Nanomaterials for electrochemical non-enzymatic glucose biosensors
Peng Sib, Youju Huangb, Taihong Wanga and Jianmin Ma*a
aKey Laboratory for Micro-Nano Optoelectronic Devices of Ministry of Education, State Key Laboratory for Chemo/Biosensing and Chemometrics, Hunan University, 410082, Changsha, P. R. China. E-mail: nanoelechem@hnu.edu.cn
bDivision of Bioengineering, School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore, 637457, Singapore
First published on 4th December 2012
Abstract
This review overviews the recent development of nanomaterials for the application of electrochemical non-enzymatic glucose biosensors. The electrocatalytic mechanism and glucose sensing performance of a variety of nanostructured materials including metallic nanoparticles, metal oxides, metal complexes, alloys and carbon nanomaterials are discussed. The merits and shortfalls of each nanomaterial as electrocatalyst for non-enzymatic biosensing are evaluated and the prospects of non-enzymatic glucose biosensors are presented.
 Peng Si | Peng Si is a final year PhD student in the School of Chemical and Biomedical Engineering at Nanyang Technological University (Singapore). He obtained his BSc degree in Biotechnology at Jilin University (China) in 2009 and he received the Nanyang Research Scholarship for his PhD program. His research interests include synthesis of functional nanomaterials, electrochemical biosensors and bio-nano interfaces. |
 Youju Huang | Youju Huang received a BSc in Polymer Science and Engineering from Anhui University in 2005, and a PhD in Synchrotron Radiation and Application with Professor Liangbin Li from University of Science and Technology of China in 2010. He is currently a postdoctoral fellow in School of Chemical and Biomedical Engineering at Nanyang Technological University, Singapore. His research interests include supramolecular self-assembly, the synthesis of noble metal nanostructures with controlled sizes and shapes, and the optical studies and applications of single noble metal nanoparticles. |
 Taihong Wang | Taihong Wang was born in November 1966 and obtained his PhD from the Graduate School of the Chinese Academy of Sciences. His main scientific interests are in the areas of sensors and lithium ion batteries. |
 Jianmin Ma | Jianmin Ma received his BSc in Chemistry at Shanxi Normal University in 2003, and PhD in Materials Physics and Chemistry with Professor Wenjun Zheng at Nankai University in 2011. He did postdoctoral research at the University of Texas at Austin and Nanyang Technological University, respectively. He joined the School of Physics and Microelectronics, Hunan University since 2012, as an associate professor. His current scientific interests are in the areas of nanomaterials, electrolytes, electrochemical devices and sensors. |
1 Introduction
Diabetes is a condition marked by the inability of the body to properly manage the level of glucose in the blood. In type 1 diabetes, the body does not make insulin, the hormone that regulates the usage of sugar in a human body. Individuals with type 1 diabetes rely on the regular monitoring of blood glucose concentration and intermittent injection of insulin to keep their blood sugar level normal. The blood glucose test is usually conducted by the patients themselves at home by pricking their fingers several times in a day to collect blood, which is then dropped on a test strip. Without strict control of glucose, type 1 diabetic individuals suffer from serious and chronic complications, such as blindness and tissue damage. Type 2 diabetes mellitus results from insulin resistance and relative deficiency of insulin secretion. Constituting 90% of the diabetes cases, type 2 diabetics may suffer from complications such as heart disease, stroke and kidney failure. Therefore, routine measurement of blood sugar level is also necessary for type 2 diabetics.The glucose biosensor developed rapidly since its invention 50 years ago, and currently occupies approximately 85% of the entire biosensor market. Besides blood glucose measurement, glucose biosensors are also widely used in bioindustrial process monitoring, quality control and fuel cells. Although commercially available glucose sensing devices are dominated by enzymatic systems, the last decade has seen an increasing research interest in non-enzymatic glucose sensors, especially those based on nanomaterials. The emerging nanotechnology has brought new opportunities and inspirations for the development of innovative non-enzymatic glucose sensors. The nanostructured electrocatalysts promise to solve the problems associated with non-enzymatic electrode such as poor selectivity and surface fouling, and nanomaterials based non-enzymatic biosensors show significantly higher sensitivity than enzymatic systems. Two comprehensive reviews on electrochemical non-enzymatic glucose sensors have been published by Park et al.1 and Toghill et al.2 in 2005 and 2010, respectively. We feel the necessity to write this review because the recent two years has seen the number of publications on nanomaterials based non-enzymatic glucose sensors increasing at a considerable rate. There are several excellent reviews on electrochemical enzymatic glucose biosensors reported by Wang3,4 and Heller et al.,5 therefore, we just briefly discussed the history of enzymatic systems herein.
1.1 Brief history of enzymatic glucose biosensor
The first-generation glucose sensors were pioneered by Clark and Lyons in the 1950s and 1960s.6 Their devices relied on a thin layer of glucose oxidase (GOx) enzyme, immobilized over an oxygen electrode (via a semipermeable dialysis membrane), and monitored the amount of oxygen consumed by the enzyme-catalyzed reaction by electrochemical method.7 The reaction could be explained by the following equation: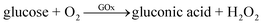 | (1) |
A negative potential was applied to the platinum cathode for a reductive detection of the oxygen consumption:
| O2 + 4H+ + 4e− → 2H2O | (2) |
The technique was further developed by Updike and Hicks,8 who employed additional oxygen working electrode (without enzyme) and measured the differential current between two working electrodes to correct for the oxygen background variation in samples. Subsequently, Guilbault and Lubrano9 introduced another enzyme electrode for blood glucose measurement based on amperometric monitoring of the hydrogen peroxide as a product:
| H2O2 → O2 + 2H+ + 2e− | (3) |
The drawbacks of first-generation glucose sensor are strong dependence of ambient oxygen and too high applied potentials for reduction of oxygen or oxidation of H2O2. Both the high negative and positive potentials may cause serious interfering reactions of electroactive compounds (e.g., ascorbate, urate and paracetamol) in the blood if a size-selective membrane is not present.
The second-generation glucose sensors are based on mediators and have been introduced in 1980s.10,11 Mediators are small, soluble redox active molecules (e.g., ferrocene derivates, ferrocyanide, conducting organic salts and quinones) capable of undergoing rapid and reversible redox reactions, shuttling the electrons between the redox center at the active site of enzyme and the electrode surface. The mechanism of the second-generation biosensors could be understood by the following equations:
| glucose + GOx(FAD) → gluconic acid + GOx(FADH2) | (4) |
| GOx(FADH2) + 2M(ox) + 2e− → GOx(FAD) + 2M(red) + 2H+ | (5) |
| 2M(red) → 2M(ox) + 2e− | (6) |
The ultimate goal of glucose sensing is to eliminate the usage of a mediator, even enzyme to lower the fabrication cost and complexity while increasing the durability of the glucose sensor. The third-generation glucose sensor enables direct electron transfer between the redox center of enzyme and electrode, leading to a very high sensitivity and reproducibility without using mediators. The system could be operated a low potential which is close to the physiological enzyme redox potential of ∼0.44 V.12 The mechanism of the third-generation of glucose biosensors can be explained by the following reactions:13,14
| glucose + GOx(FAD) → gluconic acid + GOx(FADH2) | (7) |
| GOx(FADH2) + 2e− → GOx(FAD) + 2H+ | (8) |
Due to a significantly lowered operating potential, the interferential responses of electroactive species are also greatly diminished. The third-generation glucose sensor is well suited for the in vivo monitoring of blood sugar due to its stability and biocompatibility. However, it suffers from relatively smaller linear range compared to the first-and second-generation glucose sensors. Therefore, the implantable glucose monitors on the market currently are still based on the concept of the first-generation ones. Further effort is needed to improve the performance of the third-generation of glucose sensors in order to meet the commercialization criterion.
