
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanocellulose-short peptide self-assembly for improved mechanical strength and barrier performance†
Alessandro
Marchetti‡
 a,
Elisa
Marelli‡
a,
Greta
Bergamaschi
b,
Panu
Lahtinen
c,
Arja
Paananen
c,
Markus
Linder
d,
Claudia
Pigliacelli
*a and
Pierangelo
Metrangolo
*a
a,
Elisa
Marelli‡
a,
Greta
Bergamaschi
b,
Panu
Lahtinen
c,
Arja
Paananen
c,
Markus
Linder
d,
Claudia
Pigliacelli
*a and
Pierangelo
Metrangolo
*a
aLaboratory of Supramolecular and Bio-Nanomaterials (SBNLab), Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Via L. Mancinelli 7, 20131 Milano, Italy. E-mail: claudia.pigliacelli@polimi.it; pierangelo.metrangolo@polimi.it
bIstituto di Scienze e Tecnologie Chimiche, National Research Council of Italy, Via M. Bianco 9, 20131 Milano, Italy
cVTT-Technical Research Centre of Finland Ltd, Tekniikantie 21, 02150 Espoo, Finland
dDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, Kemistintie 1, 02150 Espoo, Finland
First published on 19th August 2024
Abstract
Cellulose nanofibers (CNF) are the most abundant renewable nanoscale fibers on Earth, and their use in the design of hybrid materials is ever more acclaimed, although it has been mostly limited, to date, to CNF derivatives obtained via covalent functionalization. Herein, we propose a noncovalent approach employing a set of short peptides – DFNKF, DF(I)NKF, and DF(F5)NKF – as supramolecular additives to engineer hybrid hydrogels and films based on unfunctionalized CNF. Even at minimal concentrations (from 0.1% to 0.01% w/w), these peptides demonstrate a remarkable ability to enhance CNF rheological properties, increasing both dynamic moduli by more than an order of magnitude. Upon vacuum filtration of the hydrogels, we obtained CNF-peptide films with tailored hydrophobicity and surface wettability, modulated according to the peptide content and halogen type. Notably, the presence of fluorine in the CNF-DF(F5)NKF film, despite being minimal, strongly enhances CNF water vapor barrier properties and reduces the film water uptake. Overall, this approach offers a modular, straightforward method to create fully bio-based CNF-peptide materials, where the inclusion of DFNKF derivatives allows for facile functionalization and material property modulation, opening their potential use in the design of packaging solutions and biomedical devices.
1. Introduction
While the quest for sustainability challenges chemists and materials scientists, cellulose nanofibers (CNF) stand out as a renewable and biodegradable biomass in the design of functional nanoscale systems and composite materials.1 Such a popularity arises from the natural abundance and low-carbon footprint of CNF, which combine a substantial tunability in terms of morphology and fibril size, enabling unique mechanical, optical, and thermal properties.2–5 Given the high number of –OH groups on the CNF surface, the formation of a dense hydrogen bonding network leads to a tight entanglement of fibrils, which can potentially limit the interaction with other matrices.2 Thus, the use of CNF in the design of hybrid materials has mostly focused on custom derivatives obtained via covalent functionalization, including oxidation, phosphorylation, sulfonation, carboxymethylation and silylation.6–11 Among them, TEMPO-oxidized CNF (TO-CNF), having the hydroxyl groups functionalized with negatively charged carboxylate groups, represents the most studied and employed CNF derivative for the development of packaging solutions,12 drug delivery systems,13 optical materials,3 and energy storage devices.14 Still, the use of unfunctionalized CNF, obtained by sole mechanical fibrillation, remains, to date, seldom explored.15The use of biomolecules, including natural and/or engineered proteins, as functional additives in the design of CNF-based hybrid matrices shows promise.16,17 Indeed, some recent studies have demonstrated the effectiveness of combining TO-CNF with proteinaceous materials to achieve tuned mechanical behaviour in gels and films.18–20 Furthermore, Khatri et al. recently showed how by supplementing TO-CNF films with fibrils made by the Curli-specific gene A (CsgA) protein the surface hydrophobicity and thermal stability of the resulting films were improved with respect to the ones obtained with solely TO-CNF.21 While the use of full-length proteins has led to the above-mentioned successful results, a fine tuning of CNF features and properties has not yet been achieved employing short peptides sequences, especially when incorporated through a noncovalent approach.22,23 Thus, the design of CNF hybrid matrices bearing custom short peptides, which offer clear advantages in terms of versatility and ease of design with respect to full-length proteins, remains an unmet challenge.
