
Computationally driven design of an artificial metalloenzyme using supramolecular anchoring strategies of iridium complexes to alcohol dehydrogenase†
 *a
and
Christof M.
Jäger
*a
*a
and
Christof M.
Jäger
*a
Abstract
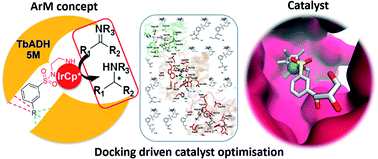
Artificial metalloenzymes (ArMs) confer non-biological reactivities to biomolecules, whilst taking advantage of the biomolecular architecture in terms of their selectivity and renewable origin. In particular, the design of ArMs by the supramolecular anchoring of metal catalysts to protein hosts provides flexible and easy to optimise systems. The use of cofactor dependent enzymes as hosts gives the advantage of both a (hydrophobic) binding site for the substrate and a cofactor pocket to accommodate the catalyst. Here, we present a computationally driven design approach of ArMs for the transfer hydrogenation reaction of cyclic imines, starting from the NADP+-dependent alcohol dehydrogenase from Thermoanaerobacter brockii (TbADH). We tested and developed a molecular docking workflow to define and optimize iridium catalysts with high affinity for the cofactor binding site of TbADH. The workflow uses high throughput docking of compound libraries to identify key structural motifs for high affinity, followed by higher accuracy docking methods on smaller, focused ligand and catalyst libraries. Iridium sulfonamide catalysts were selected and synthesised, containing either a triol, a furane, or a carboxylic acid to provide the interaction with the cofactor binding pocket. IC50 values of the resulting complexes during TbADH-catalysed alcohol oxidation were determined by competition experiments and were between 4.410 mM and 0.052 mM, demonstrating the affinity of the iridium complexes for either the substrate or the cofactor binding pocket of TbADH. The catalytic activity of the free iridium complexes in solution showed a maximal turnover number (TON) of 90 for the reduction of salsolidine by the triol-functionalised iridium catalyst, whilst in the presence of TbADH, only the iridium catalyst with the triol anchoring functionality showed activity for the same reaction (TON of 36 after 24 h). The observation that the artificial metalloenzymes developed here lacked stereoselectivity demonstrates the need for the further investigation and optimisation of the ArM. Our results serve as a starting point for the design of robust artificial metalloenzymes, exploiting supramolecular anchoring to natural NAD(P)H binding pockets.
- This article is part of the themed collection: Natural and artificial metalloenzymes
 Please wait while we load your content...
Please wait while we load your content...