1.2 Advantages of glucose sensing without enzyme
The enzymatic glucose biosensor has dominated the glucose sensor industry for more than 20 years. However, a number of critical drawbacks hinder its further development. In the first place, the enzyme-based glucose biosensor suffers from serious stability issues due to the intrinsic nature of protein. Although GOx is more stable than other enzymes, it is still constrained to pH range from 2 to 8, temperature below 44 °C and ambient humidity levels. In addition, GOx can be deactivated by a variety of detergents. In the presence of sodium n-dodecyl sulfate at low pH and hexadecyltrimethylammonium bromide at high pH, GOx quickly loses its activity.15,16 The thermal and chemical instability of GOx prevents the enzymatic biosensor from continuously monitoring fermentation process or in human bodies which requires sterilization. Ensuring the stability of enzymatic glucose biosensor requires considerate attention, with a variety of elaborate fabrication strategies including electropolymerization of enzyme within a polymer,16 covalent cross-linking of enzyme at a pre-treated electrode surface,17 sol–gel entrapment of enzyme,18 electrochemical ‘wiring’ of GOx to mediated polymer chains,19etc. These efforts ensure the short-term stability of enzymatic glucose biosensor and enable it for single-time disposable usage. Despite the aforementioned contributions having alleviated the problem of enzyme-based electrodes to a certain extent, the glucose sensor based on GOx is easily exposed to harsh thermal and chemical conditions during fabrication, storage and usage. In addition, as diabetes continues to rise in the developing countries, the glucose biosensor with a high fabrication cost and short shelf-life become less viable. All those explain why enzymeless glucose biosensors attract tremendous research interest.1.3 Mechanisms of non-enzymatic electrooxidation of glucose
Non-enzymatic glucose biosensors are based on the glucose oxidation reaction catalysed by a variety of electrocatalyts, including metals (Au, Pt, Pd, etc.), metal oxides (Co3O4, CuO, RuO2, etc.), alloys (PtPb, PtRu, etc.), complexes (cobalt phthalocyanine tetrasulfonate, nickel hexacyanoferrate, etc.) and carbon (carbon nanotubes, boron doped diamond, etc.). Except for the last category, all other catalysts listed above contain a transition metal center. There are generally two widely accepted theories that explain the electrooxidation of glucose by transition-metal containing electrocatalysts. Pletcher20 proposed activated chemisorption model which suggested that the electrocatalytic oxidation of glucose occurs via a concentrated step, i.e., the adsorption of glucose molecule on the surface of metal-containing electrocatalyst followed by the abstraction of hemiactalic hydrogen atom. The adsorption process presumably involves the formation of suitable bonds between the adsorbate and the transition-metal substrate with d-electrons and d-orbitals. The hydrogen removal process is deemed as the rate-determining step in most glucose electrooxidation experiments, and is generally considered to occur simultaneously with the chemisorption of analytes. Hence, the adjacent metal active center is occupied by a single absorbate each time, implying well-spaced adsorption sites on the surface of electrocatalysts with suitable geometry could contribute to the kinetic enhancement of glucose oxidation process. Another model, namely ‘Incipient Hydrous Oxide Adatom Mediator’ (IHOAM) proposed by Burke,21 which involved discussing the role of hydroxyl radicals in the electrocatalytic process. IHOAM was based on the observation that ‘active’ metal surface atoms undergo a pre-monolyaer oxidation step that forms an incipient hydrous oxide layer of OHads, which mediates the electrooxiation of glucose and many other organic molecules and inhibits many kinetically slow reduction process. The ‘active’ atoms on the electrode surface are considered to have a low lattice co-ordination value, and lack normal lattice stabilization energy. Polycrystalline surfaces at discontinuous areas such as grain boundaries and edges, are more easily exposed to the solution than the bulk crystal surface, thus they are more liable to undergo pre-monolayer oxidation at lower potentials. According to the IHOAM model, electrocatalytic oxidation of glucose is initiated by the formation of hydrous species on the electrode surface followed by the chemisorption of glucose molecules. After that, the hydrous pre-monolayer could mediate the electrooxidation of absorbed glucose at significantly low potential, with the regeneration of metal surface. The surface of the ‘active’ metal is then oxidized again by oxygen species at certain potential and glucose is continuously oxidized during the repetitive cycling process. Both the activated chemisorption model and IHOAM model will be discussed in the following sections regarding to different electrode materials. IHOAM model explains the enhancement of electrocatalytic performance of most noble metal-based materials. The hydroxyl group also plays important role in the electrocatalytic process of nickel- and copper-based electrodes. However, in these cases the incipient hydrous pre-monolayer is formed not to induce catalysis, but rather to change the oxidation state of the metal hydroxide.2 Nanomaterials used for electrocatalytic glucose sensing
2.1 Transition metals
Transition metals are well known to be good electrocatalysts due to either their ability to adopt multiple oxidation states and absorb other species on their surfaces to form intermediates and activate them in the reaction process. The advantages of nanostructured metallic materials are their unique physical, chemical, optical and electrical properties such as high surface-to-volume ratio, high index facets, large specific surface area, good electrical conductivity, tunable optical property and high electrocatalytic activity.22–28 Therefore, transition metal nanomaterials could serve as effective catalysts due to their high ratio of surface atoms with free valances to the cluster of total atoms and the resulting enhanced mass transport property. A wide range of transition metal nanomaterials have been studied in recent years for the electrocatalytic biosensing. According to the literatures, gold (Au), platinum (Pt), palladium (Pd), copper (Cu) and nickel (Ni) are the most intensively studied transition metal nanomaterials for non-enzymatic catalysis of glucose oxidation.| Electrode materials | Sensitivity (μA mM−1 cm−2) | Linear range (mM) | LOD (μM, S/N = 3) | Operation potential (V) | Medium | Ref. |
|---|---|---|---|---|---|---|
| a Au NPs: Au nanoparticles; Au E: Au electrode; ITO: Indium tin oxide; GCE: glassy carbon electrode. | ||||||
| Au nanowire array/glass | 309 | Up to 10 | 50 | −0.4 vs. Hg/HgO | 0.1 M NaOH | 32 |
| Porous Au/Au E | 11.8 | 2–10 | 5 | +0.35 vs. SCE | PBS (pH 7.4) | 44 |
| Au NPs/Au E | 179 | 0–8 | 0..05 | +0.16 vs. Ag/AgCl | PBS (pH 9.2) | 40 |
| Au NPs/porous silica film | 76 | 0.2–70 | 100 | — | 0.1 M NaOH | 45 |
| Au NPs/graphene paper | 52.36 | 0.01–46 | 5 | +0.08 vs. Ag/AgCl | PBS (pH 7.4) | 47 |
| Layer-by-layer Au NPs/Au E | 160 | Up to 8 | 500 | — | 0.1 M NaOH | 39 |
| Au NPs/ITO | 183.5 | 0.004–0.5 | — | +0.2 vs. SCE | 0.01 M NaOH | 41 |
| Au nanofilm | 57.5 | Up to 57.5 | 0.72 | +0.3 vs. SCE | PBS (pH 7.4) | 42 |
| Au micropillar arrays | 13.2 | 0.5–9 | 60 | — | PBS (pH 7.4) | 37 |
| Au nanotube arrays | 1.13 | 1–42.5 | 10 | +0.25 vs. SCE | PBS (pH 7.4) | 31 |
| Au nanocorals | 22.6 | 0.05–30 | 10 | +0.2 vs. Ag/AgCl | PBS (pH 7.4) | 46 |
| Au NPs/chitosan/GCE | — | 0.4–10.7 | 370 | — | PBS (pH 7.1) | 29 |
Nanoporous platinum is the most intensively studied Pt nanomaterials for non-enzymatic glucose detection because they show remarkably high sensitivity and excellent property to resist interferences due to the increased roughness factor on the electrode surface. The use of nanoporous Pt electrode for non-enzymatic glucose sensing was first investigated by Park et al.53 The roughness of nanoscopic dimension of Pt could selectively enhance the current of sluggish reaction, thus the porous Pt electrode showed much more sensitive current signals of glucose than to other electroactive compounds, such as AA and acetamidophenol. In addition, the biosensor showed excellent antifouling property. A good sensitivity of 9.6 μA mM−1 cm−2 with linear range up to 10 mM was observed reproducibly in the presence of high concentration of chloride ions. Following this study, a variety of nanoporous Pt electrodes54–59 have been fabricated for the non-enzymatic detection of glucose with enhanced sensitivity. Except nanoporous Pt electrode, Pt nanoparticles60–62 and other forms of Pt nanomaterials63–66 have also been studied. For example, highly ordered Pt nanotubule arrays were fabricated by electrodeposition and used for enzymeless glucose sensing in either 0.5 M H2SO4 solution or in PBS (pH 7.4).64 The biosensor exhibited a linear range of 2–14 mM, with a sensitivity of 0.1 μA mM−1 cm−2 and detection limit of 0.1 μM glucose in PBS. Electrode modified with 3D dendritic Pt nanostructures65 was employed for electrocatalytic glucose biosensing without enzyme in PBS solution (pH 7.4). It achieved a linear range of 1–20 mM, with a detection limit down to 1.2 μM and a sensitivity of 12.1 μA mM−1 cm−2. The glucose biosensing performance of various nanostructured Pt electrodes is compared in Table 2.
| Electrode materials | Sensitivity (μA mM−1 cm−2) | Linear range (mM) | LOD (μM, S/N = 3) | Operation potential (V) | Medium | Ref. |
|---|---|---|---|---|---|---|
| a MWCNTs: multi-walled carbon nanotubes | ||||||
| Pt mesoporous film | 9.6 | 0–10 | — | +0.4 vs. Ag/AgCl | PBS (pH 7.4) | 53 |
| 3D ordered macroporous Pt | 31.3 | 1–10 | 0.1 | +0.5 vs. SCE | PBS (pH 9.18) | 55 |
| Nanoporous Pt | 291 | 0–10 | — | +0.4 vs. Ag/AgCl | PBS (pH 7.4) | 56 |
| 3D nanoporous Pt | 642 | 0.1–1.5 | — | +0.4 vs. Ag/AgCl | PBS (pH 7.4) | 54 |
| Nanoporous Pt microsensor | 37.5 | 0.05–30 | — | +0.4 vs. Ag/AgCl | PBS (pH 7.4) | 58 |
| Nanoporous Pt thin film | 1.65 | 1–10 | 97 | +0.4 vs. Ag/AgCl | PBS (pH 7.4) | 59 |
| Ultrafine Pt NPs | 137.7 | 0.2–3.2 | 5 | +0.6 vs. Ag/AgCl | PBS (pH 6.8) | 61 |
| Pt/MWCNTsa | 11.83 | 1–23 | 50 | +0.5 vs. Ag/AgCl | 0.1 M NaOH | 62 |
| Pt nanotube arrays | 0.1 | 2–14 | 1.0 | +0.4 vs. SCE | PBS (pH 7.4) | 64 |
| 3D dendritic Pt nanostructures | 12.1 | 1–20 | 1.2 | +0.5 vs. SCE | PBS (pH 7.4) | 65 |
| Ni(OH)2 → NiOOH + H+ + e− | (9) |
Immersion of Ni electrode into alkaline solution results in the formation of Ni(OH)2, and upon electrooxidation the corresponding NiOOH species are formed. Similar to Au, Pt and Pd electrodes, the rate determining step of glucose oxidation at Ni-based electrodes is the process of abstraction of hydrogen atom at C1 carbon. Upon the formation of Ni(III) oxyhydroxide, glucose will be oxidized immediately and produce a radical intermediate, which in turn reacts rapidly with hydroxyl anions in the solution to form gluconolactone.
