
Development of polypeptide-based materials toward messenger RNA delivery
Bowen
Zhao
a,
Xiao
Zhang
 a,
Molly S.
Bickle
a,
Shiwei
Fu
a,
Qingchun
Li
a and
Fuwu
Zhang
*ab
a,
Molly S.
Bickle
a,
Shiwei
Fu
a,
Qingchun
Li
a and
Fuwu
Zhang
*ab
aDepartment of Chemistry, University of Miami, 1301 Memorial Drive, Coral Gables, Florida 33146, USA. E-mail: fxz174@miami.edu
bThe Dr John T. Macdonald Foundation Biomedical Nanotechnology Institute, University of Miami, Miami, FL 33136, USA
First published on 26th December 2023
Abstract
Messenger RNA (mRNA)-based therapeutic agents have demonstrated significant potential in recent times, particularly in the context of the COVID-19 pandemic outbreak. As a promising prophylactic and therapeutic strategy, polypeptide-based mRNA delivery systems attract significant interest because of their low cost, simple preparation, tuneable sizes and morphology, convenient large-scale production, biocompatibility, and biodegradability. In this review, we begin with a brief discussion of the synthesis of polypeptides, followed by a review of commonly used polypeptides in mRNA delivery, including classical polypeptides and cell-penetrating peptides. Then, the challenges against mRNA delivery, including extracellular, intracellular, and clinical barriers, are discussed in detail. Finally, we highlight a range of strategies for polypeptide-based mRNA delivery, offering valuable insights into the advancement of polypeptide-based mRNA carrier development.
 Fuwu Zhang | Fuwu Zhang is currently an Assistant Professor of Chemistry at the University of Miami. He received his Ph.D. degree in chemistry from Texas A&M University in 2015 and was then a postdoctoral fellow at the National Institutes of Health and Texas A&M University. Research in his group broadly lies in organic chemistry, polymer chemistry, chemical biology, drug delivery, and nanomedicine, with an emphasis on the development of innovative delivery systems for small molecules, nucleic acids, peptides, and proteins. He received the prestigious 2023 National Science Foundation CAREER Award to develop stimuli-responsive polypeptides for nucleic acid delivery. |
1. Introduction
Messenger RNA (mRNA)-based therapeutic agents are a class of “information drugs” with far-reaching therapeutic potential and have been investigated for the treatment of various diseases, including cancers, viral infections, and genetic and metabolic disorders.1–4 mRNA is a biomacromolecule carrying abundant negatively charged phosphates, holding great potential to revolutionize vaccination, protein replacement therapies, and the treatment of genetic diseases.4–7 mRNA has demonstrated significant promise for treating various diseases since the 1990s.8,9 Single-stranded DNA (ssDNA) and single-stranded RNA (ssRNA, like mRNA),8 are more flexible than double-stranded DNA (dsDNA) and double-stranded RNA (dsRNA) (Fig. 1A).10 Unlike plasmid DNA (pDNA), which must enter the cell nucleus for transcription, mRNA works through translation in the cytoplasm. This means mRNA transfection does not require the nuclear envelope to be breached and is therefore less likely to cause insertional mutagenesis.1 The use of COVID-19 mRNA vaccines has been evolving throughout the pandemic, which highlights the great potential of nucleic acid-based formulations and has caused a dramatic increase in financial support and capital investment in nucleic acid therapeutics.11–19 However, the cellular uptake of exogenous mRNA faces significant challenges, including steric hindrance effect and the electrostatic repulsion between the cell membranes and nucleic acids.20,21 The successful delivery of mRNA is hampered by its vulnerability to hydrolytic and enzymatic degradation by nucleases in the bloodstream, as well as systemic side effects arising from its lack of specificity (Fig. 1B). Thus, effective delivery remains to be the most significant barrier to the widespread use of nucleic acid therapeutics in a clinical setting.15,22 Therefore, deliberate design of effective delivery systems is essential to fully realize the potential of nucleic acid therapeutics.23,24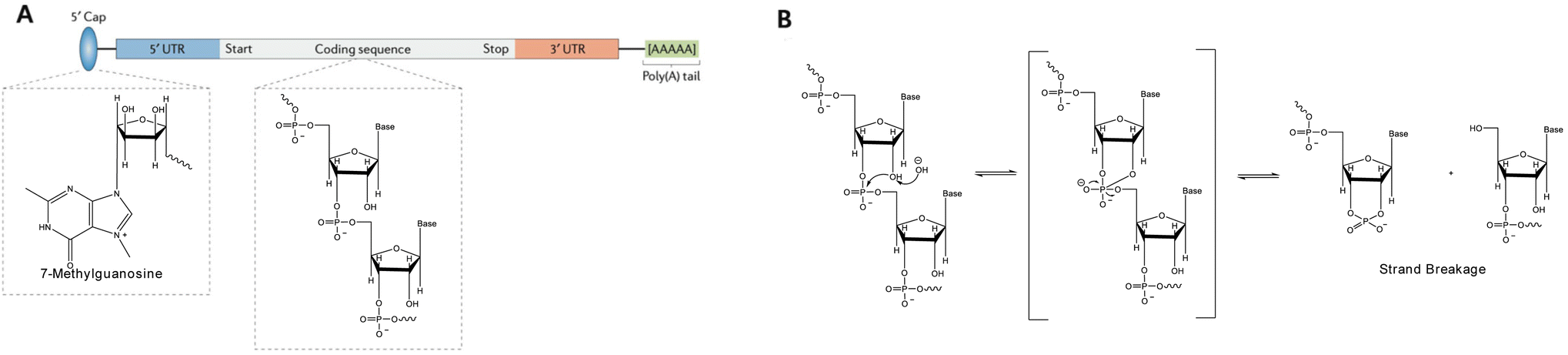 | ||
| Fig. 1 (A) Structure of native mRNA. Adapted with permission from ref. 10, Copyright 2017, Springer; (B) the mechanism of base-catalysed mRNA in-line hydrolysis. Adapted with permission from ref. 23, Copyright 2023, Multidisciplinary Digital Publishing Institute. | ||
Currently, both viral and non-viral systems have been developed to deliver nucleic acids into target cells.2,7,8,14,25,26 Viral vectors, such as adenovirus vectors, adeno-associated virus vectors, and retrovirus vectors, demonstrate high potency in delivering genetic materials to the host cells, owing to their innate ability to enter and utilize the transcription machinery.27 However, several inherent drawbacks, including immunogenicity, carcinogenicity, and insertional mutagenesis, greatly limit their applications.8,27,28 In contrast, non-viral carriers offer superior biocompatibility and, as a result, have greater potential for therapeutic applications. They also have the advantages of low cost, simple preparation, convenient large-scale production, high safety, and the ability to accommodate exogenous genes of unlimited length.8
Synthetic polypeptides are biomaterials, consisting of repeating amino acid units linked by peptide bonds, which have been widely used in drug/gene delivery and tissue engineering since they are naturally derived, biocompatible, and degradable polymers.10,29 In biological systems, there are 20 types of essential amino acids, along with over 500 non-proteinogenic amino acids that can be harnessed for designing mRNA carriers.30 Notably, poly(L-lysine) (PLL), poly(L-ornithine) (PLO), and poly(L-arginine) (PLR) have demonstrated excellent efficiency in condensing nucleic acids, making them promising candidates as mRNA carriers. Polypeptides can adopt secondary structures, such as α-helix or β-sheet, which gives them unique and versatile bio-functions that distinguish them from many other synthetic polymers.31 Various types of polypeptides are available for nucleic acid delivery, including hydrophilic and hydrophobic derivatives, which are employed in constructing diverse nanosystems, such as polyplexes, hydrogels, micelles, vesicles, supramolecular polymers, and stimuli-sensitive polymers.10,29,31–37
In this review, we begin by a brief discussion of the synthesis of polypeptides, followed by a review of commonly used polypeptides in mRNA delivery, including classical polypeptides and cell-penetrating peptides. Then, the challenges against mRNA delivery, including extracellular, intracellular, and clinical barriers, are discussed in detail. Finally, we summarize a range of strategies for polypeptide-based mRNA delivery, offering valuable insights into the advancement of polypeptide-based mRNA carrier development.
2. Synthesis of polypeptides
Polypeptides are typically synthesized by ring-opening polymerization (ROP) of N-carboxyanhydrides (NCA) of α amino acids, followed by post-polymerization modification, while short (poly)peptides are more frequently synthesized by solid phase synthesis.10,38,39 The ROP of NCAs can be initiated by dedicated initiators or catalysts themselves such as transition metal complexes, organosilicon compounds, amine, and ammonium salts, via different mechanisms.10 The most likely pathways of NCA polymerization are the so-called “amine” and the “activated monomer” (AM) mechanisms. The amine mechanism is a nucleophilic ring-opening chain growth process where the polymer could grow linearly with monomer conversion if side reactions were absent (Fig. 2A).38 The AM mechanism is initiated by deprotonation of a NCA, which then becomes the nucleophile that initiates chain growth. It is crucial to recognize that a reacting system undergoes alternating between amine and AM mechanisms multiple times during polymerization. The detailed mechanisms of synthesis of polypeptides via ROP of α-amino acid NCAs was summarized in the reviews by Jianjun Cheng and Timothy J. Deming.31 | ||
| Fig. 2 Synthesis and side-chain modification of polypeptides. (A) Synthesis of polypeptides via (a) nucleophile initiated, (b) transition metal initiated, and (c) base-initiated mechanisms ring-opening polymerization (ROP) of N-carboxyanhydrides (NCA)s. Adapted with permission from ref. 38, Copyright 2022, Elsevier. (B) Schematic showing two pathways for synthesis of side-chain modified polypeptides. Adapted with permission from ref. 40, Copyright 2016, American Chemical Society. | ||
There are two routes to prepare side-chain modification (SCM) positively charged polypeptides: (1) the functional monomer route where SCM NCA monomers are polymerized, and (2) the post-polymerization modification (PPM) route where functional groups are chemically conjugated to reactive polypeptide side-chains after polymerization(Fig. 2B).40 Polypeptides synthesized from functionalized monomers feature entirely modified side chains, enabling the controlled and easy incorporation of multiple types of modifications into individual chains.40 However, this approach necessitates additional efforts in the preparation and purification of functionalized monomers. On the other hand, PPM strategies requires high modification efficiency, typically relying on “click” reactions, to avoid incomplete functionalization due to steric hindrance and low reactivity.40
3. Classification of polypeptide-based carriers in mRNA delivery
Over the past decade, substantial progress has been made in the development of polymeric materials for mRNA delivery, such as poly(α-amino acids), polyethylenimine, poly(amino amine) dendrimers, poly(beta-amino esters), reducible poly(amino amine), and chitosan, among others. Owing to the highly negative charged characteristic, the formation of complexes by electrostatic interactions between positively charged carriers and mRNA can not only assist in their cellular uptake but also shield mRNA from hydrolytic degradation, ultimately amplifying the transfection efficiency.7,12,37,41–44Cationic polymers, also termed as polycations, represent an important category of non-viral gene carriers, which can condense nucleic acids into nano-sized polyplexes through electrostatic interactions.45 The utilization of polymeric materials has advanced the field of mRNA therapeutics, rendering it a promising strategy for both prophylactic and therapeutic purposes. Among these options, peptide-based materials exhibit remarkable potential as gene carrier systems due to their biocompatibility and biodegradability that reduce the risk of cumulative cytotoxicity in contrast to many non-degradable polymers, such as polyethylenimine (PEI) and polyethylene glycol (PEG).46 Furthermore, the tuneable size, low cytotoxicity, high transfection efficiency, unique secondary structure, and low production cost render polypeptides extremely attractive for mRNA delivery.47 Positive charged pendant groups, such as ε-amine of lysine, δ-amine of ornithine, imidazole group of histidine, and guanidine group of arginine, are all used to synthesize cationic polypeptides, including PLL,48 PLO,49 and PLR,25,50 PLH,51 as well as their derivatives (Fig. 3).
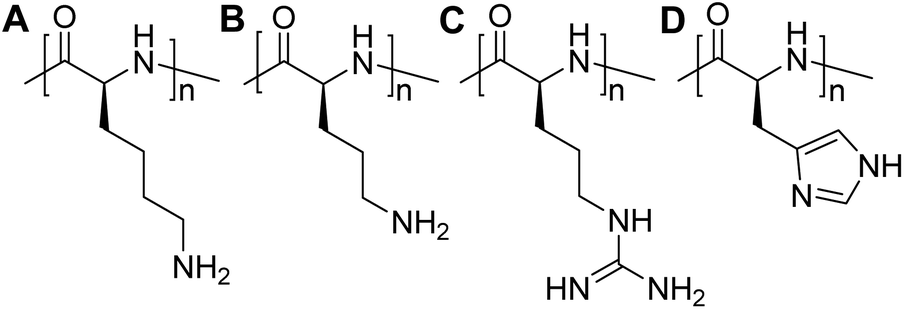 | ||
| Fig. 3 Chemical structures (A) poly(L-lysine), (B) poly(L-ornithine), (C) poly(L-arginine), (D) poly(L-histidine). | ||
3.1 Poly(L-lysine)
Poly(L-lysine) is widely used as the carrier for nucleic acids. While PLLs demonstrate effective binding to the nucleic acids, their capability is restricted due to challenges in enabling endosomal escape and facilitating cargo release within the cell, along with concerns regarding cytotoxicity.52 The remarkable capacity of polylysine to condense DNA was demonstrated by Laemmli in 1975.48 Subsequently, PLLs was employed for gene transfer both in vitro and in vivo. PLLs were synthesized via the SCM NCA approach where tert-butyloxycarbonyl (Boc) protected lysine was converted to SCM NCA monomer, followed by ROP using a primary amine initiator and Boc removal by trifluoroacetic acid. The size and molecular weight can be controlled by tuning the feed ratio of monomer to initiator and by using by different types of primary amine initiator. Lysine residues (–NH2) from PLL are easily protonated and form complexes with negatively charged nucleic acids.12 Lysine-based cationic PLLs achieve high gene transfection efficacy without significant cytotoxicity. Furthermore, Zhao et al. successfully prepared nanocomplexes of hemagglutinin gene of the H9N2 influenza virus and dendrigraft poly-L-lysines (DGLs) using electrostatic interactions.12 The encapsulation of the plasmid DNA (pDNA) within the DGLs prevented degradation and facilitated its escape from endosomes. This led to an improved antigen presentation, resulting in strong cellular and humoral immune responses. Thus, the results indicate that DGLs are an effective non-viral carrier for nucleic acid vaccine delivery.123.2 Poly(L-ornithine)
Structural factors of polypeptides, such as flexibility and charge density, could also affect their complexation with mRNA. Kataoka et al. discovered that complexing with PLO containing a shorter trimethylene spacer, as opposed to a tetramethylene spacer in PLL, offered superior protection for mRNA against RNase attack.49 Complexation with cationic polypeptides have greatly increased the stability of mRNA structure.53 Conte et al. described the divergent synthesis of biodegradable ornithine-derived oligomers and dendrimers as non-viral gene delivery carriers. Dendrimer polyplexes at an N/P ratio of 2 exhibited a much higher (up to 7 times) translated protein content compared to an optimized PEI formulation in the transfections self-amplifying RNA (saRNA).543.3 Poly(L-arginine)
In comparison to PLL, poly(L-arginine) has a higher pKa, leading to a more positively charged state under physiological conditions, thereby resulting in increased cytotoxicity.55,56 The number and density of the positive charges of PLA are positively correlated with its capacity to condense nucleic acids. Fuchs et al. identified the mechanisms through which PLA could enter mammalian cells, indicating promising prospects for cellular uptake and transfection.50 Similarly, Cheng et al. developed PLAs that exhibiting high capacity and efficiency in delivering DNA and siRNA to mammalian cells.56 These PLAs demonstrate a remarkable 1–2 orders of magnitude superiority over commercial transfection reagent Lipofectamine 2000, underscoring their exceptional effectiveness in facilitating gene transfection and silencing. They found that extending length of pendant group to polypeptide backbone would lead to augmentation of cellular uptake.563.4 Poly(L-histidine)
Among proteinogenic amino acids, histidine is notable for exhibiting some of the most fascinating physicochemical properties, which include pH-buffering capabilities, hydrogen bonding, aromaticity, the ability to form coordination bonds with transition metals, and ring alkylation, which alters the hydrophobicity of the imidazole ring.57 The imidazole ring within histidine acts as a weak base, having the capacity to become positively charged when the pH in its surroundings drops below 6, facilitating endosomal escape.51,58,59 Subsequently, various polymers and peptides rich in histidine, along with lipids featuring imidazole, imidazolinium, or imidazolium polar heads, have been reported as effective carriers for the in vitro and in vivo delivery of nucleic acids, including genes, mRNA, or siRNA.58 In the development of nucleic acid delivery systems, the incorporation of imidazoles/histidines in the carrier serves various functions, including improving the extracellular stability of polyplexes, intensifying disruption of polyplexes within acidic endosomes, and enhancing endosomal lysis through osmotic swelling. Histidine-containing carriers have displayed significant promise in facilitating the import of various forms of nucleic acids into the cytosol.57 Langer et al. demonstrated that the attachment of an increased number of imidazole groups to the polylysine template resulted in enhanced transfection. Notably, the polymer with the highest imidazole content exhibited a transfection efficiency similar to PEI but with significantly lower cytotoxicity.603.5 Cell-penetrating peptides
Cell-penetrating peptides (CPPs) usually refer to peptides consisting of 5–30 amino acids.61–63 Typically, CPPs enter cells with minimal cytotoxic effects, efficiently internalizing across cell membranes and playing a crucial role in transporting cargo into live cells.62 CPPs can be classified in different ways.64 Based on their origin, two major branches are provided: (1) protein derived peptides (e.g., transactivator of transcription (TAT) and penetratin); (2) synthetic peptides (e.g., PLL, PLA, PLO, PLH). Cell-penetrating peptides can gain entry into target cells through various mechanisms, including macropinocytosis, clathrin-dependent endocytosis, caveolae-mediated endocytosis, or clathrin-/caveolae-independent endocytosis.62,65Two primary approaches have been investigated for employing CPPs in nucleic acid delivery. The first approach involves covalently attaching CPPs to nucleic acids via chemical linkers, while the second strategy relies on electrostatic interactions and self-assembly to create noncovalent complexes between CPPs and nucleic acids.62 Peptides derived from TAT, penetratin, transportan and polyarginine have been used to enhance transfection efficiency and biological effects through covalent strategy. On the other hand, N-methylpurine-DNA glycosylase (MPG) peptide, Pep-1, and TAT, later all different kinds of cationic peptides, including PLL, PLO, PLA, PLH, have been well investigated to condense nucleic acid and form complexes via electrostatic interactions.61,62,64 For example, arginine-rich cationic CPPs are known to promote cellular internalization through hydrogen bonding and electrostatic interactions with cell membrane surfaces via the guanidinium group.47 Moreover, Kim et al. reported that amphipathic CPP/mRNA complexes with a diameter below 200 nm, exhibited substantially better cellular uptake and enhanced protein expression when compared to hydrophilic CPPs.47
4. Challenges against polypeptide-based mRNA delivery
Despite significant advancements in peptide-based carriers, numerous obstacles and challenges continue to exist in their application for mRNA delivery. Several factors can influence the nucleic acid delivery process, such as off-target effects, transfection efficiency, delivery and release mechanisms, immune response, and cytotoxicity.2,5,12,14,22,57 The complexes formed by electrostatic interaction between negatively charged mRNA and positively charged polymers suffers from poor serum stability, non-specific tissue interaction, and unsatisfactory interaction with cellular membranes. The optimization of peptide-based carriers is an essential step in addressing the complex array of chemical and biological challenges. Systematic adjustments and modification are needed to harness the full potential of these carriers to achieve more efficient and effective mRNA delivery.4.1 Obstacles in construction of polypeptide-based carriers
The construction of polypeptides faces a multitude of intricate challenges. NCA monomer purification has been one of the bottlenecks limiting the availability and scale-up of NCA monomers.31 Traditionally, recrystallization has stood out as the primary method for obtaining NCA monomers, but it falls short when dealing with monomers with complex structures. For example, Zhu et al. have developed synthesized biocompatible copolymers PEG-graft-polypeptide (PPT-g-PEG) from γ-propargyl-L-glutamate NCA monomer.66 Precipitation was used to purify the NCA monomer since it could not be recrystallized. Similarly, Barz et al. also employed precipitation to purify S-ethylsulfonyl-L-homocysteine N-carboxyanhydride (Hcy(SO2Et)-NCA) monomer.67 In fact, many designed NCA monomers could be obtained in high purity and large quantity due to the various issues in the purification process. On the other hand, post-polymerization modification (PPM) of polypeptide requires highly efficient reactions, for instance, click reactions, to install functionalities with high or complete conversion. However, incomplete conversion of functionalities is still a problem due to the steric hindrance. Additionally, the complete removal of copper species may be difficult in the classical copper catalysed azide–alkyne Huisgen cycloaddition (CuAAC).68Polypeptide is biodegradable, which is a desired feature to avoid unnecessary accumulation in the biological system and release of payload in drug and gene delivery.69 However, degradability also causes problems in the synthesis, purification, and storage. Due to their instability in the presence of proteases and peptidases, many naturally occurring peptides constructed with proteinogenic amino acids have limited therapeutic potential due to fast degradation by proteases via either lysosome or ubiquitin-proteasome approaches in the biological systems.70 Robert et al. surprisingly discovered that a slight sequence alterations could improve the stability by two orders of magnitude.70
4.2 Challenges associated with mRNA
The first successful translation of in vitro-transcribed (IVT) mRNA in mice was reported in 1990.71 Since then, subsequential reports of developing mRNA therapeutics have demonstrated immense potential.3 However, there are several challenges associated with unmodified mRNA, since exogenous mRNA is intrinsically immunogenic, triggering several innate immunogenicity. Furthermore, mRNA is susceptible to degradation by nucleases, rapid clearance by the reticular endothelial system, and low cellular uptake and translation efficiencies.72 Additionally, the therapeutic application of mRNA is significantly hindered by their relatively large size, hydrophilic nature, and highly negative charges, which collectively impede their capacity to traverse cell membranes. The resolution of these technical challenges has been partially accomplished through mRNA modifications and use of positively charged carriers.1,2 Several modified nucleotides have been introduced during the in vitro transcription of mRNA to create a synonymous modified transcript, thereby increasing stability against degradation by ribonucleases and reducing the possibilities of immune recognition and enriching the applications of mRNA therapeutics.4,73 On the other hand, different strategies of employing non-viral carriers have been used to protect and deliver mRNA drugs into targeted sites. Nowadays, the large scale production of mRNA therapeutic agents in a cost-effective manner has been achieved, as evidenced the first clinically approved mRNA vaccines against SARS-CoV-2.74 However, further advancements in carriers for nucleic acids delivery are necessary to increase storage stability and more importantly to reduce sides effects and increase therapeutic efficacy.4.3 Challenges faced by polypeptide-mRNA nanocomplexes
In addition, nanocomplexes assembled by peptides and mRNA could be affected by physical, chemical, and biological factors in the body. The stability of nanocomplexes may come into question when exposed to the complex and dynamic physiological environments. For example, the low pH level of the endosome can negatively impact the interaction between mRNA and peptides, leading to the dissociation of the vehicles and pre-release of mRNA. Additionally, the salt concentration in physiological fluid can greatly reduce the stability of nano-vehicles, especially polyplexes, and has an even stronger effect on regulating the binding affinities of polypeptides with nucleic acids than pH.10 Crosslinking of the nanocomplex may help address issues with colloid and hydrolytic stability.The targeting and accumulation within specific tissues or organs play a crucial role in achieving efficient mRNA transfection to target cells while minimizing potential side effects.75,76 Targeting strategies for mRNA delivery can be broadly categorized into two main approaches: passive and active targeting. In the case of passive targeting, the enhanced permeability and retention (EPR) effect enables mRNA delivery systems to accumulate passively in tumour tissues.75 This is achieved by leveraging the distinct features of tumour tissues, including leaky vasculature and compromised lymphatic drainage. Strategies such as surface PEGylation or decoration with polyanions can typically enhance passive targeting, thereby improving the serum stability and extending the blood circulation of gene vectors.76 On the other hand, the active targeting strategy for mRNA delivery is to use targeting molecules, such as antibody, mannose, Arg-Gly-Asp peptide (RGD),77 Cys-Lys-Lys-Lys (CKKK),78 which specifically bind to receptors that are overexpressed on the surface of target cells, enhancing the uptake of the mRNA delivery system by those cells. For example, Dong et al. demonstrated RGD modified mRNA nanocomplexes exhibited prolonged blood circulation and yielded much higher mRNA expression in tumours via intravenous administration compared to non-cRGD nanoformulation.79 Nonetheless, targeting remains a significant challenge in the development of novel mRNA carriers. Nanomaterials tend to accumulate in the liver following systemic administration, which can result in elevated toxicity and reduced therapeutic efficacy.
In terms of condensing nucleic acids, ionizable lipids and polypeptides possess different advantages. Ionizable lipids, exemplified by their success in clinically approved lipid nanoparticle formulations, offer pH responsiveness, efficient endosomal escape, and a mimicry of natural cellular membranes.3,77 On the other hand, polypeptides show advantages such as stimuli responsiveness, biodegradability, and a high degree of customization, making them valuable for applications that demand tailored and targeted nucleic acid delivery with a focus on adaptability to diverse biological environments.10,34 Further investigation is needed to develop principles and strategies for the rational design of targeted mRNA polymeric carriers that can effectively target specific cells, tissues, and organs.
4.4 Physiological barriers for the delivery of peptide-based mRNA nanocomplexes
mRNA therapy is rapidly emerging as one of the most promising approaches in gene therapy, as it leverages the cell's innate mechanisms to either supplement or enhance protein production, providing new avenues for disease treatment.25 Owing to their high molecular weights, hydrophilicity, negatively charged nature, and nuclease degradation, mRNA faces significant challenges in traversing both extracellular and intracellular barriers, which is a major obstacle to their delivery to target tissues and cells.4.4.1.1 Blood clearance. Blood clearance is a major hurdle for many gene delivery systems when administered systemically. Upon injection into the blood, nanocarriers are perceived as foreign entities and are subject to clearance by the renal filtration and the mononuclear phagocytic system (MPS). Rapid renal clearance occurs for nanocomplexes smaller than 5 nm, while larger nanocomplexes are predominantly cleared by the MPS system. Nanoparticles with sizes ranging from 5 to 500 nm are more suitable for blood circulation.10,80 To prolong the blood circulation time of nanoparticles, various strategies have been developed, such as PEGylation and charge-shielding polymeric coatings.81,82 Conjugating PEG with cationic polypeptides facilitates the formation of an outer corona. The resultant steric stabilization serves to impede protein absorption, thereby prolonging circulation time.81 Charge-shielding strategy makes the nanocomplexes avoiding being easily cleared from blood by the MPS.82 These approaches have been shown to effectively reduce the uptake and clearance of nanoparticles by the MPS system, thereby prolonging their blood circulation and enhancing their therapeutic efficacy(Fig. 4).21,75 Besides, high interstitial pressure (IFP) with sophisticated interactions with plenty of cells, including tumour cells, fibroblasts and macrophages, will impede extravasation of nano-vehicles from blood vessels.10
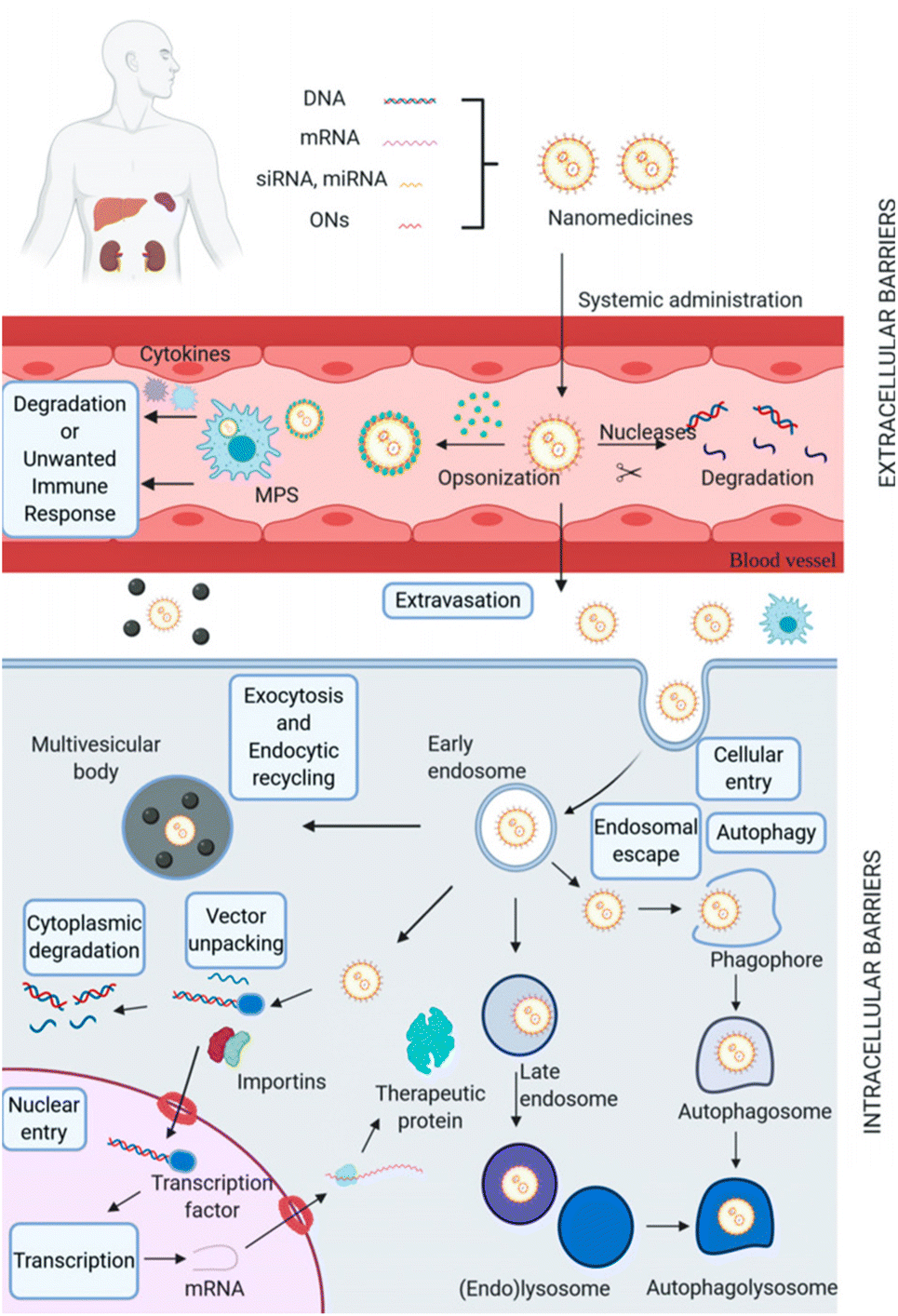 | ||
| Fig. 4 Biological barriers for non-viral gene delivery systems. Adapted with permission from ref. 75, Copyright 2021, Multidisciplinary Digital Publishing Institute. | ||
4.4.1.2 Mucus layer defence. The mucous layer is a dynamically regulated barrier, actively secreted by epithelial cells and covering the surface of the epithelium.32 Its primary function is to provide protection against pathogen invasion. Nevertheless, the presence of the mucous layer also presents a significant challenge for the effective delivery of mRNA via the mucosal route. Upon entering the mucus layer, mRNA nanocomplexes are prone to interact with mucus proteins, subsequently being entrapped by mucus fibers, leading to their rapid elimination from the body. The high abundance of negatively charged functional groups such as carboxyl and sulphate groups, and sialic acid in the mucous layer further impairs the diffusion of the positively charged nanocomplexes. The success of mRNA delivery depends on the effective crossing of the mucous layer and epithelial cells and efficient uptake by antigen-presenting cells (APCs), characterized by the presence of an epithelium and dendritic/Langerhans’ cells.83 However, the uptake of mRNA nanocomplexes is often suboptimal, resulting in reduced immune effect. One approach to improve the efficacy of mucosal mRNA is to extend the residence time of mRNA nanoparticles at the mucosal site to enhance their uptake by APCs and improve antigen presentation efficiency. Since the transport and interaction mechanisms of mucus defence in different types of mucus are not be completely same, the development of intelligent polymer delivery carriers tailored to the specific thickness, pH, protein concentration, and rheological properties of the mucous layer at different sites is crucial for the optimal design of mucosal delivery systems.84
4.4.2.1 Cellular uptake. Before reaching the target cells, mRNA nanocomplexes must navigate through the extracellular matrix, a complex network of proteins and carbohydrates, which can impede the movement of nanocomplexes and their interactions with cell membranes for uptake.1,72,85 Because of their hydrophilic nature, negative charge, and high molecular weight, mRNA experience electrostatic repulsion from the lipophilic and negatively charged cell membranes. Hence, encapsulation by cationic nanosystems appears to offer a viable solution, facilitating the binding to cell membranes and subsequent cellular internalization through electrostatic interactions. As of now, the predominant route for the internalization of nucleic acid delivery nanosystems is through the endocytosis pathway (Fig. 4).21,86
4.4.2.2 Intracellular mRNA release. As previously discussed, nano-vehicles that undergo internalization are commonly transported to late endosomes. In the cellular compartment, the pH decreases to a range of 5–6 through the activation of ATPases situated in the endosomal membrane, which actively pump protons into the endosomes.87 Subsequently, the nano-vehicles ensnared within the cellular environment could be directed towards lysosomes, characterized by a pH as low as 4.5 and the abundance of digestive enzymes. Consequently, the genetic payloads within these vehicles would undergo hydrolysis, leading to a compromise in transfection efficiency. Thus, the entrapment within endolysosomes is recognized as a significant barrier to effective nucleic acid delivery.88–90 The so-called “proton sponge effect” has been widely used to explain the endosomal escape of positively charged nanocarriers.91 Nevertheless, it is estimated only 1–2% of loaded nucleic acids can escape from endosomes.91 Endosomal escape is still a bottleneck for RNA delivery.92
Upon successfully escaping from the endolysosomes, another significant obstacle for the nano-vehicles is the cytosolic release of nucleic acids. mRNA therapeutics are more effectively condensed and internalized into cells by cationic polypeptides with high charge density and increased molecular weights.45 However, the intracellular release of nucleic acids, crucial for effective gene transfection, is impeded by the strong binding affinity between the nanocarrier and the payload. Therefore, achieving a delicate equilibrium between mRNA binding and cytoplasmic release is of paramount importance.
4.4.3.1 Scalability. Scalability is a critical clinical barrier for polypeptide-based mRNA delivery systems due to the difficulty in manufacturing these systems on a large scale. Developing scalable manufacturing processes that can produce high-quality polypeptide-based delivery systems at a reasonable cost is instrumental in overcoming this barrier. Synthetic peptide via ROP of NCA has demonstrated be one effective way to obtain tuneable peptide with narrow dispersity. However, significant challenges still exist for the large-scale preparation of advanced delivery systems, including but not limited to stimuli-responsive carriers, lipid-peptides, targeting moieties.
4.4.3.2 Efficacy and safety. Another clinical barrier that must be overcome is the demonstration of sufficient efficacy and safety of the polypeptide-based mRNA delivery system in clinical trials. These trials typically involve multiple phases, starting with small-scale trials in healthy individuals and progressing to larger-scale trials in patients with the targeted disease or condition.94 For example, polypeptides and mRNA could be recognized as foreign entities, causing inflammation or other adverse reactions after administration. Moreover, some polypeptide-based materials with relatively high cytotoxicity may cause damage to cells and tissues, limiting their overall therapeutic effects and safety profiles. The long-term storage of mRNA therapeutic agents and their distribution in areas with limited infrastructure for cold chain storage can be challenging.95 Furthermore, ensuring that polypeptide-based mRNA delivery systems are manufactured consistently with a high standard of quality is essential for their successful application in therapeutic settings. The Food and Drug Administration (FDA) requires that manufacturers adhere to current Good Manufacturing Practice (cGMP) regulations, which outline specific guidelines for the manufacturing, testing, and quality control of drugs and medical devices.96 Therefore, overcoming clinical barriers such as toxicity, stability, and scalability, is critical not only for ensuring the safety and efficacy of polypeptide-based mRNA delivery systems but also for securing FDA approval to bring these promising therapeutics to patients in need.
5. Various vectors in polypeptide-based mRNA delivery
Due to their substantial molecular weights, hydrophilic properties, negative charge, and susceptibility to nuclease degradation, mRNA encounters substantial hurdles in overcoming both extracellular and intracellular barriers. Therefore, effective delivery of mRNA to target tissues and cells requires encapsulation into vectors to achieve efficient delivery into target tissues and cells. Overcoming these barriers is a crucial aspect of designing effective mRNA delivery systems. Various strategies have been used to enhance stability, evade immune recognition, and improve the overall efficiency of mRNA delivery to target cells. The success of mRNA delivery system depends on several critical design parameters, including (i) stability to protect mRNA from enzymatic degradation, (ii) reduced non-specific interactions to avoid aggregation and subsequent accumulation in off-target organs, (iii) targeting specificity into desired tissue and cells, (iv) endosomal disruption to escape into the cytoplasm, (v) release of the mRNA cargo in the cytoplasm.975.1 Polyplexes
Polyplexes are nanoscale complexes made up of cationic polymers, such as peptide-based material, with anionic nucleic acids, for example, mRNA. The interactions between the negatively charged phosphates of nucleic acids and the cationic domains in peptides (such as amine, guanidine, or histidine) are spontaneous and driven by entropy, protecting the mRNA from degradation and helping overcome various barriers associated with mRNA delivery, such as electrostatic repulsion, enzymatic degradation, and clearance from the bloodstream.70,98 The physiochemical properties of peptide based polyplexes(e.g., size, surface charge, interaction strength, colloidal stability) may also be altered via preparation conditions, including ionic strength, pH, concentration, solvent quality, and mixing order.99In comparison with other positively charged polymers, synthetic polypeptides are capable to form stable secondary structures, such as α-helix and β-sheet, due to cooperative hydrogen-bonding, leading to unique self-assembly behaviors.29,100 PLR and PLL adopt a random coiled shape since the repulsion between charged side-chain groups hinders α-helix formation, leading to their weak membrane penetration activity.56,101,102 Polypeptides with a stable α-helical structure have demonstrated improved mRNA delivery efficiency. The helical structure facilitates stronger interactions with and destabilization of lipid bilayers, such as those found in cell and endosomal membranes.103
However, enhancing the water solubility of polypeptides by introducing pendant charge groups often conflicts with the goal of improving their helical stability. To address this challenge, Cheng et al. introduced an innovative approach to create cationic polypeptides with a stable α-helical structure. This was achieved by ensuring a minimum separation distance of 11 σ-bonds between the polypeptide backbone and pendant charge groups, which effectively minimized side-chain charge repulsion and promoted α-helix formation.104 They designed and synthesized poly(γ-(4-vinylbenzyl)-L-glutamate) (PVBLG), which served as a versatile platform for generating a range of cationic polypeptides through post-polymerization modification (PPM). The leading candidate from this library demonstrated a remarkable 12-fold enhancement over PEI (Fig. 5).104
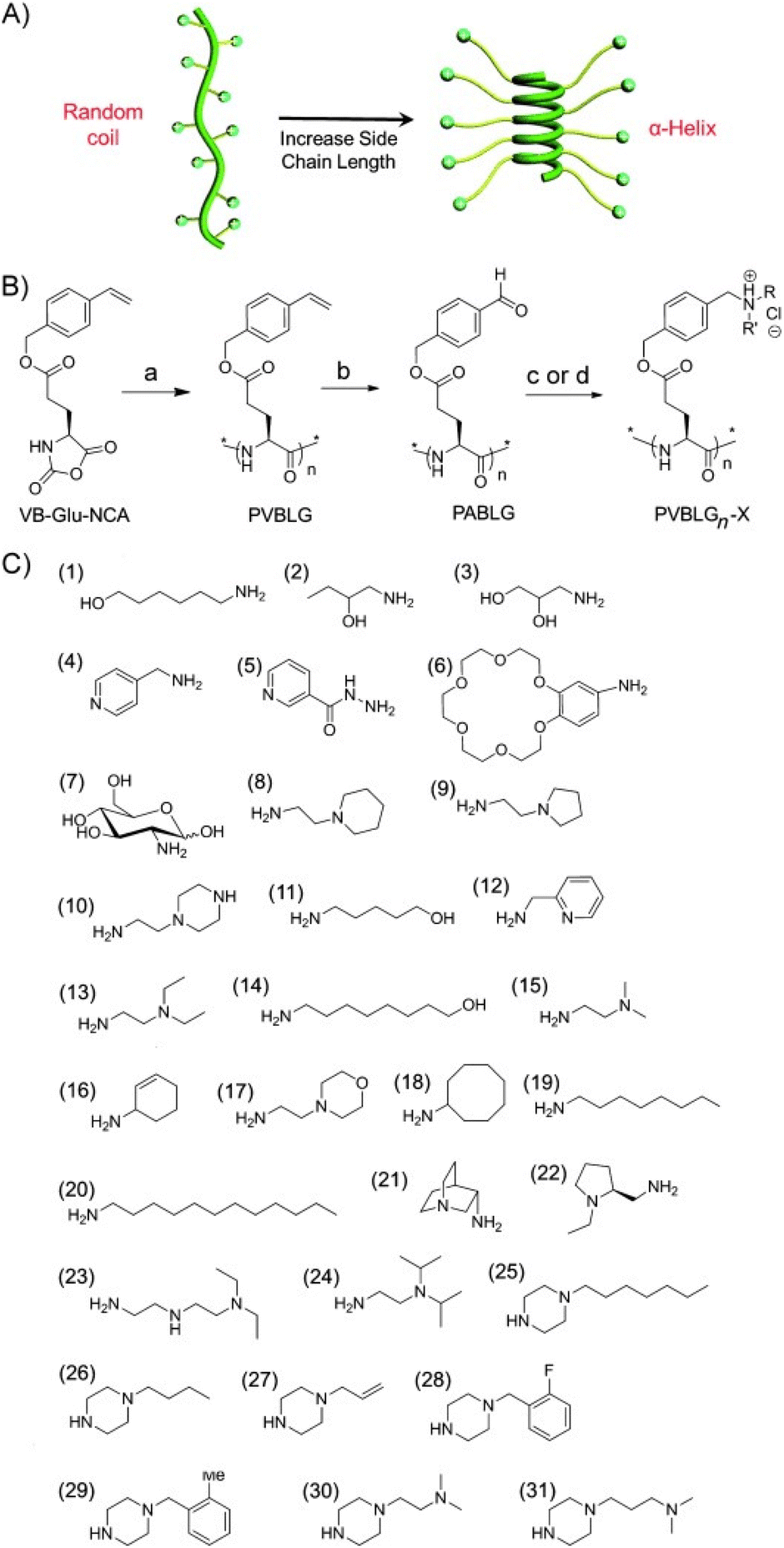 | ||
| Fig. 5 Schematic illustration of (A) polypeptide with charged side chains and the random coil to helix transformation in response to elongated side chains. (B) Reaction scheme for the synthesis of PVBLGn-X polypeptides. (C) Amine groups used to synthesize PVBLGn-X. Adapted with permission from ref. 104, Copyright 2012, Wiley. | ||
Many of these polyplexes are positively charged due to the use of excess cationic polypeptides, which enhances cellular uptake by target cells. However, this also results in toxicity to cells and instability in the presence of salt or serum.44 PLL and PLA exhibit high cytotoxicity because of their highly electropositive groups. While imidazole ring of histidine is capable of buffering the system.51,58,59 Thus, combination of different amino moieties, such as primary amines, imidazole rings, and guanidine groups, may be a viable approach to optimize the mRNA delivery.50,59 While polyplexes bound to plasma proteins could undergo rapid clearance from the bloodstream, this process might be mitigated by hydrophilic coatings, such as PEGylation or hydroxypropyl methacrylic acid. In addition, conjugation with targeting groups may also be helpful to alleviate off-target toxicities.
5.2 Micelles
Polymeric micelles, typically self-assembled from block copolymer, consist of a relatively hydrophobic inner core and a hydrophilic outer shell. Block copolymers interact with nucleic acids to form the core–shell structured polyplex micelles, where the core is constructed via the electrostatic interactions between cationic polypeptides and mRNA while the shell is composed of hydrophilic segments, such as poly (ethylene glycol) (PEG), to shield and stabilize the cores.10,81,105,106PEG-block-poly(L-lysine) (PEG-b-PLL) is one of the most widely used block copolymers for nucleic acid delivery, which form polycation complexes (PCCs) with nucleic acid through a possible two-step process.107 First, the pendant amino groups along the PEG-b-PLL couple with nucleic acid strands to form elementary complexes in a charge-stoichiometric manner (N/P = 1). Next, elementary complexes further self-assemble into micelles with normally spherical morphologies. PEGylation was confirmed to be a good strategy to develop more efficient copolymer vectors in nucleic acid delivery.81 Dong et al. developed a targeted and stable polymeric nanoformulation from PEG-b-PLL, which enhanced systemic delivery of mRNA to tumours.79 PEG-b-PLL and mRNA formed micellar complexes of 73 nm, where PEG formed the outer shell to minimize non-specific interactions with biological entities. Cabral et al. found that PEG-b-PLL and Cy5-labeled gluc mRNA formed polymeric micelles with a hydrodynamic diameter of around 70 nm. The micellar formulation demonstrated superior fluc expression in vivo when compared to both free mRNA and PEI-based polyplexes.106
You et al. have made triblock copolymers of poly(ethylene glycol)-b-poly(L-lysine)-b-poly(L-cysteine) (PEG-PLL-PCys), PCys segment with fluorocarbon can enhance the cellular uptake and the stability of the formed polyplex micelles in physiological conditions.81 Experiment results exhibit that the triblock copolypeptides have low cytotoxicity and good gene transfection efficiency even in the presence of 50% fetal bovine serum. These tuneable PEGylated-polypeptide-based polyplex micelles are widely utilized in gene therapy.108 Besides PLL and their derivatives, many polypeptide block copolymers, such as polyarginine50 and polyhistidine-based block copolymers51,57,59 also have the potential capability to form micellar structure to facilitate mRNA into cells and tissues.
5.3 Vesicles
Similar to micelles, vesicles are formed by amphiphilic molecules, but in this case, the molecules arrange themselves to form a closed lipid bilayer, resulting in an internal aqueous compartment that resembles a natural cell membrane.29,109 Vesicles are typically larger than micelles, often ranging from tens to hundreds of nanometers in diameter. Compared to the relatively hydrophobic core of polymeric micelles, polymeric vesicles can effectively enclose aqueous solutions within their bilayer membrane. This characteristic makes them excellent candidates for encapsulating hydrophilic drugs and bioactive molecules, such as mRNA. Furthermore, they provide several advantages compared to vesicles made from small molecules, including enhanced stability, the capacity to incorporate a broader spectrum of materials, and better control over size, shape, and membrane properties.10,29Positively charged polypeptides could exhibit interesting properties when self-assembled with lipids forming lipid–peptide nanocomplexes. Compared with traditional lipid nanoparticles (LNPs), lipid–peptide hybrid formulations, which amalgamate lipids with polypeptides, prove to be more effective than lipids alone by providing additional functionality to the lipocomplexes in many cases.15,25 This enables them to overcome cellular barriers more effectively, such as cell entry and endosomal escape.15,25 Multifunctional lipid–peptide nanocomplexes, composed of cationic lipids and helper lipids to enhance fusogenic properties for improved endolysosomal escape, along with cationic peptides that contribute positively charged functional groups to enhance electrostatic interactions with mRNA and potentially include an optimal targeting motif for receptor-mediated uptake, hold great promise.25 Integrating lipids with polypeptides in lipid–peptide hybrid formulations presents a promising avenue for mRNA delivery.110 These formulations have the potential to outperform lipids alone, providing enhanced functionality to lipocomplexes. This additional capability equips them to navigate cellular barriers more effectively, enabling improved cell entry and enhanced endosomal escape.
Ge et al. designed a series of arginine-rich amphiphilic peptides as cationic liposome cores for adsorbing mRNA.111 Their lipid–peptide-mRNA (LPm) NPs, was employed to deliver mRNA encoding sodium iodide symporter (NIS) to anaplastic thyroid cancer (ATC) aiming to enhance the sensitivity of ATC to radioiodine treatment, which demonstrated a substantial antitumor effect, indicating the efficacy of this approach. LP(R9H6)m nanocomplex, where R9H6 means nine-arginine peptides and six-histidine peptides, demonstrates the highest efficiency in mRNA delivery. This results in a more than 10-fold increase in NIS expression in ATC cells, as shown in in vitro studies (Fig. 6).111
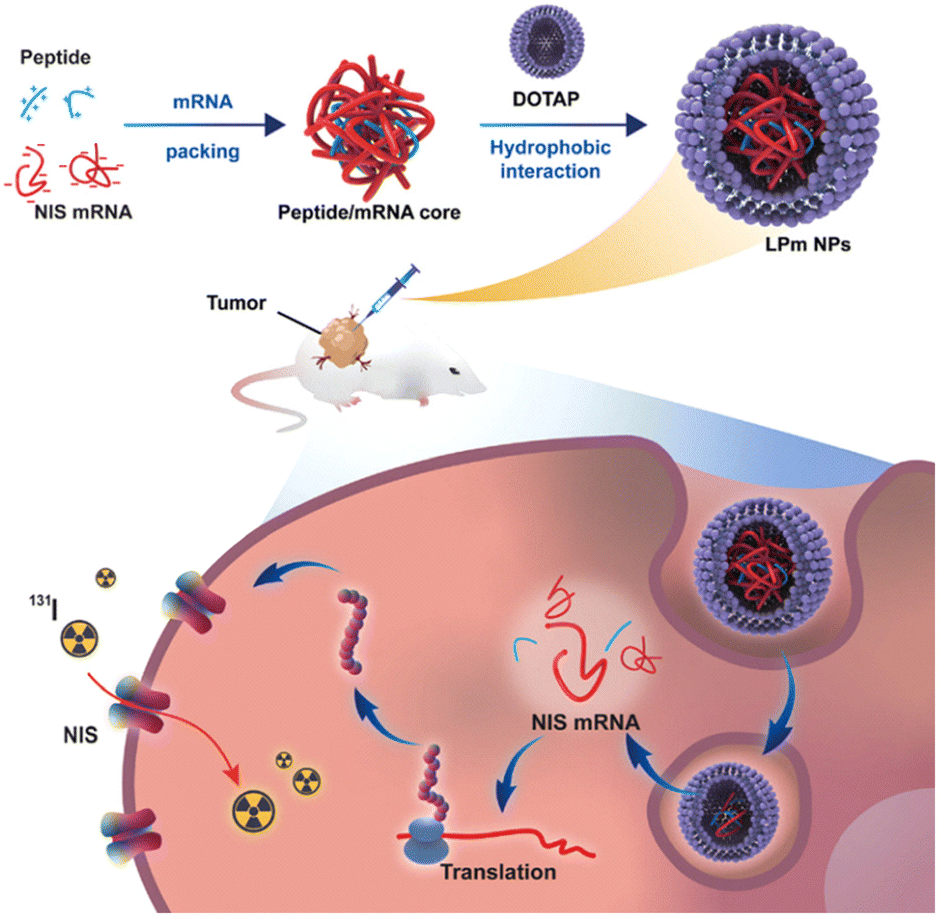 | ||
| Fig. 6 Lipid–peptide-mRNA nanoparticles augment radioiodine uptake in anaplastic thyroid cancer. Adapted with permission from ref. 111, Copyright 2023, Wiley. | ||
5.4 Hydrogel
Unlike molecular-level or nanoscale approaches, hydrogel is a water-absorbing three-dimensional network of hydrophilic polymers, which is commonly utilized in biomedical and pharmaceutical applications, especially in RNA therapy.112 Peptide-based materials have been utilized to construct scaffolds, including hydrogels, using chemical or physical crosslinking methods. For instance, the self-assembly of ethylene glycol-decorated PLG and poly(L-EG2Glu) into nanoribbons and their conformation into β-sheet structures enables the formation of hydrogels.113 However, there is a notable deficiency in relevant research on polypeptide-based hydrogels for mRNA delivery, highlighting the necessity for further investigation. One straightforward approach to integrate nucleic acid delivery systems with scaffolds is by encapsulating the polypeptide-derived polyplexes within the scaffolds for tissue engineering. For example, Artzi et al. developed an innovative platform for localized and sustained siRNA delivery, demonstrating high transfection efficiencies in vitro and in vivo in a breast cancer mice model.1146 Stimuli-responsive strategies in polypeptide-based mRNA delivery
Over the past few years, there has been a growing interest in stimuli-responsive materials capable of responding to various environmental conditions such as pH, temperature, light, oxidation/reduction, etc. Responsive polymers, in particular, are highly desirable due to their ability to address the conflicting requirements of efficient transport and controlled release in nucleic acid delivery systems.109,115–120 Ideally, the smart delivery systems should exhibit sharp phase and structure transitions in response to physiologically relevant stimuli.29 Stimuli-responsive polypeptide-based delivery system have enormous potential in mRNA therapeutics. In particular, pH-responsive and redox-responsive strategies have emerged as promising alternatives, showing great potential for overcoming these obstacles in in vivo studies.6.1 pH-responsive polypeptides
pH-responsive polypeptides can undergo structural changes in response to shifts in the surrounding pH values. The pH in normal tissues typically measures 7.4, but in tumour sites (pH 6.0–7.0), inside endosomes (pH 5.0–6.5), and within lysosomes (pH 4.0–5.0), the environment becomes more acidic. The decrease in pH values can be used as a trigger to facilitate the release of mRNA therapeutics from nanocomplexes. Kawakami et al. developed a novel pH-sensitive polypeptide known as carboxymethyl poly(L-histidine) (CM-PLH), demonstrating improved efficacy in polyplex nucleic acid delivery.59 The pH-responsive polypeptide is characterized by the presence of imidazole groups, providing a substantial capacity for proton buffering at endosomal/lysosomal pH, along with anionic carboxymethyl groups at physiological pH. Additionally, CM-PLH demonstrated hemolytic activity at endosomal/lysosomal pH, while exerting no significant impact on the rapid formation of serum protein aggregates at physiological pH. Kataoka et al. found that the nuclease stability of the mRNA/catiomer polyplexes was significantly influenced by a difference of just one methylene group. The PLO/mRNA polyplex exhibited enhanced stability in comparison to the PLL/mRNA polyplex.49 To augment the endosomal escape function, the PLO/mRNA polyplex was covered with a charge-conversion polymer (CCP). The system maintained a negative charge at extracellular pH but transitioned to a positive charge within the acidic endosomal environment, facilitating the disruption of the endosomal membrane. In comparison to the native PLO/mRNA polyplex, the incorporation of CCP significantly improved endosomal escape, resulting in an approximately 80-fold enhancement in protein expression efficiency from mRNA (Fig. 7).49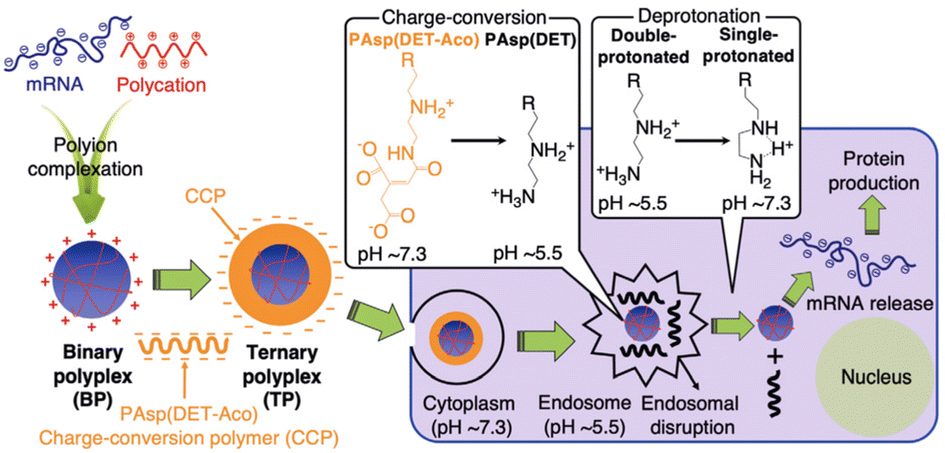 | ||
| Fig. 7 Preparation and intracellular trafficking of mRNA-loaded ternary polyplex (TP) involve coating the mRNA/polycation binary polyplex (BP) with CCP to form the TP. The CCP underwent a positive charge transition at endosomal pH, promoting efficient endosomal escape of the mRNA polyplex. Adapted with permission from ref. 49, Copyright 2022, Wiley. | ||
Recently, Cabral et al. developed a pH-sensitive micelle system for mRNA delivery, featuring a cross-linked core formed by poly(ethylene glycol)-poly(L-lysine) (PEG-pLL) block copolymers modified with cis-aconitic anhydride (CAA).106 These micelles demonstrated good stability at pH 7.4, yet exhibited complete mRNA release at endosomal pH levels (pH 5.5–4.5). PEG-pLL(CAA) mRNA complexes effectively shielded the mRNA from counter polyanion exchange and nuclease attacks. Consequently, the nanocomplex demonstrated enhanced in vivo expression in CT26 tumour-bearing mice compared to non-cross-linked micelles and even surpassed a commercially available PEI-based transfection agent (Fig. 8).106 The findings underscore the promise of pH-responsive strategy for mRNA delivery.
 | ||
| Fig. 8 Micelle formation of PEG-pLL(CAA). Adapted with permission from ref. 106, Copyright 2022, Multidisciplinary Digital Publishing Institute. | ||
6.2 Redox-responsive polypeptides
Redox-responsive polypeptides have shown significant promise in the field of mRNA delivery.121,122 The disulfide bond (–S–S–) stands out as a highly valuable functional group, prominently present in proteins and various natural products. Glutathione (GSH) is one of the most crucial intracellular antioxidants and redox balance regulators. Within the human body, the intracellular concentration of GSH ranges from 1 to 10 mM, while it is found at micromolar levels (20–40 μM) in typical extracellular fluids.29 Interestingly, cancer cells exhibit a notably higher intracellular GSH concentration compared to their normal counterparts. The stability of the disulfide bond in blood circulation and extracellular environments prevents premature drug release, mitigating systemic toxicities. Upon cellular uptake of nanocarriers, the elevated concentration of GSH in the cytoplasm triggers disulfide bond cleavage, resulting in the breakdown of nanocarriers and subsequent accelerated release of the payload.119,123 This characteristic could play a crucial role in the development of biotherapeutic, such as mRNA therapy.117Gong et al. developed a GSH-responsive cationic block copolymer, poly(aspartic acid-(2-aminoethyl disulfide)-(4-imidazolecarboxylic acid))-poly(ethylene glycol), which formed polyplexes with DNA, mRNA, and Cas9/sgRNA ribonucleoprotein (RNP) via electrostatic interactions.121 The nanoplatform exhibited efficient cellular uptake, excellent endosomal escape, and effective cytosol unpacking of the cargos (Fig. 9).121 GSH-responsive polyplexes maintained their stability in the presence of extracellular GSH concentrations, with subsequent degradation occurring when exposed to intracellular GSH concentrations, enabling payload release. Notably, these polyplexes exhibited commendable transfection efficiency and superior biocompatibility across multiple cell types, outperforming the classical cationic lipid-based delivery system, Lipo2000.121 Clearly, the GSH-responsive polypeptide holds great potential to evolve into an exceptional nanoplatform for mRNA delivery. While there have been a limited number of reports on the use of redox-responsive polypeptides for mRNA delivery, the design principles and fundamental concepts of redox-responsive polypeptides for delivering other therapeutic agents, including DNA, siRNA, small molecule drugs, and proteins, may be extended to mRNA delivery.124,125 Further exploration and in-depth investigations are warranted to fine-tune the redox-responsiveness, specifically tailored for optimizing mRNA delivery.
 | ||
| Fig. 9 Schematic depiction of the proposed intracellular pathways of the DNA polyplex, mRNA polyplex, and Cas9 RNP polyplex. In the cytosol, GSH cleaved the pendent –S–S– bonds in the polymers, thereby converting the cationic polymers to neutral polymers and thus releasing the payloads. Adapted with permission from ref. 121, Copyright 2018, American Chemical Society. | ||
6.3 Other stimuli-sensitive polypeptides
In addition to pH-responsive polypeptides and redox-responsive polypeptides, various responsive polypeptides, such as photo-responsive,69 thermal-responsive,126 enzyme-responsive,127 and adenosine triphosphate (ATP) responsive polypeptides,10 have been developed for biomedical applications. For example, Cheng et al. have utilized light-mediated de-esterification of PVBLG-8 to achieve controlled release of nucleic acids (Fig. 6C).69,104 Different quantities of the photo-sensitive 4,5-dimethoxy-2-nitrobenzyl-glutamate (DMNBLG) are integrated into PVBLG-8 to form random PDMNBLG-r-PVBLG-8 copolymers, where pendant benzyl ester bonds of PVBLG-8 are difficult to be cleaved under physiological conditions. The PDMNBLG domains would undergo a change from uncharged and hydrophobic to negatively charged by providing pendant carboxylate groups after UV or near-infrared irradiation. Thus, negatively charged nucleic acids could be disassociated from the cationic PDMNBLG-r-PVBLG-8 copolymers to promote intracellular gene release.69 Thermal-responsive polypeptides exhibit a phase transition or conformational change at a specific temperature. Wooley et al. developed amphiphilic block copolymer poly(ethylene glycol)-block-poly(DL-allylglycine) (PEG-b-PDLAG), where the hydrophilic–hydrophobic ratio could be tuned to optimize the gel transition temperature.128,129 Furthermore, enzyme-responsive polypeptides enable the selective delivery of therapeutic agents in the presence of enzymes.126 Wooley et al. synthesized a statistical terpolypeptide from L-alanine (Ala), glycine (Gly) and L-isoleucine (Ile), which was used to construct a multiple responsive hydrogel.127 The hydrogel was found to degrade much faster in the presence of enzymes, including proteinase K and matrix metalloproteinase-2 (MMP-2).127 However, most of those stimuli-sensitive polypeptides have not been used in the delivery of mRNA. Extensive research needs to be conducted to demonstrate their utility in mRNA delivery, addressing challenges including, but not limited to, complexities associated with precise temperature control, the limitations of light penetration into tissues, and structural complexity with enzyme responsive polypeptide.7 Conclusions and perspectives
mRNA-based therapeutics have made significant progress in both fundamental and clinical applications in recent decades. The deliberate design of delivery systems plays a pivotal role in unlocking the full potential of mRNA therapeutics. Polypeptides have emerged as one of the key contributors, propelling mRNA therapeutics into the spotlight as a promising prophylactic and therapeutic strategy. The process of mRNA condensation demands substantial efforts to overcome a series of intracellular and extracellular barriers. Numerous strategies have been developed, evolving from basic condensation methods to more advanced approaches. These approaches aim to ensure the serum stability of carriers, attain effective immune evasion, safeguard the payload, and optimize intracellular trafficking. The fine-tuning of physicochemical properties, including size optimization, PEGylation, and the integration of secondary structures like alpha helix and beta sheet, has been developed to enhance the encapsulation and condensation of peptide-based materials for mRNA therapeutics. Furthermore, stimuli-responsive peptide-based materials, with the ability to adapt to diverse environmental conditions such as pH, temperature, light, oxidation/reduction, etc., have been extensively investigated and hold significant promise by leveraging a sophisticated network of intermolecular interactions. In addition to these advancements, targeting strategies have been instrumental in enabling the precise delivery of mRNA drugs, thereby enhancing overall therapeutic effectiveness. Collectively, these innovative approaches underscore the dynamic landscape of mRNA-based therapeutics and their evolving role in medicine.Despite substantial progress and significant potential, numerous challenges persist, hindering the clinical translation of polypeptide-based materials for mRNA delivery. These challenges warrant further attention and extensive research. Some of the critical issues with peptide-based materials for mRNA delivery include but not limited to: (1) unsuccessful clinical applications. Until now, no polypeptide -based mRNA delivery systems have been approved by FDA. It was proposed that the current workflows employed for the biological evaluation and screening of polyplex formulations may be impeding clinical progress.43 Typically, polyplexes with low transfection efficiency or other unexpected parameters in pre-evaluation in vitro, would be excluded from future investigation, however, in vitro experiments are sometimes not good predictors of results in vivo since the experimental condition in vitro might not faithfully mimic the physiological barriers experienced by formulations within living organisms.130 Recently, alternative approaches has been proposed. For example, Dahlman et al. quantified behaviours of over 100 LNPs with the analysis of about 2000 in vivo drug delivery data point using high-throughput in vivo experimental platforms.131 Nevertheless, the approach for rapidly and efficiently screening peptide-based mRNA carriers is still in the process of development. (2) Insufficient transfection efficiency. Polypeptide-based carriers display significant potential in terms of biocompatibility and biodegradability. However, their transfection efficiency has fallen short when compared to LNPs derivatives. Extensive research is needed to better understand the structure–activity relationship of polypeptides and their delivery efficiency. Minimizing variability among the polymeric NPs within each batch and from one batch to another is also essential for reproducible results. (3) Inadequate targeting strategies. Passive and active targeting are the two primary strategies employed in the design of peptide-based mRNA carriers. Active targeting is a preferred approach for precise delivery to the desired sites. There is a critical need to develop highly specific novel targeting ligands and to understand the rules and principles involved in designing targeted mRNA polymeric carriers for specific cells, tissues, and organs.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation CAREER award under grant no. 2238812. F. Z. gratefully acknowledges 2023 Provost's Research Awards (UM PRA 2022-3284) from the University of Miami.References
- G. Han, D. Noh, H. Lee, S. Lee, S. Kim, H. Y. Yoon and S. H. Lee, Adv. Drug Delivery Rev., 2023, 199, 114973 CrossRef CAS PubMed.
- J. C. Kaczmarek, P. S. Kowalski and D. G. Anderson, Genome Med., 2017, 9, 1–16 CrossRef PubMed.
- Y. Wang, Z. Zhang, J. Luo, X. Han, Y. Wei and X. Wei, Mol. Cancer, 2021, 20, 1–33 Search PubMed.
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, Nat. Rev. Drug Discovery, 2018, 17, 261–279 CrossRef CAS PubMed.
- P. S. Kowalski, A. Rudra, L. Miao and D. G. Anderson, Mol. Ther., 2019, 27, 710–728 CrossRef CAS PubMed.
- U. Sahin, K. Kariko and O. Tureci, Nat. Rev. Drug Discovery, 2014, 13, 759–780 CrossRef CAS PubMed.
- S. Y. Tzeng and J. J. Green, Curr. Opin. Biomed. Eng., 2018, 7, 42–50 CrossRef PubMed.
- K. A. Hajj and K. A. Whitehead, Nat. Rev. Mater., 2017, 2, 17056 CrossRef CAS.
- A. Yi, D. Sim, Y. J. Lee, V. Sarangthem and R. W. Park, J. Nanobiotechnol., 2020, 18, 15 CrossRef CAS PubMed.
- Y. Liu and L. Yin, Adv. Drug Delivery Rev., 2021, 171, 139–163 CrossRef CAS PubMed.
- M. Kohli, M. Maschio, K. Joshi, A. Lee, K. Fust, E. Beck, N. Van de Velde and M. C. Weinstein, medRxiv, 2023, preprint, pp. 1–32, DOI:10.1101/2023.09.05.23295085.
- G. Chen, B. Zhao, E. F. Ruiz and F. Zhang, Theranostics, 2022, 12, 4081–4109 CrossRef CAS PubMed.
- J. Shi, M. W. Huang, Z. D. Lu, X. J. Du, S. Shen, C. F. Xu and J. Wang, J. Controlled Release, 2022, 345, 494–511 CrossRef CAS PubMed.
- B. A. S. Machado, K. V. S. Hodel, L. Fonseca, L. A. B. Mascarenhas, L. Andrade, V. P. C. Rocha, M. B. P. Soares, P. Berglund, M. S. Duthie, S. G. Reed and R. Badaro, Vaccines, 2021, 9, 9111345 Search PubMed.
- T. T. H. Thi, E. J. A. Suys, J. S. Lee, D. H. Nguyen, K. D. Park and N. P. Truong, Vaccines, 2021, 9, 9040359 Search PubMed.
- A. Hussain, H. Yang, M. Zhang, Q. Liu, G. Alotaibi, M. Irfan, H. He, J. Chang, X. J. Liang, Y. Weng and Y. Huang, J. Controlled Release, 2022, 345, 314–333 CrossRef CAS PubMed.
- X. Li, X. Guo, M. Hu, R. Cai and C. Chen, J. Mater. Chem. B, 2023, 11, 2063–2077 RSC.
- A. Ishaqat and A. Herrmann, J. Am. Chem. Soc., 2021, 143, 20529–20545 CrossRef CAS PubMed.
- M. Kohli, M. Maschio, K. Joshi, A. Lee, K. Fust, E. Beck, N. Van de Velde and M. C. Weinstein, medRxiv, 2023, preprint, DOI:10.1101/2023.09.05.23295085.
- S. Zhou, W. Chen, J. Cole and G. Zhu, Med. Drug Discovery, 2020, 6, 1–23 Search PubMed.
- J. D. Torres-Vanegas, J. C. Cruz and L. H. Reyes, Pharmaceutics, 2021, 13, 13030428 CrossRef PubMed.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef CAS PubMed.
- B. Sun, W. Wu, E. A. Narasipura, Y. Ma, C. Yu, O. S. Fenton and H. Song, Adv. Drug Delivery Rev., 2023, 200, 115042 CrossRef CAS PubMed.
- Y. Wang and C. Yu, Angew. Chem., Int. Ed., 2020, 59, 23374–23385 CrossRef CAS PubMed.
- D. Grant-Serroukh, M. R. Hunter, R. Maeshima, A. D. Tagalakis, A. M. Aldossary, N. Allahham, G. R. Williams, M. Edbrooke, A. Desai and S. L. Hart, J. Controlled Release, 2022, 348, 786–797 CrossRef CAS PubMed.
- D. van den Brand, M. A. J. Gorris, A. H. van Asbeck, E. Palmen, I. Ebisch, H. Dolstra, M. Hallbrink, L. Massuger and R. Brock, Eur. J. Pharm. Biopharm., 2019, 141, 180–190 CrossRef CAS PubMed.
- J. T. Douglas, Mol. Biotechnol., 2007, 36, 71–80 CrossRef CAS PubMed.
- T. Friedmann, Nat. Genet., 1992, 2, 93–98 CrossRef CAS PubMed.
- C. He, X. Zhuang, Z. Tang, H. Tian and X. Chen, Adv. Healthcare Mater., 2012, 1, 48–78 CrossRef CAS PubMed.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef CAS PubMed.
- J. Cheng and T. J. Deming, Top. Curr. Chem., 2012, 310, 1–26 CAS.
- C. Ge, J. Yang, S. Duan, Y. Liu, F. Meng and L. Yin, Nano Lett., 2020, 20, 1738–1746 CrossRef CAS PubMed.
- Y. Liu, Z. Song, N. Zheng, K. Nagasaka, L. Yin and J. Cheng, Nanoscale, 2018, 10, 15339–15349 RSC.
- W. A. Scott, E. G. Gharakhanian, A. G. Bell, D. Evans, E. Barun, K. N. Houk and T. J. Deming, J. Am. Chem. Soc., 2021, 143, 18196–18203 CrossRef CAS PubMed.
- F. Zhang, Q. Ni, O. Jacobson, S. Cheng, A. Liao, Z. Wang, Z. He, G. Yu, J. Song, Y. Ma, G. Niu, L. Zhang, G. Zhu and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 7066–7070 CrossRef CAS PubMed.
- C. Y. Sun, S. Shen, C. F. Xu, H. J. Li, Y. Liu, Z. T. Cao, X. Z. Yang, J. X. Xia and J. Wang, J. Am. Chem. Soc., 2015, 137, 15217–15224 CrossRef CAS PubMed.
- U. Lachelt and E. Wagner, Chem. Rev., 2015, 115, 11043–11078 CrossRef PubMed.
- N. J. Chan, S. Lentz, P. A. Gurr, T. Scheibel and G. G. Qiao, Prog. Polym. Sci., 2022, 130, 101557 CrossRef CAS.
- S. Abbasi, S. Uchida, K. Toh, T. A. Tockary, A. Dirisala, K. Hayashi, S. Fukushima and K. Kataoka, J. Controlled Release, 2021, 332, 260–268 CrossRef CAS PubMed.
- T. J. Deming, Chem. Rev., 2016, 116, 786–808 CrossRef CAS PubMed.
- Y. Cheng, H. Wei, J. K. Tan, D. J. Peeler, D. O. Maris, D. L. Sellers, P. J. Horner and S. H. Pun, Small, 2016, 12, 2750–2758 CrossRef CAS PubMed.
- M. L. Becker and J. A. Burdick, Chem. Rev., 2021, 121, 10789–10791 CrossRef CAS PubMed.
- R. Kumar, C. F. Santa Chalarca, M. R. Bockman, C. V. Bruggen, C. J. Grimme, R. J. Dalal, M. G. Hanson, J. K. Hexum and T. M. Reineke, Chem. Rev., 2021, 121, 11527–11652 CrossRef CAS PubMed.
- S. Y. Neshat, C. H. R. Chan, J. Harris, O. M. Zmily, S. Est-Witte, J. Karlsson, S. R. Shannon, M. Jain, J. C. Doloff, J. J. Green and S. Y. Tzeng, Biomaterials, 2023, 300, 122185 CrossRef CAS PubMed.
- Z. Jiang and S. Thayumanavan, Adv. Thermoelectr., 2020, 3, 1900206 Search PubMed.
- B. Demeneix and J. P. Behr, Adv. Genet., 2005, 53, 215–230 Search PubMed.
- Y. Kim, H. Kim, E. H. Kim, H. Jang, Y. Jang, S. G. Chi, Y. Yang and S. H. Kim, Pharmaceutics, 2022, 14, 14061271 Search PubMed.
- U. K. Laemmli, Proc. Natl. Acad. Sci., 1975, 72, 4288–4292 CrossRef CAS PubMed.
- A. Dirisala, S. Uchida, J. Li, J. F. R. Van Guyse, K. Hayashi, S. V. C. Vummaleti, S. Kaur, Y. Mochida, S. Fukushima and K. Kataoka, Macromol. Rapid Commun., 2022, 43, e2100754 CrossRef PubMed.
- S. M. Fuchs and R. T. Raines, Biochemistry, 2004, 2438–2444 CrossRef CAS PubMed.
- D. Putnam, A. N. Zelikin, V. A. Izumrudov and R. Langer, Biomaterials, 2003, 24, 4425–4433 CrossRef CAS PubMed.
- S. R. Little and D. S. Kohane, J. Mater. Chem., 2008, 18, 832–841 RSC.
- H. Xian, Y. Zhang, C. Yu and Y. Wang, Pharmaceutics, 2023, 15, 14061205 CrossRef PubMed.
- F. Saviano, T. Lovato, A. Russo, G. Russo, C. R. Bouton, R. J. Shattock, C. Alexander, F. Quaglia, A. K. Blakney, P. Gurnani and C. Conte, J. Mater. Chem. B, 2020, 8, 4940–4949 RSC.
- Y. Zhou, S. Han, Z. Liang, M. Zhao, G. Liu and J. Wu, J. Mater. Chem. B, 2020, 8, 5564–5577 RSC.
- H. Tang, L. Yin, K. H. Kim and J. Cheng, Chem. Sci., 2013, 4, 3839–3844 RSC.
- J. He, S. Xu and A. J. Mixson, Pharmaceutics, 2020, 12, 12080774 CrossRef PubMed.
- P. Midoux, C. Pichon, J. J. Yaouanc and P. A. Jaffres, Br. J. Pharmacol., 2009, 157, 166–178 CrossRef CAS PubMed.
- S. Asayama, M. Sudo, S. Naogaka and H. Kawakami, Mol. Pharm., 2008, 5, 898–901 CrossRef CAS PubMed.
- D. Putnam, C. A. Gentry, D. W. Pack and R. Langer, Proc. Natl. Acad. Sci., 2001, 98, 1200–1205 CrossRef CAS PubMed.
- T. Lehto, K. Kurrikoff and U. Langel, Expert Opin. Drug Delivery, 2012, 9, 823–836 CrossRef CAS PubMed.
- N. A. Alhakamy, A. S. Nigatu, C. J. Berkland and J. D. Ramsey, Ther. Delivery, 2013, 4, 741–757 CrossRef CAS PubMed.
- H. Yokoo, M. Oba and S. Uchida, Pharmaceutics, 2021, 14, 14010078 CrossRef PubMed.
- U. Langel, Pharmaceutics, 2021, 13, 13070987 CrossRef PubMed.
- H. de Jong, K. M. Bonger and D. Lowik, RSC Chem. Biol., 2020, 1, 192–203 RSC.
- G. Zhu, L. Mei, H. D. Vishwasrao, O. Jacobson, Z. Wang, Y. Liu, B. C. Yung, X. Fu, A. Jin, G. Niu, Q. Wang, F. Zhang, H. Shroff and X. Chen, Nat. Commun., 2017, 8, 1482 CrossRef PubMed.
- C. Muhl, O. Schäfer, T. Bauer, H. Räder and M. Barz, Macromolecules, 2018, 51, 8188–8196 CrossRef CAS.
- F. Zhang, S. Zhang, S. F. Pollack, R. Li, A. M. Gonzalez, J. Fan, J. Zou, S. E. Leininger, A. Pavia-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 2056–2066 CrossRef CAS PubMed.
- L. Yin, H. Tang, K. H. Kim, N. Zheng, Z. Song, N. P. Gabrielson, H. Lu and J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 9182–9186 CrossRef CAS PubMed.
- S. M. Howell, S. V. Fiacco, T. T. Takahashi, F. Jalali-Yazdi, S. W. Millward, B. Hu, P. Wang and R. W. Roberts, Sci. Rep., 2014, 4, 6008 CrossRef CAS PubMed.
- J. A. Wolff, R. W. Malcone, P. Williams, W. Chong, G. Acsadi, A. Jani and P. L. Felgner, Science, 1990, 247, 1465–1468 CrossRef CAS PubMed.
- Q. Shuai, F. Zhu, M. Zhao and Y. Yan, Int. J. Pharm., 2021, 607, 121020 CrossRef CAS PubMed.
- A. Liu and X. Wang, Front. Cell Dev. Biol., 2022, 10, 901510 CrossRef PubMed.
- E. Dolgin, Nature, 2021, 589, 189–191 CrossRef CAS PubMed.
- R. Hadianamrei and X. Zhao, J. Controlled Release, 2022, 343, 600–619 CrossRef CAS PubMed.
- Y. Xu, Y. Zheng, X. Ding, C. Wang, B. Hua, S. Hong, X. Huang, J. Lin, P. Zhang and W. Chen, Drug Delivery, 2023, 30, 2219870 CrossRef PubMed.
- J. Qin, L. Xue, N. Gong, H. Zhang, S. J. Shepherd, R. M. Haley, K. L. Swingle and M. J. Mitchell, RSC Adv., 2022, 12, 25397–25404 RSC.
- P. Huang, H. Deng, Y. Zhou and X. Chen, Matter, 2022, 5, 1670–1699 CrossRef CAS.
- Q. Chen, R. Qi, X. Chen, X. Yang, S. Wu, H. Xiao and W. Dong, Mol. Ther., 2017, 25, 92–101 CrossRef CAS PubMed.
- G. Fullstone, J. Wood, M. Holcombe and G. Battaglia, Sci. Rep., 2015, 5, 10649 CrossRef PubMed.
- T. Wu, L. Wang, S. Ding and Y. You, Macromol. Biosci., 2017, 17, 1700114 CrossRef PubMed.
- J. Di, X. Gao, Y. Du, H. Zhang, J. Gao and A. Zheng, Asian J. Pharm. Sci., 2021, 16, 444–458 CrossRef PubMed.
- E. Telemo, M. Korotkova and L. Å. Hanson, Ann. Allergy, Asthma, Immunol., 2003, 90, 28–33 CrossRef CAS PubMed.
- N. Sanders, C. Rudolph, K. Braeckmans, S. C. De Smedt and J. Demeester, Adv. Drug Delivery Rev., 2009, 61, 115–127 CrossRef CAS PubMed.
- Y. Zhong, S. Du and Y. Dong, Acta Pharm. Sin. B, 2023, 13, 1348–1357 CrossRef CAS PubMed.
- T. Bus, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2018, 6, 6904–6918 RSC.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- M. Maugeri, M. Nawaz, A. Papadimitriou, A. Angerfors, A. Camponeschi, M. Na, M. Holtta, P. Skantze, S. Johansson, M. Sundqvist, J. Lindquist, T. Kjellman, I. L. Martensson, T. Jin, P. Sunnerhagen, S. Ostman, L. Lindfors and H. Valadi, Nat. Commun., 2019, 10, 4333 CrossRef CAS PubMed.
- B. B. Mendes, J. Conniot, A. Avital, D. Yao, X. Jiang, X. Zhou, N. Sharf-Pauker, Y. Xiao, O. Adir, H. Liang, J. Shi, A. Schroeder and J. Conde, Nat. Rev. Methods Primers, 2022, 2, 1–21 CrossRef.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stoter, H. Epstein-Barash, L. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638–646 CrossRef CAS PubMed.
- B. Li, M. Zhao, W. Lai, X. Zhang, B. Yang, X. Chen and Q. Ni, Angew. Chem., Int. Ed., 2023, 62, e202302676 CrossRef CAS PubMed.
- M. E. Karim, S. T. Haque, H. Al-Busaidi, A. Bakhtiar, K. K. Tha, M. M. B. Holl and E. H. Chowdhury, Arch. Pharmacal Res., 2022, 45, 865–893 CrossRef CAS PubMed.
- E. Rohner, R. Yang, K. S. Foo, A. Goedel and K. R. Chien, Nat. Biotechnol., 2022, 40, 1586–1600 CrossRef CAS PubMed.
- D. F. Li, Q. S. Liu, M. F. Yang, H. M. Xu, M. Z. Zhu, Y. Zhang, J. Xu, C. M. Tian, J. Yao, L. S. Wang and Y. J. Liang, Bioeng. Transl. Med., 2023, 8, e10492 CrossRef CAS PubMed.
- B. G. Gouveia, P. Rijo, T. S. Goncalo and C. P. Reis, J. Pharm. BioAllied Sci., 2015, 7, 87–96 Search PubMed.
- A. B. Marciel, E. J. Chung, B. K. Brettmann and L. Leon, Adv. Colloid Interface Sci., 2017, 239, 187–198 CrossRef CAS PubMed.
- A. Yen, Y. Cheng, M. Sylvestre, H. H. Gustafson, S. Puri and S. H. Pun, Mol. Pharm., 2018, 15, 2268–2276 CrossRef CAS PubMed.
- C. Su, M. Zhao, Z. Zhu, J. Zhou, H. Wen, Y. Yin, Y. Deng, D. Qiu, B. Li and D. Liang, Macromolecules, 2015, 48, 756–763 CrossRef CAS.
- A. Carlsen and S. Lecommandoux, Curr. Opin. Biomed. Eng., 2009, 14, 329–339 CAS.
- J. Deng, N. Gao, Y. Wang, H. Yi, S. Fang, Y. Ma and L. Cai, Biomacromolecules, 2012, 13, 3795–3804 CrossRef CAS PubMed.
- F. Joubert, M. J. Munson, A. Sabirsh, R. M. England, M. Hemmerling, C. Alexander and M. B. Ashford, J. Controlled Release, 2023, 356, 580–594 CrossRef CAS PubMed.
- Z. Yang, L. Lin, Z. Guo, X. Guo, Z. Tang, H. Tian and X. Chen, Biomacromolecules, 2022, 23, 2867–2877 CrossRef CAS PubMed.
- N. P. Gabrielson, H. Lu, L. Yin, D. Li, F. Wang and J. Cheng, Angew. Chem., Int. Ed., 2012, 51, 1143–1147 CrossRef CAS PubMed.
- E. S. Lee, K. Na and Y. H. Bae, J. Controlled Release, 2003, 91, 103–113 CrossRef CAS PubMed.
- W. Yang, P. Chen, E. Boonstra, T. Hong and H. Cabral, Pharmaceutics, 2022, 14, 14061205 Search PubMed.
- K. Hayashi, H. Chaya, S. Fukushima, S. Watanabe, H. Takemoto, K. Osada, N. Nishiyama, K. Miyata and K. Kataoka, Macromol. Rapid Commun., 2016, 37, 486–493 CrossRef CAS PubMed.
- M. Qiu, J. Ouyang, Y. Wei, J. Zhang, Q. Lan, C. Deng and Z. Zhong, Adv. Healthcare Mater., 2019, 8, e1900500 CrossRef PubMed.
- Y. Zhu, B. Yang, S. Chen and J. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- A. D. Tagalakis, S. Castellaro, H. Zhou, A. Bienemann, M. M. Munye, D. McCarthy, E. A. White and S. L. Hart, Int. J. Nanomed., 2015, 10, 2673–2683 CrossRef CAS PubMed.
- Q. Li, L. Zhang, J. Lang, Z. Tan, Q. Feng, F. Zhu, G. Liu, Z. Ying, X. Yu, H. Feng, H. Yi, Q. Wen, T. Jin, K. Cheng, X. Zhao and M. Ge, Adv. Sci., 2023, 10, 2204334 CrossRef CAS PubMed.
- R. Zhong, S. Talebian, B. B. Mendes, G. Wallace, R. Langer, J. Conde and J. Shi, Nat. Mater., 2023, 22, 818–831 CrossRef CAS PubMed.
- S. Zhang, W. Fu and Z. Li, Polym. Chem., 2014, 5, 3346–3351 RSC.
- N. Segovia, M. Pont, N. Oliva, V. Ramos, S. Borros and N. Artzi, Adv. Healthcare Mater., 2015, 4, 271–280 CrossRef CAS PubMed.
- C. Alexander, Expert Opin. Drug Delivery, 2006, 3, 573–581 CrossRef CAS PubMed.
- C. Y. Ang, S. Y. Tan, C. Teh, J. M. Lee, M. F. Wong, Q. Qu, L. Q. Poh, M. Li, Y. Zhang, V. Korzh and Y. Zhao, Small, 2017, 13, 1602379 CrossRef PubMed.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- H. Ding, P. Tan, S. Fu, X. Tian, H. Zhang, X. Ma, Z. Gu and K. Luo, J. Controlled Release, 2022, 348, 206–238 CrossRef CAS PubMed.
- S. Fu, C. M. Rempson, V. Puche, B. Zhao and F. Zhang, Methods, 2022, 199, 67–79 CrossRef CAS PubMed.
- H. Wei, L. R. Volpatti, D. L. Sellers, D. O. Maris, I. W. Andrews, A. S. Hemphill, L. W. Chan, D. S. Chu, P. J. Horner and S. H. Pun, Angew. Chem., Int. Ed., 2013, 52, 5377–5381 CrossRef CAS PubMed.
- G. Chen, B. Ma, Y. Wang and S. Gong, ACS Appl. Mater. Interfaces, 2018, 10, 18515–18523 CrossRef CAS PubMed.
- N. Zheng, Z. Song, Y. Liu, R. Zhang, R. Zhang, C. Yao, F. M. Uckun, L. Yin and J. Cheng, J. Controlled Release, 2015, 205, 231–239 CrossRef CAS PubMed.
- I. Levkovskyi, S. Mochizuki, A. Zheng, X. Zhang and F. Zhang, Nano TransMed, 2023, 2, 100006 CrossRef.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- M. Gongora-Benitez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef CAS PubMed.
- K. S. S. Huang, Y. Liu, Y. Kuang, J. Li, S. An, Y. Guo, H. Ma and C. Jiang, ACS Nano, 2013, 7, 2860–2871 CrossRef PubMed.
- J. Fan, R. Li, H. Wang, X. He, T. P. Nguyen, R. A. Letteri, J. Zou and K. L. Wooley, Org. Biomol. Chem., 2017, 15, 5145–5154 RSC.
- X. He, J. Fan and K. L. Wooley, Chem.– Asian J., 2016, 11, 437–447 CrossRef CAS PubMed.
- X. He, J. Fan, F. Zhang, R. Li, K. A. Pollack, J. E. Raymond, J. Zou and K. L. Wooley, J. Mater. Chem. B, 2014, 2, 8123–8130 RSC.
- J. C. Kaczmarek, K. J. Kauffman, O. S. Fenton, K. Sadtler, A. K. Patel, M. W. Heartlein, F. DeRosa and D. G. Anderson, Nano Lett., 2018, 18, 6449–6454 CrossRef CAS PubMed.
- K. Paunovska, C. J. Gil, M. P. Lokugamage, C. D. Sago, M. Sato, G. N. Lando, M. Gamboa Castro, A. V. Bryksin and J. E. Dahlman, ACS Nano, 2018, 12, 8341–8349 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |