
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular chemistry of liquid–liquid extraction
Sourav
Pramanik
,
Abu S. M.
Islam
 ,
Iti
Ghosh
and
Pradyut
Ghosh
*
,
Iti
Ghosh
and
Pradyut
Ghosh
*
School of Chemical Sciences, Indian Association for the Cultivation of Science, Kolkata 700032, India. E-mail: icpg@iacs.res.in
First published on 30th April 2024
Abstract
Liquid–Liquid Extraction (LLE) is a venerable and widely used method for the separation of a targeted solute between two immiscible liquids. In recent years, this method has gained popularity in the supramolecular chemistry community due to the development of various types of synthetic receptors that effectively and selectively bind specific guests in an aqueous medium through different supramolecular interactions. This has eventually led to the development of state-of-the-art extraction technologies for the removal and purification of anions, cations, ion pairs, and small molecules from one liquid phase to another liquid phase, which is an industrially viable method. The focus of this perspective is to furnish a vivid picture of the current understanding of supramolecular interaction-based LLE chemistry. This will not only help to improve separation technology in the chemical, mining, nuclear waste treatment, and medicinal chemistry sectors but is also useful to address the purity issue of the extractable species, which is otherwise difficult. Thus, up-to-date knowledge on this subject will eventually provide opportunities to develop large-scale waste remediation processes and metallurgy applications that can address important real-life problems.
 Sourav Pramanik | Sourav Pramanik received his Bachelor's and Master's degrees in chemistry from Vidyasagar University, Midnapore, West Bengal. Thereafter, he joined the Indian Association for the Cultivation of Science as a Research Scholar in 2020 under the supervision of Prof. Pradyut Ghosh. He is currently working on anion chemistry, in particular, anion recognition and separation following various supramolecular interactions. |
 Abu S. M. Islam | Dr Abu S. M. Islam received his M.Sc. in inorganic chemistry from the Department of Chemistry, West Bengal State University, India. Later he obtained his PhD from Jadavpur University, under the supervision of Prof. Mahammad Ali in 2019. During his PhD, he worked on detection and bio-imaging for small biomolecules like nitric oxide (NO), HSNO (S-nitrosothiol), and thiols (S2−/SH−). Currently, he is a postdoctoral fellow at the Indian Association for the Cultivation of Science under the supervision of Prof. Pradyut Ghosh. His research interests include designing and executing chemical reactions for chalcogen bond based receptors for anion recognition as well as extraction. |
 Iti Ghosh | Iti Ghosh completed her B.Sc. in chemistry from Bethune College under the University of Calcutta. In 2022, she obtained her M.Sc. degree in chemistry from the Indian Association for the Cultivation of Science and in the same year she started her research career under the guidance of Prof. Pradyut Ghosh, working on anion recognition and separation by halogen and chalcogen bond-based receptors. |
 Pradyut Ghosh | Pradyut studied at the Indian Institute of Technology Kanpur for a PhD with Parimal K. Bharadwaj and did his first postdoctoral work in the US at Texas A&M University with Richard M. Crooks during 1998–2000. He was an Alexander von Humboldt Fellow at the University of Bonn and Freie Universität Berlin in Fritz Vögtle's and Christoph Schalley's groups. Upon his return to India, he joined CSMCRI, Bhavnagar and in 2007 he moved to the Indian Association for the Cultivation of Science (IACS), Kolkata as an Associate Professor. In 2011 he was promoted to Professor of Chemistry and he was Head, Department of Inorganic Chemistry (2012–2015), Associate Dean (2015–2018) and Chair, School of Chemical Sciences (2018–2021) at IACS. Pradyut's present research interests are recognition, extraction and chemical sensing of ions of environmental and biological relevance, catalysis and interlocked molecules. |
1. Introduction
Supramolecular chemistry is a branch of chemistry that focuses on the study of the design of molecular assemblies with smaller building units held together by non-covalent interactions.1 These interactions include hydrogen bonding, π–π stacking, van der Waals forces, and other weak forces that mainly play a pivotal role in the assembly of molecules.2 The term “supramolecular” emphasizes the scale of these structures, which is beyond the individual molecular level. Over the past few decades, supramolecular chemistry has held significance across diverse fields, including biology, medicine, materials science, and the separation and purification processes essential for environmental remediation.1,3In the realm of purification techniques, liquid–liquid extraction (LLE) stands out as a conspicuous application of supramolecular chemistry, associated with the separation of hazardous substances from the environment and the extraction of valuable metals from ores. LLE, commonly known as solvent extraction, is a separation technique that entails transferring a solute from one solvent (diluent), typically water, to an immiscible organic solvent (extractant). Such extraction is a widely employed technique in chemical labs and industry for the isolation and purification of desired compounds from mixtures.
The significance of supramolecular chemistry in LLE lies in its ability to tailor non-covalent interactions to achieve selective and efficient extraction processes. In this dynamic field, researchers explore the development of supramolecular host–guest systems, where host molecules encapsulate guests through manipulating non-covalent interactions. These host–guest interactions can be exploited in LLE processes to enhance the separation of target compounds based on size, shape, or chemical properties.4 Furthermore, the decomplexation or back-extraction of the extracted species could offer a reusable receptor. Additionally, supramolecular assemblies, by forming micelles and vesicles,5,6 can also be utilized to encapsulate and transport specific molecules through liquid phases, contributing to the efficiency of extraction processes. From the industrial aspect, solvent extraction signifies the viability of this easily accessible LLE technique in terms of lower energy consumption as well as high production capacity.7,8 Besides, in the metallurgy section, highly precious metals like gold (Au) and platinum (Pt) can be recovered from their metalate anions using solvent extraction.9
This extraction method has made significant strides in separation and purification during the past few decades, although alternative approaches based on solid–liquid extraction, selective precipitation, and crystallization are also important.4,10,11 Many research groups have reviewed the field with a focus on supramolecular interactions but these are in specific areas including (i) anion extraction by both SLE (Solid–Liquid Extraction) and LLE methods;4 (ii) extraction of ion pairs;12 and (iii) supramolecular systems along with macrocycle-based extractants for their industrial and environmental applications.12–16 However, complete coverage of LLE based on supramolecular interactions has not been discussed in other reviews published on a similar topic.
In this perspective, we will highlight the up-to-date (2023) efforts which have been made by researchers for the progress of host molecules capable of recognizing as well as extracting inorganic guests from the aqueous phase employing LLE. Particularly, the emphasis of this review will focus on the non-covalent supramolecular interactions used for the LLE process that include LLE of halides, oxo-anions, alkali metals and ion-pairs. Finally, we will discuss the problems and promises associated with the structural variety of the hosts influencing the extraction behaviour.
2. Principles of supramolecular LLE
Parallel to the simple extraction method, the fundamental principles underlying supramolecular LLE involve concepts such as the distribution coefficient and phase equilibrium.From a supramolecular aspect, after agitating two solvents, some fraction of the solute or guest molecule (G) from the aqueous phase is shifted to the organic phase by forming the host–guest complex (HG) (Fig. 1). Such shifting of the guest is driven by host–guest non-covalent interactions, for instance, hydrogen bonding, π-interactions, hydrophobic interactions, etc.17–22 Transfer of the guest by the host molecule (H) is achieved at the interface of two solvents by the following equilibrium:
| n[G]aq + n[H]org ⇌ [HnGn]org | (1) |
| E = [G]org/[G]T | (2) |
| D = [G]org/[G]aq | (3) |
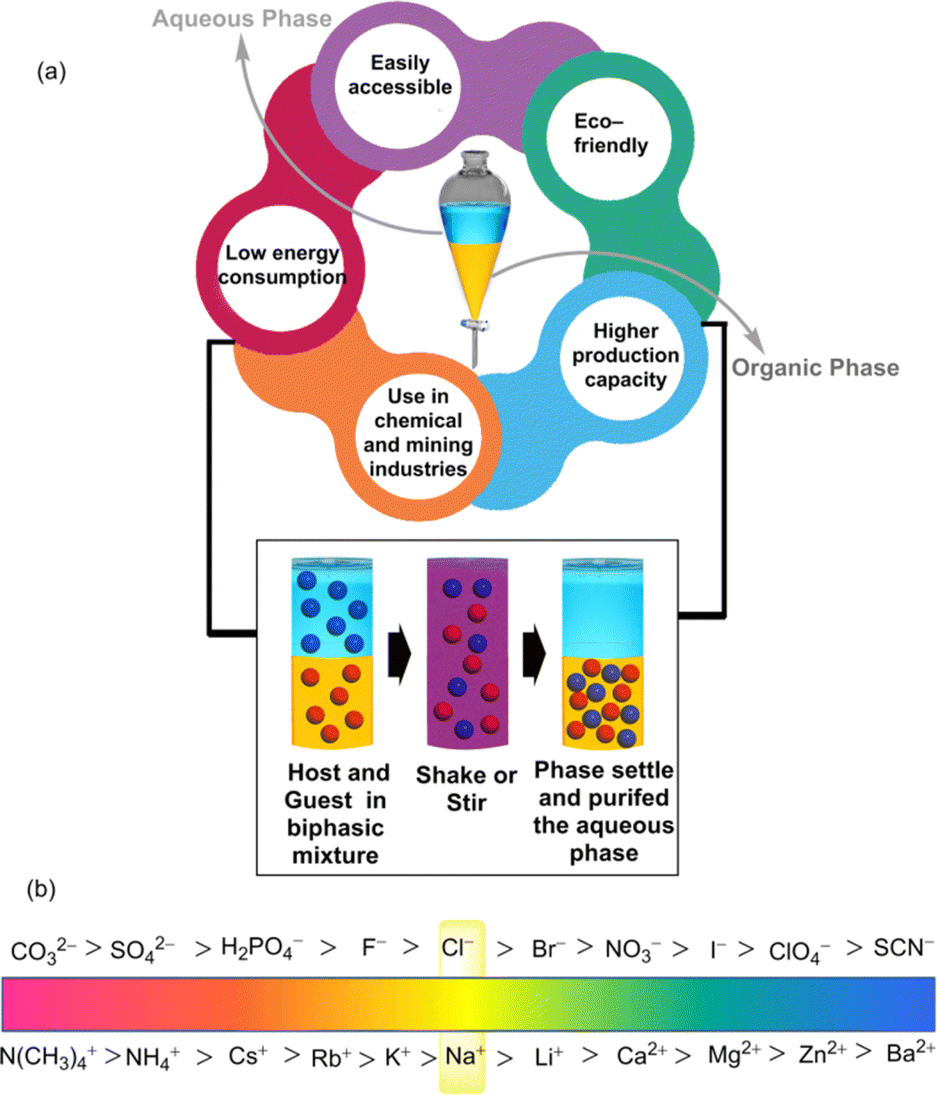 | ||
| Fig. 1 (a) Schematic illustration of the liquid–liquid extraction process. (b) Conceptual sketch of the Hofmeister series. | ||
Furthermore, assuming the interactions like hydrolysis and other complex formations with the host molecules are negligible, from eqn (1) the extraction equilibrium constant (Kex) can also be evaluated by:
| Kex = [HnGn]org/([G]aqn × [H]orgn) | (4) |
log(D) = log(Kex) + n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log[H]org log[H]org | (5) |
This equation (eqn (5)) is a useful guide for interpreting the stoichiometry for host–guest complexation during the extraction. Importantly, to achieve efficient separation, phase equilibrium plays a crucial role, where the rate of transfer of guest (G) between the two phases becomes constant, indicating that the distribution of the G between the phases has reached a balance. Hence, at equilibrium, the chemical potential of the G is the same in both phases. This equilibrium is driven by the association and dissociation kinetics of the host–guest complexes. Furthermore, during host–guest complexation, counterions have to be taken into account in the extraction process for the distribution of guests in the two immiscible aqueous and organic phases. In the LLE process, the counterions can compete with the guests for binding to the host, thereby affecting the distribution ratio and complicating the extraction process. Moreover, to maintain the electrical neutrality of the solution, the host–guest complex must be accompanied by the counterions into the organic phase during extraction. Hence, the counterions' affinity to the organic solvent impacts on the distribution ratio.23,24 Alongside, the hydrophobic ions are surface active and adsorb at the interface of two immiscible solution phases.25 Additionally, these ions exhibit a preference for partitioning into the organic phase due to their greater solubility in the organic phase compared to water. Consequently, this helps to maintain charge neutrality in the solution, facilitating the transfer of guests into the organic phase. Moreover, it also reduces the interference with the guests on extraction into the aqueous phase, leading to an enhancement in the selectivity and efficiency of the extraction process.
Supramolecular LLE relies on the above principles which are shaped by the specificities of host–guest interactions. Such interactions, governed by the design of the supramolecular hosts, dictate the selectivity and efficiency of the extraction process in these systems.
3. Supramolecular interactions in LLE
Hydrogen bonding is a common supramolecular interaction that involves the attraction between a hydrogen atom covalently bonded to an electronegative atom and a Lewis base having a lone pair of electrons. In this regard, amino (NH) and hydroxyl (OH) groups are widely employed as hydrogen bond donors.4 Moreover, the polarised CH bonds of heteroaromatic rings such as in imidazoles and triazoles can also offer excellent hydrogen bonding with the guest of interest.26 To date, the majority of supramolecular receptors comprising this non-covalent interaction have been extensively utilized for LLE.In addition, LLE may also be assisted by a Lewis base interaction with an electrophilic region or ‘σ-hole’ associated with halogen or chalcogen atoms in a molecule.27 In the last decade, these σ-hole based non-covalent interactions have attracted attention in supramolecular chemistry due to their beneficial characteristics such as high directionality and hydrophobicity compared to hydrogen bonding. Nonetheless, these interactions are scarcely utilized for the LLE process.
In supramolecular chemistry, aromatic π-systems are considered crucial building units due to their ability to non-covalently interact with several functional groups as well as ionic guests. In LLE, solutes with aromatic structures such as amino acids28 and phenols or polycyclic aromatic hydrocarbons29 can engage in π–π interactions, influencing their distribution between the aqueous and organic phases. Sometimes, electron-rich anions including SO42− and NO3− can undergo anion–π interaction with electron-deficient aromatic rings which can improve the LLE process of hydrophilic anions contrary to the Hofmeister series. On the other hand, electron-rich aromatic rings can lead to formation of cation–π interaction which allows cations typically alkali and alkaline earth metal cations to be extracted under LLE conditions.
Hence, to optimize the LLE processes, it is essential to understand these supramolecular interactions. Moreover, for the extraction process, influencing these interactions, as well as enhancing the efficiency, requires careful consideration of factors such as the choice of extraction solvent, the addition of phase modifiers, and the control of pH. Researchers often tailor these interactions to achieve the desired separation of components in a mixture during LLE.
4. Design of supramolecular extractants
In the supramolecular LLE technique, strongly hydrated inorganic cations and anions are the main solutes of interest. The partition of these ions at the interface of organo-aqueous solvents follows the Hofmeister series which orders ions according to their hydrophobicity and suggests that more hydrophobic ions are easier to extract into the apolar solvent (Fig. 1b).30 This highlights the importance of developing hydrophobic receptors with adequate recognition sites that can overcome the additional energy barrier for hydrated ions.Furthermore, for the anionic guests, suitable phase transfer is accompanied by maintaining the charge-neutrality of the LLE system under experimental conditions. Thus, positively charged receptors may give a boost to coulombic interactions with the anions, which allows the formation of a neutral anion–receptor complex that can be more intrinsically soluble in an organic phase.31 Additionally, charged receptors are also able to eliminate the necessity of transporting the counterion into the organic phase. Thus, this approach has aided in the recognition and extraction of polyvalent oxo-anions such as sulphate, phosphate, dichromate etc. Alongside, using the neutral receptors, it is difficult to transport anionic species from an aqueous environment to a nonpolar medium because strong interactions are observed between water and charge-dense anions. Therefore, receptors must be designed to consist of a large number of binding sites to overcome the hydration effects. In some cases, counterions might also be added to the neutral receptors, which can modulate the extraction process by balancing charge neutrality. Thus, it is convenient for anion extraction through a neutral receptor in terms of ion-pair recognition. Furthermore, a common challenge arises in extracting anions using monotopic receptors in the presence of counterions. This is primarily attributed to the significant energy expenditure required to separate individual anions from their respective counterions.
Therefore, such difficulties can be reduced by developing ion-pair receptors, which provide space for both cations and anions. Additionally, one prebound ion induces the binding of its counterion through a cooperative effect. Such cooperative binding is explained by electrostatic interaction between the bound ions and/or allosteric conformational changes during the binding of the ion-pair. These ion-pair receptors have widely been used as metal salt extractants and solubilizing agents as well as transporters through membranes.32
Anions commonly exist in different geometries in nature and sometimes in different forms depending on the pH of the solution (e.g., H2PO4−, HPO42−, PO43−). Hence, complementarity between the receptor and anion with regard to size and geometry needs to be addressed to stabilize the anion of interest for effective extraction. Thus, appropriately designed macrocyclic receptors with highly organised structures can efficiently bind a specific anion following geometric complementarity and facilitate LLE.13 In some cases, cyclodextrins or crown ethers may also pave the way for the selective extraction of specific guest anions or molecules from a solution.33 Specifically, these molecules can form inclusion complexes with guests, which facilitates the extraction process.
Additionally, controlling the solubility and amphiphilicity of the extractant or receptor is important for compatibility with the extraction system.34,35 Amphiphilic molecules can interact with both polar and non-polar components, allowing for extraction in various solvent environments. Hence, the design principles behind supramolecular extractants involve a thoughtful combination of structural features, and molecular interaction contributes to optimizing the chemistry of LLE. These design considerations are crucial for achieving high selectivity, efficiency, and adaptability in LLE processes.
In the next part, we will highlight and briefly cover the recent supramolecular hosts that have been developed for the LLE of inorganic anions, alkali metal cations and ion-pairs.
5. Removal of anions through LLE
Presently, on the basis of supramolecular hosts designed for the LLE of inorganic anions, two main types of receptors are developed: (a) charged and (b) neutral. In this section, notable examples of success using properly designed cationic or neutral receptor systems for the LLE of anions will be presented. Particularly, the discussion focuses on the design strategy and unique binding properties of receptors in the field in question.5.1. Removal of halides through LLE
The presence of halide ions in water resources can produce undesirable inorganic and organic halide-containing by-products when oxidative treatment is applied during drinking water production. For example, the chlorination or ozonation processes used to effectively kill harmful microorganisms may result in the production of iodoform, brominated phenols, bromate etc., which are potentially carcinogenic and may lead to health concerns.36 Thus, removal of such halide ions from water is important, where LLE is widely employed due to its usefulness in phase transfer applications.In this scenario, Chiu and Gabbaï reported the extraction of F− from water using a cationic borane R-1, in a biphasic mixture of D2O and CDCl3 (Fig. 2).39 The F− was captured through hydrogen bond interactions with the methylene proton of the ammonium group of R-1, resulting in the formation of a zwitterionic ammonium/fluoroborate complex. This interaction was further validated through solid-state crystal structure analysis and computational studies (Fig. 2). In the solution phase, the endurance of this C–H⋯F–B hydrogen bond was confirmed by 1H, 19F, and 11B-NMR studies in CDCl3. Nevertheless, in the presence of other halides, no evidence of such complex formation was observed. Furthermore, a UV titration conducted in THF/MeOH (75/25 v/v) revealed a higher binding constant (5 × 106 M−1) for F− compared to other halides. The ability of F− extraction by R-1 was assessed through NMR experiments, which revealed that the F− adducts were transferred to the organic phase with 82% efficiency. Notably, only the cationic functionality in the borane receptor was able to extract the F− because additional hydrogen bonding along with coulombic forces stabilizes the B–F bond against heterolysis. Therefore, these findings imply that cationic borane-based receptors have the capability to overcome the hydration enthalpy of F−.