Ni nanomaterials based non-enzyamtic glucose sensors has been reported in recent years with enhanced performance compared to bulk Ni electrode.74–81 Wang's group78 has reported highly ordered Ni nanowire arrays (Ni NWA) for the non-enzymatic quantification of glucose. The Ni NWA-based electrode showed a wide linear range from 0.5 μM to 7.0 mM, with a high sensitivity of 1043 μA mM−1 cm−2 and a low detection limit of 0.1 μM. Liu et al.77 reported a non-enzymatic glucose biosensor based on electrospun Ni nanoparticle-loaded carbon fibre paste electrode. The biosensor exhibited a low detection limit of 1 mM with a linear range of 2 mM to 2.5 mM. Ni nanoparticles (Ni NPs) decorated straight multi-walled carbon nanotubes (SMWNTs) has also been fabricated and used for enzymeless glucose detection.79 The Ni NPs/SMWNTs hybrid material based biosensor achieved a linear range from 1 μM to 1 mM with a detection limit of 500 nM (3σ). In addition, the nanohybrid modified electrode exhibited excellent anti-interference property and robust stability. Ni-based nanomaterials seem to be the most sensitive electrode materials for non-enzymatic glucose oxidation, with reported sensitivity as high as mA mM−1 cm−2 in many publications.78,80,81 The highest sensitivity reported is Ni nanoflakes on Ti substrate,80 which achieved a current density of 7.32 mA mM−1 cm−2. However, the most serious problem of Ni nanomaterials based non-enzymatic glucose sensors is their inability to practically detect glucose in physiological pH solution, because electrocatalytic performance of NiOOH is highly dependent on the presence of hydroxyl ions in the electrolyte medium.82 In addition, although Ni-based electrode does not have the problem of surface-fouling by chloride ions,77 it suffers from poor selectivity. According to Fleischmann's study,73 Ni-based electrode is also capable to oxidize a number of other small organic molecules at the same potential immediately after the formation of NiOOH. Most small organic molecules could oxidize on Ni electrode surface at ∼+0.5 V in alkaline solution. Except for glucose, ethanol is the major component that could easily oxidize on Ni-based electrodes.
| Electrode materials | Sensitivity (μA mM−1 cm−2) | Linear range (mM) | LOD (μM, S/N = 3) | Operation potential (V) | Medium | Ref. |
|---|---|---|---|---|---|---|
| Ni nanowire arrays | 1043 | 0.0005–7 | 0.1 | +0.55 vs. SCE | 0.1 M NaOH | 78 |
| Ni NP/carbon fiber paste | 420.4 | 0.002–2.5 | 1 | +0.6 vs. Ag/AgCl | 0.1 M NaOH | 77 |
| Ni NP/SMWNTs | 1438 | 0.001–1 | 0.5 | +0.4 vs. SCE | 0.1 M NaOH | 78 |
| Ni nanoflakes | 7320 | 0.05–0.6 | 1.2 | +0.5 vs. SCE | 0.5 M NaOH | 80 |
| Cu NPs | 3600 | 0.003–10 | 0.7 | +0.7 vs. SCE | 0.1 M NaOH | 83 |
| Cu polyhedron nanostructures | — | 0.2–4.2/4.2–32.1 | 70 | +0.5 vs. SCE | 0.1 M NaOH | 88 |
| Cu nanowires | 927 | 0.002–0.075 | 1 | +0.7 vs. SCE | 0.1 M NaOH | 91 |
| Cu nanostructures | 420 | Up to 3 | 0.035 | +0.6 vs. Ag/AgCl | 0.05 M NaOH | 92 |
| Cu nanobelts | 4433 | 0.01–1.13 | 10 | +0.6 vs. Ag/AgCl | 0.05 M NaOH | 93 |
| Cu nanocluster/MWCNTs | 253 | 0.7–3.5 | 0.21 | +0.65 vs. Ag/AgCl | 0.02 M NaOH | 94 |
| Cu nanocubes /MWCNTs | 1096 | Up to 7.5 | 1 | +0.55 vs. Ag/AgCl | 0.1 M NaOH | 89 |
| Cu NPs/MWCNTs | 714 | 0.01–0.3 | 0.5 | +0.35 vs. SCE | 0.02 M NaOH | 87 |
| Cu NPs/SWCNTs | 3657 | Up to 0.5 | 0.25 | +0.65 vs. Ag/AgCl | 0.02 M NaOH | 95 |
| Cu NPs/graphene | — | Up to 4.5 | 0.5 | +0.5 vs. SCE | 0.1 M NaOH | 84 |
| Cu NPs/graphene | 607 | 0.005–1.4 | 0.2 | +0.5 vs. SCE | 0.1 M NaOH | 85 |
2.2 Metal oxides and complexes
| 2CoO2 + C6H12O6(glucose) → 2CoOOH + C6H10O6(gluconolactone) | (10) |
The Co3O4 NFs-based glucose sensor showed a fast response within 7 s, a high sensitivity of 36.25 μA mM−1 cm−2, a detection limit of 0.97 μM with a linear range up to 2.04 mM. Followed by this Co3O4 study, Kung et al.97 reported acicular cobalt oxide nanorods (CoO NRs) synthesized by chemical bath deposition for the non-enzymatic detection of glucose. The study claimed that CoO NRs-modified electrode could selectively detect glucose without the casting of Nafion, which is a negatively charged membrane capable to exclude negatively charged interferential molecules. The sensitivity and LOD of CoO NRs-based glucose sensor are 571.8 μA mM−1 cm−2 and 0.058 μM, which is 15 times higher and 10 times lower than Ding's electrode. In addition, the biosensor also exhibited a wider linear range up to 3.5 mM and lower detection potential of 0.5 V than Ding's sensor. Simultaneously, Dong et al.98 reported a composite material of 3D graphene-Co3O4 nanowires as a freestanding electrode for non-enzymatic glucose sensing. The biosensor achieved a sensitivity of 3.39 mA mM−1 cm−2 and a LOD of 25 nM, which are nearly 6 times higher and 2 times lower than those of CoO NRs-modified electrode in Kung's study. However, the up to 80 μM linear range of 3D graphene/Co3O4 nanowire electrode is much smaller than those of both Ding's and Kung's biosensors. Very recently, a graphene/cobalt oxide-based needle electrode has been reported by the same group of Dong.99 The biosensor showed a capability for glucose detection in micro-droplets within the linear concentration range of 50–300 μM.
| NiO + OH− → Ni(OH)2 | (11) |
| Ni(OH)2 + OH− → NiOOH + H2O + e− | (12) |
| NiOOH + glucose → Ni(OH)2 + gluconolactone | (13) |
A variety of NiO nanomaterials have reported in recent years by a number of groups for the study of non-enzymatic glucose sensing performance.119–126 Mu and colleagues118 have investigated the electrochemical properties and electroanalytical performance of nano-NiO modified CPE toward the oxidation of glucose. The biosensor showed a quick response to the glucose within 5 s and a calibration plot in the concentration range of 1–110 μM, with a sensitivity of 43.9 nA μM−1 cm−2 and a LOD of 0.16 μM. NiO/MWCNTs nanocomposite-based enzymeless glucose biosensors have been reported by Shamsipur124 and Zhang,125 respectively. MWCNTs were found to improve the reactivity of NiO for glucose oxidation remarkably. Shamsipur's electrode showed a wide linear range of 0.2 mM–20 mM with a LOD of 0.16 mM, while Zhang's biosensor exhibited a high sensitivity of 1.77 mA mM−1 cm−2 with linear range up to 7 mM and a LOD of 2 μM.
| Electrode materials | Sensitivity (μA mM−1 cm−2) | Linear range (mM) | LOD (μM, S/N = 3) | Operation potential (V) | Medium | Ref. |
|---|---|---|---|---|---|---|
| Co3O4 nanofibers | 36.25 | Up to 2.04 | 0.97 | +0.59 vs. Ag/AgCl | 0.1 M NaOH | 96 |
| Co3O4/3D graphene | 3390 | Up to 0.08 | 0.025 | +0.58 vs. Ag/AgCl | 0.1 M NaOH | 98 |
| NiO/MWCNTs | 1770 | Up to 7 | 2 | +0.5 vs. Ag/AgCl | 0.12 M NaOH | 125 |
| CuO nanofibers | 431.3 | 0.006–2.5 | 0.8 | +0.4 vs. SCE | 0.1 M NaOH | 115 |
| CuXO nanowires | 1620 | Up to 4.0 | 49 | +0.5 vs. Ag/AgCl | 0.1 M NaOH | 104 |
| CuO NP/MWCNTs | 2596 | Up to 1.2 | 0.2 | +0.4 vs. Ag/AgCl | 0.1 M NaOH | 117 |
| MnO2/MWNCTs | 396 | Up to 28 | — | +0.3 vs. Ag/AgCl | 0.1 M NaOH | 127 |
| Mn3O4/3D graphene | 360 | 0.1–8 | 10 | +0.4 vs. Ag/AgCl | 0.1 M NaOH | 128 |
| ZnO nanorods | 5.6 | 0.01–10 | 0.5 | — | PBS (pH 7.4) | 129 |
| Ag2O nanowalls | — | 0.2–3.2 | 10 | +0.4 vs. Ag/AgCl | 0.1 M NaOH | 131 |
| Fe2O3 nanowire arrays | 726 | 0.015–8 | 6 | +0.52 vs. SCE | PBS (pH 7.5) | 132 |
2.3 Bimetallic systems
The bimetallic material could become a superior catalyst that exhibits desired electronic property and very high catalytic activity. Alloys, adatoms and metal (oxide)/metal oxide composites are the major forms of bimetallic electrocatalysts. Due to the synergistic effect of the two materials, the bimetallic systems can significantly enhance the electrooxidation of glucose and reduce the interference and poisoning effect of the electrode. In addition, most of the bimetallic systems were able to catalyse the glucose oxidation in physiological pH solution. Therefore, the study of bimetallic materials for non-enzymatic glucose sensing has attracted tremendous research interest in recent years.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)/MWCNTs/IL modified GCE exhibited the strongest electrocatalytic activity toward glucose oxidation in neutral pH media. The biosensor achieved a linear range up to 15 mM, with a sensitivity of 10.7 μA mM−1 cm−2 and LOD of 50 μM. The non-enzymatic glucose biosensor based on CNTs supported PtRu alloy nanoparticles has also been investigated by Li and colleagues,156 who reported a similar linear range to Xiao's work, but with a higher sensitivity of 28.26 μA mM−1 cm−2 and lower LOD of 0.25 μM. On the other hand, the catalytic performances of Cu based alloys of Ni, Fe and Mn have been investigated by Yeo.173 It was observed that the anodic response of Mn5Cu95 to glucose in 0.1 M NaOH solution is larger than other electrodes possibly due to the pre-adsorption of glucose molecules on Mn sites. NiCu alloy based non-enzymatic glucose biosensor was also reported by Jafarian et al.174 Unfortunately, the above mentioned Cu alloyed nanomaterials are not able to determine the glucose concentration in blood since they could not catalyse the glucose oxidation under physiological conditions. The recently reported a 3D Au@nanoporous Cu (Au@NPC) core-shell composite material may be promising to solve the biosensing problem associated with Cu alloyed nanomaterials.172 As was reported, the Au@NPC alloy showed strong oxidation peak of glucose in 0.1 M PBS (pH 7.4) and the nanocomposite modified GCE achieved a linear response for glucose oxidation in the range of 3–8 mM in PBS. Nevertheless, other reported Au–Cu nanoparticle alloy modified carbon electrode did not show the feature of glucose sensing in neutral pH solution.171 A more detailed comparison of the analytical performance of alloy nanomaterials based non-enzymatic glucose biosensors is listed in Table 5.
:1)/MWCNTs/IL modified GCE exhibited the strongest electrocatalytic activity toward glucose oxidation in neutral pH media. The biosensor achieved a linear range up to 15 mM, with a sensitivity of 10.7 μA mM−1 cm−2 and LOD of 50 μM. The non-enzymatic glucose biosensor based on CNTs supported PtRu alloy nanoparticles has also been investigated by Li and colleagues,156 who reported a similar linear range to Xiao's work, but with a higher sensitivity of 28.26 μA mM−1 cm−2 and lower LOD of 0.25 μM. On the other hand, the catalytic performances of Cu based alloys of Ni, Fe and Mn have been investigated by Yeo.173 It was observed that the anodic response of Mn5Cu95 to glucose in 0.1 M NaOH solution is larger than other electrodes possibly due to the pre-adsorption of glucose molecules on Mn sites. NiCu alloy based non-enzymatic glucose biosensor was also reported by Jafarian et al.174 Unfortunately, the above mentioned Cu alloyed nanomaterials are not able to determine the glucose concentration in blood since they could not catalyse the glucose oxidation under physiological conditions. The recently reported a 3D Au@nanoporous Cu (Au@NPC) core-shell composite material may be promising to solve the biosensing problem associated with Cu alloyed nanomaterials.172 As was reported, the Au@NPC alloy showed strong oxidation peak of glucose in 0.1 M PBS (pH 7.4) and the nanocomposite modified GCE achieved a linear response for glucose oxidation in the range of 3–8 mM in PBS. Nevertheless, other reported Au–Cu nanoparticle alloy modified carbon electrode did not show the feature of glucose sensing in neutral pH solution.171 A more detailed comparison of the analytical performance of alloy nanomaterials based non-enzymatic glucose biosensors is listed in Table 5.
| Electrode materials | Sensitivity (μA mM−1 cm−2) | Linear range (mM) | LOD (μM, S/N = 3) | Operation potential (V) | Medium | Ref. |
|---|---|---|---|---|---|---|
| Pt2Pb NPs | — | 0–10 | — | −0.15 vs. SCE | PBS (pH 7.4) | 153 |
| PtPb nanoporous networks | 10.8 | 1–16.9 | — | −0.08 vs. Ag/AgCl | PBS (pH 7.4) | 154 |
| PtPb nanowire arrays | 11.25 | Up to 11 | 8 | −0.2 vs. SCE | PBS (pH 7.4) | 152 |
| PtPb NPs/MWCNTs | 18 | Up to 5 | 7 | −0.15 vs. Ag/AgCl | PBS (pH 7.4) | 155 |
| PtRu/MWCNTs/IL | 10.7 | 0.2–15 | 50 | −0.1 vs. SCE | PBS (pH 7.4) | 157 |
| PtRu NPs/MWCNTs | 28.26 | 1–15 | 0.25 | +0.55 vs. Ag/AgCl | 0.1 M NaOH | 156 |
| PtNi nanowires | 920 | 0.002–2 | 1.5 | +0.45 vs. SCE | 0.1 M NaOH | 160 |
| PtNi/g–raphene | 20.42 | Up to 35 | 10 | −0.35 vs. Ag/AgCl | PBS (pH 7.4) | 159 |
| PtIr NPs | 93.7 | — | — | +0.1 vs. Ag/AgCl | PBS (pH 7.4) | 158 |
| PtAu/MWCNTs | 10.71 | Up to 24.44 | 10 | +0.3 vs. Ag/AgCl | PBS (pH 7.4) | 167 |
| PtAu/C nanocomposites | 4.7 | 0–10 | 2 | +0.35 vs. Ag/AgCl | PBS (pH 7.4) | 168 |
| AuRu NPs | 38.3 | 0–15 | 269 | −0.65 vs. Ag/AgCl | 0.1 M NaOH | 170 |
| Au@nanoporous Cu | — | 3–8 | — | +0.27 vs. Ag/AgCl | PBS (pH 7.4) | 172 |
Pletcher20 observed that partial coverage of Au and Pt electrodes with metal adatoms such as Pb could remarkably improve long-term catalytic activity of the electrode and reduce the formation of poisonous species on the electrode surface. It was proposed that Pb atom could interact with two Pt sites to form a surface of Pt atom pairs, which limited the active sites from poisoning across the electrode surface. Au electrodes with adatoms of Ag179,180 and Hg181 have also been explored. Aoun et al.179,180 investigated the electrocatalytic oxidation of glucose on single-crystalline gold electrode with a variety of adatoms including Cu, Ag, Ru, Pt, Pd, and Cd. The highest catalytic activity and largest negative shift of the overpotential for glucose oxidation was achieved by the Au electrode with 1/3 monolayer of Ag adatoms. Glucose was readily oxidized on the electrode at −0.5 V (vs. Ag/AgCl), where most of the interferential species are not reactive. In another study, Hg adatoms on Au also allows for vast enhancement of the glucose oxidation current compared to bare Au electrode in alkaline solution.181 It was supposed that the Hg adatoms increased the amount of absorbed OH− on Au electrode that results in the improvement of the oxidation current response.
| Electrode materials | Sensitivity (μA mM−1 cm−2) | Linear range (mM) | LOD (μM, S/N = 3) | Operation potential (V) | Medium | Ref. |
|---|---|---|---|---|---|---|
| Ag/NiO nanofibers | 19.3/170.2 | Up to 0.59/up to 2.63 | 1.37/0.72 | +0.1/+0.6 vs. Ag/AgCl | 0.1 M NaOH | 182 |
| Pt/NiO nanofibers | 180.8 | Up to 3.67 | 0.313 | +0.6 vs. Ag/AgCl | 0.1 M NaOH | 66 |
| Porous Cu/NiO nanocomposites | 171.8 | 0.5–5 | 0.5 | +0.4 vs. Ag/AgCl | 0.1 M NaOH | 90 |
| Cu NPs/ZnO | — | 0.001–1.5 | 0.2 | +0.8 vs. Ag/AgCl | 0.1 M NaOH | 183 |
| Cu/Cu2O hollow microspheres | 33.63 | 0.22–10.89 | 0.05 | +0.45 vs. SCE | 0.1 M NaOH | 108 |
| Cu/CuO nanowires | — | 0.1–12 | 50 | +0.3 vs. SCE | 0.1 M NaOH | 109 |
| Pd/CuO nanofibers | 1061 | 0.2–2.5 | 0.019 | +0.32 vs. SCE | 0.1 M NaOH | 185 |
| TiO2/CuO nanotube arrays | 79.79 | Up to 2.0 | 1 | +0.5 vs. SCE | 0.1 M NaOH | 105 |
| CdO/NiO nanofibers | 212.71 | Up to 6.37 | 0.35 | +0.6 vs. Ag/AgCl | 0.1 M NaOH | 123 |
2.4 Carbon
Carbon based materials are the most widely used substrates for the fabrication of electrochemical biosensors due to their electronic conductivity and electrochemical inertia. The screen printed enzymatic electrodes have been industrialized for revolutionising personal glucose monitors since 1990.186,187 Glassy carbon electrode (GCE), carbon paste electrode (CPE) and boron doped diamond (BDD) are the most widely used electrodes in laboratories for electrochemical study and biosensor research. Due to its electrochemical inactivity, bare GCE could only exhibit very small anodic current response to the addition of glucose, according to the investigation by Vassilyev et al. in 1985.51,175 CPE has been widely employed for research purposes because of its simple preparation, low cost and easily renewable surface. BDD electrodes have found increasing applications for electrocatalysis in the past 25 years. With very low capacitive current, highly inert surface, resistance to fouling and wide potential window,81,188 BDD is an ideal electrode substrate for surface modification and electrochemical biosensing.Various new carbon materials have emerged in the last decade with the rapid development of nanotechnology. Among them, nanostructured carbon materials such as fullerene, CNTs, carbon nanofibers, graphene and doped diamond-like materials have been intensively studied as enzymatic and non-enzymatic electrochemical biosensors.189 Due to their good conductivity, high surface area, ease of functionalization and good biocompatibility, the nanostructured carbon materials showed great promise to improve the performance of electrochemical glucose biosensors. In most reported biosensors they still serve as inert supporting materials for catalytic components such as enzymes and electrocatalysts, but recently a number of studies claimed that some carbon nanomaterials, such as CNTs, exhibit direct electrochemical activities. In the following subsections we explore the recent advances of non-enzymatic electrochemical glucose biosensors based on nanostructured BDD, CNTs, graphene and a number of other carbon nanomaterials.
| M(OH*) + M′(R) → M + M′(RO) + H+ + e− | (14) |
Where OH* is the absorbed hydroxyl radicals, R is the absorbed organic species (polyamines, glucose, etc), M and M′ are the different adsorption sites across the BDD electrode. The oxidation of polyamines involves the adsorption of hydroxyl radicals and polyamines on different sites of BDD followed by anodic oxygen transfer and formation of oxidized organic compound intermediate.
Zhao et al.193 has reported a nanocrystalline BDD electrode, which showed high sensitivity, low detection limit, good selectivity and large linear range toward the non-enzymatic glucose detection in sodium hydroxide solution. Similar to the BDD electrode reported by Lee, the nanocrystalline BDD electrode also underwent hydrogen flame treatment to prevent the formation of carboxyl functionalities at high anodic potentials that decrease the conductivity of BDD electrode. Although the quench of glucose signals were observed at BDD electrode and microcrystalline BDD electrode after acid treatment, the nanocrystalline BDD electrode could still maintain a good amperometric response for glucose after 30 min treatment by aqua regia. This interesting observation indicates the surface morphology with regards to the diamond dimension, grain boundaries and boron atoms distribution could strongly affect the electrochemical activity of BDD electrode.
The above proposition is further supported by a recent work by Luo et al.,194 who fabricated boron-doped diamond nanoforest (BDDNF) for the non-enzymatic detection of glucose. Compared to plane BDD, BDDNF exhibited well-defined current response and significantly improved sensitivity. The electrode also showed well distinguished peaks of AA, UA and glucose, which respond at 0.1, 0.3 and 0.6 V, respectively.
