
3D mixed ion/electron-conducting scaffolds for stable sodium metal anodes†
Xuan
Lu
ab,
Xiuxia
Zhao
c,
Shujiang
Ding
 *a and
Xiaofei
Hu
*a
*a and
Xiaofei
Hu
*a
aSchool of Chemistry, Engineering Research Center of Energy Storage Materials and Devices (Ministry of Education), State Key Laboratory of Electrical Insulation and Power Equipment, Xi'an Key Laboratory of Sustainable Energy Materials Chemistry, Xi'an Jiaotong University, Xi'an, Shaanxi 710049, People's Republic of China. E-mail: Dingsj@mail.xjtu.edu.cn; xiaofei.hu@xjtu.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin, 300071, People's Republic of China
cShaanxi Coal Chemical Industry Technology Research Institute Co., Ltd., Xi'an, Shaanxi 710100, People's Republic of China
First published on 13th December 2023
Abstract
Sodium (Na) metal batteries represent an optimal choice for the forthcoming generation of large-scale, cost-effective energy storage systems. However, Na metal anodes encounter several formidable challenges during the Na plating and stripping processes, which encompass the formation of an unstable solid electrolyte interface, uncontrollable dendrite growth, and infinite volume expansion. These issues result in a reduced Coulombic efficiency, shortened battery lifespan, and potential safety hazards, thereby constraining their commercial development. Therefore, addressing these challenges to ensure the cycling stability of Na metal anodes stands as a paramount requirement for practical applications. Among the reported strategies, three-dimensional conductive scaffolds possessing a high surface area and porous structure are acknowledged for their significant potential in stabilizing Na metal anodes. Compared with conventional electron-conducting scaffolds, emerging mixed ion/electron-conductive (MIEC) scaffolds provide rapid ion/electron transport pathways, which enable uniform Na+ flux and promote dendrite-free Na deposition, thus improving the cycle life of Na metal anodes, even at high current densities and large areal capacities. Therefore, this review primarily emphasizes the recent progress in applying MIEC scaffolds to Na metal anodes. It introduces diverse design methods, examines the electrochemical performance of MIEC scaffolds, and delves into their regulation mechanisms over Na deposition behaviour. Finally, the development prospects and research strategies for MIEC scaffolds from both fundamental research and practical application perspectives are discussed, suggesting directions for further designing high-performance Na metal batteries.
 Xuan Lu | Xuan Lu is currently an Assistant Professor at the School of Chemistry, Xi'an Jiaotong University. He received his Ph.D. degree from the School of Electrical Engineering, Xi'an Jiaotong University in 2020. His research centered around the anode protection of sodium metal batteries and the design of high-performance sodium–air/sulfur batteries. |
 Xiuxia Zhao | Xiuxia Zhao obtained her Ph.D. degree from the School of Chemistry at Nankai University in 2017. Her research interests primarily focus on the design and synthesis of carbon nanomaterials derived from metal organic frameworks for applications in electrochemical energy storage devices and photoelectrocatalysis. |
 Shujiang Ding | Shujiang Ding is a professor in the School of Chemistry at Xi'an Jiaotong University. He received his B.S. degree (2001) and M.S. degree (2004) from the School of Chemical Engineering and Technology at Xi'an Jiaotong University, followed by his Ph.D. (2007) from the Institute of Chemistry, Chinese Academy of Sciences. He then worked as a postdoctoral fellow at Nanyang Technological University, Singapore. His research involves fundamental investigations into the application of polymer/inorganic nanostructure composite materials in various electrochemical energy storage systems, including lithium/sodium-ion batteries, lithium–sulfur batteries, solid-state batteries, fuel cells, and lithium-ion battery recycling. |
 Xiaofei Hu | Xiaofei Hu is a professor in the School of Chemistry at Xi'an Jiaotong University. He received his B.S. degree (2012) from the School of Chemical Engineering and Technology at Tianjin University, followed by his Ph.D. (2017) from the School of Chemistry at Nankai University. He then joined as a postdoctoral fellow from Thayer School of Engineering at Dartmouth College (Hanover, US). His current research focuses on high-energy-density alkaline (Li, Na)–gas (air, O2, CO2) batteries and wide-temperature sodium metal batteries. |
1. Introduction
The rapid development of emerging fields, such as new energy vehicles and smart grids, has presented a formidable challenge to commercial lithium (Li)-ion batteries (LIBs) utilizing graphite anodes. These batteries struggle to meet the increasing demands of future society for higher specific capacity and greater energy density. Concurrently, the limited availability of Li resources, accounting for a mere 0.006% of the Earth's crust, serves as a further constraint in their utilization in large-scale energy storage applications, including green energy power generation and smart grids. In contrast, sodium (Na) stands as a notable abundant resource (2.64% of the Earth's crust) and is economically accessible, with electrochemical behaviours closely resembling those of Li.1–7 Therefore, Na-ion batteries (SIBs) are considered as an ideal alternative for forthcoming large-scale, low-cost energy storage systems.The commercialization of SIBs hinges on the development of low-cost, high-performance electrode materials. Although the research and development of cathode materials for SIBs have reached a relatively mature stage, the commercialization process is impeded by the sluggish progress in anode material development. Among the various anode materials, hard carbon stands out as a promising candidate due to its disordered structure and abundant sources. The numerous vacancies, edges, and defects within the hard carbon structure serve as active sites for Na storage, providing substantial Na storage capacity.8 However, the enhancement of energy density in SIBs utilizing hard carbon anodes remains constrained (approximately 200 W h kg−1).9 In comparison to hard carbon and other anodes, Na metal anodes offer a high theoretical specific capacity of 1166 mA h g−1 and a low redox potential of −2.71 V, which can significantly increase the energy density of SIBs.10–12 Besides, Na metal anodes can be paired with non-intercalated cathode materials, such as sulfur (S) and oxygen (O2). This enables the achievement of theoretical specific capacities, reaching up to 1274 W h kg−1 for room temperature Na–S batteries13–16 and 1605 W h kg−1 for Na–O2 batteries,17–20 which far surpass the current energy density levels obtained in LIBs/SIBs.
However, the utilization of Na metal anodes presents a series of challenges, including the formation of an unstable solid electrolyte interface (SEI), uncontrolled dendrite growth, and infinite volume expansion.21,22 Due to its high reactivity, Na metal anode undergoes spontaneous reactions with organic solvents in the electrolyte, forming an SEI film on its surface to impede further interactions between Na and the electrolyte (Fig. 1a). Nevertheless, the volume expansion/shrinkage induced by Na plating–stripping processes will cause the SEI to continuously crack, resulting in an uneven Na+ flux, which triggers non-uniform Na deposition and fosters dendrite growth. Simultaneously, the broken SEI fails to hinder side reactions between the electrolyte and the Na metal anode, leading to the depletion of the anode active materials and continuous electrolyte consumption, which results in a rapid decline in the coulombic efficiency (CE) of the battery. Moreover, Na dendrites are prone to fracture after growth, giving rise to the formation of “dead Na”, which significantly increases the interface resistance and ultimately leads to a marked reduction in battery performance. Additionally, the continuously growing Na dendrites may pierce the separator and cause an internal short circuit within the battery, posing severe safety risks.3 Hence, the key to the development of high-performance Na metal batteries lies in the suppression of dendrite growth and volume expansion issues in Na metal anodes to ensure uniform Na deposition and thereby maintain the stability of the electrode–electrolyte interface.
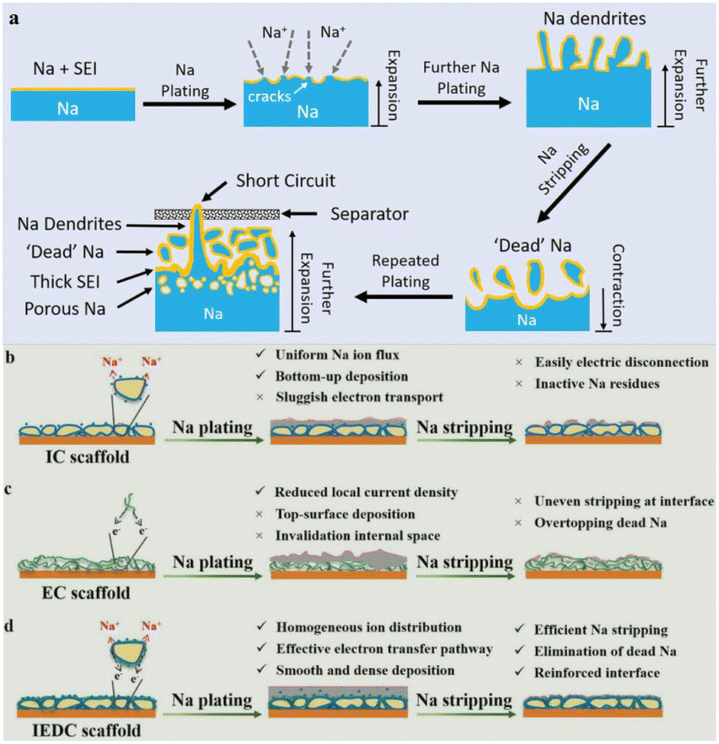 | ||
| Fig. 1 (a) Schematic diagram of the issues faced by the Na metal during the plating and stripping processes. Reproduced with permission from ref. 3 Copyright 2020, Wiley-VCH. Schematic diagrams of the deposition of the Na metal on (b) IC, (c) EC and (d) IEDC scaffolds during the plating and stripping processes. Reproduced with permission from ref. 38 Copyright 2021, Wiley-VCH. Note: The acronym IEDC refers to ion/electron dual-conducting, maintaining coherence with the concept of MIEC elucidated in this review. | ||
Currently, researchers are engaged in exploring three main strategies to solve the aforementioned issues: (I) the optimization of electrolyte composition and additives;23–25 (II) the development of artificial SEIs;26–30 (III) the utilization of three-dimensional (3D) conductive scaffolds for fabricating composite Na–scaffold anodes (CSSAs).31–36 However, the first two strategies can only sustain the prolonged cycling life of Na metal anodes at low current densities and low areal capacities, but are unable to withstand the extreme volume change and mechanical deformation induced by the Na metal during long-term cycling at high current densities required for commercial applications, which leads to the repeated rupture of the SEI film and uncontrolled dendrite growth, posing a severe threat to the reversibility and safety of Na metal anodes.37 In contrast, the third strategy exhibits significant improvements in enhancing the cycling stability of Na anodes at high current densities. The 3D conductive scaffold characterized by its porous structure has sufficient internal space to accommodate Na deposition, effectively alleviating the high degree of Na volume expansion. Additionally, its excellent electrical conductivity and high specific surface area help in reducing local current density, promoting uniform Na+ flux, and further suppressing the dendritic Na growth.
According to the characteristics of conductive scaffolds, they can be broadly categorized into three types: ion-conducting (IC) scaffolds, electronic-conducting (EC) scaffolds and mixed ion/electron-conducting (MIEC) scaffolds. The functionality of the IC scaffold resembles that of an artificial SEI or solid-state electrolyte, which possesses electronic insulation properties and features abundant ion migration channels. This enables the selective deposition of the Na metal beneath the scaffold, promoting a “bottom-up” deposition mode. However, the electrical insulation of the IC scaffold makes the deposited Na metal the sole electronic conductor within the structure. Consequently, the inside plated Na metal is susceptible to electrical disconnection during the stripping process, which reduces the reversibility of the Na metal anode (Fig. 1b).38 Commercial EC scaffolds, including carbon-based scaffolds (e.g., CNT, graphene, carbon felt) and metal-based scaffolds (e.g., Cu foam, Ni foam), are a type of scaffold that have been extensively studied.39–43 However, the plating/stripping process of the Na metal involves the simultaneous participation of both ions and electrons. For the EC scaffold, during the electrochemical process, restricted by limited ion transport, the Na+ flux participating in the charge transfer reaction at the scaffold/electrolyte interface is far higher than the Na+ diffusion flux inside the scaffold, which results in Na+ being preferentially reduced on the exterior of the scaffold (Fig. 1c).38 Consequently, metallic Na tends to accumulate and deposit outside the scaffold, rather than being uniformly distributed inside the scaffold, which leads to local overgrowth of Na dendrites. Simultaneously, the excessive accumulation of the Na metal can block the ion transport pathways inside the scaffold, further preventing ions from being transferred and deposited within the scaffold, which causes the interior of the scaffold to completely lose its buffering effect on the volume expansion of the Na metal.34,44,45 Therefore, the construction of fast ion transport pathways on the EC scaffold to enhance and balance the rapid transfer of both ions and electrons has emerged as a potential choice for realizing high-performance Na metal anodes (Fig. 1d). Herein, we choose MIEC scaffolds as the subject of this review and summarize the recent exemplary instances of designing such scaffolds. Through critical analysis and comparisons of these works, this review provides deep insights into the challenges and bottlenecks faced in the current research and provides an outlook on development directions. We hope that this will open up a new path for exploring novel approaches and ultimately achieving the commercial development of Na metal anodes.
2. Sodiophilicity and sodiophobicity
Melt infusion and electrodeposition represent the principal methodologies for fabricating CSSAs.35,46 The former leverages the low melting point of Na (97.72 °C), wherein the Na metal blocks are melted at elevated temperature and then placing the scaffold in it to allow the molten Na to infiltrate. The latter entails the direct deposition of the Na metal onto the scaffold's surface via an electrochemical process. Each approach possesses inherent advantages and drawbacks. Electrodeposition involves a complex battery assembly and disassembly process, whereas melt infusion is comparatively straightforward and does not necessitate electrochemical apparatus. However, melt infusion demands operation under a high-temperature inert atmosphere and lacks the capability to quantify molten Na, which may result in reduced active Na utilization. In contrast, electrodeposition enables precisely control over the quantity of deposited Na, thereby helps to improve the active Na utilization and energy density of the battery.Regardless of the method employed, the preparation of high-quality CSSAs requires a low surface energy between the scaffold and Na. Specifically, the scaffold should manifest favourable wettability to Na, denoted by the term “sodiophilicity” within the field of Na metal batteries. The nucleation-growth behaviours of the Na metal during the plating and stripping processes are closely linked to the sodiophilicity of the deposited scaffold. A sodiophilic scaffold provides abundant electrochemical active sites, acting as “nucleation seeds” for Na deposition. This facilitates uniform Na nucleation and promotes horizontal deposition of the Na metal instead of vertical growth, thus avoiding the formation of Na dendrites. However, a predominant trait observed in most 3D scaffolds is “sodiophobicity”, with notable Na nucleation overpotentials. This property makes them induce uneven deposition and dendrite growth of Na. To augment the sodiophilicity of 3D scaffolds and enable their effective integration with the Na metal, various sodiophilic materials have been developed, including:
(i) carbon-based materials with defects, functional groups, and heteroatom doping.47–49
Carbon-based materials serve as prominent hosts for Na metal anodes owing to their superior electronic conductivity. However, their inherent sodiophobicity poses a constraint on the uniform nucleation of Na. To address this limitation, Peng et al.50 utilized oxygen plasma treatment to modify sodiophilic oxygen functional groups on oriented carbon nanotube membranes (Fig. 2a). This modification results in the creation of a sodiophilic carbon nanotube interface (Of-CNT), effectively preventing dendrite formation during Na metal growth. First-principles calculations using density functional theory (DFT) reveal that the binding energies of Na with pristine CNTs (p-CNTs) and copper (Cu) foil are only −2.55 and −1.21 eV, respectively, while Na has stronger interactions with the Of-CNT surface (carbonyl oxygen: −3.45 eV, carboxyl oxygen: −3.32 eV, ether oxygen: −3.13 eV, and carbonyl oxygen: −2.96 eV), as shown in Fig. 2b and c. Relative to p-CNTs and Cu foil, the local charge density between oxygen functional groups in Of-CNTs is significantly increased (Fig. 2d and e). This suggests that oxygen functional groups facilitate the absorption of the surrounding Na+ flux, which create abundant and uniformly distributed active sites for initial Na nucleation and subsequent uniform Na deposition. The strong interaction between Na atoms and oxygen functional groups emerges as the crucial factor in achieving uniform Na nucleation and growth.
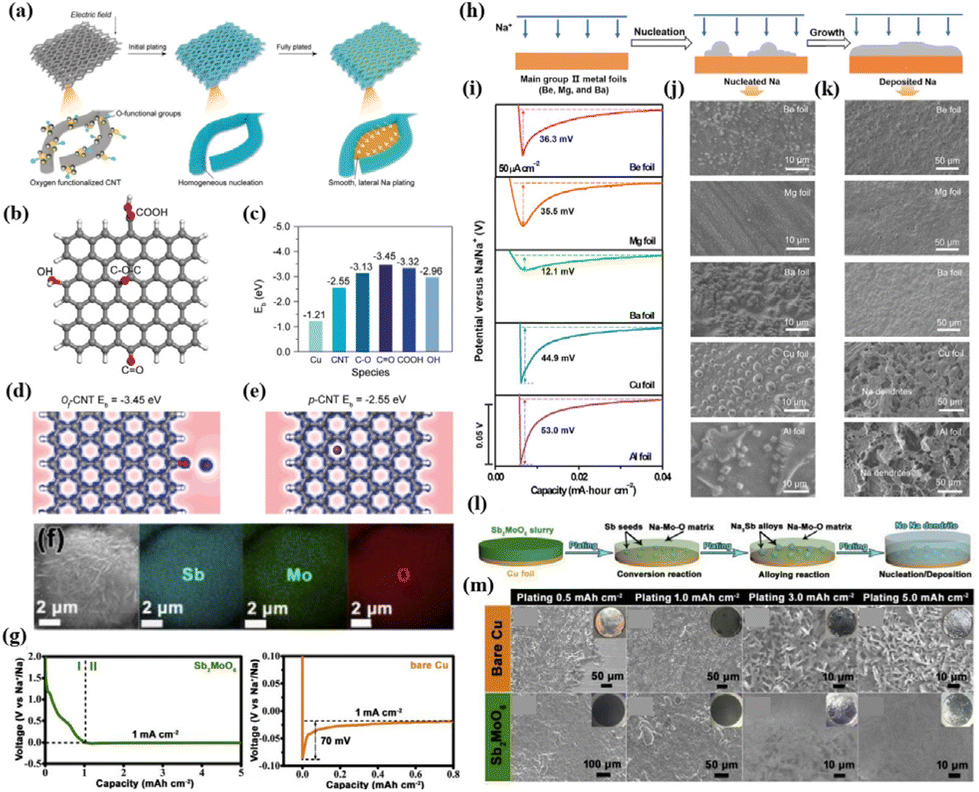 | ||
| Fig. 2 (a) Schematic diagram of “dendrite-free” Na metal deposition regulated by Of-CNTs; (b) atomic model employed in DFT calculations; (c) binding energies of Na with Cu, p-CNTs, and various functional groups on Of-CNTs; and deformation charge densities at a Na adsorption site of (d) Of-CNTs and (e) p-CNTs, respectively. Reproduced with permission from ref. 50 Copyright 2019, Wiley-VCH. (f) SEM image of the as-prepared SMO microsphere and its corresponding EDS mapping images; (g) plating curves of the Na metal on SMO and bare Cu substrates; (l) schematic diagram of the evolution of SMO during the Na plating process; and (m) SEM images of the deposition morphology of varying amounts of the Na metal on SMO and Cu substrates. Reproduced with permission from ref. 62 Copyright 2020, Elsevier. (h) Schematic diagram of the nucleation and growth of metallic Na on the main group II metal substrate; (i) nucleation overpotentials of the Na metal on different substrates at 50 μA cm−2; and SEM images of (j) nuclei and (k) growth of the Na metal on various substrates. Reproduced with permission from ref. 54 Copyright 2019, AAAS. | ||
(ii) Metals capable of alloying with Na (such as Sn, Sb, Mg).51–53
Yang et al.54 systematically investigated the nucleation process of Na on various metal foils by selecting main group II metals (Be, Mg, and Ba), aluminium (Al), and Cu as substrates for Na deposition (Fig. 2h). Notably, main group II metals can form solid solution alloys with Na, whereas the formation of such alloys is challenging for Al and Cu. These findings indicate substantial nucleation overpotentials for Al and Cu foils measured at 53.0 mV and 44.9 mV, respectively. In contrast, main group II metal foils exhibit significantly lower overpotentials (36.3, 35.5, and 12.1 mV for Be, Mg, and Ba, respectively), suggesting a reduced nucleation barrier of Na on main group II metal substrates (Fig. 2i). Scanning electron microscopy (SEM) images reveal that Na deposited on Be, Mg, and Ba substrates features a smooth surface with a uniform morphology and the absence of Na dendrite formation. Conversely, the surfaces of Na deposited on Al and Cu substrates exhibit a substantial presence of Na dendrites (Fig. 2j and k).
(iii) Metal oxides capable of undergoing redox reactions with Na (such as CuO, SnO2, ZnO).55–61
Establishing a metal oxide layer on a sodiophobic substrate has proven to significantly enhance the affinity between Na and the substrate. Typically, these metal oxides have the ability to undergo redox reactions with Na, consequently reducing the Na nucleation barrier on the substrate. Additionally, certain metal sulfides, nitrides, and phosphides also exhibit similar properties. Li et al.62 synthesized microspheres of the bimetallic oxide Sb2MoO6 (SMO) using a hydrothermal method and investigated the deposition behaviour of the Na metal on SMO (Fig. 2f). The findings reveal a two-stage deposition process of Na on SMO (Fig. 2l). Initially, Na engages in a redox reaction with SMO, resulting in the formation of Sb nanoparticles and Na–Mo–O species. The formed Sb acts as sodiophilic sites, further reacting with Na to produce a Na3Sb alloy, effectively reducing the nucleation barrier for Na. Consequently, the nucleation overpotential of Na on the SMO substrate is merely 14 mV. In contrast, the nucleation overpotential on bare Cu (foil) is higher at 70 mV due to the difficulty in alloying between Cu and Na (Fig. 2g). A comparative analysis with bare Cu demonstrates that the SMO substrate maintains a flat and smooth Na layer even after plating 5 mA h cm−2 of Na, while bare Cu exhibits aggressive dendritic Na formation with only 0.5 mA h cm−2 plating (Fig. 2m).
The interaction between Na and these materials, either through physical or chemical reactions, facilitates uniform Na nucleation and growth. This phenomenon parallels the wetting of a liquid on a solid substrate that has an absorption/reaction effect on it, known as “reactive wetting” behaviour.63 In recent years, researchers have regulated Na deposition behaviour by introducing sodiophilic sites into 3D scaffolds, which effectively improves the CE and cycle life of Na metal anodes.52,53,64,65
3. 3D electron-conducting scaffolds
Porous Cu foam (CF) functions as an excellent conductive, mechanically stable, and corrosion-resistant EC scaffold. However, given its inherent sodiophobicity nature, Li et al.57 chemically oxidized CF and infused molten Na metal into it at 500 °C. This chemical treatment results in a homogeneous infusion of molten Na into the CF (Fig. 3a), yielding a CSSA with a core–shell structure (Na@O-CF). The results demonstrate a notable enhancement in electrochemical performance when using O-CF as a host for Na metal in comparison to bare CF. The Na@O-CF symmetric cell exhibits stable cycling for over 300 h at a current density of 2 mA cm−2 and an areal capacity of 3 mA h cm−2, whereas the Na@CF anode displays substantial overpotential and limited lifespan (<150 h) (Fig. 3c). Additionally, when compared with the Na3V2(PO4)3 (NVP) cathode to assemble a full cell, it demonstrates exceptional cycling stability at a high current density of 5C (1C = 117 mA g−1), outperforming the pure Na anode (Fig. 3d). Mechanistic studies by the authors reveal two primary reasons for the performance improvement: (i) a chemical reaction occurs between Na and O-CF (Na + CuXO → Cu + Na2O) during the molten Na infusion process into the host (Fig. 3b), which reduces the Na nucleation barrier during the Na plating process and promotes uniform Na distribution within the scaffold. In contrast, only weak physical adsorption exists between Na and bare CF. (ii) O-CF exhibits unique surface characteristics that induce the deposition of Na in the pores, inhibiting Na deposition on the scaffold's top surface and thus providing protective and supportive effects.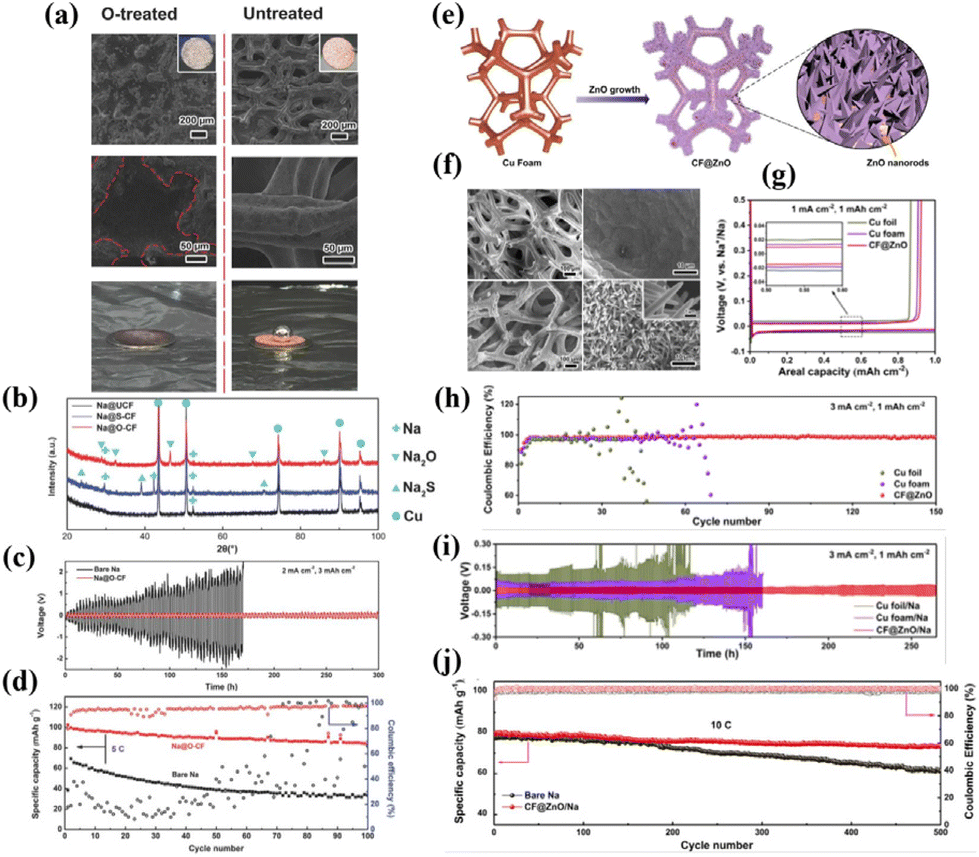 | ||
| Fig. 3 (a) Comparison of the wettability of CF to the Na metal before and after oxidation treatment; (b) reactive wetting phenomenon between the Na metal and O-CF; (c) plating–stripping curve of Na@O-CF at 2 mA cm−2 of 3 mA h cm−2; (d) cycling performance of the full cell with the Na@O-CF anode and the NVP cathode at 5C. Reproduced with permission from ref. 57 Copyright 2018, Wiley-VCH. (e) Growth diagram and (f) SEM images of the morphology of ZnO on CF; (g) nucleation overpotentials of the Na metal on Cu foil, CF, and CF@ZnO substrates; (h) CE of CF@ZnO at 3 mA cm−2; (i) plating–stripping curve of the CF@ZnO/Na anode at 3 mA cm−2; and (j) cycling performance of the full cell with the CF@ZnO/Na anode and the NVP cathode at 10C. Reproduced with permission from ref. 58 Copyright 2021, Elsevier. | ||
Different from the one-step conversion reaction of CuO, there exists another class of oxides, exemplified by ZnO and SnO2, which engage in a two-step conversion-alloying reaction with Na (Na + MxOy → Na–M alloy + Na2O), further reducing the Na nucleation barrier. As reported by Peng et al.,58 Cu foam modified with a ZnO nanorod array (CF@ZnO) efficiently modulates the nucleation-growth process of the Na metal and promotes uniform Na deposition (Fig. 3e and f), which is attributed to the “reactive wetting” behaviour between Na and ZnO (Na + ZnO → Na–Zn alloy + Na2O). When CF@ZnO serves as a scaffold for Na deposition, its voltage hysteresis at 1 mA cm−2 is approximately 25 mV, whereas bare Cu foil and CF exhibit 44 mV and 32 mV, respectively (Fig. 3g). The reduced Na nucleation overpotential implies a favourable Na affinity with ZnO, facilitating uniform Na deposition. At a current density of 3 mA cm−2, CF@ZnO keeps a stable CE at 99.0% after 150 cycles, whereas Cu foil and CF show significant decay, sustaining only a few cycles (Fig. 3h). Symmetric cell testing of CF@ZnO/Na reveals a flat voltage profile and a smaller overpotential throughout the entire cyclic period, indicative of rapid reaction kinetics. At a specific capacity of 1 mA h cm−2 and a current density of 3 mA cm−2, the CF@ZnO/Na symmetric cell demonstrates stable cycling for 400 cycles (Fig. 3i). In comparison, Cu foil/Na and CF/Na symmetric cells show pronounced voltage oscillations and large overpotentials, which are ascribed to the formation of Na dendrites during cycling. Dendritic growth consumes a considerable amount of electrolyte, resulting in a sharp increase in the electrode–electrolyte interface impedance, which hinders the transfer of ions and electrons. When combined with an NVP cathode, the CF@ZnO/Na anode exhibits outstanding cyclic stability, retaining up to 93% of its capacity after 500 cycles at a high current density of 10C (Fig. 3j).
Furthermore, the function of Na2O generated by these nucleation sites was further investigated. Zhang et al.66 fabricated a composite structure by incorporating SnO2 nanoparticles between reduced graphene oxide layers (SnO2@rGO) and found that the binding energies of the Na2O and Na15Sn4 intermediate phases formed by SnO2 during the sodiation process and metallic Na are both higher than those of rGO and Na (Fig. 4a–d). The binding energy suggests that they can simultaneously serve as sodiophilic sites to induce the nucleation of metallic Na. In addition, the nucleation overpotential of SnO2 is lower than that of Sn, further indicating that SnO2 can create dual nucleation sites (Fig. 4e and f). Notably, Na2O can assist in stabilizing the nucleation sites. As depicted in Fig. 4g, the nucleation overpotential of Sn@rGO shows a significant increase during the cycling process, while that of SnO2@rGO shows only a slight change. Transmission electron microscopy (TEM) analysis in Fig. 4n reveals that the volume expansion/shrinkage of Sn caused by the (de)alloying process coarsens the Sn nanoparticles and reduces the interface available for interaction with metallic Na, leading to a reduction in sodiophilic sites and an increase in nucleation overpotential. The robust binding energy between Na2O and rGO prevents the agglomeration of Sn nanoparticles, allowing SnO2 to maintain a low nucleation overpotential over long-term cycling. The symmetric cell with CF@ZnO/Na exhibits a stable cycle for over 3700 h at 0.5 mA cm−2 (Fig. 4h). In fact, the volume effect of alloy-type materials has always attracted much attention. For the nucleation sites that can undergo alloying reactions with Na, they themselves will undergo severe volume expansion/contraction, leading to a reduction in nucleation sites and structural failure during the continuous and repetitive processes of Na plating–stripping. Recently, Xu et al.67 proposed a micellar interface copolymerization and carbonization strategy to construct hollow carbon sphere materials with single-atom Sn confinement (At–Sn@HCN), as depicted in Fig. 4i and j. By reducing the size of the nucleation site to the single-atom scale and anchoring it with nitrogen, issues such as particle agglomeration, pulverization and deactivation resulting from volume changes in the alloy-type nucleation site are mitigated, ensuring the sustained effectiveness of the nucleation sites during cycling.68 The results show that At–Sn@HCN is highly sodiophilic, effectively lowering the nucleation barrier for metallic Na (Fig. 4k and l), achieving stable dendrite-free deposition at an ultra-high areal capacity of 16 mA h cm−2 (Fig. 4m) and maintaining stable cycling for 7000 h at 1 mA cm−2 and 1 mA h cm−2 (Fig. 4o).
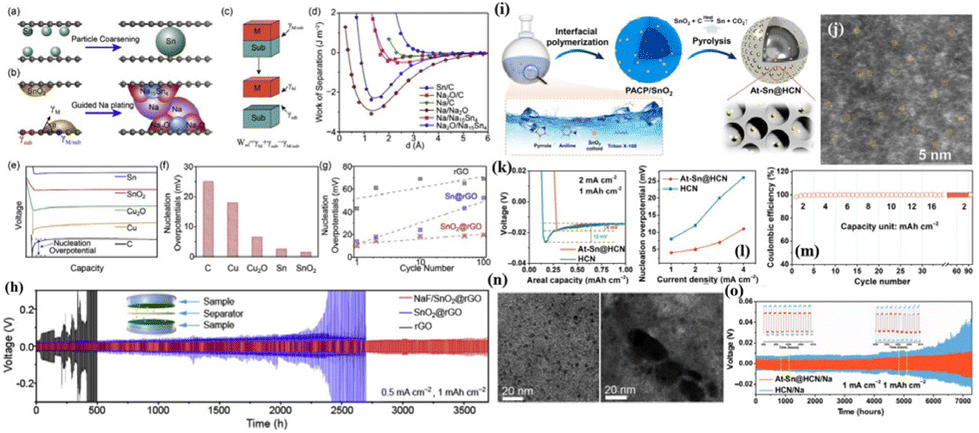 | ||
| Fig. 4 (a) Schematic diagram of the agglomeration of Sn nanoparticles during cycling; (b) schematic diagram of SnO2-induced deposition of the Na metal; (c) definition of Wad; (d) relationship between Wsep and distance; (e) voltage–capacity curves of the Na metal on different substrates and (f) the corresponding Na nucleation overpotentials; (g) changes in the nucleation overpotentials of rGO, Sn@rGO, and SnO2@rGO with the number of cycles; (h) plating–stripping curve of the NaF/SnO2@rGO anode at 0.5 mA cm−2; and (n) morphologies of SnO2 (left) and Sn (right) nanoparticles after cycling. Reproduced with permission from ref. 66 Copyright 2020, Elsevier. (i) Schematic diagram of the preparation process of At–Sn@HCN; (j) schematic diagram of the morphology of At–Sn@HCN; (k) voltage–capacity curve of At–Sn@HCN at 2 mA cm−2; (l) nucleation overpotentials of Sn@HCN at different current densities; (m) CEs of At–Sn@HCN at different areal capacities; and (o) plating–stripping curve of the At–Sn@HCN/Na anode at 1 mA cm−2–1 mA h cm−2. Reproduced with permission from ref. 67 Copyright 2022, AAAS. | ||
4. Rapid Na+-conducting interface layers
While numerous efforts have been made to stabilize Na metal anodes by introducing 3D scaffolds, the current state of CSSAs still falls short of ideal cyclic longevity at higher current densities and larger areal capacities (>5 mA cm−2, >5 mA h cm−2). This feature is pivotal for enabling fast charge and discharge characteristics in high-energy-density batteries. As previously elucidated, the redox reaction necessitates the concurrent involvement of Na+ and electrons during the Na plating–stripping processes. When the electrode undergoes rapid charging, the transfer rate of electrons within the EC scaffold significantly exceeds the diffusion rate of Na+, leading to Na+ readily accepting electrons at the top of the EC scaffold and undergoing reduction. In this scenario, the 3D scaffold paradoxically provides more active sites for the growth of Na dendrites. As the growth proceeds, Na tends to deposit more easily on itself than on the 3D scaffold, resulting in a “top deposition” mode that leads to extensive Na dendrite growth and the formation of “dead Na”. To achieve the long-term stable cycling of CSSAs at high current densities and large areal capacities, enhancing ion diffusion within the EC scaffold is crucial. This can be realized by establishing fast ion-conducting pathways on the scaffold that eliminate preferential top deposition, thereby boosting the scaffold's tolerance for high current densities.As for which materials/components can facilitate Na+ diffusion, we have to start with the solid electrolyte interlayer (SEI). Formulating the electrolyte and additive components (strategy I mentioned earlier) is an early method applied to enhance the stability of Na anodes. In 2015, Cui et al.12 presented an ether-based electrolyte consisting of sodium hexafluorophosphate (NaPF6) and diglyme, showcasing stable cycling of Na metal anodes at a current density of 0.5 mA cm−2 and an areal capacity of 1 mA h cm−2 for 300 cycles, with a CE exceeding 99.9%, and no apparent dendrite formation on the anode surface. It is noteworthy that the surface of the Na anode can form a dense and compact SEI film composed of an outer layer of the organic Na phase and an inner layer of the inorganic Na phase. Crucially, the inorganic inner layer, comprising sodium fluoride (NaF), aids in homogenizing the Na+ flux and promoting uniform Na deposition. Chen et al. further verified that NaF also has an effective role in a carbonate-based electrolyte.69 Archer's group investigated the transport ability of Na+ in NaF using a combined density functional theory study.70 The results show that NaF has extremely low diffusion barriers for Na+, which proves advantageous for the diffusion and deposition of Na+.
Despite the enhanced stability of the SEI through strategy I, achieving long-term cycling stability for Na metal anodes remains challenging due to the continuous consumption of the electrolyte/additives during cycling. Taking this into account, researchers use chemical methods or physical techniques to pre-construct an artificial SEI protective layer on the Na anode's surface (strategy II mentioned earlier) before the initiation of the electrochemical reaction, which prevents the direct contact between the anode and the electrolyte, thereby minimizing side reactions and dendrite growth. To date, researchers have successively developed multifunctional heterogeneous interface layers comprising various functional components (e.g., Na-rich alloys and NaF) to further enhance the electrochemical performance of Na metal batteries. Wang et al.71 accomplished the in situ construction of a Na3Sb/NaF interface layer on Na foil by placing it in dimethyl carbonate (DMC) containing SbF3via a simple displacement reaction of Na and SbF3 at room temperature (Fig. 5a and c). Since the Na3Sb alloy phase has a low Na nucleation barrier and the NaF phase has good ionic conductivity with strong mechanical strength, the modified Na metal anode (SAF-Na) exhibits significantly low interface impedance during cycling, ensuring an extremely stable anode–electrolyte interface (Fig. 5b). When the symmetrical cell is cycled at a current density of 2 mA cm−2, the bare Na anode shows a short lifespan of 400 h, while the SAF-Na anode achieves a cycling life exceeding 2400 h (Fig. 5d).
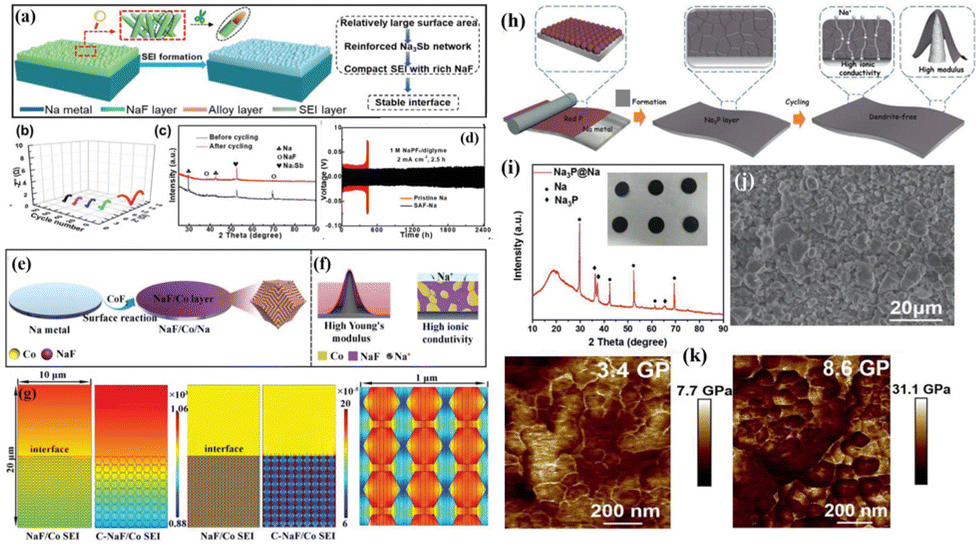 | ||
| Fig. 5 (a) Schematic diagram of a stable interface constructed using the Na3Sb alloy and NaF; (b) interface impedance of the SFA–Na anode at various cycling rates; (c) XRD pattern of the Na3Sb alloy and NaF formed using SbF3 after the Na plating process; and (d) plating–stripping curve of the SFA–Na anode at 2 mA cm−2–5 mA h cm−2. Reproduced with permission from ref. 71 Copyright 2019, Wiley-VCH. (e and f) Schematic diagram of the synthesis of NaF/Co/Na; (g) finite element simulation of Na+ concentration distribution for the NaF/Co SEI and the C-NaF/Co SEI. Reproduced with permission from ref. 72 Copyright 2022, Wiley-VCH. (h) Schematic diagram of the regulation of Na plating/stripping behaviour by the Na3P protective layer; (i and j) optical photo, XRD pattern, and SEM image of Na3P@Na; and (k) Young's modulus comparison between the bare Na and Na3P@Na anode surfaces. Reproduced with permission from ref. 73 Copyright 2021, Wiley-VCH. | ||
Yu et al. employed a similar in situ construction approach to design a series of heterogeneous interface layers, including P/Na3P (formed by red phosphorus),73 Co/NaF (formed by CoF2),72 V/Na2S (V2S3),74 and V/Na2Se (VSe2).75 These protective layers demonstrate high Na affinity, excellent ionic conductivity, and high Young's modulus, which accelerate Na+ diffusion–deposition kinetics (Fig. 5h–k and 6a–i). Excitingly, the Na metal anode modified with V/Na2Se expands the working temperature range of Na metal batteries (Fig. 6i).75Ab initio molecular dynamics (AIMD) simulations indicate that the V/Na2Se interface layer lowers the energy barrier of desolvation at −40 °C, thus enhancing low-temperature Na+ diffusion kinetics (Fig. 6f and g). Consequently, the Na@V/Na2Se symmetric cell shows a long lifespan exceeding 1500 h under 0.5 mA cm−2–0.5 mA h cm−2 conditions at −40 °C (Fig. 6h).
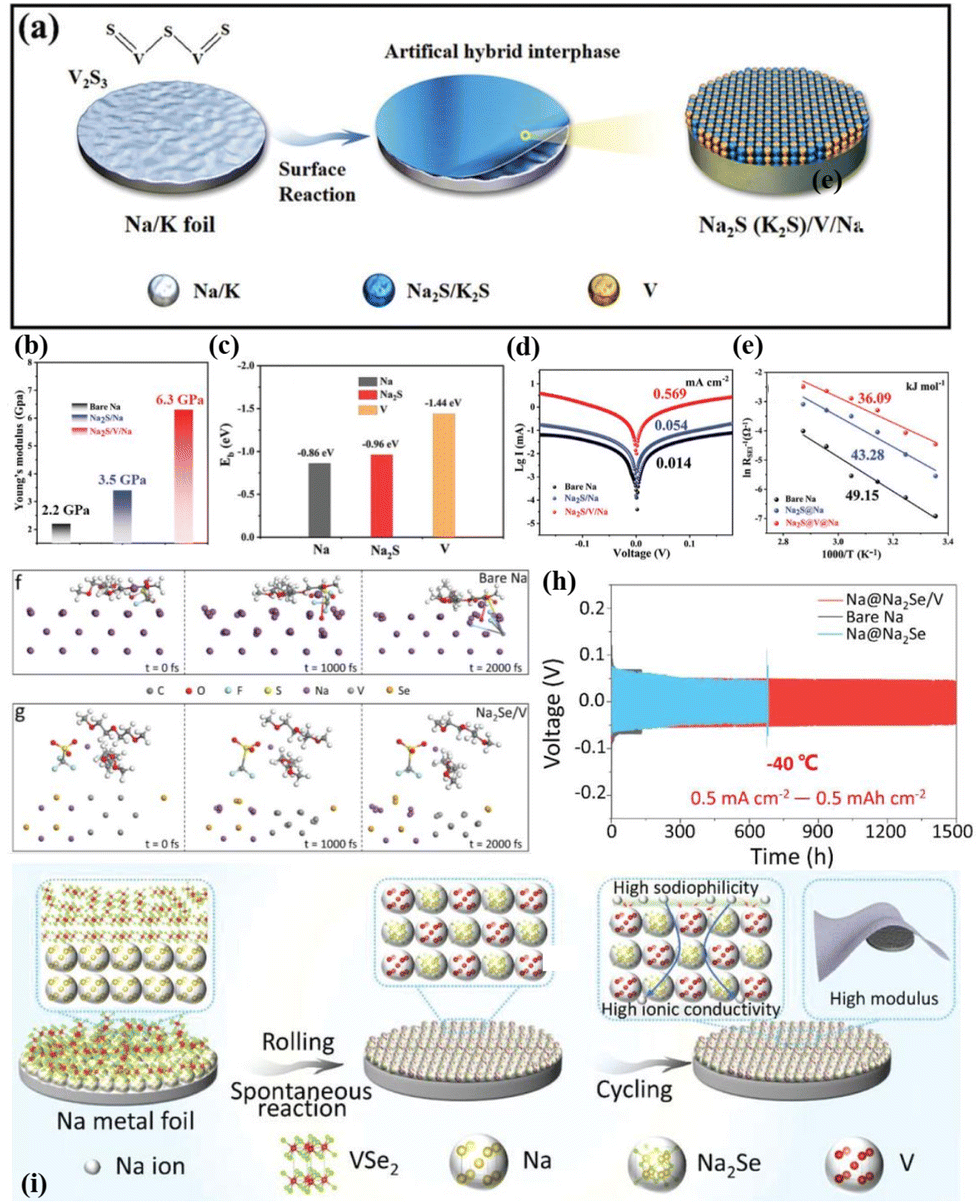 | ||
| Fig. 6 (a) Schematic diagram of a heterogeneous interface layer (Na2S/V) constructed using V2S3 after sodiation; (b) Young's modulus distribution on bare Na, Na2S/Na and Na2S/V/Na; (c) binding energies of Na with Na, Na2S, and V; (d) fitted Tafel points for bare Na, Na2S/Na and Na2S/V/Na; (e) activation energy for Na+ diffusion; reproduced with permission from ref. 74 Copyright 2022, Wiley-VCH. (f and g) AIMD simulation of the desolvation process of bare Na and Na@Na2Se/V electrode surface solvation structures at −40 °C; (h) plating–stripping curve of the Na@Na2Se/V anode −40 °C; and (i) schematic diagram of a heterogeneous interface layer (Na2Se/V) constructed using VSe2 after sodiation. Reproduced with permission from ref. 75 Copyright 2023, Wiley-VCH. | ||
5. 3D mixed ion/electron-conducting scaffolds
These endeavours have laid both theoretical and experimental foundations for the recent design of composite Na–MIEC scaffold anodes. Xie et al.76 engineered SnO2-modified 3D carbon nanofiber (SnO2-CNFs) scaffolds using a combination of electrospinning and magnetron sputtering (Fig. 7a). The in situ formation of sodiophilic Na–Sn alloys and Na2O during the initial plating process guides the uniform deposition of Na in a “bottom-up” fashion (Fig. 7b and c). Additionally, the excellent ionic conductivity of Na2O reduces Na+ diffusion barriers, promoting rapid Na+ diffusion and uniform Na deposition (Fig. 7d). These attributes play a vital role in suppressing dendrite growth and improving the spatial utilization of the 3D scaffold. Therefore, symmetric cells assembled with the designed SnO2-CNFs@Na anode demonstrate stable cycling for 1500 h at 5 mA cm−2 and 5 mA h cm−2 (Fig. 7e). When coupled with the NVP@C@CNTs cathode, the full cell delivers a capacity retention as high as 95.1% after 400 cycles at a high current density of 5C (Fig. 7f).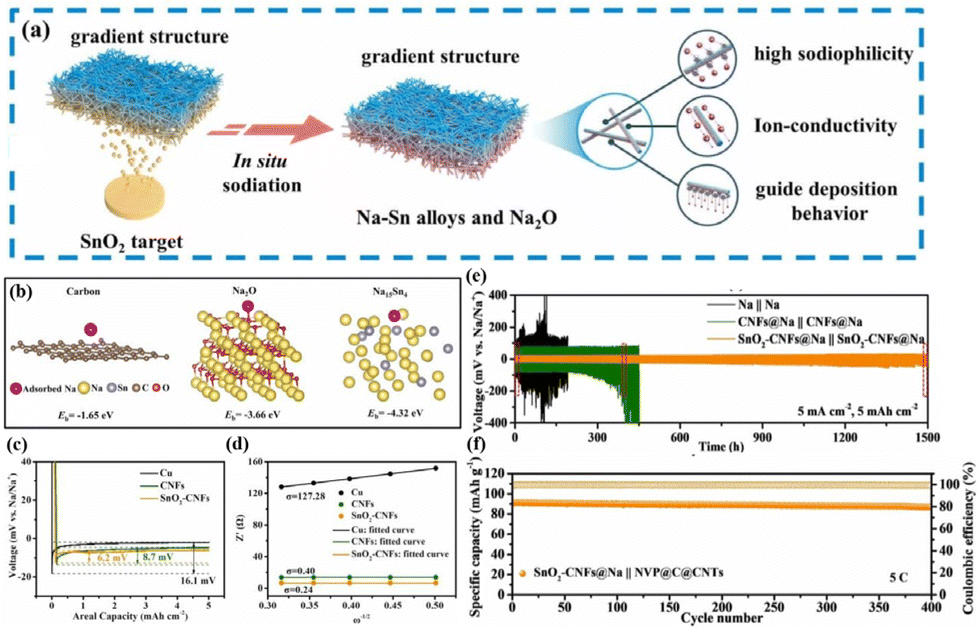 | ||
| Fig. 7 (a) Schematic diagram of a heterogeneous interface layer (Na15Sn4/Na2O) constructed using SnO2 after sodiation; (b) binding energies of Na with CNTs, Na2O, and Na15Sn4; (c) nucleation overpotentials of the Na metal on bare Cu, CNFs and SnO2-CNFs; (d) Na+ diffusion dynamics; (e) plating–stripping curve of the SnO2-CNFs@Na anode at 5 mA cm−2–5 mA h cm−2; and (f) cycling performance of the full cell with the SnO2-CNFs@Na anode and the NVP@C@CNT cathode at 5C. Reproduced with permission from ref. 76 Copyright 2022, Elsevier. | ||
Furthermore, Li et al.38 harnessed a MIEC scaffold to achieve high-performance full cells with a low negative to positive capacity (N/P) ratio. They in situ synthesized a MIEC scaffold composed of Na3P and CNTs through an alloying reaction between Na and P. The highly sodiophilic Na3P not only establishes an efficient ion-conducting network (high ionic conductivity: 0.12 mS cm−1, low Na+ diffusion barrier: 0.067 eV), but also promotes the formation of a rigid SEI layer (high Young's modulus: 8.6 GPa) (Fig. 8a).73 Moreover, CNTs provide a 3D interconnected electron transport network. Consequently, the as-prepared Na3P/CNT@Na anode shows excellent cycling stability at 3 mA cm−2 for 1200 cycles with a high CE of 99.8% (Fig. 8b). Even when the current density is increased to 5 mA cm−2, a remarkably low overpotential of 20 mV is maintained after 250 h (Fig. 8c), indicating a rapid kinetic process and a stable anode–electrolyte interface. Moreover, the full battery assembled with a Na3P/CNT@Na anode and an NVP cathode can stably cycle for 300 cycles under a low N/P ratio (1.56), high current density (1 C), and high mass loading (>20 mg cm−2 of NVP), with a high reversible capacity of 102.3 mA h g−1 and almost no fading (capacity retention: 95%, Fig. 8d). Impressively, even in the absence of a Na metal anode in a full cell (N/P = 0), it can still maintain 100 cycles with a capacity retention rate of 67.4%, which showcases significant potential for developing high-energy-density Na metal batteries (Fig. 8e).
 | ||
| Fig. 8 (a) Binding energies of Na adsorbed on Na3P, CNTs and Cu, charge difference density of Na adsorbed on Na3P, diffusion energy barrier of Na+ toward Na3P and comparison of the diffusion energy barriers of Na+ on Na3P, CNTs, and Cu; CE (b) and plating–stripping curve (c) of the IEDC anode; cycling performance of NVP-IEDC full cells under different conditions: (d) low N/P ratio of 1.56 at 1C and (e) anode free at 0.5 C. Reproduced with permission from ref. 38 Copyright 2021, Wiley-VCH. | ||
In comparison to ether-based electrolytes, carbonate-based electrolytes offer a broader electrochemical window and compatibility with most high-voltage cathode materials, which is beneficial for increasing the energy density of batteries. However, the SEI film formed by carbonate-based electrolytes exhibits inferior stability compared to its ether-based counterparts. Furthermore, the inorganic-enriched SEI film formed by ether-based electrolytes is more conducive to Na+ migration, demonstrating superior electrochemical performance compared to carbonate-based electrolytes. In response to the above problems, Chou and colleagues77 devised a graphene aerogel loaded with MgF2 nanoparticles as a host for the Na metal (Fig. 9a). This structure possesses several features: (i) MgF2 converts into Mg–Na alloys and NaF during the initial Na plating, aiding the nucleation and growth of Na on graphene sheets (Fig. 9b and c); (ii) the enriched NaF in the SEI film enhances rapid Na+ transport and promotes a uniform Na+ flux (Fig. 9b); (iii) the graphene aerogel, acting as a 3D conductive network, effectively reduces the local current density and alleviates the volume expansion effect of the Na metal (Fig. 9b). This ingenious design, combining sodiophilic nucleation sites, a stable NaF-enriched SEI film, and a 3D electronic conductive network, significantly improves the cycling performance of the Na metal anode in carbonate-based electrolytes, which achieves stable cycling for up to 1600 h at 0.5 mA cm−2 and 0.5 mA h cm−2 (Fig. 9d).
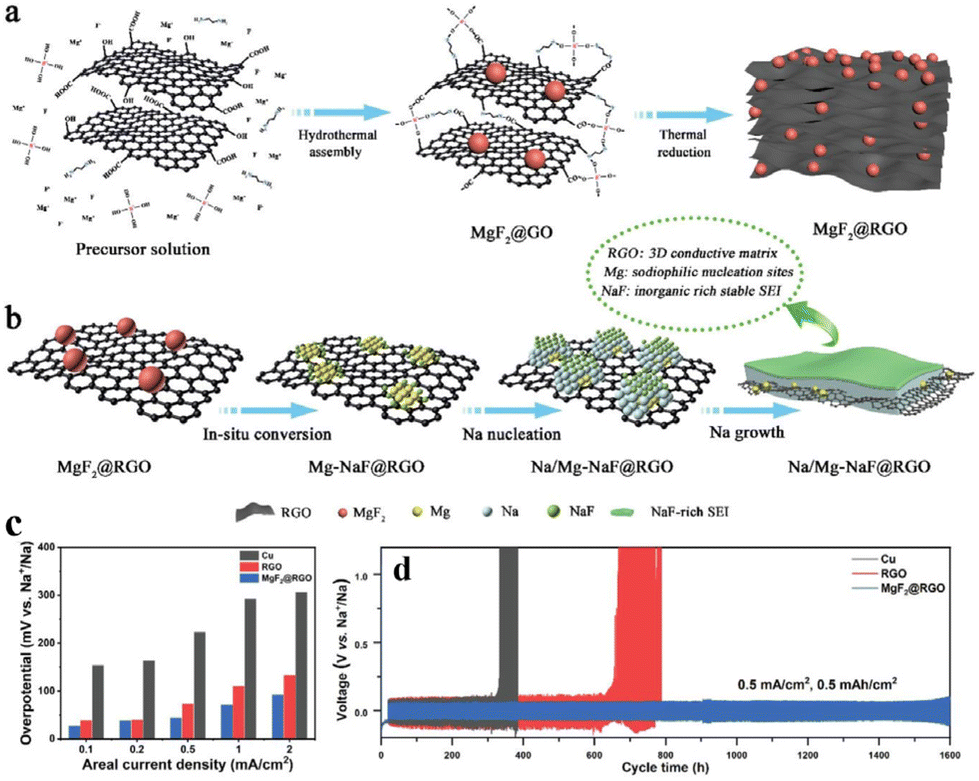 | ||
| Fig. 9 (a) Schematic diagram of the preparation of the MgF2@rGO MIEC scaffold; (b) schematic diagram of a heterogeneous interface layer (Na/Mg-NaF) constructed using MgF2 after sodiation; (c) nucleation overpotentials of the Na metal on Cu, rGO, and MgF2@rGO; (d) plating–stripping curve of the MgF2@rGO/Na anode at 0.5 mA cm−2 in the carbonate-based electrolyte. Reproduced with permission from ref. 77 Copyright 2022, Wiley-VCH. | ||
In a recent development, Li et al.34 addressed the intrinsic sodiophobic characteristics and poor ionic conductivity of commercial nickel foam (NF) through a chemical fluorination modification strategy (Fig. 10a). They constructed a MIEC scaffold (NiF2@NF) with good sodiophilic features, high electronic conductivity, and fast ion transport capability (Fig. 10b and c). Controls included oxidation-modified NiO@NF and pure NF. The study reveals that NiF2 first undergoes an electrochemical conversion reaction with Na to generate Ni nanoparticles and NaF during the plating process (Fig. 10d–f), so it has a lower Na nucleation overpotential (17 mV), while the sodiophobicity of pure NF gives it a high overpotential of 58 mV. In addition, although NiO also undergoes a similar conversion reaction (form Ni and Na2O) during the plating process, its nucleation overpotential for Na is still as high as 44 mV (Fig. 10d). Comprehensive analyses involving XPS, first-principles calculations and COMSOL simulations demonstrate that the NaF-enriched SEI film has an extremely low diffusion barrier for Na+ (0.005 eV), much lower than the 0.359 eV of the Na2O-enriched SEI (Fig. 10h and i). This enables effective regulation of ion concentration distribution throughout the 3D scaffold. Therefore, the in situ formed NaF-enriched SEI along the backbones provides a 3D interconnected fast ion transport pathway, which achieves uniform Na+ flux and enhances Na+ diffusion on the inner surface of the scaffold, thus guiding uniform Na deposition along the backbones and filling the vacancies (Fig. 10j–l). Consequently, the NiF2@NF@Na anode achieves stable cycling with a current density of up to 10 mA cm−2 and an areal capacity of up to 10 mA h cm−2 in ether-based electrolyte (Fig. 10m and n), reaching a high depth of discharge (DOD) of 83.3%. It also shows stable cycling for 400 cycles at 2 mA cm−2 in the carbonate-based electrolyte with a high-rate capability of 4 mA cm−2 (Fig. 10o and p). Furthermore, when paired with NVP, the full cell exhibits stable cycling for 300 cycles at a high current density of 5C, with a high-capacity retention of 97.8%, while the full cell using bare Na undergoes significant decay throughout the cycling period (Fig. 10q). For ease of comparison, Table 1 summarizes the electrochemical performance of representative studies on 3D composite Na–MIEC scaffold anodes (1C = 117 mA g−1).
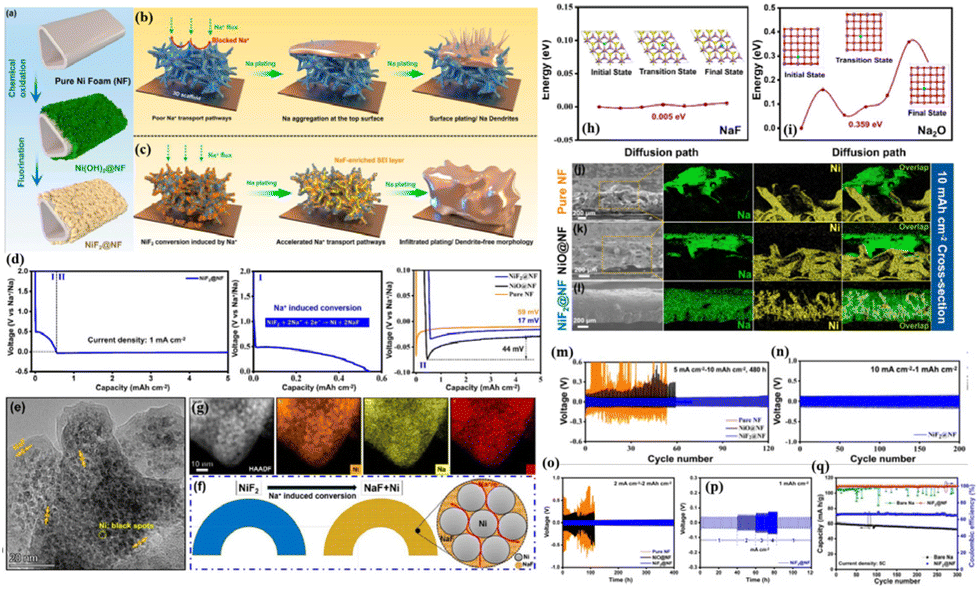 | ||
| Fig. 10 (a) Schematic diagram of the preparation of the NiF2@NF MIEC scaffold; (b and c) deposition behaviours of the Na metal on EC and MIEC scaffolds; (d) plating curve of the Na metal on NiF2@NF; (e–g) TEM images and the corresponding EDS mappings of NiF2 after sodiation; energy barrier for Na+ diffusion in NaF- (h) and Na2O- (i) enriched SEI layers; cross-sectional view of the SEM images of Na deposition on pure NF (j), NiO@NF (k) and NiF2@NF (l) after plating 10 mA h cm−2 of Na; plating–stripping curves of the NiF2@NF/Na anode under various conditions: 5 mA cm−2–10 mA h cm−2, ether-based electrolyte (m); 10 mA cm−2–1 mA h cm−2, ether-based electrolyte (n); and 2 mA cm−2–2 mA h cm−2, carbonate-based electrolyte (o); (p) rate capability of NiF2@NF@Na in the ether-based electrolyte; and (q) cycling performance of the full cell at 5C using NiF2@NF/Na. Reproduced with permission from ref. 34 Copyright 2023, American Chemical Society. | ||
| Materials | Areal capacities at current densities | Time (h) | Electrolytes | Performance in NVP-based full cells | N/P ratio |
|---|---|---|---|---|---|
| The N/P ratio is calculated based on the ratio of the areal capacity of the Na anode to that of the NVP cathode at the fully discharged state. | |||||
| SnO2-CNFs78 | 1 mA h cm−2 at 1 mA cm−2 | 3000 | 1 M NaPF6 in diglyme | 1C, 86.1 mA h cm−2, 130 cycles | 26.5 |
| 5 mA h cm−2 at 5 mA cm−2 | 1500 | 5C, 90 mA h cm−2, 400 cycles | 25 | ||
| MgF2@rGO79 | 0.5 mA h cm−2 at 0.5 mA cm−2 | 1600 | 1 M NaClO4 in EC/DEC/FEC | 1C, 79.88 mA h g−1, 200 cycles | 18.9 |
| 0.5C, 110 mA h g−1, 30 cycles | 2.5 | ||||
| IEDC80 | 1 mA h cm−2 at 5 mA cm−2 | 250 | 1 M NaPF6 in diglyme | 1C, 102.3 mA h g−1, 300 cycles | 1.56 |
| 0.5C, 90 mA h g−1, 100 cycles | 0 | ||||
| NiF2@NF81 | 2 mA h cm−2 at 2 mA cm−2 | 1600 | 1 M NaPF6 in diglyme | ||
| 4 mA h cm−2 at 4 mA cm−2 | 800 | 3C, 76.5 mA h g−1, 500 cycles | 15 | ||
| 10 mA h cm−2 at 5 mA cm−2 | 480 | 5C, 70.9 mA h g−1, 300 cycles | 16.6 | ||
| 1 mA h cm−2 at 10 mA cm−2 | 40 | ||||
NSCA-31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 82 82 |
1 mA h cm−2 at 1 mA cm−2 | 700 | 1 M NaCF3SO3 in diglyme | 1C, 86.84 mA h g−1, 300 cycles | — |
| 1 mA h cm−2 at 2 mA cm−2 | 500 | ||||
| SnSe@GF83 | 1 mA h cm−2 at 1 mA cm−2 | 2000 | 1 M NaCF3SO3 in diglyme | 1C, 100 mA h g−1, 450 cycles | 13.76 |
| 5 mA h cm−2 at 5 mA cm−2 | 900 | 5C, 70.3 mA h g−1, 100 cycles | 16.5 | ||
| h-Ti3C2/CNTs36 | 1 mA h cm−2 at 1 mA cm−2 | >4000 | 1 M NaOTF in diglyme | — | — |
| 3D-NVP84 | 1 mA h cm−2 at 1 mA cm−2 | 300 | 1 M NaClO4 in PC/FEC | 10C, 93.6 mA h g−1, 500 cycles | — |
| 2 mA h cm−2 at 1 mA cm−2 | 400 | ||||
6. Conclusions and perspectives
The incorporation of 3D scaffolds in conjunction with the Na metal to construct CSSAs plays a constructive role in overcoming challenges related to dendritic growth and extreme volume changes during the plating and stripping processes in Na metal batteries. However, conventional EC scaffolds lack effective ionic conductivity, which easily causes the preferential deposition of the Na metal on their top surfaces rather than uniform deposition across the entire scaffold. This results in top deposition of the Na metal, inducing dendritic growth and obstructing ion transport from the surface to the interior. This situation becomes more serious at high current densities. Therefore, the introduction of fast ion transport pathways on electron-conducting scaffolds to construct 3D MIEC scaffolds is considered a forward-looking strategy. The MIEC scaffolds possess abundant Na nucleation sites and 3D interconnected ion/electron transport paths, which reduce local current density and ensure uniform Na+ flux, thus enhancing Na+/e− transfer. This aids in effectively regulating Na metal nucleation-growth behaviour, achieving dendrite-free uniform Na deposition, and ensuring excellent cycling stability, which is crucial for the desired rapid charge/discharge capabilities and safety required in practical applications. This review comprehensively summarizes the latest research progress in constructing composite Na–MIEC scaffold anodes. Firstly, the differences between two commonly used construction methods and the essential characteristics that a 3D scaffold should possess for effective Na integration are discussed. After that, various types of Na nucleation sites are introduced, which have strong binding energies with Na and effectively reduce the Na nucleation barrier, facilitating Na nucleation and growth. Secondly, several typical efforts to enhance the sodiophilicity of 3D EC scaffolds are listed, emphasizing that their electrochemical performance is still restricted by their poor ionic conductivity. Finally, the structural design strategies and performance breakthroughs of various MIEC scaffolds in recent years are discussed inspired by currently reported artificial SEI protective layers with low Na diffusion barriers.Despite the significant progress in constructing 3D composite Na-MIEC scaffold anodes, practical applications of Na metal batteries still encounter numerous challenges. To better address these challenges, future research should consider the following aspects:
(i) Apply in situ characterization techniques.
The plating–stripping behaviour of Na metal anodes is a dynamic evolutionary process. Conventional ex situ characterization techniques often provide insights solely into the final state of the process, lacking crucial information about the intermediate states. This limitation impedes a comprehensive and clear understanding of the formation and evolution mechanisms of Na metal anodes. In contrast, in situ characterization technology enables real-time monitoring and dynamic tracking of Na+ from their initial nucleation to final growth.85,86 This approach facilitates a more comprehensive and accurate representation of the dynamic changes occurring at the Na anode interface, including the formation of the SEI, dendrite growth, and volume expansion.
(ii) Improve Na utilization/DOD, while reducing the N/P ratio.
Key indicators such as Na utilization, DOD and N/P ratio are inadequately considered in the most current Na metal battery research, which directly impact the practically achievable energy density of the battery. Excess metallic Na is often used in battery testing to compensate for the loss of active Na (DOD < 10%, N/P ratio > 50).87 Although the cycling stability improves, this “artificial” regulation limits the Na metal anodes from taking advantage of high specific capacity in practical applications. However, the currently reported sodiophilic scaffolds inevitably consume a large amount of active Na (e.g., conversion/alloying reactions) during the plating and stripping processes, causing severe damage to the reversible capacity and cycle life of the full battery, especially in the case of low N/P ratios or even with no anode (N/P = 0).88–91 Further exploration is needed to construct scaffolds with both high sodiophilicity and reduced Na consumption for achieving high Na utilization and DOD (>80%), a low N/P ratio (<2), a high energy density (>300 W h kg−1), and a long cycle life for Na (full) batteries.
(iii) Light weight design of 3D scaffolds.
Although 3D scaffolds play a crucial role in stabilizing and protecting Na metal anodes, they are still an inactive component in the battery, which does not contribute to the capacity of the electrochemical process. To maximize the battery energy density, the weight of introduced scaffolds in the entire electrode should be minimized. Carbon-based scaffolds are light weight but have low mechanical strength, while metal-based scaffolds have higher mechanical strength, ensuring structural stability during long-term cycling. However, metal-based scaffolds are generally more expensive and denser, somewhat diminishing the overall energy density and economic practicality of the electrode. With the development of additive manufacturing technology, 3D printing, with its characteristics of personalized manufacturing, has solved the problem of difficult molding by traditional manufacturing methods. Research efforts have been made to introduce 3D printing technology into the design of high-performance Li/Na metal anodes.92–97 Preparing a 3D scaffold through 3D printing can effectively control the size, pore diameter, porosity and structural characteristics of the scaffold, which is expected to provide both strong mechanical strength and light weight, achieving precise and personalized manufacturing.
(iv) Broaden the operating temperature range.
The battery performance deteriorates rapidly when the temperature is below 0 °C or above 40 °C. Currently, wide-temperature Li metal batteries are being explored relatively extensively. Studies have shown that at low temperatures, the desolvation energy barrier of Li+ increases, and concomitantly Li+ transport slows down, causing the reaction kinetics of Li metal batteries to be severely limited.98–100 As the temperature rises, the kinetic process improves but accelerates various side reactions at the electrode–electrolyte interface, intensifying the consumption of the electrolyte and the Li metal, thereby shortening the cycle life of Li metal batteries and easily causing thermal runaway.101,102 Currently, research on the electrochemical performance of Na metal batteries at low and high temperatures is relatively insufficient. Most reported strategies focus on addressing the challenges faced by Na metal batteries at room temperature, making it difficult to meet the practical application requirements in extreme climates worldwide. Therefore, future research should push for an in-depth exploration of Na metal batteries in extreme environments. It should focus on the effects of temperature on the Na dendrite morphology and rate, the impact of temperature on the desolvation energy of ions, and the diffusion of ions into the SEI layer. Relying on the unique advantages of composite Na-MIEC anodes, a stable electrode–electrolyte interface is co-constructed to adapt to the complex and changing climatic conditions around the world, further expanding the practical application of Na metal batteries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 52107233 and 52201278) and the China Postdoctoral Science Foundation (Grant No. 2021M690123). X. H. also acknowledges the “Young Talent Support Plan” of Xi'an Jiaotong University. The authors acknowledge the Department of Science and Technology of Shaanxi Province (QCYRCXM-2022-126).References
- L. L. Fan and X. F. Li, Nano Energy, 2018, 53, 630–642 CrossRef CAS.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS PubMed.
- B. Sun, P. Xiong, U. Maitra, D. Langsdorf, K. Yan, C. Y. Wang, J. Janek, D. Schroder and G. X. Wang, Adv. Mater., 2020, 32, 1903891 CrossRef CAS PubMed.
- B. Sun, P. Li, J. Q. Zhang, D. Wang, P. Munroe, C. Y. Wang, P. H. L. Notten and G. X. Wang, Adv. Mater., 2018, 30, 1801334 CrossRef PubMed.
- J. L. Yang, X. X. Zhao, W. H. Li, H. J. Liang, Z. Y. Gu, Y. Liu, M. Du and X. L. Wu, eScience, 2022, 2, 95–101 CrossRef.
- Y. F. Zhu, Y. Xiao, S. X. Dou, Y. M. Kang and S. L. Chou, eScience, 2021, 1, 13–27 CrossRef.
- A. M. Huang, Y. C. Ma, J. Peng, L. L. Li, S. L. Chou, S. Ramakrishna and S. J. Peng, eScience, 2021, 1, 141–162 CrossRef.
- D. Q. Chen, W. Zhang, K. Y. Luo, Y. Song, Y. J. Zhong, Y. X. Liu, G. K. Wang, B. H. Zhong, Z. G. Wu and X. D. Guo, Energy Environ. Sci., 2021, 14, 2244–2262 RSC.
- X. L. Chen, Y. H. Zheng, W. J. Liu, C. Zhang, S. Li and J. Li, Nanoscale, 2019, 11, 22196–22205 RSC.
- A. X. Wang, X. F. Hu, H. Q. Tang, C. Y. Zhang, S. Liu, Y. W. Yang, Q. H. Yang and J. Y. Luo, Angew. Chem., Int. Ed., 2017, 56, 11921–11926 CrossRef CAS PubMed.
- X. Y. Zheng, C. Bommier, W. Luo, L. H. Jiang, Y. N. Hao and Y. H. Huang, Energy Storage Mater., 2019, 16, 6–23 CrossRef.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS PubMed.
- S. Xin, Y. X. Yin, Y. G. Guo and L. J. Wan, Adv. Mater., 2014, 26, 1261–1265 CrossRef CAS PubMed.
- Y. X. Wang, B. W. Zhang, W. H. Lai, Y. F. Xu, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2017, 7, 1602829 CrossRef.
- A. Y. S. Eng, V. Kumar, Y. W. Zhang, J. M. Luo, W. Y. Wang, Y. M. Sun, W. Y. Li and Z. W. Seh, Adv. Energy Mater., 2021, 11, 2003493 CrossRef CAS.
- Y. P. Liu, F. Bettels, Z. H. Lin, Z. H. Li, Y. X. Shao, F. Ding, S. Y. Liu and L. Zhang, Adv. Funct. Mater., 2023, 2302626 CrossRef.
- H. Yadegari, Q. Sun and X. L. Sun, Adv. Mater., 2016, 28, 7065–7093 CrossRef CAS PubMed.
- K. Song, D. A. Agyeman, M. Park, J. Yang and Y. M. Kang, Adv. Mater., 2017, 29, 1606572 CrossRef PubMed.
- S. C. Wu, Y. Qiao, K. Z. Jiang, Y. B. He, S. H. Guo and H. S. Zhou, Adv. Funct. Mater., 2018, 28, 1706374 CrossRef.
- H. Yadegari and X. L. Sun, Acc. Chem. Res., 2018, 51, 1532–1540 CrossRef CAS PubMed.
- W. Liu, P. C. Liu and D. Mitlin, Chem. Soc. Rev., 2020, 49, 7284–7300 RSC.
- Q. Zhang, Y. Y. Lu, L. C. Miao, Q. Zhao, K. X. Xia, J. Liang, S. L. Chou and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 14796–14800 CrossRef CAS PubMed.
- K. N. Zhao, C. X. Wang, Y. H. Yu, M. Y. Yan, Q. L. Wei, P. He, Y. F. Dong, Z. Y. Zhang, X. D. Wang and L. Q. Mai, Adv. Mater. Interfaces, 2018, 5, 1800848 CrossRef.
- Q. W. Shi, Y. R. Zhong, M. Wu, H. Z. Wang and H. L. Wang, Angew. Chem., Int. Ed., 2018, 57, 9069–9072 CrossRef CAS PubMed.
- C. Z. Wang, Y. Zheng, Z. N. Chen, R. R. Zhang, W. He, K. X. Li, S. Yan, J. Q. Cui, X. L. Fang, J. W. Yan, G. Xu, D. L. Peng, B. Ren and N. F. Zheng, Adv. Energy Mater., 2023, 13, 2204125 CrossRef CAS.
- T. Wang, Y. B. Hua, Z. W. Xu and J. S. Yu, Small, 2022, 18, 2102250 CrossRef CAS PubMed.
- M. Zhu, L. L. Li, Y. J. Zhang, K. Wu, F. F. Yu, Z. Y. Huang, G. Y. Wang, J. Y. Li, L. Y. Wen, H. K. Liu, S. X. Dou, Y. Yu and C. Wu, Energy Storage Mater., 2021, 42, 145–153 CrossRef.
- M. J. Hou, Y. J. Zhou, F. Liang, H. P. Zhao, D. Y. Ji, D. Zhang, L. Q. Li and Y. Lei, Chem. Eng. J., 2023, 475, 146227 CrossRef CAS.
- P. Liu, L. C. Miao, Z. Q. Sun, X. C. Chen, Y. C. Si, Q. L. Wang and L. F. Jiao, Angew. Chem., Int. Ed., 2023, 62, e202312413 CrossRef CAS PubMed.
- M. Moorthy, B. Moorthy, B. K. Ganesan, A. Saha, S. Yu, D. Kim, S. Hong, S. Park, K. Kang, R. Thangavel and Y. S. Lee, Adv. Funct. Mater., 2023, 33, 2300135 CrossRef CAS.
- C. Chu, R. Li, F. Cai, Z. Bai, Y. Wang, X. Xu, N. Wang, J. Yang and S. Dou, Energy Environ. Sci., 2021, 14, 4318–4340 RSC.
- Z. Li, K. Zhu, P. Liu and L. Jiao, Adv. Energy Mater., 2021, 12, 2100359 CrossRef.
- K. Lee, Y. J. Lee, M. J. Lee, J. Han, J. Lim, K. Ryu, H. Yoon, B. H. Kim, B. J. Kim and S. W. Lee, Adv. Mater., 2022, 34, e2109767 CrossRef PubMed.
- X. Lu, H. Zhao, Y. Qin, E. Matios, J. Luo, R. Chen, H. Nan, B. Wen, Y. Zhang, Y. Li, Q. He, X. Deng, J. Lin, K. Zhang, H. Wang, K. Xi, Y. Su, X. Hu, S. Ding and W. Li, ACS Nano, 2023, 17, 10665–10676 CrossRef CAS PubMed.
- J. Y. Cui, A. X. Wang, G. J. Li, D. H. Wang, D. Shu, A. P. Dong, G. L. Zhu, J. Y. Luo and B. D. Sun, J. Mater. Chem. A, 2020, 8, 15399–15416 RSC.
- X. He, S. Jin, L. C. Miao, Y. C. Cai, Y. P. Hou, H. X. Li, K. Zhang, Z. H. Yan and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 16705–16711 CrossRef CAS PubMed.
- C. X. Chu, R. Li, F. P. Cai, Z. C. Bai, Y. X. Wang, X. Xu, N. N. Wang, J. Yang and S. X. Dou, Energy Environ. Sci., 2021, 14, 4318–4340 RSC.
- K. Lin, X. Xu, X. Qin, J. Wu, Q. Liu, Z. Tang, S. He, Y. Ye, F. Kang and B. Li, Small, 2021, 17, e2104021 CrossRef PubMed.
- S. S. Chi, X. G. Qi, Y. S. Hu and L. Z. Fan, Adv. Energy Mater., 2018, 8, 1702764 CrossRef.
- T. S. Wang, Y. C. Liu, Y. X. Lu, Y. S. Hu and L. Z. Fan, Energy Storage Mater., 2018, 15, 274–281 CrossRef.
- J. C. Sun, M. Zhang, P. Ju, Y. Hu, X. X. Chen, W. R. Z. Wang and C. C. Chen, Energy Technol., 2020, 8, 1901250 CrossRef CAS.
- D. S. Yu, Y. X. Hao, S. L. Han, S. Zhao, Q. C. Zhou, C. H. Kuo, F. Hu, L. L. Li, H. Y. Chen, J. W. Ren and S. J. Peng, ACS Nano, 2023, 17, 1701–1712 CrossRef CAS PubMed.
- S. Zhao, F. Hu, L. J. Yin, L. L. Li and S. J. Peng, Sci. Bull., 2023, 68, 1389–1398 CrossRef CAS PubMed.
- J. H. Yan, J. Y. Yu and B. Ding, Adv. Mater., 2018, 30, 1705105 CrossRef PubMed.
- W. Q. Guo, S. Liu, X. Z. Guan, X. Y. Zhang, X. J. Liu and J. Y. Luo, Adv. Energy Mater., 2019, 9, 1705105 Search PubMed.
- X. M. Xia, C. F. Du, S. E. Zhong, Y. Jiang, H. Yu, W. P. Sun, H. G. Pan, X. H. Rui and Y. Yu, Adv. Funct. Mater., 2022, 32, 2110280 CrossRef CAS.
- P. Liu, H. T. Yi, S. Y. Zheng, Z. P. Li, K. J. Zhu, Z. Q. Sun, T. Jin and L. F. Jiao, Adv. Energy Mater., 2021, 11, 2101976 CrossRef CAS.
- N. Mubarak, F. Rehman, M. Ihsan-Ul-Haq, M. Y. Xu, Y. Li, Y. H. Zhao, Z. T. Luo, B. L. Huang and J. K. Kim, Adv. Energy Mater., 2022, 12, 2103904 CrossRef CAS.
- X. Y. Cui, Y. J. Wang, H. D. Wu, X. D. Lin, S. Tang, P. Xu, H. G. Liao, M. S. Zheng and Q. F. Dong, Adv. Sci., 2021, 8, 2003178 CrossRef CAS PubMed.
- L. Ye, M. Liao, T. C. Zhao, H. Sun, Y. Zhao, X. M. Sun, B. J. Wang and H. S. Peng, Angew. Chem., Int. Ed., 2019, 58, 17054–17060 CrossRef CAS PubMed.
- G. Wang, Y. Zhang, B. Guo, L. Tang, G. Xu, Y. Zhang, M. Wu, H. K. Liu, S. X. Dou and C. Wu, Nano Lett., 2020, 20, 4464–4471 CrossRef CAS PubMed.
- G. Y. Wang, F. F. Yu, Y. Zhang, Y. J. Zhang, M. Zhu, G. Xu, M. H. Wu, H. K. Liu, S. X. Dou and C. Wu, Nano Energy, 2021, 79, 105457 CrossRef CAS.
- M. L. Mao, X. Ji, Q. Y. Wang, Z. J. Lin, M. Y. Li, T. Liu, C. L. Wang, Y. S. Hu, H. Li, X. J. Huang, L. Q. Chen and L. M. Suo, Nat. Commun., 2023, 14, 1082 CrossRef CAS PubMed.
- M. Q. Zhu, S. M. Li, B. Li, Y. J. Gong, Z. G. Du and S. B. Yang, Sci. Adv., 2019, 5, eaau6264 CrossRef CAS PubMed.
- R. C. Chen, X. Lu, Q. R. He, M. L. Yao, T. H. Yao, A. Gao, S. J. Ding, Y. H. Cheng and H. K. Wang, ACS Appl. Energy Mater., 2022, 5, 10952–10960 CrossRef CAS.
- Y. Xu, E. Matios, J. M. Luo, T. Li, X. Lu, S. H. Jiang, Q. Yue, W. Y. Li and Y. J. Kang, Nano Lett., 2021, 21, 816–822 CrossRef CAS PubMed.
- C. L. Wang, H. Wang, E. Matios, X. F. Hu and W. Y. Li, Adv. Funct. Mater., 2018, 28, 1802282 CrossRef.
- W. Yang, W. Yang, L. B. Dong, G. J. Shao, G. X. Wang and X. W. Peng, Nano Energy, 2021, 80, 105563 CrossRef CAS.
- Q. L. Chen, B. Liu, L. Zhang, Q. S. Xie, Y. G. Zhang, J. Lin, B. H. Qu, L. S. Wang, B. S. Sa and D. L. Peng, Chem. Eng. J., 2021, 404, 126469 CrossRef CAS.
- Y. X. Hao, D. S. Yu, S. Q. Zhu, C. H. Kuo, Y. M. Chang, L. Q. Wang, H. Y. Chen, M. H. Shao and S. J. Peng, Energy Environ. Sci., 2023, 16, 1100–1110 RSC.
- L. M. Deng, S. F. Hung, Z. Y. Lin, Y. Zhang, C. C. Zhang, Y. X. Hao, S. Y. Liu, C. H. Kuo, H. Y. Chen, J. Peng, J. Z. Wang and S. J. Peng, Adv. Mater., 2023, 35, 2305939 CrossRef CAS PubMed.
- X. Lu, J. M. Luo, E. Matios, Y. W. Zhang, H. Wang, X. F. Hu, C. L. Wang, H. K. Wang, J. Y. Wang and W. Y. Li, Nano Energy, 2020, 69, 104446 CrossRef CAS.
- P. C. Zou, C. Y. Wang, J. Y. Qin, R. Zhang and H. L. Xin, Energy Storage Mater., 2023, 58, 176–183 CrossRef.
- S. F. Ye, L. F. Wang, F. F. Liu, P. C. Shi and Y. Yu, eScience, 2021, 1, 75–82 CrossRef.
- Y. Zhang, C. W. Wang, G. Pastel, Y. D. Kuang, H. Xie, Y. J. Li, B. Y. Liu, W. Luo, C. J. Chen and L. B. Hu, Adv. Energy Mater., 2018, 8, 1800635 CrossRef.
- X. Jin, Y. Zhao, Z. Shen, J. Pu, X. Xu, C. Zhong, S. Zhang, J. Li and H. Zhang, Energy Storage Mater., 2020, 31, 221–229 CrossRef.
- F. Xu, C. Z. Qu, Q. Q. Lu, J. S. Meng, X. H. Zhang, X. S. Xu, Y. Q. Qiu, B. C. A. Ding, J. Y. Yang, F. R. Cao, P. H. Yang, G. S. Jiang, S. Kaskel, J. Y. Ma, L. Li, X. C. Zhang and H. Q. Wang, Sci. Adv., 2022, 8, eabm7489 CrossRef CAS PubMed.
- Y. X. Hao, S. F. Hung, W. J. Zeng, Y. Wang, C. C. Zhang, C. H. Kuo, L. Q. Wang, S. Zhao, Y. Zhang, H. Y. Chen and S. J. Peng, J. Am. Chem. Soc., 2023, 145, 23659–23669 CrossRef CAS PubMed.
- M. Han, C. B. Zhu, T. Ma, Z. Pan, Z. L. Tang and J. Chen, Chem. Commun., 2018, 54, 2381–2384 RSC.
- S. Choudhury, S. Y. Wei, Y. Ozhabes, D. Gunceler, M. J. Zachman, Z. Y. Tu, J. H. Shin, P. Nath, A. Agrawal, L. F. Kourkoutis, T. A. Arias and L. A. Archer, Nat. Commun., 2017, 8, 898 CrossRef PubMed.
- Z. Xu, J. Yang, T. Zhang, L. Sun, Y. Nuli, J. Wang and S. i. Hirano, Adv. Funct. Mater., 2019, 29, 1901924 CrossRef.
- X. Zhou, F. Liu, Y. Wang, Y. Yao, Y. Shao, X. Rui, F. Wu and Y. Yu, Adv. Energy Mater., 2022, 12, 2202323 CrossRef CAS.
- P. C. Shi, S. P. Zhang, G. X. Lu, L. F. Wang, Y. Jiang, F. F. Liu, Y. Yao, H. Yang, M. Z. Ma, S. F. Ye, X. Y. Tao, Y. Z. Feng, X. J. Wu, X. H. Rui and Y. Yu, Adv. Energy Mater., 2021, 11, 2003381 CrossRef CAS.
- Y. Jiang, Y. Yang, F. Ling, G. Lu, F. Huang, X. Tao, S. Wu, X. Cheng, F. Liu, D. Li, H. Yang, Y. Yao, P. Shi, Q. Chen, X. Rui and Y. Yu, Adv. Mater., 2022, 34, e2109439 CrossRef PubMed.
- X. Xia, S. Xu, F. Tang, Y. Yao, L. Wang, L. Liu, S. He, Y. Yang, W. Sun, C. Xu, Y. Feng, H. Pan, X. Rui and Y. Yu, Adv. Mater., 2023, 35, e2209511 CrossRef PubMed.
- Y. Zhuang, D. Deng, L. Lin, B. Liu, S. Qu, S. Li, Y. Zhang, B. Sa, L. Wang, Q. Wei, L. Mai, D.-L. Peng and Q. Xie, Nano Energy, 2022, 97, 107202 CrossRef CAS.
- L. Zhao, Z. Hu, Z. Huang, Y. Tao, W. H. Lai, A. Zhao, Q. Liu, J. Peng, Y. Lei, Y. X. Wang, Y. Cao, C. Wu, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2022, 12, 2200990 CrossRef CAS.
- Y. P. Zhuang, D. Y. Deng, L. Lin, B. Liu, S. S. Qu, S. C. Li, Y. G. Zhang, B. S. Sa, L. S. Wang, Q. L. Wei, L. Q. Mai, D. L. Peng and Q. S. Xie, Nano Energy, 2022, 97, 107202 CrossRef CAS.
- L. F. Zhao, Z. Hu, Z. Y. Huang, Y. Tao, W. H. Lai, A. L. Zhao, Q. N. Liu, J. Peng, Y. J. Lei, Y. X. Wang, Y. L. Cao, C. Wu, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2022, 12, 2200990 CrossRef CAS.
- K. Lin, X. F. Xu, X. Y. Qin, J. X. Wu, Q. Liu, Z. Y. Tang, S. He, Y. H. Ye, F. Y. Kang and B. H. Li, Small, 2021, 17, 2104021 CrossRef CAS PubMed.
- X. Lu, H. Y. Zhao, Y. Y. Qin, E. Matios, J. M. Luo, R. C. Chen, H. Nan, B. Wen, Y. A. Zhang, Y. Y. Li, Q. R. He, X. T. Deng, J. D. Lin, K. Zhang, H. K. Wang, K. Xi, Y. Q. Su, X. F. Hu, S. J. Ding and W. Y. Li, ACS Nano, 2023, 17, 10665–10676 CrossRef CAS PubMed.
- X. Y. Zheng, W. J. Yang, Z. Q. Wang, L. Q. Huang, S. Geng, J. Y. Wen, W. Luo and Y. H. Huang, Nano Energy, 2020, 69, 104387 CrossRef CAS.
- M. Y. Xu, Z. J. Liu, Y. Li, N. Mubarak, H. L. Wong, M. Tamtaji, Y. H. Zhao, Y. Y. Li, J. Wang, J. W. You, H. W. Liu, Y. T. Cai, K. A. Zhang, F. Xu, K. Amine, J. K. Kim and Z. T. Luo, Energy Storage Mater., 2023, 60, 102848 CrossRef.
- M. Guo, H. L. Dou, W. Y. Zhao, X. L. Zhao, B. X. Wan, J. H. Wang, Y. T. Yan, X. M. Wang, Z. F. Ma and X. W. Yang, Nano Energy, 2020, 70, 104479 CrossRef CAS.
- Y. C. Ji, J. M. Qiu, W. G. Zhao, T. C. Liu, Z. H. Dong, K. Yang, G. R. Zheng, G. Y. Qian, M. Yang, Q. D. Chen, K. Amine, F. Pan and L. Y. Yang, Chem, 2023, 9, 2943–2955 CAS.
- Y. X. Xiang, G. R. Zheng, Z. T. Liang, Y. T. Jin, X. S. Liu, S. J. Chen, K. Zhou, J. P. Zhu, M. Lin, H. J. He, J. J. Wan, S. S. Yu, G. M. Zhong, R. Q. Fu, Y. X. Li and Y. Yang, Nat. Nanotechnol., 2020, 15, 883–890 CrossRef CAS PubMed.
- R. Zhuang, X. H. Zhang, C. Z. Qu, X. S. Xu, J. Y. Yang, Q. Ye, Z. Liu, S. Kaskel, F. Xu and H. Q. Wang, Sci. Adv., 2023, 9, eadh8060 CrossRef CAS PubMed.
- D. Wang, J. Qiu, N. Inui, R. Hagiwara, J. Hwang and K. Matsumoto, ACS Energy Lett., 2023, 8, 5248–5252 CrossRef CAS.
- C. H. Jo, K. S. Sohn and S. T. Myung, Energy Storage Mater., 2023, 57, 471–496 CrossRef.
- C. J. Niu, D. Y. Liu, J. A. Lochala, C. S. Anderson, X. Cao, M. E. Gross, W. Xu, J. G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2021, 6, 723–732 CrossRef CAS.
- C. Heubner, S. Maletti, H. Auer, J. Hüttl, K. Voigt, O. Lohrberg, K. Nikolowski, M. Partsch and A. Michaelis, Adv. Funct. Mater., 2021, 31, 2106608 CrossRef CAS.
- Y. Y. Liu, H. Wang, H. Y. Yang, Z. X. Wang, Z. X. Huang, D. H. Pan, Z. F. Zhang, Z. Y. Duan, T. T. Xu, D. Z. Kong, X. J. Li, Y. Wang and J. Y. Sun, ACS Nano, 2023, 17, 10844–10856 CrossRef CAS PubMed.
- D. H. Pan, H. Y. Yang, Y. Y. Liu, H. Wang, T. T. Xu, D. Z. Kong, J. J. Yao, Y. M. Shi, X. J. Li, H. Y. Yang and Y. Wang, Nanoscale, 2023, 15, 17482–17493 RSC.
- H. Wang, W. L. Bai, H. Wang, D. Z. Kong, T. T. Xu, Z. F. Zhang, J. H. Zang, X. C. Wang, S. Zhang, Y. T. Tian, X. J. Li, C. S. Lee and Y. Wang, Energy Storage Mater., 2023, 55, 631–641 CrossRef.
- D. X. Cao, Y. J. Xing, K. Tantratian, X. Wang, Y. Ma, A. Mukhopadhyay, Z. Cheng, Q. Zhang, Y. C. Jiao, L. Chen and H. L. Zhu, Adv. Mater., 2019, 31, 1807313 CrossRef PubMed.
- Z. Y. Lyu, G. J. H. Lim, R. Guo, Z. H. Pan, X. Zhang, H. Zhang, Z. M. He, S. Adams, W. Chen, J. Ding and J. Wang, Energy Storage Mater., 2020, 24, 336–342 CrossRef.
- S. Zhou, I. Usman, Y. J. Wang and A. Q. Pan, Energy Storage Mater., 2021, 38, 141–156 CrossRef.
- A. J. Hu, F. Li, W. Chen, T. Y. Lei, Y. Y. Li, Y. X. Fan, M. He, F. Wang, M. J. Zhou, Y. Hu, Y. C. Yan, B. Chen, J. Zhu, J. P. Long, X. F. Wang and J. Xiong, Adv. Energy Mater., 2022, 12, 2202432 CrossRef CAS.
- T. Ma, Y. X. Ni, Q. R. Wang, W. J. Zhang, S. Jin, S. B. Zheng, X. Yang, Y. P. Hou, Z. L. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202207927 CrossRef CAS PubMed.
- Z. K. Ma, J. W. Chen, J. Vatamanu, O. Borodin, D. Bedrov, X. G. Zhou, W. G. Zhang, W. S. Li, K. Xu and L. D. Xing, Energy Storage Mater., 2022, 45, 903–910 CrossRef.
- J. Y. Wang, W. Huang, A. Pei, Y. Z. Li, F. F. Shi, X. Y. Yu and Y. Cui, Nat. Energy, 2019, 4, 664–670 CrossRef CAS.
- T. Chen, Z. K. Jin, Y. C. Liu, X. Q. Zhang, H. P. Wu, M. X. Li, W. W. Feng, Q. Zhang and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207645 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05814j |
| This journal is © The Royal Society of Chemistry 2024 |