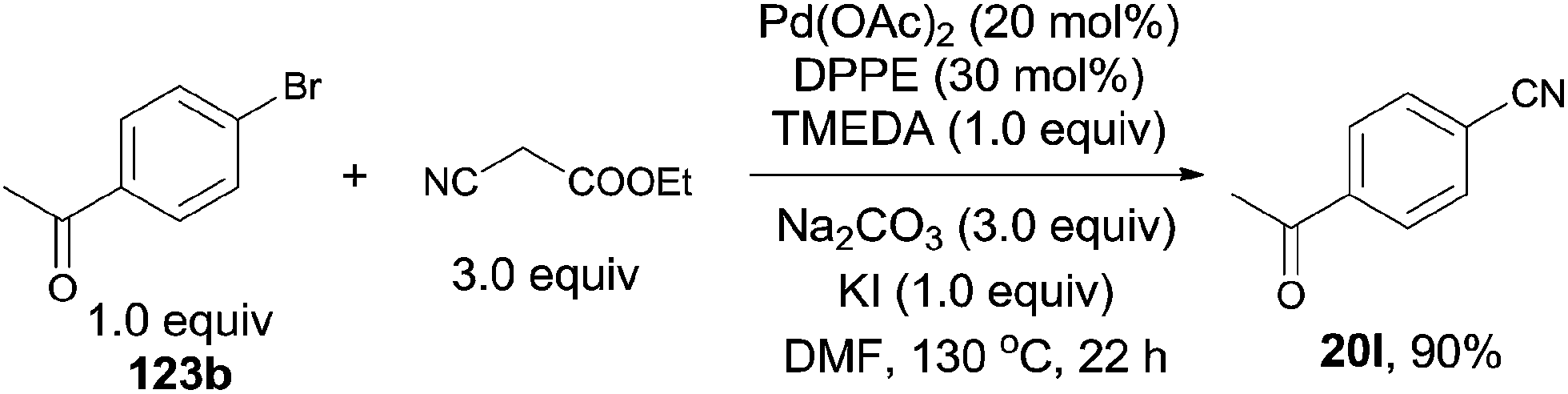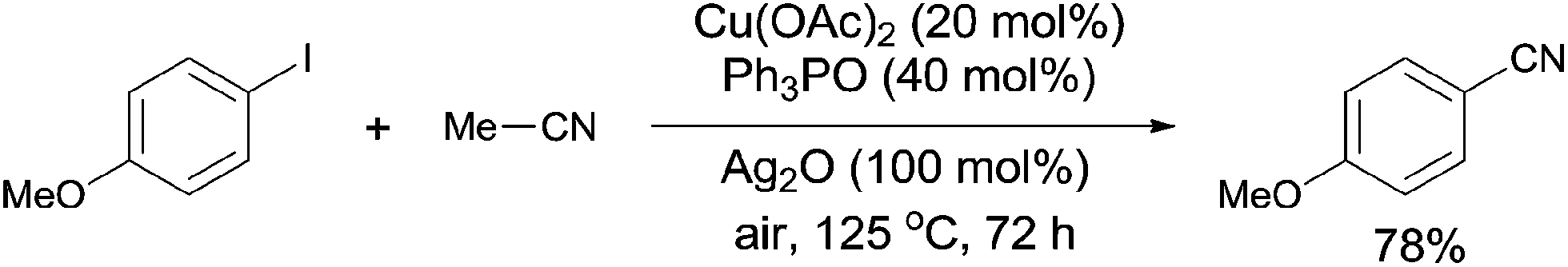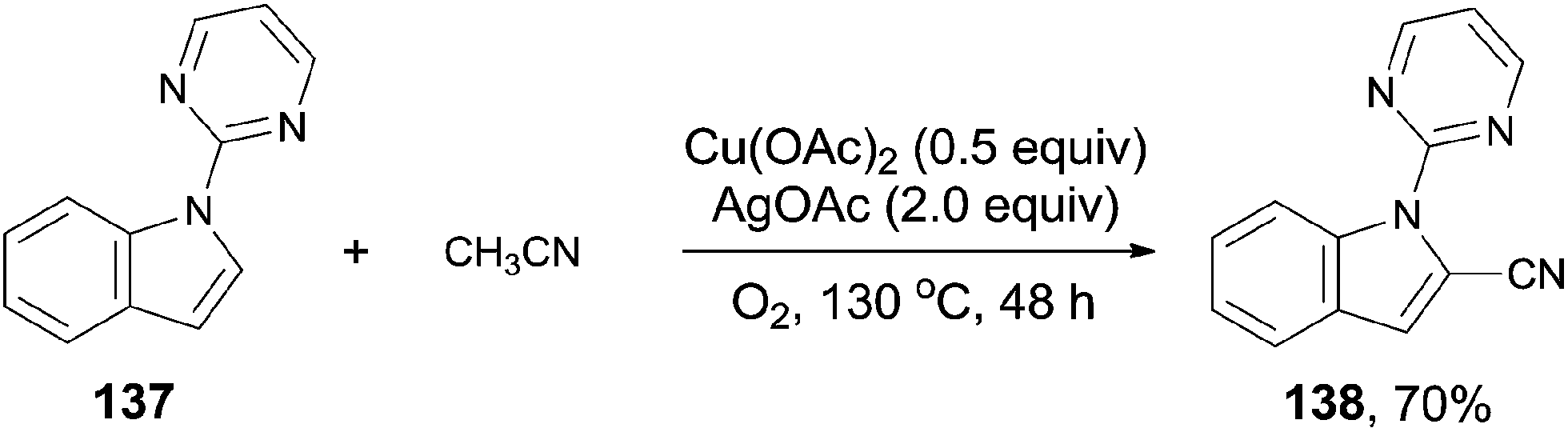DOI:
10.1039/C4RA08675A
(Review Article)
RSC Adv., 2014,
4, 47806-47826
Recent advances in transition-metal-catalyzed C–CN bond activations
Received
14th August 2014
, Accepted 23rd September 2014
First published on 23rd September 2014
Abstract
C–C bond activation and cleavage have become more and more attractive in recent years for the great challenge of breaking C–C bonds and the great advantage of constructing new chemical bonds. Among them, the activation of inert C–CN bonds by transition metals has been widely investigated and a number of improvements have been achieved by many groups. This review mainly summarizes recent advances in this field, based on different kinds of reactions derived from C–CN bond activation, including cyanofunctionalization, cross coupling reactions and cyanation. Moreover, mechanistic studies involving transition-metal-catalyzed C–CN bond activation are also mentioned.
1. Introduction
Nitriles are not only important in natural products, pharmaceuticals, agrochemicals, dyes, herbicides, and materials,1 but also can be transferred into other compounds with various functionalities, such as amines, amidines, amides, aldehydes, carboxylic acids, pyrazoles, triazoles, and tetrazoles,2 through C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N transformations. Moreover, through transition metal catalyzed C–CN bond activation, nitriles can be utilized in the reactions of cyanofunctionalization, decyanative cross-coupling, and cyanation. That is why numerous methodologies related to the making as well as the breaking of C–CN bonds have been established. In contrast to the review articles considering the construction of C–CN bonds from the aspects of various transition metals and safe cyanide sources,3 reviews about the transition-metal catalyzed C–CN bond activation are seldom reported.4 Herein, we will describe the recent progress in this field based on reaction categories and mechanistic studies.
N transformations. Moreover, through transition metal catalyzed C–CN bond activation, nitriles can be utilized in the reactions of cyanofunctionalization, decyanative cross-coupling, and cyanation. That is why numerous methodologies related to the making as well as the breaking of C–CN bonds have been established. In contrast to the review articles considering the construction of C–CN bonds from the aspects of various transition metals and safe cyanide sources,3 reviews about the transition-metal catalyzed C–CN bond activation are seldom reported.4 Herein, we will describe the recent progress in this field based on reaction categories and mechanistic studies.
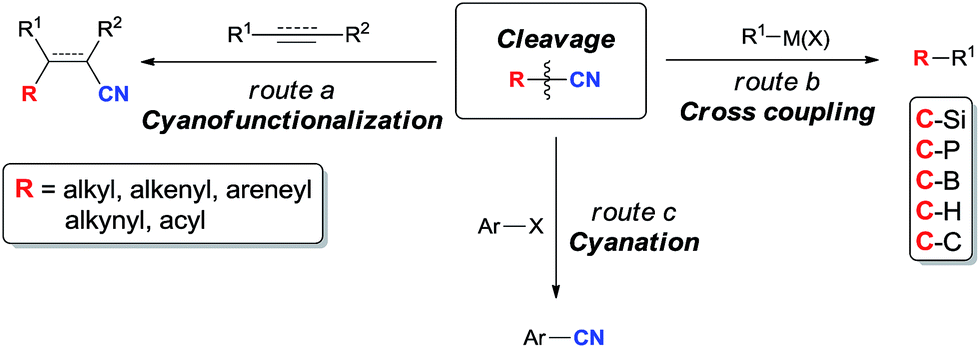
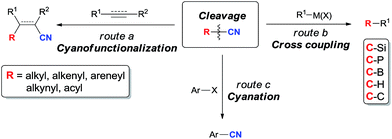
C–CN bond is relatively stronger and stable in comparison with C–X bond referring to have higher bond dissociation energy (BDE). BDEs of C–CN bonds in CH3–CN, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CN, HCC–CN, and Ph–CN are 121.1, 132.1, 152.1, and 132.7 kcal mol−1,5 respectively. In comparison with these data, BDEs of C–X bonds in Ph–F, Ph–Cl, Ph–Br, and Ph–I are 127.4, 97.3, 82.7, and 66.9 kcal mol−1, respectively.6 Thus, the selective cleavage of C–CN bonds by using transition metal catalyst is a challengeable target and of great importance in organic synthesis. Reactions involving the cleavage of C–CN bonds can be simply categorized into three types according to the utility degree of nitriles. In cyanofunctionalizations, both R and CN units are presented in products (route a, Scheme 1). When “CN” is used as the leaving group, R–CN becomes an alternative substrate of R–X/R–OTf for cross coupling reactions which have been widely applied in making C–Si, C–P, C–B, C–H, as well as C–C bonds (route b, Scheme 1). With the disclosure of the safe cyanation reagents in the recent years, R–CN has also been developed for the operational benign cyanation reagents, such as benzyl nitrile,7 and acetonitrile.8 In these reactions, “CN” unit is successfully transferred from starting materials to target molecules (route c, Scheme 1).
CH–CN, HCC–CN, and Ph–CN are 121.1, 132.1, 152.1, and 132.7 kcal mol−1,5 respectively. In comparison with these data, BDEs of C–X bonds in Ph–F, Ph–Cl, Ph–Br, and Ph–I are 127.4, 97.3, 82.7, and 66.9 kcal mol−1, respectively.6 Thus, the selective cleavage of C–CN bonds by using transition metal catalyst is a challengeable target and of great importance in organic synthesis. Reactions involving the cleavage of C–CN bonds can be simply categorized into three types according to the utility degree of nitriles. In cyanofunctionalizations, both R and CN units are presented in products (route a, Scheme 1). When “CN” is used as the leaving group, R–CN becomes an alternative substrate of R–X/R–OTf for cross coupling reactions which have been widely applied in making C–Si, C–P, C–B, C–H, as well as C–C bonds (route b, Scheme 1). With the disclosure of the safe cyanation reagents in the recent years, R–CN has also been developed for the operational benign cyanation reagents, such as benzyl nitrile,7 and acetonitrile.8 In these reactions, “CN” unit is successfully transferred from starting materials to target molecules (route c, Scheme 1).
 |
| | Scheme 1 Three patterns of the utility of RCN in organic synthesis. | |
2. Cyanofunctionalizations
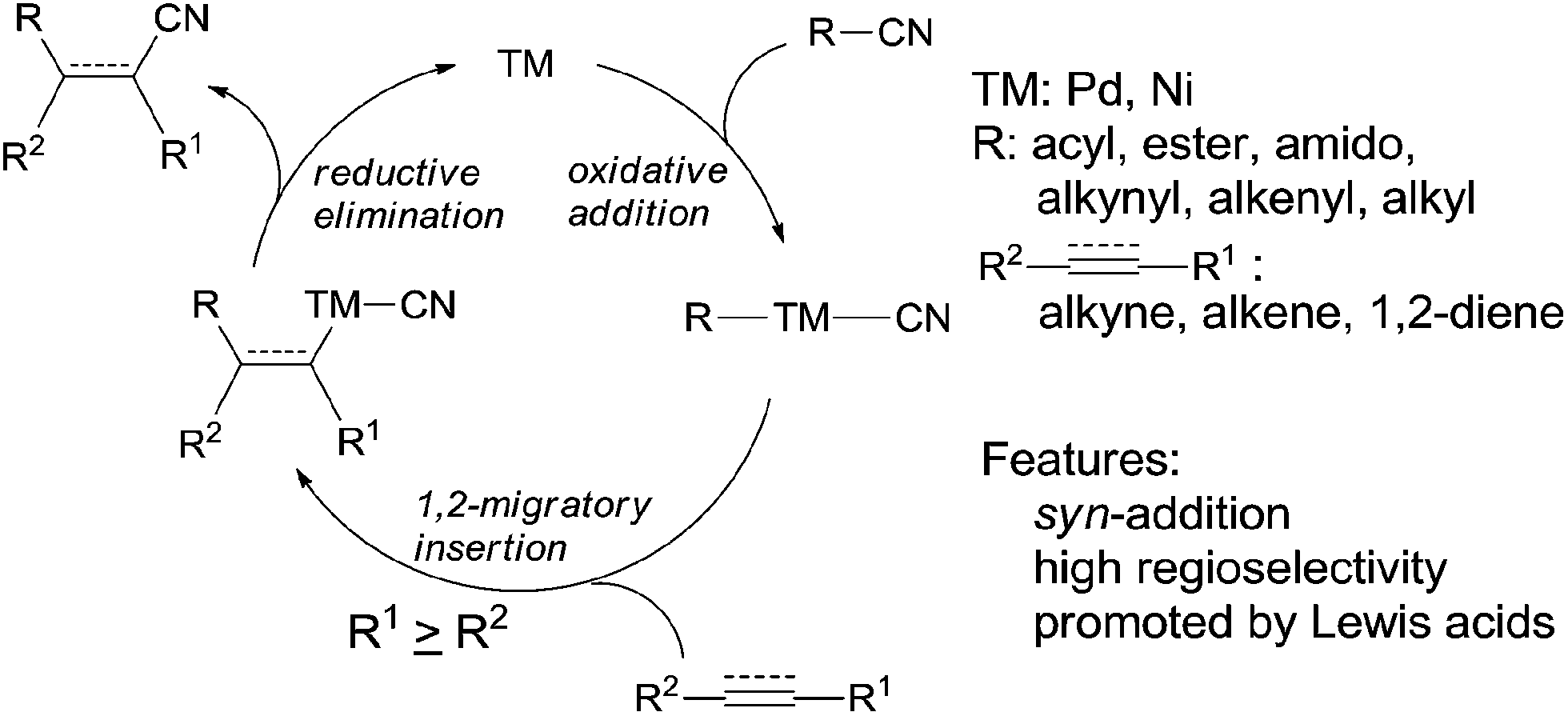
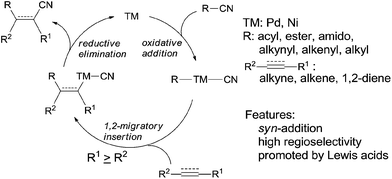
When oxidative addition of transition metal occurred on C–CN bond, an active intermediate, acyl/alkynyl/alkenyl/alkyl metal cyanide (C–TM–CN), is formed and C–CN bond is activated and finally cleaved. The subsequent 1,2-migratory insertion of C–TM–CN species into unsaturated bond, followed by reductive elimination affords a variety of addition products and regenerates transition metal for the next catalytic cycle (Scheme 2). This transition metal catalyzed reaction is called cyano functionalization or carbocyanation. Because such reaction installs two C–C bonds simultaneously onto an unsaturated bond with great atom efficiency, this reaction is surely of great synthetic values. Up to now, outstanding results have been achieved in this area, including cyanoacylation, cyanoesterification, cyanoamidation, cyanoarenylation, cyanoalkenylation, cyanoalkynylation, and cyanoalkylation, according to the carbon types where C–CN bond is activated. Most of these cyanofunctionalizations occur on alkyne, and some of them happen on alkene and 1,2-diene. All these reactions undergo exclusively in syn-addition fashion with high regioselectivities when unsymmetrical triple or double bonds are applied. Moreover, these cyanofunctionalizations could be significantly promoted by adding appropriate Lewis acid.
 |
| | Scheme 2 General mechanism of cyanofunctionalization. | |
2.1 Cyanoacylation
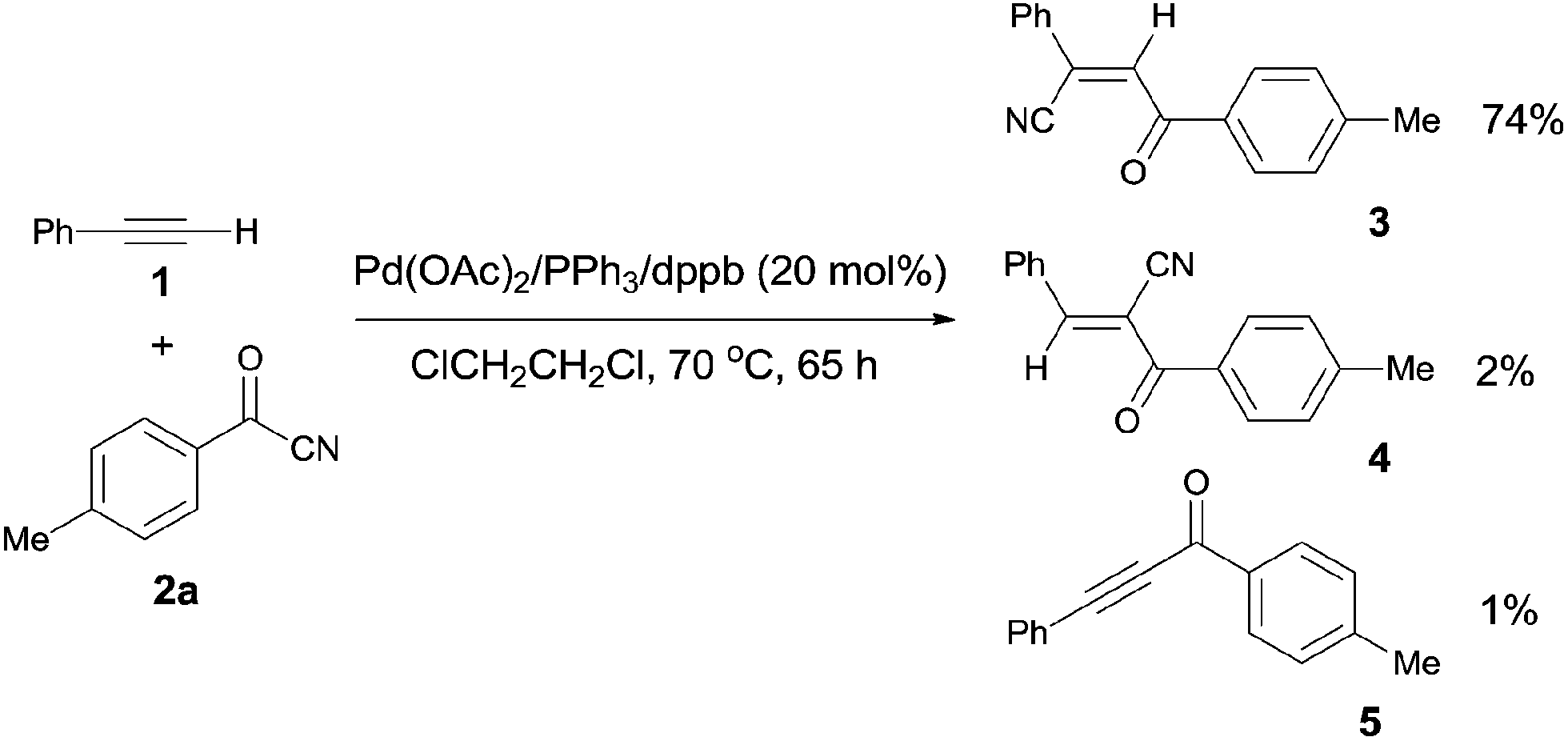
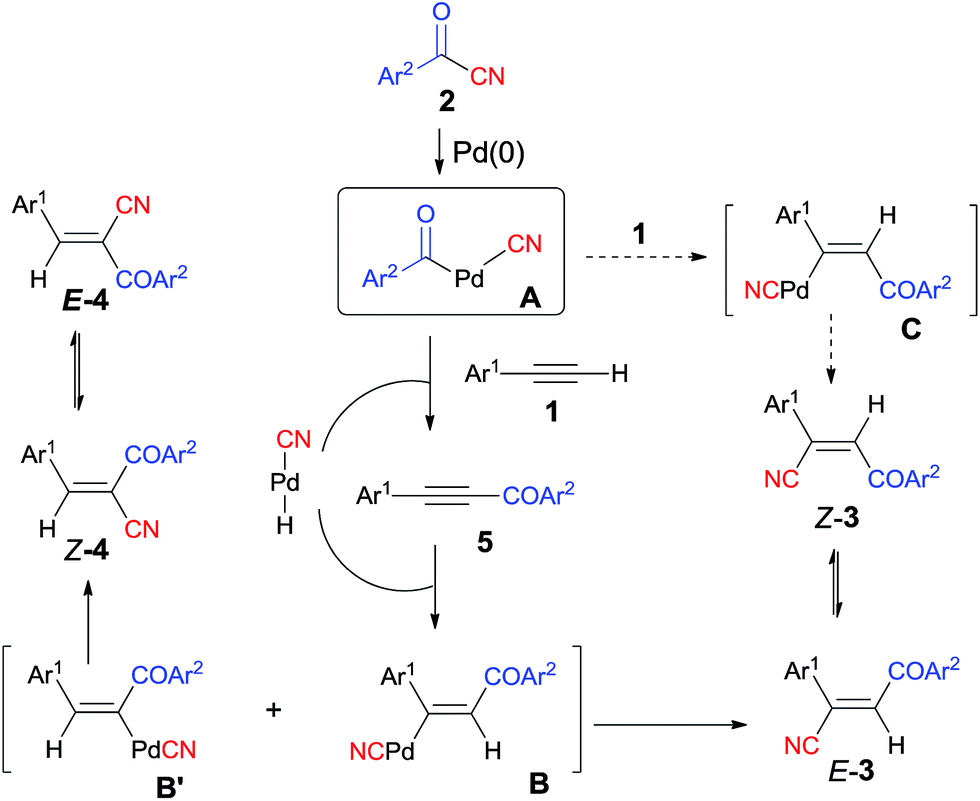
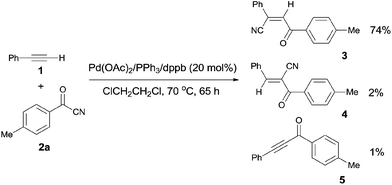
Early in 1994, Takaya reported palladium-catalyzed cyanoacylation of terminal acetylenes with aryloyl cyanides, leading to the formation of alkenes attached with two electron-withdrawing groups, acyl and cyano (Scheme 3).9a Under the designated palladium catalyst system, phenylacetylene (1) reacted with 4-methylbenzoyl cyanide (2a), giving the corresponding addition product (3), accompanied by trace amount of regio isomer (4) and acetylenic ketone (5). The amount of 5 was decreased when the reaction was carried out for a longer time. Thus acetylenic ketone (5) was proposed as a plausible intermediate during the reaction process. Through this intermediate, addition products (3) were formed by a sequence of Pd-catalyzed hydrocyanation and isomerization (Scheme 4). If the Pd-catalyzed hydrocyanation occurred in a different direction, Z-4 would be produced. Thus, E-4 was formed through isomerism. Although the formation of intermediate C was reasonable, the route through intermediate 5 was more possible. This strategy provided a route for the regioselective preparation of electron-deficient alkenes. A detail study on the role of the mixed ligands was presented in 1996.9b PPh3 was responsible for its suitable basicity and cone angle, while dppb (1,4-bis(diphenylphosphino)butane) benefitted for a higher conversion.
 |
| | Scheme 3 Pd-catalyzed cyanoacylation of phenylacetylene with 4-methylbenzoyl cyanide. | |
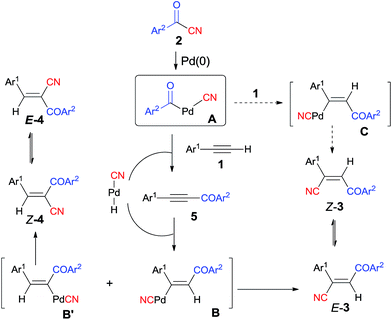 |
| | Scheme 4 Proposed mechanism of Pd-catalyzed cyanoacylation. | |
2.2 Cyanoesterification
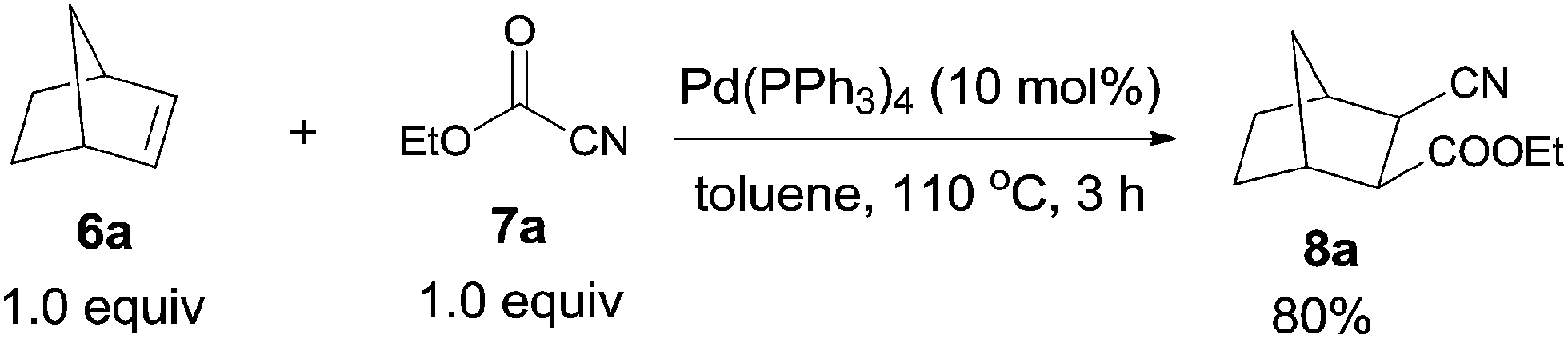
In 2006, Nishihara reported palladium-catalyzed C–CN bond cleavage of cyanoformates (7) and addition to norbornenes (6), affording various norbornane derivatives (8) with two functional groups, cyano and ester (Scheme 5).10 Benzonorbornadiene and norbornadienes were both applicable to this reaction, as well as many other cyanoformates.
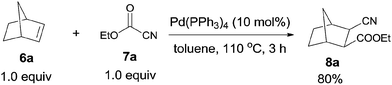 |
| | Scheme 5 Pd-catalyzed cyanoesterification of norbornene. | |
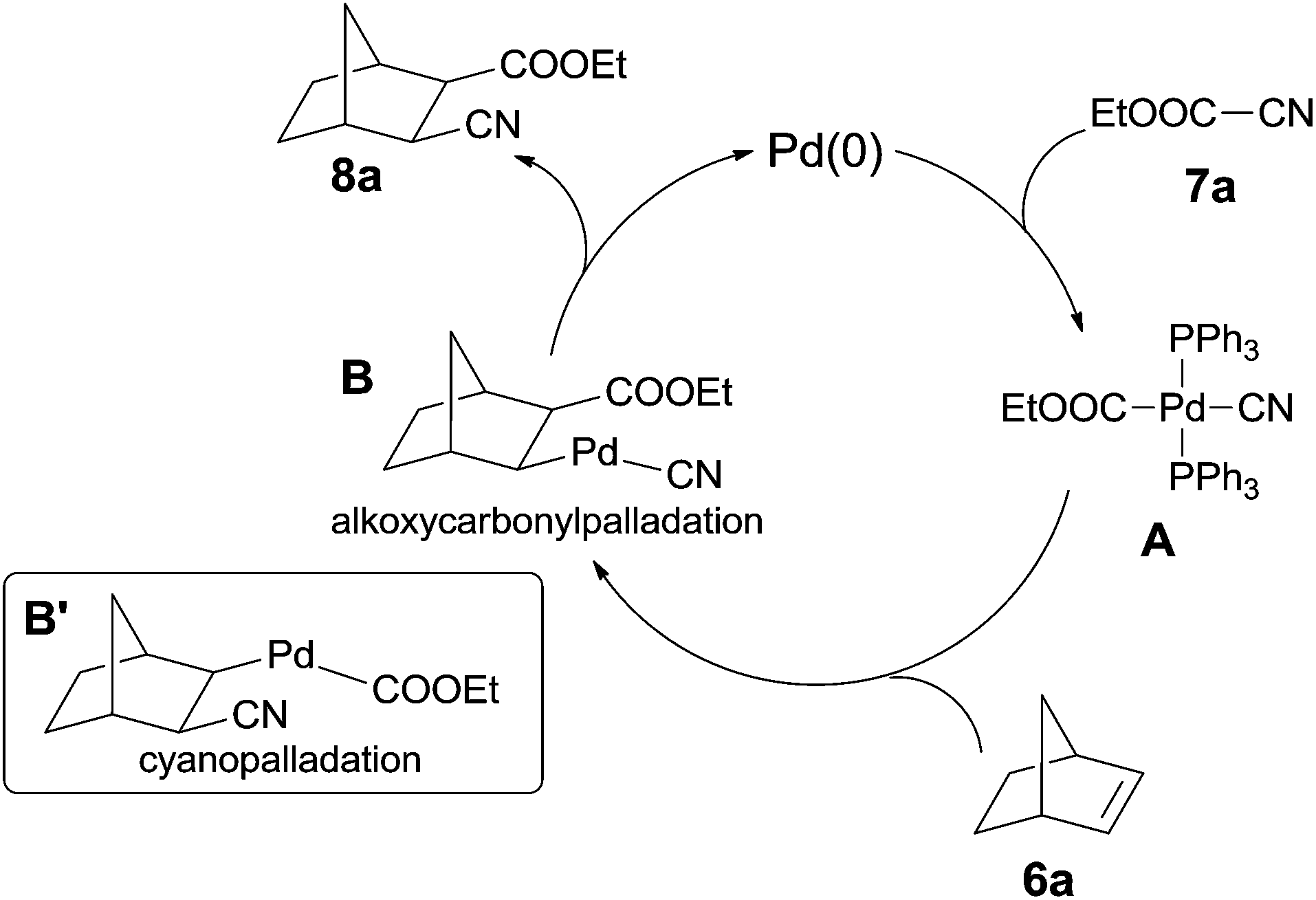
In their mechanistic studies, the trans-Pd(CN)(CO2Et)(PPh3)2 (A) was isolated and proved to be the key intermediate by the addition of Pd(PPh3)4 to ethyl cyanoformate (7a) (Scheme 6). Single crystal analysis of the Me analogue of trans-Pd(CN)(CO2Et)(PPh3)2 (A) indicated that A possessed a square planar structure with two triphenylphosphines in trans positions.10a 1,2-Migratory insertion of trans-A to the double bond of norbornene (alkoxypalladation) followed by the reductive elimination gave the cyanoesterification product (8a) in syn-addition manner. Two groups (CN and COOEt) occupied the exo-positions instead of the endo-positions. That an alternative 1,2-migratory insertion of trans-A to norbornene (cyanopalladation generating B′) was ruled out because of the high affinity of cyano to palladium center.10b
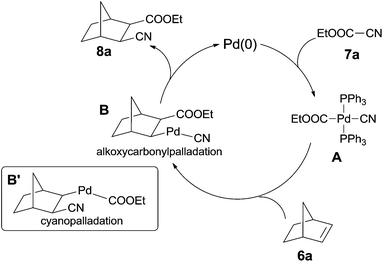 |
| | Scheme 6 Proposed mechanism of Pd-catalyzed cyanoesterification. | |
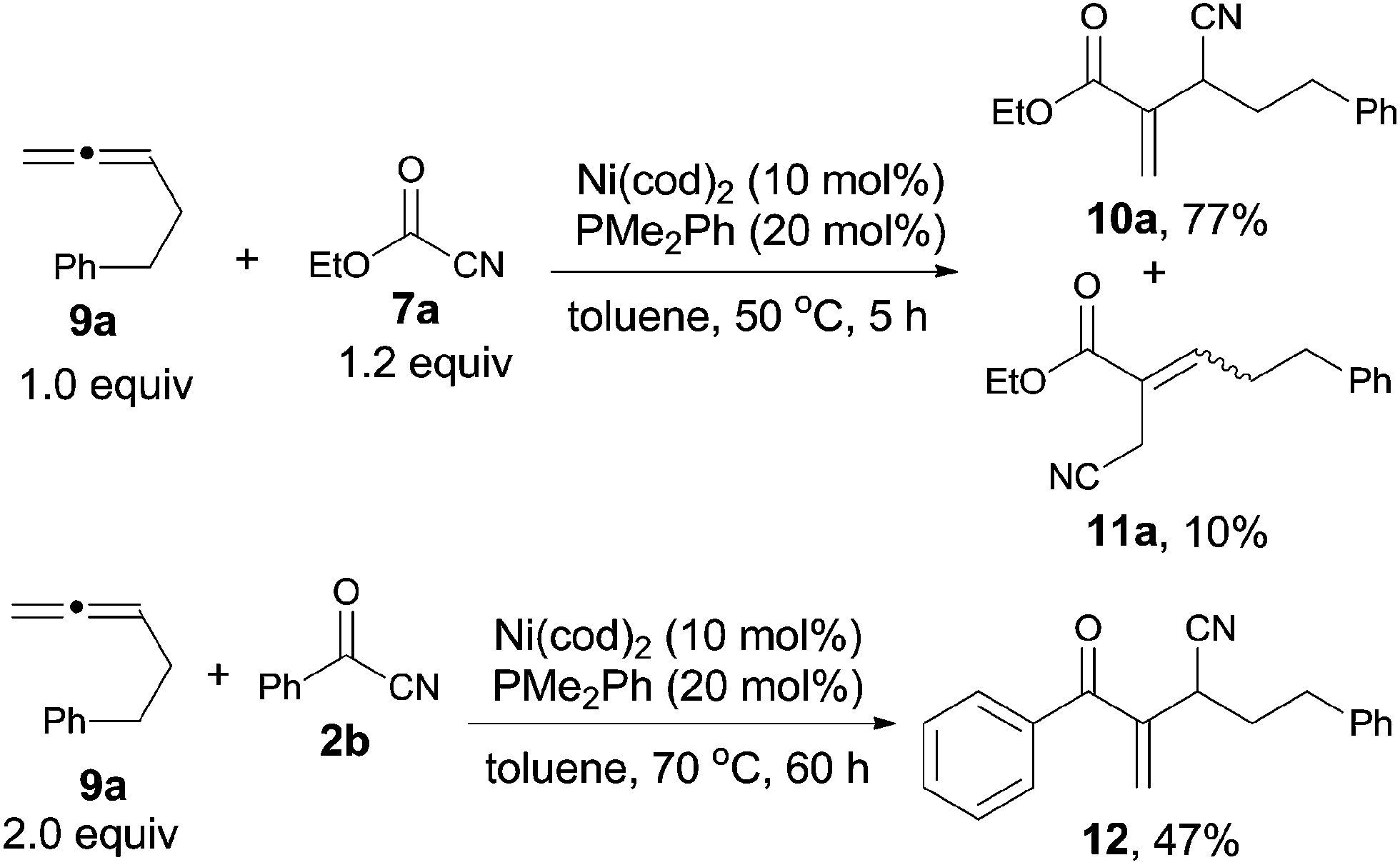
Meanwhile, Nakao and Hiyama reported a nickel-catalyzed C–CN bond cleavage of cyanoformates (7) and their sequential cyanoesterifications of cumulative 1,2-dienes (9), affording various 2-(1-cyanoalk-1-yl)acrylates (10), as well as their regioisomers (11) (Scheme 7).11a By reversing the molar ratio of 7a to 9a to 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2, 11a was formed predominantly.11b Except for cyanoformates (7), benzoyl cyanide (2b) was also suitable for this reaction under modified conditions and provided 12 with a little lower yield.
:1.2, 11a was formed predominantly.11b Except for cyanoformates (7), benzoyl cyanide (2b) was also suitable for this reaction under modified conditions and provided 12 with a little lower yield.
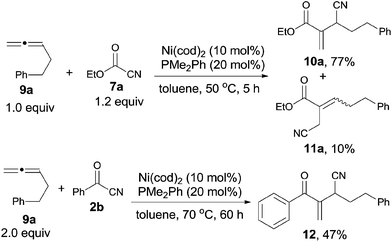 |
| | Scheme 7 Ni-catalyzed cyanoesterification/cyanoacylation on 1,2-diene. | |
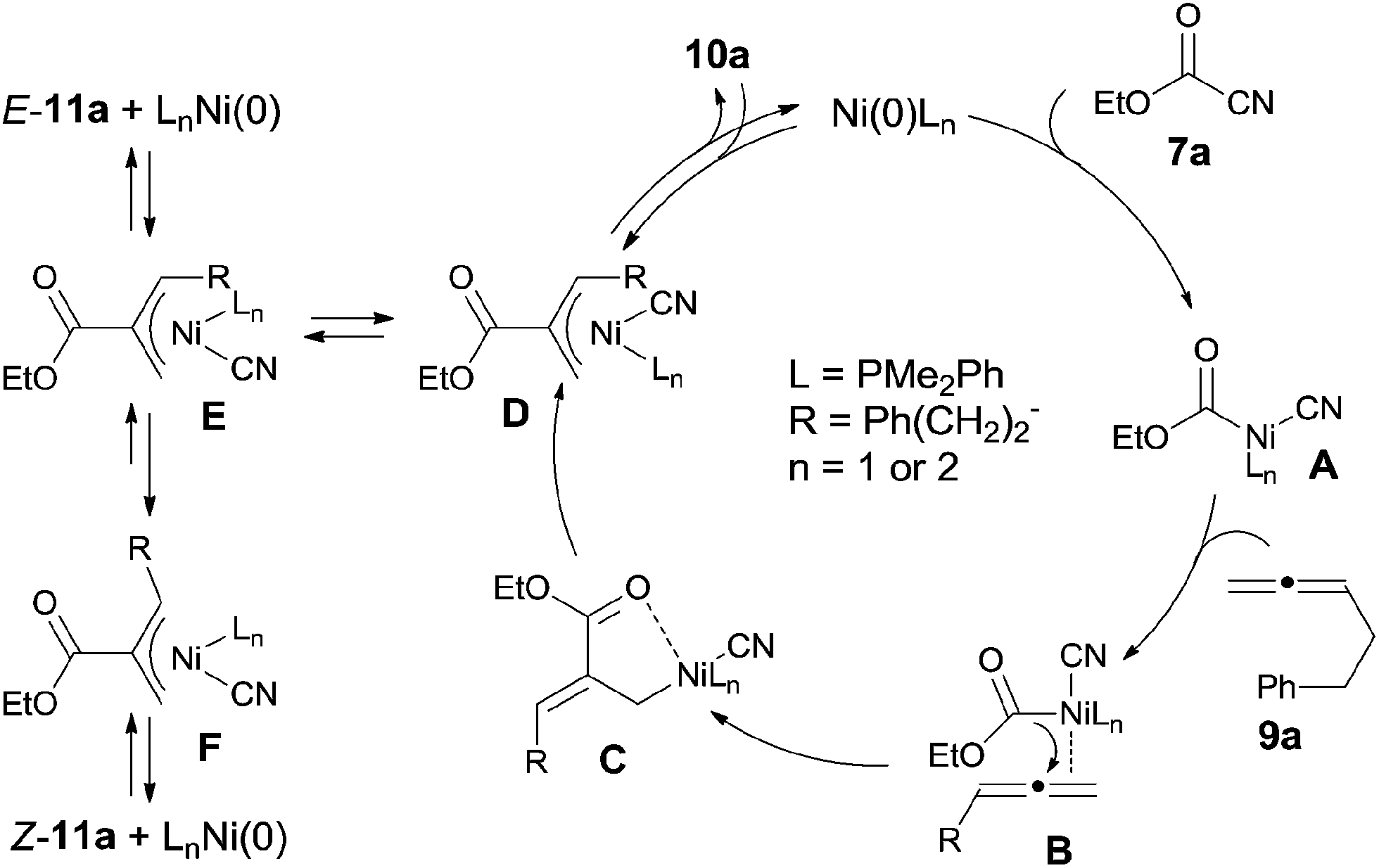
In their proposed mechanism, Ni catalyst inserted to C(sp2)–CN bond (Scheme 8). 1,2-Migratory insertion of C–Ni–CN (A) on 1,2-diene occurred on the less steric hindered terminal double bond with the ester group migrated to the central carbon of 1,2-diene. In this way, C was regioselectively formed through the possible complex B. The formed Ni(II) complex (C) could be stabilized by a π-allylnickel intermediate (D). By reductive elimination in two different types, two different products (10a and E-11a) were obtained. Z-11a was proposed to be obtained via equilibrium between intermediates E and F and followed by reductive elimination from intermediate F. The regioisomer E-11a was thought to be the thermodynamic product, whereas 10a was a kinetic product. This proposal was proved by the condition controlled experiments when the reaction temperature was increased to 100 °C and the amount of 10a was significantly decreased.
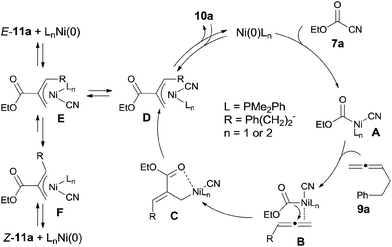 |
| | Scheme 8 Proposed mechanism for Ni-catalyzed cyanoesterification on 1,2-diene. | |
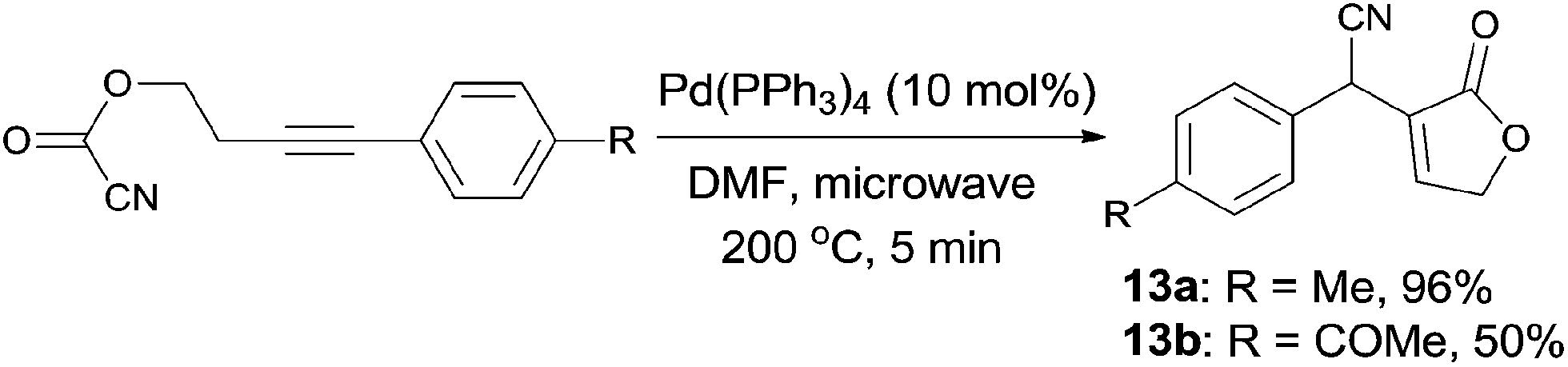
In 2011, Douglas demonstrated an intramolecular Pd-catalyzed cyanoesterification, accessing to functionalized lactones (13) in moderate to excellent yields (Scheme 9).12 The decarboxylation and decarbonylation byproducts were successfully suppressed by performing the reactions in microwave reactor at high temperature. The authors also hypothesized that the 1,2-migratory insertion was the product-determining step, rather than the step of C(sp2)–CN bond activation, because of the higher reactivity of electron-rich alkynes.
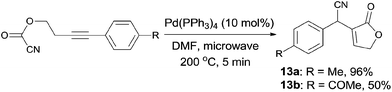 |
| | Scheme 9 Pd-catalyzed intramolecular cyanoesterification. | |
2.3 Cyanoamidation
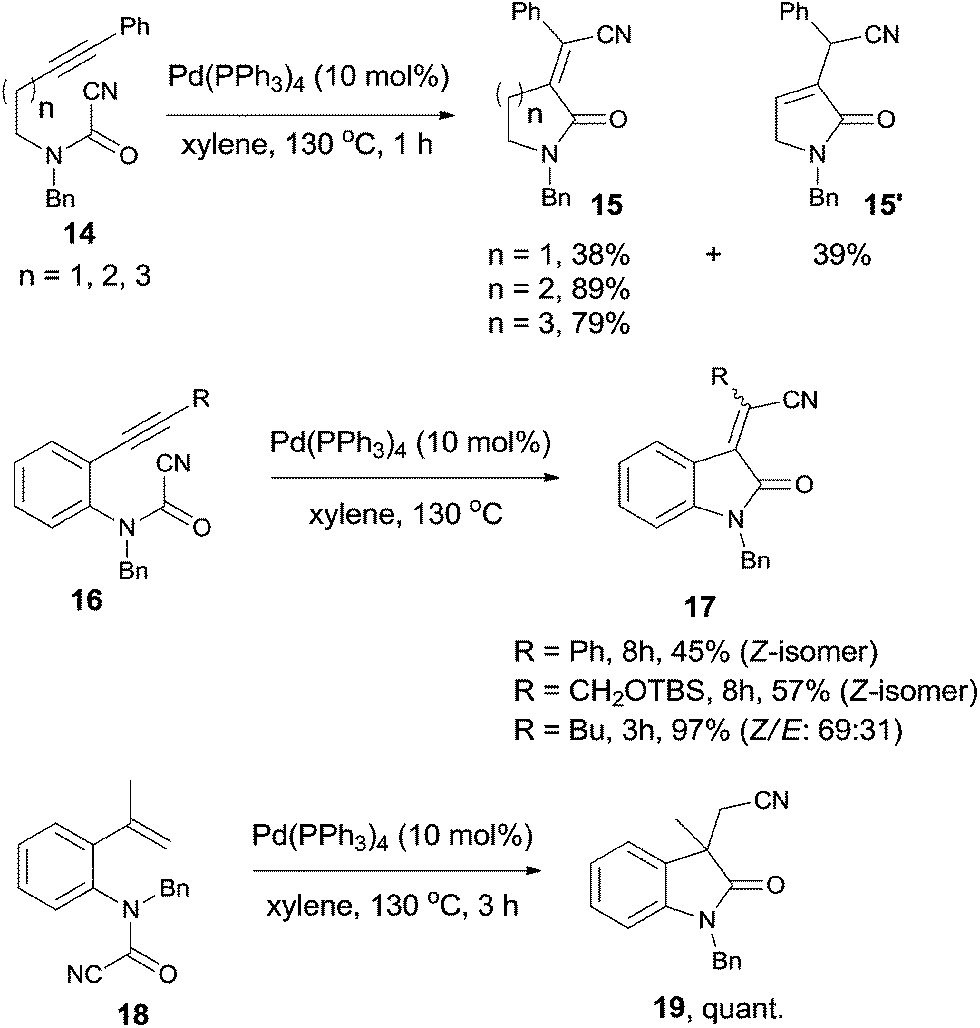
In 2006, Takemoto reported the intramolecular cyanoamidation of alkyne and alkene through the designation of substrate structures (14, 16, and 18) (Scheme 10).13 Nonaromatic substrates (14) proceeded exclusively in syn-addition manner and provided α-alkylidene lactams (15) in moderate to good yields. In case of n = 1, isomerized product 15′ was obtained in 39% yield because of the 1,3-migration of the C-4 proton.
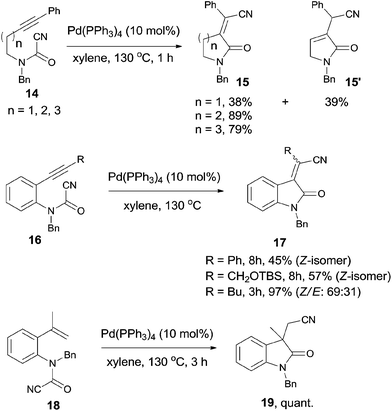 |
| | Scheme 10 Pd-catalyzed intramolecular cyanoamidations. | |
Aromatic substrate (16 and 18) provided indole derivatives 17 and 19, respectively. When cyanoamidation occurred on triple bonds, the stereoselectivity was largely depended upon the R group which was attached on the triple bonds. When R was phenyl or CH2OTBS group, yields were moderate and the products were in exclusively Z-form. When R was altered to butyl, a mixture of Z and E in a ratio of 69:31 was isolated in 97% yield. When cyanoamidation occurred on double bonds, a quaternary carbon center was created and 3,3′-disubstituted oxindole 19 was obtained almost quantitatively, which could be used as the key intermediate in the preparation of physostigmine.
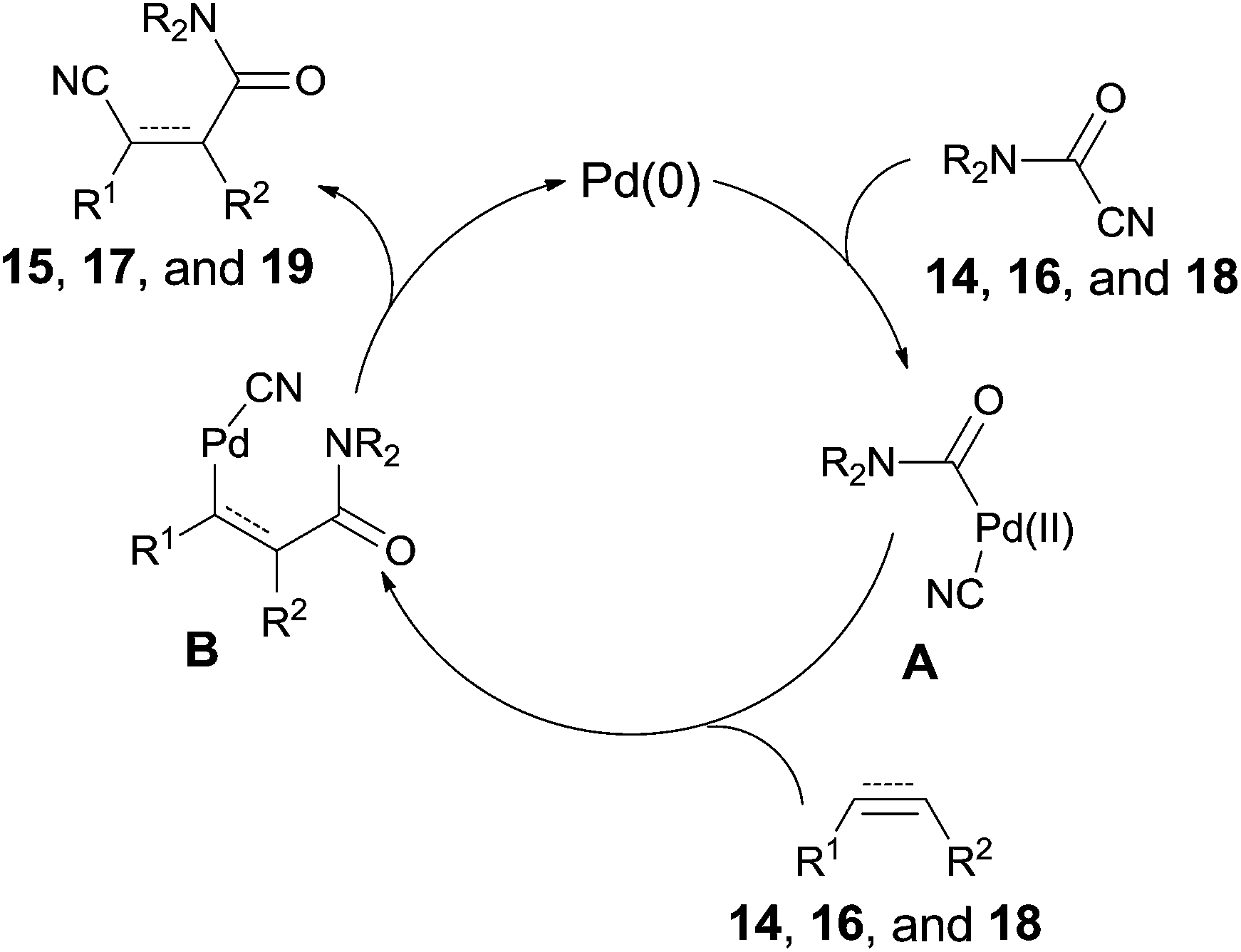
All these reactions occurred in a same manner as the previous cyanofunctionalizations. Pd(0) was oxidatively added to amido cyanides (14, 16, and 18) to form amidopalladium cyanide intermediates (A) (Scheme 11). Thus, a sequential amidopalladation occurred on triple or double bonds to generate intermediates B. Finally, amido and cyano groups were installed into both ends of triple or double bonds by reductive elimination. When two reaction sites (amido cyanide and unsaturated bonds) existed in one molecule, an intramolecular reaction happened to afford lactams successfully.
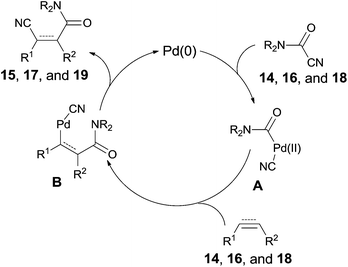 |
| | Scheme 11 Proposed mechanism for Pd-catalyzed intramolecular cyanoamidations. | |
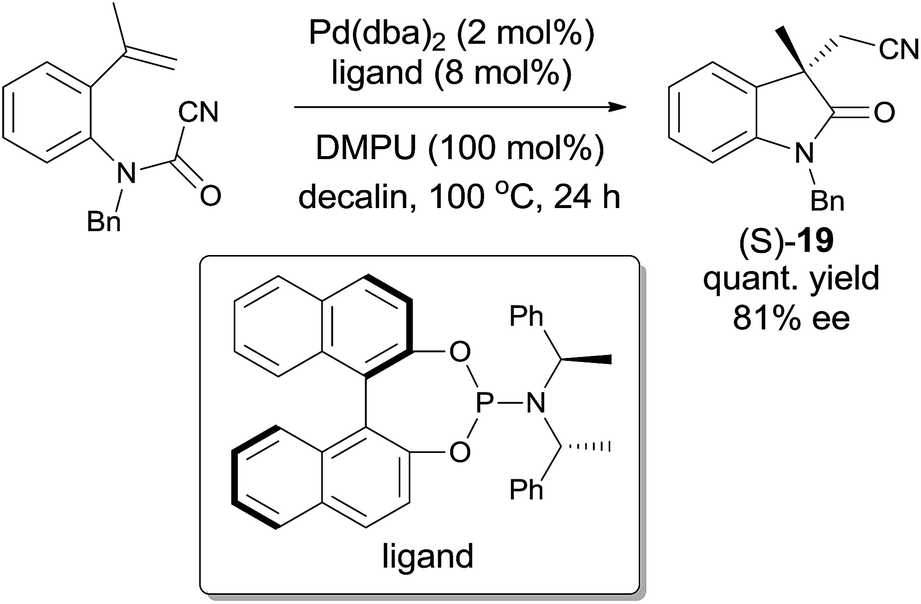
Later on, Takemoto practiced the enantioselective synthesis of 3,3′-disubstituted oxindole (S-19) by finely tuning the catalyst, chiral ligand, additive, and solvent (Scheme 12).14 By screening these factors, a combination of Pd(dba)2, an optically active phosphoramidite, and a suitable Lewis base in decalin was optimized to be the best conditions to reach the effective enantioselective synthesis of 3,3′-disubstituted oxindole (S-19). It was also noticed that this combination was effective to a broad range of substrates and might be applied in the synthesis of more complicated molecules.
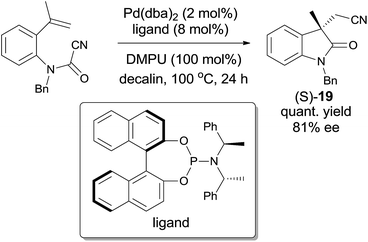 |
| | Scheme 12 Enantioselective synthesis of (S)-19 through Pd-catalyzed cyanoamidation. | |
2.4 Cyanoalkenylation/cyanoalknylation/cyanoalkylation
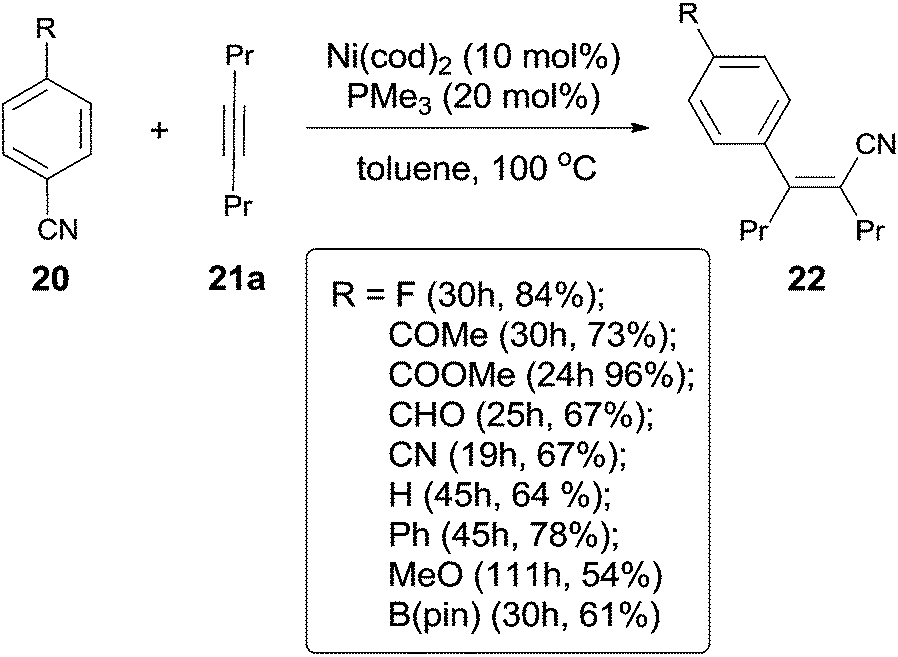
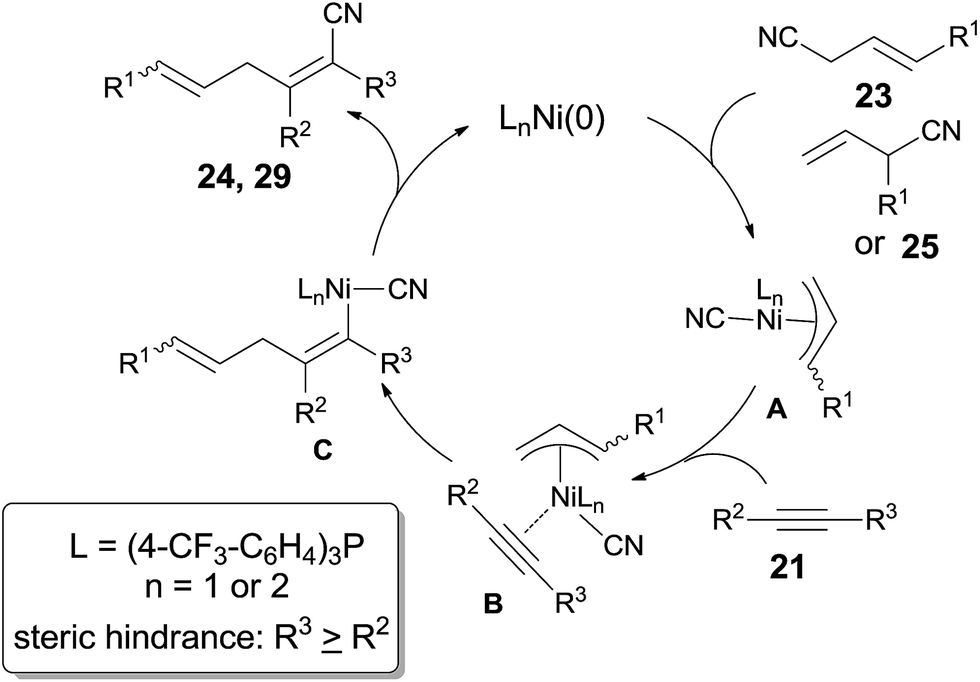
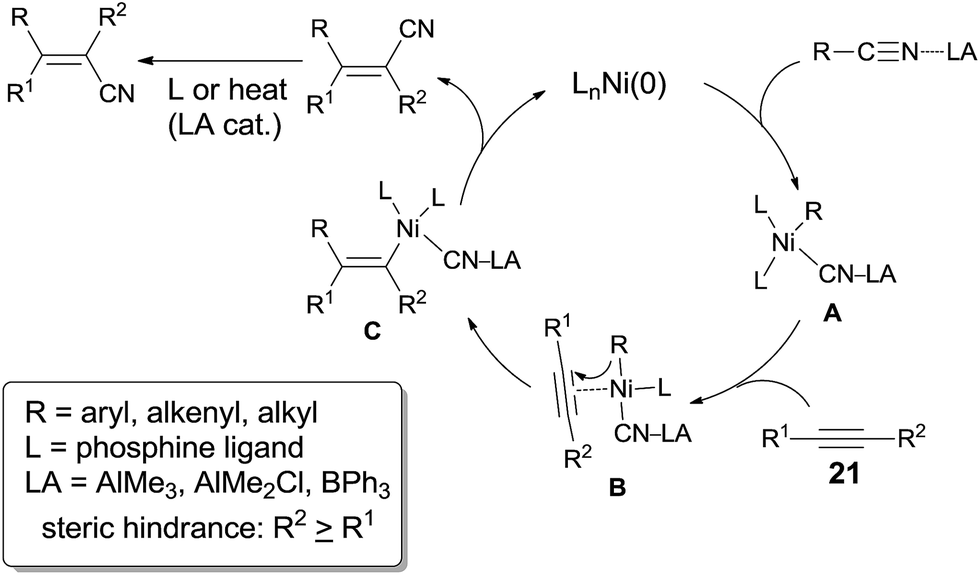
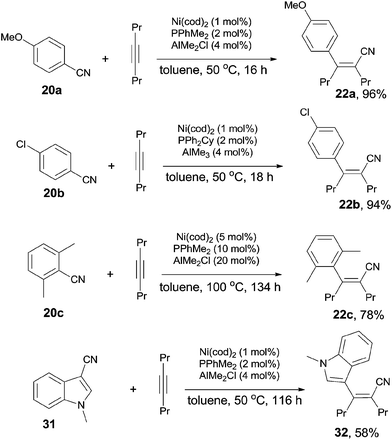
In 2004, Nakao and Hiyama developed a Ni-catalyzed cyanoarylation of different alkynes (21) through the activation of Ar–CN bonds, installing aryl and cynao in the two ends of triple bonds and providing various β-aryl substituted alkenenitriles (Scheme 13).15a Lots of functional groups on the aryl nitriles (20) tolerated the reaction conditions, such as the carbonyl groups of ketone, aldehyde and ester. Cyano group also survived after reaction if equivalent molar ratio of reactants was used. The boryl also tolerated the reaction conditions which indicated that a variety of products could be further derived from the boryl group through Suzuki–Miyaura couplings. Aryl nitriles bearing electron-neutral and electron-donating groups afforded the products with relatively longer time, which indicated the rate-determining step was oxidative addition of Ni(0) to C(sp2)–CN bond. When unsymmetrical alkynes were used, mixtures were obtained. However, a regioselective preparation could be realized when the steric hindrance of two groups on triple bond could be distinguished, such as methyl versus tert-butyl. The bulkier group and the cyano group were found to be bonded to the same carbon in the final. Unfortunately, terminal alkynes did not work under the standard reaction conditions due to its rapid trimerization or oliomerization.
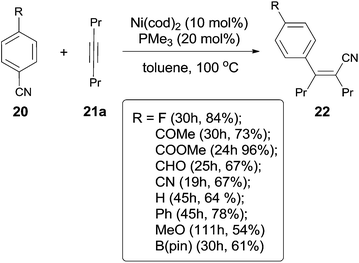 |
| | Scheme 13 Preliminary studies of Ni-catalyzed cyanoarylation on oct-4-yne. | |
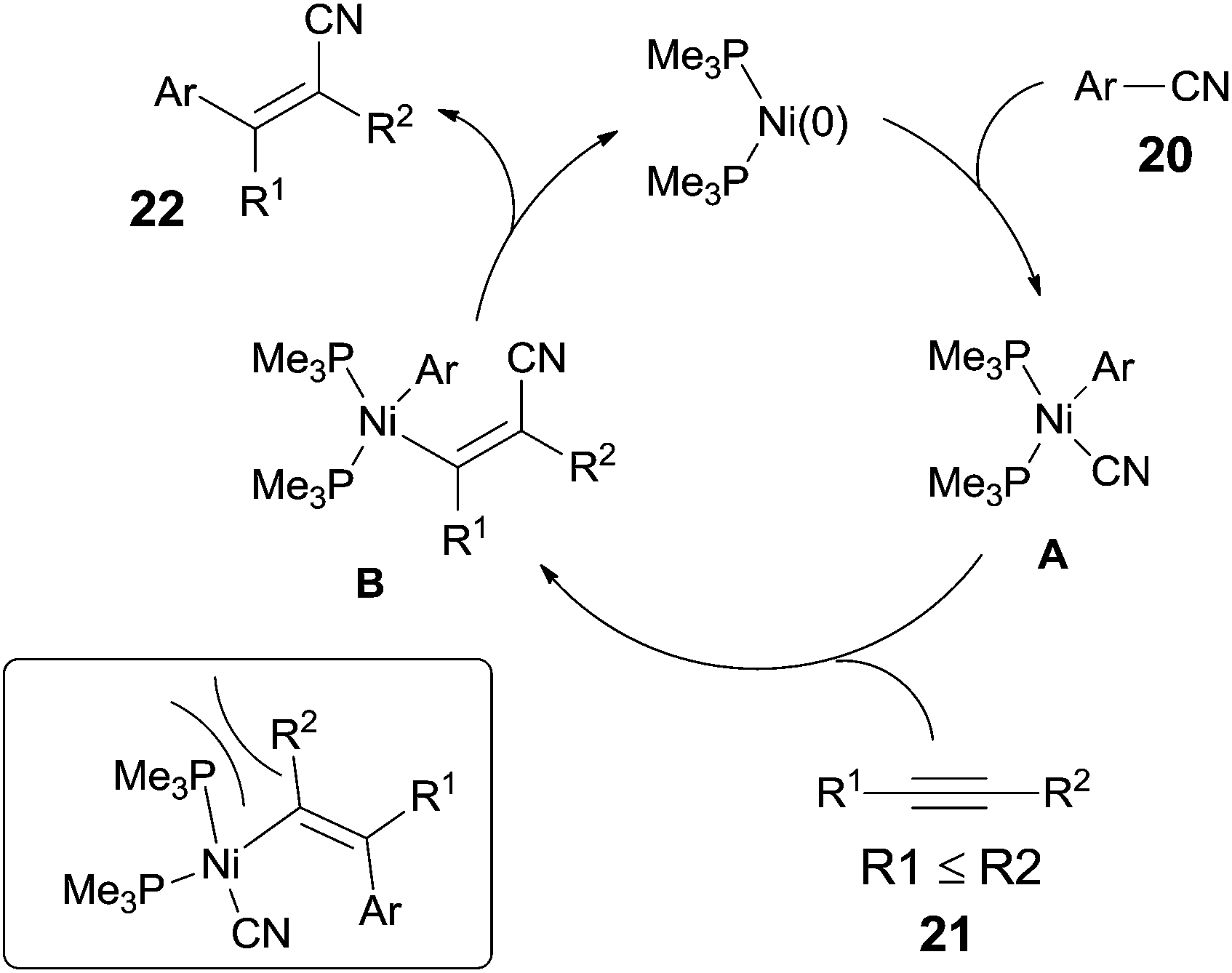
They proposed a possible mechanism (Scheme 14).15b Ni(0) catalyst inserted to Ar–CN bond of arenecarbonitrile (20) through oxidative addition, forming a Ni(II) complex (A). Then the Ni(II) complex (A) regioselectively added to the internal alkyne by 1,2-migratory insertion depending on the steric hindrance. A subsequently reductive elimination provided a cyanoarylation product (22) in syn-addition manner and regenerated the Ni(0) catalyst for the next catalytic cycle. An alternative 1,2-migratory insertion, arylnickelation instead of cyanonickelation, could not be ruled out although the repulsion between ligand and bulkier R2 group was apparent.
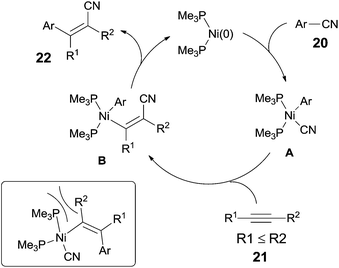 |
| | Scheme 14 Proposed mechanism for Ni-catalyzed cyanoarylation. | |
Besides aryl nitriles, allyl cyanides (23) could be used as the substrate for the oxidative addition of Ni(0), and provided the cyanoallylation products (24) when the resulting nickel species reacted with alkynes (Scheme 15).16 Thus, reaction between allyl cyanides (23) and oct-4-yne (21a) in the presence of 10 mol% Ni(cod)2 and 20 mol% P(4-CF3–C6H4)3 provided 6-substituted (2Z,5E)-2,3-dipropylhexa-2,5-dienenitriles (24) in regio- and stereoselective way. Partial isomerism of the double bond was observed. Synthetic utility of 2,5-dienenitriles was demonstrated by selective reductions. For instance, the resulting acrylonitriles could easily be reduced into acrylaldehydes and allyl alcohols by selecting suitable reductants without contamination of the reductive products derived from alkene unit.
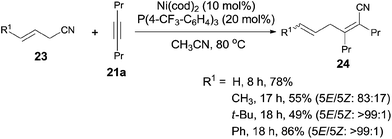 |
| | Scheme 15 Ni-catalyzed cyanoallylations with allyl cyanides. | |
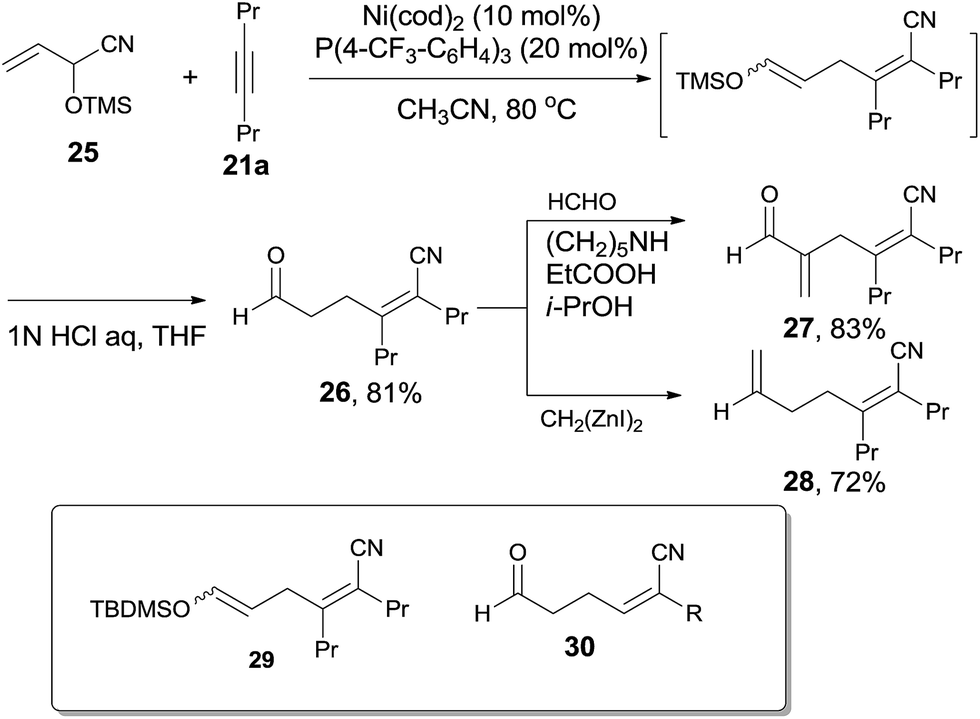
Addition of α-siloxyallyl cyanide (25), which derived from the reaction between acrolein and TMSCN, onto oct-4-yne (21a) under the aforementioned reaction conditions afforded (Z)-5-formyl-2,3-dipropylpent-2-enenitrile (26) in 81% yield (Scheme 16). The silyl enol ether (29) was isolated and indicated to be the key intermediate by using t-butyldimethylsilyl (TBDMS) instead of TMS. To these cyanoalkylation reactions, terminal alkynes worked well and provided 2-substituted (Z)-5-formyl-pent-2-enenitriles (30) in high regioselectivity and high stereoselective. Furthermore, the formyl group could perform the aldehyde reactions and provided α-functionalized acrolein (27) and hepta-2,6-dienenitrile (28) effectively.
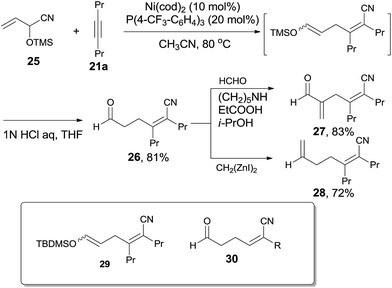 |
| | Scheme 16 Ni-catalyzed cyanoallylations with α-siloxyallyl cyanide. | |
A possible mechanism was proposed by authors (Scheme 17). Ni catalyst firstly added to C(sp3)–CN bond of allyl cyanides (23, 25) via oxidative addition, giving a π-allylnickel intermediate (A). 1,2-Migratory addition of π-allylnickel intermediate alkynes, and followed by reductive elimination, acrylonitriles (24, 29) were obtained. Here the products having the bulkier group at a cyano-substituted carbon were much favored in consideration of the steric hindrance, especially to cases of terminal alkynes in which 2-substituted (Z)-5-formyl-pent-2-enenitriles (30) were afforded in excellent regioselectivity.
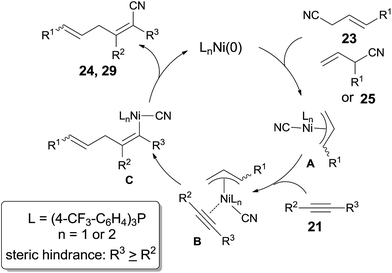 |
| | Scheme 17 Proposed mechanism for the Ni-catalyzed cyanoallylations. | |
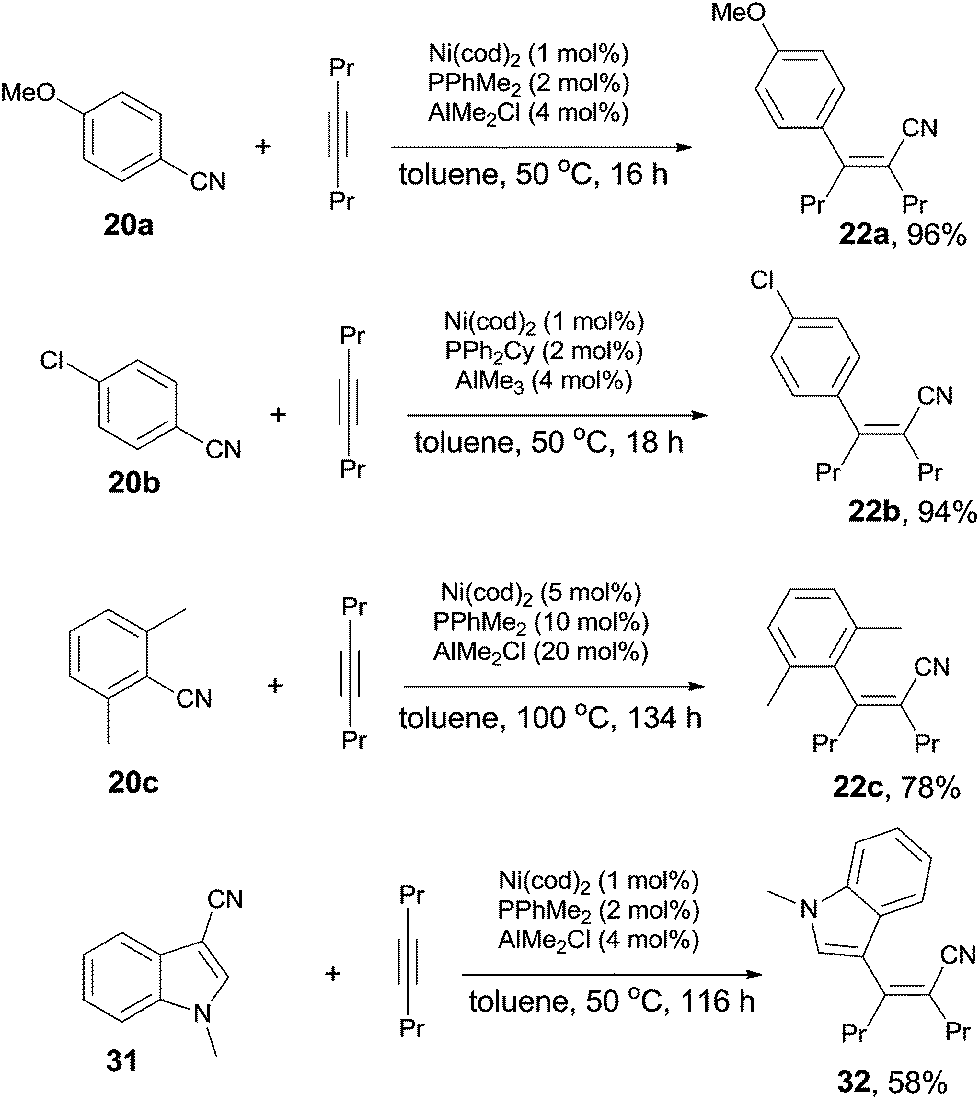
A breakthrough work was reported in 2007. Nakao and Hiyama demonstrated a Lewis-acid assisted Ni-catalyzed cyanoarylation of alkynes with nitriles, which efficiently improved the reactivities of some electron-rich aryl cyanides (20a), comparing to the previous works (Scheme 18).17a Moreover, the amount of the catalyst and the ligand was significantly decreased. By surveying the substituted group on aryl cyanides, it was indicated that selective activation of Ar–CN bond over Ar–Br/Cl was remarkable. Bromo or chloro (22b) was survived after reaction. Moreover, sterically hindered 2,6-dimethyl benzonitrile (20c) participated the reaction and the Ar-CN bond was activated if the amount of the catalyst, ligand, and Lewis acid was magnified 5 times, the reaction time was prolonged to 134 h, and the reaction temperature was raised to 100 °C. The corresponding product (22c) was obtained in 78% yield. Another interesting case was the cyanoarylation of 1-methyl-1H-indole-3-carbonitrile (31) over 21a. Activation of Ar–CN occurred prior to the activation of Ar–H at 2-position of indole in the presence of Lewis acid.18
 |
| | Scheme 18 LA-mediated Ni-catalyzed cyanoarylations. | |
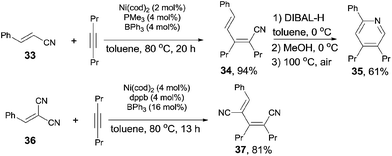
In the aid of Lewis acid, cyanoalkenylation was also successfully achieved by fine tuning ligand and Lewis acid. Thus, a series of conjugated dienenitriles were obtained in good to excellent yields (Scheme 19). Alkene configuration of trans-cinnamonitrile (33) was remained after reaction and the resulting dienenitrile (34) could be transferred into pyridine (35) by a sequence of reduction, 6π-electrocyclization, and aromatization. 2-Menzylidenemalononitrile (36) participated the reaction and provided dicyano-substituted 1,3-diene (37) in 81% yield. In this case, a selective activation of Ar–CN bond which appeared trans to phenyl group was demonstrated. Further, other cyanofunctionalizations, such as aforementioned cyanoesterification17b and cyanoamidation17c were all optimized in the aid of Lewis acid.
 |
| | Scheme 19 LA-mediated Ni-catalyzed cyanoalkenylations. | |
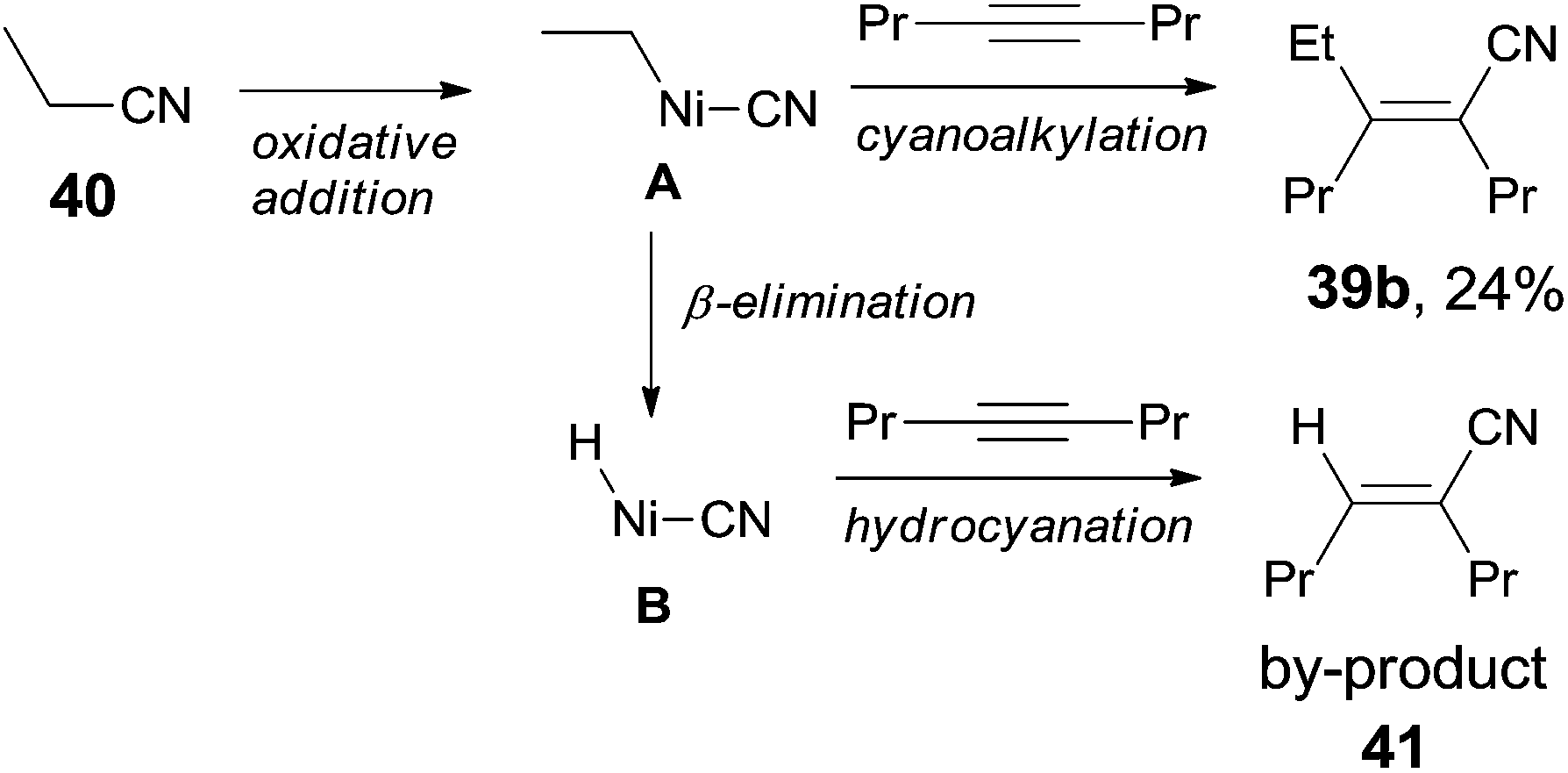
Finally, C(sp3)–CN bond in acetonitrile was activated. Reaction of acetonitrile (38) and oct-4-yne gave trisubstituted acrylonitrile (39a) in 71% yield (Scheme 20). That methyl group came from acetonitrile albeit AlMe3 was confirmed by using CH3CN-d3. Other nitriles, such as 2-(trimethylsilyl)-acetonitrile and propiononitrile, provided the corresponding addition products, but in relatively lower yields. In case of propiononitrile (40), it provided the syn-adduct (39b) in 24% yield only. One of the by-products was (E)-2-propylhex-2-enenitrile (41) (Scheme 21). Oxidative addition of Ni(0) to propiononitrile leading to the formation of ethylnickel cyanide (A), which underwent β-elimination to generate highly active species, HNiCN (B). In the presence of oct-4-yne, hydrocyanation occurred and provided the by-product 41.
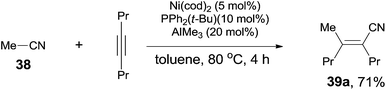 |
| | Scheme 20 LA-mediated Ni-catalyzed cyanomethylation. | |
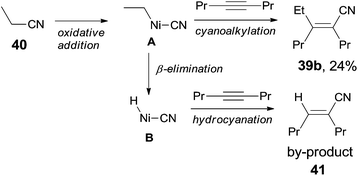 |
| | Scheme 21 Formation of the hydrocyanation byproducts in cyanoalkylations. | |
The dramatic effects of Lewis acids could be contributed to the coordination between R–CN and Lewis acid which accelerated the rate of oxidative addition of Ni(0) to R–CN bond (Scheme 22). Regioselectivity was ascribed to the repulsion between the bulkier R2 and R when the unsymmetrical alkyne and nickel species coordinated. Ultimately, the bulkier R2 group and cyano group (CN) bonded to the same carbon of alkynes. Moreover, these reactions occurred in exclusive syn-addition manner, the trans-adducts came from the isomerism of syn-adducts in the presence of Lewis acid or ligand, or under heat.
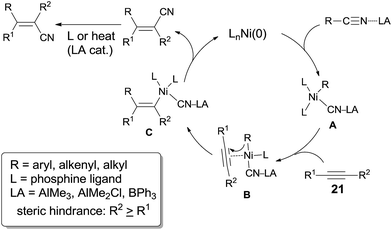 |
| | Scheme 22 Proposed mechanism for the LA-mediated Ni-catalyzed cyanoalkylation. | |
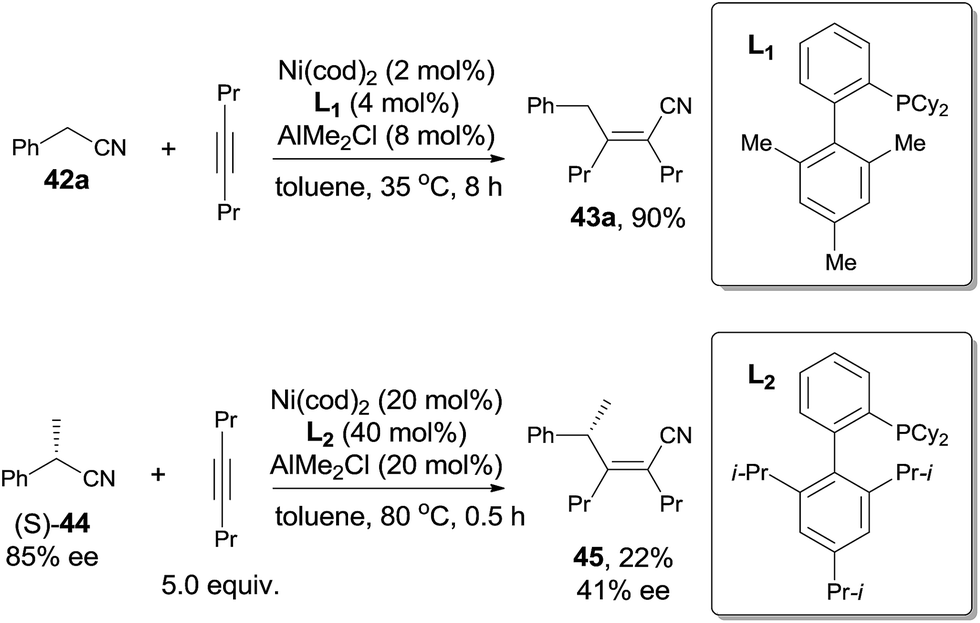
Short after, Nakao and Hiyama reported cyanoalkylation of alkynes with arylacetonitriles (42), also catalyzed by nickel and Lewis acid (Scheme 23).19 Various arylacetonitriles could undertake this reaction in good yields, including some heteroaryl derivatives. As arylacetonitriles, β-elimination resulted from the normal alkanenitriles, such as aforementioned byproduct (41), could be effectively diminished. To the symmetrical alkynes, addition occurred in an exclusive syn-fashion. However, to the unsymmetrical alkynes, poor regioselectivities were observed except for the cases where the steric hindrance could be differentiated, such as methyl versus tert-butyl, methyl versus TMS. The bulkier group always linked to carbon with CN attached in the final.
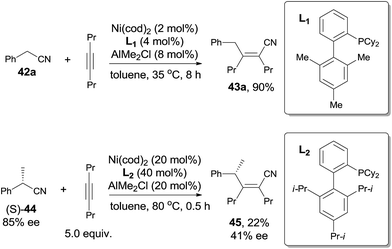 |
| | Scheme 23 LA-mediated Ni-catalyzed cyanoalkylation of alkynes with arylacetonitriles. | |
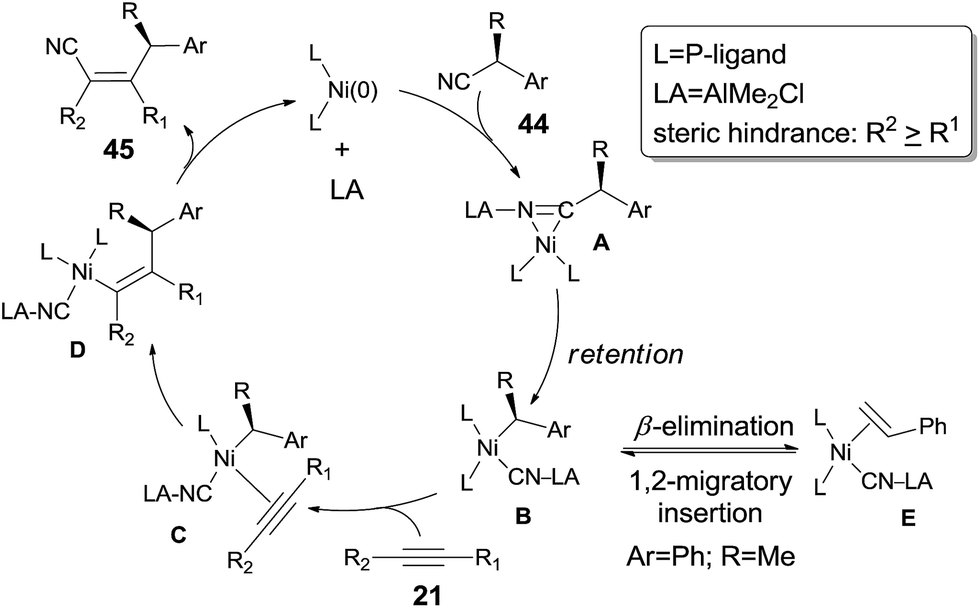
To make a clear outcome of the mechanism, the authors selected optically active (S)-α-phenylpropionitrile (44) as the substrate and detected all the byproducts. It was worth noting that the recovered starting materials showed 80% ee and the product did not undertake racemization under their conditions. These facts indicated that the absolute configuration of the chiral carbon retained after these sequential steps, including the oxidative addition, 1,2-migratory insertion, and reductive elimination. The partial loss of the optical purity could be ascribed to the equilibrium between the β-elimination and its reversed 1,2-migratory insertion as illustrated in Scheme 24.
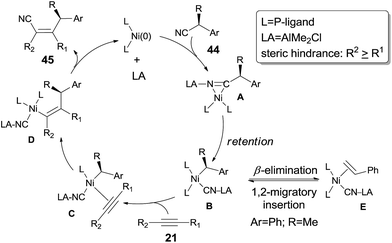 |
| | Scheme 24 Retention of configuration in LA-mediated Ni-catalyzed cyanoalkylation. | |
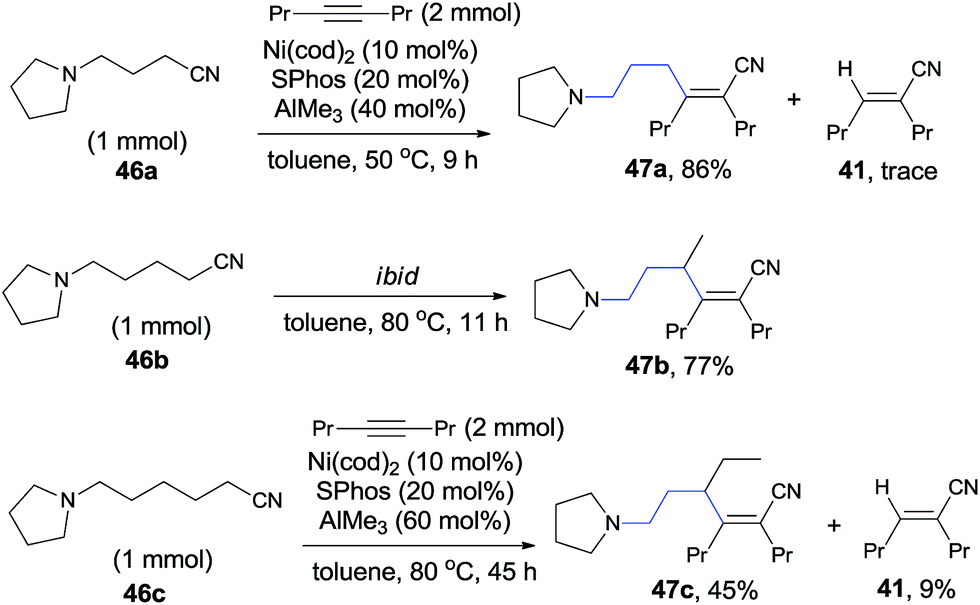
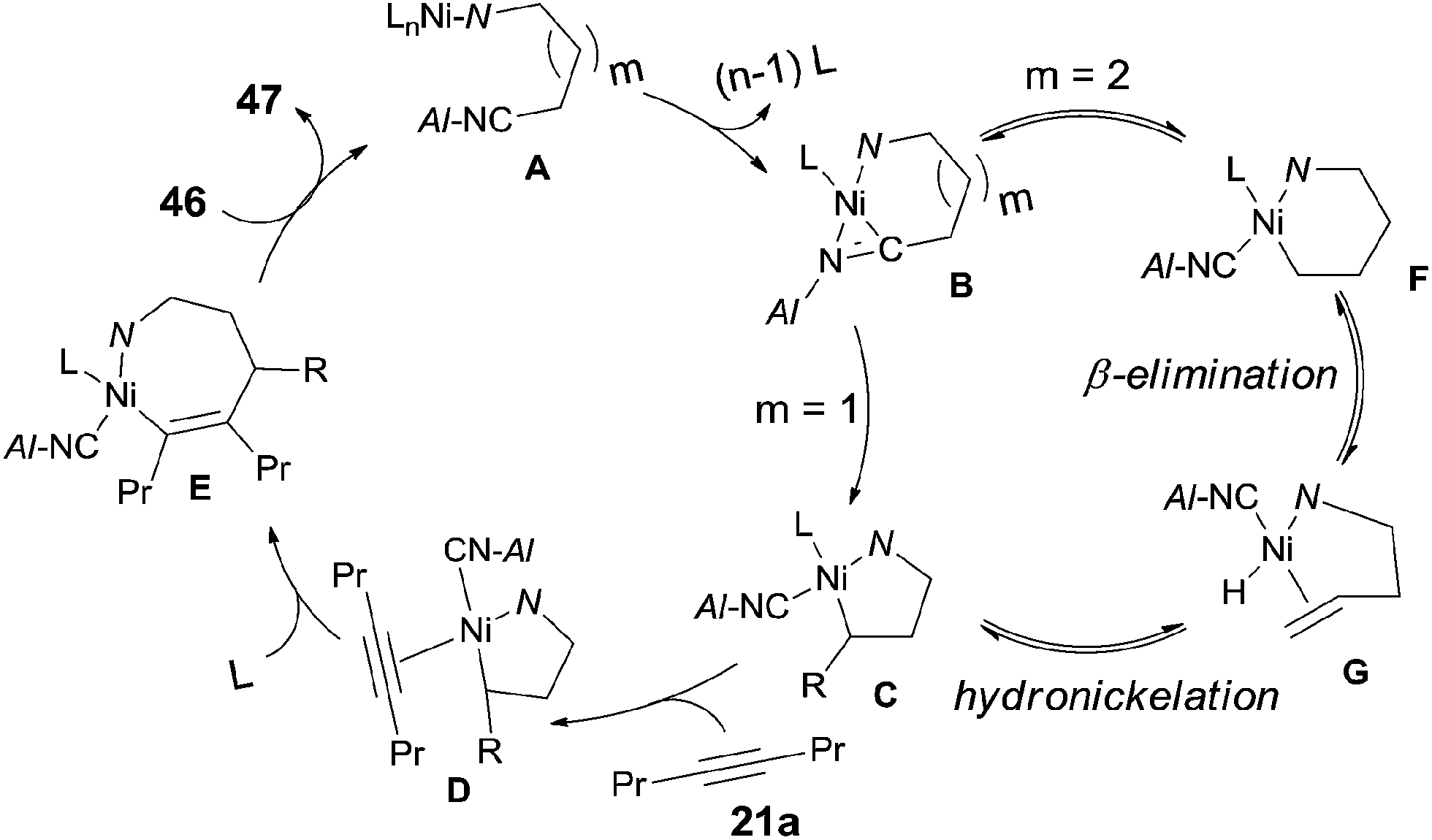
In 2010, Nakao and Hiyama reported heteroatom-directed cyanoalkylation of alkynes (Scheme 25).20 The heteroatoms, like nitrogen atom, played a significant role in suppressing the aforementioned unexpected β-hydride elimination via the intramolecular chelating of nitrogen with nickel center. The amino effect at the γ-position of butyronitrile (46a) was impressive. When the amino was on the β-position of propiononitrile, only trace amount of the syn-adduct was detected under the same reaction conditions. It was also noticeable that when the amino group was on δ-position of pentanenitrile, with one more methylene unit between pyrrolidine and cyano (46b), cyanoalkylation occurred, but on the γ-position of the pyrrolidyl group. This selectivity was also observed in case where 5-pyrrolidyl hexanenitrile (46c) was used. In this case, a longer reaction time was required and the hydrocyanation by-product (41) was isolated in 9%.
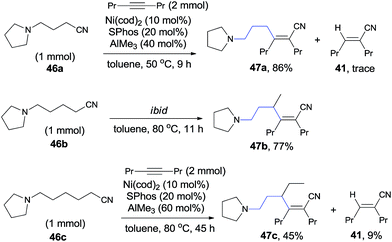 |
| | Scheme 25 Heteroatom-directed cyanoalkylation of alkyne. | |
The above experiments indicated that a 5-membered azanickelacycle (C) was the key intermediate (Scheme 26). With one more CH2, a 6-membered azanickelacycle (F) (or even 7-membered in case with two more CH2) could be converted to 5-membered azanickelacycle (C) by a sequence of β-hydrogen elimination and hydronickelation. This proposed mechanism properly explained the fact that the amino-substituted butyronitrile, valeronitrile and hexanenitrile could undertake cyanoalkylation smoothly. Similar to the above mentioned cases, the syn-adducts formed having a bulkier group and CN bonded to the same sp2-carbon when unsymmetrical alkynes were used.
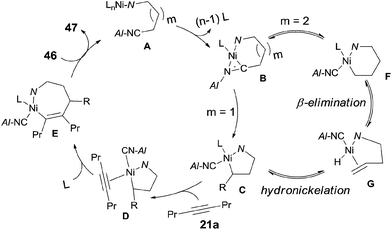 |
| | Scheme 26 Proposed mechanism for the heteroatom-directed cyanoalkylation of alkyne. | |
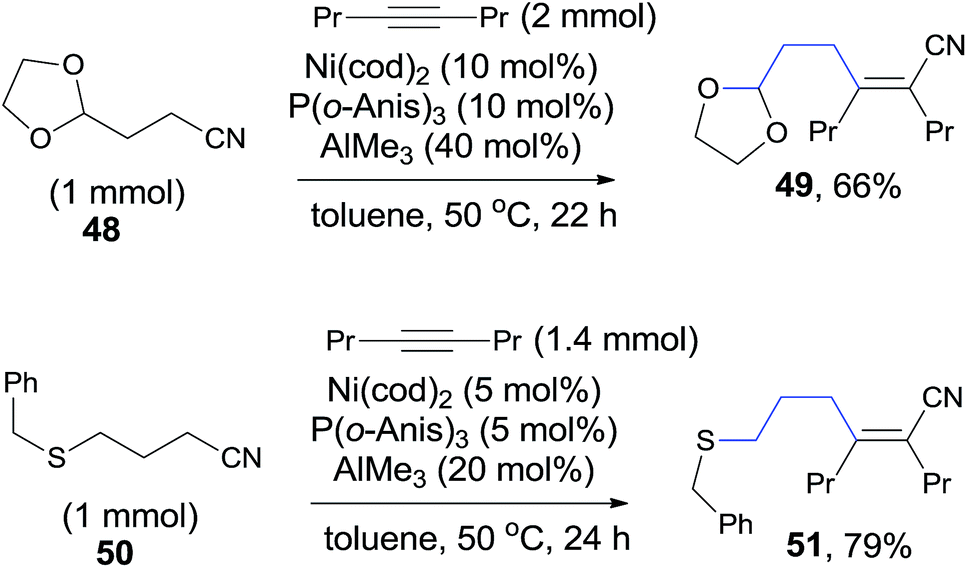
Other heteroatom (O, S) in the appropriate position of alkanenitrile (48, 50) could also function as the directing heteroatom via its coordination to nickel center and afford the corresponding cyanoalkylation products (49, 51) (Scheme 27).
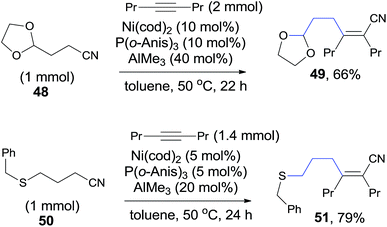 |
| | Scheme 27 Examples for O, S-directed cyanoalkylation of alkyne. | |
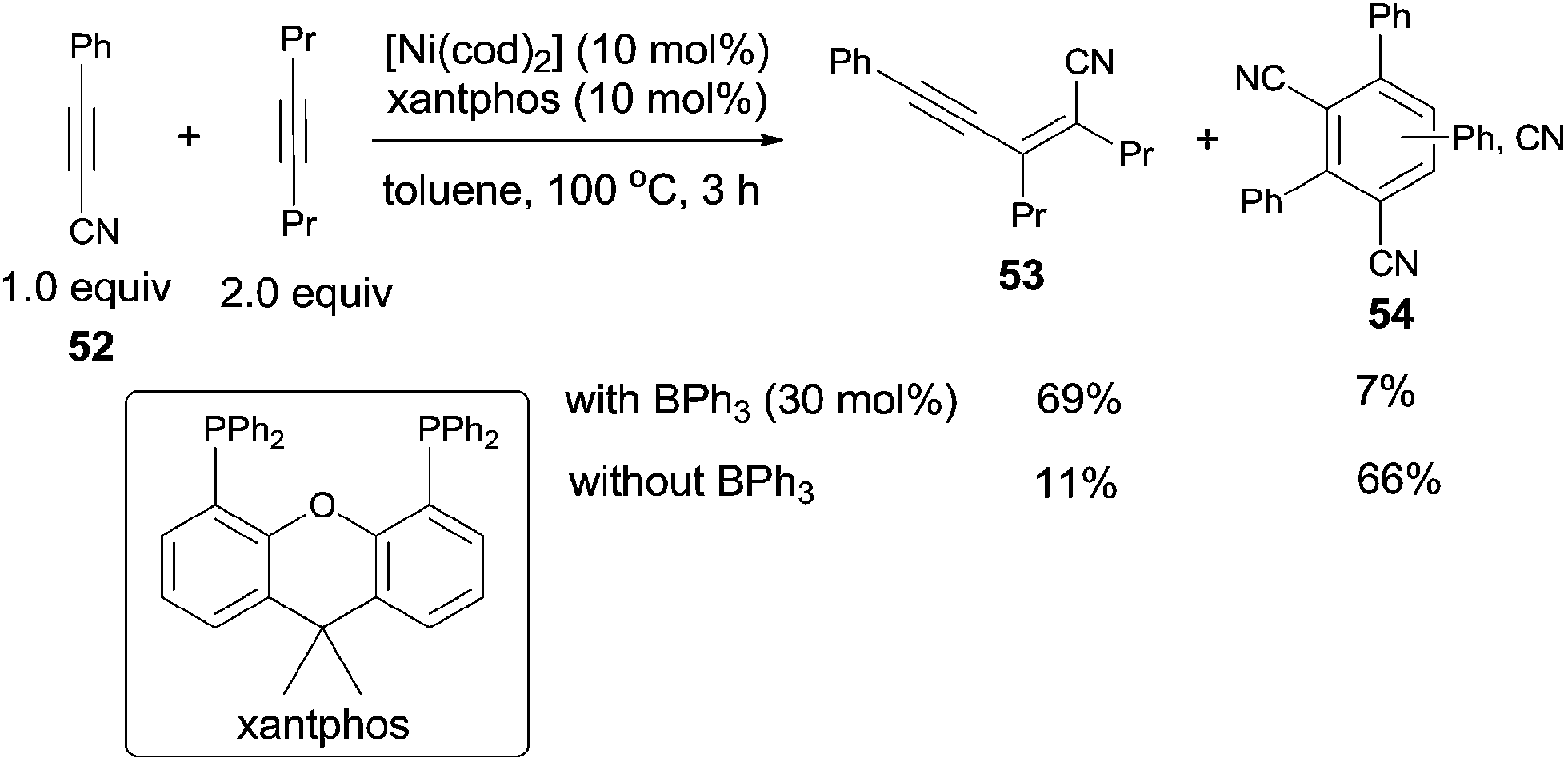
Owning to homo- and/or cross-cyclotrimerization in nickel catalyst system, it was uneasy for alkynyl cyanide (52) to undertake the catalytic insertion of Ni(0) to C(sp)–CN bond. Nevertheless, Nakao and Hiyama reported nickel/BPh3-catalyzed cyanoalkynylation of alkynes, accessing to conjugated enyne structures (53) (Scheme 28).21 With the aid of Lewis acid, the trimerization products (54) were effectively inhibited.
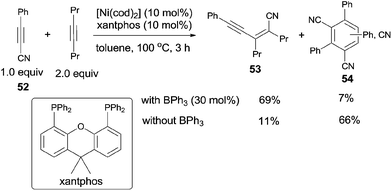 |
| | Scheme 28 BPh3-mediated Ni-catalyzed cyanoalknylation of alkyne. | |
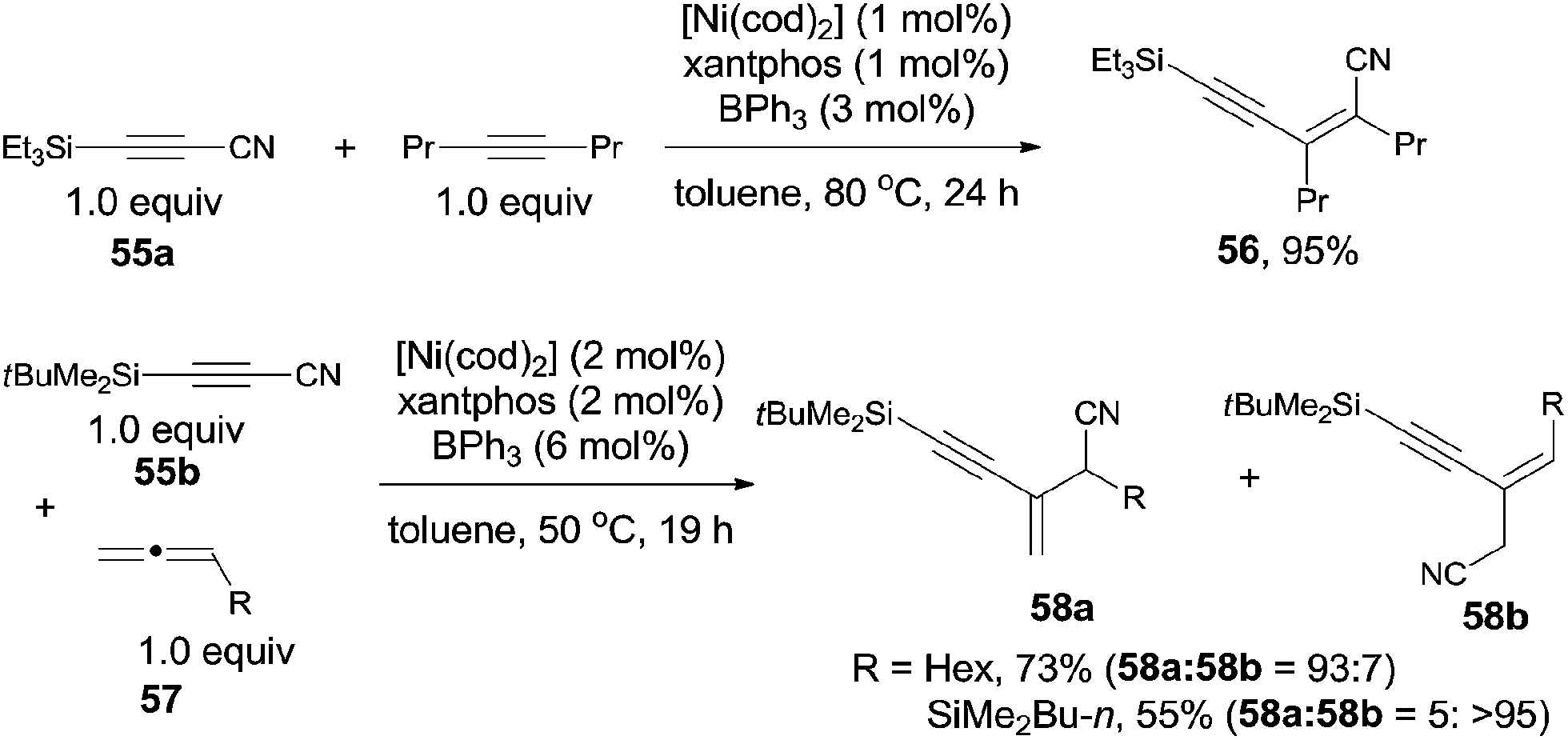
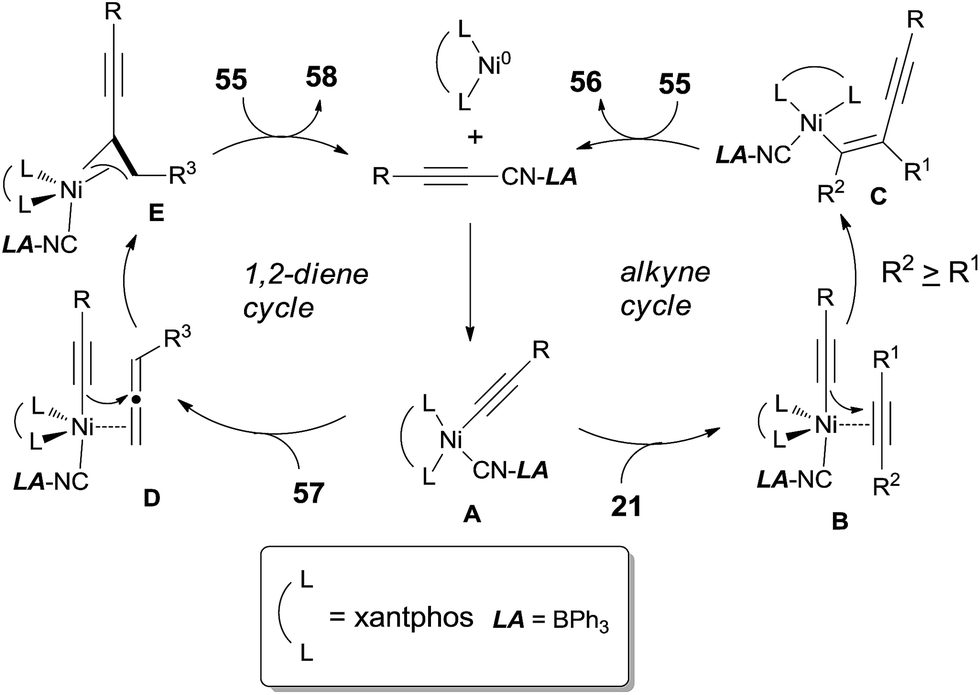
After investigating the substrate scope, they found that bulky silyl substituented alkynyl cyanides (55) might suppress the homo- and/or cross-cyclotrimerization of the nitriles, resulting in a lower requirement of catalyst amounts and approached excellent yields of 56 (Scheme 29). Diynyl cyanides were also suitable for this reaction. 1,2-Dienes (57) reacted with alkynyl cyanides (55) under the same conditions, giving conjugated enynes (58) in moderate to good yields. The regioselectivity was largely depending upon the substituted group attached on 1,2-diene.
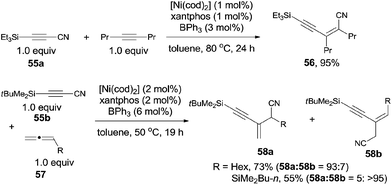 |
| | Scheme 29 Cyanoalknylation of alkyne or 1,2-diene with silyl substituented alkynyl cyanides. | |
The oxidative insertion of C(sp)–CN bond with Ni(0) catalyst assisted by BPh3 was thought to be the initial step in the proposed mechanism to form key intermediate A (Scheme 30). Followed by the coordination with alkynes or 1,2-dienes, respectively, complexes B and D were formed. In case of the alkyne as substrate, the alkynyl group migrated from the nickel center to the less hindered carbon of the coordinated alkyne to generate C. In case of 1,2-diene as substrate, the alkynyl migrated from nickel center to the central carbon of 1,2-diene to generate the allylnickel species E. Finally, the reductive elimination of C or E took place and provided the addition products, 56 and 58 respectively, with both high regioselectivity and high stereoselectivity.
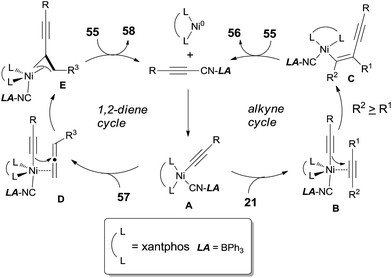 |
| | Scheme 30 Proposed mechanism for cyanoalknylation of alkyne or 1,2-diene. | |
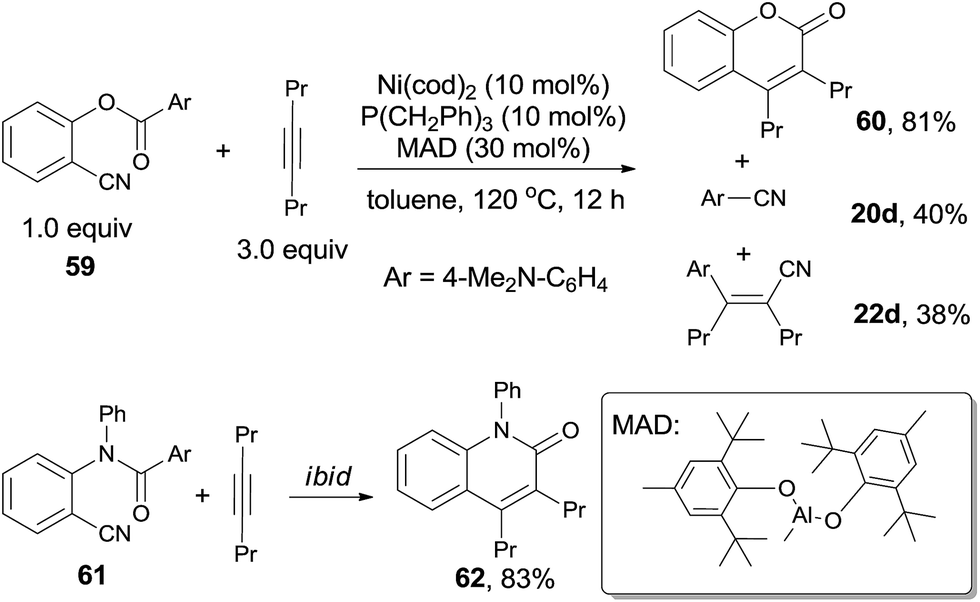
In 2011, Kurahashi and Matsubara reported an intermolecular cycloaddition of o-arylcarboxy benzonitriles (59) and alkynes (21) through Ni-catalyzed cleavage of C–CN and C–CO bond, affording coumarins (60) in good yields (Scheme 31).22 It was worth noting that aryl cyanide (20d) was isolated, as well as the compound (22d) formed from its carbocyanation with alkyne. Similarly, o-arylamide-substituted benzonitrile (61) could also undertake this reaction with oct-4-yne, which gave the quinolinone (62) in 83% yield.23
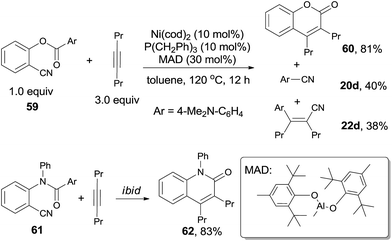 |
| | Scheme 31 Ni-catalyzed cycloadditions. | |
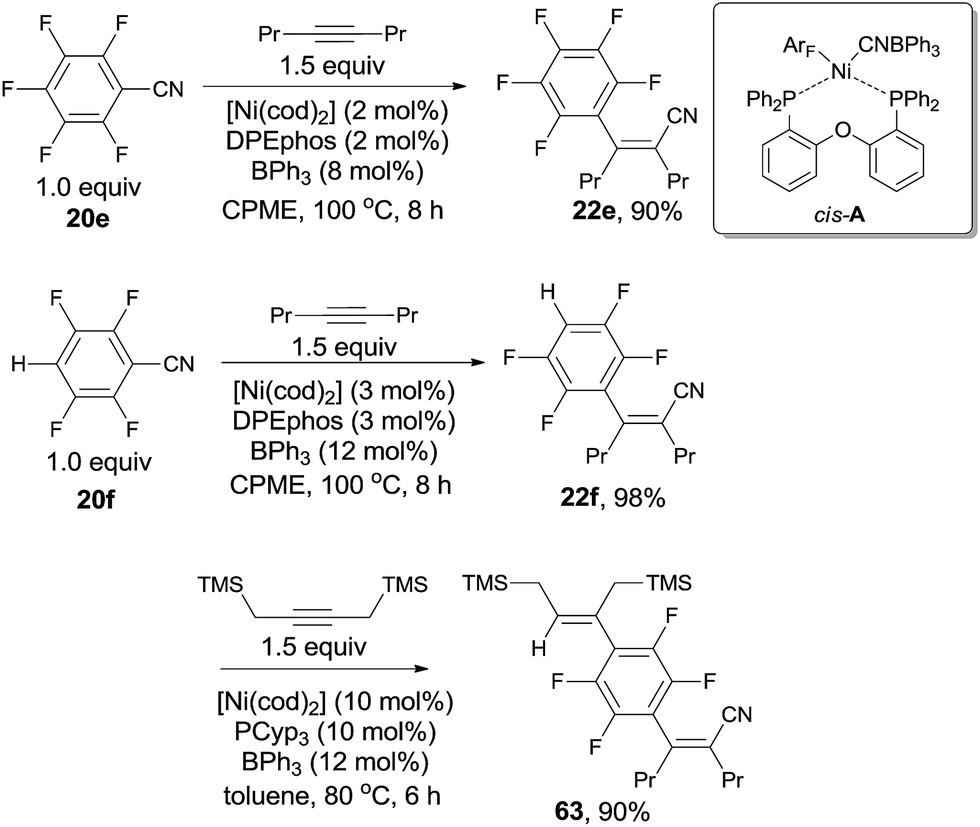
In 2013, Nakao and Hiyama chose polyfluorobenzonitriles (20e, 20f) to conduct cyanoarenylation with alkynes, affording a series of corresponding polyfluoroarylcyanation products (22e, 22f) by applying Ni(cod)2/DPEphos/BPh3 catalytic system in the solvent of cyclopentylmethylether (CPME) (Scheme 32).24 Under their reaction conditions, C–CN bond activation was prior to C–H and C–F bond activation, so the resulting cyanoarenylation products could be obtained smoothly without the interference with the byproducts derived from C–H and C–F bond activations. Of note, the existence of fluorine atoms on the arenes promoted the C–CN bond activation, driving polyfluorobenzonitriles more reactive than non-fluorine substituted aryl nitrile which was demonstrated by competition experiment between pentafluorobenzonitrile and benzonitrile. Moreover, the dramatic effect of BPh3 and DPEphos was clearly illustrated by the single crystal structure of the nickel insertion intermediate (cis-A) which was responsible for the subsequent 1,2-migratory insertion of ArF–Ni–CN on alkyne. Combination of Ni(cod)2, DPEphos, and BPh3 in CPME was also used for cyanoarenylation of terminal alkynes and alkenes. As an interesting example, selective activation of C–H bond albeit C–CN bond was achieved by using PCyp3 as ligand. Thus, compound (63) was obtained from 22f in 90% yield with high chemoselectivity.
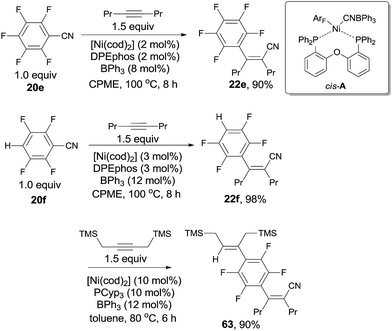 |
| | Scheme 32 Ni-catalyzed chemoselective C–CN bond activation over C–H/C–F bond activation. | |
2.5 Stereoselective cyanoarenylation
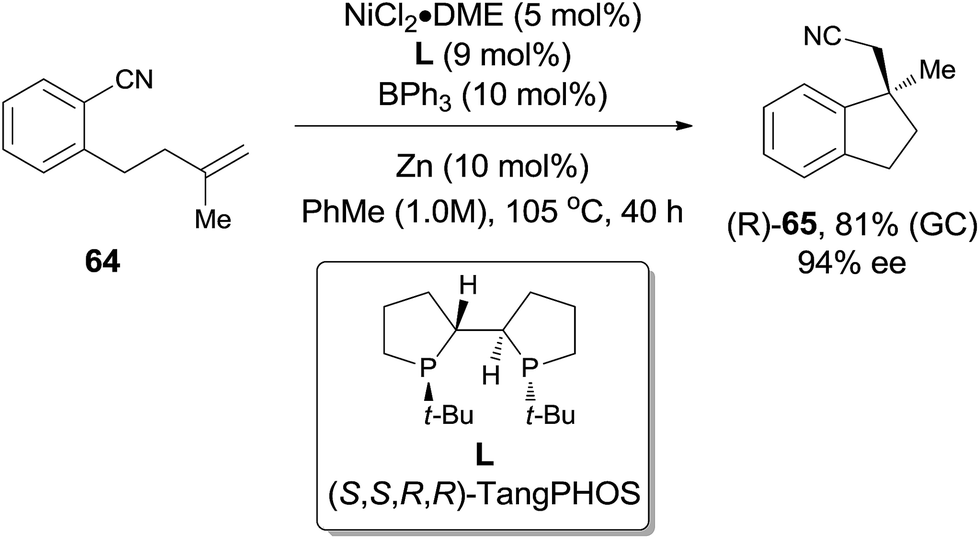
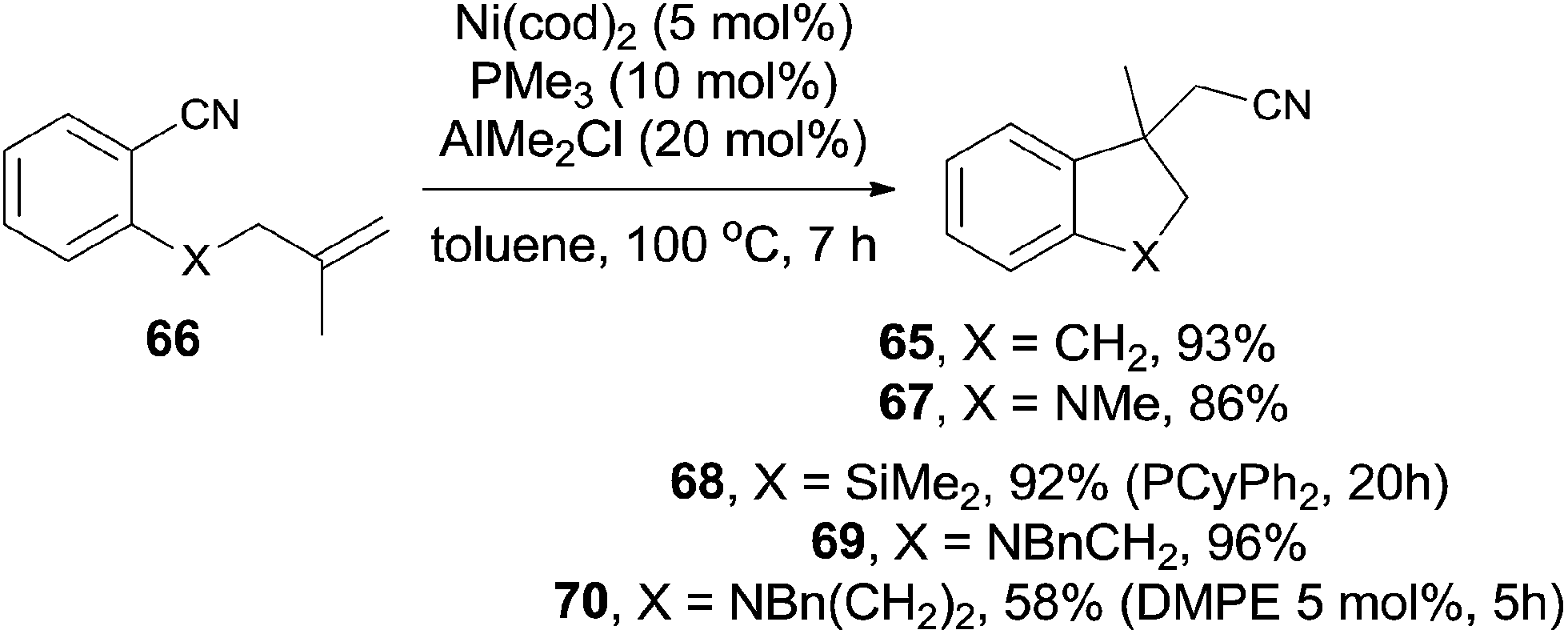
In comparison with above mentioned cyanofunctionalization of alkynes, cyanoarenylation of alkenes was seldom reported. In 2008, Jacobsen reported an enantioselectively intramolecular cyanoarenylation of unactivated olefins through Ni-catalyzed C(sp2)–CN bond activation, providing indane derivatives with a quaternary chiral center in good yield (Scheme 33).25 By screening the conditions and selecting a proper chiral ligand, the indane product (65) was obtained in high enantioselectivity. Combination of NiCl2·DME and Zn was used as the Ni(0) source, while the olefin isomerism was minimized by increasing the ratio of TangPHOS to Ni.
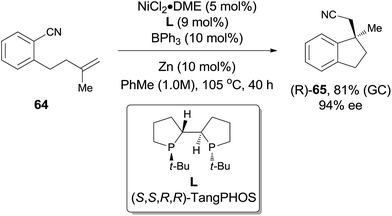 |
| | Scheme 33 Asymmetric intramolecular cyanoarenylation of inactivated olefin. | |
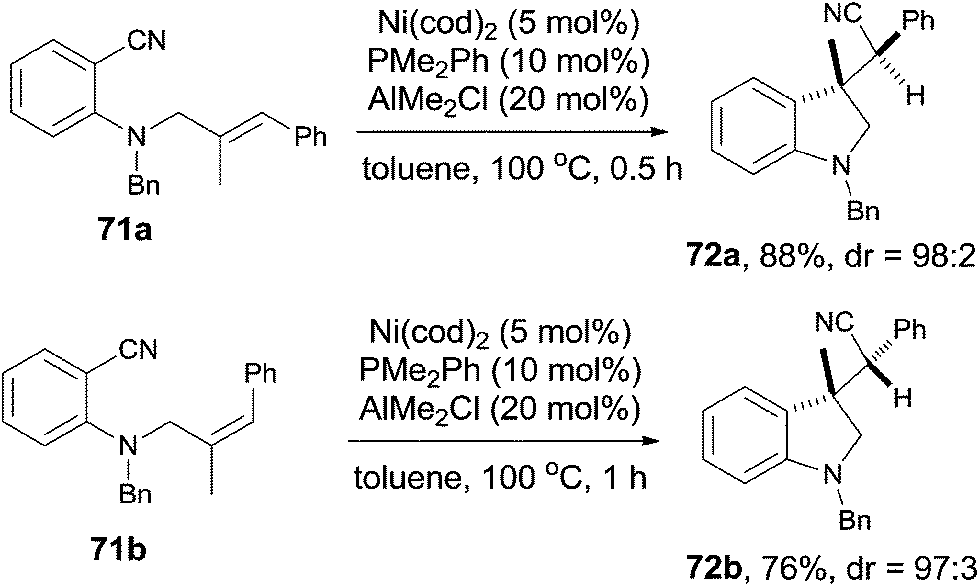
Meanwhile, Nakao and Hiyama also reported the intramolecular cyanoarenylation of various specially designed alkenes with Ni(cod)2/AlMe2Cl catalyst. A number of substrates tolerated the reaction conditions (Scheme 34).26 Amino and silyl tethers provided the corresponding cyclized products 67 and 68 in 86% and 92% yields, respectively, by choosing proper ligand. Insertion of olefin into Ar–CN bond was found to be in a 5-exo-trig fashion. The new constructed ring could be enlarged to six- or seven-membered ring, as indicated by the representative compounds 69 and 70. The cyanoarenylation reaction proceeded in a syn-addition manner which was clearly shown by the diastereoselective preparation of compounds 72a and 72b (Scheme 35).
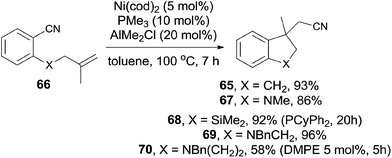 |
| | Scheme 34 Substrate diversity for the Ni-catalyzed intramolecular cyanoarenylation. | |
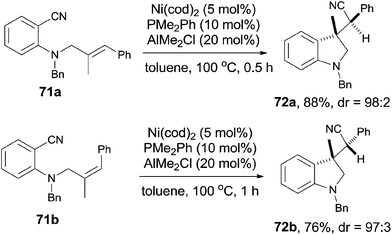 |
| | Scheme 35 Diastereoselective preparation of 72. | |
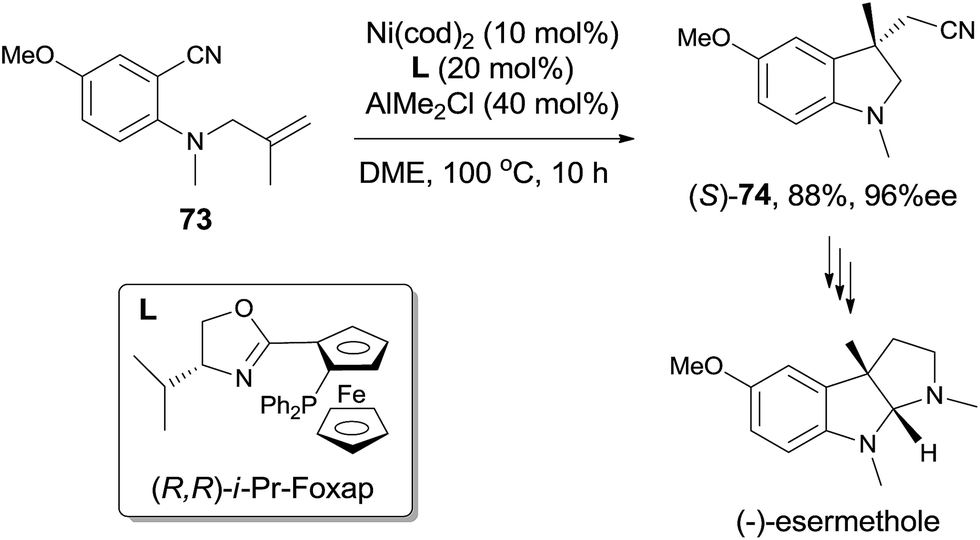
Not only the reaction could be conducted in high diastereoselectivity, but also in high enantioselectivity by using a chiral ligand, such as (R,R)-i-Pr-Foxap (Scheme 36). This asymmetric synthesis had been successfully applied in the preparation of (−)-esermethole.
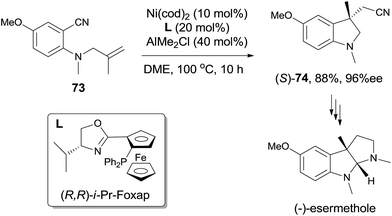 |
| | Scheme 36 Enantioselective preparation of 74. | |
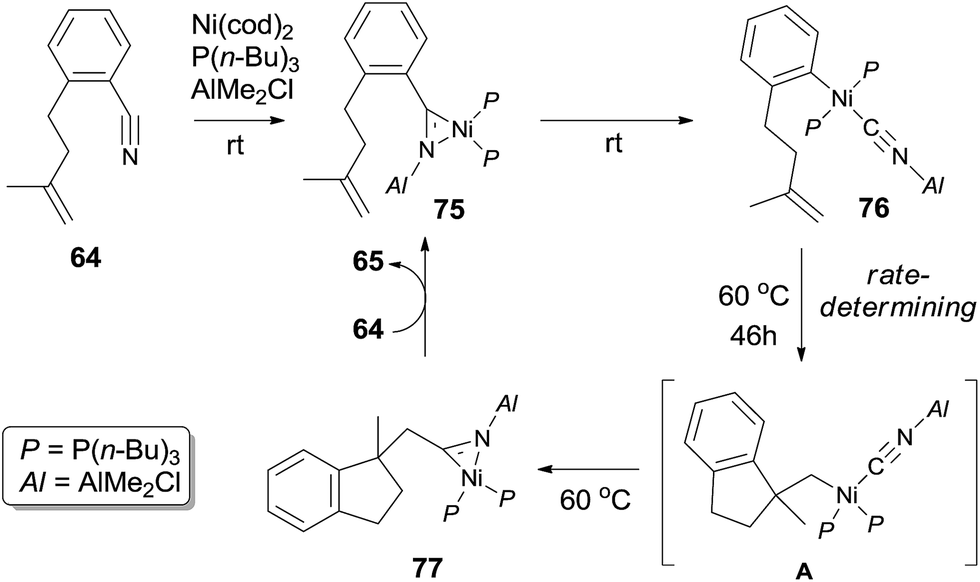
Mechanism of this intramolecular cyanoarenylation was proposed based on the isolation of key intermediates 75 and 76 (Scheme 37). Structures 75 and 76 were established by their single crystal analysis. Reaction of stoichiometric 64, Ni(cod)2, P(n-Bu)3, and AlMe2Cl at room temperature immediately provided η2-nitrile complex 75. Existence of Lewis acid facilitated the oxidative addition of Ni(0) to Ar–CN bond and afforded complex 76 after 6 h. 1,2-Migratory insertion onto intramolecular olefin provided η2-nitrile complex 77 via intermediate A. Finally, cyclized product was obtained via ligand exchange.
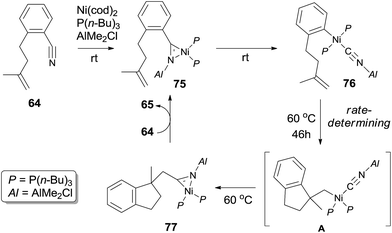 |
| | Scheme 37 Proposed mechanism of the intramolecular cyanoarenylation of olefin. | |
Above all, a number of cyanofunctionalizations on unsaturated alkynes, alkenes, 1,2-dienes were disclosed, including cyanoeserification, cyanoamidation, cyanoacylation, cyanoarenylation, cyanoalkenylation, cyanoalkylation, and cyanoalkynylation based on the carbon type bonded to CN group in the substrates. All of these reactions were of great synthetic value because they simultaneously installed two groups into one molecule with 100% atom efficiency. More noteworthy, reactions proceeded in syn-addition fashion with high stereoselectivity.
3. Cross coupling reactions
R part in nitriles (R–CN) can undertake cross coupling reactions with other components, in which CN group is regarded as a leaving group. In comparison with the typical substrates (R–X or R–OTs) for cross coupling reactions in making C–Si, C–P, C–B, C–H, as well as C–C bonds, nitrile (R–CN) provides a unique and complementary way in the preparation of functionalized compounds.
3.1 Decyanative silylation
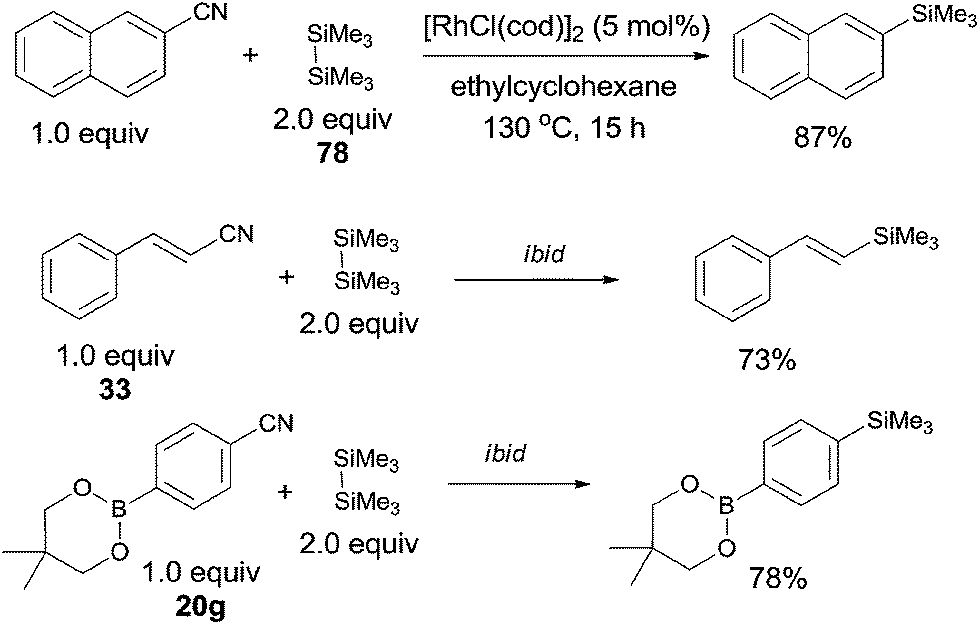
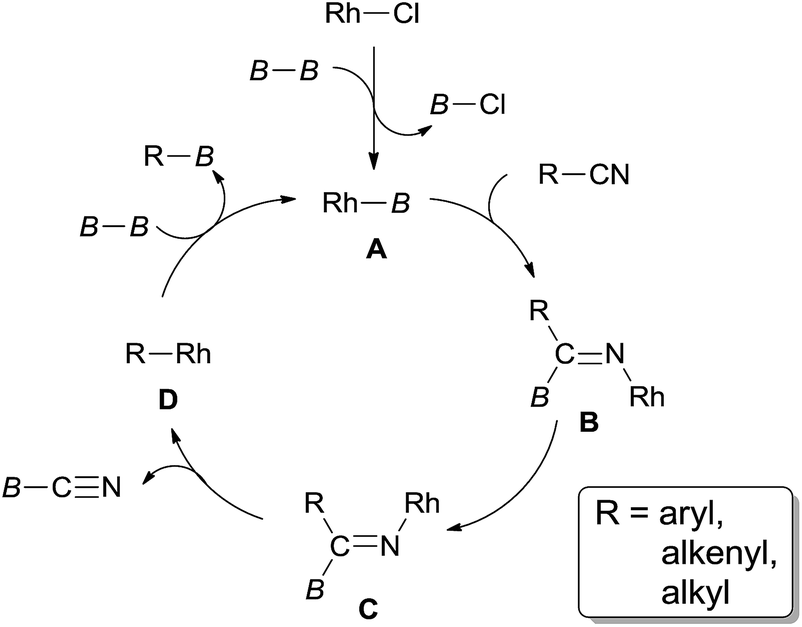
In 2006, Chatani reported a Rh(I)-catalyzed cleavage of C–CN bond and Si–Si bond, achieving the decyanative silylation of aryl and alkenyl nitriles (Scheme 38).27a They chose [RhCl(cod)]2 as the catalyst, and hexamethyldisilane (78) as the silylation agent which also functioned as the auxiliary reagent in the helping of C–CN bond cleavage. In the substrate scope, dimethylphenylsilyl and benzyldimethylsilyl groups could also be introduced under the reaction conditions established for hexamethyldisilane, although with moderate yields. Steric hindrance might influence the reaction rate and reduce the yields. Heteroaryl nitriles and alkenyl nitriles were both good substrates for this transformation by selecting suitable rhodium catalyst. It was also noteworthy that the boryl group (20g) survived after reaction which opened a window for subsequently selective cross coupling to build higher π-conjugated compounds. Although fluorine on the aryl nitriles survived after the reaction, C–Cl and C–Br bond were found to be more reactive than C–CN bond.
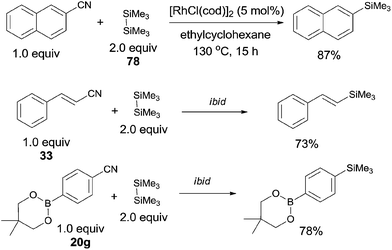 |
| | Scheme 38 Rh-catalyzed Si-assisted decyanative silylation of aryl nitriles. | |
The decyanative silylation was well extended to allyl cyanides. Thus, allyl cyanide (23a) furnished allyl silane (79) in 59% yield (Scheme 39). A relative lower reaction temperature was applied for this transformation due to the relative facile cleavage of C–CN bond and the formation of the stable π-allyl rhodium complex.28 However, when benzyl cyanide (42a) was used as the substrate, a higher reaction temperature was applied for complete conversion of benzyl cyanide. In this case, enamine formation (81) was isolated in a certain amount. The enamine byproduct (84) became dominant when 2,2-diphenylacetonitrile (82) was used.
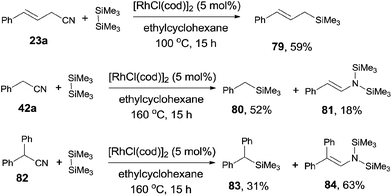 |
| | Scheme 39 Rh-catalyzed Si-assisted decyanative silylation of allyl/benzyl nitriles. | |
It was also noteworthy that even the unactivated alkyl cyanide could be used for the decyanative silylation when triisopropyl phosphite was added (Scheme 40). Thus, 4-phenylbutanenitrile (85) provided corresponding silylated product (86) in 48% yield with 34% recovery of the starting material. In this case, the byproduct resulting from the β-hydrogen elimination of the possible alkyl metal intermediate was not observed.
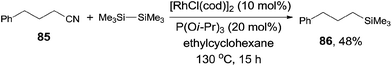 |
| | Scheme 40 Rh-catalyzed Si-assisted decyanative silylation of alkyl nitrile. | |
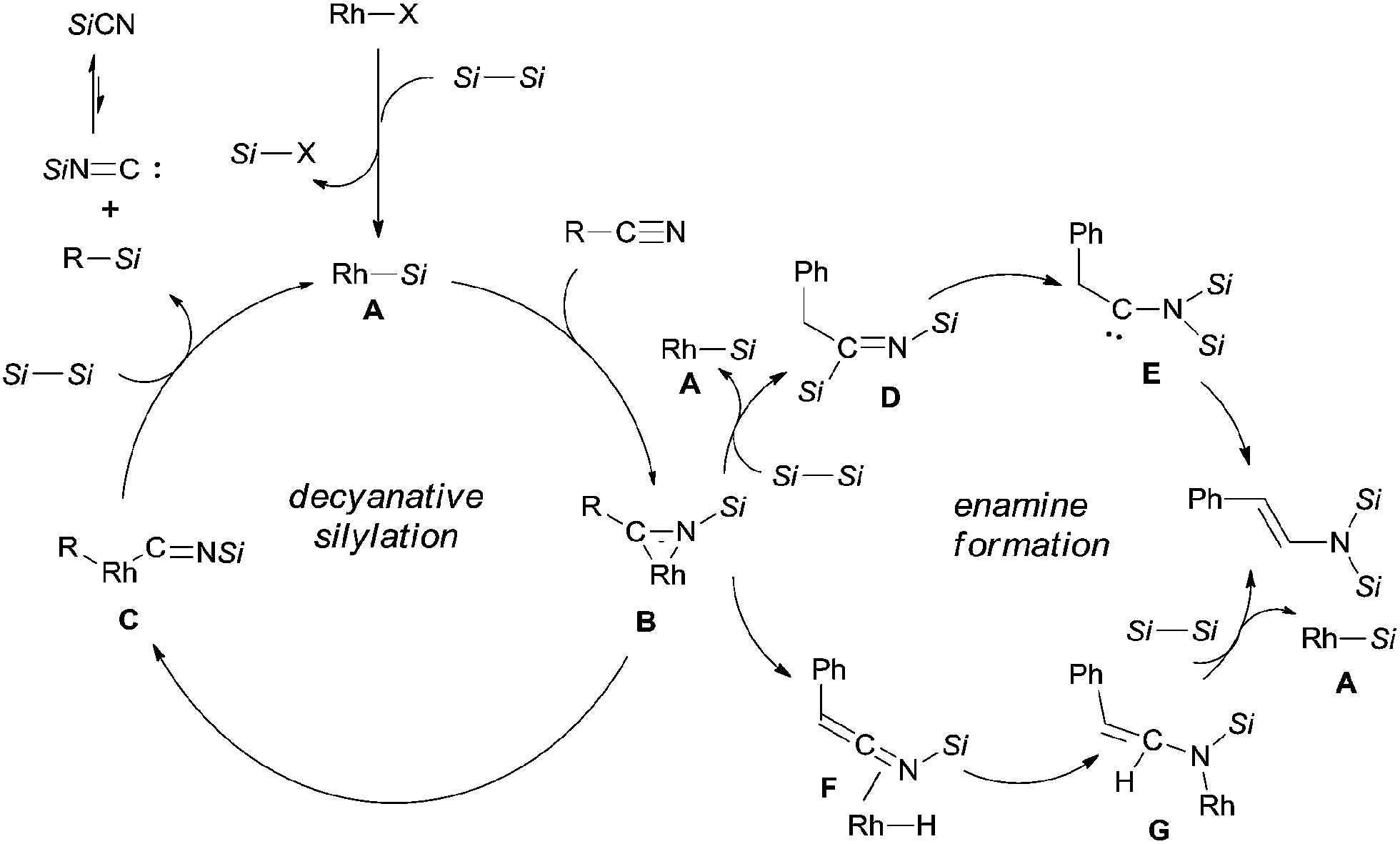
In their proposed catalytic cycle (Scheme 41), silylrhodium species (A) was generated via σ-bond metathesis between Rh–X and Si–Si bonds. Alternatively, A could be formed via an oxidative/elimination process. Migration of silyl of Rh–Si (A) to the nitrogen atom of nitrile generated η2-iminoacyl complex (B) as the key intermediate in this catalytic cycle.27b,c Subsequent migration of aryl/alkyl to rhodium center provided aryl/alkyl rhodium species (C) coordinated with silyl isocyanide. In the presence of disilane (75), σ-bond metathesis between C and disilane (75) gave rise to the silylated product (R–Si) and regenerated Rh–Si (A) for completing the catalytic cycle. Meanwhile, silyl isocyanide was excluded and isomerized into thermodynamically stable silyl cyanide in the final. When benzyl cyanide (42a) was used, two possible ways leading to the formation of enamine (81) was proposed. σ-Bond metathesis between B and disilane (75) produced D and regenerated A. Intramolecular migration of silyl from imino carbon to nitrogen via aza-Brook-type rearrangement formed aminocarbene (E). Through this path, enamine was finally obtained via the carbene insertion to adjacent C–H bond. Alternatively, β-hydrogen elimination of B, and followed by hydrorhodation across CN bond of N-silyl ketenimine (F) generated G. As a result of σ-bond metathesis between G and disilane (75), enamine (81) was afforded in a certain amount.
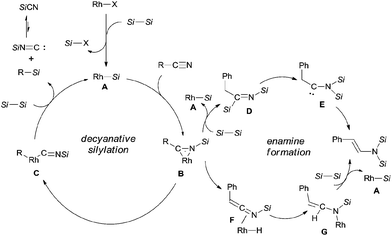 |
| | Scheme 41 Proposed mechanism for Rh-catalyzed Si-assisted decyanative silylations. | |
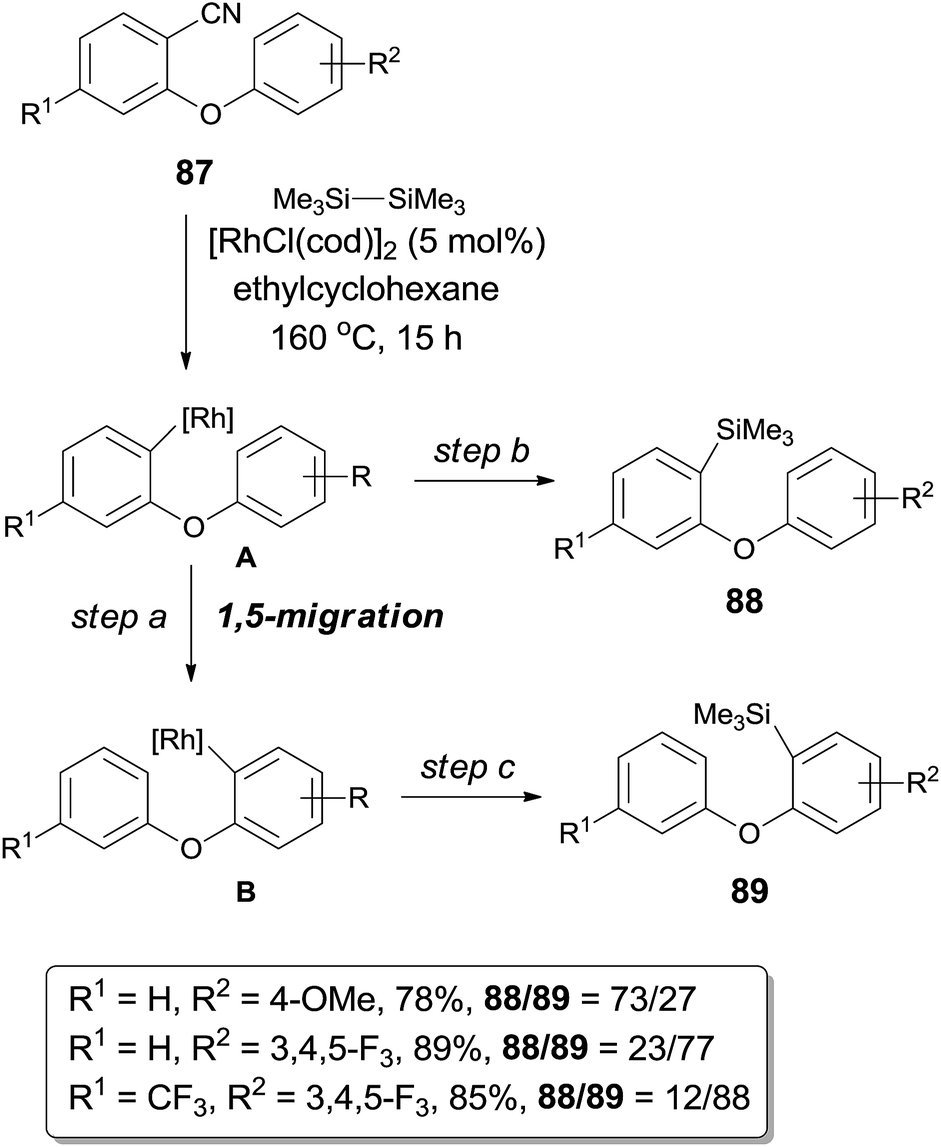
Further investigation of decyanative silylation revealed that 1,5-rhodium migration step might be involved after the Rh-catalyzed C–CN bond cleavage if the substrates (87) were rationally designed (Scheme 42).29 Thus a mixture of decyanative silyated isomers (88 and 89) was obtained after the silylation occurred on aryl rhodium species (A and B) before and after 1,5-migration. Product ratio was highly dependent on the acidity of Ar–H which was putatively to be activated. With aryl possessing the electron donating group (R2 = OMe), the direct decyanative silylation product was dominant. In a contrast, with aryl bearing the electron withdrawing group (3,4,5-trifluorophenyl), the 1,5-migrated product was isolated as a major product. 1,5-Mgiration was indicated to be irreversible by conducting a comparative experiment and the rate of 1,5-migration resulted in the product distribution. Migration distance between the metal center and the Ar–H was suitable to form the six-membered metallacyclic intermediate (or transition state), no 1,4-, 1,6- or 1,7-migration was observed. Similar 1,5-migration was observed in case when diboron was used instead of disilane in the decyanative borylation.
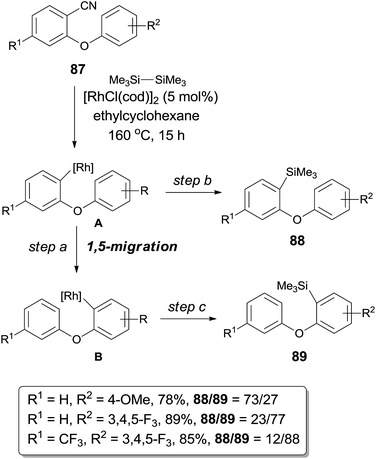 |
| | Scheme 42 1,5-Rhodium migration involved in decyanative silylation. | |
3.2 Decyanative phosphination
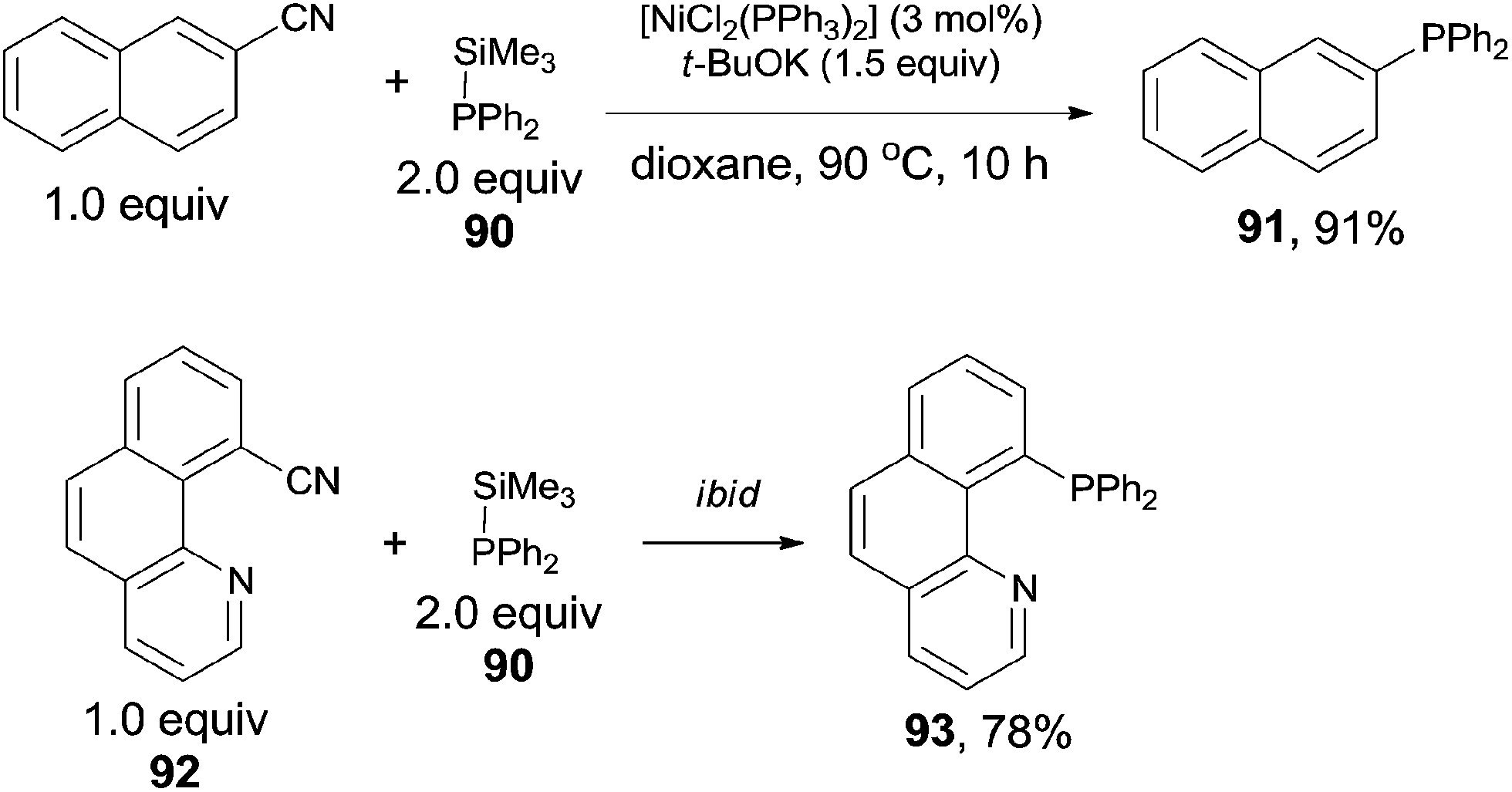
In 2011, Yang reported the phosphination of aryl nitriles with phosphate sources (90) by the Ni-catalyzed C–CN bond activation followed by C–P coupling, obtaining various phosphorus ligands (Scheme 43).30 A series of rhodium catalysts failed to catalyze this decyanative phosphination, while NiCl2(PPh3)2 and NiCl2(PCy3)2 were found to be effective. t-BuOK was crucial to this transformation. Aryl part of aryl nitriles could be transferred into the corresponding aromatic diphenylphosphines (91 and 93) in good yields, except several steric hindered aryl nitriles and pyridinyl nitriles which were obtained in relatively lower yields.
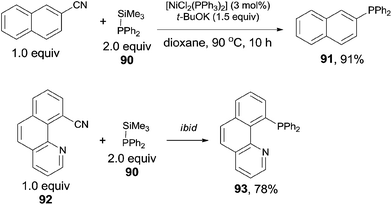 |
| | Scheme 43 Ni-catalyzed decyanative phosphination of aryl nitriles. | |
In order to clarify the mechanism, a controlled experiment was conducted (Scheme 44). By using hexamethyldisilane or tetraphenyldiphosphine instead of Me3Si–PPh2 under the same reaction conditions, the former reaction did not occur, while the later one furnished corresponding phosphinated product in 45% yield. Thus, the possible Si-assisted C–CN bond cleavage in the presence of nickel catalyst was excluded.
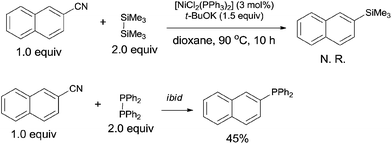 |
| | Scheme 44 Controlled experiments. | |
A plausible mechanism was proposed (Scheme 45). Ni(II) was firstly reduced to Ni(0) (A) in the presence of base for the subsequent oxidative addition to Ar–CN bond via the complexes (B and C) between aryl nitrile and nickel. As a result, aryl nickel cyanide (D) was formed. Ligand exchange through two possible routes as proposed by the authors provided intermediate (E). Subsequent reductive elimination of E produced the phosphinated product and regenerated Ni(0) (A) for next catalytic cycle.
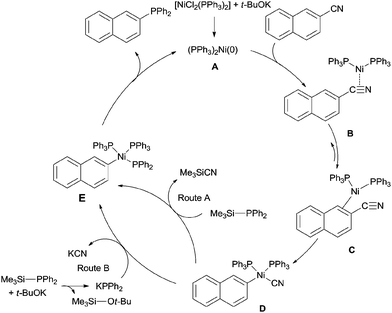 |
| | Scheme 45 Possible mechanism for the Ni-catalyzed decyanative phosphination of aryl nitriles. | |
3.3 Decyanative borylation
In 2011, Tobisu and Chatani described the borylation of nitriles with a diboron reagent (94) through Rh(I)-catalyzed C–CN bond cleavage (Scheme 46).31a,b In the optimization studies, they found the base and phosphine ligand were quite important in improving the yield, of which 1,4-diazabicyclo[2.2.2]octane (DABCO) and Xantphos were the best choices, respectively. Various nitriles, including aryl, alkenyl and benzylic nitriles, could form the corresponding boronic esters in good yields. This method provided a new strategy for the synthesis of boronic esters, which were very useful for further derivatization. Of significant note, C–Cl bond and specified C–B bond survived after reactions which respectively allowed the orthogonal and sequential designs for further cross coupling reactions.
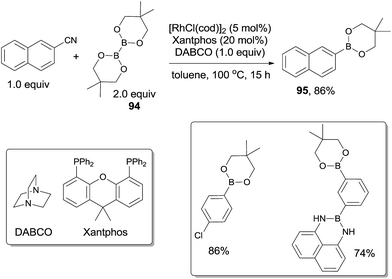 |
| | Scheme 46 Rh-catalyzed decyanative borylation of aryl cyanides. | |
The mechanistic pathway supposed that the diboron (94) played an important role in the cleavage of C–CN bond (Scheme 47). σ-Bond metathesis between Rh–Cl bond and B–B bond generated Rh–B bond and B–Cl bond which was confirmed by 11B NMR analysis. This step was the same to the previous metal-silicon bond formation. Addition of Rh–B species (A) across nitrile triple bond resulted in the formation of adduct (B) which isomerized to C for the sterically favored syn-β-aryl elimination. As a result, R–Rh (D) was formed and borylated via σ-bond metathesis with diboron. Although β-carbon elimination was proposed in Chatani's report,31a a mechanism including the sequential 2,1-insertion and deinsertion could not be excluded.31c,d
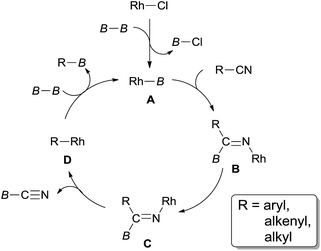 |
| | Scheme 47 Rh-catalyzed decyanative borylation of nitriles. | |
3.4 Decyanation
Besides of the cross coupling reactions with disilane, C–CN bond could also be cleaved and transfer to C–H bond, called decyanation. Such reactions were quite useful in organic synthesis because they offered the opportunity to temporally utilize the benefits from the CN group, such as the acidity of α-H, the ortho-directing effect, and the strong electron withdrawing nature of cyano group.
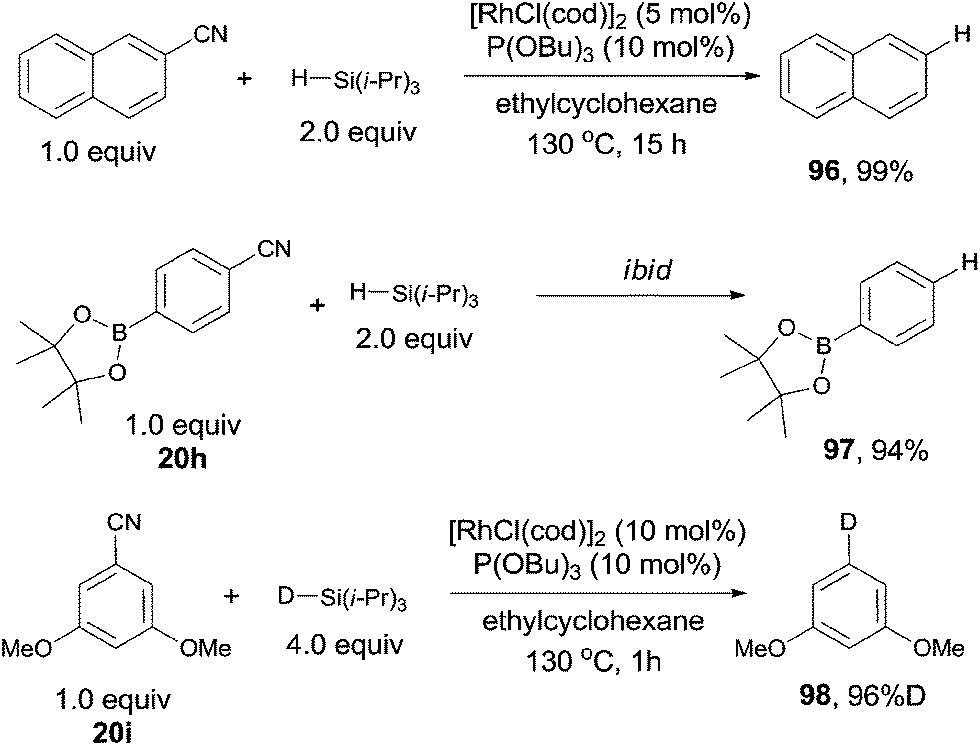
In 2009, Tobisu and Chatani reported a protocol for Rh-catalyzed reductive cleavage of C–CN bond with hydrosilane as the reducing agent, converting varying nitriles to the corresponding alkanes or arenes (Scheme 48).32 They selected [RhCl(cod)]2 as the rhodium catalyst, P(OBu)3 as the ligand and triisopropylsilane as the reducing agent, affording decyanated products with high efficiencies. A variety of functional groups tolerated the reaction conditions, such as dimethylamino, methoxy, ester, and boronic ester. A deuterated product (98) was also gained when the corresponding deuteriosilane was used in the reaction. Besides of aryl nitriles could be decyanated, benzilic nitriles as well as normal alkyl cyanides could also be decyanated in excellent yields without the contamination from β-hydrogen elimination.
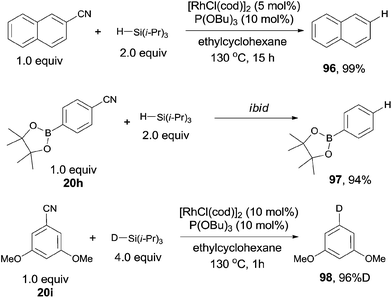 |
| | Scheme 48 Rh-catalyzed Si-assisted decyanation of aryl nitriles. | |
In 2013, Maiti demonstrated nickel catalyzed decyanation of nitriles selecting tetramethyldisiloxane (TMDSO) as the hydride source (Scheme 49).33 In the catalytic system of Ni(acac)2, PCy3, and AlMe3, aryl and aliphatic cyanides reacted with TMDSO and decyanated to arenes and aliphatic hydrocarbons, respectively. A variety of functional groups on aromatic ring suffered this reaction condition, such as methoxy, ester, and carbonyl. Cleavage of Ar–SMe bond occurred prior to the cleavage of Ar–CN bond. Heterocyclic cyanides also preceded smoothly under the reaction conditions. The deuterated product was isolated when Ph2SiD2 was used as the reductant. The trans-[Ni(PCy3)2(CN)2] was isolated from the reaction, but failed to catalyze the decyanation, indicating that it was the cause of catalyst deactivation.
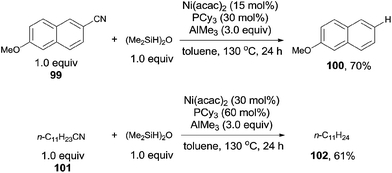 |
| | Scheme 49 Ni-catalyzed LA-assisted decyanation of aryl/alkyl nitriles with silane. | |
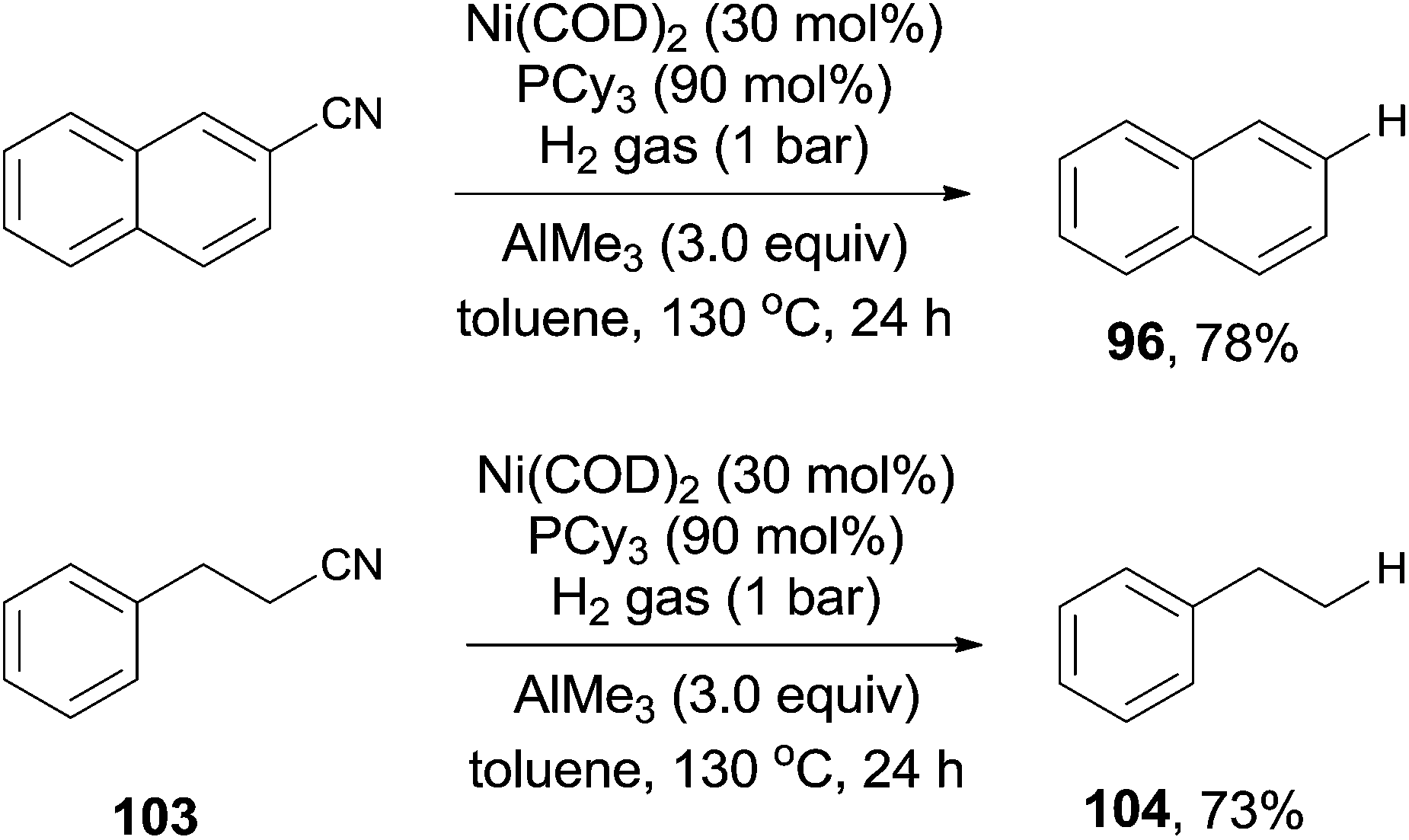
Short after, Maiti reported nickel-catalyzed decyanation of aryl and aliphatic cyanides using hydrogen gas as the hydride source (Scheme 50).34 The C–CN bond was efficiently activated in assistance of AlMe3, which was also responsible to consume the in situ produced HCN gas. It was worthy to note that aliphatic cyanides (103), possessing β-hydrogens, were also applicable for this decyanation without the formation of the byproduct arising from β-hydrogen elimination. A variety of functional groups tolerated the reaction conditions, such as MeO-, HO-, F-, ester, amide, ketone, as well as the tethered olefin.
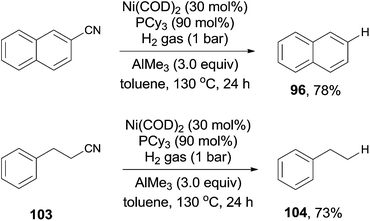 |
| | Scheme 50 Ni-catalyzed LA-assisted decyanation of aryl/alkyl nitriles with hydrogen. | |
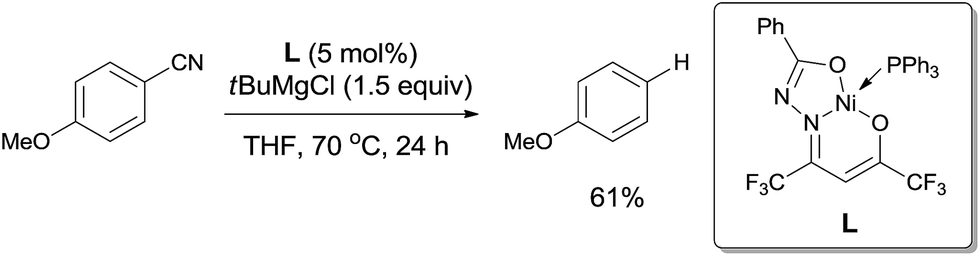
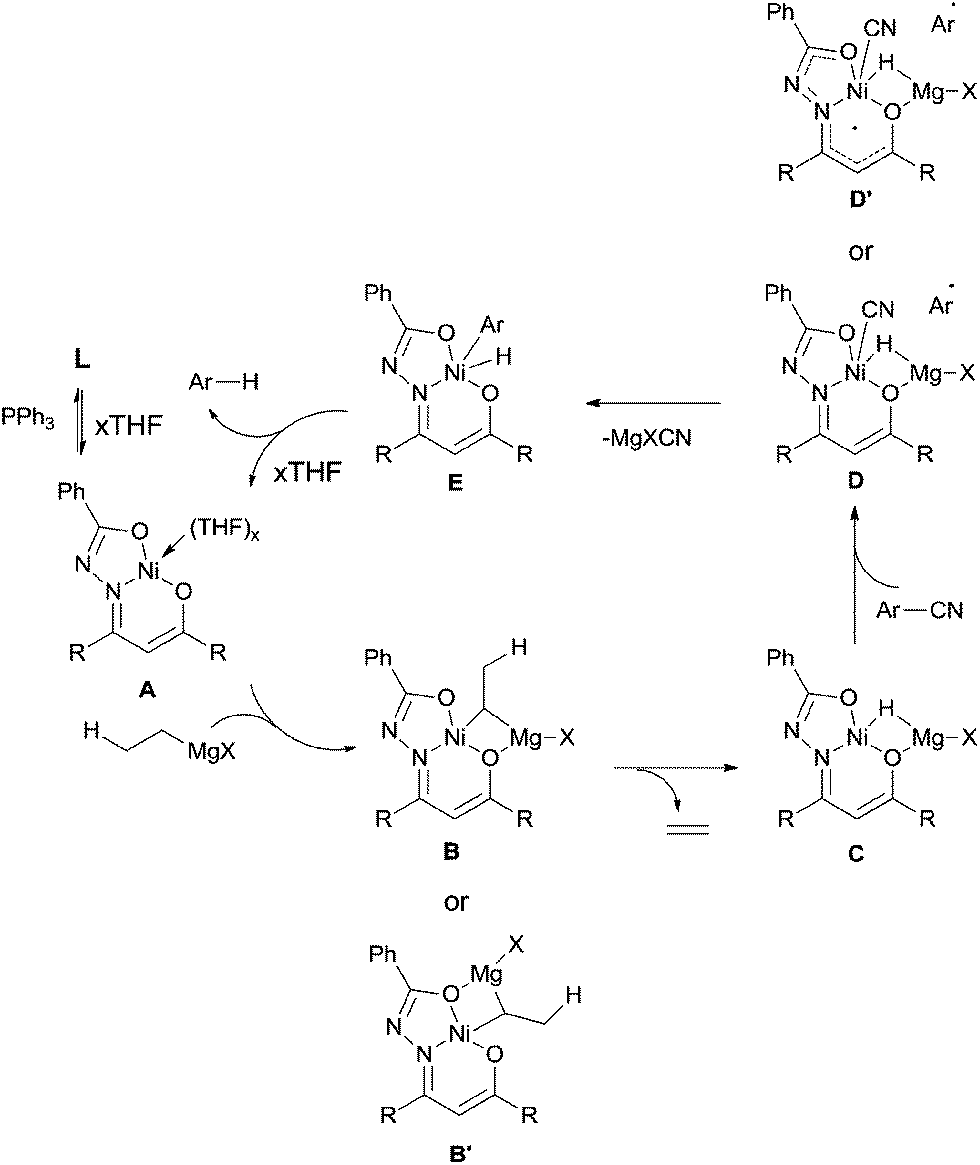
In 2013, Enthaler reported decyanation of aryl and alkyl cyanides catalyzed by their pre-prepared nickel complex and employed tert-butylmagnesium chloride as the hydride source (Scheme 51).35 Most substituent groups could tolerant the reaction conditions except for thioether and several halogens (Br and F). In these cases, the cleavage of C–S and C-X bonds was observed. Hydrogen atom in the product came from Grignard reagent which was further confirmed by using C2D5MgBr. Finally, they proposed a possible mechanism, in which C–CN bond was supposed to be cleaved through a single-electron transfer (SET) step and a radical procedure (Scheme 52).
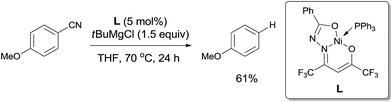 |
| | Scheme 51 Ni-catalyzed decyanation of aryl nitriles with tert-butylmagnesium chloride. | |
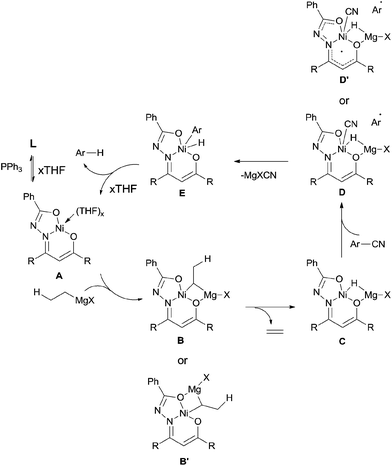 |
| | Scheme 52 Proposed mechanism for Ni-catalyzed decyanation of aryl nitriles with tert-butylmagnesium chloride. | |
3.5 Decyanative arylation
In 2001, Miller's group firstly reported the coupling of benzonitriles and aryl Grignard reagents using Me3P-based Ni catalysts, forming the corresponding cross-coupling biaryl products (105) (Scheme 53).36 This reaction involved an C–CN bond cleavage mediated by the special Cl2Ni(PMe3)2 catalyst. The yield was significantly promoted by the pretreatment of Grignard reagent with an alkoxide (t-BuOLi in most cases), which efficiently inhibited the formation of byproducts, such as 4,4′-dimethoxybiphenyl from the homocoupling, anisole from the metal-H exchange, and 4-methoxybenzophenylimine from nucleophilic addition of Grignard reagent to the cyano. By applying the combination of PhMgBr and t-BuOLi, a variety of aryl nitriles were coupled to phenyl, including the aryls either with electron donating group (MeO, Me2N, Me) or electron withdrawing group (F, COOR). Moreover, the aryl nitriles could be extended to heterocyclic, such as pyridinyl and furanyl. By using the proper lithium salt, all of three cyanopyridines, 2-, 3-, and 4-cyanopyridines, could couple to phenyl magnesium chloride.
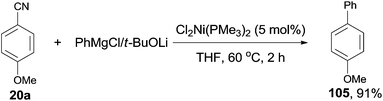 |
| | Scheme 53 Ni-catalyzed cross coupling between nitrile and aryl Grignard reagent. | |
Later in 2003, Miller and Dankwardt reported the nickel catalyzed cross-coupling of aryl nitriles with the modified alkyl and alkenyl Grignard reagents, also providing corresponding styrene and alkyl arene derivatives in good yields (Scheme 54).37 By the addition of either t-BuOLi or PhSLi to the Grignard reagents, the nucleophilic addition of Grignard to aryl nitriles could be effectively diminished. Undoubtedly, this procedure contained a nickel catalyzed activation of the C–CN bond.
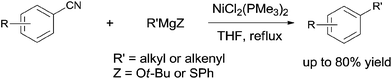 |
| | Scheme 54 Ni-catalyzed cross coupling between nitrile and alkyl/alkenyl Grignard reagent. | |
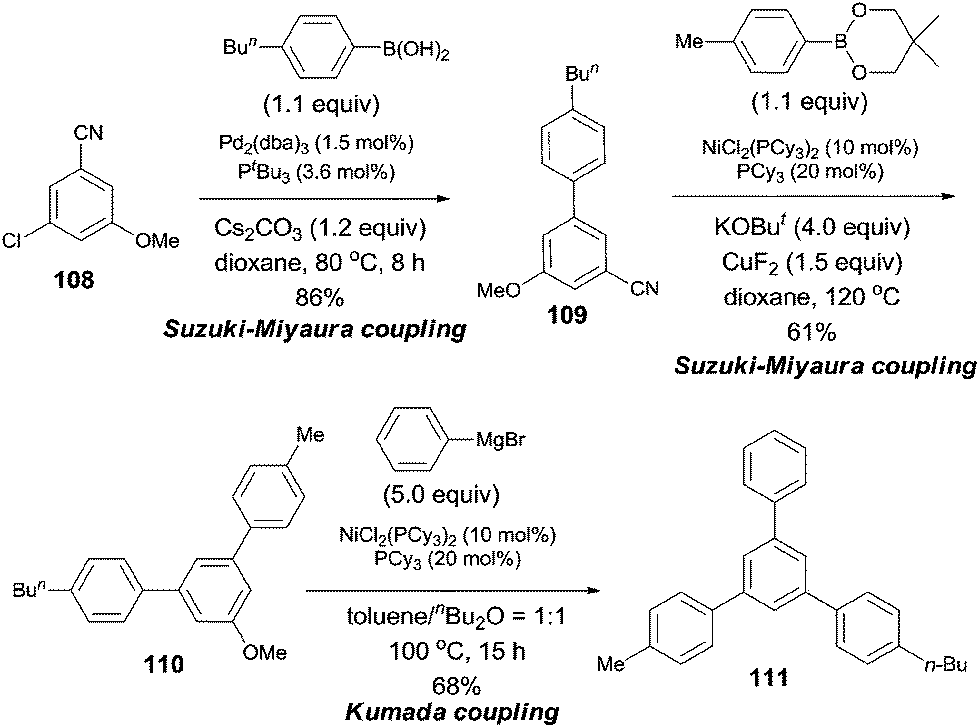
In 2009, Shi reported a cross coupling of aryl nitriles with aryl/alkenyl boronic esters through Ni-catalyzed C–CN bond cleavage (Scheme 55).38 With the aid of t-BuOK as a base and CuF2 as an additive, aryl/alkenyl boronic esters coupled with aryl nitriles and provided biaryls in moderate to good yields. A variety of aryl nitriles were tested, including the aryls with electron-withdrawing and electron-donating groups.
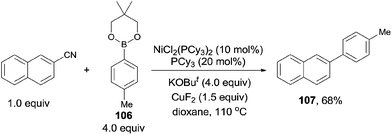 |
| | Scheme 55 Ni-catalyzed cross coupling between nitrile and boronic ester. | |
Considering the relative reactivity of C–Cl, C–CN, and C–OMe bond activations in cross-coupling reactions, the authors successfully prepared polysubstituted benzene (111) with different aryl groups by orthogonal design (Scheme 56).
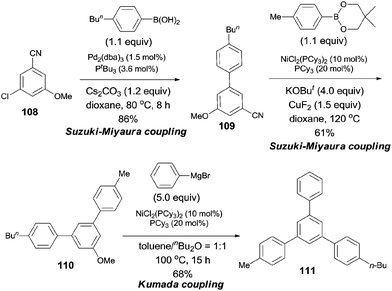 |
| | Scheme 56 Orthogonal synthesis of triarylbenzene based on relative reactivity. | |
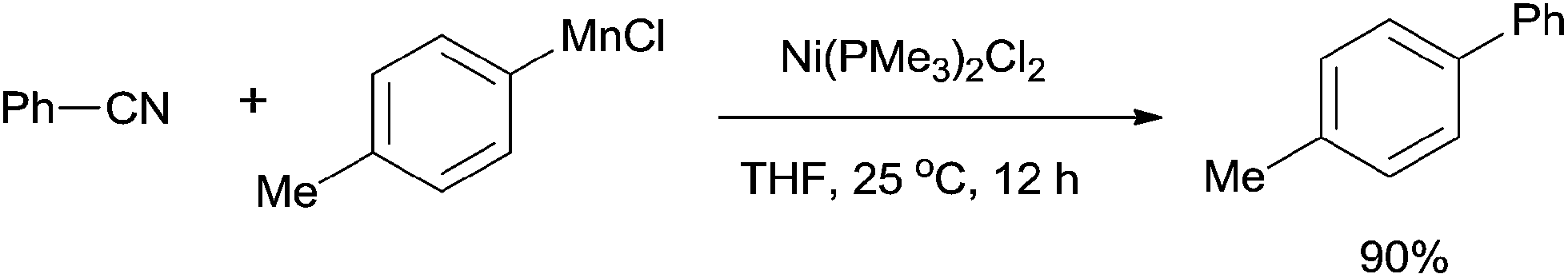
In 2012, Wang developed Ni-catalyzed cross coupling of aryl nitriles and arylmanganese reagents through the cleavage of C–CN bond (Scheme 57).39 The arylmanganese reagents, which had been commonly used in many cross-coupling reactions, showed high efficiency in these cross-coupling reactions. A number of aryl nitriles and arylmanganese chlorides could be used for coupling. In comparison with the Grignard reagents or boronic esters used above, this reaction had great advantages, such as mild conditions, and no requirement of base and additive.
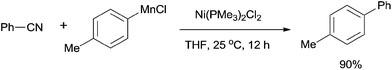 |
| | Scheme 57 Ni-catalyzed cross coupling between nitrile and arylmanganese reagent. | |
During the process of the rhodium catalyzed decyanative silylation of aryl cyanides as previously mentioned in Scheme 41,28 Ar–Rh was formed as a key intermediate in the catalytic cycle. By designation of the structure of Ar–Rh with a tethered electrophile, such as aryl chloride, C–C bond could be formed intramolecularly. Thus, substituted dibenzofurans (113) and carbazole derivatives (114) were well synthesized by the Rh-catalyzed Si-assisted C–CN bond cleavage and a sequential coupling with intramolecular Ar–Cl (Scheme 58). Besides of these, the benzylic C–CN bond could be arylated with intramolecular Ar–Cl. Thus, fluorene (116) was obtained in 60% yield, accompanying with the formation of the decyanated silylation product (117) in 15% yield. This methodology used the aryl chloride instead of organometallic reagent to realize the cross coupling between C–CN and C–Cl bonds. From this point of view, it was of synthetic value because aryl chlorides were more stable and more widely existed.
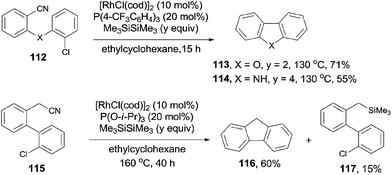 |
| | Scheme 58 Rh-catalyzed Si-assisted cross coupling between C–CN and Ar–Cl. | |
3.6 Decyanative alkenylation
In 2010, Tobisu and Chatani demonstrated a Rh-catalyzed Si-assisted C–CN bond cleavage of aryl nitrile (20j), followed by a Mizoroki–Heck type reaction with triethyl(vinyl)silane (118), achieving the alkenylation of aryl nitrile (119) as well as the silylated byproduct (120) (Scheme 59).40 All the resulting olefins were in trans-configuration. After screening the reaction conditions, ligand was proved to be a significant factor in affecting the selectivity of forming the desire alkenylated product over silylated product. As a result, P(4-FC6H4)3 was found to be optimal. The ratio of alkenylated product to silylated product largely depended on the substituted group on the silicon. Triethylvinylsilane provided alkenylated product and silylated product in 65% and 18% yields, while triisopropylvinylsilane afforded them in 11% and 79%, respectively. On the contrary, triisopropoxylvinylsilane afforded the alkenylated product as the sole product, no silylated product was detected. 4-Cyano-1-tosylindole coupled with triethylvinylsilane under the same reaction conditions and provided 4-vinyl substituted-1-tosylindole in 82% yield without the contamination from C–H bond activation of 2- or 3-position of indole. 3,3-Diphenylacrylonitrile, as an example of alkenyl cyanides, could also couple with triethylvinylsilane to afford dienylsilane, accordingly.
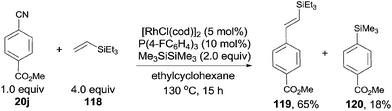 |
| | Scheme 59 Rh-catalyzed decyanative Mizoroki–Heck type coupling. | |
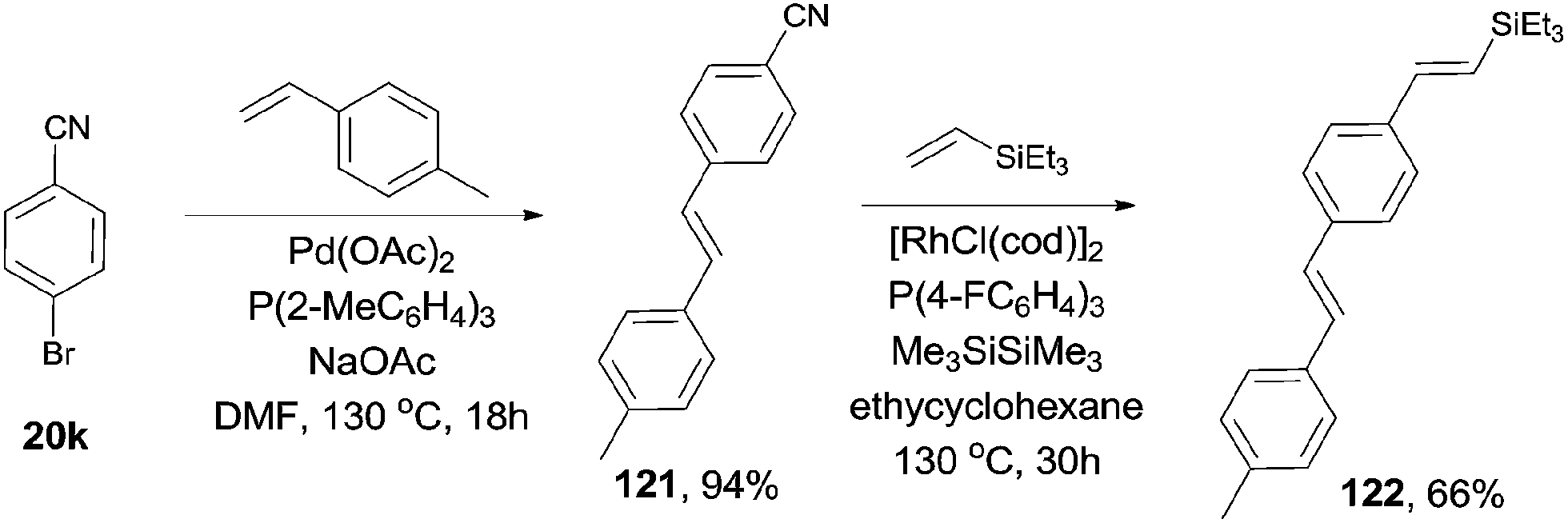
Considering the selective C–Br and C–CN bond activation by using suitable transition metal catalyst, 1,4-divinyl phenylene (122), with different substituted groups at ends could be approached (Scheme 60).
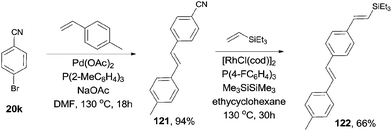 |
| | Scheme 60 Orthogonal synthesis of conjugated divinylphenylene. | |
In the proposed mechanism (Scheme 61), Rh catalyst transferred to an active silylrhodium species (A) by exclusion of TMSCl, and then Si-assisted C–CN bond cleavage occurred and arylrhodium species (C) was subsequently generated via η2-iminoacyl complex (B). During this process, silyl isocyanide was formed. Finally, a Mizoroki–Heck type reaction between intermediate (C) and vinylsilane occurred to form the desire product via addition and elimination, accompanied by the generation of rhodium hydride species (E). Subsequent reaction between rhodium hydride species (E) and disilane regenerated silylrhodium species (A) for the next catalytic cycle. In some cases, silylated product was obtained from the reaction between arylrhodium species (C) and disilane which was described in detail in the section of the coupling with heteroatoms.
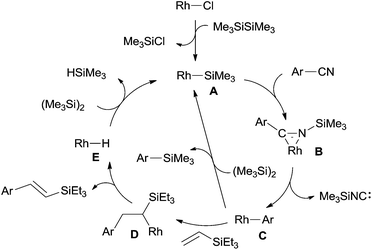 |
| | Scheme 61 Proposed mechanism for the Rh-catalyzed decyanative Mizoroki–Heck type coupling. | |
Thus, a number of σ-bonds, including C–Si, C–P, C–B, C–H, and C–C bonds, could be feasibly constructed via transition metal catalyzed C–CN bond cleavage and the subsequent cross coupling. Of particular note, advantageous of R–CN were obvious. First, they could be treated as a succedaneum for R–X/R–OTs in cross coupling. Second, based on the selectivity and the reactivity of C–CN, C–X, and C–O bonds, these protocols enabled orthogonal cross coupling possible for having higher conjugated system. Third, the decyanation reaction took the advantage of “CN” properties, such as α-C–H acidity, ortho-directing effect, and electron-withdrawing nature, and could be utilized in the synthesis of challengeable compounds.
4. Cyanation reactions
The “CN” part in nitriles can be used as cyanide source in cyanation reactions. In contrast to extremely toxic metal cyanides (MCN), organic nitriles are less toxic, soluble in normal organic solvents, and easy to handle with operational benign. They slowly release the cyanide anions through C–CN bond activation and are safe cyanation reagents. Meanwhile, the nature of slow release of cyanide anions during the reaction process effectively avoids the overloading of cyanide anions to transition metal center resulting from the thermodynamically stable TM–CN bond. With this respect, minimum amount of the catalyst and cyanide precursor are used in cyanations. In cyanation reactions, there are several ways to cleave the C–CN bonds, those are Si-assisted oxidative addition of TM, TM-assisted nucleophilic substitution, and TM-catalyzed α-C–H oxidation.
4.1 C–CN cleavage via Si-assisted TM migration insertion
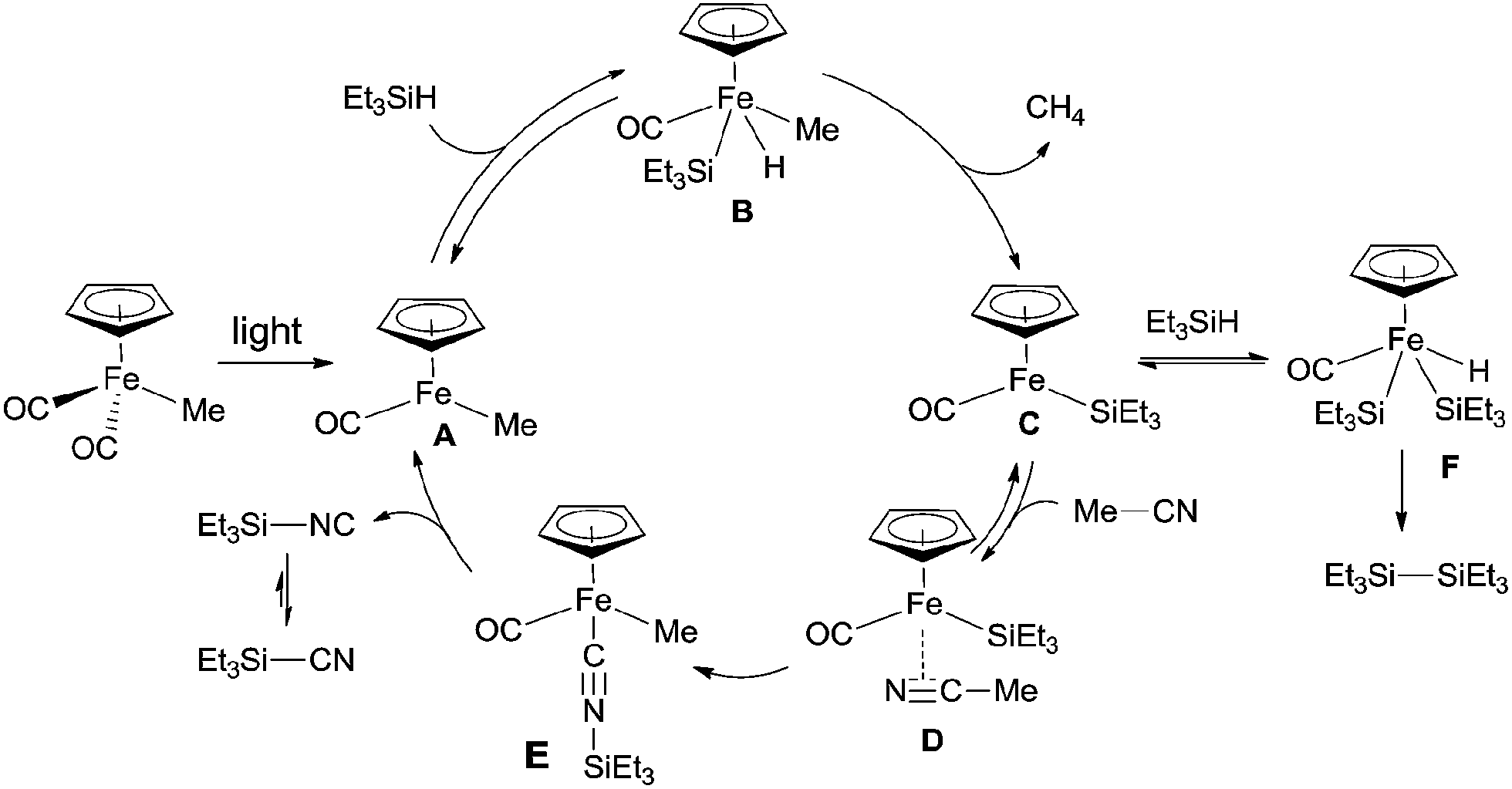
In 2005, an iron complex, Cp(CO)2FeMe was disclosed to catalyze the C–CN bond cleavage in the photoreaction of MeCN and Et3SiH. Reaction afforded Et3SiCN in 72% yield by extrusion of CH4 with a trace amount of Et3Si–SiEt3 formation (Scheme 62).41 Here Et3SiCN was thought to be thermodynamically stable product, isomerized from Et3SiNC. This catalytic system was also applicable for the cleavage of C–CN bond of aryl nitriles.
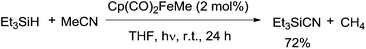 |
| | Scheme 62 Fe-catalyzed Si-assisted photocyanation of silane with acetonitrile. | |
In their proposed mechanism (Scheme 63), light triggered the exclusion of CO from Cp(CO)2FeMe and left the space for oxidative addition to Si–H bond in Et3SiH. After reductive elimination, a key intermediate (C), with 16 valence electrons, was generated. Coordinating this species with acetonitrile in η2-fashion (D), followed by silyl-assisted iron migration insertion, iron isocyanide complex (E) was formed. Exclusion of silyl isocyanide (Et3SiNC) from E regenerated A for completing the catalytic cycle. As a result, Et3SiCN was formed after isomerism, while Et3Si–SiEt3was formed from the reaction between key intermediate (C) and Et3SiH via addition/elimination sequence.
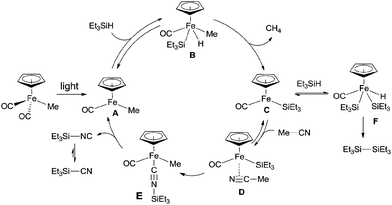 |
| | Scheme 63 Proposed mechanism for the Fe-catalyzed Si-assisted photocyanation of silane with acetonitrile. | |
4.2 C–CN cleavage via TM-assisted nucleophilic substitution
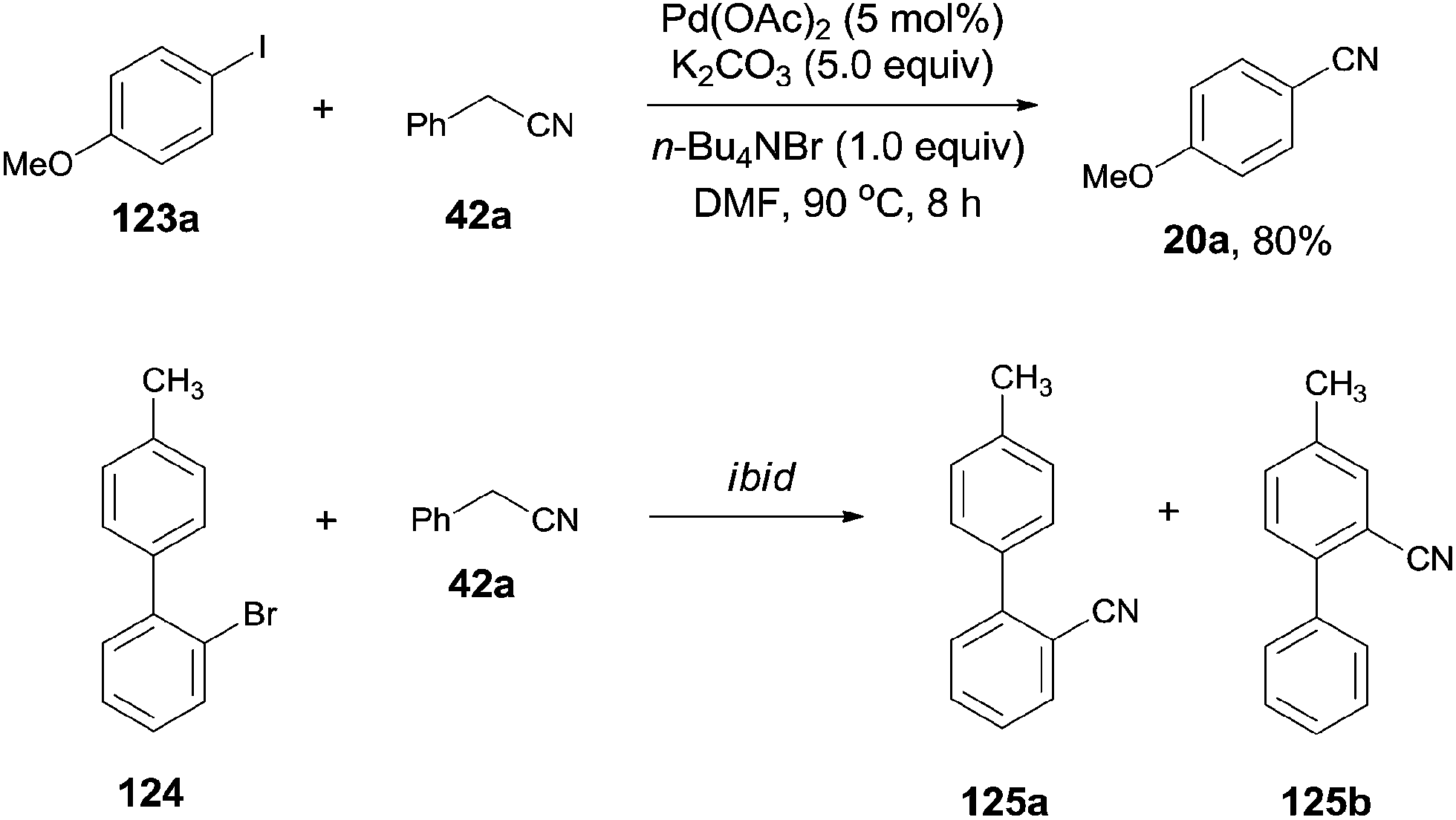
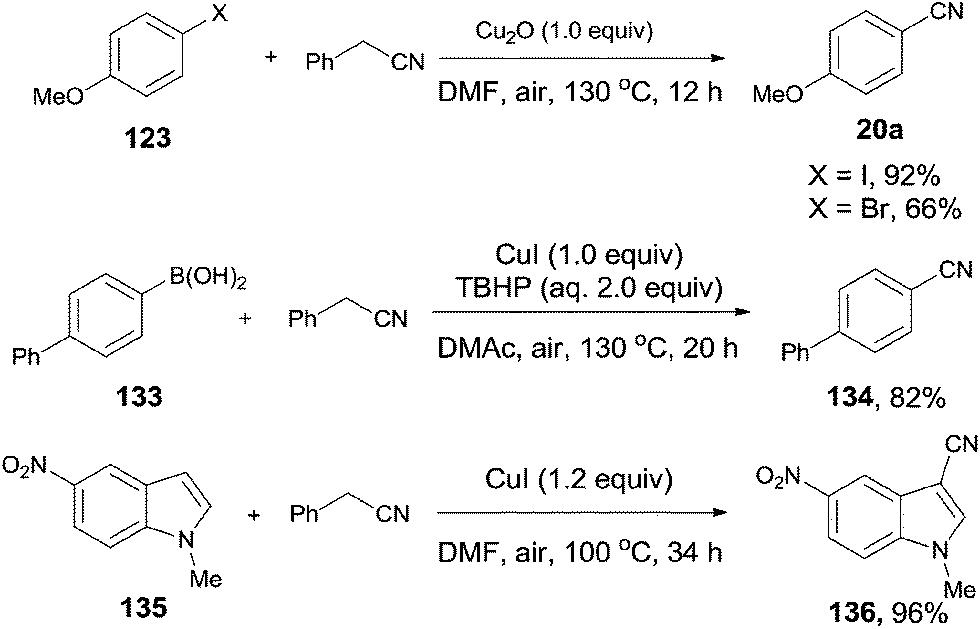
In 2012, Wang and Lu reported palladium-catalyzed cyanation of aryl halides (123) using benzyl cyanide (42a) as the cyano source (Scheme 64).7a The C–CN bond of benzyl cyanide was cleaved in the aid of palladium and transferred to aryl halides, leading to the formation of aryl nitriles. In the optimized conditions, Pd(OAc)2 was the best catalyst, and it was quite necessary for having K2CO3 as the base and n-Bu4NBr as the additive. A number of functional groups, such as amino, hydroxy, alkyloxy, and carbonyl, tolerated this reaction conditions and provided the desired aryl nitriles in moderate to good yields. 1,4-Palladium migration was observed in the designed substrate. Treatment of 124 under the standard reaction conditions, a mixture of 125a and 125b was obtained in 46% yield. The ratio of 125a and 125b was determined to be 1:1. 125a was the key intermediate for the preparation of Satan's medicine. Of particular note, this protocol was realized as a safety method because only a trace amount of cyanide anion was detected which, moreover, effectively inhibited the poison of the palladium catalyst resulting from the high affinity of palladium to cyanide anion.
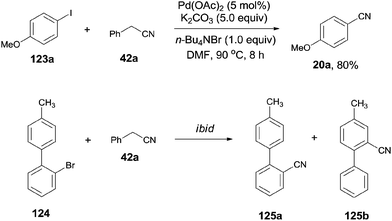 |
| | Scheme 64 Pd-catalyzed cyanation of aryl halide with benzyl cyanide. | |
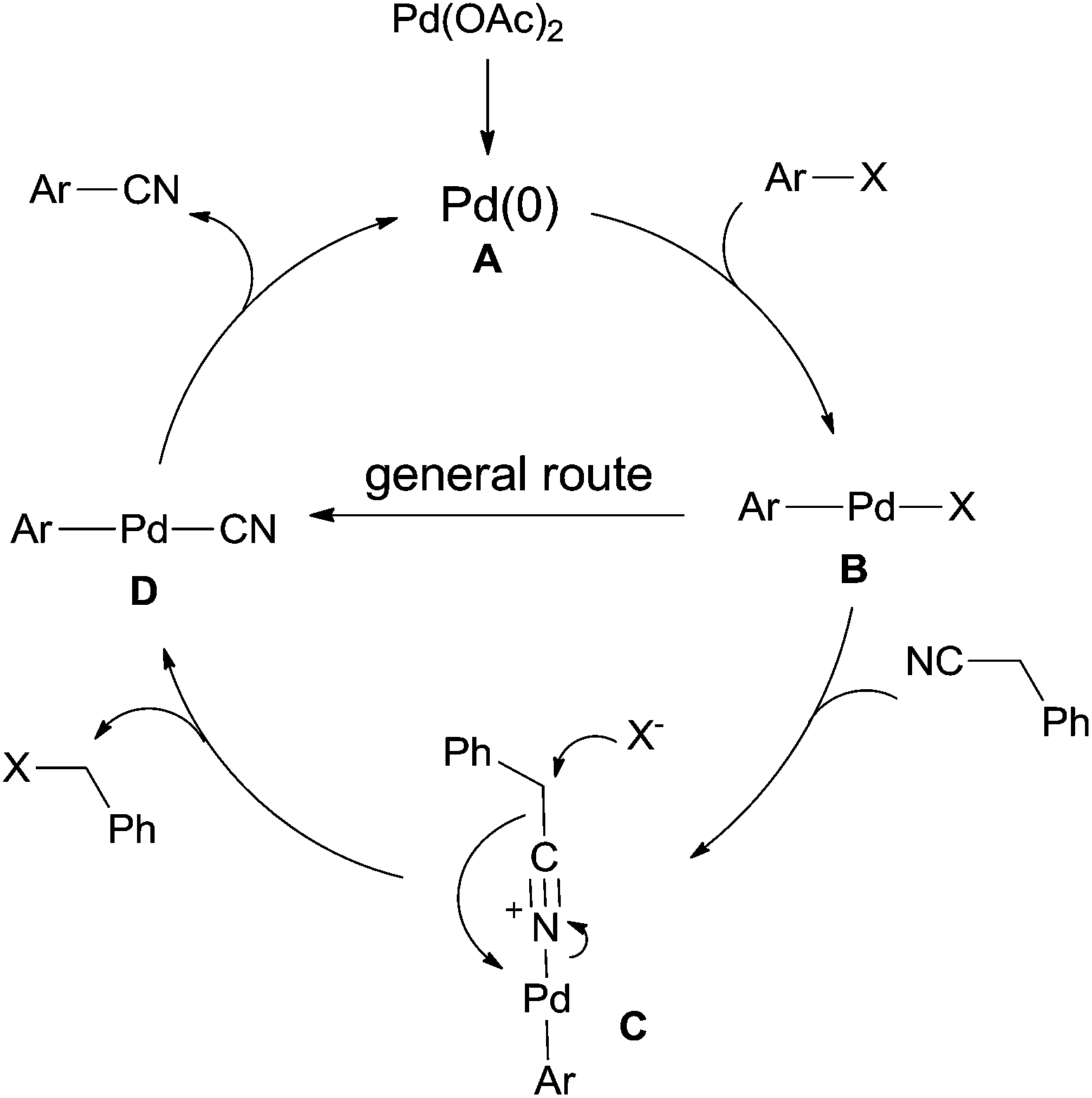
The general Pd-catalyzed cyanation of aryl halides contained three steps, oxidative addition, ligand exchange, and reductive elimination (Scheme 65). The key obstacle for this general one was the poison of the palladium catalyst which resulted from overloading of cyanide anion to palladium center. In the cyanation with benzyl cyanide, after the oxidative addition of Pd(0) to aryl halides, B was formed and subsequently coordinated with benzyl cyanide to generate key intermediate (C). Thus, C–CN bond was activated and cleaved by the assistance of halide. Finally, reductive elimination afforded aryl nitriles and regenerated Pd(0) for completing the catalytic cycle.
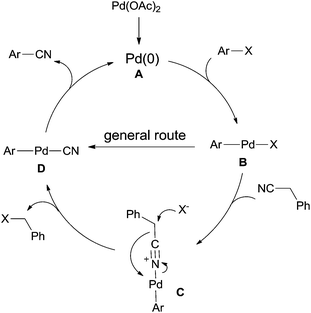 |
| | Scheme 65 Mechanism for the Pd-catalyzed cyanation of aryl halide with benzyl cyanide. | |
Subsequently, Shen developed palladium catalyzed cyanation of aryl halides, introducing ethyl cyanoacetate as the cyanating reagent (Scheme 66).42 The combination of Pd(OAc)2, TMEDA, DPPE, Na2CO3 and KI could make the best transformation from aryl halides to aryl nitriles. A broad substrate scope was presented, including the acetyl, hydroxy, amino, ester, and amide. 5-Bromo-1-mthylindole proceeded the reaction affording the desired 5-cyano-1-mthylindole in 52% yield without the byproduct resulting from the C–H activation of 3-position of indole. The major defect for this transformation was the competitive aromatic nucleophilic substitution of enolate anion on the highly electron-deficient aryl halides. It happened spontaneously in basic environment without the participation of palladium. The authors claimed the complex of the ethyl cyanoacetate enolate and the palladium was the key intermediate in this reaction. It could form the palladium-cyano species and facilitate the cleavage of C–CN bond.
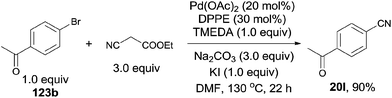 |
| | Scheme 66 Pd-catalyzed cyanation of aryl halide with ethyl cyanoacetate. | |
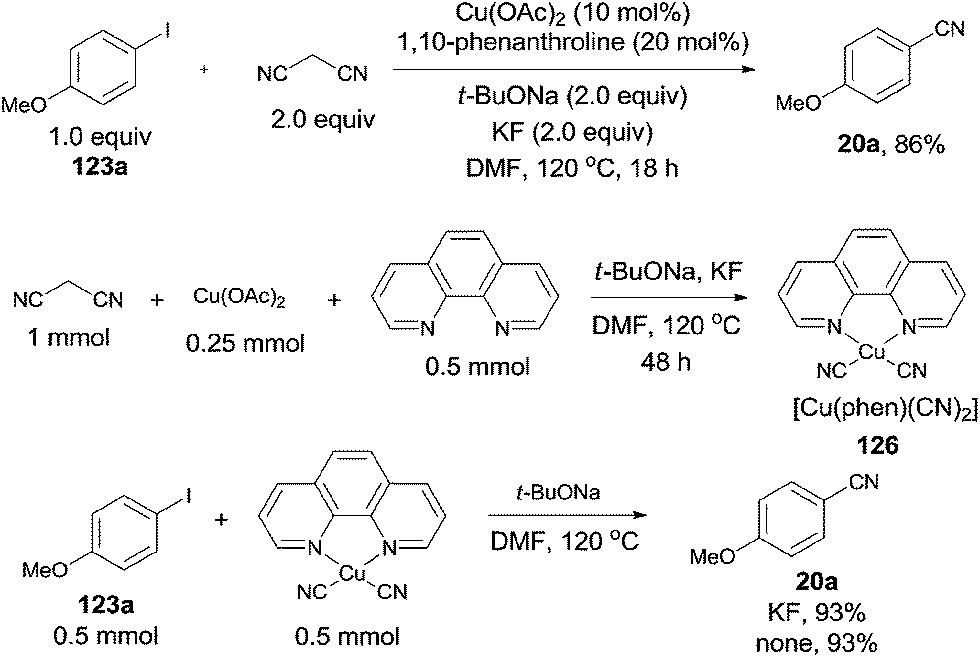
In 2012, Zhou reported Cu-catalyzed cyanation of aryl iodides using malononitrile as the cyanide source (Scheme 67).43 After screening the reaction conditions, Cu(OAc)2, 1,10-phenanthroline, t-BuONa were all the best choices, along with KF as a beneficial additive. The intermediate, a complex of [Cu(phen)(CN)2] (126), was formed by proceeding the reaction in the absence of aryl iodides. It was indicated that [Cu(phen)(CN)2] could react with the aryl iodides to form the aryl nitriles in good yields with or without KF. Thus, a copper-catalyzed C–CN bond cleavage of malononitrile assisted by KF might be involved in the reaction pathway, followed by the copper-catalyzed cyanation of aryl iodides with intermediate complex.
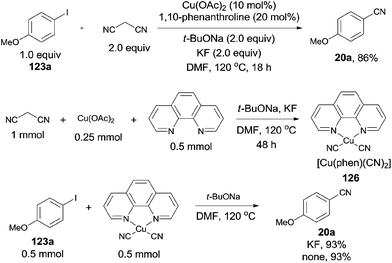 |
| | Scheme 67 Cu-catalyzed KF-assisted cyanation of aryl iodides with malononitrile. | |
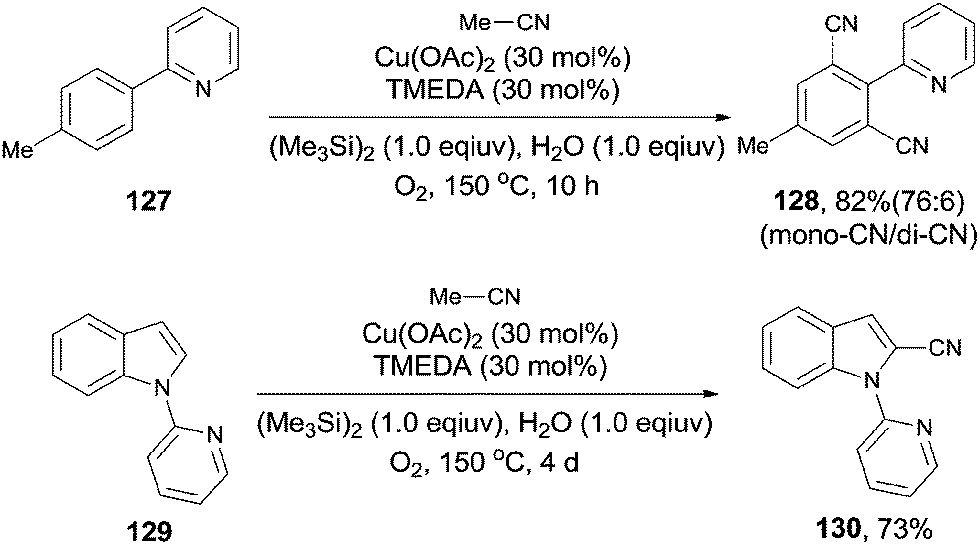
Later, Shen subsequently demonstrated a copper-catalyzed cyanation of 2-phenylpyridines and the 1-pyridinylindoles with acetonitrile (Scheme 68).8c It was hypothesized by the authors that hexamethyldisilane was employed as a Lewis acid to weaken the C–CN bond for its facile cleavage. Thus, a SN2-type nucleophilic substitution occurred on the methyl of acetonitrile and resulted in the formation of copper cyanide species for the subsequent cyanation.
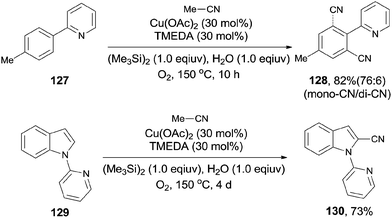 |
| | Scheme 68 Cu-catalyzed cyanation of arenes with acetonitrile using O2. | |
4.3 C–CN cleavage via TM-catalyzed α-C–H oxidation
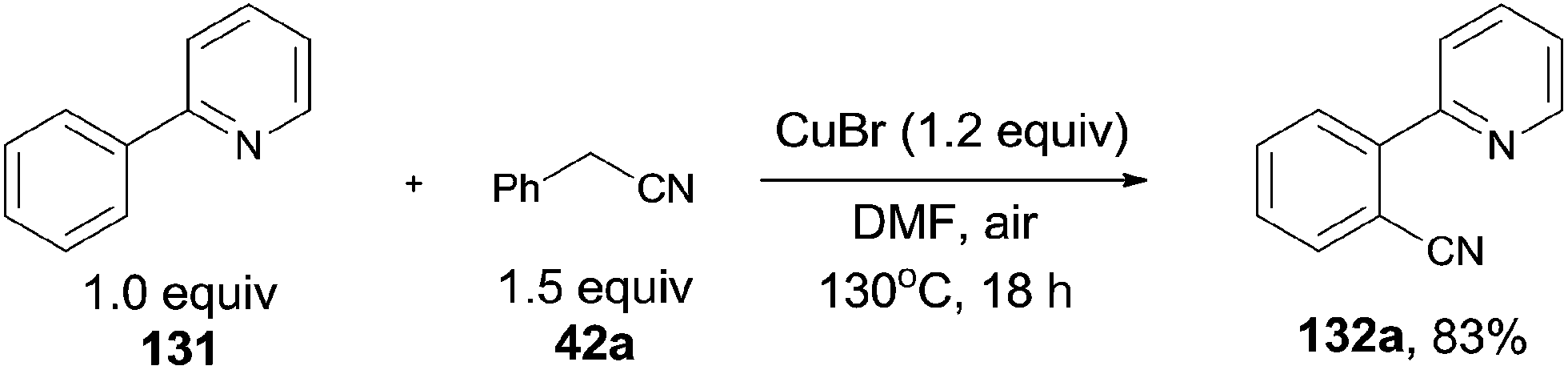
In 2012, Wang and Lu developed Cu-catalyzed cyanations of 2-phenylpyridinederivatives (131) with benzyl cyanides as the cyanation reagents (Scheme 69).7b A variety of substrates were proper for this transformation. A number of substituted groups tolerated the reaction conditions and provided desired cyanation products in excellent yields. Not only the pyridinyl group can be used as the directing group for the C–H bond activation, but pyrazolyl, pyrimidinyl also.
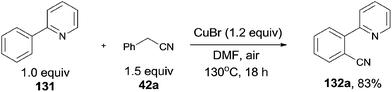 |
| | Scheme 69 Cu-catalyzed cyanation of arene via aerobic C–H oxidation of benzyl cyanide. | |
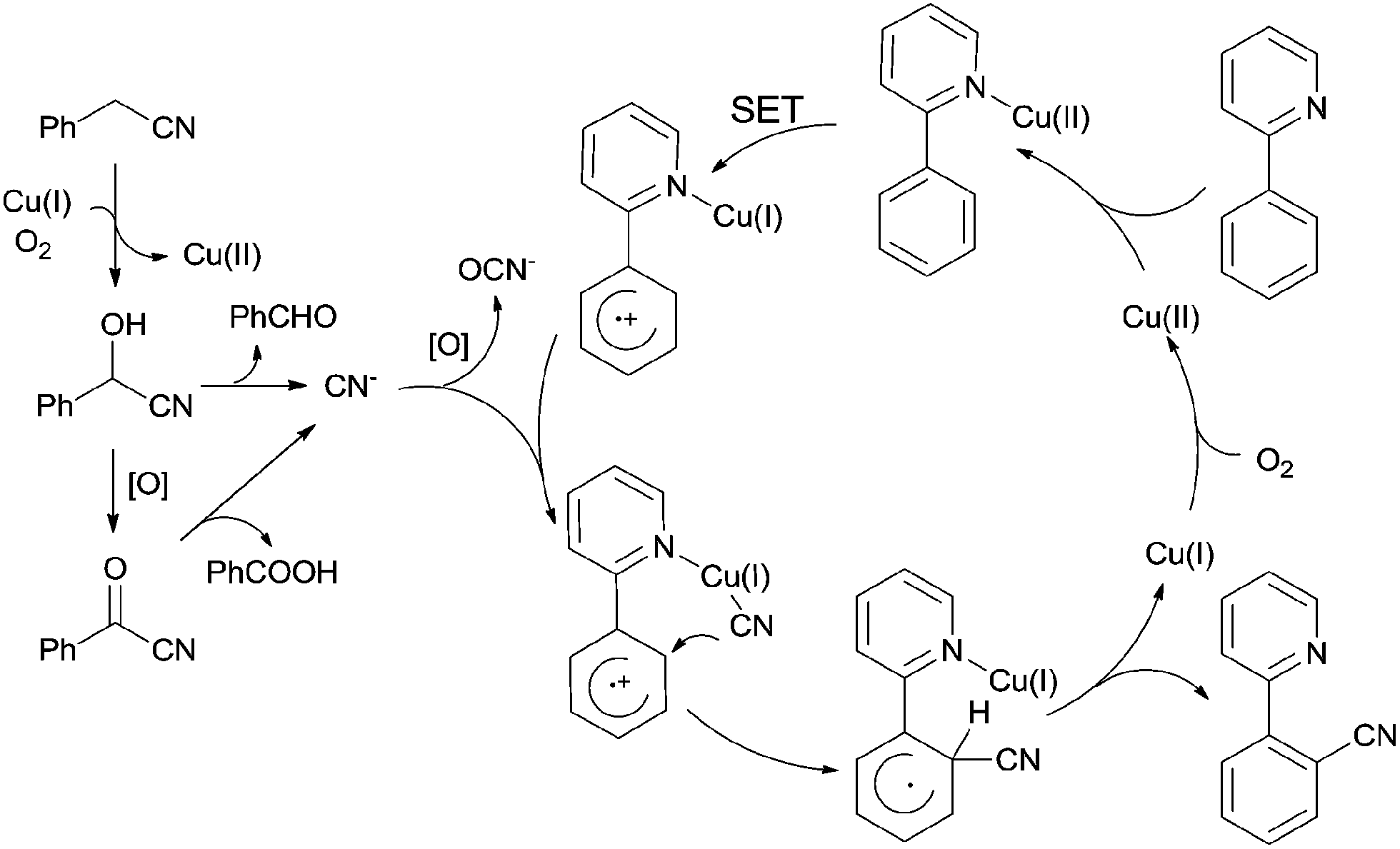
After further investigation, a different pathway was illustrated (Scheme 70) in comparison with previously Pd-catalyzed pathway (Scheme 65). An aerobic C–H oxidation of benzyl cyanide in the catalyst system was supposed to produce benzaldehyde cyanohydrin which was responsible for slowly releasing cyano anions for the subsequent Cu-catalyzed cyanation of arenes. It was pointed that matching two Cu-catalyzed reactions rates, those were Cu-catalyzed C–H aerobic oxidation of benzyl cyanide and heteroatom-directed ortho-C–H functionalization, was the key issue for approaching a successful transformation.
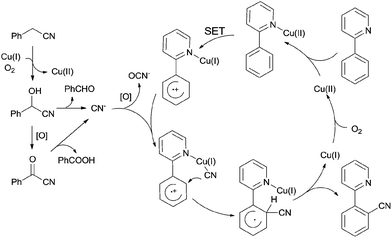 |
| | Scheme 70 Mechanism for Cu-catalyzed cyanation of arene with benzyl cyanide. | |
Afterwards, benefitted from the advantageous of benzyl cyanide as cyanation reagent and inexpensive copper salts as catalysts, cyanations of aryl halides,7c aryl boronic acids,7d and indoles7e,f were realized with nice substrate diversity and high yields (Scheme 71). Of particular note, by using the standard reaction conditions for cyanation of aryl halides, cyanation of 124 provided 125a in 50% yield without the observation of 125b.
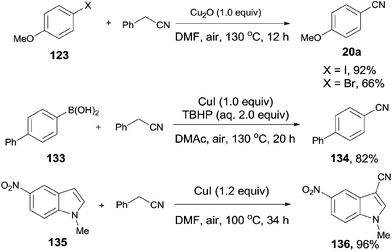 |
| | Scheme 71 Selected examples for cyanation of aryl halides, aryl boronic acids, and indoles with benzyl cyanide. | |
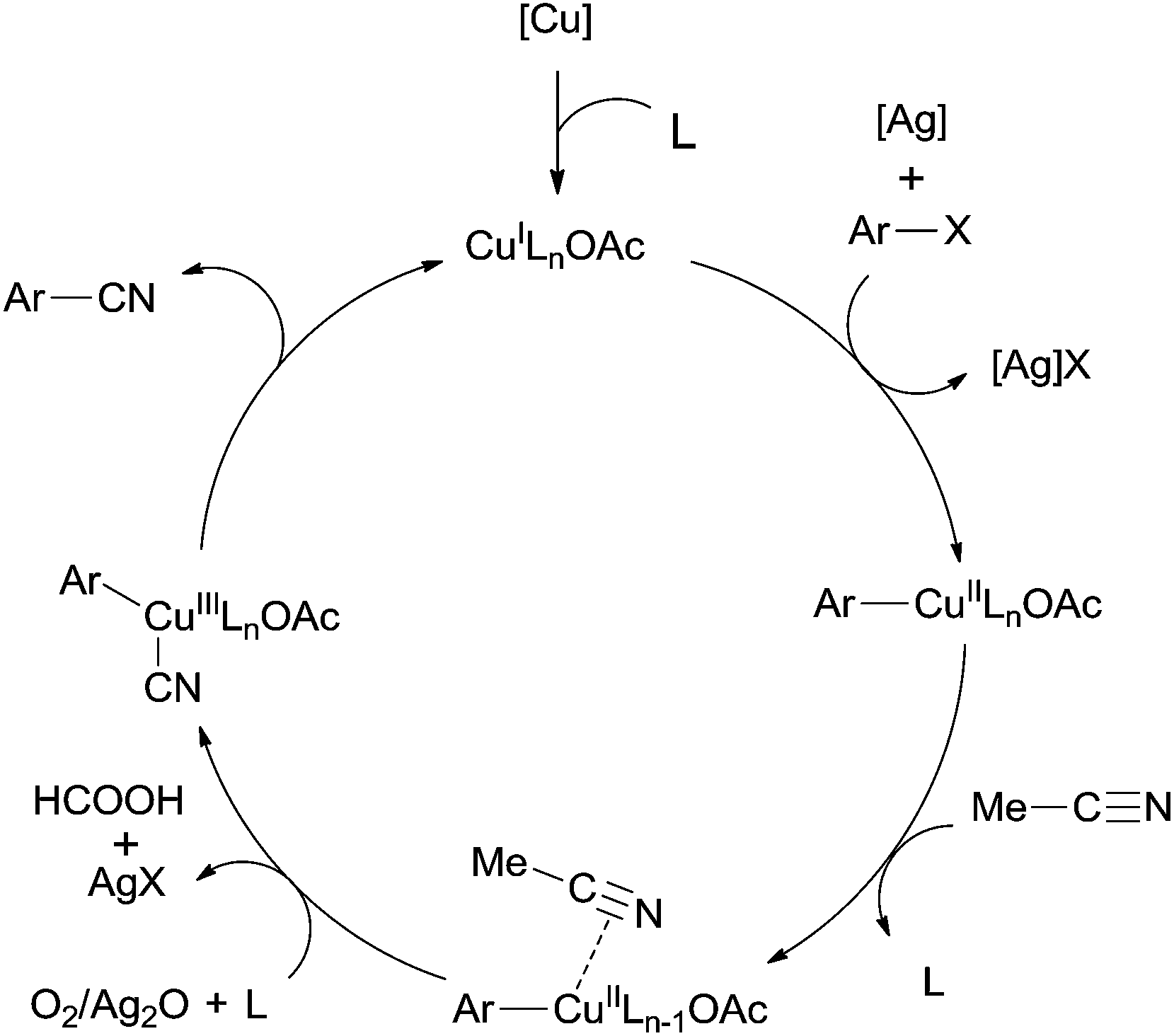
In 2012, a copper-catalyzed oxidative cyanation of aryl halides with acetonitrile as the cyanation reagent was reported by Li (Scheme 72).8a They employed Ag2O/air as the oxidant and triphenylphosphine oxide as ligand, converting various aryl iodides and bromides to aryl nitriles in moderate to good yields. Cu(I) species was thought to be responsible for the cleavage of Ar–X to form Ar–Cu species, followed by coordination with acetonitrile. In assistance of Ag2O/O2, acetonitrile was oxidized and decomposed to produce formic acid and “CN” for the following cyanation. Thus, the cleavage of C–CN of acetonitrile belonged to the Cu-catalyzed oxidative cleavage (Scheme 73).
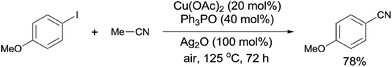 |
| | Scheme 72 Cu-catalyzed cyanation of aryl halides with acetonitrile. | |
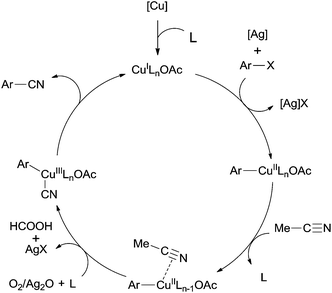 |
| | Scheme 73 Mechanism for the Cu-catalyzed cyanation of aryl halides with acetonitrile. | |
In 2013, Zhu also introduced acetonitrile as the cyanide source to the copper-mediated C2-cyanation of indoles and pyrroles without the participation of phosphine ligand (Scheme 74).8b With the pyrimidyl or pyridinyl on the N atom as the directing group, cyanation occurred selectively at C2-position. AgOAc and the O2 atmosphere were used as oxidants in this reaction, also involving copper-mediated C–CN cleavage of acetonitrile via α-C–H oxidation.
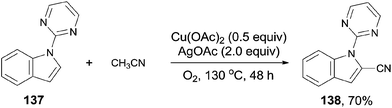 |
| | Scheme 74 Cu-catalyzed cyanation of arenes with acetonitrile. | |
Throughout the cyanation reactions by using organic nitriles as “CN” surrogates, both cleavage and formation of C–CN bond were involved. Although organic sources had significantly advantageous over traditional used metal cyanides, such as non-toxic and soluble in common organic solvents, only few organic nitriles had been hitherto examined and developed. Further investigation in this area for easy synthesis of aryl nitriles is still highly demanded.
5. Conclusions
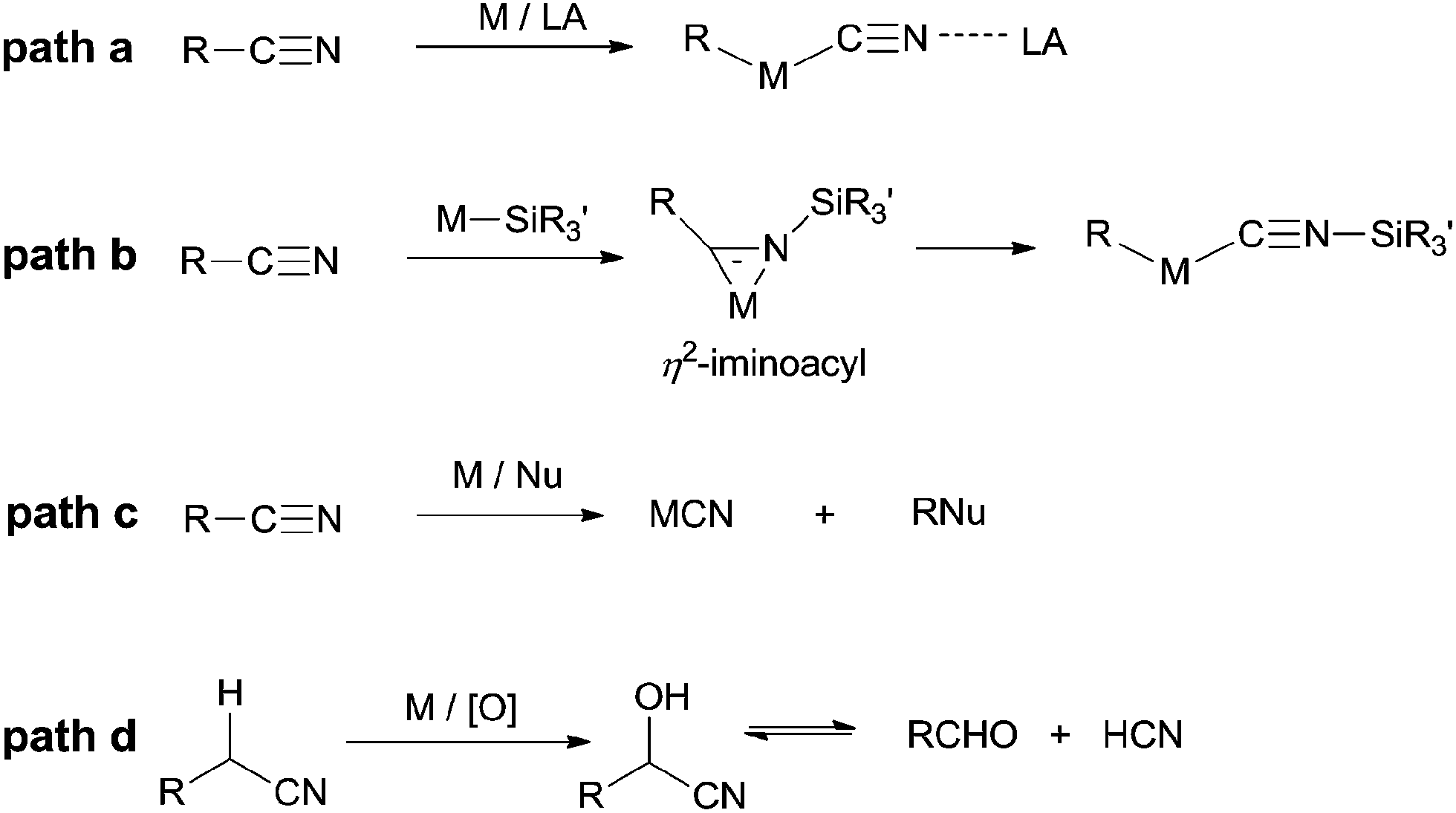
In summary, C–C σ-bond cleavage, catalyzed by transition metals with high selectivity and high efficiency, has gained lots of interests in recent years for its great usage in constructing new chemical bond. However, the high BDE of C–CN makes the C–CN bond inert and brings a huge challenge to organic chemists. Based on the aforementioned research works, it is noticed that transition metal plays a key role in activating C–CN bonds and can be categorized into four principles (Scheme 75). Most of mechanisms involve an oxidative addition of low-valent TM to C–CN bond which is greatly facilitated by adding of Lewis acid (Scheme 75, path a). In some cases, insertion of cyano into metal-silyl bond forms η2-iminoacyl complex which subsequently undergoes de-insertion of silyl isocyanide and results in the cleavage of C–CN bond (Scheme 75, path b). In others, transition metals function as Lewis acid which weakens C–CN bond for its sequential nucleophilic substitution (Scheme 75, path c). Besides of these, by applying the α-H acidity of nitrile, transition metal catalyzes C–H oxidation and leads to the formation of unstable cyanohydrins which releases cyanide anion by retro-cyanohydrination (Scheme 75, path d). Although some functions of transition metal have been clearly shown in C–CN bond activations, some of others still remain unclear, which drives researchers devoting more endeavours in mechanistic studies.
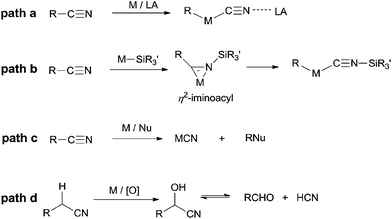 |
| | Scheme 75 Representative pathways of transition metal catalyzed C–CN cleavage. | |
Although above mentioned cyanofunctionalizations, cross-coupling reactions, and cyanations have well presented the utilities of nitriles in organic synthesis in recent years, the huge potentials of using the abundant, stable, and low-cost R–CN in feasibly making various C–C, C–H, as well as C-heteroatom bonds in organic synthesis still need to be explored and developed.
Acknowledgements
The authors thank the National Nature Science Foundation of China (NSFC) (21032005, 21272203).
Notes and references
-
(a) A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substance: Synthesis, Patents, Applications, Georg Thieme, Stuttgart, 4th edn, 2001 Search PubMed;
(b) R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, Weinheim, Germany, 1989, pp. 819–995 Search PubMed.
-
(a) A. J. Fatiadi, Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and Z. Rappoport, Wiley-VCH, New York, NY, 1983 Search PubMed;
(b) Z. Rappoport, Chemistry of the Cyano Group, John Wiley & Sons, London, UK, 1970, pp. 121–312 Search PubMed.
- For reviews, see:
(a) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779–794 CrossRef CAS;
(b) M. Sundermeier, A. Zapf and M. Beller, Eur. J. Inorg. Chem., 2003, 3513–3526 CrossRef CAS;
(c) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049–5067 RSC;
(d) J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948–11959 CrossRef CAS PubMed;
(e) G. Yan, J. Yu and L. Zhang, Chin. J. Org. Chem., 2012, 32, 294–303 CrossRef CAS;
(f) Q. Wen, J. Jin, L. Zhang, Y. Luo, P. Lu and Y. Wang, Tetrahedron Lett., 2014, 55, 1271–1280 CrossRef CAS PubMed.
- For recent reviews, see:
(a) M. Tobisub and N. Chatani, Chem. Soc. Rev., 2008, 37, 300–307 RSC;
(b) C. Nájera and J. M. Sansano, Angew. Chem., Int. Ed., 2009, 48, 2452–2456 CrossRef PubMed;
(c) X. Kou, J. Fan, X. Tong and Z. Shen, Chin. J. Org. Chem., 2013, 33, 1407–1422 CrossRef CAS;
(d) R. Wang and J. R. Falck, RSC Adv., 2014, 4, 1062–1066 RSC.
- A. A. Zavitsas, J. Phys. Chem. A, 2003, 107, 897–898 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. R. D. Lide, CRC Press, Boca Raton, FL, 82nd edn, 2001, pp. 9–74 Search PubMed.
-
(a) Q. Wen, J. Jin, B. Hu, P. Lu and Y. Wang, RSC Adv., 2012, 2, 6167–6169 RSC;
(b) J. Jin, Q. Wen, P. Lu and Y. Wang, Chem. Commun., 2012, 48, 9933–9935 RSC;
(c) Q. Wen, J. Jin, Y. Mei, P. Lu and Y. Wang, Eur. J. Org. Chem., 2013, 4032–4036 CrossRef CAS;
(d) Y. Luo, Q. Wen, Z. Wu, J. Jin, P. Lu and Y. Wang, Tetrahedron, 2013, 69, 8400–8404 CrossRef CAS PubMed;
(e) L. Zhang, Q. Wen, J. Jin, C. Wang, P. Lu and Y. Wang, Tetrahedron, 2013, 69, 4236–4240 CrossRef CAS PubMed;
(f) O. Y. Yuen, P. Y. Choy, W. K. Chow, W. T. Wong and F. Y. Kwong, J. Org. Chem., 2013, 78, 3374–3378 CrossRef CAS PubMed.
-
(a) R.-J. Song, J.-C. Wu, Y. Liu, G.-B. Deng, C.-Y. Wu, W.-T. Wei and J.-H. Li, Synlett, 2012, 23, 2491–2496 CrossRef CAS PubMed;
(b) C. Pan, H. Jin, P. Xu, X. Liu, Y. Cheng and C. Zhu, J. Org. Chem., 2013, 78, 9494–9498 CrossRef CAS PubMed;
(c) X. Kou, M. Zhao, X. Qiao, Y. Zhu, X. Tong and Z. Shen, Chem.–Eur. J., 2013, 19, 16880–16886 CrossRef CAS PubMed.
-
(a) K. Nozaki, N. Sato and H. Takaya, J. Org. Chem., 1994, 59, 2679–2681 CrossRef CAS;
(b) K. Nozaki, N. Sato and H. Takaya, Bull. Chem. Soc. Jpn., 1996, 59, 1629–1637 CrossRef.
-
(a) Y. Nishihara, Y. Inoue, M. Itazaki and K. Takagi, Org. Lett., 2005, 7, 2639–2641 CrossRef CAS PubMed;
(b) Y. Nishihara, Y. Inoue, S. Izawa, M. Miyasaka, K. Tanemura, K. Nakajimab and K. Takagi, Tetrahedron, 2006, 62, 9872–9882 CrossRef CAS PubMed.
-
(a) Y. Nakao, Y. Hirata and T. Hiyama, J. Am. Chem. Soc., 2006, 128, 7420–7421 CrossRef CAS PubMed;
(b) Y. Hirata, T. Inui, Y. Nakao and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 6624–6631 CrossRef CAS PubMed.
- N. R. Rondla, S. M. Levi, J. M. Ryss, R. A. V. Berg and C. J. Douglas, Org. Lett., 2011, 13, 1940–1943 CrossRef CAS PubMed.
- Y. Kobayashi, H. Kamisaki, R. Yanada and Y. Takemoto, Org. Lett., 2006, 8, 2711–2713 CrossRef CAS PubMed.
- Y. Yasui, H. Kamisaki and Y. Takemoto, Org. Lett., 2008, 10, 3303–3306 CrossRef CAS PubMed.
-
(a) Y. Nakao, S. Od and T. Hiyama, J. Am. Chem. Soc., 2004, 126, 13904–13905 CrossRef CAS PubMed;
(b) Y. Nakao and T. Hiyama, Pure Appl. Chem., 2008, 80, 1097–1107 CrossRef CAS.
- Y. Nakao, T. Yukawa, Y. Hirata, S. Oda, J. Sato and T. Hiyama, J. Am. Chem. Soc., 2006, 128, 7116–7117 CrossRef CAS PubMed.
-
(a) Y. Nakao, A. Yada, S. Ebata and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 2428–2429 CrossRef CAS PubMed;
(b) Y. Hirata, A. Yada, E. Morita, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2010, 132, 10070–10077 CrossRef CAS PubMed;
(c) Y. Yasui, H. Kamisaki, T. Ishida and Y. Takemoto, Tetrahedron, 2010, 66, 1980–1989 CrossRef CAS PubMed.
- Y. Nakao, K. S. Kanyiva, S. Oda and T. Hiyama, J. Am. Chem. Soc., 2006, 128, 8146–8147 CrossRef CAS PubMed.
- A. Yada, T. Yukawa, Y. Nakao and T. Hiyama, Chem. Commun., 2009, 3931–3933 RSC.
- Y. Nakao, A. Yada and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 10024–10026 CrossRef CAS PubMed.
- Y. Nakao, Y. Hirata, M. Tanaka and T. Hiyama, Angew. Chem., Int. Ed., 2008, 47, 385–387 CrossRef CAS PubMed.
- K. Nakai, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2011, 133, 11066–11068 CrossRef CAS PubMed.
- For a further study on this topic, see: K. Nakai, T. Kurahashi and S. Matsubara, Org. Lett., 2013, 15, 856–859 CrossRef CAS PubMed.
- Y. Minami, H. Yoshiyasu, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2013, 52, 883–887 CrossRef CAS PubMed.
- M. P. Watson and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 12594–12595 CrossRef CAS PubMed.
- Y. Nakao, S. Ebata, A. Yada, T. Hiyama, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2008, 130, 12874–12875 CrossRef CAS PubMed.
-
(a) M. Tobisu, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2006, 128, 8152–8153 CrossRef CAS PubMed;
(b) F. L. Taw, P. S. White, R. G. Bergman and M. Brookhart, J. Am. Chem. Soc., 2002, 124, 4192–4193 CrossRef CAS PubMed;
(c) F. L. Taw, A. H. Mueller, R. G. Bergman and M. Brookhart, J. Am. Chem. Soc., 2003, 125, 9808–9813 CrossRef CAS PubMed.
- M. Tobisu, Y. Kita, Y. Ano and N. Chatani, J. Am. Chem. Soc., 2008, 130, 15982–15989 CrossRef CAS PubMed.
- M. Tobisu, J. Hasegawa, Y. Kita, H. Kinutab and N. Chatani, Chem. Commun., 2012, 48, 11437–11439 RSC.
- M. Sun, H.-Y. Zhang, Q. Han, K. Yang and S.-D. Yang, Chem.–Eur. J., 2011, 17, 9566–9570 CrossRef CAS PubMed.
-
(a) M. Tobisu, H. Kinuta, Y. Kita, E. Rémond and N. Chatani, J. Am. Chem. Soc., 2012, 134, 115–118 CrossRef CAS PubMed;
(b) H. Kinuta, Y. Kita, E. Rémond, M. Tobisu and N. Chatani, Synthesis, 2012, 44, 2999–3002 CrossRef CAS PubMed;
(c) Y.-Y. Jiang, H.-Z. Yu and Y. Fu, Organometallics, 2013, 32, 926–936 CrossRef CAS;
(d) H. Kinuta, H. Takahashi, M. Tobisu, S. Mori and N. Chatani, Bull. Chem. Soc. Jpn., 2014, 87, 655–669 CrossRef CAS.
-
(a) M. Tobisu, R. Nakamura, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2009, 131, 3174–3175 CrossRef CAS PubMed;
(b) M. Tobisu, R. Nakamura, Y. Kita and N. Chatani, Bull. Korean Chem. Soc., 2010, 31, 582–587 CrossRef CAS.
- T. Patra, S. Agasti, Akanksha and D. Maiti, Chem. Commun., 2013, 49, 69–71 RSC.
- T. Patra, S. Agasti, A. Modak and D. Maiti, Chem. Commun., 2013, 49, 8362–8364 RSC.
- M. Weidauer, C. I. Someya, E. Irran and S. Enthaler, Asian J. Org. Chem., 2013, 2, 150–156 CrossRef CAS.
- J. A. Miller, Tetrahedron Lett., 2001, 42, 6991–6993 CrossRef CAS.
- J. A. Miller and J. W. Dankwardt, Tetrahedron Lett., 2003, 44, 1907–1910 CrossRef CAS.
- D.-G. Yu, M. Yu, B.-T. Guan, B.-J. Li, Y. Zheng, Z.-H. Wu and Z.-J. Shi, Org. Lett., 2009, 11, 3374–3377 CrossRef CAS PubMed.
- N. Liu and Z. Wang, Adv. Synth. Catal., 2012, 354, 1641–1645 CrossRef CAS.
- Y. Kita, M. Tobisu and N. Chatani, Org. Lett., 2010, 12, 1864–1867 CrossRef CAS PubMed.
- H. Nakazawa, K. Kamata and M. Itazaki, Chem. Commun., 2005, 4004–4006 RSC.
- S. Zheng, C. Yu and Z. Shen, Org. Lett., 2012, 14, 3644–3647 CrossRef CAS PubMed.
- Z. Jiang, Q. Huang, S. Chen, L. Long and X. Zhou, Adv. Synth. Catal., 2012, 354, 589–592 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.