Despite the enhanced sensitivity by tailoring the nanostructure of BDD, the linear range of glucose sensors in all three publications are not above 10 mM, which is the major limitation for their practical usage. In addition, the sensitivities of these electrodes are also lower than some metal modified BDD electrodes. For example, nickel nanoparticles modified BDD achieved a sensitivity of 101.9 μA mM−1 cm−2,81 while the value of BDDNF is only 8.1 μA mM−1 cm−2. However, nanocrystalline and nanostructured BDD electrodes still hold great potential for the development of viable and reusable glucose biosensors.
The first CNTs-based non-enzymatic glucose sensor was based on well aligned MWCNTs grown on tantalum substrate, as reported by Ye et al.205 The electrode showed a high sensitivity for glucose oxidation at low overpotential of +0.2 V (vs. Ag/AgCl) in 0.1 M NaOH, with a large linear range up to 11 mM. In addition, the MWCNTs-based non-enzymatic glucose sensor exhibited reproducible current response for glucose in the presence of high concentration of chloride ions, indicating excellent anti-poisoning property of the electrode. However, the interferential species such as AA and UA had a significant impact on glucose detection, possibly due to the overlapped oxidation potentials of these species with glucose. SWCNTs-based non-enzymatic glucose biosensor has been reported by Wang et al.,206 who fabricated freestanding SWCNTs films with large area which were bound to the glass surface by Nafion. The SWCNTs film electrode showed direct oxidation of glucose in alkaline solution at the potential of 0.5 V (vs. Ag/AgCl), and exhibited a fast response less than 10 s to glucose even in the presence of 0.2 M chloride ions. The fabricated biosensor achieved a reproducible high sensitivity of 248.6 μA mM−1 cm−2, which is significantly larger than the value of MWCNTs based electrode, ca. 4.36 μA mM−1 cm−2. However, the linear range of SWCNTs film electrode was limited up to 2.16 mM.
The non-enzymatic glucose biosensors based on CNTs decorated with various other electrocatalysts including metallic nanoparticles, metal oxides, and alloys have been already discussed in the previous sections, which are not repeated here.
Carbon nanofibers (CNFs) are another class of carbon nanomaterials which find many interesting applications in chemical and biosensors. CNFs comprise of well-arranged graphite layers organized into cylindrical structures. Unlike CNTs, CNFs are not hollow in the core. In addition, they expose the edge plane rather than the basal plane of graphene on their surfaces, which provide them with very large surface area and extremely high conductivity.212 Non-enzymatic glucose biosensors based on CNFs have been reported recently. For example, Rathod et al.213 investigated the electrocatalytic performance of CNFs/Pt NPs toward the glucose oxidation under physiological pH conditions. Liu et al.77 developed a non-enzymatic glucose biosensor based on CNFs/Ni NPs, which exhibited a high sensitivity of 420.4 μA mM−1 cm−2 with a wide linear range of 2 μM–2.5 mM.
3 Conclusions and future prospects
Nanomaterials based non-enzymatic glucose biosensors have been continuously and increasingly reported in recent years. With the help of nanotechnology, many problems associated with non-enzymatic glucose sensors have been alleviated or solved. For example, the surface poisoning of Pt electrode by chloride ions was successfully solved by nanoporous Pt electrode and PtPb nanoparticle alloy modified electrode. In addition, some nanostructured catalysts could significantly reduce the overpotential for glucose oxidation, which effectively avoided the interferences by electroactive species in the blood. More importantly, the sensitivity of non-enzymatic glucose sensor is significantly improved by nanomaterials modified electrode, which usually achieved a value of more than 100 μA mM−1 cm−2. The sensitivity is considerably higher than those of non-enzymatic glucose sensors based bulk electrode or most enzymatic glucose biosensors, which are usually less than 10 μA mM−1 cm−2. Nevertheless, the major obstacle that prevents many non-enzymatic glucose biosensors working in human blood is that they are not able to catalyse the glucose oxidation under physiological conditions. This is especially the case for Ni, Cu, metal oxide and carbon nanomaterials based electrodes, in spite of their extraordinarily high sensitivity achieved. The most promising non-enzymatic glucose sensors should be nanoporous Pt electrode and Pt based alloys modified electrode, since they not only effectively resist the surface fouling by chloride ions and avoid the various interferences, but also allow the catalysis of glucose in neural pH solution. The major concern of non-enzymatic electrodes toward commercialization would be the fabrication cost. In addition to blood glucose sensing, nanomaterials based non-enzymatic glucose sensors may also find extensive applications in bioindustrial process monitoring due to their sterilization compatibility. Besides oxygen and pH, we might also be able to monitor the glucose level in a fermentation plant or bioreactor in real time manner. Furthermore, although some non-enzymatic glucose catalysts are not applicable for blood glucose monitoring, they are well suited for application in glucose fuel cells which do not require physiological conditions. In contrast to enzymatic systems which could merely oxidize glucose to gluconolactone, many nanostructured non-enzymatic electrocatalysts are capable to oxidize glucose completely to CO2 and H2O at low potentials, significantly enhancing the energy conversion efficiency. With new inspirations suggested by novel characteristics of nanostructured materials and vast opportunities brought by nanotechnology to innovative non-enzymatic glucose biosensors, it is reasonable to expect the significant advances that revolutionize the glucose biosensor industry in the near future.Acknowledgements
This work was financially supported by the “973” National Key Basic Research Program of China (Grant 2007CB310500), the National Natural Science Foundation of China (Grant No. 21003041 and 21103046) and the Hunan Provincial Natural Science Foundation of China (Grant No. 10JJ1011 and 11JJ7004). Dr Ma gratefully acknowledges the Young Teachers' Growth Plan of Hunan University (No. 2012-118).References
- S. Park, H. Boo and T. D. Chung, Anal. Chim. Acta, 2006, 556, 46–57 CrossRef CAS.
- K. E. Toghill and R. G. Compton, Int. J. Electrochem. Soc., 2010, 5, 1246–1301 CAS.
- J. Wang, Electroanalysis, 2001, 13, 983–988 CrossRef CAS.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS.
- A. Heller and B. Feldman, Chem. Rev., 2008, 108, 2482–2505 CrossRef CAS.
- L. C. Clark Jr and C. Lyons, Ann. N. Y. Acad. Sci., 1962, 102, 29–45 CrossRef CAS.
- S. R. Mikkelsen and E. Cortón, Bioanalytical chemistry, LibreDigital, 2004 Search PubMed.
- S. J. Updike and G. P. Hicks, Nature, 1967, 214, 986–988 CrossRef CAS.
- G. G. Guilbault and G. J. Lubrano, Anal. Chim. Acta, 1973, 64, 439–455 CrossRef CAS.
- A. E. G. Cass, G. Davis, G. D. Francis, H. A. O. Hill, W. J. Aston, I. J. Higgins, E. V. Plotkin, L. D. L. Scott and A. P. F. Turner, Anal. Chem., 1984, 56, 667–671 CrossRef CAS.
- P. D. Hale, T. Inagaki, H. I. Karan, Y. Okamoto and T. A. Skotheim, J. Am. Chem. Soc., 1989, 111, 3482–3484 CrossRef CAS.
- J. Wang, Chem. Rev., 2007, 108, 814–825 CrossRef.
- J. P. Liu, C. X. Guo, C. M. Li, Y. Y. Li, Q. B. Chi, X. T. Huang, L. Liao and T. Yu, Electrochem. Commun., 2009, 11, 202–205 CrossRef CAS.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS.
- R. Wilson and A. P. F. Turner, Biosens. Bioelectron., 1992, 7, 165–185 CrossRef CAS.
- J. Li and X. Q. Lin, Biosens. Bioelectron., 2007, 22, 2898–2905 CrossRef CAS.
- B. L. Wu, G. M. Zhang, S. M. Shuang and M. M. F. Choi, Talanta, 2004, 64, 546–553 CrossRef CAS.
- K. C. Han, Z. J. Wu, J. Lee, I. S. Ahn, J. W. Park, B. R. Min and K. T. Lee, Biochem. Eng. J., 2005, 22, 161–166 CrossRef CAS.
- T. J. Ohara, R. Rajagopalan and A. Heller, Anal. Chem., 1994, 66, 2451–2457 CrossRef CAS.
- D. Pletcher, J. Appl. Electrochem., 1984, 14, 403–415 CrossRef CAS.
- L. D. Burke, Electrochim. Acta, 1994, 39, 1841–1848 CrossRef CAS.
- Y. Huang and D. H. Kim, Langmuir, 2011, 27, 13861–13867 CrossRef CAS.
- Y. Huang and D. H. Kim, Nanoscale, 2011, 3, 3228–3232 RSC.
- Y. Huang and D. H. Kim, Nanoscale, 2012, 4, 6312–6317 RSC.
- L. Wei, Y. J. Fan, H. H. Wang, N. Tian, Z. Y. Zhou and S. G. Sun, Electrochim. Acta, 2012, 76, 468–474 CrossRef CAS.
- L. Wei, Y. J. Fan, N. Tian, Z. Y. Zhou, X. Q. Zhao, B. W. Mao and S. G. Sun, J. Phys. Chem. C, 2012, 116, 2040–2044 CAS.
- J. H. Zeng, J. Mater. Chem., 2012, 22, 3170–3176 RSC.
- Z. L. Xiao, C. Y. Han, W. K. Kwok, H. W. Wang, U. Welp, J. Wang and G. W. Crabtree, J. Am. Chem. Soc., 2004, 126, 2316–2317 CrossRef CAS.
- D. Feng, F. Wang and Z. L. Chen, Sens. Actuators, B, 2009, 138, 539–544 CrossRef.
- H. Ciftci and U. Tamer, React. Funct. Polym., 2012, 72, 127–132 CrossRef CAS.
- Y. G. Zhou, S. Yang, Q. Y. Qian and X. H. Xia, Electrochem. Commun., 2009, 11, 216–219 CrossRef CAS.
- S. Cherevko and C. H. Chung, Sens. Actuators, B, 2009, 142, 216–223 CrossRef.
- F. Y. Kong, X. R. Li, W. W. Zhao, J. J. Xu and H. Y. Chen, Electrochem. Commun., 2012, 14, 59–62 CrossRef CAS.
- A. P. Liu, Q. H. Ren, T. Xu, M. Yuan and W. H. Tang, Sens. Actuators, B, 2012, 162, 135–142 CrossRef CAS.
- F. H. Meng, X. L. Yan, J. G. Liu, J. Gu and Z. G. Zou, Electrochim. Acta, 2011, 56, 4657–4662 CrossRef CAS.
- A. P. Liu, E. J. Liu, G. C. Yang, N. W. Khun and W. G. Ma, Pure Appl. Chem., 2010, 82, 2217–2229 CrossRef CAS.
- R. Prehn, M. Cortina-Puig and F. X. Munoz, J. Electrochem. Soc., 2012, 159, F134–F139 CrossRef CAS.
- T. S. Li, K. Zhu, S. He, X. Xia, S. Q. Liu, Z. Wang and X. Y. Jiang, Analyst, 2011, 136, 2893–2896 RSC.
- F. Kurniawan, V. Tsakova and V. M. Mirsky, Electroanalysis, 2006, 18, 1937–1942 CrossRef CAS.
- B. K. Jena and C. R. Raj, Chem.–Eur. J., 2006, 12, 2702–2708 CrossRef CAS.
- Y. T. Ma, J. W. Di, X. Yan, M. L. Zhao, Z. J. Lu and Y. F. Tu, Biosens. Bioelectron., 2009, 24, 1480–1483 CrossRef CAS.
- W. Zhao, J. J. Xu, C. G. Shi and H. Y. Chen, Electrochem. Commun., 2006, 8, 773–778 CrossRef CAS.
- F. Xiao, Y. Q. Li, X. L. Zan, K. Liao, R. Xu and H. W. Duan, Adv. Funct. Mater., 2012, 22, 2487–2494 CrossRef CAS.
- Y. Li, Y. Y. Song, C. Yang and X. H. Xia, Electrochem. Commun., 2007, 9, 981–988 CrossRef CAS.
- J. J. Yu, S. Lu, J. W. Li, F. Q. Zhao and B. Z. Zeng, J. Solid State Electrochem., 2007, 11, 1211–1219 CrossRef CAS.
- T. M. Cheng, T. K. Huang, H. K. Lin, S. P. Tung, Y. L. Chen, C. Y. Lee and H. T. Chiu, ACS Appl. Mater. Interfaces, 2010, 2, 2773–2780 CAS.
- F. Xiao, J. B. Song, H. C. Gao, X. L. Zan, R. Xu and H. W. Duan, ACS Nano, 2012, 6, 100–110 CrossRef CAS.
- E. M. Skou, Acta Chem. Scand., 1973, 27, 2239–2241 CrossRef CAS.
- D. A. Gough, F. L. Anderson, J. Giner, C. K. Colton and J. S. Soeldner, Anal. Chem., 1978, 50, 941–944 CrossRef CAS.
- U. Gebhardt, G. Luft, G. J. Richter and F. V. Sturm, Bioelectrochem. Bioenerg., 1978, 5, 607–624 CrossRef CAS.
- Y. B. Vassilyev, O. A. Khazova and N. N. Nikolaeva, J. Electroanal. Chem., 1985, 196, 105–125 CrossRef.
- E. S. McLamore, J. Shi, D. Jaroch, C. Barcus, J. Osbourne, A. Uchida, Y. Jiang, K. K. Buhman, M. K. Banks, D. Teegarden, J. L. Rickus and D. M. Porterfield, Biosens. Bioelectron., 2011, 26, 2237–2245 CrossRef CAS.
- S. Park, T. D. Chung and H. C. Kim, Anal. Chem., 2003, 75, 3046–3049 CrossRef CAS.
- J. F. Huang, Electroanalysis, 2008, 20, 2229–2234 CrossRef CAS.
- Y. Y. Song, D. Zhang, W. Gao and X. H. Xia, Chem.–Eur. J., 2005, 11, 2177–2182 CrossRef CAS.
- C. H. Chou, J. C. Chen, C. C. Tai, I. W. Sun and J. M. Zen, Electroanalysis, 2008, 20, 771–775 CrossRef CAS.
- Y. J. Lee, D. J. Park and J. Y. Park, IEEE Sens. J., 2008, 8, 1922–1927 CrossRef CAS.
- Y. J. Lee, D. J. Park, J. Y. Park and Y. Kim, Sensors, 2008, 8, 6154–6164 CrossRef CAS.
- S. Joo, S. Park, T. D. Chung and H. C. Kim, Anal. Sci., 2007, 23, 277–281 CrossRef.
- J. Lu, I. Do, L. T. Drzal, R. M. Worden and I. Lee, ACS Nano, 2008, 2, 1825–1832 CrossRef CAS.
- Z. X. Cao, Y. J. Zou, C. L. Xiang, L. X. Sun and F. Xu, Anal. Lett., 2007, 40, 2116–2127 CrossRef CAS.
- L. H. Li and W. D. Zhang, Microchim. Acta, 2008, 163, 305–311 CrossRef CAS.
- Y. Myung, D. M. Jang, Y. J. Cho, H. S. Kim, J. Park, J. U. Kim, Y. Choi and C. J. Lee, J. Phys. Chem. C, 2009, 113, 1251–1259 CAS.
- J. H. Yuan, K. Wang and X. H. Xia, Adv. Funct. Mater., 2005, 15, 803–809 CrossRef CAS.
- Q. M. Shen, L. P. Jiang, H. Zhang, Q. H. Min, W. H. Hou and J. J. Zhu, J. Phys. Chem. C, 2008, 112, 16385–16392 CAS.
- Y. Ding, Y. X. Liu, L. C. Zhang, Y. Wang, M. Bellagamba, J. Parisi, C. M. Li and Y. Lei, Electrochim. Acta, 2011, 58, 209–214 CrossRef CAS.
- F. L. Qu, H. Y. Sun, Y. Zhang, H. M. Lu and M. H. Yang, Sens. Actuators, B, 2012, 166–167, 837–841 CrossRef CAS.
- Q. Y. Wang, X. Q. Cui, J. L. Chen, X. L. Zheng, C. Liu, T. Y. Xue, H. T. Wang, Z. Jin, L. Qiao and W. T. Zheng, RSC Adv., 2012, 2, 6245–6249 RSC.
- H. Y. Bai, M. Han, Y. Z. Du, J. C. Bao and Z. H. Dai, Chem. Commun., 2010, 46, 1739–1741 RSC.
- L. Meng, J. Jin, G. X. Yang, T. H. Lu, H. Zhang and C. X. Cai, Anal. Chem., 2009, 81, 7271–7280 CrossRef CAS.
- X. M. Chen, Z. J. Lin, D. J. Chen, T. T. Jia, Z. M. Cai, X. R. Wang, X. Chen, G. N. Chen and M. Oyama, Biosens. Bioelectron., 2010, 25, 1803–1808 CrossRef CAS.
- L. M. Lu, H. B. Li, F. L. Qu, X. B. Zhang, G. L. Shen and R. Q. Yu, Biosens. Bioelectron., 2011, 26, 3500–3504 CrossRef CAS.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem., 1971, 31, 39–49 CrossRef CAS.
- J. H. Zhu, J. Jiang, J. P. Liu, R. M. Ding, Y. Y. Li, H. Ding, Y. M. Feng, G. M. Wei and X. T. Huang, RSC Adv., 2011, 1, 1020–1025 RSC.
- T. Watanabe and Y. Einaga, Biosens. Bioelectron., 2009, 24, 2684–2689 CrossRef CAS.
- X. J. Zhang, G. F. Wang, Y. Huang, L. T. Yu and B. Fang, Micro Nano Lett., 2012, 7, 168–170 CAS.
- Y. Liu, H. Teng, H. Q. Hou and T. Y. You, Biosens. Bioelectron., 2009, 24, 3329–3334 CrossRef CAS.
- L. M. Lu, L. Zhang, F. L. Qu, H. X. Lu, X. B. Zhang, Z. S. Wu, S. Y. Huan, Q. A. Wang, G. L. Shen and R. Q. Yu, Biosens. Bioelectron., 2009, 25, 218–223 CrossRef CAS.
- H. G. Nie, Z. Yao, X. M. Zhou, Z. Yang and S. M. Huang, Biosens. Bioelectron., 2011, 30, 28–34 CrossRef CAS.
- Q. F. Yi, W. Huang, W. Q. Yu, L. Li and X. P. Liu, Electroanalysis, 2008, 20, 2016–2022 CrossRef CAS.
- K. E. Toghill, L. Xiao, M. A. Phillips and R. G. Compton, Sens. Actuators, B, 2010, 147, 642–652 CrossRef.
- K. E. Toghill, L. Xiao, N. R. Stradiotto and R. G. Compton, Electroanalysis, 2010, 22, 491–500 CrossRef CAS.
- J. Zhao, F. Wang, J. J. Yu and S. S. Hu, Talanta, 2006, 70, 449–454 CrossRef CAS.
- J. Luo, S. S. Jiang, H. Y. Zhang, J. Q. Jiang and X. Y. Liu, Anal. Chim. Acta, 2012, 709, 47–53 CrossRef CAS.
- J. Luo, H. Y. Zhang, S. S. Jiang, J. Q. Jiang and X. Y. Liu, Microchim. Acta, 2012, 177, 485–490 CrossRef CAS.
- F. Sun, P. Liu, L. Li and Y. F. Lian, Chem. Res. Chinese U., 2011, 27, 1049–1054 CAS.
- H. X. Wu, W. M. Cao, Y. Li, G. Liu, Y. Wen, H. F. Yang and S. P. Yang, Electrochim. Acta, 2010, 55, 3734–3740 CrossRef CAS.
- L. Xu, J. X. Xia, H. M. Li, H. N. Li, K. Wang and S. Yin, Eur. J. Inorg. Chem., 2011, 1361–1365 CrossRef CAS.
- J. A. Yang, W. D. Zhang and S. Gunasekaran, Biosens. Bioelectron., 2010, 26, 279–284 CrossRef CAS.
- X. J. Zhang, A. X. Gu, G. F. Wang, Y. Huang, H. Q. Ji and B. Fang, Analyst, 2011, 136, 5175–5180 RSC.
- Y. C. Zhang, L. Su, D. Manuzzi, H. V. E. de los Monteros, W. Z. Jia, D. Q. Huo, C. J. Hou and Y. Lei, Biosens. Bioelectron., 2012, 31, 426–432 CrossRef CAS.
- Y. Zhao, J. Z. Zhao, D. C. Ma, Y. L. Li, X. L. Hao, L. Z. Li, C. Z. Yu, L. Zhang, Y. Lu and Z. C. Wang, Colloids Surf., A, 2012, 409, 105–111 CrossRef CAS.
- T. K. Huang, K. W. Lin, S. P. Tung, T. M. Cheng, I. C. Chang, Y. Z. Hsieh, C. Y. Lee and H. T. Chiu, J. Electroanal. Chem., 2009, 636, 123–127 CrossRef CAS.
- X. H. Kang, Z. B. Mai, X. Y. Zou, P. X. Cai and J. Y. Mo, Anal. Biochem., 2007, 363, 143–150 CrossRef CAS.
- K. B. Male, S. Hrapovic, Y. L. Liu, D. S. Wang and J. H. T. Luong, Anal. Chim. Acta, 2004, 516, 35–41 CrossRef CAS.
- Y. Ding, Y. Wang, L. Su, M. Bellagamba, H. Zhang and Y. Lei, Biosens. Bioelectron., 2010, 26, 542–548 CrossRef CAS.
- C. W. Kung, C. Y. Lin, Y. H. Lai, R. Vittal and K. C. Ho, Biosens. Bioelectron., 2011, 27, 125–131 CrossRef CAS.
- X. C. Dong, H. Xu, X. W. Wang, Y. X. Huang, M. B. Chan-Park, H. Zhang, L. H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206–3213 CrossRef CAS.
- X. W. Wang, X. C. Dong, Y. Q. Wen, C. M. Li, Q. H. Xiong and P. Chen, Chem. Commun., 2012, 48, 6490–6492 RSC.
- S. Cherevko and C. H. Chung, Talanta, 2010, 80, 1371–1377 CrossRef CAS.
- N. A. Choudhry, D. K. Kampouris, R. O. Kadara, N. Jenkinson and C. E. Banks, Anal. Methods, 2009, 1, 183–187 RSC.
- K. M. El Khatib and R. M. A. Hameed, Biosens. Bioelectron., 2011, 26, 3542–3548 CrossRef CAS.
- K. K. Lee, P. Y. Loh, C. H. Sow and W. S. Chin, Electrochem. Commun., 2012, 20, 128–132 CrossRef CAS.
- C. L. Li, Y. Su, S. W. Zhang, X. Y. Lv, H. L. Xia and Y. J. Wang, Biosens. Bioelectron., 2010, 26, 903–907 CrossRef CAS.
- S. L. Luo, F. Su, C. B. Liu, J. X. Li, R. H. Liu, Y. Xiao, Y. Li, X. N. Liu and Q. Y. Cai, Talanta, 2011, 86, 157–163 CrossRef CAS.
- M. U. A. Prathap, B. Kaur and R. Srivastava, J. Colloid Interface Sci., 2012, 370, 144–154 CrossRef.
- S. Priya and S. Berchmans, J. Electrochem. Soc., 2012, 159, F73–F80 CrossRef CAS.
- A. J. Wang, J. J. Feng, Z. H. Li, Q. C. Liao, Z. Z. Wang and J. R. Chen, CrystEngComm, 2012, 14, 1289–1295 RSC.
- G. Wang, Y. Wei, W. Zhang, X. Zhang, B. Fang and L. Wang, Microchim. Acta, 2010, 168, 87–92 CrossRef CAS.
- J. Yang, L. C. Jiang, W. D. Zhang and S. Gunasekaran, Talanta, 2010, 82, 25–33 CrossRef CAS.
- Y. J. Yang, W. K. Li and X. H. Chen, J. Solid State Electrochem., 2012, 16, 2877–2881 CrossRef CAS.
- P. Zhang, L. Zhang, G. C. Zhao and F. Feng, Microchim. Acta, 2012, 176, 411–417 CrossRef CAS.
- X. J. Zhang, G. F. Wang, W. Zhang, Y. Wei and B. Fang, Biosens. Bioelectron., 2009, 24, 3395–3398 CrossRef CAS.
- Z. J. Zhuang, X. D. Su, H. Y. Yuan, Q. Sun, D. Xiao and M. M. F. Choi, Analyst, 2008, 133, 126–132 RSC.
- W. Wang, L. L. Zhang, S. F. Tong, X. Li and W. B. Song, Biosens. Bioelectron., 2009, 25, 708–714 CrossRef CAS.
- E. Reitz, W. Z. Jia, M. Gentile, Y. Wang and Y. Lei, Electroanalysis, 2008, 20, 2482–2486 CrossRef CAS.
- L. C. Jiang and W. D. Zhang, Biosens. Bioelectron., 2010, 25, 1402–1407 CrossRef CAS.
- Y. Mu, D. L. Jia, Y. Y. He, Y. Q. Miao and H. L. Wu, Biosens. Bioelectron., 2011, 26, 2948–2952 CrossRef CAS.
- Y. Zhang, F. G. Xu, Y. J. Sun, Y. Shi, Z. W. Wen and Z. Li, J. Mater. Chem., 2011, 21, 16949–16954 RSC.
- W. Lv, F. M. Jin, Q. G. Guo, Q. H. Yang and F. Y. Kang, Electrochim. Acta, 2012, 73, 129–135 CrossRef CAS.
- G. M. Wang, X. H. Lu, T. Zhai, Y. C. Ling, H. Y. Wang, Y. X. Tong and Y. Li, Nanoscale, 2012, 4, 3123–3127 RSC.
- W. Wang, Z. Y. Li, W. Zheng, B. Dong, S. Y. Li and C. Wang, J. Nanosci. Nanotechnol., 2010, 10, 7537–7540 CrossRef CAS.
- Y. Ding, Y. Wang, L. C. Zhang, H. Zhang and Y. Lei, J. Mater. Chem., 2012, 22, 980–986 RSC.
- M. Shamsipur, M. Najafi and M. R. M. Hosseini, Bioelectrochemistry, 2010, 77, 120–124 CrossRef CAS.
- W. D. Zhang, J. Chen, L. C. Jiang, Y. X. Yu and J. Q. Zhang, Microchim. Acta, 2010, 168, 259–265 CrossRef CAS.
- C. C. Li, Y. L. Liu, L. M. Li, Z. F. Du, S. J. Xu, M. Zhang, X. M. Yin and T. H. Wang, Talanta, 2008, 77, 455–459 CrossRef CAS.
- J. Chen, W. D. Zhang and J. S. Ye, Electrochem. Commun., 2008, 10, 1268–1271 CrossRef CAS.
- P. Si, X. Dong, P. Chen and D. Kim, J. Mater. Chem. B, 2013, 1, 110 RSC.
- G. N. Dar, A. Umar, S. A. Zaidi, S. Baskoutas, S. H. Kim, M. Abaker, A. Al-Hajry and S. A. Al-Sayari, Sci. Adv. Mater., 2011, 3, 901–906 CrossRef CAS.
- T. T. Baby and S. Ramaprabhu, J. Nanosci. Nanotechnol., 2011, 11, 4684–4691 CrossRef CAS.
- B. Fang, A. X. Gu, G. F. Wang, W. Wang, Y. H. Feng, C. H. Zhang and X. J. Zhang, ACS Appl. Mater. Interfaces, 2009, 1, 2829–2834 CAS.
- X. Cao and N. Wang, Analyst, 2011, 136, 4241–4246 RSC.
- C. Xia and W. Ning, Electrochem. Commun., 2010, 12, 1581–1584 CrossRef CAS.
- V. Dharuman and K. C. Pillai, J. Solid State Electrochem., 2006, 10, 967–979 CrossRef CAS.
- M. E. G. Lyons, C. A. Fitzgerald and M. R. Smyth, Analyst, 1994, 119, 855–861 RSC.
- L. C. Jiang and W. D. Zhang, Electroanalysis, 2009, 21, 1811–1815 CrossRef CAS.
- J. M. Zen, A. S. Kumar and J. C. Chen, Electroanalysis, 2001, 13, 457–464 CrossRef CAS.
- C. Barrera, I. Zhukov, E. Villagra, F. Bedioui, M. A. Paez, J. Costamagna and J. H. Zagal, J. Electroanal. Chem., 2006, 589, 212–218 CrossRef CAS.
- L. Ozcan, Y. Sahin and H. Turk, Biosens. Bioelectron., 2008, 24, 512–517 CrossRef CAS.
- L. M. Santos and R. P. Baldwin, Anal. Chem., 1987, 59, 1766–1770 CrossRef CAS.
- P. N. Mashazi, K. I. Ozoemena and T. Nyokong, Electrochim. Acta, 2006, 52, 177–186 CrossRef CAS.
- K. I. Ozoemena and T. Nyokong, Electrochim. Acta, 2006, 51, 5131–5136 CrossRef CAS.
- Z. S. Sun and H. Tachikawa, Anal. Chem., 1992, 64, 1112–1117 CrossRef CAS.
- K. Wang, J. J. Xu and H. Y. Chen, Biosens. Bioelectron., 2005, 20, 1388–1396 CrossRef CAS.
- J. S. Ye, Y. Wen, W. D. Zhang, H. F. Cui, G. Q. Xu and F. S. Sheu, Electroanalysis, 2005, 17, 89–96 CrossRef CAS.
- J. Wang, X. J. Zhang and L. Chen, Electroanalysis, 2000, 12, 1277–1281 CrossRef CAS.
- G. L. D. Gonzalez, H. Kahlert and F. Scholz, Electrochim. Acta, 2007, 52, 1968–1974 CrossRef.
- Q. L. Sheng, Y. Shen, H. F. Zhang and J. B. Zheng, Electrochim. Acta, 2008, 53, 4687–4692 CrossRef CAS.
- S. M. Chen, C. Y. Liou and R. Thangamuthu, Electroanalysis, 2007, 19, 2457–2464 CrossRef CAS.
- M. Yang, J. Jiang, Y. Lu, Y. He, G. Shen and R. Yu, Biomaterials, 2007, 28, 3408–3417 CrossRef CAS.
- X. Y. Wang, Y. Zhang, C. E. Banks, Q. Y. Chen and X. B. Ji, Colloids Surf., B, 2010, 78, 363–366 CrossRef CAS.
- Y. Bai, Y. Y. Sun and C. Q. Sun, Biosens. Bioelectron., 2008, 24, 579–585 CrossRef CAS.
- Y. P. Sun, H. Buck and T. E. Mallouk, Anal. Chem., 2001, 73, 1599–1604 CrossRef CAS.
- J. Wang, D. F. Thomas and A. Chen, Anal. Chem., 2008, 80, 997–1004 CrossRef CAS.
- H. F. Cui, J. S. Ye, W. D. Zhang, C. M. Li, J. H. T. Luong and F. S. Sheu, Anal. Chim. Acta, 2007, 594, 175–183 CrossRef CAS.
- L. H. Li, W. D. Zhang and J. S. Ye, Electroanalysis, 2008, 20, 2212–2216 CrossRef CAS.
- F. Xiao, F. Q. Zhao, D. P. Mei, Z. R. Mo and B. Z. Zeng, Biosens. Bioelectron., 2009, 24, 3481–3486 CrossRef CAS.
- P. Holt-Hindle, S. Nigro, M. Asmussen and A. C. Chen, Electrochem. Commun., 2008, 10, 1438–1441 CrossRef CAS.
- H. C. Gao, F. Xiao, C. B. Ching and H. W. Duan, ACS Appl. Mater. Interfaces, 2011, 3, 3049–3057 CAS.
- S. S. Mahshid, S. Mahshid, A. Dolati, M. Ghorbani, L. X. Yang, S. L. Luo and Q. Y. Cai, Electrochim. Acta, 2011, 58, 551–555 CrossRef CAS.
- S. S. Mahshid, S. L. Luo, L. X. Yang, S. Mahshid, A. Dolati, M. Ghorbani and Q. Y. Cai, Sens. Lett., 2011, 9, 1598–1605 CrossRef CAS.
- B. Lim, M. J. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. N. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS.
- M. Khalid, N. Wasio, T. Chase and K. Bandyopadhyay, Nanoscale Res. Lett., 2010, 5, 61–67 CrossRef CAS.
- S. J. Guo, L. Wang, S. J. Dong and E. K. Wang, J. Phys. Chem. C, 2008, 112, 13510–13515 CAS.
- F. Xiao, Z. R. Mo, F. Q. Zhao and B. Z. Zeng, Electrochem. Commun., 2008, 10, 1740–1743 CrossRef CAS.
- F. Xiao, F. Q. Zhao, Y. F. Zhang, G. P. Guo and B. Z. Zeng, J. Phys. Chem. C, 2009, 113, 849–855 CAS.
- J. Ryu, K. Kim, H. S. Kim, H. T. Hahn and D. Lashmore, Biosens. Bioelectron., 2010, 26, 602–607 CrossRef CAS.
- B. Singh, F. Laffir, T. McCormac and E. Dempsey, Sens. Actuators, B, 2010, 150, 80–92 CrossRef.
- M. Tominaga, T. Shimazoe, M. Nagashima and I. Taniguchi, J. Electroanal. Chem., 2008, 615, 51–61 CrossRef CAS.
- Q. F. Yi, W. Q. Yu and F. J. Niu, Electroanalysis, 2010, 22, 556–563 CrossRef CAS.
- M. Tominaga, Y. Taema and I. Taniguchi, J. Electroanal. Chem., 2008, 624, 1–8 CrossRef CAS.
- L. Y. Chen, T. Fujita, Y. Ding and M. W. Chen, Adv. Funct. Mater., 2010, 20, 2279–2285 CrossRef CAS.
- I. H. Yeo and D. C. Johnson, J. Electroanal. Chem., 2000, 484, 157–163 CrossRef CAS.
- M. Jafarian, F. Forouzandeh, I. Danaee, F. Gobal and M. G. Mahjani, J. Solid State Electrochem., 2009, 13, 1171–1179 CrossRef CAS.
- Y. B. Vassilyev, O. A. Khazova and N. N. Nikolaeva, J. Electroanal. Chem., 1985, 196, 127–144 CrossRef.
- G. Kokkinidis and N. Xonoglou, Bioelectrochem. Bioenerg., 1985, 14, 375–387 CrossRef CAS.
- N. Xonoglou and G. Kokkinidis, Bioelectrochem. Bioenerg., 1984, 12, 485–498 CrossRef CAS.
- M. Sakamoto and K. Takamura, Bioelectrochem. Bioenerg., 1982, 9, 571–582 CrossRef CAS.
- S. B. Aoun, G. S. Bang, T. Koga, Y. Nonaka, T. Sotomura and I. Taniguchi, Electrochem. Commun., 2003, 5, 317–320 CrossRef CAS.
- S. B. Aoun, Z. Dursun, T. Koga, G. S. Bang, T. Sotomura and I. Taniguchi, J. Electroanal. Chem., 2004, 567, 175–183 CrossRef.
- F. Matsumoto, M. Harada, N. Koura and S. Uesugi, Electrochem. Commun., 2003, 5, 42–46 CrossRef CAS.
- Y. Ding, Y. Wang, L. A. Su, H. Zhang and Y. Lei, J. Mater. Chem., 2010, 20, 9918–9926 RSC.
- S. A. Kumar, H. W. Cheng, S. M. Chen and S. F. Wang, Mater. Sci. Eng., C, 2010, 30, 86–91 CrossRef CAS.
- G. F. Wang, Y. Wei, W. Zhang, X. J. Zhang, B. Fang and L. Wang, Microchim. Acta, 2010, 168, 87–92 CrossRef CAS.
- W. Wang, Z. Y. Li, W. Zheng, J. Yang, H. N. Zhang and C. Wang, Electrochem. Commun., 2009, 11, 1811–1814 CrossRef CAS.
- S. A. Wring, J. P. Hart, L. Bracey and B. J. Birch, Anal. Chim. Acta, 1990, 231, 203–212 CrossRef CAS.
- J. D. Newman, A. P. F. Turner and G. Marrazza, Anal. Chim. Acta, 1992, 262, 13–17 CrossRef CAS.
- R. Uchikado, T. N. Rao, D. A. Tryk and A. Fujishima, Chem. Lett., 2001, 144–145 CrossRef CAS.
- L. Zhang, G. C. Zhao, X. W. Wei and Z. S. Yang, Electroanalysis, 2005, 17, 630–634 CrossRef CAS.
- T. Watanabe, T. A. Ivandini, Y. Makide, A. Fujishima and Y. Einaga, Anal. Chem., 2006, 78, 7857–7860 CrossRef CAS.
- T. A. Ivandini, R. Sato, Y. Makide, A. Fujishima and Y. Einaga, Diamond Relat. Mater., 2004, 13, 2003–2008 CrossRef CAS.
- J. Lee and S. M. Park, Anal. Chim. Acta, 2005, 545, 27–32 CrossRef CAS.
- J. W. Zhao, L. Z. Wu and J. F. Zhi, Analyst, 2009, 134, 794–799 RSC.
- D. B. Luo, L. Z. Wu and J. F. Zhi, ACS Nano, 2009, 3, 2121–2128 CrossRef CAS.
- M. D. Koppang, M. Witek, J. Blau and G. M. Swain, Anal. Chem., 1999, 71, 1188–1195 CrossRef CAS.
- S. G. Wang, Q. Zhang, R. L. Wang, S. F. Yoon, J. Ahn, D. J. Yang, J. Z. Tian, J. Q. Li and Q. Zhou, Electrochem. Commun., 2003, 5, 800–803 CrossRef CAS.
- B. Y. Wu, S. H. Hou, F. Yin, Z. X. Zhao, Y. Y. Wang, X. S. Wang and Q. Chen, Biosens. Bioelectron., 2007, 22, 2854–2860 CrossRef CAS.
- P. Si, P. Kannan, L. H. Guo, H. S. Son and D. H. Kim, Biosens. Bioelectron., 2011, 26, 3845–3851 CrossRef CAS.
- X. B. Yan, X. J. Chen, B. K. Tay and K. A. Khor, Electrochem. Commun., 2007, 9, 1269–1275 CrossRef CAS.
- J. Shi, J. C. Claussen, E. S. McLamore, A. Ul Haque, D. Jaroch, A. R. Diggs, P. Calvo-Marzal, J. L. Rickus and D. M. Porterfield, Nanotechnology, 2011, 22, 355502 CrossRef.
- J. Shi, T. Cha, J. C. Claussen, A. R. Diggs, J. H. Choi and D. M. Porterfield, Analyst, 2011, 136, 4916–4924 RSC.
- S. Sotiropoulou and N. A. Chaniotakis, Anal. Bioanal. Chem., 2003, 375, 103–105 CAS.
- S. G. Wang, Q. Zhang, R. L. Wang and S. F. Yoon, Biochem. Biophys. Res. Commun., 2003, 311, 572–576 CrossRef CAS.
- C. Y. Deng, J. H. Chen, Z. Nie and S. H. Si, Biosens. Bioelectron., 2010, 26, 213–219 CrossRef CAS.
- J. S. Ye, Y. Wen, W. D. Zhang, L. M. Gan, G. Q. Xu and F. S. Sheu, Electrochem. Commun., 2004, 6, 66–70 CrossRef CAS.
- J. X. Wang, X. W. Sun, X. P. Cai, Y. Lei, L. Song and S. S. Xie, Electrochem. Solid-State Lett., 2007, 10, J58–J60 CrossRef CAS.
- S. Hrapovic, Y. L. Liu, K. B. Male and J. H. T. Luong, Anal. Chem., 2004, 76, 1083–1088 CrossRef CAS.
- F. L. Qu, M. H. Yang, G. L. Shen and R. Q. Yu, Biosens. Bioelectron., 2007, 22, 1749–1755 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- J. Shi, H. Y. Zhang, A. Snyder, M. X. Wang, J. Xie, D. M. Porterfield and L. A. Stanciu, Biosens. Bioelectron., 2012, 38, 314–320 CrossRef CAS.
- M. Mallesha, R. Manjunatha, G. S. Suresh, J. S. Melo, S. F. D'Souza and T. V. Venkatesha, J. Solid State Electrochem., 2012, 16, 2675–2681 CrossRef CAS.
- V. Stavyiannoudaki, V. Vamvakaki and N. Chaniotakis, Anal. Bioanal. Chem., 2009, 395, 429–435 CrossRef CAS.
- D. Rathod, C. Dickinson, D. Egan and E. Dempsey, Sens. Actuators, B, 2010, 143, 547–554 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |