
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
‘Green’ synthesis of amines from renewable resources? A detailed analysis of case studies using the CHEM21 green metrics toolkit†
Anastasiia M.
Afanasenko
 *a,
Noemi
Deak
b,
Jacquin
October
c,
Roberto
Sole
c and
Katalin
Barta
*c
*a,
Noemi
Deak
b,
Jacquin
October
c,
Roberto
Sole
c and
Katalin
Barta
*c
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 3, 9747 AG Groningen, the Netherlands. E-mail: anastasiia.afanasenko@mail.mcgill.ca
bFaculty of Chemistry and Chemical Engineering, Babeş-Bolyai University, str. Arany Janos nr.11, 400028, Cluj-Napoca, Romania
cInstitute for Chemistry, University of Graz, Heinrichstrasse 28/II, 8010 Graz, Austria. E-mail: katalin.barta@uni-graz.at
First published on 6th March 2025
Abstract
Amines play pivotal roles in both chemical industry processes and various biological functions, necessitating efficient and sustainable synthesis methodologies. Despite the emergence of greener catalytic methods in the past decades, assessing the environmental impact of these processes remains a challenge. While bio-based amine synthesis from renewable sources appears inherently green, comprehensive evaluation across diverse sustainability metrics is imperative. This tutorial review explores the methodology of such assessments, focusing on the systematic evaluation of amine synthesis pathways using the CHEM21 green metrics toolkit. Targeting early career researchers, we provide a step-by-step demonstration to integrate this toolkit into laboratory practices, empowering researchers to evaluate the environmental footprint of their chemical transformations. By promoting the use of green metrics, we aim to foster a greater understanding of sustainability in chemical research and encourage environmentally conscious decision-making.
Green foundation1. Amine-containing compounds find application in various fields ranging from pharmaceuticals, polymers, surfactants and detergents, just to name a few. In this tutorial review, we show the sustainable production of these compounds via hydrogen borrowing, reductive amination and hydroamination, directly from renewable resources. We further evaluate the greenness and sustainability of these processes using the CHEM21 green metrics toolkit.2. Shifting to sustainable synthetic methods is becoming more and more important. To facilitate this shift, this review will help early-career researchers to incorporate this toolkit into laboratory practices. It also provides a basic evaluation of the feasibility of incorporating renewable resources into the synthesis of value-added chemicals. 3. As the availability of renewable resources on a large scale grows, these synthetic and catalytic methods have the potential to substitute the current petrochemical routes. However, further insights regarding their environmental impact are necessary, and this review provides a first glimpse into the effectiveness and sustainability of these approaches. |
1. Introduction
Nitrogen-containing compounds play a central role in all fields of chemistry; among them, amine and amide derivatives (nucleotides and amino acids) are the most fundamental building blocks that Nature has chosen in the construction of Life.1,2 Besides biochemistry, amines are centrally important compounds in the bulk and fine chemical industry since they are omnipresent scaffolds in agrochemicals, dyes, natural products, pharmaceutically active compounds, and polymers.3,4 The vast majority of these products originate from depleting fossil resources and are frequently manufactured through environmentally unfriendly routes, multistep syntheses, and generating large amounts of waste.5,6 A change of unsustainable practices is highly desired: we should be able to obtain the needed chemicals from renewable resources, through clean catalytic routes, without generating large amounts of waste. Lignocellulosic biomass and derived platform chemicals are highly oxygenated, hence ideally positioned for the development of such zero-waste, catalytic pathways.Terrestrial biomass, a complex natural renewable material, has a potent carbon-capture capability (est. 200 Gt at 4.4 × 109 ha canopy cover potential),7 combined with enormous chemical variability. Unlike fossil fuels, biomass is replenished in time, which makes it a promising alternative feedstock for the sustainable production of chemicals. While the structural complexity of biomass represents challenges in terms of processing and conversion/depolymerization,8 it also offers unique possibilities for developing entirely new, energy-efficient, and waste-free pathways to obtain valuable chemicals.9,10 Recently, specific effort has been devoted to the development of strategies for the synthesis of amines from renewables via waste-minimized catalytic routes.9–11 The main objective of this review is to summarize recent progress and guide the readers through the evaluation of these developments in a green chemistry context.
Amines are highly important and their synthesis is well established in the chemical industry. Classical synthetic methods to access amines include the Gabriel synthesis of alkyl halides12 and reductive alkylation processes.13 However, these approaches suffer from low atom economy due to the formation of stoichiometric amounts of waste, low selectivity, and encounter some restrictions in substrate cost and availability. Catalytic methods such as Buchwald–Hartwig,14,15 Ullmann reactions16,17 have emerged to be efficient and powerful alternatives, albeit these methods typically utilize halides as substrates, which negatively affects atom economy and are not readily applicable to the conversion of renewable platform chemicals.
Biomass and derived platform chemicals contain specific functional groups (alcohols, aldehydes, acids) that represent valuable precursors, easing the synthetic pathways and potentially reducing the number of steps needed for synthesizing targeted products.18,19 With regard to the mission of developing efficient catalytic pathways from biomass without the formation of side products, several catalytic methods can be envisioned. These approaches, which have – independently of biomass conversion – gained intensive interest due to their sustainability, are (i) ‘hydrogen borrowing’ amination of alcohols;20–23 (ii) reductive amination of aldehydes and ketones;24–26 (iii) hydroamination of unsaturated alkanes.27–29 In this review, all three research areas will be thoroughly discussed.
But how to properly evaluate the ‘greenness’ of a chemical reaction and process? This is a non-trivial question, which has been engaging the ‘green chemistry’ community over the past decades. The 12 principles of Green Chemistry are a set of design guidelines, established to facilitate the fundamental discovery process, where holistic ‘green chemistry’ thinking rests at the core of the research questions, prioritizing pollution prevention and the development of clean transformations. There are specific green metrics (as detailed below) describing a chemical reaction, which are directly connected to the principles of Green Chemistry. These are related to the amount of waste generated (E factor, 1st principle) or the percentage of atoms incorporated in the final products (atom economy, 2nd principle). Calculation of these relatively simple numerical values is therefore an important first step of evaluation, yet not entirely comprehensive without a full life-cycle analysis. In this tutorial review, we will show the application of these metrics to specific transformations. To take this further, more comprehensive evaluation methods have been developed, taking into account a broader set of parameters and specific aspects of the entire process. For example, the CHEM21 green metrics toolkit, developed by Clark and coworkers,30 was introduced to provide a practical and relatively easy-to-use tool for objective evaluation and quantification of a process, aligning with the Twelve Principles of green chemistry. This method incorporates resource efficiency (waste, atom economy, energy), environmental, health, and safety considerations of the reaction and related purification protocols.31 Thus, the CHEM21 green metrics toolkit assists in the calculation and assessment of key quantitative and qualitative parameters, giving a suitable starting point for reaction development. Subsequently, it is desired to perform a full life cycle analysis (LCA), including feedstock sourcing and product end-of-life considerations, to derive final conclusions about the environmental impact and feasibility of a process.32,33 Since LCA is resource and time-intensive and requires specialized knowledge, it typically cannot be routinely performed in every laboratory. Therefore, the main focus of this review is devoted to the calculation of metrics and the use of the CHEM21 toolkit, which can be easily performed in any laboratory by students and post-doctoral researchers.
The goals of this tutorial review are multifold. First, we give an overview of the types and classes of renewable starting materials and platform chemicals, which can be used to produce bio-based amines. Next, we discuss three specific types of catalytic reactions, namely (i) catalytic N-alkylation of amines with alcohols via ‘hydrogen borrowing’ methodology, (ii) reductive amination, (iii) hydroamination, including recent developments in catalyst development in these domains (Fig. 1). The attention will be focused on heterogenous and homogenous metal catalysts. Then we demonstrate the use of these catalytic transformations for the development of specific bio-based amines on selected examples. And finally, we demonstrate the use of green metrics and the CHEM21 green metrics toolkit for evaluating selected reactions. These calculations will reflect upon the combination of mild biomass depolymerization and clean amination strategies towards the sustainable synthesis of amines from renewables, compared to petroleum.
 | ||
| Fig. 1 Overview of the present review: synthesis of green amines starting from renewable resources using atom-economic transformations following the principles of green chemistry. | ||
2. Green metrics
The prime goal of green chemistry is to minimize the formation of waste in the design and manufacturing of chemical processes and products.18 Over the past years, metrics for measuring/quantifying the green credentials of a process, such as atom-economy (AE), reaction mass efficiency (RME), process mass intensity (PMI) were introduced. The list of green metrics will be discussed in detail in this chapter.34 Green metrics should be used to calculate improvements in the greenness of new synthetic pathways by comparing them to current methodologies.As earlier mentioned, the CHEM21 green metrics toolkit35 serves as a practical guide for the evaluation of chemical processes from a green chemistry perspective. Considering the researcher's goals, the toolkit is divided into four passes, from zero pass to third pass, with different parameters included in each. While the zero and first pass toolkits are designed to be used on a bench scale, the second and third pass ones are to be employed on an industrial level. Reactions that were shown to be promising on the zero and first pass are subjected to more in-depth analysis studying their scalability, therefore life cycle assessment (LCA) is not incorporated until the second pass CHEM21 metrics toolkit. The reason behind such a structure of passes with a level of complexity corresponding to the scale of the experiment is that the system of green metrics should primarily give a meaningful assessment, being simple to utilize in everyday laboratory practice.
In this chapter, we will detail green metrics that are calculated using the first pass CHEM21 metrics toolkit to provide a preliminary assessment of new reactions performed on the laboratory scale, at the discovery level. In the following chapters, we are going to use these parameters to evaluate the greenness of selected catalytic reactions and identify the potential areas for improvement.
2.1 Quantitative parameters
The basic parameters that describe a chemical transformation are yield, conversion, and selectivity. The equations for calculating these parameters are presented in Fig. 2, from a green chemistry point of view, a reaction with high yield, conversion, and selectivity is highly desirable. Here it is worth clarifying the difference between a reactant and a reagent since we will operate with these terms further. A reactant is defined as a compound containing at least one atom that is incorporated into the final product, while a reagent is a compound that is consumed during the reaction but not included in the product. | ||
| Fig. 2 Overview of the first pass CHEM21 green metrics toolkit: quantitative (inner circle) and qualitative parameters (outer circle). Abbreviations: R – reactant; Rl – limiting reactant; P – product; nP – moles of product formed; ni, Rl – initial moles of limiting reactant; nf, Rl – moles of limiting reactant left; m – mass; Mw – molecular weight. | ||
With the help of such metrics as atom economy (AE)36 and reaction mass efficiency (RME), chemists gain insight into the extent to which the reactants are converted into the target product (Fig. 2). AE is calculated as the ratio of the molecular weight of the product to the sum of the molecular weights of all reactants. Notably, AE, being a fully theoretical parameter, inherent to the reaction and does not incorporate the excesses of the used reactants. The latter drawback is successfully implemented in the RME.
In contrast to the mass-based metrics mentioned above, the process mass intensity (PMI), defined as “the mass of all chemicals used in the reaction divided by the mass of the isolated product”, includes all mass-based inputs: yield, stoichiometry, solvents, reagents, and work-up (Fig. 2). Later in this review, this parameter will be divided into three categories in order to simplify its assessment. As such, PMI reactants, reagents and catalysts (PMI RRC), which includes, as it is named, reactants, reagents, and catalysts; PMI reaction (PMI Rxn), besides the mentioned parameters involves reaction solvents; PMI work-up (PMI WU) considers only the auxiliary materials and solvents for the work-up. Recently, new metrics such as feedstock intensity (FI) and circular process feedstock intensity (CPFI) were developed.37 Being calculated in a similar way as PMI, these parameters are taken into account besides the desired product formation the production of valuable co-products (FI) as well as recycling of the material (CPFI). However, in this tutorial review, we will not implement these additional parameters in our analysis.
2.2 Qualitative parameters
At the First Pass level, the quantitative parameters such as mass-based metrics are calculated, and the obtained numerical values are compared, while qualitative metrics such as solvent, energy consumption, environmental concerns, are measured with a color code. This way, the results are flagged green (preferred), yellow (acceptable, but with some issues), and red (undesirable), indicating how well that parameter fits into the green and sustainable scoring system and/or pointing to an issue associated with the certain parameter. The following is a brief overview of qualitative parameters to be considered in a green metrics analysis at the first pass level.The use of stoichiometric or superstoichiometric amounts of reagents instead of using a catalyst/enzyme receives yellow or red flags, respectively. The possibility to regenerate and reuse the catalyst is scored with a green flag. The presence of critical elements38 with the risk of depletion of less than 50 years is marked in red, in a time interval between 50 and 100 years – yellow, and more than 100 years – green flag. This is interesting to keep in mind – even though a reaction is catalytic which is desired by the 9th principle of Green Chemistry, the use of rare metals for these processes, especially without appropriate recycling, markedly impacts the sustainability of the developed approach. The work-up method is evaluated by assigning a green flag to procedures with lower solvent consumption such as evaporation, filtration, centrifugation; in this case, column chromatography commonly used in organic synthesis receives a red flag. The same idea underlies the choice of batch or flow set-up: less solvent and energy-consuming flow set-up is marked with a green flag, the batch one – with a yellow flag.
The assessment of the energy use is significantly simplified for the discovery phase (first pass level) and considers the absolute temperature of the reaction: between 0 °C and 70 °C receives a green flag, lower than −20 °C or higher than 140 °C – red flag, and intermediate values – yellow flag. Furthermore, the reaction temperature is compared with the boiling point of the reaction solvent used, obtaining a red flag if the reaction is carried out under the reflux conditions. On the contrary, a green flag is assigned to a reaction whose temperature is lower than the boiling point of the solvent by 5 °C or more.
To evaluate potential health and safety concerns, the respective MSDS files of all chemicals involved in a reaction should be analysed. In this case, the colour code depends on how hazardous the used materials are and is assigned with red, yellow, and green flags, respectively. The open-access CHEM21 Solvent Selection guide, based on the solvent's physical properties, peroxability, resistivity, possible impact on the environment, and health, should be considered for scoring the utilized solvents.39
3. Catalytic approaches for the synthesis of bio-based amines
3.1 N-Alkylation of amines with alcohols via the ‘hydrogen borrowing’ strategy
This atom-economic transformation represents a prime example in green chemistry since it only produces water as a by-product and does not require any additional external hydrogen source since the parent alcohol acts as a hydrogen donor. Moreover, this methodology utilizes widely available inexpensive alcohol reaction partners that can be potentially derived from renewable resources.41–43 More specifically, the ‘hydrogen borrowing’ strategy begins with the metal-catalysed dehydrogenation of the alcohol substrate. Further, the formed carbonyl compound reacts with the amine to form the corresponding imine, which is reduced to the alkylated amine by means of the metal hydride generated during the first dehydrogenation step (Fig. 3).40 It is no surprise therefore, that the development of an appropriate catalytic species able to efficiently catalyse both the dehydrogenation and imine reduction steps, is key to achieving desired efficiency and selectivity in the overall transformation.
 | ||
| Fig. 3 (A) General mechanism of the hydrogen borrowing strategy; (B) Shvo's catalyst and Knölker's complex. | ||
In 1981, Watanabe44 and Grigg45 reported the first homogeneous catalysts, namely iridium, rhodium, and ruthenium-based phosphine complexes, for the N-alkylation of amines with alcohols (Fig. 4). Following these pioneering works after three decades of discovery, Fujita46 disclosed an efficient catalytic system consisting of [Cp*IrCl2]2/K2CO3 (Cp* = pentamethylcyclopentadienyl) for the selective mono-alkylation of primary amines to produce secondary amines. With regard to ruthenium, Beller's47 and Williams's48 groups reported on the use of Ru3(CO)12 with bulky phosphines and [Ru(p-cymene)Cl2]2 with the bidentate phosphines dppf or DPEphos as the catalyst, respectively, capable of alkylating amines with alcohols. The latter catalyst system has proven its efficiency for the synthesis of some simple pharmaceuticals, as well as for cyclization reactions and the N-alkylation of sulfonamides. While a large number of protocols have been developed in this field using precious metals20,49,50 for N-alkylation of amines with alcohols, since 2014 there have been more reports about the use of base metal catalysts for this transformation. Thus, almost simultaneously, Feringa and Barta with the Wills group demonstrated the C–N bond formation between alcohols and amines using iron-based homogeneous Knölker's complex51 and iron-tetraphenylcyclopentadienone tricarbonyl complex,52 respectively. Later, Kirchner and co-workers reported the use of complexes [Fe(PNP)Br2] and [Fe(PNP)(CO)Br2] comprising PNP pincer ligands based on triazine and pyridine backbones for the same type of transformation.53
 | ||
| Fig. 4 N-Alkylation of amines with alcohols using the ‘hydrogen borrowing’ strategy: history and key milestones. | ||
Besides iron and cobalt,54,55 manganese pincer56,57 complexes were successfully applied for the selective N-alkylation of amines with alcohols. Remarkably, as reported by Beller,58 manganese-based catalytic protocol allowed the N-methylation of various amines with methanol, which constituted the first examples of this transformation using non-noble metal complexes under mild reaction conditions. The first homogeneous nickel-based catalyst systems as a combination of nickel bromide and 1,10-phenanthroline and a well-defined and bench-stable azo-phenolate ligand-coordinated nickel catalyst capable of N-alkylation of a variety of anilines with alcohols were described by Banerjee59 and Adhikari60 groups, respectively. Soon after, efficient protocols emerged using Ni(II) pincer-type complex61 and nickel nanoparticles62 for the same type of reaction. Recently, the major focus of the field has shifted towards the asymmetric version of amination via the ‘hydrogen borrowing’ strategy.63 Thus, Donohoe group disclosed an elegant iridium-catalysed annulation strategy for the stereo-controlled synthesis of substituted saturated azaheterocycles as well as the synthesis of enantioenriched C3-substituted pyrrolidines and piperidines from enantiopure 1,4- and 1,5-diols.64
The recent breakthroughs in the usage of earth-abundant metals in homogeneous ‘hydrogen borrowing’ catalysis have been extensively reviewed.21–23,65 However, despite the achievements in the field, there is still tremendous demand for novel methods using precious or non-precious metals with improved scope, higher stability, higher TON, and applicability to bio-derived substrates.
Nonetheless, the literature offers several cases where hydrogen borrowing was utilized for Kg-scale pharmaceutical synthesis. For example, in 2011 Pfizer developed the [Cp*IrCl2]2 catalysed synthesis of a GlyT1 Inhibitor by using a hydrogen borrowing reaction as the key step.66 The optimized alcohol amination step allowed to improve the step economy, enabling the use of a catalyst loading as low as 0.05 mol% while resulting in the isolation of 4.81 kg of the amine product (76% yield). Similarly, Gonzales-Bobes from Bristol Myers Squibb recently reported the 2.8 kg synthesis (>70% yield) of an N-alkylated pyridine-2-amine via Ir-catalysed hydrogen borrowing.67 Although these reports do not involve the use of biomass-derived precursors, they underscore the increasing use of this attractive approach to minimize the economic and environmental impact of industrially relevant amines synthesis.
Taking into account that biomass is a complex natural renewable material with enormous chemical variability, hereinafter each type of biomass as a potential precursor for amines synthesis is separately described. Due to the large number of reports on the amination of bio-based alcohols in recent years, only selected examples have been discussed in this tutorial review (for more examples and details, see the recent review on this topic68).
Carbohydrate derivatives. Lignocellulosic biomass is a robust complex biopolymer that is primarily composed of cellulose, hemicellulose, and the aromatic polymer lignin. (Hemi)cellulosic carbohydrates are the largest fraction of lignocellulose; processes for the production of a number of platform chemicals by chemo- or biocatalytic methods are well-established.89 Some of these chemicals are termed ‘top value-added platform chemicals from carbohydrates’.90 Among these, there are several examples that feature an alcohol or carbonyl moiety. Herein the conversion of (hemi)cellulose-derived alcohols into amines via the ‘hydrogen borrowing’ approach is discussed (Table 1).
| Type of biomass | Platform chemical (PC) | Reaction conditions (selected examples) | Amine product | Amount of PC required | Scale | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| (Hemi)cellulose |

|
[IrH2Cl{(iPr2PC2H4)2NH}], amine, 140 °C, 20 hr |

|
0.062 g–0.090 g (1 mmol) | 1 mmol | C2–C4: 77–97% | 69 |
| [Cp*IrCl2(bmim)], K2CO3, toluene or N1,8,8,8NTf2, 115 °C, 24–48 hr | 0.152 g (2 mmol) | 2 mmol | C3: 89–99% | 70 | |||
| Knölker cat., Me3NO, CPME, 130 °C, 18–44 hr | 0.090 g–0.118 g (1 mmol) | 0.5 mmol | C4–C6: 60–85% | 51 | |||

|
Ru/C, aq ammonia, 10 bar H2, 170 °C, 6 hr |

|
1 g (6.84 mmol) | 6.84 mmol | 45% | 71 | |
| [Ru(CO)ClH(PPh3)3]/Xantphos, ammonia, t-amyl alcohol, 170 °C, 20 hr | 0.146 g (1 mmol) | 1 mmol | 96% | 72 | |||
| [Ru3(CO)12], A-iPr-PNP, aq ammonia, tert-amyl alcohol, 170 °C, 21 hr | 1.461 g (10 mmol) | 10 mmol | 96% | 73 | |||
| Ni/Al2O3-SiO2, aq ammonia, t-amyl alcohol, 160 °C, 18 hr | 0.073 g (0.5 mmol) | 0.5 mmol | 51% | 74 | |||
| [Ir] cat., (HO)P(O)(OPh)2, toluene, 3 Å mol. sieves, 120 °C, 24 hr | 0.060 g–0.080 g (0.25–0.5 mmol) | 0.25–0.5 mmol | 11–83% | 75 and 76 | |||

|
Ru/CNT, aq ammonia, 10 bar H2, 220 °C, 2 hr |
|
0.045 g–0.067 g (0.5 mmol) | 0.5 mmol | 62% 27% | 77 | |

|
Shvo cat., DPP, amine, toluene, 120 °C, 18 hr |

|
0.118 g–0.146 g (1 mmol) | 0.5 mmol | 45–72% | 78 | |

|
Nanosized zeolite Beta, aniline, 135 °C, 4 hr |
|
0.589 g (6 mmol) | 2 mmol | 52% | 79 | |
| RuCl3 3H2O, dppf, amine, PhCl, 145 °C, 15 hr | 0.059 g (0.6 mmol) | 0.2 mmol | 68% | 80 | |||
| [Ru3(CO)12], CataCXium PCy, ammonia, 150 °C, 20 hr | 0.098 g (1 mmol) | 1 mmol | 71% | 81 | |||
| [RuHCl(A-iPr-PNP)(CO)], ammonia, toluene, 110 °C, 12 hr | 0.981 g (10 mmol) | 10 mmol | 95% | 82 | |||
| [Mn(iPr2PC2H4)2NH)(CO)2Br], t-BuOK, amine, toluene, 80 °C, 24 hr | 0.118 g (1.2 mmol) | 1 mmol | 37–51% | 56 | |||
| Ni/Al2O3-SiO2, aq ammonia, t-amyl alcohol, 160 °C, 18 hr | 0.049 g (0.5 mmol) | 0.5 mmol | 52% | 74 | |||

|
Knölker cat., Me3NO, amine, toluene, mol. sieves, 135 °C, 18–24 hr |
|
0.224 g–0.256 g (2 mmol) | 0.5 mmol | 59–60% | 83 | |
| [RuHCl(A-iPr-PNP)(CO)], ammonia, t-amyl alcohol, 150 °C, 7 hr | 0.640 g (5 mmol) | 5 mmol | 65% | 84 | |||
| Ru-20MgO/TiO2, ammonia, toluene, 110 °C, 20 hr | 0.064 g (0.5 mmol) | 0.5 mmol | 86% | 85 | |||
| Lignin |
|
Shvo catalyst, amine, CPME, 130 °C, 20 hr |
|
0.087 g–0.102 g (0.48 mmol) | 0.4 mmol | 43–98% | 11 |
| RANEY® Ni, ammonia gas, t-amyl alcohol, 160–180 °C, 18 hr | 0.091 g–0.106 g (0.5 mmol) | 0.5 mmol | 65–76% | 86 | |||
| Cu-SiO2, DMAn, 3 bar H2, o-xylene, 210 °C, 16 hr | 0.182 g–0.212 g (1 mmol) | 1 mmol | 83–84% | 87 | |||
|
Ni/Al2O3-SiO2, (NH4)2CO3, t-amyl alcohol, 140 °C, 18 hr |

|
0.077 g (0.5 mmol) | 0.5 mmol | 40% | 74 | |
| Au/TiO2-VS, urea, toluene, 140 °C, 5 bar N2, 18 hr |

|
0.092 g (0.6 mmol) | 0.1 mmol | 80% | 88 |
Alcohols are important building blocks, inherently contained in and readily sourced from biomass. Perhaps the most simple and prominent primary alcohol, typically derived from syngas originating from thermal biomass conversion methods, is methanol.91 The other important short-chain alcohol, ethanol, could be obtained from cellulose via the fermentation of glucose.92 Furthermore, ethylene glycol can be subsequently obtained by dehydration of ethanol to ethene followed by conventional routes or alternatively by sugar or sorbitol/polyols hydrogenolysis pathways.91 Several methods have been developed for the conversion of cellulose-derived 5-(hydroxymethyl)furfural (5-HMF) or hemicellulose-derived furfural into various diols such as 1,6-hexanediol, 1,4-butanediol.93 On the other hand, the hydrogenation of glucose results in the formation of sorbitol.94
Direct amination of short-chain alcohols such as methanol, ethanol, propanol as well as diols is a well-established fundamental process producing alkylated amines that are highly essential in the chemical industry.95 Myriads of heterogeneous catalyst systems are known to carry out such transformation, therefore herein we will mention only selected ones. Baiker96 reported the gas phase amination of ethylene glycol (EG), potentially produced from cellulose, with dimethylamine (DMA) in a continuous fixed-bed reactor at atmospheric pressure over an Al2O3-supported Cu catalyst allowing the selective production of both ethanolamine, an important building block for detergents,97 and ethylenediamine, a common bidentate ligand in organometallic catalysts,98 products. The same group later discovered the Co–Fe system for the amination of 1,3-propanediol with supercritical ammonia.99 Other heterogeneous catalytic systems for the amination of aliphatic bio-derived diols, namely 1,2-propanediol, 1,4-butanediol, and 1,6-hexanediol have been described as well.100 Some selected homogeneous catalyst systems, namely N-heterocyclic carbene iridium complex,70 as well as iridium pincer metal species,69 were found to be efficient for the amination of short aliphatic chain alcohols and diols. Remarkably, Knölker's complex was successfully applied for the amination of diols furnishing the formation of various saturated azaheterocycles.51
Another essential type of diols is dianhydrohexitols, which could be obtained by double dehydration of the corresponding hexitols. Rose71 developed selective mono- and diamination of isosorbide, in the presence of a Ru/C catalyst; the obtained products could be used as polymer building blocks in polyamide101 and polyimide102 synthesis. Our group has reported the synthesis of the mono-aminated isosorbide product using Ni/Al2O3-SiO2 as a catalyst.74 Besides the mentioned heterogeneous catalysts, Beller reported the amination of isosorbide with ammonia using commercially available [Ru(CO)ClH(PPh3)3]/Xantphos catalyst.72 Similar example of a Ru-based catalytic system was described by Vogt.73 Interestingly, this robust catalytic system could be applied for the amination of natural alcohols as well, such as nerol, geraniol, farnesol, apart from the diols. Stereoselective aminations of partially protected isohexides, namely exo-monobenzylated isosorbides,75 as well as unprotected ones76 were established by Popowycz and co-workers applying Ir-based catalyst system along with diphenyl phosphate as an additive.
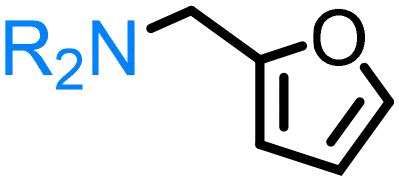
Multiple examples of conversion of hemicellulose-derived furfuryl alcohol to the corresponding furfuryl amine are established over heterogeneous catalysts (nanosized zeolite beta,79 Ni/Al2O3-SiO2,74 Ru-20MgO/TiO2,85 Ru nanoparticles103etc.). Among known homogeneous systems, Milstein disclosed the selective synthesis of furfuryl amine directly from furfuryl alcohol and ammonia in the presence of acridine-based pincer complex [RuHCl(A-iPr-PNP)(CO)].82 Following this study, other ruthenium-based catalyst systems for the direct amination of alcohols, including bio-based furfuryl alcohol, with aliphatic tertiary amines80 and with ammonia81 were developed. A few examples of the amination of furfuryl alcohol in the presence of manganese pincer complexes under strikingly mild reaction conditions were demonstrated by Beller.56
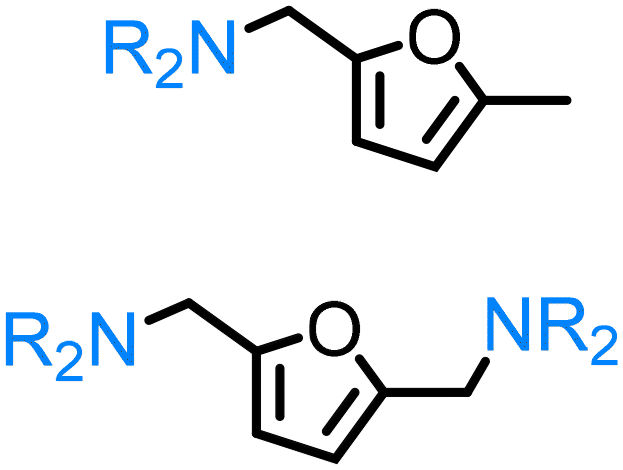
Regarding the cellulose-derived 5-HMF, it could also serve as a starting material for the construction of bio-based amines. Our group reported the highly selective hydrogenation of 5-HMF to 2,5-furan-dimethanol using Cu/Zn alloy nanopowder104 and Cu20-PMO catalysts105 under mild conditions (up to 100 °C) and the subsequent selective amination of the obtained diol using Knölker's complex.83 One homogeneous84 and one heterogeneous85 catalyst system is known in the literature for the synthesis of 2,5-bis(aminomethyl)furan (BAMF) using ammonia as a nitrogen source.
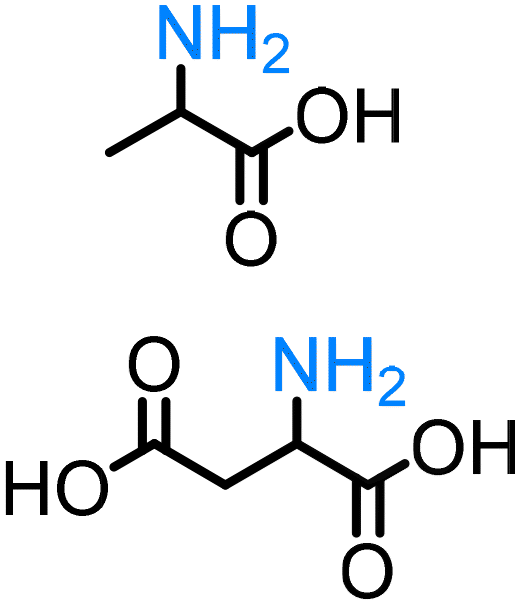
α-Hydroxy acids (e.g. lactic acid, malic acid) are important, primary platform chemicals readily accessible from renewable carbohydrates and, notably, several examples belong to the Top 12 value-added biomass-derived platform chemicals.90 To the best of our knowledge, the direct amination of α-hydroxy acids remains a rarity. Yan reported the first example of the amination of lactic acid with ammonia catalysed by a heterogeneous ruthenium-based catalyst.77,106 This pioneering work represents an alternative, chemocatalytic approach for the synthesis of amino acids, enabling for the first time the production of valuable proteins from lignocellulosic biomass. Our group has developed a homogeneous catalyst system based on a combination of Shvo's catalyst and Brønsted acid additive for the amination of 3-hydroxypropionic acid esters (3-HPe) via the ‘hydrogen borrowing’ strategy.78 This work demonstrates a very important pathway for easy access to beta-amino acid scaffolds from biomass.
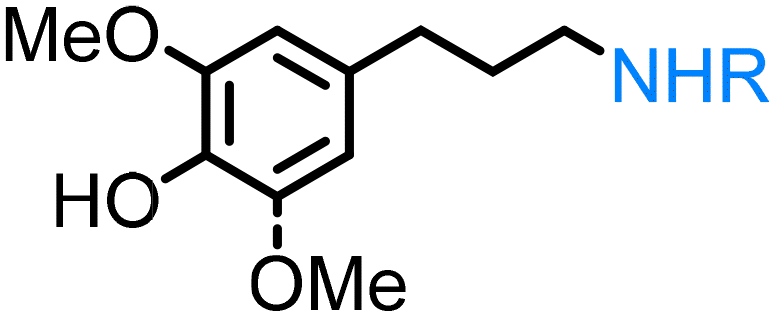
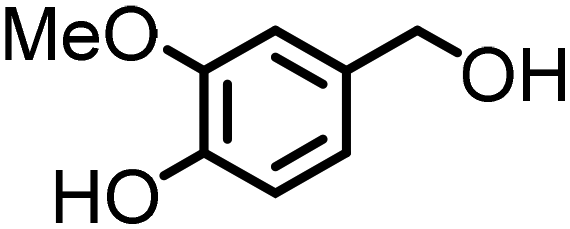
Lignin derivatives. Lignin is the largest source of bio-renewable aromatics. Its chemical structure consists of phenylpropane units, originating from three aromatic alcohols: p-coumaryl, coniferyl, and sinapyl alcohol, and therefore it is a promising feedstock for producing bio-aromatic alcohols and further functionalized derivatives. It is worth mentioning that before the recent work of our group11 there were no reports on the direct synthesis of amines from lignin-derived monomers by the ‘borrowing hydrogen’ method: reported protocols focused on lignin oligomers or reductive amination of vanillin.43 The reason for limited studies on lignin monomers was the limited availability of lignin-derived platform chemicals due to the recurrent depolymerization challenge.
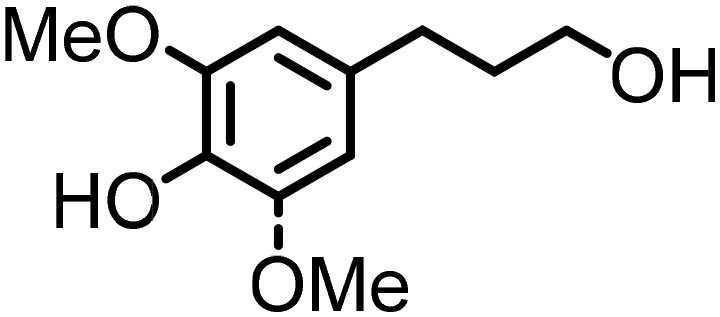
Lignin oxidation is a classical way of lignin depolymerization, though also associated with lower monomer yields.107 Recently, methods that deliver higher yields of vanillin or syringaldehyde have been developed,108 therefore, the hydrogenation of these building blocks to the corresponding alcohols (and their subsequent amination) becomes a feasible alternative. Indeed, there are few reports of applying ‘hydrogen borrowing’ methodologies to vanillyl alcohol, syringyl alcohol, or even the simpler p-hydroxybenzyl alcohol as a substrate (Table 1). Cao reported an example of the direct synthesis of tertiary amine from vanillyl alcohol and urea in the presence of the heterogeneous gold-mediated system.88 Our group demonstrated the direct amination of vanillyl alcohol using Ni/Al2O3-SiO2 as a catalyst and (NH4)2CO3 as an ammonia source.74 Furthermore, we developed a RANEY® Ni-based amination methodology (with ammonia) capable of converting lignin-derived phenolic primary alcohols, such as dihydroconiferyl alcohol, to corresponding primary amines.86
The introduction of the concept of stabilization of reactive intermediates109 during lignin depolymerization, the so-called ‘lignin first’ strategies110 have significantly shaped the possibilities to suppress undesired recondensation phenomena and obtain monophenolic compounds, for example, propyl- or ethyl-guaiacol and syringol or propanol-guaiacol or syringol in high yield from organosolv lignin or lignocellulose.108 These compounds may become potential platform chemicals in the future. It was shown in the recent works of our group, that employing homogeneous Shvo's catalyst leads to the first example of catalytic amination of lignin-derived dihydroconiferyl and dihydrosinapyl alcohols11 demonstrating the feasibility of this complex for the synthesis of N-alkyl amino-phenols. Our group also reported a catalytic strategy for producing industrially relevant 4,4′-methylenebiscyclohexamine from lignin oxidation mixtures in two steps; this occurs via the catalytic funneling of lignin-derived bisphenol mixtures into 4,4′-methylenebiscyclohexanol (MBC) and direct amination of MBC with ammonia.111 Efficient Cu-catalysed hydrogen borrowing strategy was developed by Sels and co-workers to functionalize aliphatic alcohols in aromatic monomers and dimers, derived from lignin by the reductive catalytic fraction (RCF), into tertiary dimethylamines.87 Very recently, Yan disclosed an elegant single-step catalytic strategy for directly producing phenolic amines from wood lignin via RCF in an aqueous ammonia–alcohol mixture over Ru catalyst.112 Remarkably, all amine products obtained through the above-mentioned strategies can be further upgraded into products with enhanced functions, thereby increasing the economic feasibility of lignocellulosic biorefineries.
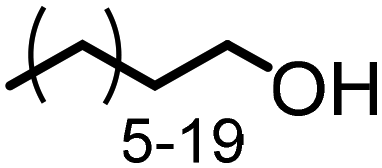
Triglycerides. Oils and fats, chemically composed of mainly triglycerides, are plant-based, and typically used for the food industry. Recently, large amounts of waste cooking oils are generated composed of specific triglycerides, which should find better utilization as a renewable feedstock for the chemical industry. Triglycerides are esters of glycerol composed of various linear saturated or unsaturated long–chain carboxylic acids (C8–C22). The most prominent and larger-scale application of triglycerides is the production of fatty acid methyl esters (FAME) by transesterification with methanol, leaving glycerol as a by-product in significant quantities. Both glycerol, as well as FAME or the derived fatty alcohols (FAO), obtained upon hydrogenation of FAME113 have been investigated as renewable starting materials to produce a range of bio-based products.114
The amination of fatty alcohols (Table 2) with dimethylamine is one of the main commercial processes for the production of dimethylalkylamines, and one of the key reactions for the oleo and detergents industries.115 Employing various heterogeneous catalysts (Cu–Ni,116 Cu–Cr,117 Cu–Ni–Ba118) this reaction was extensively studied. Apart from the mentioned examples, there are multiple efficient heterogeneous catalyst systems known for the amination of long-chain aliphatic alcohols (some selected Ni62,74,119,120 and Ru121-based catalyst systems are depicted in Table 2).
| Type of biomass | Platform chemical (PC) | Reaction conditions (selected examples) | Amine product | Amount of PC required | Scale | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| Triglycerides |
|
RANEY® Ni, aq ammonia, t-amyl alcohol, 180 °C, 18 hr |

|
0.065 g–0.079 g (0.5 mmol) | 0.5 mmol | C8–C10: 41–47% | 74 |
| Ni(COD)2, KOH, amine, CPME, 140 °C, 18 hr | 0.195 g–0.406 g (1.5 mmol) | 1 mmol | C8–C18: 85–90% | 62 | |||
| NiCuFeOx, amine, xylene, 140 °C, 24 hr | 0.130 g–0.186 g (1 mmol) | 1 mmol | C8–C12: 61–95% | 119 | |||
| Ni/Al2O3, amine, o-xylene, 144 °C, 24 hr | 0.156 g–0.190 g (1.2 mmol) | 0.5–1 mmol | C8–C10: 88–99% | 120 | |||
| Ru(OH)x/TiO2, urea, mesitylene, 1 atm Ar, 141 °C, 15 hr | 0.326 g (2.5 mmol) | 0.25 mmol | C8: 87% | 121 | |||
| [Ru(CO)ClH(DPEphos)(PPh3)], liq. ammonia, toluene, 140 °C, 24 hr | 1.302 g (10 mmol) | 10 mmol | C8: 71% | 122 | |||
| Knölker-CH3CN cat., amino acid, CF3CH2OH, 110 °C, 24 hr | 0.827 g–0.830 g (5.2–6.3 mmol for C9-C10) 0.429 g–0.541 g (2 mmol for C14-C18) | 0.5 mmol | C9–C18: 32–69% | 123 | |||
| [Mn(iPr2PC2H4)2NH)(CO)2Br], t-BuOK, amine, toluene, 80 °C, 24 hr | 0.156 g (1.2 mmol) | 1 mmol | C8: 85% | 56 | |||
| [Co(TriazNHC3H5-iPr)Cl2], t-BuOK, amine, toluene, 80 °C, 24 hr | 0.327 g (1 mmol) | 1 mmol | C22: 69–86% | 54 | |||
| NiBr2, 1,10-phenanthroline, t-BuOK, amine, toluene, 130 °C, 48 hr | 0.145 g–0.198 g (1.25 mmol) | 0.25 mmol | C7–C10: 59–76% | 59 | |||
|
[Ru(p-cymene)Cl2]2, DPEphos, amine, toluene or tert-amyl alcohol or neat, 130 °C, 48 hr |
|
0.264 g (2 mmol) | 2 mmol | 52–92% | 124 | |

|
[Cp*IrCl2]2, amine, base, H2O or toluene or neat, 80–140 °C, 6–96 hr |

|
0.100 g (1.1 mmol) | 1.1 mmol | 2–42% | 125 | |
| CuNiAlOx, K2CO3, amine, 1,4-dioxane, 150 °C, 24–36 hr | 0.460 g (5 mmol) | 1 mmol | 17–94% | 126 | |||
| Ru1Ni7/MgO, NaOH, aq ammonia, 10 bar H2, 220 °C, 4 hr | 0.100 g (1.1 mmol) | 1.1 mmol | Ala: 43% | 127 | |||
| Amino acids |

|
Pd/C or Pd(OH)2/C, alcohol, RT, 24–88 hr |

|
0.1 mmol | 0.1 mmol | R,R′: Alk 72–96% | 128 |
| hν, Ag/TiO2, alcohol, H2O, 25 °C, 10 hr | 2 mmol | 2 mmol | 94% (SM: L-Pro) | 129 | |||
| Shvo or Knölker-CH3CN cat., alcohol, 60–110 °C, 18–47 hr | 0.2 mmol | 0.2 mmol | R,R′: Alk 35–99% | 123 | |||
| Pd(OAc)2, TPPMS, alcohol, AcONa, H2O, 120 °C, 16–24 hr | 0.5 mmol | 0.5 mmol | R,R′: Alk, Allyl 43–86% | 130 | |||
| Ir(III)-NHC cat, alcohol, TFE, 90 °C, 20 hr | 0.25 mmol | 0.25 mmol | R,R′: Alk >95% | 131 |
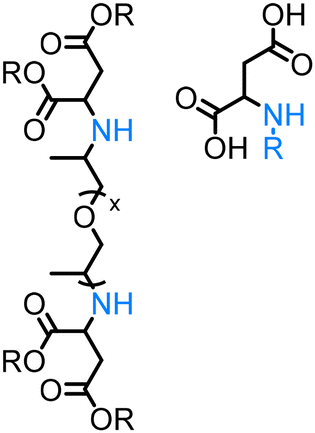
Significantly less homogeneous catalytic methods using long-chain aliphatic alcohols as coupling partners are reported.54,56,59,122 Among them a suitable protocol was developed by our group – the N-alkylation of natural α-amino acids with long-chain fatty alcohols using Knölker's complex as a catalyst.123 This methodology uses unprotected amino acids as coupling partners and discusses the possibility of the fully sustainable synthesis of surfactants entirely derived from biomass.
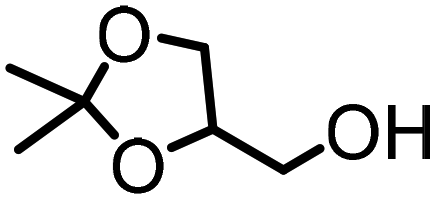
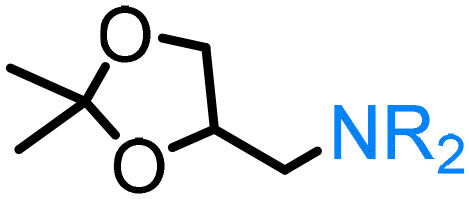
As mentioned above, glycerol – an unavoidable by-product of triglyceride transesterification or hydrolysis – is another important substrate in the field of biomass valorisation.132 However, the presence of three hydroxyl groups in its structure causes considerable hurdles regarding the selective transformation of one single functional group. In the course of research on the amination of glycerol via the ‘hydrogen borrowing’ approach, BASF reported RANEY® Ni/RANEY® Co system133 that allows selectively obtaining the 1,2,3-triaminopropane and 1,2-diamino-3-propanol as major reaction products. Crotti125 established the first example of the formation of N-heterocyclic products using glycerol and cis- and trans-1,2-diaminocyclohexane as starting materials catalysed by the simple and versatile iridium derivative [Cp*IrCl2]2 in water. Interestingly, the hydrogenated piperazines and benzimidazoles synthesised via this procedure are valuable building blocks to produce fine chemicals and pharmaceuticals. The direct amination of solketal, a 1,2-hydroxy-protected derivative of glycerol, via ruthenium-catalysed ‘hydrogen borrowing’ was extensively investigated by Kann.124 The formation of the highly valuable prochiral aminoketones, the key intermediate for the synthesis of chiral amino alcohols, was demonstrated by Shi126 over CuNiAlOx catalyst; the latter is formed via dehydration of glycerol followed by the amination of the hydroxyacetone intermediate.
Recently, an elegant protocol to convert ‘waste’ glycerol from the biodiesel industry via lactic acid to alanine in a single step over a Ru1Ni7/MgO catalyst was disclosed by Yan.127 It was established that a multifunctional Ni-doped Ru catalyst system remarkably promoted the amination of lactic acid, the key intermediate of the reaction formed via sequential dehydrogenation, dehydration, and rearrangement. This methodology is capable of providing access to another class of valuable chemicals.
Amino acids. Being a widespread class of nitrogen-containing compounds in nature, α-amino acids may serve as prominent sustainable alternatives to amines derived from petrochemicals via the energy-intensive Haber–Bosch process.134 Remarkably, nature ‘manufactures’ amino acids with significantly less energy demand through the biocatalytic nitrogen fixation process.135
However, notwithstanding its abundance and availability the further functionalization of the natural amino acids, in our present case – the N-alkylation of the amino acids with alcohols via the ‘hydrogen borrowing’ approach – is a very challenging task. Firstly, most amino acids are pH sensitive and have limited solubility in non-polar organic solvents, a typical reaction medium for N-alkylation reactions. Secondly, competing esterification of the amino acid could take place instead of the desired N-alkylation. Thirdly, due to the addition of a strong base, typically required for most ‘hydrogen borrowing’ systems, the racemization of the substrate or the formed N-alkylated product could occur. Moreover, amino acid could potentially reduce the catalyst activity by means of coordination to the metal centre blocking the vacant coordination site essential for efficient catalysis.
To the best of our knowledge, in literature, there are few examples of direct N-alkylation of free amino acids with alcohols (Table 2) justifying the difficulty of such an approach. Thus, Huang128 developed a mild and efficient Pd/C-catalysed monoalkylation of amino acids using alcohols as alkylating agents. Following this pioneering work, Hikawa130 reported a methodology for mono-N-allylation of unprotected amino acids with 1,1-dimethylallyl alcohol applying the homogeneous palladium-catalysed system in water as a reaction medium. Another work, described by Saito,129 demonstrated the only example of the photocatalytic N-methylation of proline with methanol in the presence of a silver-loaded titanium dioxide (Ag/TiO2). Our group disclosed the direct amination of the unprotected natural α-amino acids123 and α-amino acid esters/amides136 with alcohols (including long-chain fatty alcohols) in the presence of both Shvo's and Knölker's catalysts. Very recently, Ir(III)-NHC catalyst system was reported by Martín–Matute group that shows remarkable activity and broad scope in the N-alkylation of unprotected α-amino acids.131
3.2 Reductive amination
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond into a C–N one. It involves the reaction of an aldehyde or a ketone with an amine (including the industrially important ammonia). For a better understanding of the importance of this method and the challenges that still need to be tackled to transform this catalytic process into a cost-effective and elegant pathway towards bio-based amines, first one must have an insight into the mechanism. The general mechanism of this process is presented below in Fig. 5 showing the complexity and some challenges of this transformation. Detailed studies can be found in relevant reviews.24,25,137 In the first step of the reductive amination, the carbonyl group is subjected to nucleophilic attack by the amine to form a hemi-aminal intermediate, which dehydrates to afford the desired imine. In the second step, the resulting imine is reduced to the final amine product.
O bond into a C–N one. It involves the reaction of an aldehyde or a ketone with an amine (including the industrially important ammonia). For a better understanding of the importance of this method and the challenges that still need to be tackled to transform this catalytic process into a cost-effective and elegant pathway towards bio-based amines, first one must have an insight into the mechanism. The general mechanism of this process is presented below in Fig. 5 showing the complexity and some challenges of this transformation. Detailed studies can be found in relevant reviews.24,25,137 In the first step of the reductive amination, the carbonyl group is subjected to nucleophilic attack by the amine to form a hemi-aminal intermediate, which dehydrates to afford the desired imine. In the second step, the resulting imine is reduced to the final amine product.
 | ||
| Fig. 5 General mechanism of reductive amination. | ||
It is worth mentioning that reductive amination using ammonia would be an excellent method to form important primary amines, however, this transformation is typically highly challenging. This is not only due to the relatively sluggish imine formation step, but also due to the fact that the formed primary amines are generally better nucleophiles than ammonia itself, leading to the competing formation of secondary and tertiary amines, resulting in loss of selectivity. Mechanistically, with primary amine substrates, giving secondary amine products, the situation is similar to the case of ammonia. Differences occur when a secondary amine is used as substrate: instead of imine, an enamine is formed, which then is reduced to the desired tertiary amine. A further limitation may be that CO reduction is more favoured than CN reduction, leading to the competing hydrogenation of the substrate. Also, in the case of sluggish imine formation or imine reduction, the carbonyl compound itself can undergo aldol condensation reactions, diminishing product selectivity. Additionally, some reactive imine intermediates can also undergo condensation reactions, further compromising product selectivity. These side reactions can be avoided with the right selection of the catalytic system and optimization of reaction conditions to have the selective formation of the imine or enamine instead of the hydrogenation of the carbonyl substrate and hydrogenation of the imine (or enamine) at a high rate, preventing side reactions.24,25 Thus, the key rate-limiting steps in reductive amination are: (a) imine formation, (b) imine reduction, which depends on the catalyst system utilized.
As described below, the most atom-economic and desired way to perform reductive amination is with molecular hydrogen (or hydrogen donor), which requires carefully designed catalyst systems for efficient hydrogen activation.
Among the heterogeneous catalysts used for reductive amination reactions, RANEY® Ni, different supported ruthenium, cobalt, nickel catalysts, carbon-supported Ru, Rh, Pd, Pt are known to be effective for the formation of the target amine products.24,25,142,144,145
The case of using ammonia as a coupling partner to obtain primary amines deserves special mention. When developing a catalytic system for such a transformation, a number of difficulties must be taken into account. For the homogeneous catalyst system, the deactivation of the metal catalyst can take place as the ammonia coordinates to the metal, forming stable Warner-type complexes;24,25 for the heterogeneous systems – irreversible adsorption of reagents on its surface or leaching of the active metal or support, as well as sintering. Fortunately, all these challenges can be solved by proper selection of the catalyst system, namely the catalyst itself and reaction conditions.24,25,142,143
| Type of biomass | Platform chemical (PC) | Reaction conditions (selected examples) | Amine product | Amount of PC required | Scale | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| (Hemi)cellulose |

|
Ru/ZrO2, H2 30 bar, aq ammonia, 75 °C, 12 hr |

|
0.120 g (2 mmol) | 2 mmol | R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H, 93% :H, 93% |
146 |
| Ni-6458P (56 wt.%) or RANEY® Co-2725, 70 bar H2, aq ammonia, 100 °C, 1 hr | 0.500 g (8.3 mmol) | 8.3 mmol | R:H, 35–45% |
148 | |||
| Pd/C, 70 bar H2, aq ammonia/amine, 100 °C, 1 hr |

|
0.500 g (8.3 mmol) | 8.3 mmol | R:H, Me, 28–91% |
148 | ||

|
Ru/γ-Al2O3, 30 bar H2, 4 bar NH3, MeOH, 80 °C, 2 hr |

|
0.634 g (6.6 mmol) | 6.6 mmol | R:H, 75% |
153 | |
| Ru/Nb2O5, 40 bar H2, 1 bar NH3, MeOH, 90 °C, 2 hr | 0.048 g (0.5 mmol) | 0.5 mmol | R:H, 99% |
154 | |||
| Au/SiO2-SO3H, Pt/SiO2-SO3H, Ir/SiO2-SO3H, 0.1M sol of amine, 50 bar H2, EtOAc, r.t., 8 hr | 0.577 g (6 mmol) | 5 mmol | R:H, 3–21% |
161 | |||
| Ru1/NC-900-800NH3, 20 bar H2, 5 bar NH3, MeOH, 100 °C, 10 hr | 0.192 g (2 mmol) | 2 mmol | R:H, 94% |
160 | |||
| NiAl-10, MeOH, 20 bar H2, 5 bar NH3, 80 °C, 1.5 hr | 1.16 g (12 mmol) | 12 mmol | R:H, 99% |
155 | |||
|
Co-DABCO-TPA@C-800, 40 bar H2, 5 to 7 bar NH3, t-BuOH, 120 °C, 15 hr |
|
0.063 g (0.5 mmol) | 0.5 mmol | R:H, 89% |
162 | |
| Ru/Nb2O5, 1 bar NH3, 40 bar H2, MeOH, 90 °C, 4 hr | 0.063 g (0.5 mmol) | 0.5 mmol | R:H, 96% |
154 | |||
| Ni6AlOx, 21:1 NH3/HMF, 10 bar H2, H2O, 90 °C, 12 hr |
3 g (23.8 mmol) | 23.8 mmol | R:H, 81% |
163 | |||
| 1%Pd/C, 3 bar H2, trifluorotoluene, 100 °C, 1–3 hr | 0.126 g (1 mmol) | 1 mmol | R:Alk, Ar 76–100% |
164 | |||
| Ru(DMP)2Cl2, amine, 12 bar H2, EtOH, 60 °C, 5–24 hr | 0.063 g (0.5 mmol) | 0.5 mmol | R:Alk, Ar 43–95% |
165 | |||

|
Pt-MoOx/TiO2, amine, 3 bar of H2, solvent-free, 100 °C, 20 hr |
|
0.116 g (1 mmol) | 1 mmol | R:Alk, Ar 77–95% |
166 | |
| Pd/ZrO2, amine, 5 bar of H2, 90 °C, 12 hr | 0.581 g (5 mmol) | 5 mmol | R:Alk, Ar 80–94% |
167 | |||
| Ru/NMC, amine, 15 bar H2, ethanol, 120 °C, 4 hr | 1.16 g (10 mmol) | 10 mmol | R:Alk, Ar 80–99% |
168 | |||
| (R)-[RuCl(p-cymene)(BINAP)]Cl, NH4OAc, Ti(Oi-Pr)4, TFE, 60 bar H2, 90 °C, 22 hr | 0.058 g (0.5 mmol) | 0.5 mmol | R:H, 89% |
169 | |||
| Lignin |
|
Rh/Al2O3, aq ammonia sol, 20 bar H2, 80 °C |
|
0.2 g (1.3 mmol) | 1.3 mmol | 91% | 170 |
| Co-DABCO-TPA@C-800, 40 bar H2, 5 to 7 bar NH3, t-BuOH, 120 °C, 15 hr | 0.076 g (0.5 mmol) | 0.5 mmol | 88% (SM: isovanillin) | 162 | |||
| Ni6AlOx, 49:1 NH3/aldehyde, 2 bar H2, H2O, 100 °C, 10 hr |
0.152 g (1 mmol) | 1 mmol | 82% | 163 | |||
| NiAl-10, THF, 30 bar H2, 5 bar NH3, 90 °C, 1 hr | 0.152 g (1 mmol) | 1 mmol | 96% | 155 | |||
| Ni(BF4)2·6H2O, triphos, 5–7 bar NH3, 40 bar H2, TFE, 100 °C, 24 hr | 0.076 g (0.5 mmol) | 0.5 mmol | 90% (SM: isovanillin) | 171 |
Carbohydrate derivatives. Glycolaldehyde is an important bio-derived substrate that can be sourced from cellulose or sugars through retro–aldol reactions or from pyrolysis oil.148 It constitutes a C2 platform chemical and can lead to ethylenediamine, ethanolamine (monoethanolamine), diethanolamine, or piperazine through reductive amination pathways in the presence of NH3, H2, and a suitable catalyst. These products are industrially relevant: ethanolamine is used as an intermediate for pharmaceuticals, surfactants, or cosmetics.148 Its industrial synthetic route typically involves chlorination or epoxidation of petrol-based ethylene utilizing ethylene oxide as an intermediate, which is a toxic, highly reactive compound. In this context, obtaining ethanolamine from bio-derived glycolaldehyde by reductive amination is a safer and more sustainable route. Different catalytic systems are known for this transformation.42,106 For example, using a Ru/ZrO2 catalyst Zhang and coworkers prepared amines from bio-based aldehydes or ketones in aqueous ammonia.146 In these conditions (water) ethanolamine was synthesized with 93% yield when starting from glycolaldehyde and with 10% yield directly from cellulose in a two-step process.146 Later, Sels and his team148 underlined the importance of the choice of solvent, catalyst, and amine to glycolaldehyde ratio in the reductive amination of glycolaldehyde for producing alkanolamines. They developed various effective catalytic routes to several C2 amines (ethyleneamine, alkanolamines, ethylene diamines, higher alkanolamines) starting from glycolaldehyde using heterogeneous catalysts (Ni, Co, Pd, Ru – based), aqueous ammonia or corresponding amines and pressurized H2 gas.
Other versatile important platform chemicals derived from cellulosic biomass or sugars through established dehydration pathways are furfural and 5-hydroxymethyl furfural (5-HMF).89,107,109,149,151,152
Selective reductive amination of furfural, with NH3, leads to furfurylamine.24,42,145,147 For example, with heterogeneous Ru/γ-Al2O3 under mild conditions, Dong et al. reported the production of primary amines from furfurylamine with 75% yield.153 The key to high product selectivity was the choice of amphoteric alumina γ-Al2O3 support. The importance of the support was also shown by Hara and coworkers:154 they observed that Nb2O5-supported ruthenium catalysts were the most efficient for the selective transformation of furfural to furfurylamine with 99% yield. Also, a nickel-containing catalyst was used for the switchable transformation of 5-HMF, presented by Wang.155 The particularity of the catalyst is that by adjusting the particle size of the carbon-doped Ni nanoparticles grafted on hollow polymer nanospheres, the product of the reductive amination can be switched between the primary amine or the secondary imine, in both cases with excellent yield. Besides furfural, the catalyst could be used on other biomass-derived substrates such as 5-HMF or vanillin. The selective synthesis of furfurylamine from furfural has been investigated by several research groups, leading to the development of various heterogeneous catalysts based on ruthenium156,157 as well as earth-abundant metals. For instance, single-crystal cobalt phosphide nanorods (Co2P NRs),158 working in aqueous ammonia and low H2 pressure, or RANEY® cobalt.159 The latter catalyst, under mild reaction conditions, gave the target product with good selectivity, which is explained by its efficiency at the hydrogenolysis step. Recently, Zhang reported a highly active N-doped carbon-supported ruthenium single-atom catalyst Ru1/NC-900-800NH3 for the selective production of primary amines. The catalyst exhibits good reuse stability and can be applied to other biomass-derived aldehydes (furfural, 5-HMF, glycolaldehyde).160
Equally well studied is the reaction of furfural with various primary or secondary amines leading to the formation of secondary or tertiary amines using heterogeneous catalytic systems,24,25,145 such as sulfonic acid-functionalized silica-supported Au, Pt, and Ir catalyst systems shown by Martinez et al.161
5-HMF, one of the most important and versatile sugar-derived platform chemicals, has the potential to serve as a precursor for various amines. Interestingly, the selective reductive amination of the aldehyde functionality can be accomplished with ammonia in the presence of a suitable catalyst.149 For example, with the catalytic system mentioned earlier for furfural, namely the Ru/Nb2O5 catalyst, Hara and coworkers showed the formation of 5-aminomethyl-2-furylmethanol with 96% yield.154 MOF-derived cobalt nanoparticles Co-DABCO-TPA@C-800 prepared by Beller and his team have been successfully used for the amination of various aldehydes, including 5-HMF.162 A nickel-based catalyst Ni6AlOx was developed by Pera-Titus and Shi with coworkers, delivering 5-aminomethyl-2-furylmethanol with excellent selectivity.163
When 5-HMF reacts with a primary or secondary amine, instead of ammonia, the corresponding N-substituted-5-(hydroxymethyl)-2-furfuryl amines are formed.147,163–165 For example, Zhang and team described a homogeneous ruthenium complex, dichlorobis(2,9-dimethyl-1,10-phenanthroline)-ruthenium(II) (Ru(DMP)2Cl2), that gave the desired secondary or tertiary amines from 5-HMF in good yields (60–95%).165 Corma and Iborra demonstrated a highly selective catalytic system, supported Pd nanoparticles (Pd/C), in the reaction of 5-HMF with aniline, which delivered N-phenyl-5-(hydroxymethyl)-2-furfuryl amine in quantitative yield.164 A series of secondary and tertiary amines derived from 5-HMF were prepared by Pera-Titus and Shi, with substrates such as benzylamine, aniline, morpholine, 1-butylamine, and ethanolamine, using a Ni6AlOx catalyst.163 Several other catalytic systems were presented for the transformation of 5-HMF into amines, both with noble (Pd, Ru) and earth-abundant metals (Ni, Co, Cu),149,152 highlighting the significance of different methods for upgrading this biomass-derived precursor.
An interesting example of reductive amination of carbohydrate-derived chemicals can be found in the case of levulinic acid. In the first step, reductive amination of the carbonyl group takes place, followed by dehydrative cyclization of the amine and carboxylic groups, leading to the corresponding N-substituted 5-methyl-2-pyrrolidones. Shimizu et al. used a Pt-MoOx/TiO2 catalyst for the transformation of levulinic acid into a series of 5-methyl-2-pyrrolidones under mild, solvent-free conditions.166 The reusable heterogenous catalytic system was successfully employed with a wide variety of amines leading to very good yields (>80%). Later, Zheng and Xiao et al.167 emphasized that the catalyst plays an important role in the reaction to obtain the pyrrolidone rather than gamma-valerolactone, which is usually formed from levulinic acid by hydrogenation. Using ZrO2-supported Pd nanoparticles, pyrrolidines could be produced from levulinic acid and various amines, and the high chemoselectivity of the catalyst was attributed to the strong Lewis acidity of the ZrO2 support. Excellent yields (80% to 99.8%) could be achieved using a highly stable and recyclable N-doped mesoporous carbon (NMC)-supported Ru catalyst as well.168 Recently, de Vries and coworkers synthesized chiral 5-methyl-2-pyrrolidinone from levulinic acid through asymmetric reductive amination employing a commercially available homogeneous ruthenium-bisphosphine catalyst. The (R)-[RuCl(p-cymene)(BINAP)]Cl catalyst using ammonium acetate as an amine source afforded the target product with 96% ee in 89% yield.169
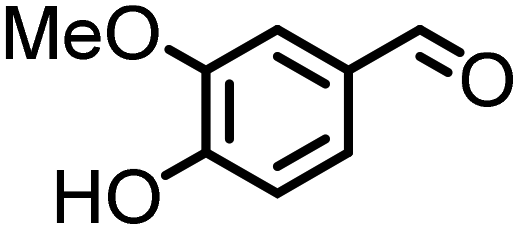
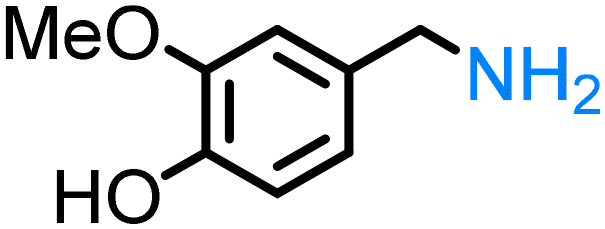
Lignin derivatives. Reductive amination can also be employed on lignin-derived monomers. As such, vanillin (4-hydroxy-3-methoxybenzaldehyde) in an aqueous ammonia solution over a Rh/Al2O3 catalyst forms vanillylamine ((3-hydroxy-4-methoxyphenyl)methanamine) in good yield.170 Other catalysts such as Ni6AlOx
163 or Co-DABCO-TPA@C-800162 have been described for the conversion of vanillin to the corresponding primary amines.
Reductive amination of vanillin presents a few examples of homogeneous catalytic systems for the production of bio-derived amines. Such systems are based on Ru, Fe, Cu, Mn, Mo, Ir, Co complexes supported by different types of ligands.24,25,142 Thus, with a nickel-based complex with triphos(bis(diphenylphosphinoethyl)phenylphosphine) ligand, vanillylamine was obtained with 90% yield in trifluoroethanol.171
Similar results were observed with a cobalt triphos catalyst (95% yield)172 and RuCl2(PPh3)3 complex (88% yield).173
3.3. Hydroamination of alkenes
C bond forming a C–N bond, either in an intermolecular or intramolecular fashion (Fig. 6).174–176 As the simplest amine, ammonia can also be used.29 One of the key benefits from a green chemistry perspective is that the reaction proceeds with a complete atom economy, enabling the waste-free production of alkylamines from inexpensive and widely available starting materials.42,177
 | ||
| Fig. 6 General scheme of the hydroamination of alkenes and overview of mechanisms: (a) the amido-mechanism typical of alkali and alkaline metals, lanthanides and actinides; (b) the C–C multiple bond mechanism and (c) the amine activation mechanism that are typical of late transition metals. All mechanisms shown lead to the anti-Markovnikov product. The same pathways apply to the Markovnikov product, but are not depicted due to space constraints. | ||
Hydroamination reactions are nearly energy-neutral processes (ΔG ∼ 0 kcal mol−1).178,179 Due to the thermoneutral nature of the reaction, the use of activated amines or alkenes is typically necessary.180,181 Hydroamination of non-activated CC bonds is more favorable in an intramolecular fashion, driven by ring-closing, and leading to nitrogen-containing heterocycles.182
Hurdles that so far limited the development of efficient hydroamination reactions derive from the number of side reactions, such as isomerization and telomerization of alkenes, as well as oxidative amination.183
In the presence of metal-catalysed hydroamination reactions, the high affinity of σ donor amines with the metal centers employed as catalysts may inhibit their activity by preventing the coordination of the alkenes.28,184 When ammonia is considered, high bond dissociation energy (415 kJ mol−1), strong affinity with metal centres and high activation energy barrier to initiate the reaction are the major challenges that prevent it from being employed in alkene hydroamination reactions.29
The regioselective addition of the amine to the CC bond also represents a key factor to consider (Fig. 6). Achieving anti-Markovnikov regioselectivity is highly preferred for obtaining industrially appealing linear primary amines.
Several catalytic systems have been explored over the last decades. Early studies predominantly employed alkali (Li, K, Cs), alkaline earth (Ca, Sr, Mg), actinides (U) and lanthanide (Nd, La, Lu, Sm, Y, Eu, Yb) metals as catalysts. It is generally accepted that reactions involving the use of these metals proceed via the so-called amido mechanism (Fig. 6a),185 whereby the active metal–amido complex (A) is generated by protonolysis of the initial ligand by the amine substrate. This is followed by the insertion of the alkene into the metal–amido bond and the protonolysis of the M-alkyl species (B) by a new amine molecule to ultimately release the alkylamine and regenerate the catalytically active metal–amido species.
Later, group 4 metals (Ti and Zr) were also used as catalysts. Although these effectively facilitate amine addition to a wide range of alkenes, their high oxophilicity makes them sensitive to oxygen and moisture, restricting their use to controlled atmospheres and oxygen-free functional groups. These drawbacks prompted researchers to switch their focus to late-transition metals (Rh, Au, Cu, Pd, Ru, Pt, Ir).28 Catalysts based on these metals display a C–C multiple bond activation mechanism (Fig. 6b).186 This mechanism relies on the ability of cationic metal complexes with poor d-electrons to activate C–C multiple bonds by withdrawing electron density. This way, the nucleophilic amine attacks the metal-coordinated alkene (B) followed by protonolysis of M–C bond of the formed intermediate (C) giving rise to the hydroaminated product.
Importantly, late transition metals have been proposed to exhibit an amine activation mechanism as well (Fig. 6c). This involves the oxidative addition of an amine to generate an intermediate containing both a hydride and an amido group (B), followed by the insertion of a CC bond into the M–N bond, resulting in intermediate (C), and final reductive elimination.186,187
For more detailed mechanistic and catalytic considerations, one may refer to the numerous excellent reviews.28,174–176,183,184
C bonds suitable for upgrading through hydroamination (Table 4).
| Type of biomass | Platform chemical (PC) | Reaction conditions (selected examples) | Amine product | Amount of PC required | Scale | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| (Hemi)cellulose |
|
Na metal, 175–200 °C n-heptane, ammonia |

|
800–1000 bar | 2.94 moles | 70% | 188 |
| H-clinoptilolite, 370 °C, ammonia | 11 bar | Not given | 13% | 189 | |||
| Lithium diethylamide, TMEDA, 80 °C, toluene, 18 hr | 40 bar | 0.03 mol amine | 93% | 191 | |||

|
[Fe(Cy2PC2H4)2NH)COBr2], NaHBEt3, K3PO4, cyclohexane, 80 °C, N2, 12–24 hr |

|
0.044–0.087 g (0.75–1.5 mmol) | 0.5 mmol | 53–99% | 194 | |
| [Mn(Et2PC2H4)2NH)(CO)2Br], NaHBEt3, K2CO3, cyclohexane, 60 °C, Ar, 24 hr | 0.058 g (1 mmol) | 0.5 mmol | 13–96% | 196 | |||
| [Mn(Ph-SMe)-Py)(CH2)2NH)(CO)3Br], K2CO3, toluene, 100 °C, Ar, 24 hr | 1.196 g (21.5 mmol) | 5.37 mmol | 74% | 195 | |||

|
Ni(NO3)2 on SiO2, neat, RT, 2–24 hr |
|
5.2 g (30 mmol) | 15 mmol | 76–84% | 197 | |
| NiBr2, AgOTf, toluene, 100 °C, 16 hr | 0.43–0.86 g (3–6 mmol) | 2 mmol | 42% | 198 | |||
| Ln(OTf)3, toluene, 100 °C, 24 hr | 0.22–0.43 g (1.5–3 mmol) | 1 mmol | 33% | 199 | |||

|
Cp2′LaCH(TMS), pentane, RT, Ar, 24 hr |
|
1.69 g (20 mmol) | 20 mmol | 86% | 202 | |
| Y[H2(Dip2BINO)][N(SiHMe2)2](THF)2, benzene-d6, 100 °C, Ar, 16 hr | 0.032 g (0.38 mmol) | 0.38 mmol | ∼99% | 205 | |||
| [(4R,5S)-Ph2Box]La[N(TMS)2]2, benzene-d6, 23 °C, Ar | 0.012 g (0.14 mmol) | 0.14 mmol | ∼98% | 206 | |||
| Y[Bis(thiolate)][N(TMS)2], thiophene, benzene-d6, 60 °C, Ar, 8 hr | 0.027 g (0.32 mmol) | 0.32 mmol | 81% | 207 | |||
| {PhB(η5-C5H4)(OxMe2)2}Zr(NMe2)2, benzene-d6, 23 °C, Ar, 33 hr | 0.009 g (0.1 mmol) | 0.1 mmol | ∼62% | 208 | |||
| (R,R)-(−)-N,N′-Bis(3,5-di-tert-butylsalicyliden)-1,2-cyclohexandiaminocobalt(II) tosylate, TMDSO, NBu4OTf, O2, neat, RT, 6 hr | 0.026 g (0.3 mmol) | 0.3 mmol | 78% | 203 | |||
| Y[H2XN2](CH2SiMe3)3(THF)2, benzene-d6, 60 °C, Ar, 45 min | Not given | Not given | ∼99% | 209 | |||

|
Cs2.5H0.5PMo12O40 on SiO2, 290 °C, water, N2, 3 hr |

|
Not given | Not given | 5% | 210 | |
| Lignin |
|
[Ir[(S)-DTBM-SEGPHOS](ethylene)(Cl)], NaBArF, 1,4-dioxane, 60 °C, N2, 18–19 hr Ru(PEt3)3NTf2, 1,2-dichlorobenzene, 80 °C, 48 hr |

|
0.013–0.036 g (0.1–0.2 mmol) | 0.1–0.2 mmol | 52–95% | 211 and 212 |
| Terpenes |
|
Pd(tfa)2, DPEphos, DMF:heptane (40:60), 110 °C, Ar, 3 hr |
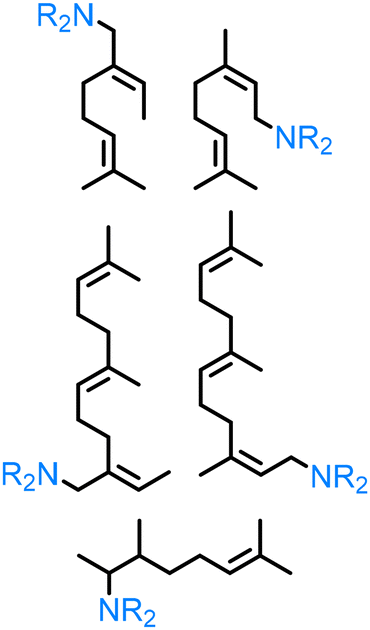
|
Flow | Flow 0.1667 mol L−1 | 80% | 213 |
| Pd(tfa)2, DPEphos, DMF, 100 °C, Ar, 5 hr | 0.87 g (4 mmol) | 4 mmol | 94–99% | 214 | |||
| Ru(PEt3)3NTf2, 1,2-dichlorobenzene, 100 °C, 48 hr | 0.022 g (0.2 mmol) | 0.2 mmol | 57% | 211 | |||
| Fatty alcohol & fatty acids |

|
Cu(OAc)2, (±)-DTBM-SEGPHOS, diethoxymethylsilane, 40 °C, Ar, 36 hr |

|
0.381 g (1.2 mmol) | 1 mmol | 95% | 215 |
| Ru(PEt3)3NTf2, 1,2-dichlorobenzene, 80 °C, 48 hr | 0.022 g (0.2 mmol) | 0.2 mmol | 63% | 211 | |||
| [Ir[(S)-DTBM-SEGPHOS](ethylene)(Cl), NaBArF, 1,4-dioxane, 60 °C, N2, 18–19 hr | 0.011 g (0.1 mmol) | 0.1 mmol | 81% | 212 |
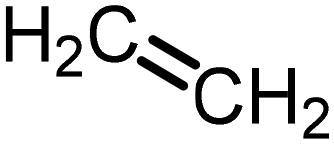
Carbohydrate derivatives. The first hydroamination of ethylene, as the simplest alkene now accessible from bioethanol, dates back to 1954. In that year, Howk et al.188 reported the sodium-catalysed addition of ammonia to ethylene under harsh conditions (175–200 °C, 800–1000 bar), producing nearly equimolar amounts of ethylamine, diethylamine, and triethylamine, with a total yield of 70%. Afterwards, Deeba and Ford189,190 investigated the use of Zeolites, achieving a 13% yield of ethylamine. In 2005, Beller191 reported a milder approach via base-catalysed hydroamination with diethylamine in the presence of LiNEt2 at 80 °C and 40 bar, achieving up to 90% yield of the hydroaminated product.
Allyl alcohol is naturally present in garlic but it can also be obtained by hydrogenation of acrolein, which can in turn be derived from the dehydration of glycerol.192,193 The anti-Markovnikov iron-catalysed hydroamination of allyl alcohols with aryl and aliphatic amines was demonstrated to access the γ-amino alcohols up to 99% yield by using Fe194 as well as Mn catalysts. For example, Das et al.195 investigated the use of Mn complexes bearing a soft sulfur donor side-arm in the N,N ligand backbone. Similarly, a PNP pincer complex was used by Duarte de Almeida et al.196 for the hydroamination of allyl alcohol with N-aryl amines, leading to high product yield.
Tompos and colleagues197 reacted diamines (Jeffamine polyether amines) with maleate and fumarate diesters, both with and without catalysts. Catalysts used included heterogeneous catalysts, such as Ni(NO3)2 on SiO2, and organocatalysts like imidazole. However, the presence of catalysts resulted in only a modest increase in conversion rates, with yields improving by just around 10%.
The hydroamination of dimethyl maleate with aniline has been demonstrated using NiBr2 and AgOTf, at 100 °C for 16 h, producing the corresponding N-aryl dimethylmaleate in 42% isolated yield.198 Kunjanpillai and co-workers recently expanded the catalyst scope to Ln(OTf)3, with the product isolated in 33% yield, at 100 °C for 24 h.199
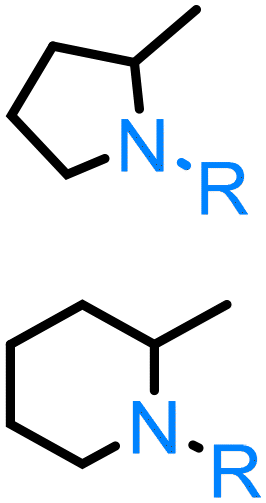
As regards to amino alkenes, the lanthanocene-catalysed intramolecular hydroamination of 5-amino-1-pentene, which can be obtained from the dehydration of bio-based 5-amino-1-pentanol,200 can form 2-methylpyrrolidine, as demonstrated by the Marks group.201,202 Later, Ye and Zhu203 investigated the intermolecular hydroamination of N-tosylated 5-amino-1-pentene by employing a cobalt catalyst, to produce N-tosylated 2-methylpyrrolidine derivatives. Recently, Hennecke and co-workers204 reported the hydroamination of N-nosyl 5-amino-1-pentene. Hultzsch et al.205 synthesized chiral rare-earth-metal (yttrium and lanthanide) complexes containing biphenolate and binaphtholate amido complexes, which were utilized for the hydroamination/cyclization of 5-amino-1-pentene. Complete conversion was achieved at 100 °C and a reaction time of 16 h, with a moderate enantiomeric excess (ee) of 52% for the R-product. Marks206 obtained a medium ee 40%, R-product, employing lanthanide-based bis(oxazolinato) complexes, under ambient conditions. A significant improvement was achieved by Kim and Livinghouse207 who utilized yttrium complexes of axially chiral bis(thiolate) ligands, achieving 81% ee. Sadow and co-workers208 reported on the use of zwitterionic cyclopentadienyl-bis(2-oxazolinyl)borate diamidozirconium complexes as pre-catalyst for the hydroamination/cyclization of 5-amino-1-pentene at room temperature with a 62% yield.
In a similar manner, 1-amino-5-hexene, a compound which can be obtained through the dehydration of 6-amino-1-hexenol, the corresponding 6-membered N-heterocycle can be obtained through different rare earth, yttrium or aluminum catalysed processes.209
While most alkenes used in hydroamination are derived from bio-based alcohols, a straightforward alternative would be to use the said alcohols directly. In one case, (dimethylamino)acetone was obtained from glycerol using a Cs2.5H0.5PMo12O40 supported on a mesoporous silica.210
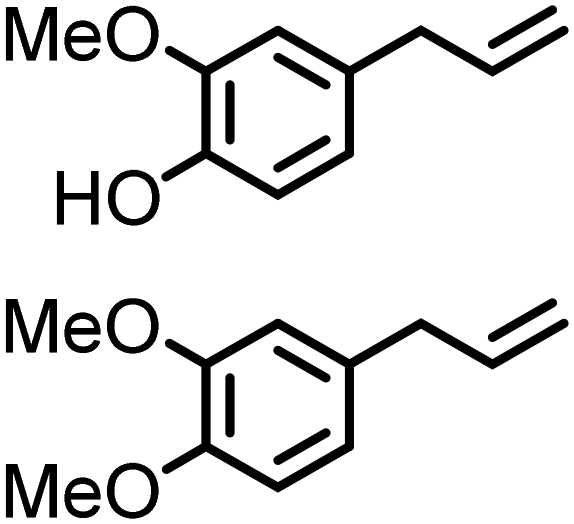
Lignin derivatives. A few examples of lignin-derived compounds were successfully employed as renewable platform chemicals for the synthesis of amines via hydroamination. Hartwig and co-workers211 studied the hydroamination of various unactivated terminal alkenes with ammonia surrogates (2-amino-5-methylpyridine) in the presence of Ru(PEt3)3NTf2. In this regard, they evaluated the hydroamination of 4-allyl-1,2-dimethoxybenzene, which can be obtained from lignin-derived eugenol, with 2-amino-5-methylpyridine at 80 °C for 48 h, isolating the Markovnikov product in 52% yield. The pyridyl moiety can be removed in the presence of PtO2, H2 and NaBH4, to afford the corresponding primary amine product. In a similar study, the same group also reported on the use of a chiral (S)-Ir catalyst for the hydroamination of estragole (1-allyl-p-methoxybenzene), chavicol (p-allylphenol) and eugenol.212 The corresponding aminated products were isolated in excellent enantioselectivity and 86%, 92% and 95% yield, respectively.
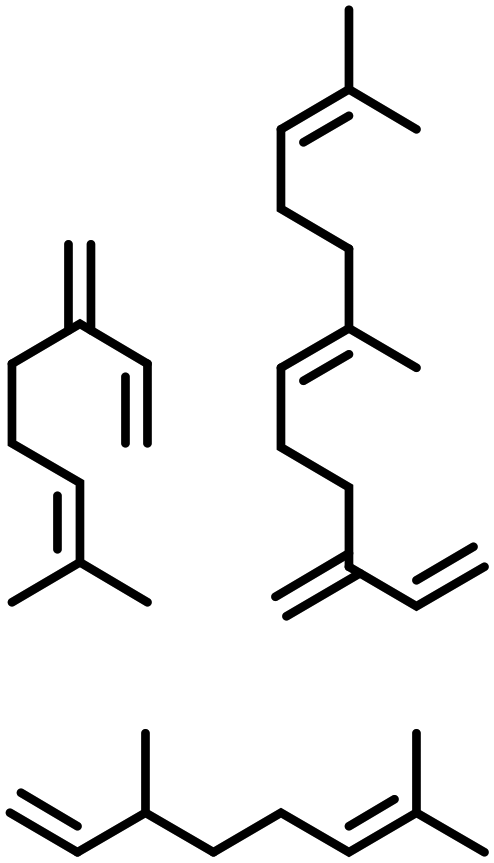
Terpene derivatives. As naturally available unsaturated compounds, terpenes are a valuable source to obtain renewable amines via hydroamination. In this frame, Vorholt and co-workers213 investigated the hydroamination of β-myrcene with morpholine, in the presence of a Pd(tfa)2/Bis[(2-diphenylphosphino)phenyl]ether catalyst system. They have also expanded this to the hydroamination β-farnesene with dimethyl and diethyl amine, piperidine, morpholine and diethanolamine, using the same catalyst system as reported for β-myrcene.214 The corresponding amines were isolated in moderate (48% in the case of diethanolamine) to excellent (94–99% for the other amines) yields at 100 °C for 5 h. Hartwig212 studied the hydroamination of (+)-β-Citronellene, which can be obtained from citronellol, a monoterpenoid, using Ru(PEt3)3NTf2 at 80 °C for 48 h. The hydroamination was performed in the presence of an ammonia surrogate (2-amino-5-methylpyridine), which can be removed in the presence of PtO2, H2 and NaBH4, in order to produce the corresponding primary amine product in 57% isolated yield.
Fatty acids derivatives. The internal double of fatty acids/esters can also undergo hydroamination, resulting in renewable compounds which are of interest in the polymer industry. In this regard, the hydroamination of 1-dodecene, which can be obtained via the dehydration of 1-dodecanol, itself derived from bio-based lauric acid, has been investigated by Buchwald and co-workers.215 In their work, they utilized copper phosphine catalysts, which were able to regioselectively catalyse the hydroamination of terminal alkenes to afford the anti-Markovnikov product. In the case of 1-dodecene with O-benzoylhydroxylamine, the corresponding aminated product was isolated in 94% yield, at 40 °C and 36 h reaction time. In contrast, the Markovnikov product derived from the hydroamination of 1-dodecene with 2-amino-5-methylpyridine, used as ammonia surrogate, was obtained by Ma et al.211 in 63% yield. Ru(PEt3)3NTf2 was utilized as catalyst, at 80 °C for 48 h. This was later expanded to the use of a chiral (S)-Ir catalyst for the hydroamination of the same substrates, where the product could be isolated in 81% and an er of 95
:5 (S-product).212
4. Evaluation of the green credentials for the synthesis of amines: a comparative analysis
Having established the foundation of various catalytic amination approaches as applied to renewables, we now turn the focus on a comparative analysis between traditional petrochemical routes and bio-based synthesis methods for selected case studies. By employing the CHEM21 green metrics toolkit, we assess the environmental and operational sustainability of these pathways.However, it is important to acknowledge the inherent differences between mature, well-established petrochemical processes and emerging bio-based technologies, whereby the developments regarding biorefineries and innovative solutions to access renewable platform chemicals are naturally lagging behind. Biorefinery approaches, especially those for processing complex non-edible lignocellulosic biomass are at the discovery or pilot stage, nonetheless a rapid progress is expected in the development of biorefineries towards higher TRL levels and commercial feasibility.
This presents a unique challenge when comparing the fossil-based versus bio-based approaches during our analysis. Therefore, our comparative analysis (sections 4.2–4.5) focuses on comparing the value chains even at laboratory stage development, but excludes raw material sourcing. A specific discussion on bio-based platform chemical sourcing is provided in section 4.1 below.
This means that a cradle-to-grave analysis of the entire process goes beyond the scope of this tutorial review.
4.1 Feedstock sourcing
Herein we provide a discussion on the starting reagents employed for the comparative analysis outlined afterwards. Except for cardanol (Scheme 3), all of the compounds selected as starting reagents for the bio-based synthetic routes are derived from lignocellulosic biomass. Specifically, dihydroconiferyl alcohol (Scheme 1) is derived from lignin, while 5-(chloromethyl)furfural (Scheme 2) and 5-hydroxymethyl furfural (Scheme 4) are derived from cellulose. | ||
| Scheme 1 Synthesis of N-substituted tetrahydro-2-benzazepines via a classical route (Steps 1–13) starting from benzene vs. a sustainable route (Steps A–B) starting from lignin-derived dihydroconiferyl alcohol. | ||
 | ||
| Scheme 2 Synthesis of ranitidine by the classical pathway (Steps 1–4) vs. the alternative pathway (Steps A–D). | ||
 | ||
| Scheme 3 Synthesis of norfenefrine by the classical pathway (Steps 1–5) vs. the bio-based pathway (Steps A–C). | ||
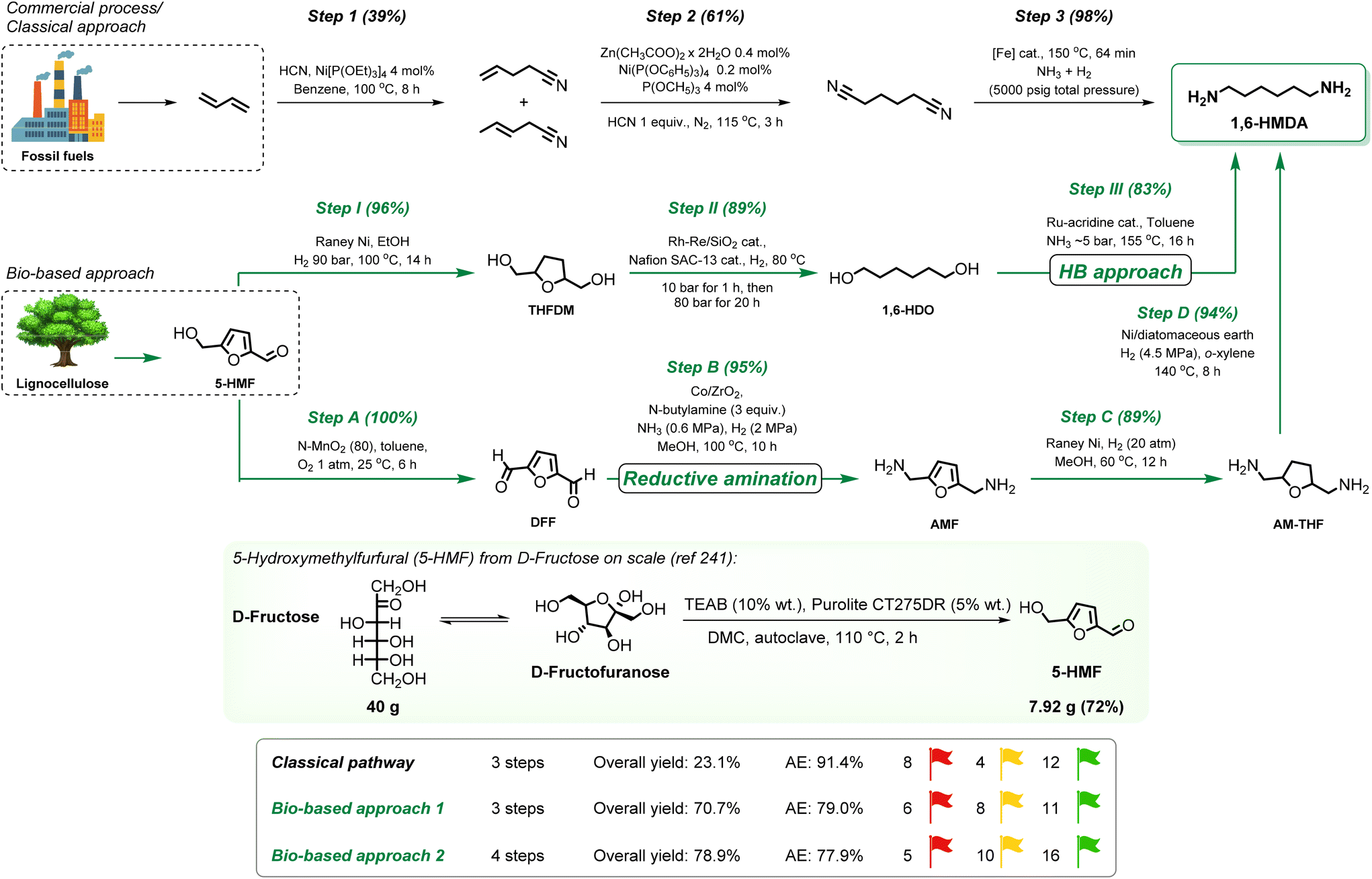 | ||
| Scheme 4 Synthesis of HMDA: comparison of the commercial route with sustainable approaches based on reductive amination and ‘hydrogen borrowing’ atom-economic transformations. | ||
These reports reveal the feasibility of obtaining dihydroconiferyl alcohol from lignocellulose, positioning it as the ideal starting material for the pathways showcased in Scheme 1.
Although the production of fossil-based benzene may be assumed as more cost-effective, several LCA studies related to RCF exist in the literature.
Researchers have made remarkable strides, advancing RCF biorefining, by optimizing reaction conditions, screening catalysts, and enhancing experimental setups. However, recent techno-economic analysis (TEA) and life cycle assessment (LCA) have flagged the necessity of further developments to make RCF more economically viable.219–221
Tschulkow et al.222 recently reported that woody biomass conversion into bleached pulp, phenolic monomers and oligomers currently generates higher greenhouse gas (GHG) emissions compared to alternative market options (unbleached sulfate pulp, phenol, and bisphenol A), largely due to by-products and gaseous waste streams. The authors provided alternatives that could reduce the emissions by up to 50%, such as incinerating gaseous waste streams for energy recovery. An additional challenge associated with the RCF process is the cost and recovery of pure solvents. Arts et al.220 demonstrated that using solvent mixtures derived from RCF effluent improved lignin extraction while significantly reducing costs and environmental impact in terms of lignin oil minimum selling price (MSP-RLO) and global warming potential (GWP-RLO), making possible comparison with fossil-based routes. Beckham and Román-Leshkov groups221 further explored the role of RCF recycled oil as a solvent for the RCF process, demonstrating that a concentration of lignin oil in the solvent up to 100 wt% is feasible while achieving similar performances of a methanol–water mixture.
An additional recommendation is to develop hydrogen-free RCF processes, as lowering reactors’ operating pressures could reduce the RCF oil selling price by 32%. In this frame, encouraging results were disclosed by Kenny et al.223 when using Pd/C and Pt/C on poplar lignin, as high monomer yields were retained while using these catalysts in the absence of H2. Debecker224 and coworkers investigated the role of different hydrogen donors for straw RCF. Notably, to prevent (hemi)cellulose decomposition due to the high temperatures (250 °C), the use of H3PO4 boosted the generation of monophenolic compounds, enabling monomer yields to reach approximately 18 wt% in hydrogen-free conditions.
The synthesis of furfural from cellulose is a well-established process involving the H2SO4-promoted hydrolysis of pentosan sugars to monosaccharides (mainly xylose), followed by dehydration to furfural.225,226 The produced furfural is recovered through steam distillation and fractionation. This process was first developed by Quaker Oats,227 which started commercialising furfural in 1921, later followed by other companies that adopted similar methods based on the same process.228 Side reactions occurring during the synthesis, e.g. formation of humins, are responsible for the typically low yields (∼50%), which is acceptable for an industrial operation. Much research is currently devoted to optimizing furfural synthesis mainly focusing on the use of solid catalysts and biphasic systems.229
The Mascal group230,231 has pioneered the synthesis of CMF from cellulose, which was found to be synthesized from corn stover with 72% yield in a biphasic reactor, under mild conditions, using aqueous HCl as the sole reagent. The main drawback in this case is related to the use of hazardous 1,2-dichloroethane solvent, which is used to create a phase separation that is not compromised by aqueous acid. Breeden et al.232 investigated CMF synthesis via microwave heating using cyclopentyl methyl ether (CPME) and cyclohexane. Recently, Bueno Moron et al. integrated CMF production with selective lignocellulose saccharification (DAWN technology, Avantium), optimizing a biphasic system to enhance yields (>90%) and scalability.233
Nowadays, bio-CMF is commercialized by Origin Materials,234–236 making it a suitable candidate for the sustainable synthesis of ranitidine.
5-HMF is an ideal building block for producing bio-HMDA given its facile access from carbohydrates via catalytic dehydration of pentoses. Having multiple functional groups, such as carbonyl and hydroxyl, within its structure, this compound provides opportunities for further functionalization in an atom- and step-efficient manner.93,238 Nevertheless, the challenge persists in achieving the selective conversion of 5-HMF into the desired product, given its heightened reactivity. For example, Dros et al.239 highlighted that based on current technologies, the petrochemical-based synthesis of hexamethylenediamine (HMDA) is more economically and environmentally favourable than bio-based alternatives due to lower manufacturing costs (1.84 € per kg vs. 2.13–2.55 € per kg) and high impact on eutrophication derived from the use of fertilizer-intensive feedstocks. The main issues are related to the feedstock used for fructose production and the high energy demand needed to dry the high-fructose syrup (HFS) used in the production of HMF. Therefore, it could seem impractical to compare the HMF-based route with a conventional one based on a bulk petrol-based chemical such as butadiene. However, the authors highlighted promising alternatives which should focus on optimizing synthetic strategies to produce fructose (and HMF) directly from agricultural lignocellulose and on using aqueous or biphasic systems to reduce the need for costly and environmentally impacting drying procedures and extensive use of organic solvents. Several studies have already shown encouraging results in this context.240–243
4.2 Evaluation of the green credentials for the synthesis of N-substituted tetrahydro-2-benzazepines
In order to demonstrate the efficiency and potential of the ‘hydrogen borrowing’ methodology, herein we perform a comparative analysis in terms of the greenness of a classical, literature-based, and sustainable protocol based on the ‘hydrogen borrowing’ approach for the synthesis of N-substituted tetrahydro-2-benzazepines.We selected two examples, namely 2-benzyl-8-methoxy-2,3,4,5-tetrahydro-1H-benzo[c]azepin-7-ol and 2-(4-chloro-phenyl)-7-methoxy-2,3,4,5-tetrahydro-1H-benzo[c]azepin-8-ol since for these compounds well-described and detailed experimental petroleum-based244–249 and bio-based11 synthetic procedures are available in the literature. These literature pathways are at the discovery level – first pass CHEM21 green metrics toolkit.
The values of quantitative (calculated for each step and overall route) and qualitative metrics for both the classical and sustainable approaches are summarised in Scheme 1 and Tables S2 and S3 (see ESI for more details†). On the whole, the results of the analysis of the quantitative and qualitative metrics demonstrate a clear advantage of the sustainable approach over the classical one; below the obtained results and each evaluated parameter are discussed in detail.
When comparing mass-based metrics of the two approaches toward N-substituted tetrahydro-2-benzazepines (Scheme 1 and Table S2†), it is obvious that the step economy of the sustainable one is higher: only 2 steps are required to obtain N-substituted 7-membered heterocycle starting from lignin-derived dihydroconiferyl alcohol, compared to 13 steps in the classical approach from benzene. As such, the yield of the former is almost 11 times higher (84.4% vs. 7.8%)! The same trend is observed for AE and RME: for the green path, both parameters are increased to 89% and 67%, correspondingly, compared to 31% and 5% for the classical route. Fewer reaction steps significantly reduce waste production: PMI tot reduced by 72% (3866 g g−1vs. 1082.3 g g−1). The latter is promising, given the fact that the sustainable route entirely consists of steps for which only data on the discovery level are available. Moreover, it can be expected that further optimization of the reaction conditions, the solvent used in each step as well as the solvent recycling, and the work-up method when moving toward the pilot scale will make the sustainable route even greener compared to the classical one. It is worth mentioning that cumulative PMI values contain the PMIs WU that were not optimized at the discovery level, as such comparison and interpretation of cumulative overall PMIs should be performed with caution.
Looking at the qualitative parameters (Scheme 1 and Table S3†), it can be seen that the sustainable pathway uses a biomass-derived substrate, dihydroconiferyl alcohol, which scores higher in the ‘health and safety’ category than the reactants benzene and cumene. Both petrochemical substrates, benzene, and cumene, receive red flags (benzene: H340, H350, H372, H410; cumene: H411), while lignin-derived dihydroconiferyl alcohol receives a yellow label (H351). In the analysis of other chemicals used in the green route, only three red flags were received for the ‘health and safety’, namely p-chloroaniline, paraformaldehyde, and pentane, while the classical method requires the use of catechol, CuCl2 2H2O, LiAlH4, benzylamine, etc., resulting in many additional red flags.
Regarding the reaction solvent used, yellow and green flags are obtained (CPME and DES in Steps A and B) in the green path, in contrast to the literature-based approach which is scored with a red flag (DCM in Step 8) and 3 yellow flags (THF, CH3CN in Steps 10, 12, 13).
For critical elements, a red flag is obtained due to the use of a Ru-based Shvo's catalyst in Step A of the alternative route. In the classical pathway, 10 yellow labels are obtained using Al, S, Cu, Li, Pd, and P-containing reagents.
Concerning the chemical requirements, it can be noted that in the alternative route, the amination of the dihydroconiferyl alcohol is a catalytic transformation that utilizes a small loading of ruthenium-based Shvo's catalyst (green flag), while the acid-catalysed Pictet–Spengler cyclization takes place in presence of the stoichiometric amount of deep eutectic solvent (yellow flag). In fact, this class of promising solvents displays the behaviour of a non-innocent, low vapour-pressure reaction media, acting both as a catalyst and a solvent, resulting in milder reaction conditions and improved reaction selectivity. This recyclable reaction media has proven itself as a benign replacement for conventional organic solvents and mineral corrosive acids. In a classical route, most of the reaction steps (9 out of 13) require excess or stoichiometric amounts of reagents (red and yellow flags, respectively).
Considering energy requirements, in the new path, Step A obtains a red flag due to the reflux conditions (130 °C) using CPME as a solvent (boiling point of CPME is 106 °C); Step B receives a green flag using DES as a reagent and a solvent at 70 °C. The classical route gets six red flags for energy requirement since all of these steps require harsh reaction conditions (under reflux). Also, given that the number of steps for the alternative route is significantly less compared to the literature-based approach, the overall energy consumption for the former one is more beneficial. Considering the work-up, the bio-based approach involves the use of column chromatography in both steps, resulting in two red flags. However, as it was mentioned above, in this case, work-up methods were not optimized at the discovery level, which opens up the possibility of further improvements of this approach, in particular, with regard to efficient isolation (such as precipitation, crystallization, or extraction) of the target amine product. This way the emerging route would significantly reduce solvent and energy consumption, resulting in a decrease in the overall PMI value. In the classical route, Steps 1–4 are well-established industrially-used procedures utilizing simple techniques such as distillation and filtration (green flags), while the remaining steps require the use of column chromatography and extraction, leading to red and yellow labels, respectively.
4.3 Evaluation of the green credentials for the synthesis of ranitidine
As in the previous chapter, herein we attempted to compare two main synthetic routes for the production of ranitidine, a drug currently used to treat peptic ulcer and gastroesophageal reflux disease, in terms of greenness. Although both paths start with biomass-derived platform chemicals, namely furfural and 5-(chloromethyl)furfural (CMF), choosing these examples we primarily aimed to emphasize the advantage of using reductive amination transformation in a synthetic sequence over classical organic stoichiometric reactions.As such, for the comparative analysis of quantitative and qualitative green credentials, the following four-step pathways were selected (Scheme 2). For both routes, the last step, condensation/nucleophilic substitution with N-methyl-1-(methylthio)-2-nitroethen-1-amine, is the same, whereas the first three transformations are significantly different. The classical route proceeds through (1) furfural reduction;250 (2) aminomethylation of furfuryl alcohol by a Mannich-type reaction251 and (3) nucleophilic substitution with cysteamine.252 In contrast, the alternative path begins with (a) nucleophilic substitution of the alkyl halide with cysteamine; followed by (b) reductive amination step to convert the aldehyde functionality of CMF to a tertiary amine group and (c) acetyl deprotection.253
Data obtained using the CHEM21 green metrics toolkit for both the classical and alternative routes are presented in Scheme 2 (for more details see ESI, Tables S4 and S5†). Although in this particular case neither of the two paths shows significant overall benefits, the alternative path has some promising improvements in individual steps.
Thus, when comparing the mass-based metrics, it can be clearly seen that the use of a reductive amination step in the reaction sequence significantly improves the yield of the entire route. The yield of Step B in the alternative route is 90% compared to 43% in the classical approach, which leads to a fourfold increase in the yield of the target product (68% vs. 18%). A similar trend is true for AE: the classical Mannich reaction used in Step 2 is somewhat inferior to reductive amination in terms of atom-efficiency (74% vs. 83%). Although looking at other metrics, such as PMI and WU, the alternative route seems to have a clear disadvantage compared to the classical method, showing significantly higher values for these parameters at each individual step, it is worth noting that this process is at the development stage. Therefore, comparison and evaluation of these parameters should be made with caution.
Considering the qualitative parameters, both approaches received relatively equal numbers of red and yellow flags based on the toxicity of the solvent, ‘health and safety’ considerations, critical elements used, energy consumption, and work-up methods.
Overall, although the comparison of these two approaches to the synthesis of ranitidine is less straightforward than in the previous chapter using the example of N-substituted tetrahydro-2-benzazepines, here we have sought to draw the reader's attention to the importance of analyzing each step of the synthesis and its contribution to the overall greenness of the process. The alternative path for the synthesis of ranitidine clearly indicates the benefits of employing atom-economic transformation such as reductive amination in the synthetic sequence. This conclusion becomes more obvious when taking into account the quantitative and qualitative values obtained using the CHEM21 toolkit for each step, as well as cumulative metrics.
4.4 Evaluation of the green credentials for the synthesis of norfenefrine
Norfenefrine is a sympathomimetic pharmaceutical used to treat hypotension regulating blood pressure by acting as a minor neurotransmitter in the brain. In this section, we analyze two approaches to synthesise norfenfrine. The classical pathway254–258 starts from petrol-based benzene, while the bio-based route237 involves cardanol as the starting reagent. Data obtained via the CHEM21 green metrics toolkit are depicted in Scheme 3 (see ESI, Tables S6 and S7†).In terms of quantitative green credentials, the bio-based pathway shows a higher overall yield compared to the classical pathway (52% vs. 17%). It should be noted that the urotropine-mediated amination in Step 5 is particularly limiting as it allows to obtain norfenerine in only 22% yield. In contrast, the aminohydroxylation performed in Step C yields the target drug at 73% yield. A similar trend is observed for RME, where Step C shows a higher efficiency (13.8% vs. 7.3%, Table S6†), confirming the advantages of a catalytic amination approach over traditional organic stochiometric reactions. However, the overall RME is relatively low in both routes. For the bio-based pathway, this is largely due to the high solvent usage in synthetic and purification steps. The PMI data, however, highlights a key difference between the two processes: the bio-based route achieves a much lower PMI (6036 g g−1vs. 47588 g g−1) compared to the classical pathway. This is attributed to the fewer reaction steps which exclusively involves catalytic transformations.
As the qualitative parameters are concerned (Scheme 3 and Table S7†) the bio-based route shows a slightly lower amount of red and yellow flags. The classical pathway accounts for three red flags (DCM, CHCl3, 1,4-dioxane) as regards the reaction solvents, while only yellow (2-Me-THF, THF, AcCN) and green labels (water) are observed for the bio-based pathway. However, both pathways have similar disadvantages when considering the solvents used for the work-up procedures. Three red flags arise from each route. As a consequence, ‘health & safety’ data are similar for both routes. In this regard, petrol-based benzene (H340, H350, H372, H410) is utilized as the starting reagent in Step 1 and as the solvent in Step 4's work-up, while most bio-based route's red flags derive from the use of DCM (H351, H 373) and petroleum ether (H411) during work-up. Only green labels are observed for the reagent use (catalyst) and energy use (RT to 50 °C) utilized in each step of the bio-based pathway. It should be noted that the bio-based approach accounts for two red flags concerning the metals employed since Ru is used in Steps A and B. It is not surprising that the work-up procedures display only red flags for this route, given the extensive use of flash-chromatography protocols, typically employed in lab-scale procedures when low amounts of product need to be isolated. On the other hand, as previously stated, this opens up the way for further improvements to deliver the target products while utilizing less impactful work-up methods.
Overall, it is clear from the processes herein described that catalytic aminohydroxylation helps to reduce the impact of the chemical synthesis of norfenefrine.
However, further optimization is needed in areas such as the use of non-noble catalysts and the development of efficient work-up procedures with non-hazardous, environmentally friendly solvents.
4.5 Evaluation of the green credentials for the synthesis of hexane-1,6-diamine (1,6-HMDA)
Renowned as nylon-6,6, poly(hexamethylene adipamide) is a polymer with widespread applications and significant market value. It is manufactured on an industrial scale through the polycondensation of hexamethylene diamine (1,6-HMDA) and adipic acid.259,260 Although the actual coupling reaction of these starting materials does not pose any difficulties, the synthesis of both starting materials 1,6-HMDA as well as adipic acid has been subject to scrutiny in terms of environmental impact.The primary method for obtaining hexamethylene diamine is the hydrocyanation of fossil-based butadiene, followed by hydrogenation of the resulting adiponitrile. Despite its effectiveness, this classical approach suffers from serious limitations, including the petrochemical nature of the starting materials, the high toxicity of reagents (hydrogen cyanide – HCN), as well as the requirement for high temperature and pressure conditions. In the pursuit of transitioning towards a bio-based economy and recognizing the significance of nylon-6,6, extensive research has been dedicated to formulating a more sustainable reaction scheme rooted in renewable resources.239 This has led to the development of alternative bio-based pathways for HMDA production, starting with high fructose syrup derived from maize and potato-based starch, with 5-(hydroxymethyl)furfural (5-HMF) serving as an intermediate building block.93,238
Numerous research groups reported various approaches for the conversion of 5-HMF into the desired HMDA, and these efforts can be categorized into two primary approaches: through the intermediates of 1,6-hexanediol (1,6-HDO) and 2,5-bis(aminomethyl)tetra-hydrofuran (AM-THF).
Route from 5-HMF to 1,6-HDO involves catalytic hydrogenation and hydrodeoxygenation transformations and can be carried out in four different ways: (1) the direct hydrogenation of HMF to 1,6-HDO; (2) a two-step sequence via 2,5-THF-dimethanol (THFDM); (3) a three-step synthesis via THFDM and 1,2,6-hexanetriol (1,2,6-HT); and (4) a four-step synthesis via THFDM, 1,2,6-HT, and tetrahydro-2H-pyran-2-ylmethanol (2-THPM). It is worth noting that although one-step, one-pot approaches were realised to directly convert 5-HMF to 1,6-HDO, poor yields of the final product and low selectivity remained a drawback for these protocols.261,262 In fact, hydrogenolytic THFDM ring opening with the selective formation of 1,6-HDO presents the main challenge in this sequence, making it difficult to overcome in a one-pot process. This issue can be addressed in stepwise processes through a careful design of the catalyst system. For example, employing the RANEY® nickel catalyst 5-HMF is first reduced to THFDM and then further hydrogenated over a Rh–Re/SiO2 catalyst with the addition of Nafion SAC-13 to the desired 1,6-HDO.263 The final amination of 1,6-HDO to produce HMDA poses its own set of challenges. Achieving the selective synthesis of a primary α,ω-amine from the corresponding diol is a complex task, given the potential for undesired cyclization and over-alkylation. To overcome these issues, the hydrogen borrowing methodology has proven to be the most efficient. As an example, 1,6-HMDA can be generated through an amination reaction using the Ru-acridine complex as a catalyst within this approach.264
An alternative synthetic route to obtain HMDA involves (a) the oxidation of 5-HMF to 2,5-diformylfuran (DFF), (b) reductive amination/hydrogenation of DFF to AM-THF, and subsequent (c) hydrodeoxygenation of the latter to produce the desired product 1,6-HMDA. The first step of the sequence, namely selective oxidation or dehydrogenation of the hydroxymethyl group of 5-HMF, is frequently associated with multiple undesired side reactions such as over-oxidation, decarboxylation, ring-opening, and cross-polymerization. As an example, the over-oxidation process could lead to the formation of 5-hydroxymethylfuran-2-carboxylic acid (HMFCA) and furan dicarboxylic acid (FDCA).265–267 Although dehydrogenation eliminates concerns about over-oxidation, nevertheless, this process being endothermic requires higher temperatures, making side reactions more probable. In the subsequent step, the conversion of DFF to AM-THF also remains a challenge despite recent advancements in reductive amination. To prevent the formation of by-products such as secondary, tertiary, and polymeric amine species resulting from the condensation and/or hydrogenation of the reactive di-aldehyde groups and highly nucleophilic amine groups of the product, highly selective and delicate approaches are required. The final step of this sequence is the opening of the AM-THF molecular ring by reaction over a hydrodeoxygenation catalyst leading to the formation of HMDA.
Adipic acid (ADP), another crucial chemical in the synthesis of nylon-6,6, is predominantly manufactured through a multi-step sequence.268–271 This involves the hydrogenation of benzene obtained from petroleum to produce cyclohexane, subsequent cyclohexane oxidation to yield a mixture of cyclohexanone and cyclohexanol known as ketone-alcohol oil (KA oil), and further oxidation of KA oil using nitric acid to generate adipic acid.272 This process, being highly energy inefficient, faces the problems of emission of nitrous oxide (N2O), a potent greenhouse and ozone-depletion gas, and low efficiency largely attributed to the meager single-pass yield (<10%) of ketone-alcohol oil (KA oil) in the oxidation of cyclohexane.
The production of adipic acid, one of the most important industrial chemicals, through a wasteful energy-inefficient multistep process involving an aggressive oxidant (HNO3) has raised many concerns. As a result, over the last decades, significant efforts were devoted to the development of alternative oxidants269 to enable NOx free process, and more sustainable routes273 for the synthesis of adipic acid starting from petrochemical or, ideally, renewable feedstocks, capable of reducing the capital cost investments.274 Multiple studies on the use of various feedstocks including cyclohexane, phenol, butadiene, and adiponitrile for ADP synthesis have been reported; however, most of these materials are produced from petroleum-derived benzene, leading to a comparable cost structure. Recently, advances in the sustainable preparation of ADP from biomass-based platform molecules, including 5-hydroxymethylfufural, glucose, γ-valerolactone, and phenolic compounds, through biocatalysis, chemocatalysis, and the combination of both, have emerged.275,276
A major component of lignocellulosic biomass, cellulose,277–280 which consists of glucose units connected by β-1,4-glycosidic linkage, has proven to be a promising starting material for the synthesis of adipic acid or adipate esters through catalytic transformations of glucose-derived C6 dicarboxylic acids such as 2,5-furandicarboxylic acid (FDCA)281,282 and mucic acid.283–285 However, despite the effectiveness of these methodologies, the production of these acids is not straightforward, demanding multiple chemical reactions277,279 or biological fermentation processes.284,285 As an attempt to overcome these issues, very recently, a fascinating two-step transformation of cellulose-derived glucose into adipic acid via glucaric acid was reported.286 In this approach, glucose is first oxidized into glucaric acid over a Pt/CNT catalyst, which undergoes deoxydehydration reaction to form adipic acid after the removal of four OH groups in the presence of a bifunctional Pd–ReOx/AC catalyst.
5-Hydroxymethylfufural (5-HMF) is another renewable platform molecule that could be extracted from cellulose and used as a feedstock for ADP production.93 As an example, catalytic hydrolysis of lignocellulose to HMF under acidic conditions (H2SO4), followed by oxidative conversion to 2,5-furandicarboxylic acid (FDCA), and a subsequent deoxygenation reaction yields the final product, ADP. The alternative path is based on catalytic hydrogenation of HMF to 1,6-hexanediol (1,6-HD) followed by biocatalytic oxidation of two hydroxy groups of 1,6-HD to carboxylic acid groups to obtain ADP. It is worth noting that the first step of the latter route poses a significant challenge as identifying effective catalysts capable of simultaneously saturating the CC bond and facilitating the ring opening of cyclic ethers proves to be difficult.
Besides glucose and 5-hydroxymethylfufural, γ-valerolactone (GVL)287 and lignin-derived phenolic compounds288 represent other renewable starting materials with great potential for the synthesis of ADP. It is noteworthy that γ-valerolactone (GVL), in turn, can be produced by hydrogenating levulinic acid (LA), which is obtained through the acid-catalysed hydrolysis of biomass feedstocks.289 Thus, the general catalytic approach for the synthesis of ADP from GVL includes (a) GVL acid-catalysed hydrolysis and reactive distillation to form pentenoic acid isomers (PEAs) and (b) the carbonylation of PEAs to ADP.290,291 As another contrasting example, an integrated strategy for the biochemical conversion of lignin-derived monomers to adipic acid was recently disclosed.284 In this process, the oxidative ring-opening reactions of phenolic compounds catalysed by biocatalysts (Pseudomonas putida KT2440) obtain muconate, which is further hydrogenated to ADP over a Pd/C catalyst.292,293
Based on the obtained data, it is clear that bio-based approaches have the potential to be considered as a future replacement for the current industrial process after appropriate optimization. Considering that bio-based pathways are at the discovery level (first pass CHEM21 green metrics toolkit), it is noteworthy that the yield of the target HMDA is almost three times higher for bio-based approaches 1 and 2 than for commercial process (71% and 79% vs. 23%, respectively). Although the PMI and RME values are still considerably more advantageous for the industrial process, careful optimization of the isolation procedure as well as catalyst/solvent recovery at pilot and industrial scales might significantly improve current values for the bio-based routes. The benefits of bio-based approaches become more apparent when comparing the qualitative parameters of commercial and sustainable routes, as evidenced by fewer red flags. Both bio-based pathways (1) utilize renewable feedstocks such as cellulose-derived 5-HMF instead of petroleum-based butadiene; (2) avoid the use of toxic and hazardous materials and solvents such as hydrogen cyanide and benzene; (3) require less energy consumption – reaction temperatures at most stages are below the boiling point of the solvent/between 0 and 70 °C.
When comparing bio-based pathways with each other (Table S8†), the reductive amination-based route (bio-based approach 2) despite the increased number of steps (4 steps vs. 3 steps) leads to a higher overall yield (79% vs. 71%). The opposite is true for the PMI: fewer reaction steps and efficient work-up procedures (distillation) for hydrogen-borrowing-based approach (bio-based approach 1) result in reduced waste generation; thus, PMI Tot for bio-based approach 1 is six times less than for Bio-based approach 2 (144 g g−1vs. 876 g g−1). In fact, the reductive amination step (Step B in Bio-based approach 2) negatively contributed to the overall PMI value due to the inefficient isolation procedure and the uneconomic amount of the solvent utilized. While AE values are comparable for both approaches (86% vs. 88%), RME for Step III, bio-based approach 1 is significantly higher than for Step B, bio-based approach 2 (56% vs. 9%). This parameter, considering the stoichiometry of the reaction, clearly indicates the excess of the reactants used in the reductive amination step, which negatively affects the overall RME of the entire process. Overall, despite some minor differences in values of quantitative and qualitative parameters, both atom-economic routes have proven to be effective for introducing amine functionality into the target product, 1,6-HMDA.
In their invaluable contribution, Pera-Titus and coworkers have presented239 an economic and LCA comparative study which revealed that the commercial process for HMDA synthesis is more cost-effective and environmentally favourable than existing bio-based alternatives, mainly due to costs related to feedstock sourcing and energy demands (see section 4.1). This points out the need for more efficient biorefinery approaches. Nonetheless, the green credentials calculated here give an immediate comparison of the synthetic routes and sustainability of laboratory processes, decoupled from feedstock sourcing.
5. Conclusions
The transition to a circular, bio-based economy requires the development of novel catalytic approaches, which enable alternative pathways to the production of industrially important chemicals from renewable carbonaceous feedstocks;301 chief among these are amines. Due to the structural mismatch between renewable and fossil-based feedstocks, catalyst development should embrace the inherent structural features of renewable starting materials to allow markedly more sustainable and efficient routes to amines.9 This is important to ensure the competitiveness of the newly developed routes since the established petrochemical production pathways have been optimized – literally – for decades, to maximize performance. However, fossil-based pathways to industrial products display a fundamental drawback: they were developed with the sole focus on performance without consideration of principles of sustainability and green chemistry as design criteria.302 Moreover, the distinctly simple nature of petrochemical building blocks typically leads to multi-step or harsh conditions to access amines. Therefore, any new bio-based route should aim for higher step, atom, and energy efficiency and sustainability from renewable platform chemical to product to allow for competitive benefits. However, it is not sufficient to simply state “The new route is ‘greener’ because it comes from renewables”. Any newly developed route should be closely scrutinized in terms of reaction performance metrics, and green metrics to understand future substitution potential and possible industrial importance.303,304Herein, we illustrate the application of green metrics incorporated in the CHEM21 toolkit for a first basis comparison of novel (renewables-based) and classical (petrol-based) pathways through four selected case studies. The first case study considers the synthesis of N-substituted tetrahydro-2-benzazepines, compounds with pharmaceutical importance, one starting from petrol-based benzene through classical synthesis, and one from lignin-derived dihydroconiferyl alcohol via the hydrogen borrowing methodology. It can be clearly seen that the classical route requires 13 steps to obtain the targeted compound, while the new pathway delivers the final product in only 3 steps from woody biomass. The advantages of this latter approach in terms of yield, biomass utilization and energy efficiency, are immediately apparent.
A second example considers the synthesis of ranitidine from biomass, namely from furfural and 5-(chloromethyl)furfural (CMF) important cellulose-derived platform chemicals, involving a reductive amination approach and stoichiometric classical transformations, respectively. This case demonstrates that simply being bio-based is not enough to achieve a marked difference compared to a well-optimized classical route. The advantages and disadvantages of both routes are elaborated, and possibilities for improvement of the new route are proposed.
As a third example, the synthesis of norfenefrine is illustrated via a comparison between the classical pathway starting from benzene and a bio-based alternative route starting from cardanol. This example highlights the advantages of catalytic processes over traditional organic reactions in terms of overall yield and process mass intensity (PMI). However, it also shows the need to optimize work-up procedures for synthetic procedures from renewables. These are often performed at the lab scale and require high solvent loads for tedious work-ups, such as chromatography columns.
Finally, very important nylon-6,6 monomers were illustrated, namely 1,6-HMDA and adipic acid. The importance of this example lies in its significant market value, nylon-6,6 being produced on a scale of >8 million tons a year,305,306 and its classical production is environmentally largely problematic. Therefore, the development of a sustainable and preferably bio-based pathway will be of major significance. Here our analysis outlines several promising possibilities for the production of nylon-6,6, starting from renewable resources. In particular, we have cross-compared two alternative approaches based on cellulose-derived 5-HMF and with the current industrial process based on petroleum-derived butadiene. Given the quantitative and qualitative values of the key green metrics, it was concluded that bio-based routes could be considered a viable alternative to the current industrial process, potentially offering a safer and energy-efficient approach with a higher overall yield after appropriate optimization. Comparison of atom-economic routes with each other, namely reductive amination and ‘hydrogen borrowing’, revealed that these approaches are equally effective in introducing amine functionality into the 1,6-HMDA structure, as expected.
Overall, in this contribution we have demonstrated the development of ‘waste-free’ catalytic methods for the selective amination of biomass-derived platform chemicals, to furnish industrially important amines. A detailed description of available catalytic systems was presented for reductive amination and alcohol-to-amine transformations via the hydrogen borrowing methodology, as applied to substrates that (potentially) originate from renewable resources. While many of these fundamental research results represent impressive scientific progress, the reported processes are typically at lower TRL levels, where process intensification has not yet been performed. Nonetheless, it is expected that such developments will happen towards integrating such methods into industrial processes.
In this review article, our focus was on the demonstration and use of numerical and qualitative green chemistry metrics, included in the CHEM21 toolkit, as an important first step towards the comparison of fossil-based and renewables-based routes, at the reaction and process level. Of course, this approach alone is not sufficient to derive final conclusions about the specific routes and processes and a more elaborate life cycle analysis will be necessary to estimate the overall sustainability of the processes in question. Full life cycle analysis, cradle to grave, will consider potential challenges with regard to feedstock sourcing, global warming potential as well as the fate of the final products.32,33 While fossil-based pathways already possess a well-established raw material supply chain, the non-renewable nature of petroleum-based starting materials will negatively impact the carbon footprint of related products.239,243,293 On the other hand, the processing, sourcing, and availability of specific renewable resources are ongoing developments and success will depend on future overall progress in the development of biorefinery approaches at scale. While several frontrunners have emerged over the past decade, future research and development should allow efficient access to specific platform chemicals at scale and competitive price points.307,308 Other factors, such as environmental impact related to land use, production capacity, and availability of diverse renewable feedstocks within planetary boundaries, will also have an influence on overall sustainability.309–311
It is our strong conviction that progress toward a circular and bio-based economy depends on our capacity to deliver elegant fundamental scientific solutions. The relatively simple metrics discussed in the review provide easy-to-use tools in the hands of laboratory chemists to evaluate, compare, and scrutinize their fundamental innovations early in the discovery process. Such early assessment may guide catalyst design and method development, and illuminate immediate advantages of a newly developed synthetic route, which will ultimately provide a road ahead for future industrial development.
Author contributions
Conceptualization: AMA, KB; visualization: AMA; project administration: KB; funding acquisition: KB; writing – original draft: AMA, ND, JO, RS, KB; writing – review & editing: AMA, ND, JO, RS, KB.Data availability
The data supporting this article have been included as part of the ESI.†A separate dataset detailing specific calculations has been provided as additional dataset.
This review does not contain specific experimental data.
Conflicts of interest
There are no conflicts to declare.References
- Chemical Biology, ed. H. Waldmann and P. Janning, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed.
- Chemical Biology: From Small Molecules to Systems Biology and Drug Design, ed. S. L. Schreiber, T. Kapoor and G. Wess, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- S. A. Lawerence, Amines: Synthesis Properties, and Applications, Cambridge University, 2004 Search PubMed.
- A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- P. G. Levi and J. M. Cullen, Environ. Sci. Technol., 2018, 52, 1725–1734 CrossRef CAS PubMed.
- P. Friedlingstein, M. Allen, J. G. Canadell, G. P. Peters and S. I. Seneviratne, Science, 2019, 366, 76–79 CrossRef PubMed.
- Biomass Utilization: Conversion Strategies, ed. N. P. Nghiem, T. H. Kim and C. G. Yoo, Springer Nature, 2022 Search PubMed.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- Z. Sun and K. Barta, Chem. Commun., 2018, 54, 7725–7745 RSC.
- S. Elangovan, A. Afanasenko, J. Haupenthal, Z. Sun, Y. Liu, A. K. H. Hirsch and K. Barta, ACS Cent. Sci., 2019, 5, 1707–1716 CrossRef CAS PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, Wiley-Interscience, New York, 2001 Search PubMed.
- G. W. Gribble, Chem. Soc. Rev., 1998, 27, 395 RSC.
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed.
- E. Sperotto, G. P. M. van Klink, G. van Koten and J. G. de Vries, Dalton Trans., 2010, 39, 10352 RSC.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- T. Irrgang and R. Kempe, Chem. Rev., 2018, 118, 1410–1459 CrossRef PubMed.
- B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Org. Biomol. Chem., 2018, 17, 1595–1607 RSC.
- B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS PubMed.
- K. Murugesan, T. Senthamarai, V. G. Chandrashekhar, K. Natte, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed.
- J. Liu, Y. Song and L. Ma, Chem. – Asian J., 2021, 16, 2371–2391 CrossRef CAS PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- L. Huang, M. Arndt, K. Gooben, H. Heydt and L. J. Gooben, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed.
- S. Streiff and F. Jérôme, Chem. Soc. Rev., 2021, 50, 1512–1521 RSC.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- H. C. Erythropel, J. B. Zimmerman, T. M. De Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- M. A. Curran, Life cycle assessment handbook, Wiley-Scrivener, Salem, MA, 2012 Search PubMed.
- S. Hellweg and L. M. I. Canals, Science, 2014, 344, 1109–1113 CrossRef CAS PubMed.
- Green Chemistry Metrics: Measuring and Monitoring Sustainable Processes, ed. A. Lapkin and D. J. C. Constable, Wiley-Blackwell, 2008 Search PubMed.
- CHEM21 Metrics Toolkit, https://learning.acsgcipr.org/guides-and-metrics/metrics/the-chem21-metrics-toolkit/.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- K. Lokesh, A. S. Matharu, I. K. Kookos, D. Ladakis, A. Koutinas, P. Morone and J. Clark, Green Chem., 2020, 22, 803–813 RSC.
- Element Recovery and Sustainability, ed. A. J. Hunt, RSC Publishing, 2013 Search PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635–636 CrossRef CAS PubMed.
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512 CrossRef CAS PubMed.
- M. Pelckmans, T. Renders, S. Van De Vyver and B. F. Sels, Green Chem., 2017, 19, 5303–5331 RSC.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667–2670 CrossRef CAS.
- R. Grigg, T. R. B. Mitchell, N. Sutthivaiyakit and S. Tongpenyai, J. Chem. Soc., 1981, 611–612 CAS.
- K. I. Fujita, Z. Li, N. Ozeki and R. Yamaguchi, Tetrahedron Lett., 2003, 44, 2687–2690 CrossRef CAS.
- A. Tillack, D. Hollmann, D. Michalik and M. Beller, Tetrahedron Lett., 2006, 47, 8881–8885 CrossRef CAS.
- A. C. Maxwell, A. J. A. Watson, M. H. S. A. Hamid, J. M. J. Williams, H. C. Maytum, G. W. Lamb and C. L. Allen, J. Am. Chem. Soc., 2009, 40, 1766–1774 Search PubMed.
- S. Bahn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef.
- G. Guillena, D. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed.
- A. J. Rawlings, L. J. Diorazio and M. Wills, Org. Lett., 2015, 17, 1086–1089 CrossRef CAS PubMed.
- M. Mastalir, B. Stöger, E. Pittenauer, M. Puchberger, G. Allmaier and K. Kirchner, Adv. Synth. Catal., 2016, 358, 3824–3831 CrossRef CAS.
- S. Rösler, M. Ertl, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2015, 54, 15046–15050 CrossRef PubMed.
- Z. Yin, H. Zeng, J. Wu, S. Zheng and G. Zhang, ACS Catal., 2016, 6, 6546–6550 CrossRef CAS.
- S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J. B. Sortais, J. Catal., 2017, 347, 57–62 CrossRef CAS.
- J. Neumann, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, Chem. – Eur. J., 2017, 23, 5410–5413 CrossRef CAS PubMed.
- M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS.
- A. K. Bains, A. Kundu, S. Yadav and D. Adhikari, ACS Catal., 2019, 9, 9051–9059 CrossRef CAS.
- G. Balamurugan, R. Ramesh and J. G. Malecki, J. Org. Chem., 2020, 85, 7125–7135 CrossRef CAS PubMed.
- A. Afanasenko, S. Elangovan, M. C. A. Stuart, G. Bonura, F. Frusteri and K. Barta, Catal. Sci. Technol., 2018, 8, 5498–5505 RSC.
- T. Kwok, O. Hoff, R. J. Armstrong and T. J. Donohoe, Chem. – Eur. J., 2020, 26, 12912–12926 CrossRef CAS PubMed.
- A. E. R. Chamberlain, K. J. Paterson, R. J. Armstrong, H. C. Twin and T. J. Donohoe, Chem. Commun., 2020, 56, 3563–3566 RSC.
- R. Kempe and F. Kallmeier, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef PubMed.
- M. A. Berliner, S. P. A. Dubant, T. Makowski, K. Ng, B. Sitter, C. Wager and Y. Zhang, Org. Process Res. Dev., 2011, 15, 1052–1062 CrossRef CAS.
- S. K. Nimmagadda, S. Kalidindi, S. S. Bondigela, S. Korapati, D. Dasgupta, N. A. Malik, P. Rao, P. Maity, J. R. Coombs, M. Hay, E. M. Simmons, S. Mukherjee, R. Vaidyanathan, M. D. Eastgate and F. González-Bobes, Org. Process Res. Dev., 2024, 28, 3414–3422 CrossRef CAS.
- S. Hameury, H. Bensalem and K. De Oliveira Vigier, Catalysts, 2022, 12, 1306 CrossRef CAS.
- N. Andrushko, V. Andrushko, P. Roose, K. Moonen and A. Börner, ChemCatChem, 2010, 2, 640–643 CrossRef CAS.
- S. D. Lacroix, A. Pennycook, S. Liu, T. T. Eisenhart and A. C. Marr, Catal. Sci. Technol., 2012, 2, 288–290 RSC.
- J. Niemeier, R. V. Engel and M. Rose, Green Chem., 2017, 19, 2839–2845 RSC.
- S. Imm, S. Bähn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599–7603 CrossRef CAS PubMed.
- D. Pingen, O. Diebolt and D. Vogt, ChemCatChem, 2013, 5, 2905–2912 CrossRef CAS.
- Y. Liu, A. Afanasenko, S. Elangovan, Z. Sun and K. Barta, ACS Sustainable Chem. Eng., 2019, 7, 11267–11274 CrossRef CAS PubMed.
- M. Jacolot, S. Moebs-Sanchez and F. Popowycz, J. Org. Chem., 2018, 83, 9456–9463 CrossRef CAS PubMed.
- F. Bahé, L. Grand, E. Cartier, M. Jacolot, S. Moebs-Sanchez, D. Portinha, E. Fleury and F. Popowycz, Eur. J. Org. Chem., 2020, 599–608 CrossRef.
- W. Deng, Y. Wang, S. Zhang, K. M. Gupta, M. J. Hülsey, H. Asakura, L. Liu, Y. Han, E. M. Karp, G. T. Beckham, P. J. Dyson, J. Jiang, T. Tanaka, Y. Wang and N. Yan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5093–5098 CrossRef CAS PubMed.
- A. Afanasenko, T. Yan and K. Barta, Commun. Chem., 2019, 2, 127 CrossRef.
- M. M. Reddy, M. A. Kumar, P. Swamy, M. Naresh, K. Srujana, L. Satyanarayana, A. Venugopal and N. Narender, Green Chem., 2013, 15, 3474–3483 Search PubMed.
- J. Luo, M. Wu, F. Xiao and G. Deng, Tetrahedron Lett., 2011, 52, 2706–2709 CrossRef CAS.
- S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 3, 8126–8129 Search PubMed.
- C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS PubMed.
- T. Yan, B. L. Feringa and K. Barta, ACS Catal., 2016, 6, 381–388 CrossRef CAS.
- A. D. Pingen, J. B. Schwaderer, J. Walter, J. Wen, G. Murray, D. Vogt and S. Mecking, ChemCatChem, 2018, 10, 3027–3033 CrossRef.
- Y. Kita, M. Kuwabara, S. Yamadera and K. Kamata, Chem. Sci., 2020, 11, 9884–9890 RSC.
- X. Wu, M. De Bruyn and K. Barta, Catal. Sci. Technol., 2022, 12, 5908–5916 Search PubMed.
- D. Ruijten, T. Narmon, K. Van Aelst, H. De Weer, R. van der Zweep, T. Hendrickx, C. Poleunis, L. Li, K. M. Van Geem, D. P. Debecker and B. F. Sels, ACS Sustainable Chem. Eng., 2023, 11, 4776–4788 CrossRef CAS.
- L. He, Y. Qian, R. S. Ding, Y. M. Liu, H. Y. He, K. N. Fan and Y. Cao, ChemSusChem, 2012, 5, 621–624 Search PubMed.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 Search PubMed.
- T. Werpy and G. Petersen, in Report no. DOE/GO-102004-1992, National Renewable Energy Lab., Golden, CO, 2004.
- P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, ACS Catal., 2012, 2, 1487–1499 Search PubMed.
- R. M. Ceballos, Bioethanol and Natural Resources Substrates, Chemistry and Engineered Systems, CRC Press, Boca Raton, FL, USA, 2018 Search PubMed.
- R.-J. van Putten, J. G. de Vries, J. C. van der Waal, E. de Jong, C. B. Rasrendra and H. J. Heeres, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876–2924 RSC.
- A. B. van Gysel and W. Musin, Methylamines in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- J. Runeberg, A. Baiker and J. Kijenski, Appl. Catal., 1985, 17, 309–319 CrossRef CAS.
- M. Frauenkron, J.-P. Melder, G. Ruider, R. Rossbacher and H. Höke, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2012 Search PubMed.
- J. Xia and K. Matyjaszewski, Macromolecules, 1997, 30, 7697–7700 Search PubMed.
- A. Fischer, M. Maciejewski, T. Bürgi, T. Mallat and A. Baiker, J. Catal., 1999, 183, 373–383 Search PubMed.
- A. Fischer, Heterogeneous transition metal catalyzed amination of aliphatic diols, ETH Zürich, 1998 Search PubMed.
- L. Jasinska-Walc, D. Dudenko, A. Rozanski, S. Thiyagarajan, P. Sowinski, D. Van Es, J. Shu, M. R. Hansen and C. E. Koning, Macromolecules, 2012, 45, 5653–5666 CrossRef CAS.
- X. Ji, Z. Wang, J. Yan and Z. Wang, Polymer, 2015, 74, 38–45 CrossRef CAS.
- G. Liang, Y. Zhou, J. Zhao, A. Y. Khodakov and V. V. Ordomsky, ACS Catal., 2018, 8, 11226–11234 CrossRef CAS.
- G. Bottari, A. J. Kumalaputri, K. K. Krawczyk, B. L. Feringa, H. J. Heeres and K. Barta, ChemSusChem, 2015, 8, 1323–1327 CrossRef CAS PubMed.
- A. J. Kumalaputri, G. Bottari, P. M. Erne, H. J. Heeres and K. Barta, ChemSusChem, 2014, 7, 2266–2275 CrossRef CAS PubMed.
- X. Chen, S. Song, H. Li, G. Gözaydln and N. Yan, Acc. Chem. Res., 2021, 54, 1711–1722 CrossRef CAS PubMed.
- P. J. Deuss, K. Barta and J. G. De Vries, Catal. Sci. Technol., 2014, 4, 1174–1196 RSC.
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS PubMed.
- T. Renders, S. Van Den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 Search PubMed.
- X. Wu, M. V. Galkin and K. Barta, Chem. Catal., 2021, 1, 1466–1479 Search PubMed.
- J. Ma, D. Le and N. Yan, Chem, 2023, 9, 2869–2880 CAS.
- A. Philippaerts, P. Jacobs and B. Sels, Catalytic Hydrogenation for Biomass Valorization, RSC Energy and Environmental Series, 2015 Search PubMed.
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- H. Kimura, Catal. Rev. - Sci. Eng., 2011, 53, 1–90 CrossRef CAS.
- L. Qiuxiao, Z. Gaoyong and P. Shaoyi, J. Surfactants Deterg., 2002, 5, 229–233 Search PubMed.
- A. Baiker and W. Richarz, Ind. Eng. Chem. Prod. Res. Dev., 1977, 16, 261–265 Search PubMed.
- H. Kimura, K. Matsutani, S. I. Tsutsumi, S. Nomura, K. Ishikawa, Y. Hattori, M. Itahashi and H. Hoshino, Catal. Lett., 2005, 99, 119–131 Search PubMed.
- X. Cui, X. Dai, Y. Deng and F. Shi, Chem. – Eur. J., 2013, 19, 3665–3675 Search PubMed.
- K. I. Shimizu, N. Imaiida, K. Kon, S. M. A. Hakim Siddiki and A. Satsuma, ACS Catal., 2013, 3, 998–1005 CrossRef CAS.
- J. He, J. W. Kim, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 9888–9891 CrossRef CAS PubMed.
- W. Baumann, A. Spannenberg, J. Pfeffer, T. Haas, A. Köckritz, A. Martin and J. Deutsch, Chem. – Eur. J., 2013, 19, 17702–17706 CrossRef CAS PubMed.
- T. Yan, B. L. Feringa and K. Barta, Sci. Adv., 2017, 3, eaao6494 CrossRef PubMed.
- A. Said Stålsmeden, J. L. Belmonte Vázquez, K. Van Weerdenburg, R. Rae, P. O. Norrby and N. Kann, ACS Sustainable Chem. Eng., 2016, 4, 5730–5736 CrossRef.
- C. Crotti, E. Farnetti, S. Licen, P. Barbieri and G. Pitacco, J. Mol. Catal. A: Chem., 2014, 382, 64–70 CrossRef CAS.
- X. Dai, J. Rabeah, H. Yuan, A. Brückner, X. Cui and F. Shi, ChemSusChem, 2016, 9, 3133–3138 CrossRef CAS PubMed.
- Y. Wang, S. Furukawa, S. Song, Q. He, H. Asakura and N. Yan, Angew. Chem., Int. Ed., 2020, 59, 2289–2293 CrossRef CAS PubMed.
- C. P. Xu, Z. H. Xiao, B. Q. Zhuo, Y. H. Wang and P. Q. Huang, Chem. Commun., 2010, 46, 7834–7836 RSC.
- V. N. Tsarev, Y. Morioka, J. Caner, Q. Wang, R. Ushimaru, A. Kudo, H. Naka and S. Saito, Org. Lett., 2015, 17, 2530–2533 CrossRef CAS PubMed.
- H. Hikawa and Y. Yokoyama, Org. Biomol. Chem., 2011, 9, 4044–4050 RSC.
- A. Bermejo-López, M. Raeder, E. Martínez-Castro and B. Martín-Matute, Chem, 2022, 8, 3302–3323 Search PubMed.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- M. Ernst, B. W. Hoffer and J.-P. Melder, Method for producing amines from glycerin, US 2010/0240894 A1, 2010 Search PubMed.
- T. M. Lammens, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Biomass Bioenergy, 2012, 44, 168–181 CrossRef CAS.
- Biological Nitrogen Fixation, ed. F. J. de Bruijn, John Wiley & Sons, Inc., 2015 Search PubMed.
- T. Yan, B. L. Feringa and K. Barta, ChemSusChem, 2021, 14, 2303–2307 CrossRef CAS PubMed.
- S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- K. S. Hayes, Appl. Catal., A, 2001, 221, 187–195 CrossRef CAS.
- C. J. Elsevier and J. G. de Vries, The Handbook of Homogeneous Hydrogenation, Wiley, Weinheim, 2006 Search PubMed.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- N. U. D. Reshi, V. B. Saptal, M. Beller and J. K. Bera, ACS Catal., 2021, 11, 13809–13837 CrossRef CAS.
- S. Raoufmoghaddam, Org. Biomol. Chem., 2014, 12, 7179 RSC.
- J. Jose, E. J. Diana, U. S. Kanchana and T. V. Mathew, Eur. J. Org. Chem., 2023, 26, e202300035 CrossRef CAS.
- P. Mäki-Arvela, I. L. Simakova and D. Y. Murzin, Catal. Rev., 2023, 65, 501–568 CrossRef.
- G. Liang, A. Wang, L. Li, G. Xu, N. Yan and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3050–3054 CrossRef CAS PubMed.
- J. He, L. Chen, S. Liu, K. Song, S. Yang and A. Riisager, Green Chem., 2020, 22, 6714–6747 RSC.
- W. Faveere, T. Mihaylov, M. Pelckmans, K. Moonen, F. Gillis-D'Hamers, R. Bosschaerts, K. Pierloot and B. F. Sels, ACS Catal., 2020, 10, 391–404 CrossRef CAS.
- C. C. Truong, D. K. Mishra and Y. Suh, ChemSusChem, 2023, 16, e202201846 CrossRef CAS PubMed.
- N. K. Gupta, P. Reif, P. Palenicek and M. Rose, ACS Catal., 2022, 12, 10400–10440 CrossRef CAS.
- K. Gupta, R. K. Rai and S. K. Singh, ChemCatChem, 2018, 10, 2326–2349 CrossRef CAS.
- J. Zhang, J. Yang, X. Li, H. Liu, X. Yao, C. Xia and Z. Huang, Catalysts, 2023, 13, 528 CrossRef.
- B. Dong, X. Guo, B. Zhang, X. Chen, J. Guan, Y. Qi, S. Han and X. Mu, Catalysts, 2015, 5, 2258–2270 CrossRef CAS.
- T. Komanoya, T. Kinemura, Y. Kita, K. Kamata and M. Hara, J. Am. Chem. Soc., 2017, 139, 11493–11499 CrossRef CAS PubMed.
- Z. Pan, Q. Zhang, W. Wang, L. Wang and G. H. Wang, ACS Sustainable Chem. Eng., 2022, 10, 3777–3786 CrossRef CAS.
- H. Zou and J. Chen, Appl. Catal., B, 2022, 309, 121262 CrossRef CAS.
- D. Chandra, Y. Inoue, M. Sasase, M. Kitano, A. Bhaumik, K. Kamata, H. Hosono and M. Hara, Chem. Sci., 2018, 9, 5949–5956 RSC.
- M. Sheng, S. Fujita, S. Yamaguchi, J. Yamasaki, K. Nakajima, S. Yamazoe, T. Mizugaki and T. Mitsudome, JACS Au, 2021, 1, 501–507 CrossRef CAS PubMed.
- K. Zhou, B. Chen, X. Zhou, S. Kang, Y. Xu and J. Wei, ChemCatChem, 2019, 11, 5562–5569 CrossRef CAS.
- H. Qi, J. Yang, F. Liu, L. Zhang, J. Yang, X. Liu, L. Li, Y. Su, Y. Liu, R. Hao, A. Wang and T. Zhang, Nat. Commun., 2021, 12, 3295 CrossRef CAS PubMed.
- J. J. Martínez, E. Nope, H. Rojas, M. H. Brijaldo, F. Passos and G. Romanelli, J. Mol. Catal. A: Chem., 2014, 392, 235–240 CrossRef.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M. M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- H. Yuan, J.-P. Li, F. Su, Z. Yan, B. T. Kusema, S. Streiff, Y. Huang, M. Pera-Titus and F. Shi, ACS Omega, 2019, 4, 2510–2516 CrossRef CAS PubMed.
- A. García-Ortiz, J. D. Vidal, M. J. Climent, P. Concepción, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2019, 7, 6243–6250 CrossRef.
- Z. Xu, P. Yan, W. Xu, S. Jia, Z. Xia, B. Chung and Z. C. Zhang, RSC Adv., 2014, 4, 59083–59087 RSC.
- A. S. Touchy, S. M. A. Hakim Siddiki, K. Kon and K. Shimizu, ACS Catal., 2014, 4, 3045–3050 CrossRef CAS.
- J. Zhang, B. Xie, L. Wang, X. Yi, C. Wang, G. Wang, Z. Dai, A. Zheng and F.-S. Xiao, ChemCatChem, 2017, 9, 2661–2667 CrossRef CAS.
- Y. Wang, H. Wu, J. Wang, K. Zhang, Y. Liu and Z. Wei, ACS Sustainable Chem. Eng., 2022, 10, 17274–17285 Search PubMed.
- S. Chakrabortty, S. Zheng, F. Kallmeier, E. Baráth, S. Tin and J. G. de Vries, ChemSusChem, 2023, 16, e202202353 CrossRef CAS PubMed.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 Search PubMed.
- K. Murugesan, Z. Wei, V. G. Chandrashekhar, H. Jiao, M. Beller and R. V. Jagadeesh, Chem. Sci., 2020, 11, 4332–4339 RSC.
- K. Murugesan, Z. Wei, V. G. Chandrashekhar, H. Neumann, A. Spannenberg, H. Jiao, M. Beller and R. V. Jagadeesh, Nat. Commun., 2019, 10, 5443 CrossRef PubMed.
- T. Senthamarai, K. Murugesan, J. Schneidewind, N. V. Kalevaru, W. Baumann, H. Neumann, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Nat. Commun., 2018, 9, 4123 CrossRef PubMed.
- T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675–703 CrossRef PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368–3398 CrossRef CAS PubMed.
- A. L. Reznichenko and K. C. Hultzsch, Organic Reactions, 2016, vol. 88, pp. 1–554 Search PubMed.
- J. S. Ryu, G. Y. Li and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 12584–12605 CrossRef CAS PubMed.
- H. M. Senn, P. E. Blöchl and A. Togni, J. Am. Chem. Soc., 2000, 122, 4098–4107 CrossRef CAS.
- A. M. Johns, N. Sakai, A. Ridder and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9306–9307 CrossRef CAS PubMed.
- S. Doye, Sci. Synth., 2009, 40, 241–304 Search PubMed.
- T. E. Müller, M. Grosche, E. Herdtweck, A. K. Pleier, E. Walter and Y. K. Yan, Organometallics, 2000, 19, 170–183 Search PubMed.
- S. Ma and J. F. Hartwig, Acc. Chem. Res., 2023, 56, 1565–1577 CrossRef CAS PubMed.
- J. Escorihuela, A. Lledós and G. Ujaque, Chem. Rev., 2023, 123, 9139–9203 CrossRef CAS PubMed.
- A. L. Reznichenko and K. C. Hultzsch, Hydrofunctionalization. Topics in Organometallic Chemistry, ed. V. Ananikov and M. Tanaka, Springer, Berlin, Heidelberg, 2011, vol. 43, pp. 51–114 Search PubMed.
- N. Nishina and Y. Yamamoto, Top. Organomet. Chem., 2013, 43, 115–144 CrossRef CAS.
- J. Takaya and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 5756–5757 CrossRef CAS PubMed.
- B. W. Howk, E. L. Little, S. L. Scott and G. M. Whitman, J. Am. Chem. Soc., 1954, 76, 1899–1902 CrossRef CAS.
- M. Deeba, M. E. Ford and T. A. Johnson, J. Chem. Soc., Chem. Commun., 1987, 562–563 RSC.
- M. Deeba and M. E. Ford, Zeolites, 1990, 10, 794–797 CrossRef CAS.
- V. Khedkar, A. Tillack, C. Benisch, J. P. Melder and M. Beller, J. Mol. Catal. A: Chem., 2005, 241, 175–183 CrossRef CAS.
- K. M. Lemar, O. Passa, M. A. Aon, S. Cortassa, C. T. Müller, S. Plummer, B. O'Rourke and D. Lloyd, Microbiology, 2005, 151, 3257–3265 CrossRef CAS PubMed.
- W. Bühler, E. Dinjus, H. J. Ederer, A. Kruse and C. Mas, J. Supercrit. Fluids, 2002, 22, 37–53 CrossRef.
- W. Ma, X. Zhang, J. Fan, Y. Liu, W. Tang, D. Xue, C. Li, J. Xiao and C. Wang, J. Am. Chem. Soc., 2019, 141, 13506–13515 Search PubMed.
- K. Das, K. Sarkar and B. Maji, ACS Catal., 2021, 11, 7060–7069 Search PubMed.
- L. Duarte de Almeida, F. Bourriquen, K. Junge and M. Beller, Adv. Synth. Catal., 2021, 363, 4177–4181 Search PubMed.
- E. Tálas, G. P. Szíjjártó and A. Tompos, React. Kinet., Mech. Catal., 2015, 115, 431–447 CrossRef.
- R. Rajesh, J. A. Garg, P. Thiruvengetam and R. Kunjanpillai, Asian J. Org. Chem., 2022, 11, e202200440 CrossRef CAS.
- R. Rajesh, O. K. Sandhya, S. P. Narayanan and R. Kunjanpillai, Synthesis, 2024, 2047–2058 CAS.
- X. Li, J. Tian, H. Liu, X. Hu, J. Zhang, C. Xia, J. Chen, H. Liu and Z. Huang, ACS Sustainable Chem. Eng., 2020, 8, 6352–6362 CrossRef CAS.
- M. R. Gagné and T. J. Marks, J. Am. Chem. Soc., 1989, 111, 4108–4109 CrossRef.
- M. R. Gagné, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275–294 Search PubMed.
- W. T. Ye and R. Zhu, Chem. Catal., 2022, 2, 345–357 CrossRef CAS.
- S. Guria, A. N. Volkov, R. Khudaverdyan, R. Van Lommel, R. Pan, C. G. Daniliuc, F. De Proft and U. Hennecke, J. Am. Chem. Soc., 2024, 146, 17180–17188 Search PubMed.
- D. V. Gribkov, K. C. Hultzsch and F. Hampel, Chem. – Eur. J., 2003, 9, 4796–4810 CrossRef CAS PubMed.
- S. Hong, S. Tian, M. V. Metz and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 14768–14783 CrossRef CAS PubMed.
- J. Y. Kim and T. Livinghouse, Org. Lett., 2005, 7, 1737–1739 CrossRef CAS PubMed.
- K. Manna, A. Ellern and A. D. Sadow, Chem. Commun., 2010, 46, 339–341 RSC.
- K. S. A. Motolko, D. J. H. Emslie and H. A. Jenkins, Organometallics, 2017, 36, 1601–1608 CrossRef CAS.
- M. Safariamin, S. Paul, K. Moonen, D. Ulrichts, F. Dumeignil and B. Katryniok, Catal. Sci. Technol., 2016, 6, 2129–2135 RSC.
- S. Ma, C. K. Hill, C. L. Olen and J. F. Hartwig, J. Am. Chem. Soc., 2021, 143, 359–368 CrossRef CAS PubMed.
- S. Ma, Y. Xi, H. Fan, S. Roediger and J. F. Hartwig, Chem, 2022, 8, 532–542 CAS.
- T. Färber, O. Riechert, T. Zeiner, G. Sadowski, A. Behr and A. J. Vorholt, Chem. Eng. Res. Des., 2016, 112, 263–273 CrossRef.
- T. A. Faßbach, N. Gösser, F. O. Sommer, A. Behr, X. Guo, S. Romanski, D. Leinweber and A. J. Vorholt, Appl. Catal., A, 2017, 543, 173–179 CrossRef.
- S. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746–15749 CrossRef CAS PubMed.
- S. Van Den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- T. Renders, W. Schutyser, S. Van Den Bosch, S. F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- W. Lan, M. T. Amiri, C. M. Hunston and J. S. Luterbacher, Angew. Chem., Int. Ed., 2018, 57, 1356–1360 CrossRef CAS PubMed.
- Y. Liao, S. F. Koelewijn, G. van den Bossche, J. van Aelst, S. van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- W. Arts, K. Van Aelst, E. Cooreman, J. Van Aelst, S. Van den Bosch and B. F. Sels, Energy Environ. Sci., 2023, 16, 2518–2539 RSC.
- A. W. Bartling, M. L. Stone, R. J. Hanes, A. Bhatt, Y. Zhang, M. J. Biddy, R. Davis, J. S. Kruger, N. E. Thornburg, J. S. Luterbacher, R. Rinaldi, J. S. M. Samec, B. F. Sels, Y. Román-Leshkov and G. T. Beckham, Energy Environ. Sci., 2021, 14, 4147–4168 RSC.
- M. Tschulkow, M. Pizzol, T. Compernolle, S. Van den Bosch, B. Sels and S. Van Passel, Resour., Conserv. Recycl., 2024, 204, 107466 CrossRef CAS.
- J. K. Kenny, D. G. Brandner, S. R. Neefe, W. E. Michener, Y. Román-Leshkov, G. T. Beckham and J. W. Medlin, React. Chem. Eng., 2022, 7, 2527–2533 Search PubMed.
- F. Brienza, K. Van Aelst, F. Devred, D. Magnin, B. F. Sels, P. Gerin, I. Cybulska and D. P. Debecker, ChemSusChem, 2023, 16, e202300103 CrossRef CAS PubMed.
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640 CrossRef CAS.
- H. Y. Zheng, Z. L. Zhao, L. Q. Xiao, W. S. Zhao, X. B. Liang, Y. F. Xue, H. Yang, Y. L. Niu and Y. L. Zhu, J. Fuel Chem. Technol., 2021, 49, 1261–1269 CrossRef CAS.
- H. J. Brownlee and C. S. Miner, Ind. Eng. Chem., 1948, 40, 201–204 CrossRef CAS.
- Y. Bao, Z. Du, X. Liu, H. Liu, J. Tang, C. Qin, C. Liang, C. Huang and S. Yao, Green Chem., 2024, 26, 6318–6338 RSC.
- A. Kumar, A. S. Chauhan, R. Bains and P. Das, Green Chem., 2022, 25, 849–870 RSC.
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 859–861 CrossRef CAS PubMed.
- M. Mascal, ACS Sustainable Chem. Eng., 2019, 7, 5588–5601 CrossRef CAS.
- S. W. Breeden, J. H. Clark, T. J. Farmer, D. J. MacQuarrie, J. S. Meimoun, Y. Nonne and J. E. S. J. Reid, Green Chem., 2013, 15, 72–75 RSC.
- J. Bueno Moron, G. van Klink and G. J. M. Gruter, ACS Sustainable Chem. Eng., 2023, 11, 17492–17509 CrossRef CAS PubMed.
- M. Mascal, ChemSusChem, 2015, 8, 3391–3395 CrossRef CAS PubMed.
- D. Soukup-Carne, F. S. Bragagnolo, C. Soleo Funari and J. Esteban, Catalysts, 2024, 14, 117 CrossRef CAS.
- A. B. Wood, M. N. Masuno, R. L. Smith, J. Bissell, D. A. Hirsch-Weil, R. J. Araiza, D. R. Henton and J. H. Plonka, 2020, US10710970B2.
- Y. Shi, P. C. J. Kamer and D. J. Cole-Hamilton, Green Chem., 2019, 21, 1043–1053 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- A. B. Dros, O. Larue, A. Reimond, F. De Campo and M. Pera-Titus, Green Chem., 2015, 17, 4760–4772 RSC.
- C. Thoma, J. Konnerth, W. Sailer-Kronlachner, P. Solt, T. Rosenau and H. W. G. van Herwijnen, ChemSusChem, 2020, 13, 3544–3564 CrossRef CAS PubMed.
- G. Trapasso, G. Mazzi, B. Chícharo, M. Annatelli, D. Dalla Torre and F. Aricò, Org. Process Res. Dev., 2022, 26, 2830–2838 CrossRef CAS PubMed.
- Z. Wang, S. Xia, X. Wang, Y. Fan, K. Zhao, S. Wang, Z. Zhao and A. Zheng, Renewable Sustainable Energy Rev., 2024, 196, 114332 CrossRef CAS.
- M. G. Davidson, S. Elgie, S. Parsons and T. J. Young, Green Chem., 2021, 23, 3154–3171 RSC.
- G. Spano, S. Ramello, G. Girotti, F. Rivetti and A. Carati, Catalysts for Alkylated Aromatic Hydrocarbon Preparation through Alkylation And/or Transalkylation of Aromatic Compounds, 2006, WO2006002805A1 Search PubMed.
- H. Bartkowiak, B. Haase, R. Hofmann, H. J. Naumann and B. Raue, Process for Cumene Hydroperoxide Manufacture by Cumene Oxidation with Reduced Energy Consumption by Efficient Heat of Formation Exchange, 1988, DD258531A3 Search PubMed.
- M. S. Kharasch, A. Fono and W. Nudenberg, J. Org. Chem., 1950, 14, 748–752 CrossRef.
- H. R. Bjørsvik, L. Liguori and F. Minisci, Org. Process Res. Dev., 2000, 4, 534–543 CrossRef.
- M. Stein, A. Breit, T. Fehrentz, T. Gudermann and D. Trauner, Angew. Chem., Int. Ed., 2013, 52, 9845–9848 CrossRef CAS PubMed.
- S. Umemura, N. Takamitsu, T. Hamamoto and N. Kuroda, Process for Preparing Dihydric Phenol Derivatives, 1978, US4078006A Search PubMed.
- L. J. Frainier and H. H. Fineberg, Copper Chromite Catalyst for Preparation of Furfuryl Alcohol from Furfural, 1980, US4251396A Search PubMed.
- S. Hirai, H. Hirano, H. Arai, Y. Kiba, H. Shibata, Y. Kusayanagi, M. Yotsuji, K. Hashiba and K. Tanada, Intermediates for Urea and Thiourea Derivatives, 1987, US4643849 Search PubMed.
- B. J. Price, J. W. Clitherow and J. Bradshaw, Aminoalkyl Furan Derivatives, 1976, US4128658A Search PubMed.
- M. Mascal and S. Dutta, Green Chem., 2011, 13, 3101–3102 RSC.
- S. R. D. George, T. D. H. Frith, D. S. Thomas and J. B. Harper, Org. Biomol. Chem., 2015, 13, 9035–9041 RSC.
- S. Rozen, M. Brand and R. Lidor, J. Org. Chem., 1988, 5545–5547 CrossRef CAS.
- K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694–10695 CrossRef CAS PubMed.
- V. R. Uchil and V. Joshi, Indian J. Chem., 2003, 42, 408–411 Search PubMed.
- A. Chatterjee, S. K. Srimany and B. Chaudhury, J. Chem. Soc., 1961, 4576–4579 RSC.
- B. D. Herzog and R. A. Smiley, Ullmann's Encyclopedia of Industrial Chemistry, WileyVCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- P. Sriram, Hexamethylenediamine/Adiponitrile, in Chemical Economics Handbook, 2013 Search PubMed.
- B. Xiao, M. Zheng, X. Li, J. Pang, R. Sun, H. Wang, X. Pang, A. Wang, X. Wang and T. Zhang, Green Chem., 2016, 18, 2175–2184 RSC.
- J. Tuteja, H. Choudhary, S. Nishimura and K. Ebitani, ChemSusChem, 2014, 7, 96–100 CrossRef CAS PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. De Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- J. Mormul, J. Breitenfeld, O. Trapp, R. Paciello, T. Schaub and P. Hofmann, ACS Catal., 2016, 6, 2802–2810 CrossRef CAS.
- D. X. Martínez-Vargas, J. Rivera De La Rosa, L. Sandoval-Rangel, J. L. Guzmán-Mar, M. A. Garza-Navarro, C. J. Lucio-Ortiz and D. A. De Haro-Del Río, Appl. Catal., A, 2017, 547, 132–145 CrossRef.
- Y. Yan, K. Li, J. Zhao, W. Cai, Y. Yang and J. M. Lee, Appl. Catal., B, 2017, 207, 358–365 CrossRef CAS.
- Z. Yang, B. Zhu, Y. He, G. Zhang, P. Cui and J. He, New J. Chem., 2021, 45, 16482–16489 RSC.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1648 CrossRef CAS PubMed.
- K. C. Hwang and A. Sagadevan, Science, 2014, 346, 1495–1498 CrossRef CAS PubMed.
- J. Yang, J. Liu, H. Neumann, R. Franke, R. Jackstell and M. Beller, Science, 2019, 366, 1514–1517 CrossRef CAS PubMed.
- J. C. J. Bart and S. Cavallaro, Ind. Eng. Chem. Res., 2015, 54, 1–46 CrossRef CAS.
- S. Van De Vyver and Y. Román-Leshkov, Catal. Sci. Technol., 2013, 3, 1465–1479 RSC.
- Z. Li, X. Li, H. Zhou, Y. Xu, S. M. Xu, Y. Ren, Y. Yan, J. Yang, K. Ji, L. Li, M. Xu, M. Shao, X. Kong, X. Sun and H. Duan, Nat. Commun., 2022, 13, 5009 CrossRef CAS PubMed.
- J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 RSC.
- J. C. J. Bart and S. Cavallaro, Ind. Eng. Chem. Res., 2015, 54, 567–576 CrossRef CAS.
- M. Lang and H. Li, ChemSusChem, 2022, 15, e202101531 CrossRef CAS PubMed.
- S. VandeVyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- S. Li, W. Deng, S. Wang, P. Wang, D. An, Y. Li, Q. Zhang and Y. Wang, ChemSusChem, 2018, 11, 1995–2028 CrossRef CAS PubMed.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761–768 CrossRef CAS PubMed.
- Y. Jing, Y. Guo, Q. Xia, X. Liu and Y. Wang, Chem, 2019, 5, 2520–2546 CAS.
- T. Asano, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2016, 4, 6253–6257 CrossRef CAS.
- L. Wei, J. Zhang, W. Deng, S. Xie, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 8013–8016 RSC.
- X. Li, D. Wu, T. Lu, G. Yi, H. Su and Y. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4200–4204 CrossRef CAS PubMed.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 RSC.
- H. Zhang, X. Li, X. Su, E. L. Ang, Y. Zhang and H. Zhao, ChemCatChem, 2016, 8, 1500–1506 CrossRef CAS.
- W. Deng, L. Yan, B. Wang, Q. Zhang, H. Song, S. Wang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 4712–4719 CrossRef CAS PubMed.
- M. Choi, J. Byun, H. Park, K. Jeong, S. M. Kim and J. Han, Biomass Bioenergy, 2022, 162, 106503 CrossRef CAS.
- J. B. J. H. van Duuren, P. J. de Wild, S. Starck, C. Bradtmöller, M. Selzer, K. Mehlmann, R. Schneider, M. Kohlstedt, I. Poblete-Castro, J. Stolzenberger, N. Barton, M. Fritz, S. Scholl, J. Venus and C. Wittmann, Biotechnol. Bioeng., 2020, 117, 1381–1393 CrossRef CAS PubMed.
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- Y. Yang, X. Wei, F. Zeng and L. Deng, Green Chem., 2016, 18, 691–694 RSC.
- A. Marckwordt, F. El Ouahabi, H. Amani, S. Tin, N. V. Kalevaru, P. C. J. Kamer, S. Wohlrab and J. G. de Vries, Angew. Chem., Int. Ed., 2019, 58, 3486–3490 CrossRef CAS PubMed.
- D. R. Vardon, N. A. Rorrer, D. Salvachúa, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC.
- A. Corona, M. J. Biddy, D. R. Vardon, M. Birkved, M. Z. Hauschild and G. T. Beckham, Green Chem., 2018, 20, 3857–3866 RSC.
- W. C. Drinkard and R. V. Lindsey, Hydrocyanation of Olefins Using Selected Nickel Phosphite Catalysts, 1970, US3496215(A) Search PubMed.
- Y. Chia, W. C. Drinkard and E. N. Squire, Hydrocyanation of Olefins, 1973, US3766237A Search PubMed.
- D. B. Bivens, L. W. Patton and W. E. Thomas, Hydrogenation of Adiponitrile, 1973, US3758584A Search PubMed.
- Q. Ke, Y. Jin, F. Ruan, M. N. Ha, D. Li, Y. Cao, H. Wang, T. Wang, V. N. Nguyen, X. Han, X. Wang, P. Cui and P. Cui, Green Chem., 2019, 21, 4313–4318 RSC.
- H. Qi, F. Liu, L. Zhang, L. Li, Y. Su, J. Yang, R. Hao, A. Wang and T. Zhang, Green Chem., 2020, 22, 6897–6901 RSC.
- P. Li and A. T. Liebens, A Process for Producing a Tetrahydrofuran Compound Comprising at Least Two Amine Functional Groups, 2017, WO2018113599A1 Search PubMed.
- J. Xu, Y. Xu, J. Ma, X. Jia, J. Gao, H. Miao and F. Xia, Method for Preparing Aliphatic Amine Compound from Furfurylamine Compound by Catalytic Hydrogenolysis, 2020, CN 111100015A Search PubMed.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- E. T. C. Vogt and B. M. Weckhuysen, Nature, 2024, 629, 295–306 CrossRef CAS PubMed.
- B. Subramaniam, R. K. Helling and C. J. Bode, ACS Sustainable Chem. Eng., 2016, 4, 5859–5865 CrossRef CAS.
- P. G. Jessop and A. R. MacDonald, Green Chem., 2023, 25, 9457–9462 RSC.
- Statista. Market volume of polyamide industry worldwide from 2017 to 2018 with a forecast until 2025.
- Grand View Research, Global Nylon Market Size, Share & Trends Report, 2030.
- D. B. Sulis, N. Lavoine, H. Sederoff, X. Jiang, B. M. Marques, K. Lan, C. Cofre-vega, R. Barrangou and J. P. Wang, Nat. Commun., 2025, 16, 1244 CrossRef CAS PubMed.
- E. Cooreman, T. Nicolaï, W. Arts, K. Van Aelst, T. Vangeel, S. V. Van den Bosch, J. Van Aelst, B. Lagrain, K. Thiele, J. Thevelein and B. F. Sels, ACS Sustainable Chem. Eng., 2023, 11, 5440–5450 CrossRef CAS.
- Ó. Ögmundarson, M. J. Herrgård, J. Forster, M. Z. Hauschild and P. Fantke, Nat. Sustain., 2020, 3, 167–174 CrossRef.
- H. H. Khoo, W. L. Ee and V. Isoni, Green Chem., 2016, 18, 1912–1922 RSC.
- E. A. R. Zuiderveen, K. J. J. Kuipers, C. Caldeira, S. V. Hanssen, M. K. van der Hulst, M. M. J. de Jonge, A. Vlysidis, R. van Zelm, S. Sala and M. A. J. Huijbregts, Nat. Commun., 2023, 14, 8521 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00924c |
| This journal is © The Royal Society of Chemistry 2025 |