Pursuing a noncovalent design strategy, here we employ a series of short peptides to tune the rheological and functional properties of unmodified CNF and engineer hybrid hydrogels and films thereof. We selected DFNKF, the core fragment of the human calcitonin hormone, known for its self-assembly and fibrillogenic propensity,24 as a model peptide sequence. Self-assembling peptides represent ideal partners for CNF, owing to their intrinsic fibril-forming tendency and the possible presence of charged residues engageable in electrostatic interactions, features that, we reasoned, could favour the entanglement between CNF and peptide fibrils.25 Indeed, the presence of a lysine (Lys) residue in the DFNKF sequence facilitates the interaction with CNF, which bears a net negative charge due to hemicellulose residues within CNF fibrils.26 Moreover, the two phenylalanine (Phe) residues in DFNKF offers opportunities for facile functionalization. Building upon this feature, we employed two halogenated derivatives, DF(I)NKF and DF(F5)NKF (see Fig. S1 and S2, ESI†), as supramolecular additives to enhance and tune CNF hydrophobicity, in addition to their mechanical properties. Our results demonstrate that even minimal quantities of these peptides, ranging from 0.1% to 0.01% w/w, significantly strengthen the CNF rheological properties through electrostatic interactions. These quantities are extremely low, especially when compared to the protein-to-CNF ratios used in previous works,22,27 which were used with up to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) ratio, yet still allowed for remarkable mechanical enhancement. Furthermore, the tunability given by varying the halogen type and number in Phe residues of the peptides offers exciting prospects for tailoring the functionality of the resulting materials. Indeed, while iodine (I) acts as a labelling function and potential site for post-processing functionalization, the presence of fluorine (F) decreases the wettability of the CNF-fluorinated peptide films, which exhibited excellent water vapor barrier properties, high hydrophobicity, and low water uptake capability.
:1 (w/w) ratio, yet still allowed for remarkable mechanical enhancement. Furthermore, the tunability given by varying the halogen type and number in Phe residues of the peptides offers exciting prospects for tailoring the functionality of the resulting materials. Indeed, while iodine (I) acts as a labelling function and potential site for post-processing functionalization, the presence of fluorine (F) decreases the wettability of the CNF-fluorinated peptide films, which exhibited excellent water vapor barrier properties, high hydrophobicity, and low water uptake capability.
2. Results and discussion
2.1. Formation of CNF-peptide hydrogels
At first, a simple and quick procedure to trigger CNF gelation was designed. The high dry matter content of the stock CNF dispersion, namely 1.6% w/w, rendered it highly viscous and locally aggregated, potentially preventing homogeneous peptide inclusion. Thus, we combined dilution and mechanical agitation to exploit the shear-thinning behaviour of CNF28,29 and successively incorporate peptides within the CNF matrix. We began by performing flow curve measurements of a series of dilutions (Fig. S3, ESI†), identifying 0.8% w/w as the optimal concentration. Under such conditions, tip sonication can effectively favour fiber disentanglement (Fig. S4a, ESI†), allowing the formation of a stronger gel to be easily achieved.30 As gelation is a concentration-dependent phenomenon,31 the sonication impact was more noticeable with 0.8% CNF content rather than lower. Despite sonication being a widespread method to promote CNF separation, especially for surface-functionalized fibers, the prolonged exposure of CNF to ultrasound is known to potentially deteriorate them, leading to fiber fragmentation.32 In our case, neither fibers breakage nor crystallinity index (CI) variations were observed upon sonication (CI = 48.9% before and CI = 49.2% after, Fig. S5, ESI†), suggesting that the CNF morphology is mostly preserved after the ultrasonic treatment.32–34 After identifying the ideal conditions to trigger CNF gelation, we incorporated the peptides within the CNF matrix to obtain hybrid hydrogels. Solutions of DFNKF and its two halogenated analogues, DF(I)NKF and DF(F5)NKF (Fig. 1), were used to dilute the 1.6% w/w CNF dispersion. The hydrogels, namely CNF-H, CNF-I, CNF-F5 in accordance with the peptide used, were prepared (Fig. 2a) with varying peptide amounts ranging from 0.1% (1.25 mM) to 0.01% w/w (0.125 mM), meaning CNF:peptide ratios comprised between 8:1 to 80:1 (Table S2, ESI†). Such peptides, especially DF(I)NKF, are known to possess strong self-assembly properties.35 However, at such a low content, gelation cannot be driven by peptide self-assembly, as the minimum gelation concentration of the best gelator in the series was reported to be around 0.2% w/w.35
 | ||
| Fig. 1 Schematic illustration of the hybrid network based on CNF and DFNKF, highlighting the residues exploited for functionalization and interaction with CNF. The halogenated derivatives, DF(I)NKF and DF(F5)NKF, were obtained substituting the Phe in the second position with the corresponding halogenated amino acids. | ||
 | ||
| Fig. 2 (a) Schematic representation of hybrid hydrogels preparation protocol; (b) and (c) Vial inversion tests of the CNF-based hydrogels right after 0.05% w/w DF(I)NKF addition (b) and after sonication (c); TEM images of the (d) neat CNF hydrogel and (e) CNF-I at 0.05% peptide concentration; (f) ξ-potential measurements of neat CNF, of the peptides alone, and of the hybrid hydrogels at the different CNF:peptide ratios. | ||
Nevertheless, peptide inclusion deeply affected CNF networks by thickening the dispersion and stiffening the gel after sonication (Fig. 2b and c). Importantly, such an effect was observed in all of the tested hydrogels, regardless of the peptide sequence used. Microscopic characterization revealed a long and well-dispersed network of individual CNF fibers with a size of 18.25 ± 8.32 nm, which, upon peptide addition, appeared to be increased in thickness, and coated by an amorphous matrix (Fig. 2d and e). Such a morphology was not observed in either peptide or CNF alone, confirming that the interaction between the two components deeply affects the assembly pattern of the system. Overall, the aspect of the network turned less regular, with respect to the network composed of sole CNF, with the peptide matrix tightly binding to pre-existing fibers. As the CNF is unfunctionalized, the surface of individual nanofibers should lack charged groups. However, untreated CNF can contain slight impurities, as hemicellulose or pectin,26,36 leading to the presence of some sugar acids (e.g., uronic acids) on the CNF surface. These negatively charged groups are considered responsible for the observed CNF negative surface potential (Fig. 2f), favouring the complexation with the peptides, which, owing to their Lys residue, are positively charged (pI = 6.8) under the conditions tested (pH = 5.7). By increasing peptide content, the net ξ-potential of hydrogels progressively increased from highly negative values (ξ = −30 mV) to less negative ones (ξ = −15 to −20 mV), despite the minimal content, confirming the electrostatic complexation between the two components. Moreover, the protonated NH2 group of the Lys residue, besides providing a positive charge, may enable the interaction with carboxylate anions present on the CNF surface via hydrogen bonding. Fourier-transform infrared spectroscopic analysis provided an indication of such modifications in the hydrogen bonding network of the amine groups, as suggested by the blueshifts of the N–H bands after complexation with CNF (Fig. S6 and S7, ESI†).
The morphology of the hybrid samples slightly changed also in their dry state, where peptide addition led to the appearance of local bundles of densely fibrillated patches, which could not be observed in pure CNF samples (Fig. 3a and b). To better probe the peptide presence within the observed network, we exploited their halogenated derivatives by coupling scanning electron microscopy (SEM) with energy dispersive X-ray spectroscopy (EDS) to track their location within the matrix. In fact, halogens and heavy atoms have historically been utilized as labels for structure phasing of complex biomolecules such as proteins37 or, more recently, for inferring conformational parameters of halogen-labelled amino acids.38,39 By selecting nitrogen, iodine, and fluorine as exclusive labels for CNF-H, CNF-I and CNF-F5, respectively, we obtained elemental maps (Fig. 3c and d) that showed an even peptide distribution throughout the whole sample, confirming a successful blending, i.e., co-assembly of the two constituents in the systems. For CNF-I and CNF-F5, the results were further corroborated by the nitrogen channel, utilized as a secondary label, which provided an almost superimposable map with respect to the halogen channel. The lack of segregation or self-sorting in N, I, and F signals confirmed, once more, the inability of peptides to self-assemble into fully mature amyloid fibrils when incorporated in a CNF network. This finding supports the hypothesis of a combination of two competitive mechanisms for peptide self-assembly. On the one hand, noncovalent peptide–peptide interactions – mainly π-stacking between aromatic residues, ionic interactions between charged residues, and salt bridges between free termini and hydrogen bonds – drive the self-assembly towards the fibrillar state and the formation of small protofibrils. On the other, CNF could engage peptide monomers and small oligomers in electrostatic interactions, hindering the development of mature fibrils and leading to the formation of amorphous aggregates diffused all over the surface of cellulose nanofibrils.
 | ||
| Fig. 3 SEM images of lyophilized (a) neat CNF hydrogels and (b) CNF-I containing 0.05% w/w of peptide. EDS maps with the corresponding SEM images of (c) CNF-I and (d) CNF-F5 systems at 0.05% w/w peptide concentration. Colour code: I, violet; N, yellow; F, green. | ||
2.2. Rheology enhancement and insights into interactions between peptides and CNF
After assessing the homogeneous inclusion of the peptides in the hydrogels, we reasoned that the noncovalent conjugation of small peptides may alter the mechanical properties of the system. Indeed, Sharma et al.23 and Kaur et al.22 demonstrated that when short bioactive peptides were added to TEMPO-oxidized CNF (TO-CNF) hydrogels, their mechanical response was tailored in accordance with the peptide content. In such a way, they were able to modulate the storage moduli of the hydrogels to achieve intermediate properties between TO-CNF hydrogels and peptide hydrogels, exploited to design biocompatible scaffolds for cellular growth.22,23 On the other hand, when large proteins (e.g., lysozyme or β-lactoglobulin) were combined with nanocellulose materials, it was possible to obtain self-conjugating systems where complexation led to a synergistic improvement of the mechanical properties of the gels.17,27In our study, we first probed the rheological properties of neat CNF and of the obtained hydrogels via oscillatory rheology studies. The 0.8% w/w CNF dispersion behaved as a weak percolating network possessing a single plateau region of the dynamic moduli, with the storage modulus (G′) dominating over the loss modulus (G′′), indicative of a gel/solid-with like behaviour, and a linear viscoelastic region (LVER) with up to ∼4% strain, after which the structure started to break down (Fig. 4). Overall, G′ and G′′ values were relatively low (G′ = 40.5 ± 5.4 Pa, G′′ = 5.9 ± 0.8 Pa), but remarkably improved upon sonication treatment (G′ = 110 ± 13 Pa, G′′ = 17 ± 2 Pa) (Fig. S8, ESI†). Even at the lowest concentration (0.01% w/w, 0.125 mM), the introduction of the peptides could lead to appreciably improved rheological response of the CNF network (Fig. 4a and b). At higher peptide concentrations, between 0.025% and 0.1% w/w (0.5 and 1.25 mM), all the tested hybrid systems (CNF-H, CNF-I, and CNF-F5) shared a more marked change with similar viscoelastic responses, showing both dynamic moduli rising up by one order of magnitude (e.g., G′ = 1124 ± 152 Pa for 0.1% w/w CNF-F5 hydrogels) (Fig. S9 and S10, ESI†). Peptide inclusion did not affect CNF crossover frequency (G′ = G′′), which remained constant (∼30 Hz) at all the tested conditions and equal to the one of neat CNF, thus not impacting CNF relaxation behaviour (Fig. 4c). Amplitude strain sweeps showed that the composite samples were able to withstand slightly smaller strains (LVER ∼2% strain), indicating more connections in the CNF network and an easily flowing system (Fig. 4d). Additionally, at lower peptide contents (≤0.025%, i.e., 0.36 mM) there was a rise in the G′′ in the proximity of the yield point, indicating a temporary internal structuring prior to network collapse (Fig. 4e). This rheological response is similar to what is typically observed when ionic solutes are added to cellulose networks, and their impact on CNF mechanical properties is largely documented in the literature.29,36,40 At increased ionic strength, the repulsion between cellulose fibrils is screened, leading to aggregation and stronger networks. Similarly, in our systems, CNF electrostatic screening occurs upon peptide addition, as evidenced by the gradual increase in the ξ-potential value. The peptides, acting as physical crosslinks, enable stronger and more robust connections between cellulose fibers, resulting in enhanced rheological properties. Aside from local interactions, the CNF-peptide interaction can also be favoured by the entropic gain resulting from the release of structured water molecules present on the CNF surface before peptide adsorption, as suggested by the decreased ability of the composite hydrogels to retain water (Fig. S11, ESI†). This agrees with the works by Nyström et al.,18,27,41 who have also previously described how the interaction of nanocellulose-based materials with proteinaceous components, such as amyloid proteins, can be driven by the gain of system entropy due to the release of counter-ions and water previously bound to cellulose, rather than relying solely on short-range ionic interactions. Notably, without sonication, no change in rheological properties was observed for any CNF-peptide combination (Fig. S12, ESI†), underscoring the crucial role of sonication in making CNF surface accessible for peptide insertion and enabling the formation of the hybrid material. Sonication, while promoting CNF–peptide complexation, may also induce the formation of more extensive supramolecular peptide structures (e.g., protofibrils), as observed when applying ultrasounds to peptides as single components (Fig. S3b, ESI†).41,42 These larger structures have an increased capacity to bind with CNF20 and could provide more entanglement points, ultimately resulting in improved rheological properties. This possibility, together with the positive net charge carried by the peptides, may also account for the observed rheological synergistic enhancement obtained with such short peptides and at such low contents, which contrasts with other studies reporting no substantial improvements when using non-assembled (monomeric and/or prefibrillar states) proteins.21,43
 | ||
| Fig. 4 (a) and (b) Frequency sweep measurements of CNF-I hydrogels compared with neat CNF. (c) Frequency sweep measurements in small deformation oscillatory regime of CNF-I hydrogels, highlighting the crossover of G′ (solid lines) and G′′ (dotted lines). (d) and (e) Amplitude sweep measurements for linear viscoelastic region identification of CNF-I hydrogels. | ||
2.3. Hydrophobic film formation
CNF films offer a green and sustainable alternative to non-renewable resource-based membranes in various applications ranging from packaging and electronic displays to biomedical devices. Nonetheless, these films exhibit a detrimental response when exposed to water or highly humid environments due to the strong hygroscopic nature of CNF.44 Many of the strategies proposed, to date, to tailor the CNF surface properties, rely on chemical hydrophobization involving the use of potentially hazardous chemicals and solvents, leading to high processing costs and environmental concerns. As an example, a traditional way to increase CNF barrier properties is to surface coat with per- and polyfluorinated substances (PFAS). However, the use of PFAS in consumer and medical products is raising significant environmental and health concerns due to their persistence, bioaccumulation, and potential toxicity.45,46 In this context, the CNF-peptide hybrid materials presented here provide a more straightforward and sustainable alternative to enhance and tune hydrophobicity of CNF, considering that the different variants of the DFNKF sequence have been reported to possess different hydrophobicity according to the halogenated moieties introduced on the Phe residue.35 Moreover, by avoiding covalent functionalization of CNF with hydrophobic groups, the inherent properties of cellulose are mostly preserved, including its biodegradability.47Accordingly, vacuum filtration of CNF-H, CNF-I, and CNF-F5 hydrogels was performed (Fig. 5a), to obtain self-standing membranes with varying CNF:peptide ratios. Peptide inclusion in the final films was verified by IR spectroscopy, highlighted by the presence of distinguishable amide peaks at ∼1665 cm−1, ∼1635 cm−1 and ∼1545 cm−1 (Fig. S13, ESI†), and by elemental analysis for CNF-F5 (Fig. S14b, ESI†). As shown by the SEM images (Fig. S13a, ESI†), peptides addition did not alter the CNF film morphology significantly, resulting in a homogeneous film with a relatively low porosity. In order to evaluate the impact of peptide addition on CNF hydrophobicity, film surface wettability was first investigated through contact angle measurements. As expected, pristine CNF films showed a water contact angle (wCA) of around 41 ± 2°, in agreement with some literature reports confirming that CNF-based films are vulnerable to water.48,49 Upon peptide introduction, wCA on average increased under all tested conditions (Fig. 5b), as a result of the peptides adsorption on the CNF surface, partially hindering the interaction between CNF hydroxyl groups and water. Notably, a significant increase in wCA was observed at 0.1% wt peptide concentration, with DF(F5)NKF addition resulting in a wCA increment to above 90° (93 ± 10°), thus achieving hydrophobic wCA values. CNF-F5 reduced wettability was followed by CNF-I (67 ± 11°) and CNF-H (53 ± 1°) films. The observed trend is easily attributable to the different hydrophobicity of the slightly modified Phe residues, showing the great impact provided by the type, number, and position of halogen atoms used. Despite the possibility of further reducing the wettability of CNF-based systems with more common hydrophobic additives, it is essential to note that the overall peptide content added to these systems is remarkably low, with a maximum CNF:peptide ratio of 8:1 and an overall peptide content of milligram fractions. Moreover, peptide inclusion enriches the system with additional functional groups, which could potentially be further exploited for biomolecule detection and bio-diagnostics purposes.50
 | ||
| Fig. 5 (a) Hybrid films production procedure by vacuum filtration of the CNF-peptides hydrogels. (b) Water contact angles and (c) n-C12 contact angles of neat CNF, CNF-H, CNF-I and CNF-F5 at different peptide content. (d) Water uptake after 24 hours of immersion of neat CNF and CNF-F5. (e) Water vapor transmission rates (WVTR) of neat CNF and CNF-F5 (insert: WTVR of CNF-F5 compared with those of cellulose acetate (CA) and cellulose acetate propionate (CAP)51,52). Colour code: neat CNF, black; CNF-H, blue; CNF-I, violet; CNF-F5, green. | ||
Complementarily to water contact angles, n-dodecane (C12) contact angle (dCA) measurements were performed. dCA progressively decreased by peptide addition until dropping to almost negligible values at sufficiently high contents (Fig. 5c), resulting in CNF films with enhanced water resistance and increased affinity to oils. Given the remarkable decrease of the CNF hydrophilic character upon addition of 0.1 wt% DF(F5)NKF, CNF-F5 films with the highest peptide content were further studied in terms of water uptake and water vapor permeability. As shown by Fig. 5d, neat CNF films absorbed around 175 ± 17% of water after 24 hours of immersion, a value that decreased to 95 ± 19.9% for the CNF-F5 films, demonstrating their reduced water uptake capacity. Subsequently, the films water permeability was determined in terms of water vapor transmission rate (WVTR). CNF-F5 films exhibited a WVTR of 2046 ± 169 g m−2 day−1, which was twofold lower than the 5360 ± 1077 g m−2 day−1 observed for pristine CNF films (Fig. 5e). This value remained stable throughout the entire monitoring period (up to 24 hours). More importantly, CNF-F5 films demonstrated a WVTR value that was comparable to, if not superior to, other commonly used and industrially relevant cellulose materials, including cellulose acetate (CA), cellulose acetate propionate (CAP) and other fluorinated cellulose esters (Fig. S15, ESI†).51–53 These findings underscore, once again, the significant impact of even small quantities of DFNKF and its halogenated variants on the surface and barrier properties of CNF, providing a new paradigm for potentially valuable alternative for waterproofing cellulose-based materials.
3. Conclusions
In summary, we designed hybrid hydrogels and films based on unfunctionalized cellulose nanofibers and short peptides, some of which bear halogenated residues. Despite the minimal peptide content introduced in the system (from 0.1% to 0.01% by weight), the electrostatic interactions and physical entanglement taking place between the positively charged peptides and negatively charged CNF allowed CNF rheological properties to be readily strengthened, resulting in a more than tenfold increase in both dynamic moduli. The presence of halogen atoms on the Phe residue did not alter the core features of the system, but allowed for the easy tracking of their distribution within the matrix by means of elemental analysis, confirming a homogeneous distribution. Vacuum filtration of the hydrogels yielded hybrid CNF-peptide films with enhanced hydrodynamic properties. Notably, the addition of a minimal peptide content enabled the modulation of film wettability without the need of prior chemical modifications, achieving hydrophobicity when a fluorinated residue is introduced within the amino acid sequence. We believe our supramolecular approach may pave the way to totally new, modular, and straightforward design strategies for obtaining cellulose nanofiber materials with tuned properties. In fact, the obtained CNF-fluorinated peptide films exhibited excellent water vapor barrier properties, high hydrophobicity, and low water uptake capability, features that are potentially useful in several applications, e.g., moisture-resistant wound dressings, high-performance biosensors or antifouling coatings, where the use of natural materials with tuneable surface properties is highly desired but still limited.4. Materials and methods
4.1. Materials
Nanocellulose was provided by VTT Technical Research Centre (Espoo, Finland). It was obtained from bleached birch pulp, which has been Na-washed and fluidized six times to make nanocellulose. The final nanocellulose suspension is characterized by a dry matter content of 1.6 wt%. The peptides (DFNKF, DF(I)NKF and DF(F5)NKF) were synthesized through classical solid-phase synthesis. Details on the synthetic procedure and purification are reported in the ESI,† (Fig. S1 and S2).4.2. Methods
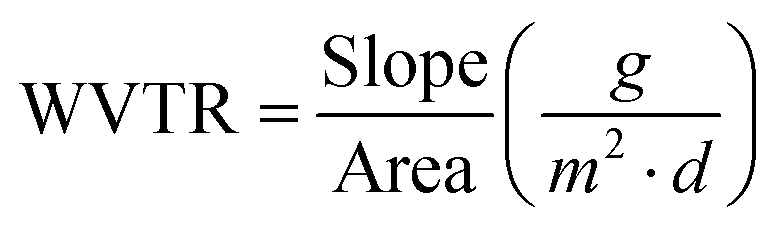
where mw is the wet film mass and md is the dry film mass. Measurements were performed in triplicates and average results are reported.

Author contributions
P. M. designed the project and secured funding. P. M. and C. P. supervised project realization and share corresponding authorship. A. P. and P. L. provided CNF sample and related expertise. M. L. provided expertise in CNF self-assembly. G. B. provided peptide samples and rheology expertise. All authors contributed to article writing and revising, A. M., E. M., and C. P., in particular, and approved it for publication.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declareAcknowledgements
P. M. is grateful to the European Research Council (ERC) for the Starting Grant ERC-2012-StG_20111012 FOLDHALO (Grant Agreement no. 307108) and the Proof-of-Concept Grant ERC-2017-PoC MINIRES (Grant Agreement no. 789815). A. M. and P. M. are thankful to the projects Hydrogex (grant no. 2018-1720) and KARATE (grant no. 2022-0437) funded by Cariplo Foundation. M. L. is grateful to the Research Council of Finland for funding the Liber project (no. 346105). We also acknowledge the provision of facilities and technical support by Aalto University at the OtaNano Nanomicroscopy Center.References
- T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 590, 47–56 CrossRef CAS PubMed.
- C. Salas, T. Nypelö, C. Rodriguez-Abreu, C. Carrillo and O. J. Rojas, Curr. Opin. Colloid Interface Sci., 2014, 19, 383–396 CrossRef CAS.
- I. Niskanen, K. Zhang, M. Karzarjeddi, H. Liimatainen, S. Shibata, N. Hagen, R. Heikkilä, H. Yoda and Y. Otani, J. Polym. Res., 2022, 29, 1–11 CrossRef.
- Y. Dong, C. Rossner and A. Fery, ACS Appl. Opt. Mater., 2023, 12, 1862–1878 CrossRef.
- E. Kontturi, P. Laaksonen, M. B. Linder, A. H. Nonappa, O. J. Gröschel, O. Rojas and O. Ikkala, Adv. Mater., 2018, 30, 1703779 CrossRef PubMed.
- A. Blasi-Romero, M. Ångström, A. Franconetti, T. Muhammad, J. Jiménez-Barbero, U. Göransson, C. Palo-Nieto and N. Ferraz, ACS Appl. Mater. Interfaces, 2023, 15, 24186–24196 CrossRef CAS PubMed.
- L. T. Juan, S. H. Lin, C. W. Wong, U. S. Jeng, C. F. Huang and S. H. Hsu, ACS Appl. Mater. Interfaces, 2022, 14, 36353–36365 CrossRef CAS PubMed.
- H. Kono, E. Tsukamoto and K. Tajima, ACS Omega, 2021, 6, 34107–34114 CrossRef CAS PubMed.
- M. Ghanadpour, F. Carosio, P. T. Larsson and L. Wågberg, Biomacromolecules, 2015, 16, 3399–3410 CrossRef CAS PubMed.
- A. Oberlintner, B. Likozar and U. Novak, Carbohydr. Polym., 2021, 259, 117742 CrossRef CAS PubMed.
- H. Sehaqui, T. Zimmermann and P. Tingaut, Cellulose, 2014, 21, 367–382 CrossRef CAS.
- B. Soni, E. B. Hassan, M. W. Schilling and B. Mahmoud, Carbohydr. Polym., 2016, 151, 779–789 CrossRef CAS PubMed.
- M. Mujtaba, A. Negi, A. W. T. King, M. Zare and J. Kuncova-Kallio, Curr. Opin. Biomed. Eng., 2023, 28, 100475 CrossRef CAS.
- Z. Wang, Y. H. Lee, S. W. Kim, J. Y. Seo, S. Y. Lee and L. Nyholm, Adv. Mater., 2021, 33, 1–18 Search PubMed.
- K. Li, L. N. Skolrood, T. Aytug, H. Tekinalp and S. Ozcan, ACS Sustainable Chem. Eng., 2019, 7, 14341–14346 CrossRef CAS.
- A. C. Q. Silva, A. J. D. Silvestre, C. Vilela and C. S. R. Freire, Front. Bioeng. Biotechnol., 2022, 10, 1059097 CrossRef PubMed.
- N. H. C. S. Silva, P. Garrido-Pascual, C. Moreirinha, A. Almeida, T. Palomares, A. Alonso-Varona, C. Vilela and C. S. R. Freire, Int. J. Biol. Macromol., 2020, 165, 1198–1210 CrossRef CAS PubMed.
- L. Severini, K. J. De France, D. Sivaraman, N. Kummer and G. Nyström, ACS Omega, 2022, 7, 578–586 CrossRef CAS PubMed.
- E. Trovatti, H. Tang, A. Hajian, Q. Meng and A. Gandini, Carbohydr. Polym., 2018, 181, 256–263 CrossRef CAS PubMed.
- S. Lombardo, S. Eyley, C. Schütz, H. Van Gorp, S. Rosenfeldt, G. Van Den Mooter and W. Thielemans, Langmuir, 2017, 33, 5473–5481 CrossRef CAS PubMed.
- V. Khatri, M. Jafari, R. Gaudreault, M. Beauregard, M. Siaj, D. Archambault, É. Loranger and S. Bourgault, Biomacromolecules, 2023, 24, 5290–5302 CrossRef CAS PubMed.
- H. Kaur, P. Sharma, V. K. Pal, S. Sen and S. Roy, ACS Biomater. Sci. Eng., 2023, 9, 1422–1436 CrossRef CAS PubMed.
- P. Sharma, V. K. Pal, H. Kaur and S. Roy, Biomacromolecules, 2022, 23, 2496–2511 CrossRef CAS PubMed.
- M. Reches, Y. Porat and E. Gazit, J. Biol. Chem., 2002, 277, 35475–35480 CrossRef CAS PubMed.
- D. T. Seroski, X. Dong, K. M. Wong, R. Liu, Q. Shao, A. K. Paravastu, C. K. Hall and G. A. Hudalla, Commun. Chem., 2020, 3, 172 CrossRef CAS PubMed.
- A. Kumagai and T. Endo, Cellulose, 2021, 28, 259–271 CrossRef CAS.
- N. Kummer, C. E. Giacomin, P. Fischer, S. Campioni and G. Nyström, J. Colloid Interface Sci., 2023, 641, 338–347 CrossRef CAS PubMed.
- E. J. Foster, R. J. Moon, U. P. Agarwal, M. J. Bortner, J. Bras, S. Camarero-Espinosa, K. J. Chan, M. J. D. Clift, E. D. Cranston, S. J. Eichhorn, D. M. Fox, W. Y. Hamad, L. Heux, B. Jean, M. Korey, W. Nieh, K. J. Ong, M. S. Reid, S. Renneckar, R. Roberts, J. A. Shatkin, J. Simonsen, K. Stinson-Bagby, N. Wanasekara and J. Youngblood, Chem. Soc. Rev., 2018, 47, 2609–2679 RSC.
- S. Arola, Z. Kou, B. J. M. Rooijakkers, R. Velagapudi, M. Sammalkorpi and M. B. Linder, Cellulose, 2022, 29, 6109–6121 CrossRef CAS.
- B. Nazari, V. Kumar, D. W. Bousfield and M. Toivakka, J. Rheol., 2016, 60, 1151–1159 CrossRef CAS.
- L. Geng, X. Peng, C. Zhan, A. Naderi, P. R. Sharma, Y. Mao and B. S. Hsiao, Cellulose, 2017, 24, 5417–5429 CrossRef CAS.
- S. Sumari, A. Roesyadi and S. Sumarno, Scientific Study and Research: Chemistry and Chemical Engineering, Biotechnology, Food Industry, 2013, 14, 229–239.
- B. Mazela, W. Perdoch, B. Peplińska and M. Zieliński, Materials, 2020, 13, 5274 CrossRef CAS PubMed.
- D. Y. Hoo, Z. L. Low, D. Y. S. Low, S. Y. Tang, S. Manickam, K. W. Tan and Z. H. Ban, Ultrason. Sonochem., 2022, 90, 106176 CrossRef CAS PubMed.
- A. Bertolani, L. Pirrie, L. Stefan, N. Houbenov, J. S. Haataja, L. Catalano, G. Terraneo, G. Giancane, L. Valli, R. Milani, O. Ikkala, G. Resnati and P. Metrangolo, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- M. C. Li, Q. Wu, R. J. Moon, M. A. Hubbe and M. J. Bortner, Adv. Mater., 2021, 33, 2006052 CrossRef CAS PubMed.
- A. A. Kermani and A. A. Kermani, FEBS J., 2021, 288, 5788–5804 CrossRef CAS PubMed.
- A. Marchetti, A. Pizzi, G. Bergamaschi, N. Demitri, U. Stollberg, U. Diederichsen, C. Pigliacelli and P. Metrangolo, Chem. – A Eur. J., 2022, 28, e202104089 CrossRef CAS PubMed.
- P. E. Schneggenburger, A. Beerlink, B. Worbs, T. Salditt and U. Diederichsen, Chem. Phys. Chem., 2009, 10, 1567–1576 CrossRef CAS PubMed.
- A. Fall, M. Henriksson, A. Karppinen, A. Opstad, E. B. Heggset and K. Syverud, Cellulose, 2022, 29, 7649–7662 CrossRef CAS.
- K. J. De France, N. Kummer, Q. Ren, S. Campioni and G. Nyström, Biomacromolecules, 2020, 21, 5139–5147 CrossRef CAS PubMed.
- S. Maity, P. Kumar and D. Haldar, Soft Matter, 2011, 7, 5239 RSC.
- N. H. C. S. Silva, P. Garrido-Pascual, C. Moreirinha, A. Almeida, T. Palomares, A. Alonso-Varona, C. Vilela and C. S. R. Freire, Int. J. Biol. Macromol., 2020, 165, 1198–1210 CrossRef CAS PubMed.
- H. Nadeem, M. Dehghani, S. Miri, M. Pazirofteh, G. Garnier and W. Batchelor, Cellulose, 2023, 30, 5157–5170 CrossRef CAS.
- J. Zhang and J. P. Youngblood, ACS App. Polym. Mater., 2023, 5, 5696–5706 CrossRef CAS.
- Z. Habib, M. Song, S. Ikram and Z. Zahra, Pollutants, 2024, 4, 136–152 CrossRef.
- N. B. Erdal and M. Hakkarainen, Biomacromolecules, 2022, 23, 2713–2729 CrossRef CAS PubMed.
- A. Cherpinski, S. Torres-Giner, J. Vartiainen, M. S. Peresin, P. Lahtinen and J. M. Lagaron, Cellulose, 2018, 25, 1291–1307 CrossRef CAS.
- W. Li, S. Wang, W. Wang, C. Qin and M. Wu, Cellulose, 2019, 26, 3271–3284 CrossRef CAS.
- R. T. Mackin, K. R. Fontenot, J. V. Edwards, N. T. Prevost, C. Grimm, B. D. Condon, F. Liebner, J. H. Jordan, M. W. Easson and A. D. French, Cellulose, 2022, 29, 1293–1305 CrossRef CAS.
- R. Shogren, J. Environ. Polym. Degrad., 1997, 5, 91–95 CrossRef CAS.
- P. K. Kunam, D. Ramakanth, K. Akhila and K. K. Gaikwad, Biomass Conv. Bioref., 2022, 12637–12652 Search PubMed.
- G. Tedeschi, S. Guzman-Puyol, L. Ceseracciu, J. J. Benitez, L. Goldoni, A. Koschella, T. Heinze, G. Cavallo, V. Dichiarante, G. Terraneo, A. Athanassiou, P. Metrangolo and J. A. Heredia-Guerrero, Carbohydr. Polym., 2021, 271, 118031 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01359j |
| ‡ A. M. and E. M. contributed equally to this work by realizing all experiments. |
| This journal is © The Royal Society of Chemistry 2024 |