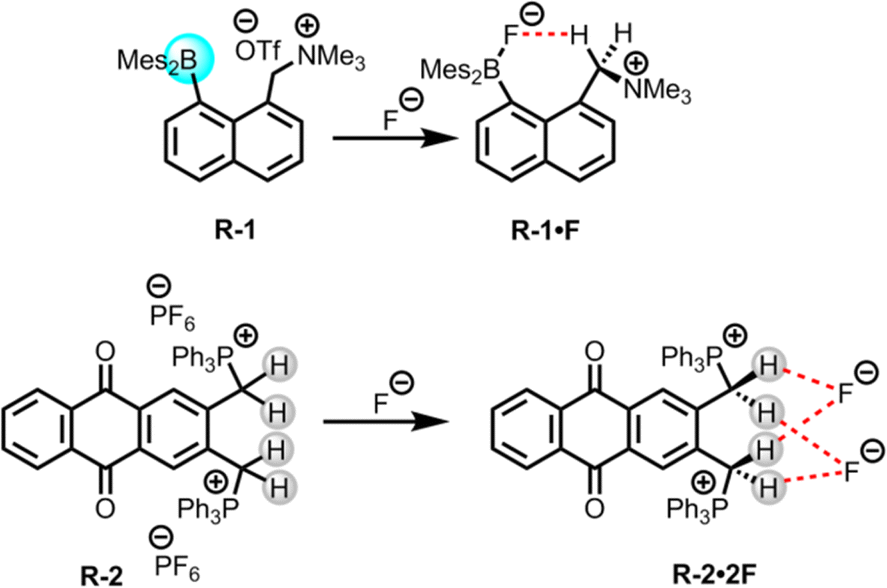 | ||
| Fig. 2 Chemical structure of the receptors R-1 and R-2. | ||
Additionally, in 2011 Das, Ganguly, and co-workers synthesized a phosphonium (PPh3+) incorporated anthraquinone-based receptor, R-2, for the selective colorimetric sensing and extraction of F− (Fig. 2).401H and 31P NMR studies of R-2 with different anions revealed that only F− was able to form a 1:2 complex with the receptor. This complex formation was assisted by the robust hydrogen bond interaction with the acidic methylene CH2 protons along with an electrostatic interaction with the cationic PPh3+ groups of the receptor, rather than via a deprotonation mechanism. Some weak interactions were also found for chloride (Cl−), bromide (Br−), and hydrogen sulphate (HSO4−), but dihydrogen phosphate (H2PO4−) exhibited somewhat stronger interactions. The calculated association constants for F− and H2PO4− were 2.24 × 106 and 8.98 × 104 M−1 respectively in acetonitrile. Hence, with a notable binding constant for F−, the receptor exhibited the capability to extract it from water into CH2Cl2 or CHCl3 with 99.3% efficiency, even at F− concentration as low as 2.3 × 10−7 M. Furthermore, the receptor also demonstrated a similar capability for colorimetric detection of F− as compared to the WHO-reported method utilizing sulphonylazodihydroxy naphthalene disulphonic acid, with a lower detection limit of 3.8 × 10−7 M. In addition, the authors successfully employed the receptor for a practical application, to extract the F− from seawater (Arabian Sea, and Sambhar Lake in India), even in the presence of high concentrations of competing anions.
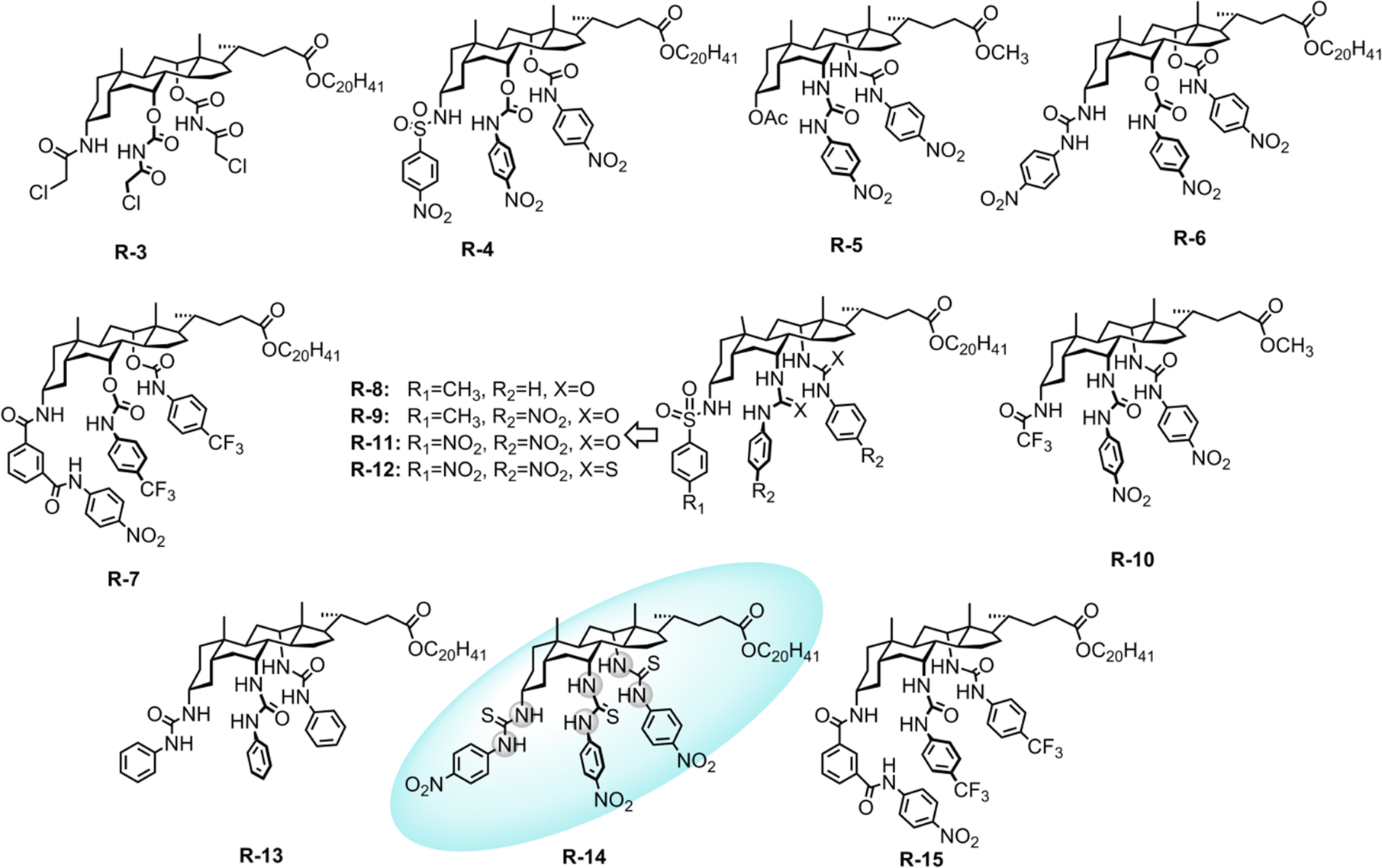 | ||
| Fig. 3 Chemical structure of the receptors R-3 to R-15. | ||
In a later study, Davis, Gale, and co-workers modified the cholapod receptors with squaramide-functionalization to increase the extraction selectivity and efficiency.43 The inclusion of highly acidic squaramide NH groups in receptor R-16 bestowed elevated affinities to anions compared to previously reported urea or thiourea-based cholapods (Fig. 4).44–48 In the case of R-16a, the apparent stability constant for Cl− was measured to be 1.2 × 1014 M−1 employing Cram's extraction method in water-saturated CDCl3. In contrast, Br−, I−, NO3−, ClO4−, and EtSO3− were seen in the range of 1010 to 1013 M−1. Expectedly, this value is even higher compared to that of R-14 for selective Cl− recognition by employing cholapod-based receptors. Therefore, the TBA salt of Cl− was readily extracted in the presence of competing monovalent anions from water and transported to the chloroform solvent. Hence, enhancing the effectiveness of hydrogen bonding in receptors can also be achieved without incorporating electrostatic interactions or Lewis-acidic metals.
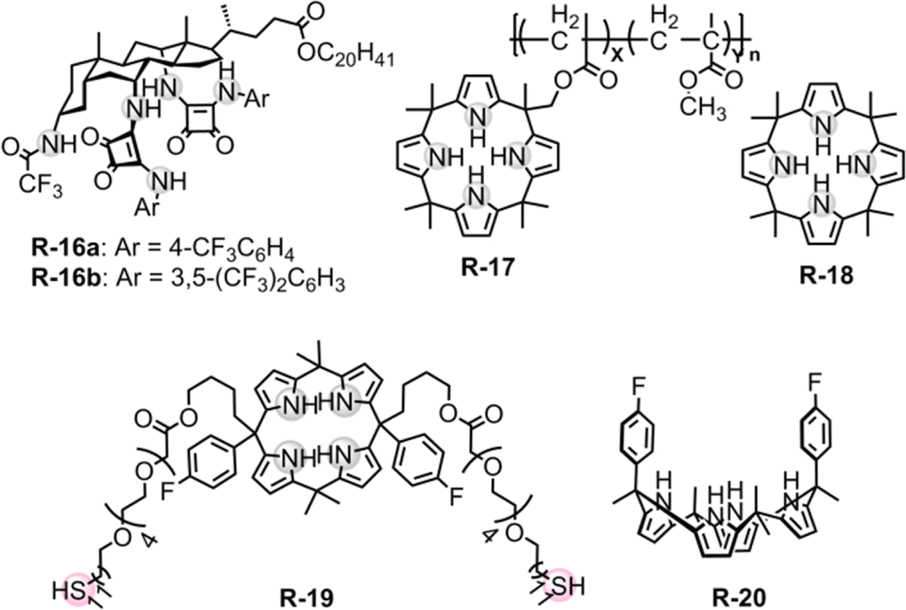 | ||
| Fig. 4 Chemical structure of the receptors R-16 and R-20. | ||
To develop soluble multivalency receptors for the LLE of halides, Sessler's group reported a calixpyrrole-functionalized organo-soluble polymeric receptor R-17 (Fig. 4).49 Prototypical copolymer R-17 was effectively able to extract Cl− and F− from D2O to CD2Cl2. The extraction ability was assessed through 1H-NMR studies and thermogravimetric analysis. The 1H-NMR studies revealed a downfield chemical shift of the pyrrole NH protons upon the addition of Cl− and F−, indicating strong hydrogen bond formation. For R-17, extraction efficiency was higher for Cl− than F− or H2PO4−, which is in accord with Hofmeister bias. Interestingly, octamethylcalix[4]pyrrole R-18 failed to extract F− from water, and Cl− extraction efficiency was also considerably lower compared to that of R-17. These results indicate that polymeric receptors enhance the capability for anion extraction through multivalency, while also enabling the tuning of the solubility, stability, and other properties of the extracting agent.
Inspired by the above results Lee et al. constructed a gold (Au) nanoparticle decorated with double-armed calix[4]pyrrole receptor R-19, which selectively recognized F− with an association constant of (1.43 ± 0.16) × 108 M−1 in CH2Cl2 (Fig. 4).50 The core anion recognition subunit of the receptor was calix[4]pyrrole. Additionally, for gold binding, a flexible double anchoring with two thiol-bearing alkyl chains was incorporated. The effective binding between R-19 and F− was established by the 1H-NMR studies. A substantial chemical shift of the pyrrole-NH protons occurred, indicative of the formation of N–H⋯F hydrogen bonds. The advantage of multivalency of the receptor further enhanced the effective extraction of highly hydrated F− from water into CH2Cl2. In contrast, the analogous monomeric calixpyrrole (R-20) failed to extract anions under comparable conditions.
A similar result was reported by Akar and Aydogan, based on the tricalix[4]pyrrole and pentacalix[4]pyrrole receptors R-21 to R-24 (Fig. 5).51 These oligomeric calixpyrrole receptors were capable of extracting the TBA salt of halide ions from a water medium. The extraction capacity was found to be dependent on the number of donor sites present in the receptors. Both R-21 and R-22 exhibited the ability to selectively extract Cl− from an aqueous phase into CH2Cl2. In contrast, pentacalix[4]pyrrole receptors R-23 and R-24 not only extracted the Cl− but were also capable of extracting F−. To visualize the Cl− extraction by the receptors under interfacial aqueous–organic conditions a water-soluble dye, Basic Blue 17, was utilized where Cl− was present as a counter ion (Fig. 6).52 The result revealed that R-24 achieved the highest efficiency, with a remarkable 94% extraction compared to other receptors. Furthermore, the enhanced extraction ability of R-24 was attributed to the cooperative effect of the number of individual calixpyrrole units within its structure, which was established through NMR and thermogravimetric analyses.
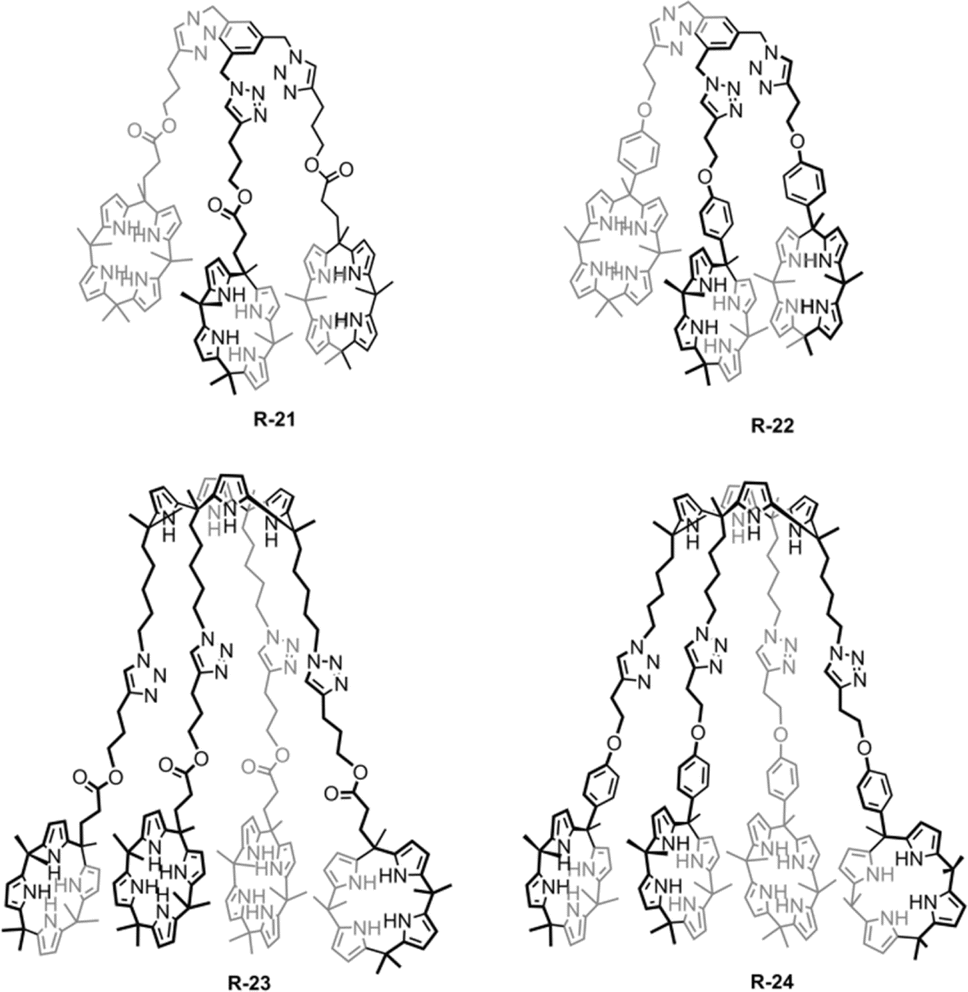 | ||
| Fig. 5 Chemical structure of the receptors R-21 and R-24. | ||
 | ||
| Fig. 6 Visualization of the Cl− extraction by the receptors R-21 to R-24 using Basic Blue 17 (top layer: aqueous solution, bottom layer: CH2Cl2). (Reproduced with permission from ref. 51. Copyright 2018, Wiley.) | ||
Along these lines, in 2018, Jang et al. demonstrated triazole containing calix[4]pyrrole based cis and trans receptors R-25 and R-26 respectively (Fig. 7).53 In comparison to the parent calix[4]pyrrole, both the receptors have effective Cl− binding affinities relative to Br− and I− with binding constants of 1.1 × 105 and 2.3 × 104 M−1 for cis and trans respectively in CDCl3/CD3CN (9/1, v/v) solution. The high binding affinity was rationalised by robust hydrogen bond formation between four pyrrolic N–H and two triazole C–H protons of the receptors. Notably, upon binding with Cl−, 1H-NMR studies demonstrated that the contribution of the C–H⋯X hydrogen bonding interaction was significantly greater in R-26 compared to R-25. This led to a higher binding constant for the trans receptor. The DFT study further unveiled that both receptors interacted with Cl− through cone-shaped conformations. Parenthetically, it is worth noting that both the receptors were able to transfer Cl− into CH2Cl2 from water-soluble Basic Blue 17 solutions.
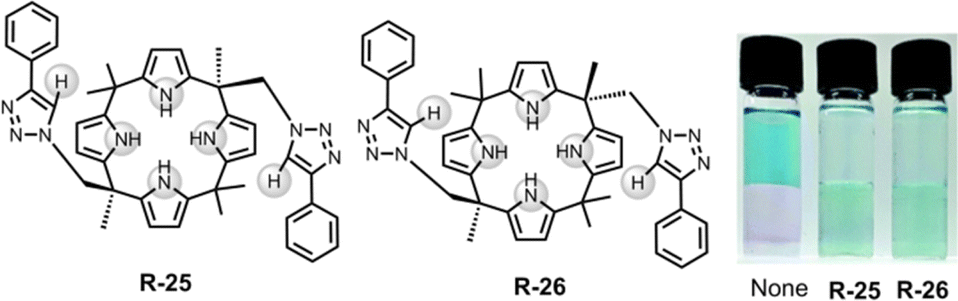 | ||
| Fig. 7 Chemical structure of the receptors R-25 and R-26 and visualization of the CH2Cl2 phase after extraction of Cl− using Basic Blue 17 through R-25 and R-26. (Reproduced with permission from ref. 53. Copyright 2018, The Royal Society of Chemistry.) | ||
Flood and co-workers reported the utility of a polymeric backbone containing receptors R-27 and R-28 for the extraction of Cl− from water through C–H⋯Cl hydrogen bond formation (Fig. 8).54 The two triazole units of R-27 adopted both syn and anti-conformations across the benzene ring, making R-27 an unorganised receptor. However, R-28 exhibited a single dominant conformation, which is attributed to intramolecular hydrogen bond interactions. This led to a preorganized binding site for the anion (Fig. 8). Consequently, 1H-NMR revealed that the Cl− binding affinity was higher for R-28 compared to R-27. Furthermore, R-28 was able to extract Cl− from water into CH2Cl2. It was inferred that the high binding and extraction ability was achieved through the effective hydrogen bond formation with the strongly polarized triazole C–H protons.
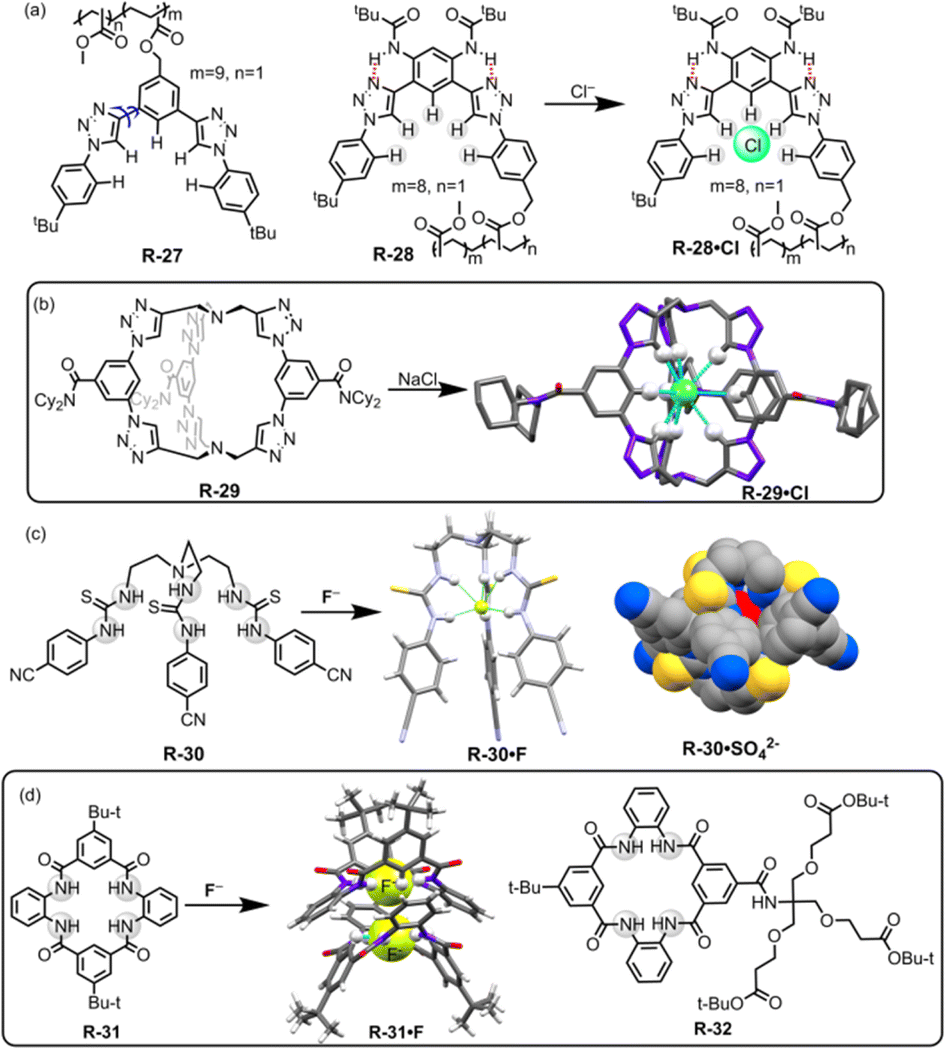 | ||
| Fig. 8 (a) Chemical structures of R-27 and R-28 (blue arrows indicate the bond rotation). (b) Cl− encapsulation inside the receptor R-29. (Data from the Cambridge Crystallographic Data Centre [CCDC # 1533500].26) (c) Chemical structures of the receptors R-30 and crystal structures of R-30·F and R-30·SO42−. (Data from the Cambridge Crystallographic Data Centre [CCDC # 875998 and 875999 respectively].55) (d) Chemical structures of R-31 and 32 and crystal structure of R-31·F. (Data from the Cambridge Crystallographic Data Centre [CCDC # 1887996].56) | ||
In continuation of this work, recently Flood, Liu, and co-workers developed a bicyclic cryptand R-29 with six CH hydrogen-bond donors of triazole groups, which specifically recognized Cl− with a record-breaking attomolar binding affinity (1017 M−1) in wet CH2Cl2 and high nanomolar affinity (108 M−1) in DMSO.26 The X-ray crystal structure of the receptor anion complex (R-29·Cl) suggested that the Cl− is encapsulated in the cavity of the R-29 and stabilized by the nine C–H⋯Cl hydrogen bonds (Fig. 8). Therefore R-29 was employed for the extraction of Cl− from water into CH2Cl2, achieving up to ∼90% efficiency. In addition, larger anions such as Br−, NO3−, and I− showed lower binding affinity towards R-29, indicating that the receptor displayed anti-Hofmeister behaviour. The receptor was further applied for corrosion inhibition and the result revealed that a film of R-29 could protect mild steel from corrosion in a brine solution (5.6 M NaCl). This report is undoubtedly a turning point in supramolecular chemistry for the extraction of Cl− that may provide insights into industrial waste purification.
Our group introduced thiourea-based 4-cyanophenyl-substituted tripodal receptor R-30 for binding of anions both in solution and solid phase and further exploited it for LLE from water (Fig. 8).55 Isothermal titration calorimetric (ITC) studies revealed that R-30 exhibited high binding affinity towards spherical F− with a binding constant of 1.1 × 107 M−1 in CH3CN compared to other tested tetrahedral oxo-anions (H2PO4− and HSO4−). NMR and crystal structure analysis indicated that the F− was encapsulated inside the cavity of the tripodal receptor through the formation of six N–H⋯F hydrogen-bonding interactions, resulting in a 1:1 complex with distorted trigonal-prismatic geometry. In typical LLE experiments, a CHCl3 solution of R-30 was layered with an aqueous solution of KF containing TBAI as a phase transfer agent, giving rise to an extraction efficiency of ∼70% based on the pure extracted mass. Additionally, R-30 was also able to extract tetrahedral sulphate with an efficiency of ∼42% by forming a 2:1 tight dimeric capsular assembly. Hence, encapsulating the anions, suitably designed receptors can form a capsular assembly via a supramolecular self-assembly process and thereby enhance the extraction efficacy of hydrated anions from competing bulk media.
Recently, Smith and co-workers reported symmetric, neutral tetralactam macrocycle R-31 containing four amide groups that exhibited anion selectivity towards F− (Fig. 8).56 In the solution phase, 1H-NMR titration for F− demonstrated the formation of a 2:1 (host:guest) complex formed with high association constants (K1 = (5.3 ± 0.8) × 104 M−1, K2 = (3.1 ± 0.7) × 104 M−1 in DMSO-d6) compared to other tested anions. The solid-state structure of the F− complex (R-31·F) disclosed that the F− occupied the tetralactam cavity through the formation of hydrogen bonds with the four amidic –NH and two –CH protons. Moreover, the complex also formed a “saddle-shaped” lattice packing as a self-complementary dimer. Inspired by such effective interactions of F− inside the tetralactam cavity, the authors incorporated highly lipophilic groups into that macrocycle to enhance the extraction efficiency of F− from an aqueous solution. Therefore, the macrocycle R-32 was able to extract F− from water to the CH2Cl2 phase, even at a low millimolar concentration. Nevertheless, when a mixture of equimolar concentrations of F− and Cl− was present, 1H and 19F NMR indicated that the extraction efficiency of F− was only 4% while for Cl− it became 96%. This discrepancy was attributed to the significant hydration energy difference between F− and Cl−. Therefore, in an environment where there is a highly common occurrence of Cl− the extraction of F− becomes a limitation but the receptors may be used in 18F radiolabelling technology.57 F− encapsulation with high binding affinity in the tetralactam cavity could also significantly contribute to the development of fluoride ion batteries (FIBs).58
5.2. Removal of oxo-anions through LLE
In this section, we will discuss the progress in LLE for oxo-anions through supramolecular approaches. Inorganic oxo-anions such as phosphate, sulphate, pertechnetate, and dichromate are highly toxic in the environment.59 These oxo-anions contaminate water and soils, mainly through industrial operations and from electroplating. Hence, removal of such ions from the environment is very important but on account of the presence of single/double charge and high hydration enthalpies, extraction from water is difficult.60–63 Charged and neutral receptors recently reported for oxo-anion extraction are discussed below.Hence, to remove such metallic oxo-anions from a water medium, Bayrakci et al. designed tetra-substituted calix[4]arene receptors R-33 and R-34 (Fig. 9).65 These receptors demonstrated the ability to extract arsenic (H2AsO4−) from water under acidic conditions, achieving an extraction efficiency of 81.6% at pH 3.5. R-33 exhibited high extraction capabilities (Kex = 8.9 × 102 M−1), because in an acidic medium, all four pyridyl rings became protonated. This protonation facilitated the formation of a stable host–guest complex via electrostatic interactions. Additionally, the amide NH protons formed an effective hydrogen bond with the oxygen atoms of arsenate (H2AsO4−). At highly acidic pH the stability of H2AsO4− is higher than that of H2AsO42− in water. Therefore, at pH 3.5, R-33 was capable of facilitating the extraction of H2AsO4− having lower hydration energy than H2AsO42−. In the case of R-34, a relatively low 17.7% NaH2AsO4 extraction ability was observed, which was attributed to the absence of protonatable amine groups and the more electron-donating nature of the furyl groups. Indeed, R-34 behaved as a ditopic extractant and its H2AsO4− extraction ability was dependent on the binding of Na+ within the calixarene cavity. Thus, the formation of an ion-pair complex with the receptor governed the extraction of H2AsO4−. Consequently, these types of receptors offer the opportunity to design the tetra-substituted calix[4]arene architecture for anion-binding with phase-transfer catalysis.
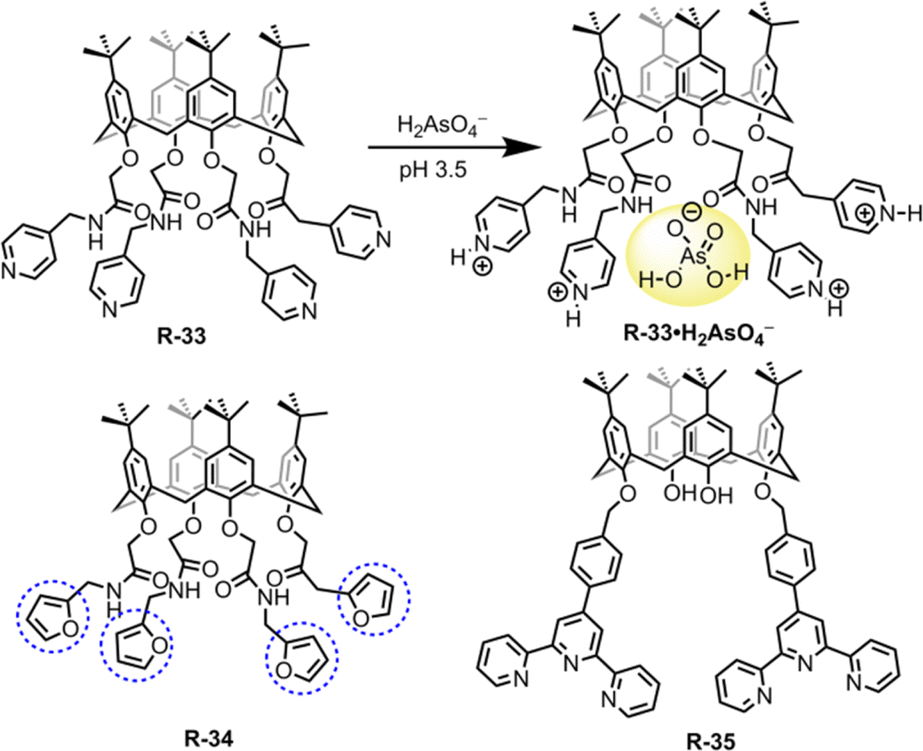 | ||
| Fig. 9 Chemical structure of the receptors R-33 to R-35. | ||
Drawing upon the aforementioned proton-switchable mechanism, Yilmaz et al. explored the terpyridin-conjugated calix[4]arene based receptor R-35 for dichromate extraction (Fig. 9).66 In the extraction process, a CH2Cl2 solution of the receptor was agitated with the aqueous solution of Na2Cr2O7 at different pH (1.5–4.5). Upon phase separation, the amount of dichromate (HCr2O7−) extracted in the organic medium was calculated using UV-Vis analysis. The obtained results revealed that the extraction efficiency depends on the acidity of the solution. Effective extraction of HCr2O7− was not observed at solution pH > 2.5. However, at pH 1.5, the extraction efficiency became ∼65% and the HCr2O7− extraction constant Kex was found to be 2.3 × 106 M−1. Furthermore, the extraction of HCr2O7− by the receptors was not affected in the presence of other competitive anions (Cl−, SO4−, and NO3−). At low pH, the selectivity and overall effectiveness of R-35 as an extractant for HCr2O7− were attributed to the presence of proton-switchable pyridine moieties (Fig. 10). This led to the formation of a cationic receptor that formed a 1:1 complex with HCr2O7− through electrostatic and hydrogen bonding interactions.
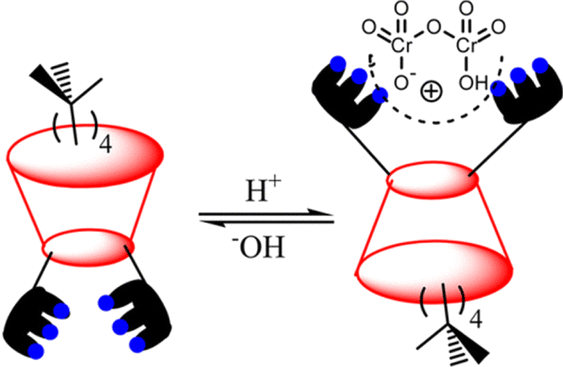 | ||
| Fig. 10 Schematic representation of interactions between the terpyridin-conjugated calix[4]arene (R-35) and HCr2O7−. (Reproduced with permission from ref. 66. Copyright 2014, ACS.) | ||
Removing the superhydrophilic sulphate (SO42−) from water is notoriously difficult due to its high hydration enthalpy (−1080 kJ mol−1).60 In this regard, Moyer et al. reported simple guanidinium-based receptors R-36 and R-37 for the separation of SO42− from an aqueous medium containing NaCl into 1,2-dichloroethane or isopar L (Fig. 11).67,68 Noticeably, isopar L is an isoparaffinic hydrocarbon solvent mainly used in industrial extraction processes. During extraction, this low cost diluent plays a significant role in adjusting the receptors' solubility. Moreover, its superior physical properties, such as lower volatility and higher flash point compared to other solvents, make it suitable for use in a radioactive waste processing environment.69,70 Furthermore, during the extraction experiment the authors utilized a phase-transfer catalyst, a lipophilic quaternary ammonium salt such as Aliquat 336 (methyltrialkyl-(C8–10)ammonium chloride), which increased the SO42− extraction efficiency by 28000-fold. It is noteworthy that when utilizing the isopar L solvent for extraction, the separation factor (SF) for SO42− over Cl− was found to be >1, which is rarely observed in the literature. The main driving force for effective binding and separation of SO42− was the coulombic interaction with the guanidinium-based receptors, along with the formation of a 1:2 complex. The SO42− distribution ratio for such a stoichiometric complex hinged on the extractant concentration which was influenced by the charge discrepancy between the SO42− and Cl− during the ion exchange process. Additionally, R-37 showed better selectivity and high extraction power compared to R-36. This was attributed to the formation of reverse-micelles of the sulphate complex of R-37 in solution, which was further supported by Karl Fischer titrations and small angle X-ray scattering experiments.
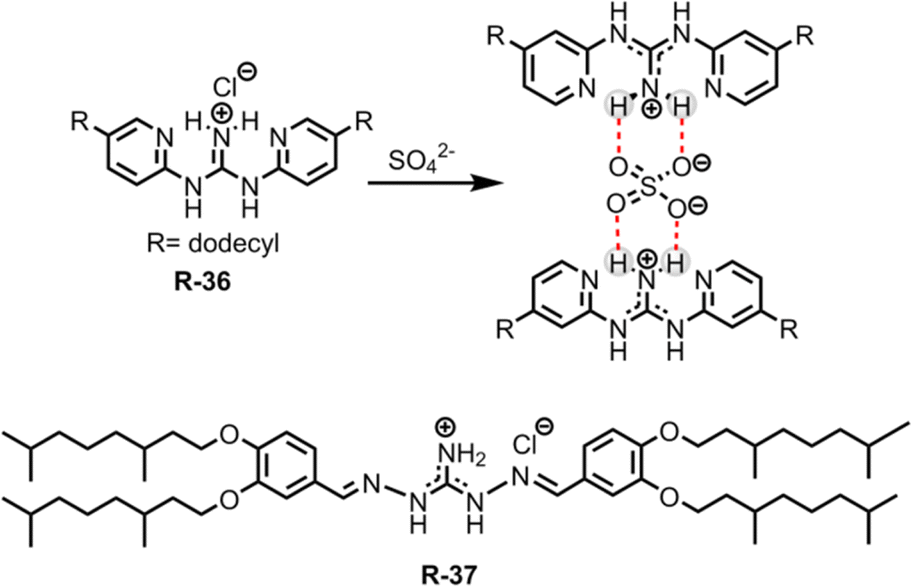 | ||
| Fig. 11 Chemical structures and SO42− binding with R-36 and R-37. | ||
The aforementioned extraction of SO42− through LLE clearly illustrated that phase-transfer catalysts such as Aliquat 336 abbreviated to A336N can play a pivotal role in overcoming the Hofmeister bias. The primary function of this catalyst involves an anion exchange mechanism between Cl− of the extractant and the targeted anion (SO42−, HPO42−etc.) of the feed solution. The structure of Aliquat 336 and reaction involved during salt exchange are depicted in Fig. 12. In addition, methyl(n-octyl,n-decyl)ammonium nitrate and N,N-bis(2-ethylhexyl)guanidine (LIX 79) also behaved as classic phase-transfer catalysts.71,72 Particularly, in the case of highly hydrated anions, like sulphate and phosphate, during LLE, a third phase is developed by this catalyst. At the critical point of phase splitting, the amount of water in the organic phase is correlated with the targeted anion's hydration energy, thereby facilitating the extraction process. Furthermore, phase transfer catalysts not only maintain the charge neutrality but also improve the stability of the targeted anion–receptor complex in less polar solvents. Nevertheless, they enhanced the exchange kinetics of LLE. Using such a phase-transfer catalyst approach Sessler and Moyer successfully reported several macrocyclic-based receptors for anion extraction.15,16,73
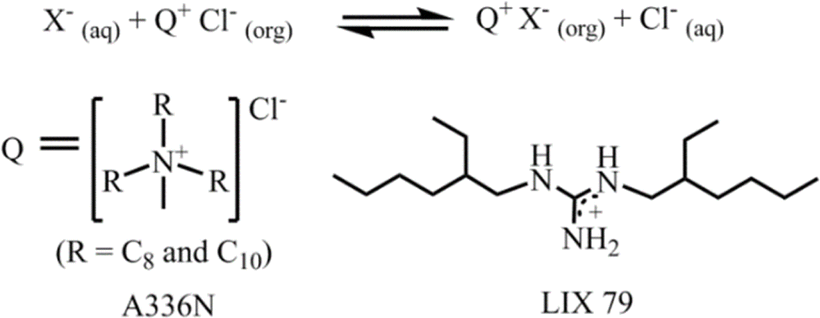 | ||
| Fig. 12 Representation of the anion exchange mechanism for univalent anions with chloride and the structures of A336N and LIX 79. | ||
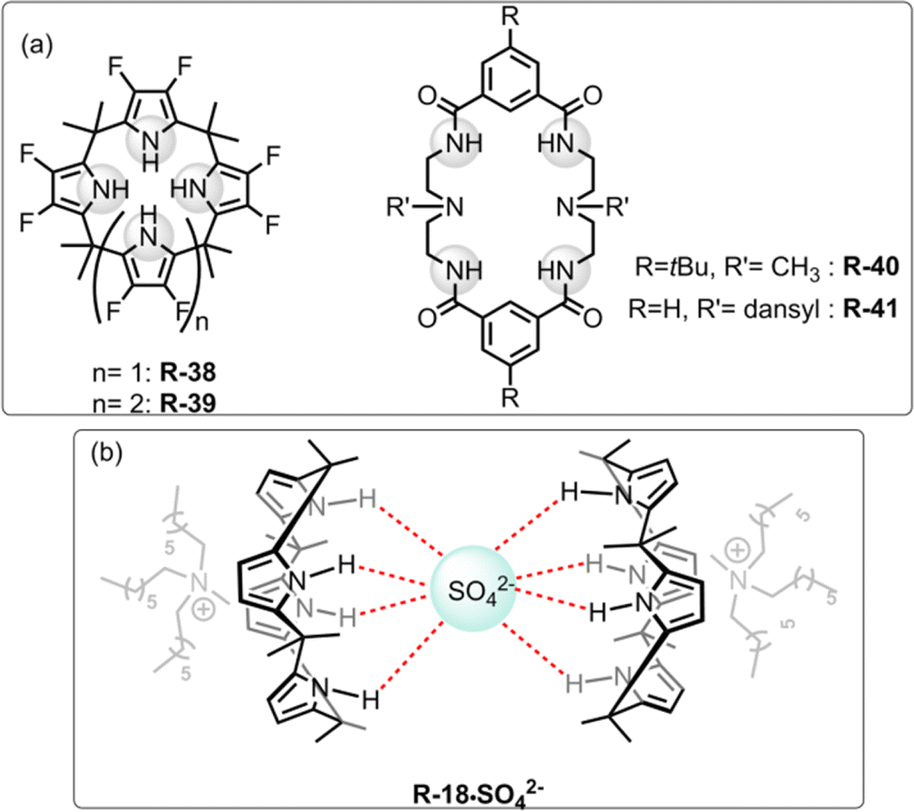 | ||
| Fig. 13 (a) Chemical structure of the receptors R-38 to R-41. (b) Representation of SO42− binding with R-18. | ||
In subsequent works,75,76 the authors described the effectiveness of adding Aliquat 336 to aid SO42− extraction by the receptor R-18, even when NaNO3 was present in excess. Interestingly, it was found that despite the higher binding capacity, R-38 showed lower efficiency for removing SO42− than R-18. This finding was observed as in the presence of Aliquat 336, R-18 acted as a ditopic receptor which helped to stabilize the R-18·SO42− complex (Fig. 13). The solid-state structure revealed that SO42− bound to the cavity through the oxygen atom while the cationic part of A336N concurrently bound within the cup-like portion of R-18. In addition, if the size of the long-chain quaternary ammonium increased the extraction efficiency decreased due to a poor fit with the “cup”.
In a further report, the same group demonstrated bipyrrole-strapped calix[4]pyrrole-based receptors R-42 and R-43 with enhanced extraction efficiency of SO42− from water (Fig. 14).77 It was also found that the SO42− extraction capacity of the receptors was significantly increased in the presence of excess NaCl, as well as upon using the co-extractant A336N. In fact, A336N stabilized the sulphate–receptor complex by the formation of C–H⋯π interactions between one of the methyl groups of A336N and the π-faces of the pyrroles. These methyl groups also concomitantly bound to the SO42− which significantly improved the ion pairing effects (Fig. 14). Hence, the formation of a higher-order complex improved the stability of SO42− in the organic phase. R-43 exhibited a higher extraction efficiency compared to R-42, with distribution ratios of 0.013 and 0.00061 respectively. The higher performance of R-43 was attributed to the fact that the divergent NH vectors of the bipyrrole subunit were able to better encapsulate the SO42− by generating strong hydrogen bond interactions with the two oxygen atoms.
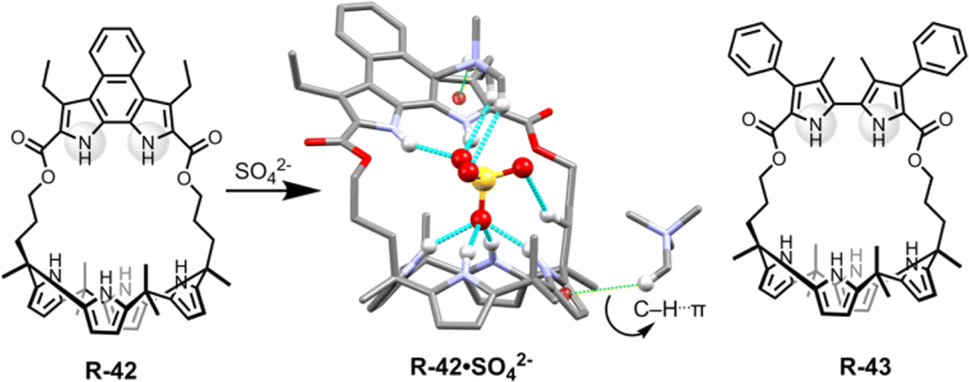 | ||
| Fig. 14 Chemical structure of the receptors R-42 and R-43 and crystal structure of R-42·SO42−. (Data from the Cambridge Crystallographic Data Centre [CCDC # 1017884 (ref. 77)].) | ||
Theoretical analysis has established that the binding of SO42− can be stabilized by the saturated 12 coordinated tetrahedral cavity within the receptor, facilitated by organized hydrogen bonds.78 In this regard, Li, Wu, and co-workers achieved hexaurea-based tripodal receptor R-44 for the effective extraction of SO42− in the presence of nitrate-rich mixtures (Fig. 15).79 The X-ray crystal structure demonstrated that the receptor was capable of encapsulating SO42− through effective hydrogen-bonding interactions with NH protons, resulting in a saturated twelve-coordinate tetrahedral geometry. Therefore, the SO42− binding ability became quite high (Ka > 104 M−1) compared to other tested anions. This finding encouraged the authors to investigate the LLE of SO42− using NMR and gravimetric techniques. 1H-NMR studies revealed that R-44 efficiently extracted the SO42− into the organic phase (CDCl3) from a nitrate-rich aqueous solution by an ion exchange method with 95% extraction efficiency. Furthermore, once extracted, BaCl2 was loaded to the organic phase for the precipitation of SO42− as BaSO4 allowing the extractant to be recycled for reuse.
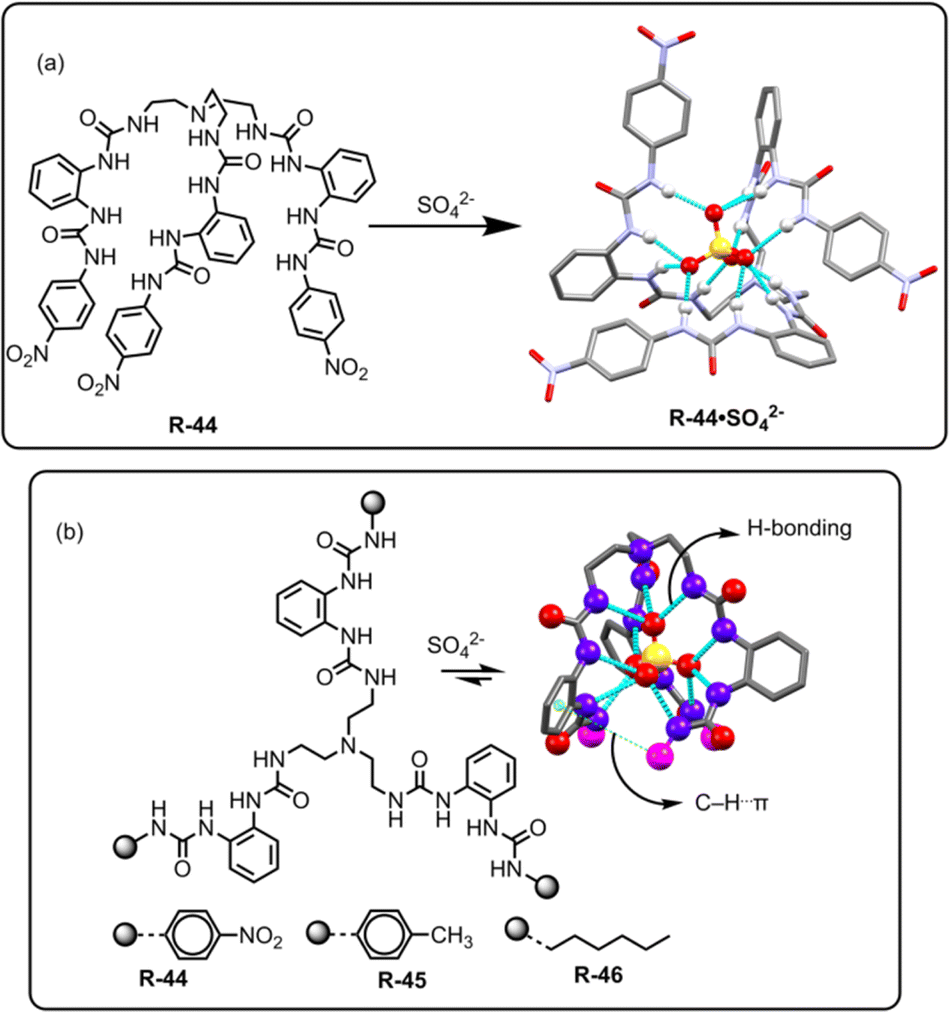 | ||
| Fig. 15 (a) Chemical structure of the receptor R-44 and crystal structure of R-44·SO42−. (Data from the Cambridge Crystallographic Data Centre [CCDC # 784518 (ref. 79)].) (b) Chemical structures of the receptors R-44 to R-46 and SO42− binding crystal structure of R-46. (Data from the Cambridge Crystallographic Data Centre [CCDC # 2206147].80) | ||
In another recent report, to explore the SO42− binding capability, the same research group of Zhao, Wu, and co-workers elucidated a class of C3 symmetric tripodal hexaurea-based receptors R-44 to R-46 for the separation of SO42− from water by the LLE method (Fig. 15).80 The receptor's affinity for SO42− binding was controlled by altering the terminal substituents present in the receptor, which contributed to the formation of robust hydrogen bonds and secondary C–H⋯π interactions. Therefore, the tendency for stronger binding as well as the better potential for SO42− removal followed the order R-44 > R-45 > R-46. To enhance the SO42− extraction efficiency of the receptors, A336N was used instead of TBACl. Furthermore, 1H-NMR and ICP-MS studies revealed that the extraction efficiency for R-44 became ∼95% while it was 86% and 78% for R-45 and R-46, respectively. The extraction equilibrium constants (Kex) were also calculated for the receptors and were found to be 2.4 × 109, 1.5 × 109, and 7.8 × 108 M−1 for R-44, R-45, and R-46 respectively. A weak C–H⋯π interaction between the phenyl spacers and terminal hexyl groups was observed for the hydrophobic receptor R-46 which enabled the receptor to be repeatedly used for the extraction and release of the SO42− in an acidic medium by converting SO42− to HSO4−. Moreover, Cram's U-tube experiments employing R-46 illustrated the dynamic transport of SO42− and ∼70% of sulphate was transported from the source phase to the receiving phase in 3 days. The authors asserted that, to date, this represents the most efficient report on sulphate transport efficiency. Eventually, the designing principles of such receptors represent a paradigm that can aid in enhancing our understanding and tailoring the receptors for selective SO42− separation.
In 2012, our group used a urea-based receptor R-47 for the LLE of SO42− from an aqueous medium (Fig. 16).81 It is noteworthy to mention that the receptor itself was not able to extract SO42− due to solubility issues. However, pre-encapsulation of CO32− with R-47 solubilised the receptor, enabling it to effectively extract SO42− through an anion exchange method. This finding can be explained by considering that SO42− has a lower hydration energy (ΔGh = −1080 kJ mol−1) as well as a higher association constant for R-47 compared to CO32− (Ka = 4.68 × 104 and 1.17 × 104 M−1 respectively, in dry DMSO). Despite the presence of other competing oxo-anions of TBA salts like H2PO4− and NO3−, the R-47·CO32− complex demonstrated selective extraction of SO42− from aqueous media. In the extraction process, a CHCl3 solution of R-47·CO32− was agitated with an aqueous solution of K2SO4. The salt exchange mechanism was elucidated using a phenolphthalein indicator, and the pink coloration of the aqueous phase indicated the exchange of CO32− in the water medium. Additionally, NMR analyses of the organic phase suggested 99% SO42− complexation with R-47. Moreover, for the gravimetric analysis and to facilitate the reuse of the receptor, BaCl2 was added to the organic phase to precipitate the extracted SO42− as BaSO4, confirming the extraction of SO42−.
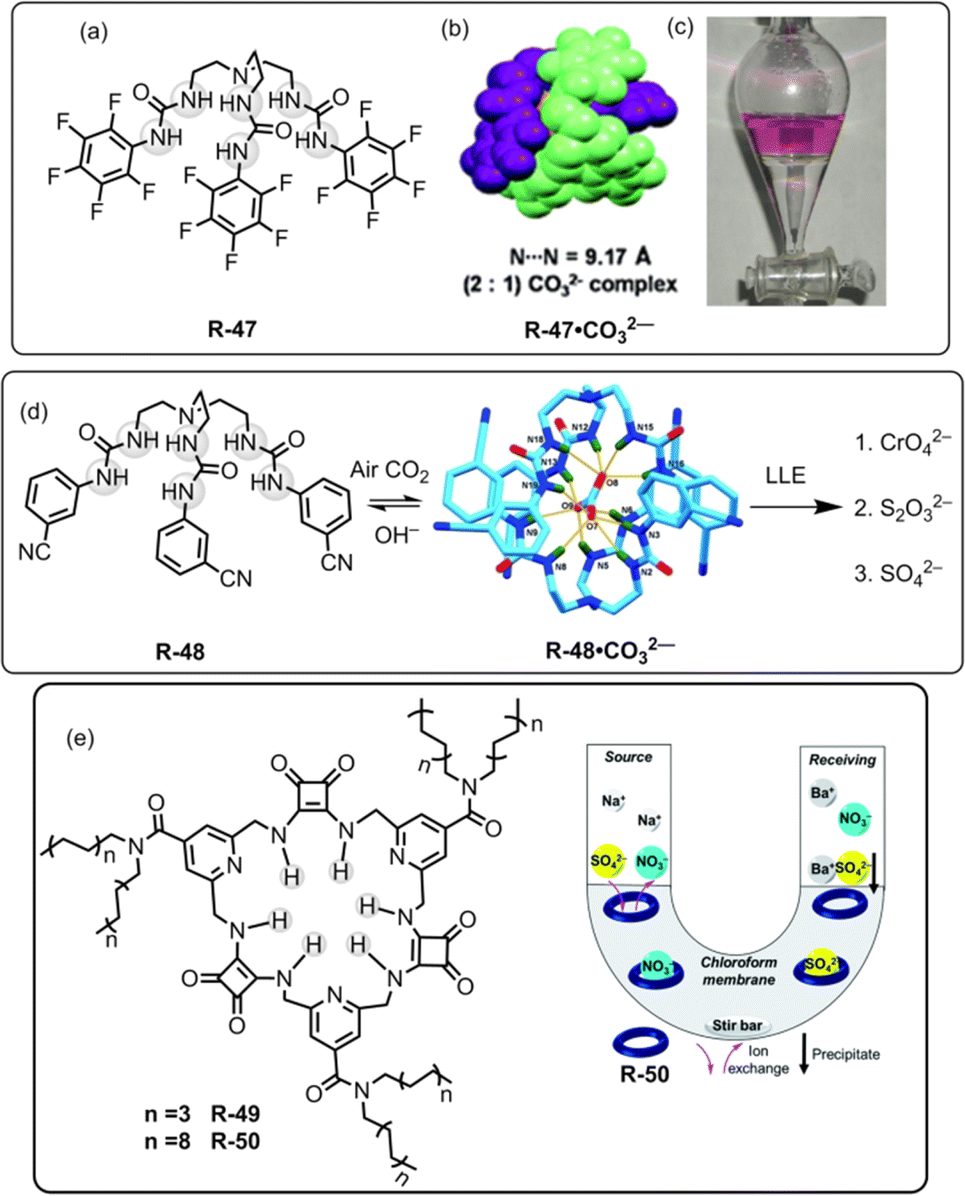 | ||
| Fig. 16 (a) Chemical structure of the receptor R-47. (b) Representation of the assembly of R-47·CO32−. (c) Visualization of SO42− extraction using phenolphthalein through the R-47·CO32− complex. (Reproduced from ref. 81. Copyright 2012, The Royal Society of Chemistry.) (d) Schematic representation of LLE by R-48 (Reproduced from ref. 82. Copyright 2014, Wiley.) (e) Chemical structures of the receptors R-49 and R-50. Illustration of the general setup for the U-tube experiments used to test the ability of R-50 to act as a carrier for SO42−. (Reproduced from ref. 83. Copyright 2020, The Royal Society of Chemistry). | ||
In a subsequent report, our research team introduced another tripodal scaffold with the 3-cyanophenyl-substituted receptor R-48 (Fig. 16).82 This receptor also formed a 2:1 dimeric capsular assembly of CO32− by capturing atmospheric carbon dioxide (CO2) from a basic DMSO solution. The outstanding solubility of R-48·CO32− in CHCl3 had been harnessed for the selective LLE of CrO42−, SO42−, and S2O32− ions from water through an anion-exchange metathesis process. All of these three anions were capable of forming an almost identical 2:1 (H–G) dimeric capsular assembly. However, the R-48·CO32− complex failed to extract H2PO4−, HAsO42− and F− ions. The CrO42− extraction was confirmed by the 53Cr-NMR and UV-Vis studies. Moreover, the extraction of the aforementioned anions by the carbonate complex was supported by the NMR, IR, PXRD, and X-ray diffraction analyses. Quantification through weighing the extracted complexes revealed an extraction efficiency of ∼90% for the three anions. Furthermore, quantitative estimations through gravimetric analysis, by treating BaCl2 to form BaCrO4 and BaSO4, demonstrated an efficiency of >90% for CrO42− and SO42−. Particularly, during the extraction of CrO42− in the presence of SO42− ions, the UV-Vis spectra analysis indicated ∼50% extraction capacity for CrO42− ions. Moreover, in the separation of SO42− for the nuclear waste treatment, the pH of the medium is a crucial factor. Consequently, in the presence of 5 equiv. of NO3−, the 48·CO32− complex demonstrated excellent SO42− extraction capability at pH 12.5, whereas R-47·CO32− could only achieve extraction up to pH 10.5. Interestingly, this marks the inaugural report on the LLE of CrO42− and S2O32− ions from water using an anion-exchange technique.
Most recently, Jolliffe et al. developed squaramide-based macrocycles R-49 and R-50, which were able to extract SO42− from the aqueous phase into CHCl3, via a nitrate ion exchange mechanism with the assistance of lipophilic tetraalkylammonium cations (Fig. 16).831H-NMR titrations in CDCl3 revealed that R-50 can bind to TBASO4 with Ka > 104 M−1. Therefore, in a wide pH range (pH 3.2–9.4), R-50 was able to transport the SO42− across a bulk CHCl3 membrane in a Cram U-tube experiment. Furthermore, to recycle the receptor, the authors employed the aforementioned method for the precipitation of SO42− as BaSO4. The amount of sulphate extracted from the precipitated BaSO4 was calculated by gravimetric analysis and inductively coupled plasma mass spectrometry (ICP-MS) experiments. In contrast to sodium salts, the lipophilic tetrabutylammonium salt of SO42− had been extracted by R-50. The extraction ability for the SO42− was also evaluated in the presence of nitrate ions by the metathesis method. NMR analysis of the organic phase indicated that the NH protons of R-50 were involved in the extraction of SO42−. Moreover, in acidic medium (pH 3.2), the SO42− extraction affinity was increased due to the protonation of the isonicotinamide part of the receptor. Moreover, R-50 possesses the capability to transport SO42−, signifying its potential for usefulness for nuclear-waste treatment.
For the extraction of polyvalent oxo-anions, urea-based neutral tripodal receptor R-51 was reported by Dey and co-workers, which exhibited selective encapsulation and extraction properties for HPO42− from water (Fig. 17).841H- and 31P-NMR, and crystal structure analysis revealed that encapsulation of HPO42− involved the formation of six hydrogen bond interactions with the three urea groups present in the receptor. The formation of a hydrogen-bonded capsule by the dimeric association between receptors and HPO42− was also a strong reason for the selectivity (Fig. 17). Therefore, receptors can efficiently extract HPO42− from water to CH2Cl2 even in the presence of different competitive anions (F−, CN−, CH3COO−, and HSO4−) as indicated by 1H and 31P-NMR analysis. The authors also suggested that at neutral pH H2PO4− is in equilibrium with HPO42− (pKa 7.21) whereas at high pH only PO43− is present. Thus, K3PO4 was used in the extraction experiment. Moreover, another tris-amide-based receptor R-52 was unable to extract the anions due to the absence of sufficient hydrogen bond donors inside the receptor cavity to stabilise an anion.
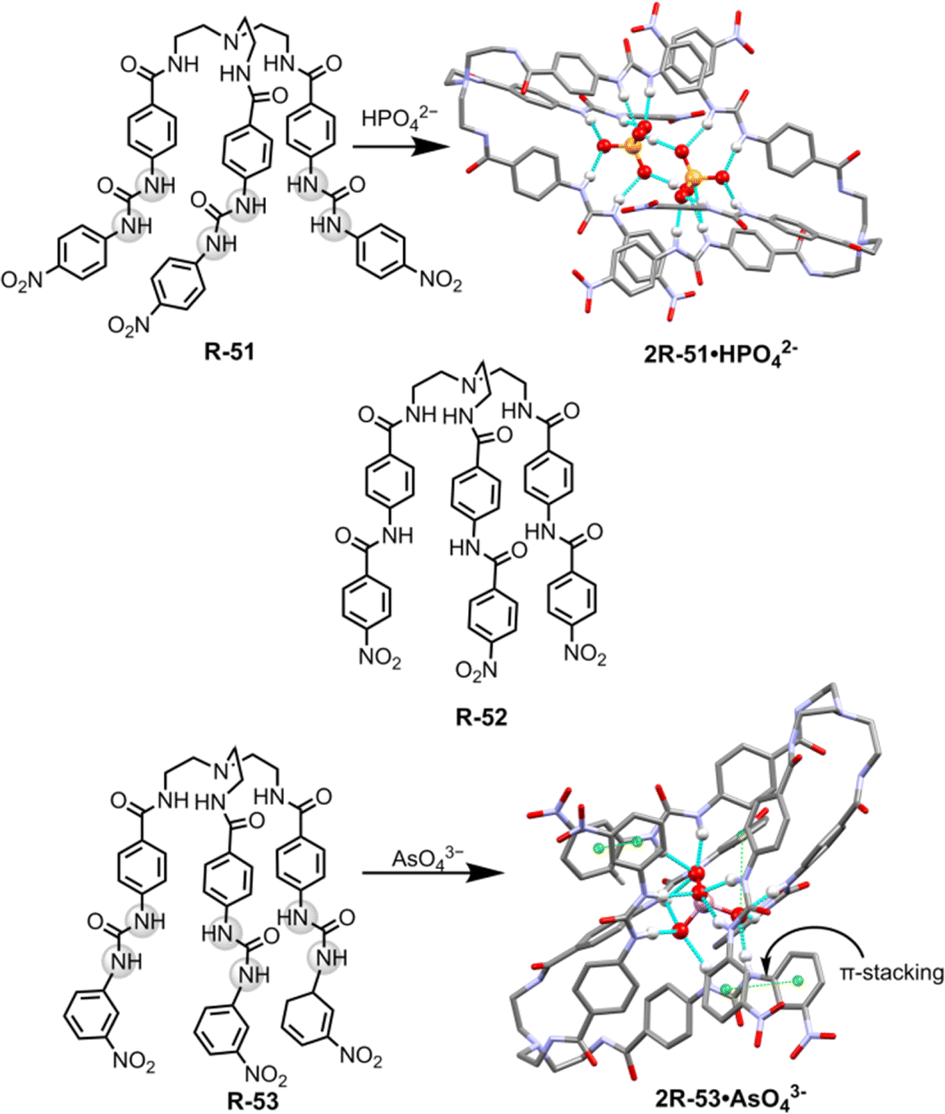 | ||
| Fig. 17 Chemical structure of the receptors R-51 to R-53 and crystal structures of 2R-51·HPO42− and 2R-53·AsO43− (data from the Cambridge Crystallographic Data Centre [CCDC # 2008261 and 2172773 respectively]84,85). | ||
Additionally, in 2022, the same group crafted an N-bridged urea-based tripodal receptor R-53 which was able to selectively extract arsenate (AsO43−) from water in the presence of phosphate, despite having close similarities in ionic charge, thermodynamic radii, pKa values, and hydration energies (Fig. 17).85 The high selectivity for arsenate was exhibited through its encapsulation inside the receptor, facilitated by eleven hydrogen bond interactions and the formation of a π-stacked rigid capsular assembly. The extraction experiments were executed at different pH (5.5, 7.4, 9.5, and 11). Notably, only under basic pH conditions (7.4, 9.5, and 11) ∼90% of arsenate was successfully extracted in the organic phase by R-53, employing an anion exchange method using (n-Bu4N+)OH−. Nevertheless, arsenate extraction was not possible at acidic pH 5.5. Thus, the major disadvantage of the charged receptor R-33, i.e., extraction ability only in an acidic medium, can be overcome by the neutral receptor R-53. However, there remains a need for the development of pH-independent receptors to facilitate arsenic extraction.
Radioactive wastewater treatment is one of the most pressing issues in current research relating to the LLE of anions. Therefore, researchers have become interested in the efficient removal of radioactive and toxic inorganic metal oxo-anions such as 99TcO4−/ReO4− from wastewater.86–88 In this context, recently, Gale and co-workers developed a superphane receptor R-54 which contains six amide-NH and six secondary NH-units that selectively encapsulated and efficiently extracted ReO4− from water (Fig. 18).891H-NMR studies with ReO4− revealed that ReO4− was able to form 18 strong hydrogen bonds leading to a binding constant (Ka) of (5.62 × 0.15) × 103 M−1 in CDCl3/CD3OD (1:1, v/v). In addition, the single crystal structure of the ReO4− complex suggested the encapsulation of ReO4− inside the cavity of the receptor by 1:1 complexation. Most importantly, in LLE studies within a CHCl3− water medium, by employing R-54, a remarkable 99.99% ReO4− extraction was accomplished, where the initial perrhenate concentration was ∼0.003 M. Additionally, after careful separation of the aqueous phase, further consecutive multiple extractions (1 to 7 cycles) of that aqueous solution were conducted. ICP-MS analysis revealed a sharp decrease in ReO4− concentration, reaching 4.40 × 10−6 M. Astonishingly, with an increase in the number of extraction cycles up to 10, there was a notable improvement in extraction efficiency, ultimately reducing the perrhenate concentration to less than ∼3.67 × 10−8 M. Even in the presence of high concentrations of other competing anions such as SO42−, H2PO4−, and MoO4−, the ReO4− extraction ability was not significantly affected. Interestingly, at lower pH the receptor showed high extraction ability but at higher pH the extraction ability was reduced because ReO4− was extracted in the form of [R-54·2H+·ReO4−]. Therefore, this study will be expected to boost the designing of the receptors in the future for controlled binding, release, and transport of the targeted anion for biological and industrial purposes.
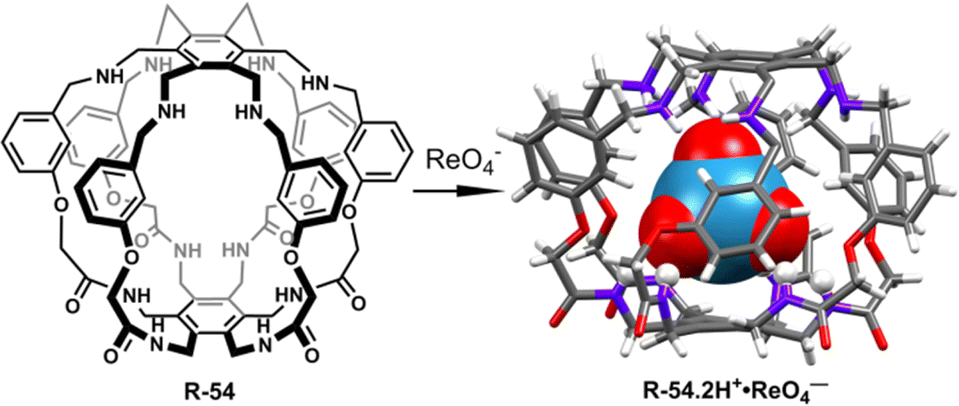 | ||
| Fig. 18 Structure of receptor R-54 and the corresponding ReO4− binding crystal. (Data from the Cambridge Crystallographic Data Centre [CCDC # 2111255].89) | ||
The Nitschke group exploited the extraction capability of a coordination cage under an interfacial state. In this regard, they adopted the azaphosphatrane unit, which was able to bind anions through hydrogen bonding along with electrostatic interaction. The Fe4IIL4 cage R-55 was synthesized via an anionic template-directed self-assembly process (Fig. 19).90 For R-55 with lipophilic counterion BArF4−, the relatively weakly bound template nBuBF3− (n-butyltrifluoroborate) was able to exchange easily with ReO4−. As a consequence, 97% of ReO4− was extracted from water to nitromethane in the presence of competing anions (F−, Cl−, Br−, I−, SO42−, ClO4−, NO3−, BF4−, H2PO4−, and AcO−) which was confirmed via1H-NMR. In relatively less polar ethyl acetate, the ReO4− ⊂ R-55 was disassembled to release the extracted ReO4− which could be reassembled again in acetonitrile with the help of the nBuBF3− template. Furthermore, a reverse extraction of ReO4− from the organic to the aqueous phase was also achieved with a water-soluble receptor with the SO42− counterion. Therefore, for repeated oxo-anion extraction through the assembly and disassembly process in different solvents, these metallacages may be advantageous compared to conventionally used covalent receptors.
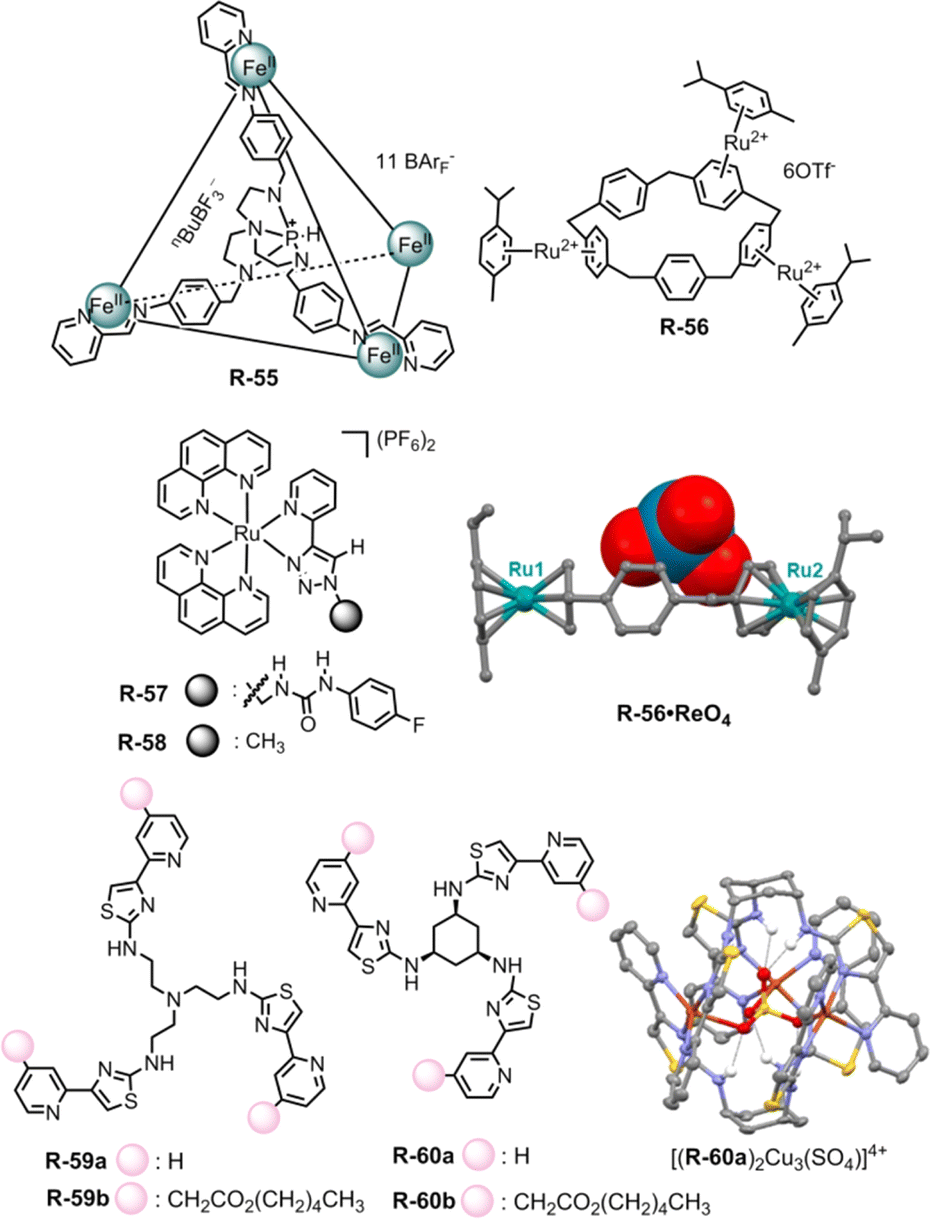 | ||
| Fig. 19 Chemical structure of the receptors R-55 to R-60, crystal structure of the R-56·ReO4 complex (data from the Cambridge Crystallographic Data Centre [CCDC # 1951588]91) and self-assembled structure of [(R-60a)2Cu3(SO4)]4+. (Reproduced from ref. 93. Copyright 2020, Wiley-VCH GmbH.) | ||
On the other hand, Huang et al. reported ReO4− extraction with functionalized electron-deficient paracyclophane (PCP) supramolecular host R-56. In contrast to the electron-rich PCP moiety, the embedding of the ruthenium unit via π-metallation generated a polymetalated PCP which exhibited better regioselectivity as well as efficiency for anion binding (Fig. 19).91 As evident from 1H-NMR, R-56 preferentially bound ReO4− over other tested anions both in D2O and CD3NO2 solvents, further supporting the fact that the extraction of ReO4− was not significantly hampered by other oxo-anions including SO42−, NO3−, ClO4−, etc. At the interface, about 79% of ReO4− had been extracted from D2O to CD3NO2, when the receptor concentration was maintained at 0.004 M. Furthermore, the tri-metalated species R-56 bound ReO4− in a tweezers-like manner through anion–π interaction as evidenced from the solid state structure. On that account, this study could also be expanded to anion encapsulation to facilitate the extraction process by increasing the number of benzene rings in PCP with further modification in π-metalation through the group-VIII transition elements.
For the extraction of oxo-anions like phosphate and carboxylate our group has utilized the ruthenium-coordinated receptor R-57 (Fig. 19).92 Considerable hydrogen bonding was observed in the case of phosphates, involving the urea –NH and the triazole CH protons. For carboxylates, only the urea –NH participated, as evidenced by a significant downfield shift in the 1H-NMR spectrum in DMSO-d6. Further investigations on LLE following several NMR approaches manifested that R-57 could extract H2PO4−, CH3COO− and PhCOO− from water to CHCl3 with 28%, 74%, and 80% efficiency respectively. The notable role of urea in the extraction of CH3COO− and PhCOO− was demonstrated through the negative extraction results when using the receptor R-58 without urea connectivity.
Recently, Rice and co-workers reported the LLE of phosphate selectively by self-assembly driven copper complexes. In this regard, R-59a and R-60a were synthesized and can form trinuclear copper complexes.93 This trinuclear assembly in host–guest complexation was confirmed by a single crystal of a SO42− encapsulated complex [(R-60a)2Cu3(SO4)]4+. Further, R-59b and R-60b with aliphatic hexyl esters were prepared to increase the solubility of the assemblies in an organic solvent. The initial dark-yellow or pale-yellow CH2Cl2 solution of Cu(OTf)2 and R-59b or R-60b changed to lime green upon exposure to an aqueous NaH2PO4 solution indicating phosphate binding in the cavity which was further supported by UV-Vis studies (Fig. 19). Based on ion chromatography, a stoichiometric amount of hosts [(R-59b)2Cu3]6+ and [(R-60b)2Cu3]6+ can extract around 82 and 76% of H2PO4− respectively from water. In addition, such assemblies were formed selectively in the case of sulphate and phosphate rather than chloride and nitrate salts. While using Na2HPO4, a slightly higher phosphate preference was observed with [(R-60b)2Cu3]6+ compared to [(R-59b)2Cu3]6+. The rigid small cyclohexyl spacer in R-60 induced shorter Cu⋯anion and NH⋯anion distances, thus creating a cavity that was favourable for selective phosphate binding over other anions.
6. Removal of alkali metals through LLE
Contamination of water with highly concentrated alkali metal salts makes it undrinkable. In particular, the diffusion of radioactive caesium (Cs) from the testing of weapons and the nuclear industry has caused a major environmental problem.94 To avoid these environmental as well as health problems, it is important to remove alkali metal ions from an aqueous solution.In this context, Akkus et al. prepared a series of calix[4]-oxa-crown and calix[4]-thia-crown derivatives R-61 to R-64, where nitrile, amino, and oxime groups attached at the lower rim of the calixcrown (Fig. 20).95 Among the receptors, R-61a exhibited a preference for extracting Na+ over K+ owing to a perfect fit in the cavity. Meanwhile R-62a demonstrated strong affinity for both Cs+ and Na+. The more polarizable Cs+ binding to R-62a was rationalized by the presence of nitrile groups which acted as π-donors. Furthermore, the high extractability and selectivity of R-61b and R-62b towards soft Hg2+ can be attributed to the soft nature of the sulphur atom in calix[4]-thia-crown. However, oxime derivatives R-64 did not exhibit any selectivity even though all cations were extracted through cation–π interactions with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N ligating groups.
N ligating groups.
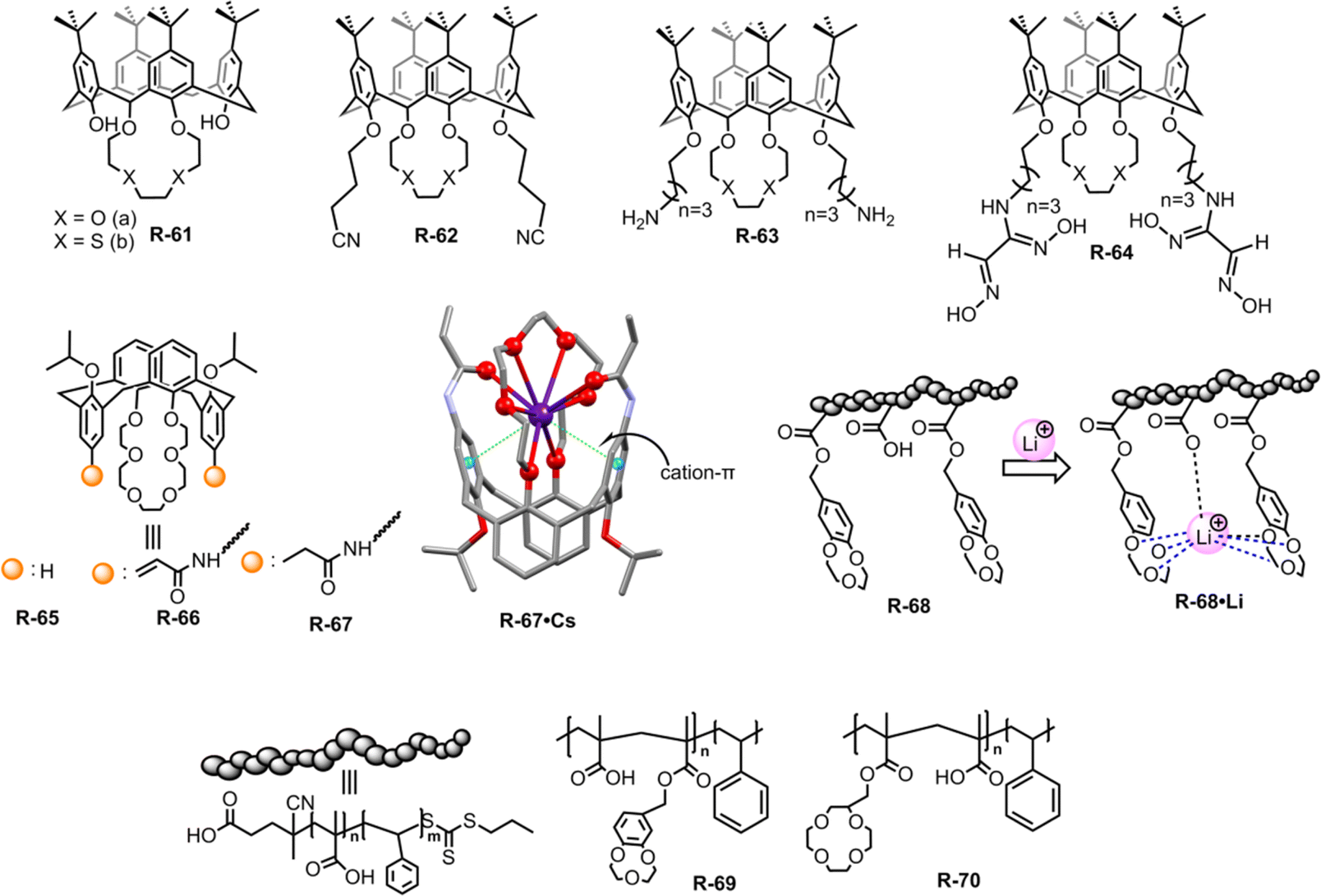 | ||
| Fig. 20 Chemical structure of the receptors R-61 to R-70 and crystal structures of R-67·Cs (data from the Cambridge Crystallographic Data Centre [CCDC #1569925]96). | ||
Then, in 2018, Chen and Sun et al. employed acrylamido and propionamido substituents in calix[4]arene-crown-6-ether derivatives R-65 to R-67 for the selective extraction of Cs+ (Fig. 20).96 In the LLE experiment, the distribution ratios of the receptors toward Cs+ were as follows: propionamido derivative (R-67) > acrylamido (R-66) > R-65. The deformation density of the Cs-complexes, as calculated from DFT using extended transition state coupled with natural orbitals for chemical valence (ETS-NOCV) analysis, indicated that the charge transfer occurred through the crown ether groups and the two rotated phenyl rings. Furthermore, the highest Cs+ affinity towards R-66 and R-67 was attributed to three intramolecular cooperative effects: (i) high affinity of the crown ether part for Cs+, (ii) cation–π interactions with the rotated phenyl rings, and (iii) interaction with the –CO groups of the acrylamido or propionamido units. These triply cooperative effects were also supported by the solid-state structure. Based on the above consequence, the 1,3-alternate calix[4]arene crown-6 derivatives can improve the Cs+ extraction. Therefore, the aforementioned receptors elucidated that the well-organized arm of the calix[4]arene can provide harmony between the receptors and alkali metals which can further accelerate the ion-pair receptor design.
In recent decades, lithium ion (Li+) separation has become increasingly attractive due to its many application in our daily life, such as Li-ion batteries, medicines, catalysis, and in alloy industries.97–100 However, selective extraction of Li+ is challenging over its other family members due to its larger hydration energy (−475 kJ mol−1).38 In this regard, just recently Abetz et al. developed a crown ether functionalized diblock copolymer R-68via RAFT polymerization by employing styrene and methacrylic acid as monomer units for the selective extraction of Li+ over other interfering alkali metal ions (Na+, Cs+, and K+) (Fig. 20).101 Moreover, in the LLE experiment, various crown ethers, including benzo-9-crown-3, benzo-12-crown-4, and benzo-15-crown-5, were employed along with the UV-responsive 2′,4′,5′,7′-tetrabromoeosin ethyl ester salt of Li+, Na+ & K+. The finding illustrated that the selective complexation of alkali metal ions depends on the cavity size of the crown ether and the stability of the complexes.102,103 Hence, the polymer R-68 based on benzo-9-crown-3 exhibited significant selectivity towards Li+ over Na+ and K+ attributed to the matching ionic size with the cavity along with enthalpically driven sandwich-type complex formation. Furthermore, R-68 displayed 104 times higher complex stability for Li+ compared to free benzo-9-crown-3. Importantly, the bound Li+ could easily be released from the polymer by raising the temperature. Hence, this polymer material holds promise for potential industrial applications in lithium recovery. Building upon the Li+ binding mechanism illustrated earlier, the same authors synthesized another two polymers R-69 and R-70 to improve the Li+ extraction from an aqueous medium. R-69 exhibited a higher complexation constant (K) (3.9 × 1010 M−1) compared to R-70 (3.8 × 107 M−1) towards Li+ (Fig. 20).104 Astonishingly, even in the presence of high concentrations of Na+ and K+, R-69 successfully extracted Li+ from seawater by forming a higher-order sandwich-type complex structure with Li+.
Very recently, Weigand et al. investigated LLE by utilizing 4-phosphoryl pyrazolones R-71 to R-73 in the presence of various co-ligands (TOPO, TBPO and TBP) for the selective extraction of Li+ from the aqueous phase (Fig. 21).105 In the presence of TOPO at pH 8.2, R-73 exhibited maximum 78% Li+ extraction efficiency while it was 70 and 61% for R-72 and R-71 respectively. Surprisingly, in the absence of TOPO, the receptors demonstrated negligible (<5%) Li+ extraction efficiency. Hence, highly hydrophobic co-ligand TOPO played a pivotal role in extraction studies. Additionally, after Li+ extraction a noticeable visual colour change and chemical shifting of TOPO in the 31P-NMR spectrum suggested that the three Li+ ions were attached to the O-donor atom of TOPO forming a tetrahedral [Li3O]-core which was the key factor for effective Li+ extraction. Thus, a new class of N, O, and P-donor-based receptors may aid in the investigation of coordination as well as extraction of s-block elements, mainly alkali metals, which are rarely observed.
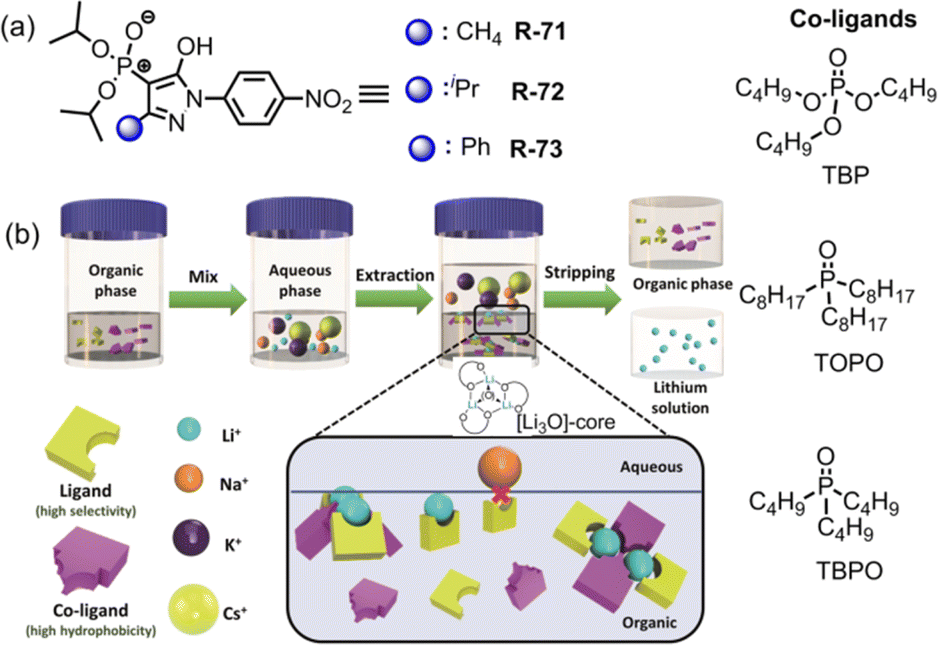 | ||
| Fig. 21 (a) Chemical structure of the receptors R-71 to R-73. (b) Schematic illustration of the Li+ extraction process with the presence of receptor and co-ligand. Reproduced with permission from ref. 105. Copyright 2022, Chemistry – A European Journal published by Wiley-VCH GmbH. | ||
7. LLE of ion-pairs using ditopic receptors
In LLE, ion pair receptors play a crucial role in facilitating the transfer of cations along with anions from one phase to another. In this context, the ion pair receptor design usually involves incorporating hydrogen bond donor functional groups, as well as Lewis acidic sites or positively charged groups for the recognition of anions. For cations Lewis basic sites and π-electron moieties are integrated in the receptor. One of the first reports of an ion-pair receptor was made in 1991 by Reetz and co-workers.106 Over the course of time, designing of such heteroditopic receptors was gradually developed. Several reviews have summarized this early progress on ion-pair binding.12,13,107–109 Ion pair binding with different receptors can be categorized into three modes: (a) contact, (b) solvent-bridged and (c) host–guest separated ion pair (Fig. 22).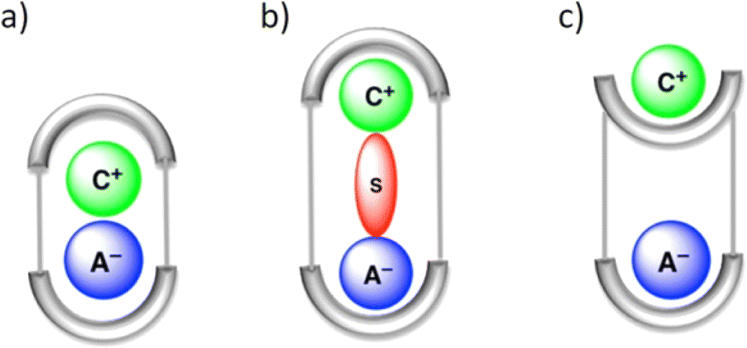 | ||
| Fig. 22 Schematic representation of the binding modes for an ion pair receptor: (a) contact, (b) solvent-bridged, and (c) host–guest separated ion pair. (Reproduced from ref. 32. Copyright 2010 Royal Society of Chemistry.) | ||
7.1. Metal halide salt extraction by ion-pair receptors
In 2018, Kim, Moyer, and Sessler et al. designed receptors R-74 and R-75via an anion-selective calix[4]pyrrole unit strapped with a cationic domain hemispherand and phenanthroline unit respectively (Fig. 23).110 From the crystal structure of the LiCl complex, the compact space within the receptor binds an alkali metal chloride salt through contact with a 1:1 stoichiometry, which maximises the coulombic interaction between the ion-pairs. Based on 1H-NMR studies, R-74 was able to extract LiCl, NaCl, and KCl from D2O to C6D5NO2 with selectivity order: KCl > NaCl > LiCl. The above selectivity can be described in terms of the hydration energies of the respective cations. In contrast to R-74, R-75 showed ∼100% selectivity towards LiCl extraction from D2O to CDCl3. The latter observation reflected on the relatively small cavity size formed by the strapping unit in R-75 which favoured the LiCl binding.
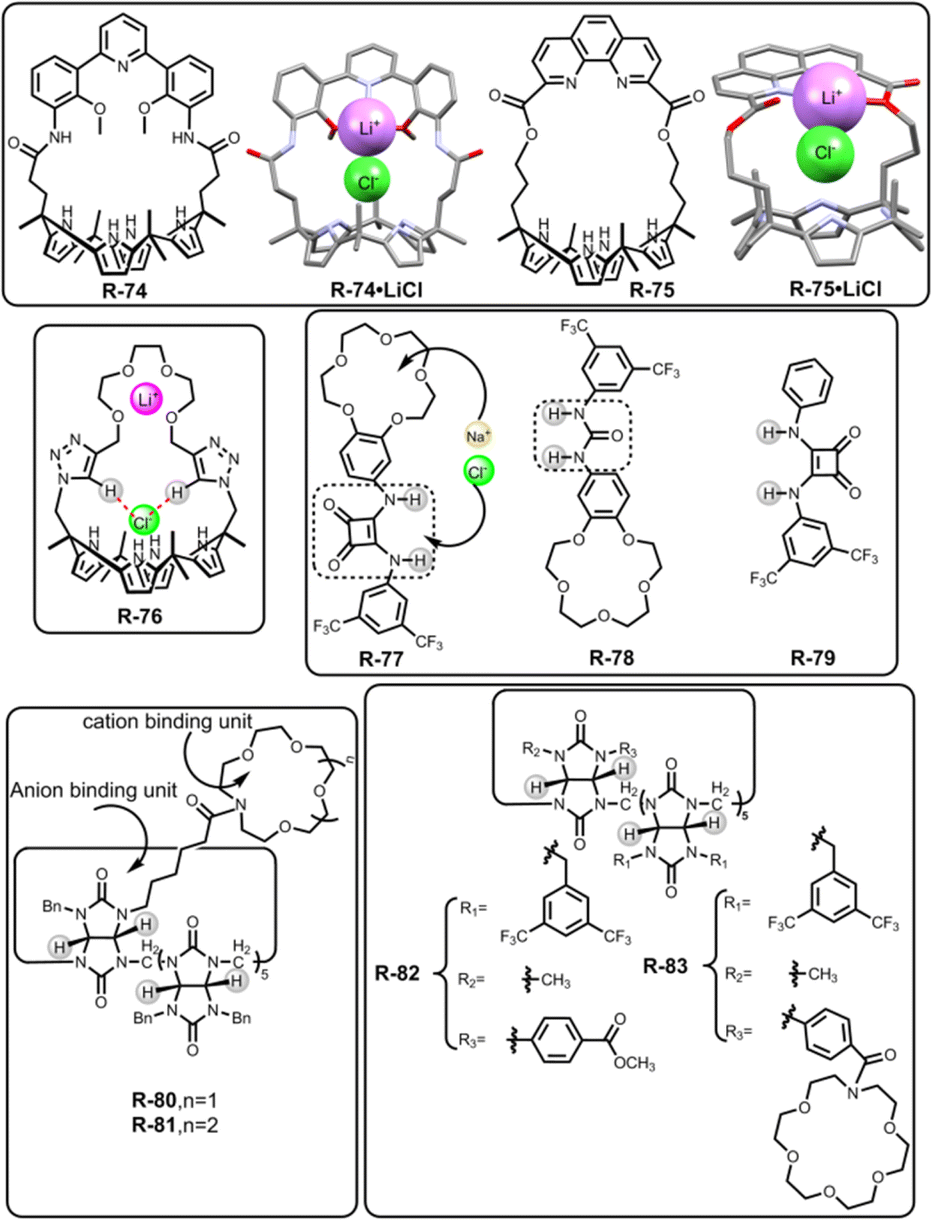 | ||
| Fig. 23 Chemical structure of the receptors R-74 to R-83 and crystal structures of R-74·LiCl and R-75·LiCl (data from the Cambridge Crystallographic Data Centre [CCDC # 1821811 and 1821810 respectively]).110 | ||
Later, Jang and co-workers contributed to the extraction of LiCl salts by synthesizing R-76 (Fig. 23).111 Inside the receptor, oligoethylene glycol was chosen as a Li-binding site while calix[4]pyrrole along with spacer triazole protons interacted with the counter halide ion. As indicated by the solid-state structure, R-76 formed a 2:2 host–guest complex with LiCl. However, unlike the previous LiCl receptors, R-76 exhibited a separated ion-pair binding. From the 1H-NMR in 1:10 CD3OD and CD2Cl2 solvent, it was established that R-76 selectively bound LiCl and LiBr. Furthermore, R-76 was able to extract LiCl from water to an organic solvent (CD2Cl2) in the presence of more than 80% saturated solution of LiCl. This extracted LiCl solution was further isolated as Li2CO3 upon reacting with CO2 gas or solid K2CO3. Moreover, the amount of precipitated Li2CO3 was calculated through acid–base titration using methyl orange as an indicator.
To extract NaCl, Romanski and co-workers prepared a heteroditopic receptor R-77 by integrating a 15-crown-5 moiety and anion-selective squaramide unit (Fig. 23).112 This receptor cooperatively interacted with Cl− in the presence of Na+ as directed by the Cl− binding constant of the cation-free (1.15 × 105 M−1) and cation-bound receptor (1.97 × 105 M−1) derived from UV studies in CH3CN. This enhancement of the association constant was rationalized by the increasing acidity of the squaramide protons induced by cation binding. Under LLE conditions, NaCl was transferred by the host from water to the C6D5NO2 solvent, as indicated by the large chemical shifts (Δδ = 0.72 and 0.79 ppm) of the two squaramide NH-protons. Additionally, by back-extracting the NaCl into the water, R-77 could also be freed easily. However, receptors R-78 and R-79 failed to extract NaCl, which signified the lower acidity of urea –NHs over squaramide protons and the superiority of heteroditopic receptors for ion-pair binding, respectively.
In 2020, the extraction of inorganic salts was reported by Sindelar et al. with aza-crown ether functionalized bambus[6]uril receptors R-80 and R-81 (Fig. 23).113 The typical anion receptor bambusurils effectively bind anions into the cavity with 12 methine–H⋯A− hydrogen bond interactions. Phase separation between the CHCl3 solution of receptors and the aqueous solution of inorganic salts allowed the extraction of inorganic salts including NaCl to the receiving organic phase. Both the methine and crown-CH2 protons were shifted in 1H-NMR implying a successful extraction process, whereas the methine proton integration allowed the extraction efficiency to be determined. For both the receptors, the higher extraction ability for MI and MBr salts (M = Li, Na, K) was justified by the larger binding affinities of these anions toward bambusuril, while for MCl salt extraction depended on the counter cation and crown ether size of the receptor. Thus, R-81 extracted ∼100% NaBr and NaI from water, while only 74% efficiency was noted for NaCl.
In later studies, the same group reported two bambus[6]uril derivatives R-82 and R-83, and demonstrated their anion extraction and transport ability (Fig. 23).114 Herein, they introduced an electron withdrawing as well as lipophilic substituent into the receptor to improve interaction with small hydrated anions. Both receptors were capable of quantitatively extracting the tetrabutylammonium salts of Cl−, NO3−, and H2PO4− from water to the organic nitrobenzene phase. However, among the inorganic salts, R-82 could only extract NaNO3 with 68% efficiency due to the receptor having a strong affinity towards smaller NO3−. In contrast, for R-83, inorganic salt extraction depended on the cavity size of the aza-crown ether. Thus, a preferable KCl (41%) extraction over NaCl (36%) was observed, whereas, in the case of NaNO3 salt, R-83 showed quantitative extraction. These two studies established the efficacy of bambusuril-based receptors for extraction. Moreover, the incorporation of squaramide and calix[4]pyrrole units in the receptors also played a crucial role in binding hard-to-capture ion pairs.
The selective separation of highly hydrated KF and KCl salts from the water medium is of fundamental importance in chemistry. In this context, our group for the first time reported solvent extraction of KF from water to CHCl3 using a ‘dual host’ strategy. Besides ditopic receptors, ion pair binding through a dual host system (binary mixture of two receptors) has also proved to be promising. This dual host strategy could be more versatile and economical instead of designing complex receptors for salt extraction application. In our approach, 18-crown-6 (R-84a) and a tripodal amide receptor R-84b were used as dual hosts. The later tripodal receptor effectively bound halide through 3 acidic ‘NH’ protons (Fig. 24).115 The crystal structure clearly revealed the formation of a 1D coordination polymer with the KF guest where K+ and F− were encapsulated in the respective domains. Such encapsulation was also confirmed from the downfield chemical shift of the NH protons in the 1H-NMR in CDCl3. An extraction enhancement of anions (F− and Cl−) with the K+-bound receptor defined the dual host synergistic extraction of KF and KCl. At pH ∼6.0, 48% and 44% extraction efficiency were observed for KF and KCl respectively.
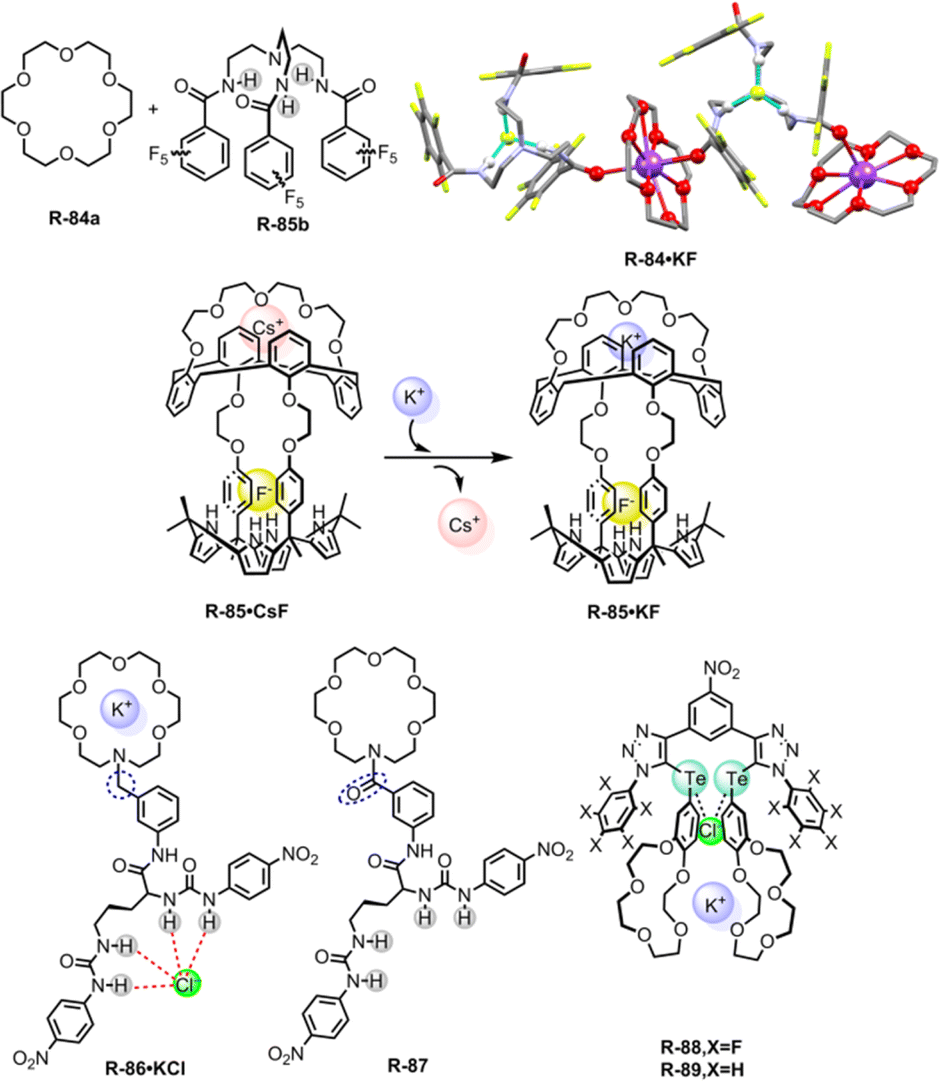 | ||
| Fig. 24 Chemical structure of the receptors R-84 to R-89 with ion-pair binding and crystal structures of R-84·KF (data from the Cambridge Crystallographic Data Centre [CCDC # 732760]).115 | ||
Later, Sessler and co-workers reported multitopic receptor R-85 for KF extraction. Anion selective calix[4]pyrrole and cation selective crown-linked calix[4]arene were connected through ethylene glycol spacers, thus forming multiple recognition zones inside the receptor (Fig. 24).116 An 1H-NMR study in a 1:9 CD3OD/CDCl3 solvent mixture revealed that R-85 was capable of binding both KF and CsF salts. However, distinct binding modes and complexation processes were observed for each. In the mentioned solvent system, a relatively stronger binding of K+ (6.5 × 106 M−1) to the upper crown pocket was observed compared to Cs+ (3.3 × 104 M−1). This robust binding led to the selective extraction of KF salt from water to the nitrobenzene phase through the formation of the 1:1 [R-85·KF] complex. KF extraction was also well supported by the large chemical shift of the calix –NH protons (Δδ = 5.1 ppm) of the extracted organic phase. In contrast, CsF could not significantly alter the proton signals, indicating lower efficiency of the receptor under the LLE conditions. Additionally, the strong binding of K+ to the upper crown pocket displaced the weakly bound Cs+ from the receptor through the ‘cation metathesis’ process. Thus, R-85 demonstrated a new idea of recyclable receptor designing, where cation metathesis could play a significant role.
A notable study on the cooperative effects during ion-pair binding was performed by Piątek and co-workers. In 2017, they constructed two ion-pair receptors, R-86 and R-87, via linking aza-18-crown-6 with urea-functionalized amino acid L-ornithine (Fig. 24).117 The only difference between R-86 and R-87 was the spacer unit (benzyl vs. benzoyl) which played a vital role in anion recognition. In R-87, the presence of a less conformationally flexible amide spacer near the cation domain caused less cooperative ion-pair binding. In contrast, the flexible benzyl version R-86 showed significant cooperation during ion-pair binding specially for KCl and NH4Cl. UV-Vis spectroscopic analysis in CH3CN demonstrated almost 8 times higher Cl− binding ability to the R-86 over R-87 in the presence of the K+ ion with binding constant values of 3.7 × 105 and 4.4 × 104 M−1 respectively. The interaction mechanism was established by 1H-NMR studies in CD3CN which revealed anion binding involving all the urea protons and cation encapsulation to the crown ether moiety. Furthermore, R-86 showed efficient extraction capacity for KCl (97%) as well as NH4Cl (93%) under LLE conditions while R-87 failed to extract the same from H2O to CHCl3.
In parallel to hydrogen bonding, Beer and his colleagues recently synthesized a heteroditopic receptor R-88 by employing σ-hole-based chalcogen bonding (ChB) interaction (Fig. 24).27 Binding of a cation in the crown moiety induced the formation of strongly polarised σ-holes on Te-atoms. Electron-withdrawing pentafluorobenzene substituents were also involved in such polarization. NMR titration in 1:1 CDCl3/CD3CN in the presence of equimolar KPF6 exhibited relatively higher association constants for Cl− over other common anions. Under LLE conditions, almost 71% KCl was extracted from D2O to CDCl3 when the KCl concentration was 4 M. Moreover, nonfluorinated receptor R-89 failed to extract KCl under identical conditions, indicating a lower effect of electron polarization on chalcogen bonding. Therefore, appropriately designed ChB receptors are able to bind a harder Lewis base through a polarized σ-hole, offering a potential alternative for selective salt extraction from water.
It has been found that the supramolecular self-assembly procedure is efficient for the recognition and extraction of ion pairs. In these cases, a properly designed molecule interacts with the guests via non-covalent interactions and thereby aggregates to form various supramolecular self-assembly structures (such as micelles, vesicles).118
In 2017, Sessler and colleagues designed a receptor R-90, in which the calix[4]pyrrole subunit and long-tailed anthracene moiety were connected by a triazole spacer (Fig. 25).119 This long-tailed host undergoes self-assembly in an aqueous medium upon binding Fe2+ and F− into the respective recognition sites, forming cross-linkable micelles at the interface. The change in polarity upon guest binding resulted in the formation of micelles, as confirmed by microscopic analyses such as scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Moreover, by forming such assemblies, the receptor R-90 demonstrated notable efficiency in extracting FeF2, as evidenced by approximately 75% extraction yield, as indicated by 19F-NMR.
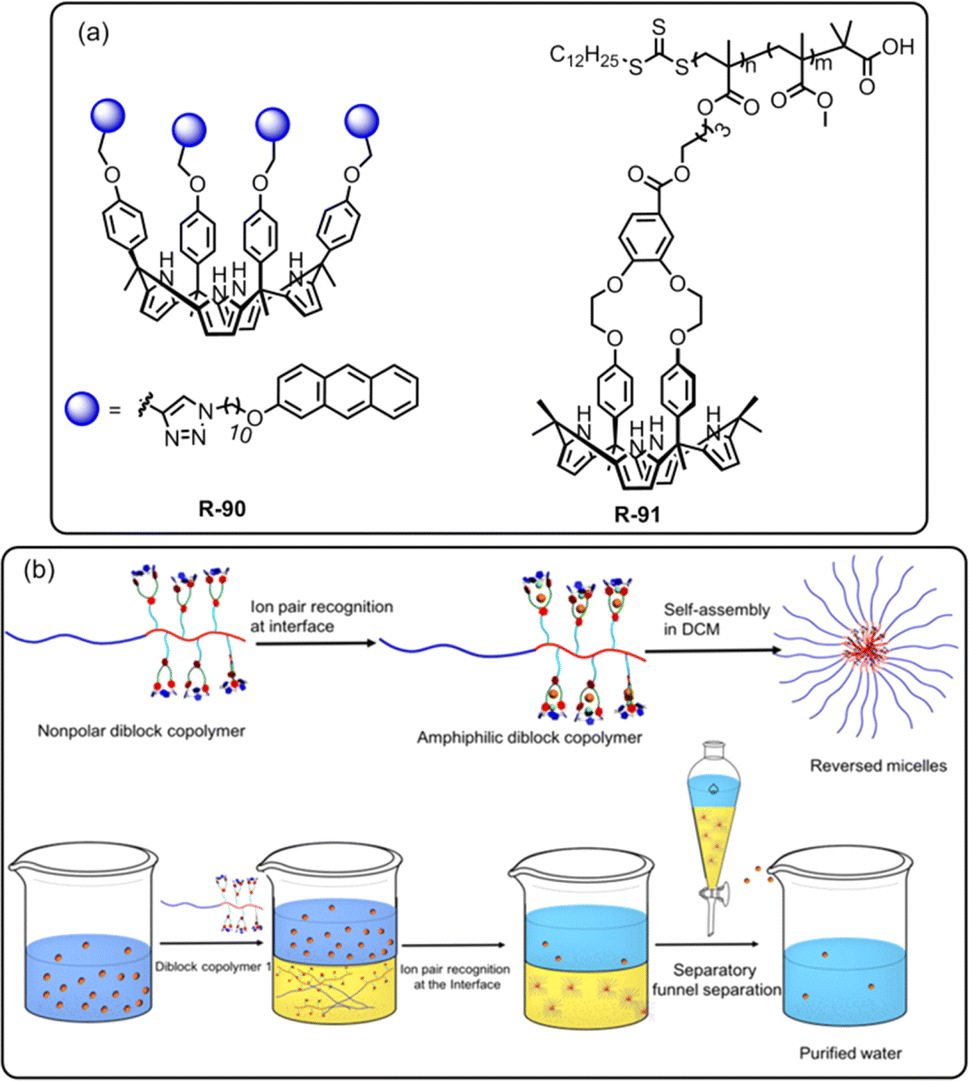 | ||
| Fig. 25 (a) Chemical structure of the receptors R-90 and R-91. (b) Self-assembly driven micelle formation at the interface by R-91. (Reproduced with permission from ref. 120. Copyright 2018, ACS Publication.) | ||
In a later report, the same group found a separation method for CsF salt under LLE conditions via a diblock copolymer receptor R-91 (Fig. 25).120 In CH2Cl2 solvent, receptor R-91 formed reversed micelles via aggregation (Fig. 25b) after binding to Cs+ and F−, which was confirmed by TEM, dynamic light scattering (DLS), and energy-dispersive X-ray analysis. Solvent extraction of CsF salt from water to CH2Cl2 unequivocally indicated the self-assembly process to be a good driving force for solvent extraction.
7.2. Metal oxo-anion salt extraction by ion-pair receptors
Sessler and his group prepared R-92 by connecting calix[4]pyrrole with the cationic domain hemispherand unit. This tritopic receptor can serve as an extractant for both MX and M2X type metal salts. In 2016, this receptor was utilized for the recognition of exceptionally hydrated lithium salts (Fig. 26).121R-92 demonstrated the capability to bind various Li salts, including LiCl, LiBr, LiI, LiNO2, and LiNO3, as confirmed by crystal structures. 1H-NMR analysis in 1:9 (v/v) CD3OD/CD2Cl2 revealed a preferential binding of Li+ salt over Na+, K+, and Rb+ metals salts. Moreover, R-92 selectively extracted LiNO2 over NaNO2 and KNO2 from water to the organic phase, which appeared to be significant for corrosion inhibition in the construction industry.
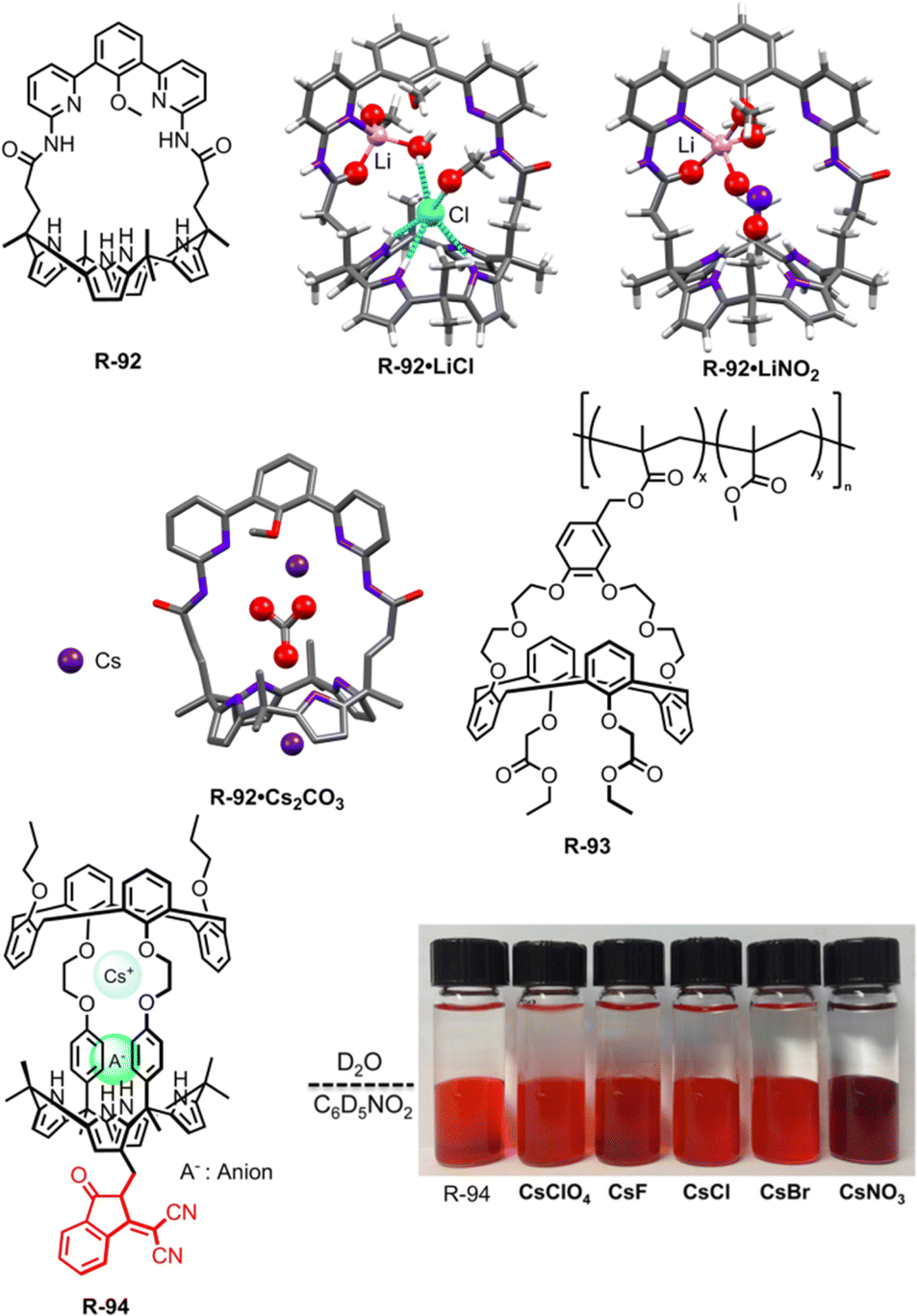 | ||
| Fig. 26 Chemical structure of the receptors R-92 to R-94 (data from the Cambridge Crystallographic Data Centre [CCDC # 1483286, 1483283, 1549914].121,122) and visible colour change observed for R-94 during the LLE experiment. (Reproduced with permission from ref. 124. Copyright 2016, ACS Publication.) | ||
Later, R-92 was also proved to bind both MX type Cs-salts, namely, halides and hydroxide as well as M2X type Cs2CO3 (Fig. 26).122 For Cs2CO3, one Cs+ was bound to the cavity offered by the hemispherand unit, while the other Cs+ was bound ‘in the cup’ formed by the combination of pyrrolic units to finally make a 1:1 R-92·Cs2CO3 complex. From 1H-NMR and ICP-MS investigations, it was revealed that R-92 can extract Cs2CO3 and CsOH over CsX (X = halide) salts from the highly basic aqueous phase to the organic CDCl3 phase. This result suggested that R-92 may overcome the large hydration enthalpies of the anions OH− (−430 kJ mol−1) and CO32− (−1315 kJ mol−1). Furthermore, the U-tube transport experiment was also carried out using R-92, to confirm the transport of CsOH through the organic CHCl3 layer.
An early example of selective Cs salt extraction was reported in 2010 by the same group. A co-polymeric receptor, R-93, comprising benzocrown-6-calix[4]arene as the core recognition unit was found to be selective towards Cs+ (Fig. 26).1231H-NMR revealed that R-93 was a selective extractor for CsNO3 over other Cs-salts like CsX (X = halide), CsSO4 and other nitrate salts like KNO3 and NaNO3. Thus, a CD2Cl2 solution of R-93 extracted CsNO3 from an aqueous solution in the presence of the above-mentioned salts. Additional evidence of CsNO3 extraction was obtained from ICP-MS studies which showed almost 60% Cs+ complexation with respect to the receptor. Furthermore, the extracted host–guest complex was isolated by adding cold hexane resulting in the formation of a precipitate, which gave 10% more mass than the free receptor indicating guest binding.
Salt extraction by colorimetric receptors provides an additional advantage because they can induce rapid detection, leading to a visual colour change in the organic phase. Specifically, this array provides an attractive alternative compared to other existing techniques due to its efficiency and lack of complexity, making it less time-consuming and presumptive. In this regard, Sessler and co-workers synthesized receptor R-94 by connecting a chromophore (3-(dicyanomethylidene) indan-1-one) functionalised calix[4]pyrrole with calix[4]arene through an ethylene glycol spacer (Fig. 26).124 In 1:10 CH3OH/CHCl3, UV-Vis spectroscopic analysis of R-94 indicated selective interaction with Cs-salts including CsF, CsCl and CsNO3 with the formation of a 1:1 complex. Furthermore, the recognition ability of R-94 was observed only in the presence of CsX (X = F, Cl, Br, NO3, ClO4) salts rather than individual Cs+ or X− ions. Therefore, R-94 acted as a chemical AND logic gate for ion-pair binding. 1H-NMR spectral changes of –NH protons in R-94 supported the selective extraction of CsNO3 from D2O to C6D5NO2. Such ion-pair binding to the receptor caused an alteration of the electronic environment within the chromophore unit leading to a visual colour change of the organic solution.
Another example of CsNO3 salt extraction from the aqueous to organic nitrobenzene phase was reported with the previously mentioned receptor R-85 (Fig. 24).125 In this case, CsNO3 complexed with the receptor in a contact mode, where Cs+ bound to the defined cavity of the ethylene glycol spacer along with nitrate binding to the calix-pyrrole unit. However, the strong affinity of the upper 1,3-alternate calix-crown system towards K+ initiated cation metathesis by replacing Cs+ with K+ within the receptor. Thus, addition of a KClO4 solution in 1:9 CD3OD/CDCl3 to a CsNO3 complex in the same solvent leads to the formation of [R-85·K+]ClO4− as a white precipitate. As indicated by 1H-NMR, R-85 could extract CsNO3 from water to nitrobenzene even in the presence of competing NaNO3. Moreover, this extracted CsNO3 could be released back to the aqueous phase by adding an aq. KClO4 solution. Ultimately the free receptor could be recovered after H2O/CHCl3 contact (Fig. 27). Thus, as mentioned earlier, the metathesis process within the ion-pair receptor provided the preliminary idea of the recyclable receptor design as well as the on–off switching mechanism of extraction.
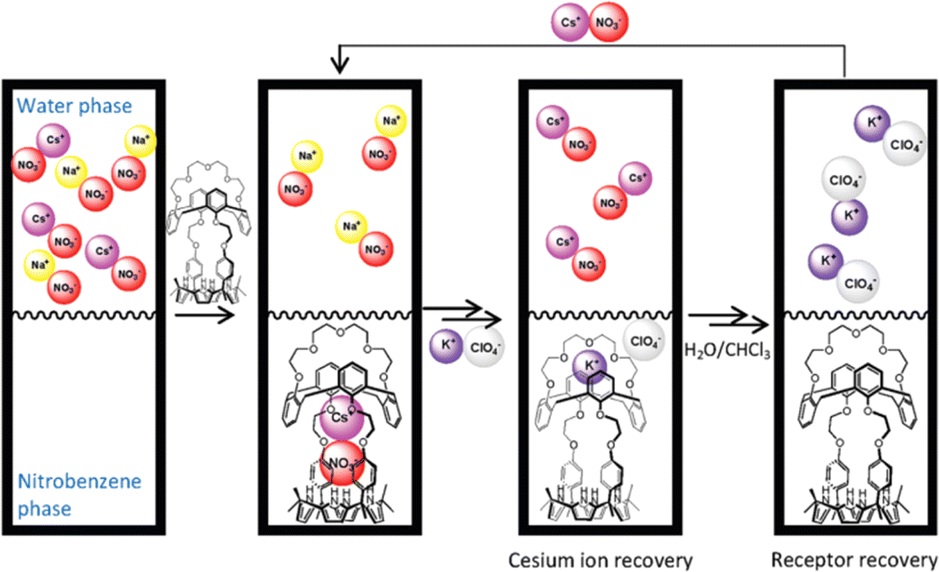 | ||
| Fig. 27 Schematic illustration of the two-phase extraction through the cation metathesis process by R-85. (Reprinted from ref. 125. Copyright 2012, ACS Publication.) | ||
In 2012, Romanski and Piątek prepared heteroditopic monomeric receptor R-95 and its polymer R-96 using an L-ornithine scaffold (Fig. 28).126 Solution phase studies in CH3CN indicated effective binding of Na+ into the aza-crown moiety which in turn enhanced the nitrate (NO3−) binding to the thiourea section. However, negative cooperativity was observed for Cl− and CH3COO− in the presence of Na+ indicating ion-pairing outside the receptor. Therefore, solvent extraction of NaNO3 from water to CHCl3 was performed. However, extraction of nitrate with R-95 appeared negligible, while the polymeric receptor R-96 could accomplish the same extraction with 32% efficiency as determined by the colorimetric nitrite–nitrate technique. Additionally, co-extraction of Na+ was confirmed by atomic emission spectroscopy which measured ∼44% sodium content by mass of the organic phase after the extraction. Furthermore, analogous extraction experiments for NaCl by R-96 resulted in 19% sodium extraction. This report emphasized that a properly designed polymeric system could be a better salt extractant than its monomeric analogue.
 | ||
| Fig. 28 Chemical structure of the receptors R-95 to R-112. | ||
The same group had synthesized a series of heteroditopic receptors R-97 to R-101 and studied their cooperative ion-pair interaction with respect to the geometrical as well as electronic environment of the receptors (Fig. 28).127 For R-97 to R-99, control 1H-NMR in CD3CN established the fact that the co-bound Na+ enhanced the nitrite (NO2−) binding rather than the non-coordinating TBA+ cation that indicates cooperative interaction for the former case. The urea oxygen atom upon coordinating with the bound Na+ increased the acidity of the urea protons resulting in such positive cooperativity. In addition to urea NH, the functionalised amide –NH substituent interacted with the anion through a hydrogen bond. Interestingly, R-99 showed the highest cooperativity compared to other receptors including R-101. This highlighted a stronger effect of the favourably positioned electron-withdrawing –CF3 group on polarizing NH protons for anion recognition. In fact, in R-101 the exceptionally strong influence of –SO2CF3 resulted in the subsequent deprotonation of the urea protons upon binding with the anion. However, in the LLE experiment, R-99 could extract NaNO2 to the receiving organic phase with 3.1% efficiency as proved by NMR analysis.
In 2018, the same group constructed receptor R-102 based on an L-valine platform (Fig. 28).128 Unlike L-ornithine amino acid, L-valine lacks the extra anion binding amine group. However, in R-102, acid functionalised 18-crown-6 strongly bound cations and enhanced the anion binding ability. Thus, in the presence of K+, the anion association constant for CH3COO− binding exceeded 107 orders as obtained from UV-Vis studies in CH3CN. Discernible chemical shifts of the NH protons (∼2 ppm) and crown –OCH2 protons indicated ∼100% CH3COOK salt extraction from water to the CDCl3 phase.
In 2021, they prepared a series of receptors R-103 to R-107 by changing the gap between core cation and anion binding groups (Fig. 28).129 The amide –NH proton, in addition to urea protons, played a significant role in CH3COO− binding. In R-104 and R-105, the increased conformational flexibility enabled greater involvement of the amide-NH proton in anion binding. Meanwhile, in R-106 and R-107 less involvement of amide –NH protons for anion binding was justified in terms of the larger gap between the urea group and amide –NH protons. In R-103, conformational rigidity restricted the amide –NH protons for anion binding. In accordance with the design features of the receptors, the increasing distance between the cation and anion binding group allows a more flexible and lipophilic host resulting in a more polar host–guest complex formation at the interface ultimately reducing the extraction capability. However, due to less conformational freedom, the most rigid receptor R-103 (76%) was less efficient than R-104 (85%) in the extraction of CH3COOK salt.
The critical feature of receptors R-95 to R-107 is the urea or thiourea connectivity involved in anion recognition. Romański and co-workers, instead, use squaramide as it provides more polarised protons compared to urea or thiourea groups. The increased aromaticity of the squaramide ring following anion complexation emphasises the defined polarisation of its protons. Accordingly, in 2019, heteroditopic receptors R-108 and R-109 were synthesized. Binding of K+ in the crown moiety which induced SO42− binding with the squaramide protons eventually overcame the dominating hydration energy for SO42− recognition in aqueous solution (Fig. 28).130 The solid-state structures revealed 4:1 receptor–SO42− complex formation through 8 hydrogen bonds, while a 1:1 stoichiometry was observed for KNO3. Such larger stoichiometry resulted in receptor–SO42− assembly which was confirmed by a reduction of the diffusion coefficient in DOSY NMR. Furthermore, such strong interaction associated with the receptor–SO42− complex preferentially extracted K2SO4 from the aqueous to the CDCl3 phase even in the presence of KNO3. Thus, from the solution containing 10 times higher KNO3, the receptor extracted K2SO4 with 64% efficiency.
Interestingly, receptor R-109 acted as a colorimetric sensor for SO42− extraction which was described in terms of –NH deprotonation of the extremely acidic squaramide.
Similarly, large stoichiometry (4:1) was observed for host–SO42− binding with receptors R-110 to R-112. Among them, R-111 showed better cooperativity for ion-pair binding as indicated by the initial UV-Vis studies in CH3CN.131 This could be attributed to the interplay of favourable allosteric and electrostatic factors between the receptor and the ion-pair. R-112, without the cationic domain, was less efficient for anion recognition (Fig. 28). For R-111, the formation of a similar host–K2SO4 core–shell assembly was confirmed and assumed to be the functional driving force for the selective extraction of K2SO4 to the CHCl3 phase. Atomic emission spectroscopy measured potassium content of 51%, 58%, 61%, and 90% by mass of the organic phase after extraction from 0.5 M aqueous solutions of KCl, KBr, KI, and K2SO4, respectively. Moreover, the control U-tube experiment established the relatively higher transportability of KNO3 and KCl salts by the R-111 over K2SO4, representing stronger receptor–K2SO4 complex formation.
8. Outlook and conclusions
Supramolecular non-covalent interactions have been used for LLE of anions as well as cations. Over the past few decades, this field of chemistry has evolved from almost nothing into a rapidly maturing field of research. In this perspective, we illustrate the progress of LLE by highlighting, in particular, receptor engineering for selective guest binding following the supramolecular approach. Moreover, the breadth of practical applications of this extraction principle in water treatment, environmental cleanup, and corrosion inhibition is also addressed. In these extraction processes, success is accomplished by leveraging relatively weak interactions. Separating ionic species, of great importance for environmental concern, is often fraught with difficulties due to their high hydration energies and this emphasizes the need for appropriate receptor design with sufficient binding sources. In this regard, we categorized the perspective based on an ionic guest and covered the liquid–liquid extractants for halides, oxo-anions, cations, and ion-pairs. This may pave the way for designing more receptors that can overcome the Hofmeister bias to extract more hydrated ions and cooperatively bind the guests to ensure ion-pair extraction. Besides, we envisioned that receptors having XB and ChB donor motifs instead of an HB domain could be a better option for LLE of selective anions due to their directional and hydrophobic character. Soluble polymeric receptor designing could be another fruitful way to conduct LLE which is also limited in number. Additionally, there is a need to investigate supramolecular self-assembly-driven LLE processes for the potential separation of highly solvated anions. This approach could also enhance the reusability of the receptors. In order to keep pace with the ever-expanding industrial demands specifically in the petrochemical, pharmaceutical, and metallurgical industry sectors, extensive research should be committed to the development of receptors capable of separating and purifying industrial guests via LLE. Nevertheless, this technique provides an economical and energy-efficient process for purifying wastewater, making it a promising and effective alternative in industry.In summary, the current instances of supramolecular receptors in the context of LLE presented in this perspective will catch the attention of chemists, biochemists, environmental scientists, and materials and metallurgy scientists along with the chemical and pharmaceutical industries. Furthermore, they will also attract the interest of undergraduate and graduate students who are interested in application-oriented research related to industrial and environmental issues.
Data availability
X-ray crystallography structural files are available free of charge from the CCDC viahttps://www.ccdc.cam.ac.uk/.Author contributions
S. P., A. S. M. I., I. G. and P. G. conceived and collaboratively drafted the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Science and Engineering Research Board (SERB; CRG/2019/002236; PDF/2019/000353) and J. C. Bose National Fellowship (JCB/2021/000032). A. S. M. I. acknowledged SERB for NPDF. S. P. and I. G. thanks CSIR and Inspire, respectively for fellowship.Notes and references
- A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. Ó'Máille and T. Gunnlaugsson, Chem, 2017, 3, 764–811 CAS.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, 2nd edn, 2009 Search PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed., 1988, 27, 89–112 CrossRef.
- N. Busschaert, C. Caltagirone, W. V. Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- E. D. Doidge, I. Carson, P. A. Tasker, R. J. Ellis, C. A. Morrison and J. B. Love, Angew. Chem., Int. Ed., 2016, 55, 12436–12439 CrossRef CAS.
- E. D. Doidge, L. M. M. Kinsman, Y. Ji, I. Carson, A. J. Duffy, I. A. Kordas, E. Shao, P. A. Tasker, B. T. Ngwenya, C. A. Morrison and J. B. Love, ACS Sustainable Chem. Eng., 2019, 7, 15019–15029 CrossRef CAS.
- M. Sharma, P. Sharma and J. N. Kim, RSC Adv., 2013, 3, 10103–10126 RSC.
- G. Zhang, Y. Ding, A. Hashem, A. Fakim and N. M. Khashab, Cell Rep. Phys. Sci., 2021, 2, 100470 CrossRef CAS.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- R. Ghosh, T. K. Ghosh, S. Pramanik, A. S. M. Islam and P. Ghosh, ACS Appl. Mater. Interfaces, 2023, 15, 25184–25192 CrossRef CAS.
- R. Ghosh, T. K. Ghosh and P. Ghosh, Dalton Trans., 2020, 49, 3093–3097 RSC.
- Q. He, G. I. Vargas-Zuniga, S. H. Kim, S. K. Kim and J. L. Sessler, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed.
- A. Li, H. Zhai, J. Li and Q. He, Chem. Lett., 2020, 49, 1125–1135 CrossRef CAS.
- Q. Zhang, Y. Zhou, M. Ahmed, N. M. Khashab, W. Han, H. Wang, Z. A. Page and J. L. Sessler, J. Mater. Chem. A, 2022, 10, 15297–15308 RSC.
- G. I. Vargas-Zúñiga, H. Qing and J. L. Sessler, Ion Exchange and Solvent Extraction, CRC Press, 2019, vol. 23, pp. 45–82 Search PubMed.
- G. I. Vargas-Zúñiga and J. L. Sessler, Comprehensive Supramolecular Chemistry II, ed. Atwood, J. L., Elsevier, Oxford, U.K., 2017, pp. 161–189 Search PubMed.
- B. A. Moyer and P. V. Bonnesen, Physical Factors in Anion Separations, in The Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. García-España, VCH, Weinheim, Germany, 1997, ch. 1, pp. 1–44 Search PubMed.
- K. Gloe, K. Gloe, M. Wenzel, L. F. Lindoy and F. Li, Supramolecular chemistry in solvent extraction: toward highly selective extractants and a better understanding of phase-transfer phenomena, CRC, U.S.A., 2013 Search PubMed.
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC.
- I. A. Rather, S. A. Wagay and R. Ali, Coord. Chem. Rev., 2020, 415, 213327 CrossRef CAS.
- L. J. Chen, S. N. Berry, X. Wu, E. N. W. Howe and P. A. Gale, Chem, 2020, 6, 61–141 CAS.
- M. Wenzel, J. J. Weigand and J. Inclusion Phenom, Macrocyclic Chem., 2017, 89, 247–251 CrossRef CAS.
- J. Rydberg, Solvent extraction principles and practice, revised and expanded, CRC press, 2nd edn, 2004 Search PubMed.
- V. S. Kislik, Solvent extraction: classical and novel approaches, Elsevier, 2011 Search PubMed.
- B. Schnell, R. Schurhammer and G. Wipff, J. Phys. Chem. B, 2004, 108, 2285–2294 CrossRef CAS.
- Y. Liu, W. Zhao, C. H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CrossRef CAS.
- A. Docker, I. Marques, H. Kuhn, Z. Zhang, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2022, 144(32), 14778–14789 CrossRef CAS.
- B. Schuur, B. J. V. Verkuijl, A. J. Minnaard, J. G. De Vries, H. J. Heeres and B. L. Feringa, Org. Biomol. Chem., 2011, 9, 36–51 RSC.
- X. M. Wu and W. J. Wu, Food Chem., 2012, 134, 597–601 CrossRef.
- X. Wu, A. M. Gilchrist and P. A. Gale, Chem, 2020, 6, 1296–1309 CAS.
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed.
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784–3809 RSC.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- X. Zhang and C. Wang, Chem. Soc. Rev., 2011, 40, 94–101 RSC.
- C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS PubMed.
- U. V. Gunten, Water Res., 2003, 37, 1469–1487 CrossRef PubMed.
- N. R. Johnston and S. A. Strobel, Arch. Toxicol., 2020, 94, 1051 CrossRef CAS.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- C. W. Chiu and F. P. Gabbaï, J. Am. Chem. Soc., 2006, 128, 14248–14249 CrossRef CAS PubMed.
- P. Das, A. K. Mandal, M. K. Kesharwani, E. Suresh, B. Ganguly and A. Das, Chem. Commun., 2011, 47, 7398–7400 RSC.
- P. R. Brotherhood and A. P. Davis, Chem. Soc. Rev., 2010, 39, 3633–3647 RSC.
- J. P. Clare, A. J. Ayling, J. B. Joos, A. L. Sisson, G. Magro, M. N. Pérez-Payán, T. N. Lambert, R. Shukla, B. D. Smith and A. P. Davis, J. Am. Chem. Soc., 2005, 127, 10739–10746 CrossRef CAS PubMed.
- S. J. Edwards, H. Valkenier, N. Busschaert, P. A. Gale and A. P. Davis, Angew. Chem., 2015, 127, 4675–4679 CrossRef.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and M. Milani, Chem.–Eur. J., 2010, 16, 4368–4380 CrossRef CAS.
- V. Amendola, L. Fabbrizzi, L. Mosca and F. P. Schmidtchen, Chem.–Eur. J., 2011, 17, 5972–5981 CrossRef CAS.
- A. Rostami, C. J. Wei, G. Guérin and M. S. Taylor, Angew. Chem., 2011, 123, 2107–2110 CrossRef.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., 2012, 124, 4502–4506 CrossRef.
- N. Busschaert, R. B. P. Elmes, D. D. Czech, X. Wu, I. L. Kirby, E. M. Peck, K. D. Hendzel, S. K. Shaw, B. Chan, B. D. Smith, K. A. Jolliffe and P. A. Gale, Chem. Sci., 2014, 5, 3617 RSC.
- A. Aydogan, D. J. Coady, V. M. Lynch, A. Akar, M. Marquez, C. W. Bielawski and J. L. Sessler, Chem. Commun., 2008, 12, 1455–1457 RSC.
- P. Sokkalingam, S. J. Hong, A. Aydogan, J. L. Sessler and C. H. Lee, Chem.–Eur. J., 2013, 19, 5860–5867 CrossRef CAS PubMed.
- A. Aydogan and A. Akar, Chem.–Eur. J., 2012, 18, 1999–2005 CrossRef CAS PubMed.
- A. Aydogan, D. J. Coady, S. K. Kim, A. Akar, C. W. Bielawski, M. Marquez and J. L. Sessler, Angew. Chem., Int. Ed., 2008, 47, 9648–9652 CrossRef CAS.
- H. Kim, K. I. Hong, J. H. Lee, P. Kang, M. G. Choi and W. D. Jang, Chem. Commun., 2018, 54, 10863–10865 RSC.
- K. P. McDonald, B. Qiao, E. B. Twum, S. Lee, P. J. Gamache, C. H. Chen, Y. Yi and A. H. Flood, Chem. Commun., 2014, 50, 13285–13288 RSC.
- P. Bose, R. Dutta, S. Santra, B. Chowdhury and P. Ghosh, Eur. J. Inorg. Chem., 2012, 35, 5791–5801 CrossRef.
- W. Liu, A. G. Oliver and B. D. Smith, J. Org. Chem., 2019, 84, 4050–4057 CrossRef CAS PubMed.
- L. H. Bowen and T. R. Rood, J. Inorg. Nucl. Chem., 1966, 28, 1985–1990 CrossRef CAS.
- V. K. Davis, C. M. Bates, K. Omichi, B. M. Savoie, N. Momcilovic, Q. Xu, W. J. Wolf, M. A. Webb, K. J. Billings, N. H. Chou, S. Alayoglu, R. Mckenney, I. M. Darolles, N. G. Nair, A. Hightower, D. Rosenberg, M. Ahmed, C. J. Brooks, T. F. Miller III, R. H. Grubbs and S. C. Jones, Science, 2018, 362, 1144–1148 CrossRef CAS.
- P. R. Wittbrodt and C. D. Palmer, Environ. Sci. Technol., 1995, 29, 255–263 CrossRef CAS.
- I. Ravikumar and P. Ghosh, Chem. Soc. Rev., 2012, 41, 3077–3098 RSC.
- B. A. Moyer, R. Custelcean, B. P. Hay, J. L. Sessler, K. Bowman-James, V. W. Day and S. O. Kang, Inorg. Chem., 2013, 52, 3473–3490 CrossRef CAS PubMed.
- S. Q. Chen, W. Zhao and B. Wu, Front. Chem., 2022, 10, 905563 CrossRef CAS PubMed.
- H. Kaur, S. Sinha, V. Krishnan and R. R. Koner, Dalton Trans., 2021, 50, 8273–8291 RSC.
- M. D. Cohen, B. Kargacin, C. B. Klein and M. Costa, Crit. Rev. Toxicol., 1993, 23, 255–281 CrossRef CAS PubMed.
- M. Bayrakcı and Ş. Yiğiter, Tetrahedron, 2013, 69, 3218–3224 CrossRef.
- S. Sayin, S. Eymur and M. Yilmaz, Ind. Eng. Chem. Res., 2014, 53, 2396–2402 CrossRef CAS.
- C. A. Seipp, N. J. Williams, V. S. Bryantsev and B. A. Moyer, Sep. Sci. Technol., 2018, 53, 1864–1873 CrossRef CAS.
- N. J. Williams, C. A. Seipp, K. A. Garrabrant, R. Custelcean, E. Holguin, J. K. Keum, R. J. Ellis and B. A. Moyer, Chem. Commun., 2018, 54, 10048–10051 RSC.
- D. J. Wood and J. D. Law, Sep. Sci. Technol., 1997, 32, 241–253 CrossRef CAS.
- M. L. Dietz, E. P. Horwitz and R. D. Rogers, Solvent Extr. Ion Exch., 1995, 13, 1–17 CrossRef CAS.
- K. Gloe, H. Stephan and M. Grotjahn, Chem. Eng. Technol., 2003, 26, 1107–1117 CrossRef CAS.
- C. J. Borman, R. Custelcean, B. P. Hay, N. L. Bill, J. L. Sessler and B. A. Moyer, Chem. Commun., 2011, 47, 7611–7613 RSC.
- L. R. Eller, M. Stępień, C. J. Fowler, J. T. Lee, J. L. Sessler and B. A. Moyer, J. Am. Chem. Soc., 2007, 129, 11020–11021 CrossRef CAS PubMed.
- C. J. Fowler, T. J. Haverlock, B. A. Moyer, J. A. Shriver, D. E. Gross, M. Marquez, J. L. Sessler, A. Hossain and K. Bowman-James, J. Am. Chem. Soc., 2008, 130, 14386–14387 CrossRef CAS PubMed.
- C. J. Borman, R. Custelcean, B. P. Hay, N. L. Bill, J. L. Sessler and B. A. Moyer, Chem. Commun., 2011, 47, 7611–7613 RSC.
- B. A. Moyer, F. V. Sloop Jr, C. J. Fowler, T. J. Haverlock, H. A. Kang, L. H. Delmau, D. M. Bau, M. A. Hossain, K. Bowman-James, J. A. Shriver, N. L. Bill, D. E. Gross, M. Marquez, V. M. Lynch and J. L. Sessler, Supramol. Chem., 2010, 22, 653–671 CrossRef CAS.
- S. K. Kim, J. Lee, N. J. Williams, V. M. Lynch, B. P. Hay, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 15079–15085 CrossRef CAS PubMed.
- R. Custelan, Chem. Commun., 2013, 49, 2173–2182 RSC.
- C. Jia, B. Wu, S. Li, X. Huang, Q. Zhao, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 486–490 CrossRef CAS PubMed.
- S. Q. Chen, S. N. Yu, W. Zhao, L. Liang, Y. Gong, L. Yuan, J. Tang, X. J. Yang and B. Wu, Inorg. Chem. Front., 2022, 9, 6091–6101 RSC.
- B. Akhuli, I. Ravikumar and P. Ghosh, Chem. Sci., 2012, 3, 1522–1530 RSC.
- R. Dutta, S. Chakraborty, P. Bose and P. Ghosh, Eur. J. Inorg. Chem., 2014, 25, 4134–4143 CrossRef.
- L. Qin, S. J. N. Vervuurt, R. B. P. Elmes, S. N. Berry, N. Proschogo and K. A. Jolliffe, Chem. Sci., 2020, 11, 201–207 RSC.
- S. K. Dey, S. Pereira, S. S. Harmalkar, S. N. Mhaldar, V. V. Gobrea and C. Janiak, CrystEngComm, 2020, 22, 6152–6160 RSC.
- S. K. Dey, B. G. Hernández, V. V. Gobre, D. Woschko, S. S. Harmalkar, F. R. Gayen, B. Saha, R. L. Goswamee and C. Janiak, Dalton Trans., 2022, 51, 15239 RSC.
- R. Alberto, G. Bergamaschi, H. Braband, T. Fox and V. Amendola, Angew. Chem., 2012, 124, 9910–9914 CrossRef.
- V. Amendola, G. Alberti, G. Bergamaschi, R. Biesuz, M. Boiocchi, S. Ferrito and F.-P. Schmidtchen, Eur. J. Inorg. Chem., 2012, 21, 3410–3417 CrossRef.
- G. V. Kolesnikov, K. E. German, G. Kirakosyan, I. G. Tananaev, Y. A. Ustynyuk, V. N. Khrustalev and E. A. Katayev, Org. Biomol. Chem., 2011, 9, 7358–7364 RSC.
- W. Zhou, A. Li, P. A. Gale and Q. He, Cell Rep., 2022, 3, 100875 CAS.
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez and J. R. Nitschke, Angew. Chem., Int. Ed., 2018, 57, 3717–3721 CrossRef CAS PubMed.
- H. Zeng, P. Liu, G. Feng and F. Huang, J. Am. Chem. Soc., 2019, 141, 16501–16511 CrossRef.
- T. K. Ghosh, S. Chakraborty, B. Chowdhury and P. Ghosh, Inorg. Chem., 2017, 56, 5371–5382 CrossRef CAS.
- R. Andrews, S. Begum, C. J. Clemett, R. A. Faulkner, M. L. Ginger, J. Harmer, M. Molinari, G. M. B. Parkes, Z. M. H. Qureshi, C. R. Rice, M. D. Ward, H. M. Williams and P. B. Wilson, Angew. Chem., Int. Ed., 2020, 132, 20660–20664 CrossRef.
- S. Dubchak, Impact of Caesium on Plants and the Environment, ed. D. K. Gupta and C. Walther, Springer International Publishing, Cham, 2017; pp. pp. 1–18 Search PubMed.
- G. U. Akkuş and S. Aslan, Int. J. Org. Chem., 2017, 7, 301–311 CrossRef.
- R. Yi, C. Xu, T. Sun, Y. Wang, G. Ye, S. Wang and J. Chen, Sep. Purif. Technol., 2018, 199, 97–104 CrossRef CAS.
- L. Gupta, A. C. Hoepker, K. J. Singh and D. B. Collum, J. Org. Chem., 2009, 74, 2231–2233 CrossRef CAS PubMed.
- H. Haferkamp, M. Niemeyer, R. Boehm, U. Holzkamp, C. Jaschik and V. Kaese, Mater. Sci. Forum, 2000, 350, 31–42 Search PubMed.
- C. Grosjean, P. Herrera Miranda, M. Perrin and P. Poggi, Renewable Sustainable Energy Rev., 2012, 16, 1735–1744 CrossRef.
- O. A. Avramchik, E. I. Korotkova, E. V. Plotnikov, A. N. Lukina and Y. A. Karbainov, J. Pharm. Biomed. Anal., 2005, 37, 1149–1154 CrossRef CAS PubMed.
- I. Oral and V. Abetz, Macromol. Rapid Commun., 2021, 42, 2000746 CrossRef CAS PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- C. J. Pederson, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef.
- I. Oral and V. Abetz, Soft Matter, 2022, 18, 934–937 RSC.
- J. Zhang, M. Wenzel, J. Steup, G. Schaper, F. Hennersdorf, H. Du, S. Zheng, L. F. Lindoy and J. J. Weigand, Chem.–Eur. J., 2022, 28, e2021036 Search PubMed.
- M. T. Reetz, C. M. Niemeyer and K. Harms, Angew. Chem., Int. Ed., 1991, 30, 1472–1474 CrossRef.
- A. J. McConnell and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 5052–5061 CrossRef CAS.
- A. McConnell, A. Docker and P. Beer, ChemPlusChem, 2020, 85, 1824–1841 CrossRef CAS PubMed.
- S. A. Wagay, L. Khan and R. Ali, Chem.–Asian J., 2022, e202201080 Search PubMed.
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS.
- K. I. Hong, H. Kim, Y. Kim, M. G. Choi and W.-D. Jang, Chem. Commun., 2020, 56, 10541–10544 RSC.
- D. Jagleniec, S. Siennicka, Ł. Dobrzycki, M. Karbarz and J. Romański, Inorg. Chem., 2018, 57, 12941–12952 CrossRef CAS PubMed.
- K. Maršálek and V. Šindelář, Org. Lett., 2020, 22, 1633–1637 CrossRef.
- N. A. D. Simone, M. Chvojka, J. Lapešová, L. Martínez-Crespo, P. Slávik, J. Sokolov, S. J. Butler, H. Valkenier and V. Šindelář, J. Org. Chem., 2022, 87, 9829–9838 CrossRef PubMed.
- I. Ravikumar, S. Saha and P. Ghosh, Chem. Commun., 2011, 47, 4721–4723 RSC.
- S. K. Kim, V. M. Lynch, N. J. Young, B. P. Hay, C.-H. Lee, J. S. Kim, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 20837–20843 CrossRef CAS PubMed.
- M. Zakrzewski, D. Załubiniak and P. Piątek, Dalton Trans., 2018, 47, 323–330 RSC.
- C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS.
- X. Chi, G. M. Peters, F. Hammel, C. Brockman and J. L. Sessler, J. Am. Chem. Soc., 2017, 139, 9124–9127 CrossRef CAS.
- X. Chi, G. M. Peters, C. Brockman, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 13219–13222 CrossRef CAS PubMed.
- Q. He, Z. Zhang, J. T. Brewster, V. M. Lynch, S. K. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 9779–9782 CrossRef CAS PubMed.
- Q. He, G. M. Peters, V. M. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2017, 56, 13396–13400 CrossRef CAS.
- B. M. Rambo, S. K. Kim, J. S. Kim, C. W. Bielawski and J. L. Sessler, Chem. Sci., 2010, 1, 716–722 RSC.
- Y. Yeon, S. Leem, C. Wagen, V. M. Lynch, S. K. Kim and J. L. Sessler, Org. Lett., 2016, 18, 4396–4399 CrossRef CAS.
- S. K. Kim, G. I. Vargas-Zúñiga, B. P. Hay, N. J. Young, L. H. Delmau, C. Masselin, C.-H. Lee, J. S. Kim, V. M. Lynch, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 1782–1792 CrossRef CAS.
- J. Romański and P. Piątek, Chem. Commun., 2012, 48, 11346–11348 RSC.
- P. Piątek, M. Karbarz and J. Romański, Dalton Trans., 2014, 43, 8515–8522 RSC.
- M. Zakrzewski, N. Kwietniewska, W. Walczaka and P. Piątek, Chem. Commun., 2018, 54, 7018–7021 RSC.
- M. Zakrzewski and P. Piątek, New J. Chem., 2021, 45, 18635–18640 RSC.
- D. Jagleniec, L. Dobrzycki, M. Karbarz and J. Romański, Chem. Sci., 2019, 10, 9542–9547 RSC.
- M. Zaleskaya, M. Karbarz, M. Wilczek, Ł. Dobrzycki and J. Romański, Inorg. Chem., 2020, 59, 13749–13759 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |