
Recent advances in therapeutic engineered extracellular vesicles
Chenlu
Yao
a,
Hong
Zhang
*b and
Chao
Wang
 *a
*a
aLaboratory for Biomaterial and ImmunoEngineering, Institute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou, Jiangsu 215123, China. E-mail: cwang@suda.edu.cn
bDepartment of Obstetrics and Gynecology, The Second Affiliated Hospital of Soochow University, Suzhou, China. E-mail: szzhanghong@suda.edu.cn
First published on 27th March 2024
Abstract
Extracellular vesicles (EVs) are natural particles secreted by living cells, which hold significant potential for various therapeutic applications. Native EVs have specific components and structures, allowing them to cross biological barriers, and circulate in vivo for a long time. Native EVs have also been bioengineered to enhance their therapeutic efficacy and targeting affinity. Recently, the therapeutic potential of surface-engineered EVs has been explored in the treatment of tumors, autoimmune diseases, infections and other diseases by ongoing research and clinical trials. In this review, we will introduce the modified methods of engineered EVs, summarize the application of engineered EVs in preclinical and clinical trials, and discuss the opportunities and challenges for the clinical translation of surface-engineered EVs.
 Chao Wang | Dr Chao Wang is a professor at Soochow University in China. Dr Wang focuses on the research field of development of cell-derived biomaterials and immunoengineering strategies for immunotherapies of cancer, infectious diseases, autoimmune disorders, etc. In the past five years, Dr Wang has published 40 papers as the corresponding author. Dr Wang has authored over 100 peer-reviewed papers, which have received a total citation of >13 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 times and given him an H-index of 76. Dr Wang has been selected as a “TR35” by the Asia-Pacific MIT Technology Review (2021) and has been included in Clarivate's “Highly Cited Scientists” (2018–2023).
500 times and given him an H-index of 76. Dr Wang has been selected as a “TR35” by the Asia-Pacific MIT Technology Review (2021) and has been included in Clarivate's “Highly Cited Scientists” (2018–2023).1. Introduction
Extracellular vesicles (EVs), small particles derived from cells, were first discovered in the 1980s, when they were regarded as cell dust.1 Subsequently, researchers investigated the structures and biological functions of EVs in intracellular communication and the transport of various biomolecules, including RNAs, proteins, and lipids, to recipient cells or tissues.2,3 In the 2010s, scientists gained a better understanding of the preparation, isolation and functions of EVs, particularly in the fields of cancer treatment and immunology, revealing their critical roles in immune responses and tumor development.4,5 In recent years, scientists further developed EVs as cell-free therapeutics for drug delivery, immunomodulation, and therapy. EV-based therapies offer the potential for scalable production, improved storage and transport, reduced adverse effects, and overcoming the biological barriers due to their nanosize.6,7The surface engineering of extracellular vesicles could achieve many additional goals. One dominating purpose is to endow EVs with targeting affinity. Even though some EVs have been reported to intrinsically target specific cells, previous literature has revealed that it is insufficient to achieve accumulation in vivo.8,9 Another motivation for EV surface engineering is the introduction of therapeutic molecules. Compared with loading drugs, especially for large molecules, modification on the surface may be more accessible. To achieve the above purposes, researchers have utilized mainly genetic and chemical methods to modify the surface of extracellular vesicles and apply these extracellular vesicles in different disease therapies.
2. Source of EVs
Compared with parental cells, EVs are reported to lack certain structural proteins, such as actin, but can inherit surface proteins and some secreted proteins from the mother cells.10,11 This inherent property simplifies the development of EVs for potential applications, eliminating the need for additional procedures to introduce specific proteins of interest. Moreover, multiple proteins may collectively contribute to specific functions of EVs, enhancing their efficacy in these roles.12 However, EVs secreted from mother cells exhibit heterogeneity, resulting in variations in the abundance of proteins of interest among individual EVs. Furthermore, the quality of EVs is greatly influenced by the conditions of the mother cells. EVs can be derived from various cell types and sources, each of which may have specific characteristics and functions.2.1. Blood cell-derived EVs
Extracellular vesicles derived from blood cells, including Red Blood Cells (RBCs) and platelets could be produced in an easier protocol, because of the lack of nuclei and composite organelles.13–17 Additionally, as RBCs and platelets are the most two abundant cells in the blood, the output of EVs of RBCs and PEVs could be produced on a large scale.18 However, as these blood cells cannot reproduce in vitro, it is difficult to insert interested genes into these EVs.RBC-derived EVs are released during various physiological and pathological conditions, reflecting the state of their parent cells. These vesicles carry a cargo rich in proteins, lipids, and microRNAs, including cytoskeletal proteins and antioxidant enzymes, which are also widely utilized for drug delivery.16 Platelet-derived EVs are key players in hemostasis, thrombosis, and inflammation. Laden with platelet-specific molecules like glycoprotein IIb/IIIa and P-selectin, as well as bioactive molecules such as growth factors and cytokines, these EVs orchestrate various physiological responses.19,20
2.2. Immune cell-derived EVs
Extracellular vesicles from immune cells, including macrophages,21–23 dendritic cells,24,25 and T cells,26,27 are also utilized in disease therapy because of their immunomodulation capacity. For instance, T cell-derived EVs carry surface markers and cytokines characteristic of T cell subtypes, facilitating antigen presentation, immune activation, and regulation.28 B cell-derived EVs may contain immunoglobulins and antigens, participating in humoral immune responses and antibody-mediated immunity.29,30 Similarly, NK cell-derived EVs modulate target cell recognition and immune responses through the transfer of cytotoxic molecules and regulatory proteins.31,32Macrophages and dendritic cells, as key antigen-presenting cells, release EVs with significant immunomodulatory functions. These EVs carry antigen-presenting molecules such as MHC molecules and co-stimulatory molecules, facilitating antigen presentation and T cell activation.21,33 Furthermore, macrophage-derived EVs may contain inflammatory mediators such as cytokines and chemokines, regulating local immune responses and inflammation.34 Dendritic cell-derived EVs play crucial roles in immune tolerance and suppression by promoting regulatory T cell differentiation and suppressing effector T cell responses.35
2.3. Tumor-derived EVs
Tumor-derived EVs (TEXs) play crucial roles in various stages of cancer progression, including tumor growth, angiogenesis, metastasis, immune evasion, and drug resistance. They facilitate intercellular communication by transferring oncogenic molecules, promoting tumor–stroma interactions, and modulating the behavior of recipient cells within the tumor microenvironment.5 TEXs have inherited specific surface proteins from tumor cells, leading to the tumor-targeting ability.36 Moreover, TEXs have emerged as promising biomarkers for cancer diagnosis, prognosis, and treatment response prediction, offering non-invasive avenues for monitoring disease progression and guiding therapeutic interventions.372.4. Stem cell-derived EVs
Extracellular vesicles derived from stem cells are also strong candidates for the next generation of drugs.38,39 These EVs play crucial roles in intercellular communication, tissue repair, immune modulation, and regeneration. They exert their effects by transferring regulatory molecules to recipient cells, influencing cellular processes such as proliferation, differentiation, apoptosis, and inflammation.40 Stem cell-derived EVs offer several advantages over stem cell-based therapies, including reduced risk of tumorigenicity, immunogenicity, and ethical concerns associated with cell transplantation. MSC EVs have been reported to play a vital role in tissue repair,41 and therapeutics of COVID-19.42 That may contribute to the large scale of MSC reproduction.2.5. Bacteria-derived EVs
Outer membrane vesicles (OMVs) carry a variety of proteins from the parent bacterial cell surface, playing crucial roles in bacterial physiology, virulence, and host–pathogen interactions.43 These proteins may include adhesins such as coagulase, adhesion factors, and lectins, facilitating bacterial attachment to host cell surfaces and establishment of infection.44,45 Additionally, OMVs may harbor virulence factors such as toxins, lytic enzymes, and lipopolysaccharides (LPSs), which, when released into host cells via OMVs, can induce cell damage and inflammation, and promote pathogen invasion and disease progression. OMVs, especially from E. coli., are utilized in tumor therapy, as LPSs on the surface of OMVs are immunomodulators to activate an immune response.46,472.6. Plant-derived EVs
Plant EVs play crucial roles in regulating plant growth and development, responding to stress, and mediating intercellular communication. They are released from various cellular compartments such as chloroplasts, mitochondria, endoplasmic reticulum, and Golgi apparatus into the extracellular space, facilitating long-distance communication within and outside the plant organism and influencing the physiological and metabolic states of recipient cells.48,49 Moreover, plant EVs are strong candidates for the next generation of drug delivery systems because of the high output of nanovesicles.50,512.7. Isolation methods of EVs
Various methods exist for isolating EVs from complex biological samples. These methods include ultracentrifugation, size exclusion chromatography, ultrafiltration, immunoaffinity capture, precipitation, and microfluidic devices.52,53 Each method has its advantages and limitations, and the choice depends on factors such as sample type, desired purity, yield, and downstream applications. Integration of multiple isolation techniques often enhances the purity and yield of isolated EVs, facilitating their functional characterization and enabling their use in research and clinical settings.3. Surface engineering strategies on EVs
Researchers are trying to introduce interesting peptides or other molecules on the surface of EVs to broaden the applications of EVs. Some molecules including aptamers, nanobodies, or specific cell-targeted peptides could endow EVs with targeted capacity, solving the off-target problem. Some molecules like maleimide can capture antigens after being modified on the surface of EVs, boosting the immune response through interactions with antigen-presenting cells. Other molecules such as SIRP-α and aPD-L1 could modulate the immune microenvironment of tumor sites, enhancing the therapeutic efficacy of EVs. To modify EVs with these molecules, the most two common strategies are genetic modification and chemical modification (Fig. 1). | ||
| Fig. 1 Schematic diagram of surface-engineered extracellular vesicles. | ||
3.1. Genetic modification on EVs
To fuse the interested proteins on the membranes of EVs, researchers introduce the target gene through plasmid or lentivirus into the parental cells, inverting the target gene into the gene of parental cells. Then, EVs secreted by these cells will carry these proteins. Typically, the interested protein is fused with specific membrane surface proteins of the parent cells, such as Lamp2b, CD63, and CD9. For example, Wang et al. ligated a cardiomyocyte specific peptide (CMP) to the extra-exosomal N-terminus of Lamp2b on cardiosphere-derived cells.54 Additionally, Scott et al. fused CD63 of HEK293T cells with an anti-CoV-2 nanobody to neutralize SARS-CoV-2 in vitro.55 Kim and colleagues also truncated CD9 to display sACE-2 variants on the surface of extracellular vesicles.56 This fusion allows these proteins to effectively localize to the surface of EVs. The choice of fusion protein depends on specific application requirements.Gene modification techniques offer robust tools for engineering EVs and can be used to achieve various purposes, including targeted delivery, imaging and therapeutics. In detail, by introducing specific targeting proteins, EVs can selectively deliver their cargo to specific cells or tissues, enabling efficient targeted therapy. For instance, IL-3 was introduced into EVs derived from HEK293 cells to enhance the binding affinity of EVs in Chronic Myeloid Leukemia (CML) blasts, where IL-3 receptors were overexpressed.57 The fusion of fluorescent or other imaging-labeled proteins to the surface of EVs also allows real-time tracking of EV distribution and delivery in vivo.
Through the incorporation of therapeutic proteins or RNA, EVs can be engineered into effective drug carriers for the treatment of conditions such as cancer, infections, and various diseases. For example, EV overexpressed SIRP-α could increase cancer cell phagocytosis, boosting the immune response.58
3.2. Chemical modification on the surface of EVs
Unlike genetic modifications which usually display proteins on EVs, modifications based on chemical methods could display peptides or other molecules on EVs.59 Chemical modification strategies can be categorized into two distinct approaches based on the timing of the modifications: pre-isolation modifications and post-isolation modifications. Chemical modifications encompass several techniques, including electrostatic incorporation, click chemistry and covalent conjugation. As the membrane surface of the cells is negatively charged, the positively charged molecules could simply absorb on the surface by electrostatic incorporation. For instance, Qin and colleagues displayed positive tumor antigens on the surface of OMVs through electrostatic incorporation.60Additionally, click chemistry also plays a crucial role in the modification of extracellular vesicles (EVs). Click chemistry is typically an efficient method that allows for rapid surface modification of EVs.61,62 It could also be used to introduce a wide range of molecular modifications, such as drugs, targeting molecules, and fluorescent labels. Both fluorescent dyes and targeting peptides conjugated with azide groups could simply modify the NHS-engineered EV surface by click chemistry.63
Moreover, covalent conjugation is another widely utilized technique for tailoring EVs’ surface properties, offering several key advantages. This approach involves the attachment of various molecules to the EV surface through covalent bonds, resulting in a functionalized EV platform. For instance, DSPE–PEG, phospholipid–polyethylene glycol, could typically react with hydroxyl or amine groups on the surface of membranes, to form covalent bonds. Wang et al. functionalized targeting peptides with DSPE–PEG and modified them on the surface of donor cells.64 The advantages of covalent conjugation include: (1) robust stability to the modified EVs, preventing the detachment of surface molecules and ensuring their persistence during storage and circulation in biological environments; (2) fine control over the degree and nature of modification, tailoring EVs to meet diverse research and clinical needs; and (3) minimizing non-specific protein adsorption to EV surfaces, maintaining their purity.
These chemical modification strategies can be utilized to endow EVs with specific functionalities, such as targeted drug delivery, real-time imaging, and therapeutic capabilities. For instance, cardiac homing peptides were conjugated on the surface of EVs, endowing EVs with affinity towards the infracted hearts.65 The insertion of fluorescent molecules labeled on the surface of EVs also helps to identify the location of the infraction.66
3.3. Physical modification on the surface of EVs
Physical modification strategies offer versatile approaches for surface engineering of extracellular vesicles (EVs), enabling precise control over their surface properties and functionalities.67,68 These strategies involve the manipulation of EVs through physical means without directly altering their genetic or biochemical compositions.One common physical modification strategy is membrane permeabilization, which involves techniques such as sonication, freeze–thaw cycles, or electroporation. These methods disrupt the integrity of the EV membrane, allowing for the insertion or attachment of exogenous molecules onto the EV surface.69 This enables the introduction of targeting ligands, therapeutic agents, or imaging probes onto EVs, thereby enhancing their targeting specificity or therapeutic efficacy. Rayamajhi et al. hybridized macrophage-derived EVs with gadolinium-infused liposomes using sonication and membrane extrusion, achieving magnetic resonance imaging.70
Another physical modification approach is membrane fusion, where EVs are fused with liposomes or synthetic nanoparticles carrying desired surface modifications.67 This enables the transfer of surface proteins, lipids, or other molecules from the synthetic carriers to EVs, leading to the engineering of EVs with customized surface functionalities. Li et al. modified MSC EVs with platelet membranes based on extrusion, endowing EVs with the binding ability to monocytes.71 This strategy successfully targeted the infracted heart and realized cardiac repair.
3.4. Metabolic engineering on the surface of EVs
Metabolic engineering strategies involve manipulating the cellular metabolism of EV-producing cells to modulate the biosynthesis and incorporation of specific molecules into EV membranes. One approach is to engineer donor cells to overexpress or knockdown genes involved in lipid metabolism, such as enzymes responsible for lipid synthesis or modification.72 This can lead to the production of EVs with altered lipid compositions, which in turn influence their surface properties and interactions with target cells or tissues.Another metabolic engineering strategy involves the modulation of glycosylation pathways in donor cells to control the glycosylation patterns on EV surfaces.67 Glycosylation plays crucial roles in EV biogenesis, cargo sorting, and intercellular communication, making it an attractive target for surface engineering.73 By manipulating the expression of glycosyltransferases or glycosidases, researchers can engineer EVs with specific glycan structures on their surfaces, which can modulate their targeting, uptake, and immunogenicity profiles. For instance, Lim et al. modified EVs with an exogenous azide group based on metabolic glycoengineering to enhance the targeting affinity of EVs towards target tissues.74
3.5. Lipid-based engineering on the surface of EVs
Lipid-based engineering strategies involve the manipulation of lipid composition and structure on the EV surface. One common approach is lipid insertion or incorporation, where lipids with desired functionalities, such as targeting ligands or therapeutic molecules, are inserted into the EV membrane.75 This can be achieved through various techniques, including membrane fusion, incubation with lipid-modified molecules, and extrusion through lipid bilayers. For instance, Jhan et al. fused EVs with synthetic lipids to realize highly efficient gene delivery.68 Lipid exchange is another strategy, where the native lipids of EVs are replaced with synthetic or modified lipids carrying specific functionalities.76 This can be accomplished using methods such as solvent-based lipid exchange or membrane extrusion. Additionally, lipid conjugation involves the covalent attachment of molecules of interest, such as targeting ligands or imaging agents, to lipid molecules, which are then incorporated into the EV membrane.75 The choice of different modification techniques depends on the specific goals of the study and the intended applications. Overall, chemical modifications of EVs provide a versatile toolkit for tailoring these vesicles to meet various research and clinical needs.4. EVs for different disease therapy and clinical translation
4.1. Engineered EVs for cancer therapy
The therapeutic potential of EVs for tumors has been evaluated in recent years with encouraging outcomes. Drug delivery systems and tumor vaccines based on EVs could inhibit tumor growth in pre-clinical trials.77–81 Compared with their parent cells, EVs have lower immunogenicity and cytotoxicity. When loading drugs on EVs, these natural nanoscale carriers could enhance the targeting ability, and prolong the circulation time compared with free drugs. Researchers have also modified the surface of EVs derived from various cells and utilized engineered EVs for targeted therapy and immunotherapy (Table 1).| Therapy | Parent cells | Surface engineering | Outcomes | Ref. |
|---|---|---|---|---|
| Targeted therapy | MPA–MB-231; HCT-116 | PHA | Enhanced binding affinity of EVs | 74 |
| Targeted therapy | MSCs | Targeting peptides | Improved uptake of EVs | 83 |
| Targeted therapy | Neuro2A | Anti-EGFR nanobodies | 88 | |
| Targeted therapy | HEK293T | IL-3 | Enhanced targeting ability | 57 |
| Targeted therapy | HEK293T | CDH17nanobody | Targeted delivery of drugs | 90 |
| Targeted therapy | imDCs | iRGD | Enhanced binding ability | 85 |
| Targeted therapy | Grapefruits | Aptamers | Enhanced targeting ability | 91 |
| Immunotherapy | 4T1; B16F10 | SIRP-α and PD-1 | Boosting multi-targeted ICB therapy | 98 |
| Immunotherapy | HEK293T | PD-1 | Boosting anti-tumor immunity | 99 |
| Immunotherapy | HEK293T | SIRP-α | Increased cancer cell phagocytosis | 58 |
| Immunotherapy | HEK293T | CD64 | Carrying aPD–L1; introducing ICB therapy | 100 |
| Immunotherapy | HEK293T | aPD–L1 | Strengthen ICB therapy | 131 |
| Immunotherapy | Jurkat T | IL-2 | Improving anti-cancer efficacy | 132 |
| Immunotherapy | CAR-T | Gene engineering | Potent antitumor effects and low toxicity | 133 |
| Immunotherapy | imDCs | aPD–L1 | Introducing ICB therapy | 103 |
| Immunotherapy | DC2.4 | Anti-CD19, PD-1 | Targeting tumor cells; enhancing ICB therapy | 104 |
| Immunotherapy | E. coli | PD-1 | Increasing anti-tumor efficacy | 134 |
| Immunotherapy | E. coli | TAAs | Triggering potential antitumor immunity | 60 |
| Immunotherapy | E. coli | TAAs | In situ controllable production of tumor antigens | 107 |
| Immunotherapy | E. coli | TAAs | Display antigens | 106 |
| Immunotherapy | E. coli | Maleimide group | Capture of TAAs | 108 |
| Immunotherapy | E. coli | Maleimide group | Capture of TAAs after PTT | 109 |
PEGylated hyaluronic acid (PHA) was reported to have strong affinity to CD44-overexpressing cells,82 which are usually utilized in the surface engineering of nanomaterials. Considering that the tumor site is also a CD44-abundant tissue, Lim et al. modified MDA–MB-231 cells and HCT-116 cells with PHA, based on biorthogonal copper-free click chemistry (BCC) and metabolic glycoengineering (MGE).74 This strategy could successfully increase the tumor targeting of EVs derived from these two cell lines, as the fluorescence signal of PHA-EVs in the tumor site was about 2-fold higher than that of bare EVs.
Apart from targeted small molecules, targeted peptides are also utilized to enhance the targeting affinity of EVs. Sajeesh and colleagues have engineered adult human BM–MSCs with azide-modified cathepsin K-targeting peptide sequence based on copper-free click chemistry.83 Cathepsin K is overexpressed in the wall of abdominal aortic aneurysm (AAA).84 The cellular uptake of EVs increased sharply after the medication. The ex vivo binding of engineered EVs was about 10 times higher than that of bare EVs, indicating enhanced binding affinity of EVs towards the tumor site.83 Tian et al. modified immature DCs with the iRGD targeting peptide and isolated EVs from engineered cells.85 These EVs were loaded with doxorubicin via electroporation, which could enhance tumor binding affinity by about 3 times, compared with unmodified EVs. This strategy could inhibit MDA–MB-231 tumor growth and reduce cardiac damage compared with free DOX.
Single-chain variable antibodies (scFv) have significant applications in achieving tumor-targeting of EVs as they could bind with specific tumor-associated antigens. Human Epidermal Growth Factor Receptor 2 (HER2) is a membrane-bound receptor protein, which has become an important target in breast cancer.86 Longatti and colleagues transfected HEK293 cells with the vector of anti-Her2-scFv and confirmed the high affinity of the engineered exosomes to HER2-positive cells.87 The epidermal growth factor receptor (EGFR) is another target of some tumor cells, which is overexpressed on tumor cells. Kooijmans et al. modified Neutro2A cell lines with anti-EGFR nanobodies and collected EVs derived from these cells, evaluating their association with tumor cells.88 This Cadherin 17 (CDH17) is reported as one of the most upregulated genes in gastric cancers (GC).89 Hence Xia et al. developed a delivery platform based on EVs from HEK293 cells modified with CDH17 nanobodies to deliver imaging probes and other drugs.90 This smart strategy was confirmed to accumulate more than three times in the tumor site compared with unmodified EVs, showing higher therapeutic efficacy in GC mouse models.
The above-mentioned EVs are derived from animal cells. Plant-derived extracellular vesicles, however, could be produced with mass production, and after simple modification, they could also be endowed with high affinity.50 Moon et al. engineered EVs derived from grapefruits with a targeted aptamer.91 These engineered EVs could have 3 times higher affinity towards blood–brain barrier endothelial cells, compared with unmodified EVs, indicating their potential in targeting brain tumors.
Extracellular vesicles derived from tumor cells (TEXs) offer advantages in tumor immunotherapy, including their natural antigen presentation, immune stimulation, targeting capabilities, drug delivery potential, and immune monitoring ability. These attributes make TEXs a promising tool for enhancing immune therapy and improving immune responses. To further enhance the effectiveness of tumor treatment, SIRPα was edited on the surface of breast cancer cells and carried on the extracellular vesicles derived from these cancer cells.98 SIRPα EVs were fused with PD-1 EVs, boosting both the innate immune response and the adaptive immune response towards tumors (Fig. 2A). Consequently, these fused EVs could successfully inhibit tumor growth and protect the mice from tumor recurrence and metastasis.
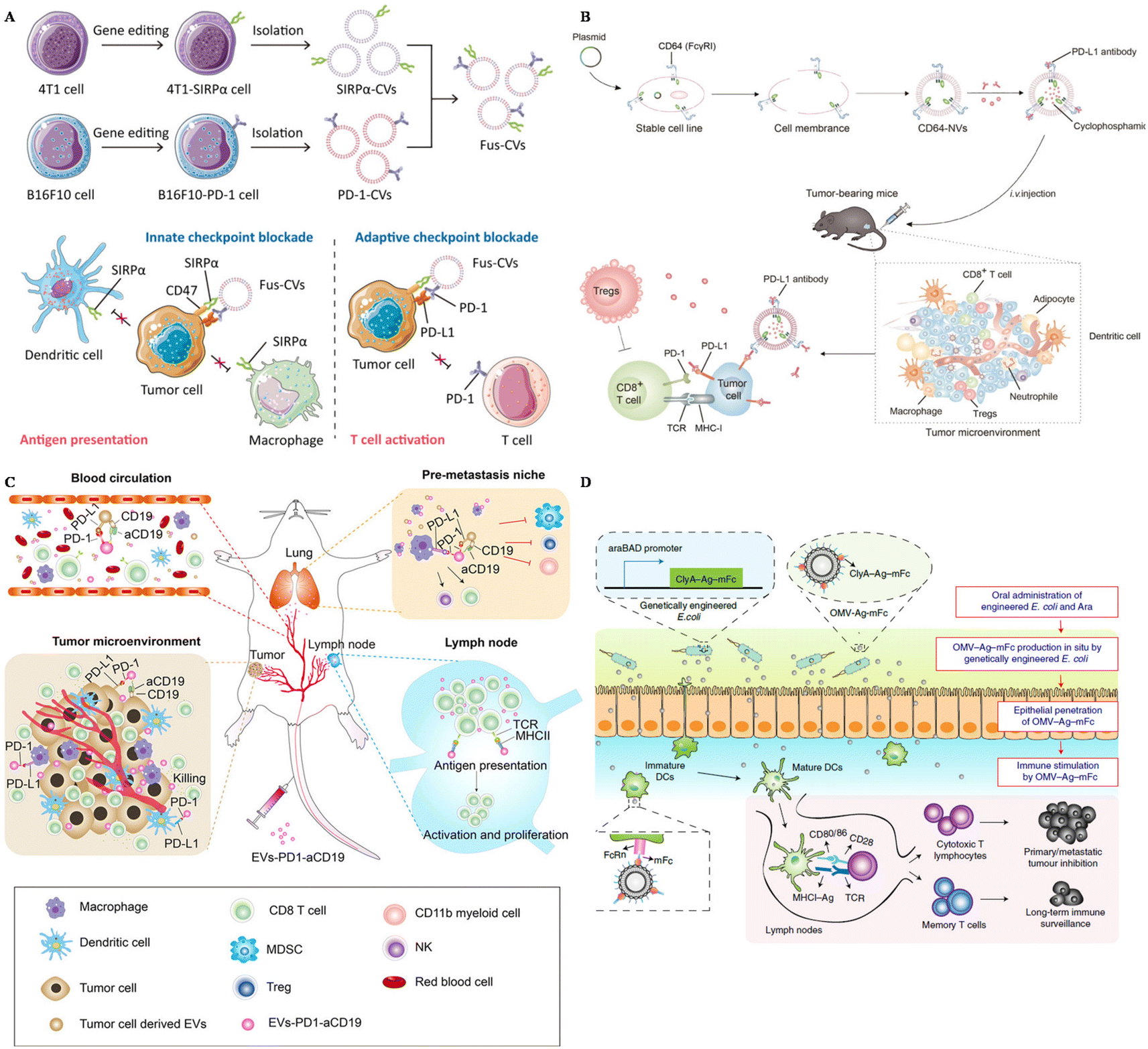 | ||
| Fig. 2 The design of surface-engineered EVs for tumor therapy. (A) The preparation of multi-targeted extracellular vesicles.98 Reproduced with permission. Copyright 2021, Wiley. (B) Scheme of the preparation of CD64–NVs–aPD–L1–CP and mechanisms.100 Reproduced with permission. Copyright, 2021, Ivyspring International. (C) Scheme of the immune-boosting mechanism of EVs–PD-1–aCD19.104 Reproduced with permission. Copyright, 2023, Elsevier. (D) Genetically engineered bacteria-derived-OMV-based oral tumor vaccine.107 Reproduced with permission. Copyright, 2022, Springer. | ||
HEK293 (Human Embryonic Kidney 293) cells are widely utilized EV-producers in tumor immunotherapy. For example, Koh et al. developed exosomes derived from HEK293 cells overexpressing SIRPα on the surface to intervene in the CD47–SIRPα axis.58 These SIRPα-EVs could enhance the phagocytosis of bone marrow-derived macrophages in vitro two times compared with unmodified EVs. In vivo experiments, SIRPα EVs could selectively target tumor sites, further inhibiting the tumor growth significantly, even compared with CD47 antibody. Additionally, Li and colleagues modified HEK293 cells with the overexpression of PD-1 and utilized PD-1-EVs to treat cancers.99 These PD-1-EVs could induce apoptosis of tumor cells in vitro via the expression of Fasl and GramB. The antitumor efficacy of PD-1-EVs was even more significant compared with anti-PD–L1. To carry PD–L1 antibodies, Li et al. genetically engineered EVs from HEK293T cells with the display of CD64, working as a linker to catch antibodies (Fig. 2B).100 This strategy could block PD–L1 of tumor cells in vivo, showing great therapeutic efficacy loaded with cyclophosphamide in the engineered EVs. Besides, Shi et al. transfected Expi 293 cells with αCD3 and αHER2 scFv, achieving dual targeting to T cells and breast cancer cells.101 These αCD3–αHER2-EVs could inhibit HER-2 expressing tumor growth and upregulate the percentage of T cells in the tumor site.
Apart from the primary investigation, HEK293 cells are also employed for the large-scale production of EVs. Their high proliferation rate and stability make them suitable for the mass production of EVs, meeting the quantity requirements for research and applications. Codiak Biosciences evaluated the therapeutic efficacy of intratumorally-administered HEK293-exosomes overexpressed PTGFRN on the surface to enhance the targeting delivery of the STING activator to antigen-presenting cells in solid tumors (NCT04592484). They also proposed to treat patients suffering from cutaneous T-cell lymphoma with HEK293-exosomes displayed IL-12 on the surface (NCT05156229). Moreover, Codiak Biosciences registered the use of HEK293-derived exosomes, the STAT6 anti-sense oligonucleotide (ASO) in tumor treatment. This strategy was proposed for the targeted delivery of drugs towards the myeloid, repolarizing macrophages from the immune suppressive M2 to the proinflammatory M1 phenotype (NCT05375604).
Dendritic cells (DCs) are professional antigen-presenting cells, originating mainly from bone marrow. They also play an important role in the anti-tumor immune response, as they can capture tumor-associated antigens, and process and present these antigens via MHC molecules, subsequently activating naïve T cells.102 Considering the complex interaction between DCs and other immune cells and the capacity to display antigens, EVs derived from DCs are also utilized in tumor immunotherapy. Dai et al. developed antigens including EVs derived from DCs and these EVs were also armed with aPD–L1, to prevent immune escape through the PD-1–PD–L1 axis.103 This strategy could remarkably inhibit tumor growth and protect mice from tumor recurrence by boosting the immune response in the tumor site and lung tissues. To reprogram the immune microenvironment and enhance the tumor affinity, Xu and colleagues genetically engineered DC EVs with aCD19 and PD-1, loaded with TAAs and CPG (Fig. 2C).104 This smart strategy could be used to interact with tumor cells, DCs, and macrophages because of the overexpression of PD-1, and modulate T cells due to MHCII and CD80 expressed on the surface of EVs. Engineered EVs were confirmed to accumulate more than four times compared with unmodified EVs. In general, these multifunctional EVs could reverse not only the immunosuppressive microenvironment in situ, but also the lung premetastatic niches in mouse models.
Outer membrane vesicles (OMVs) are secreted by most bacteria, containing a broad range of pathogen-associated molecular patterns (PAMPs), regarded as ideal vaccine adjuvants as they could boost the innate immune response.105 Because of this intrinsic characteristic, OMVs has become a candidate to display tumor-associated antigens (TAAs) to activate the immune response towards tumors. Qin et al. simply displayed TAAs on the surface of OMVs by electrostatic interactions.60 This strategy could successfully elicit DC maturation and activation in vitro and further inhibit the metastasis of melanoma in mouse models with negligible toxicity. Moreover, Cheng and colleagues simplified the antigen display process with a plug-and-display system, and they modified the surface of OMVs with different antigen peptides.106 Featuring different antigen peptides, these OMVs showed remarkable therapeutic efficacy in mouse melanoma and colorectal cancer models. They could also protect mice in the long term, with immune memory 80 days after the last vaccination, indicating the application potential of prophylactic vaccines. Apart from the preparation of OMVs in vitro, in situ produced OMVs could also boost the maturation of DCs. Yue et al. orally administered gene-engineered E. coli with TAAs (Fig. 2D).107 This engineered E. coli could then produce OMVs with antigens in the intestine and OMVs could penetrate epithelial barriers, captured by DCs, followed by the rapid maturation of DCs. This strategy could also inhibit subcutaneous colorectal cancers, and elicit long-term immune memory in mouse models. Apart from directly displaying the antigens on the surface of OMVs, researchers also modified OMVs with the maleimide group to enhance the antigen capture capacity of DCs.108,109 Maleimide endows OMVs with high affinity to antigens, and after migration to the draining lymph nodes, OMV–Mal carried with TAAs could be taken up by immature DCs, boosting the maturation of DCs. OMVs modified with maleimide and loaded with 1-methyl-tryptophan (1-MT) could modulate both regulatory T cells and DCs after photothermal therapy.109 1-MT is an inhibitor of IDO, which could lead to a decrease of Treg, while the modification of the Mal group endows OMVs with carrying TAAs to DCs. This strategy could notably prolong the life span of tumor-bearing mice.
4.2. Engineered EVs for inflammatory disease therapy
Inflammatory diseases, including autoimmune diseases and infectious diseases, are disturbing people's health globally. Autoimmune diseases are usually related to uncontrollable immune responses, such as rheumatoid arthritis (RA), type I diabetes and ulcerative colitis.110 You et al. developed MSC EVs conjugated with dextran sulfate on the surface, achieving the targeted reprogramming macrophages in inflamed joints (Table 2).111 An upregulated expression of PD-1 was confirmed in inflamed tissue in autoimmune patients.112 To suppress the overwhelmed immune response and also increase the targeting affinity of EVs, the PD-1/L1 axis has been introduced into the treatment of autoimmune diseases. Becker modified a lymphoblast cell line, K562, with the expression of PD–L1 and utilized PD–L1 EVs to treat type I diabetes (Fig. 3A).113 This strategy could successfully suppress T cell activation in vitro, nearly the same level as healthy controls, further inducing the reduction of T cell cytotoxicity towards targeted cells in vitro.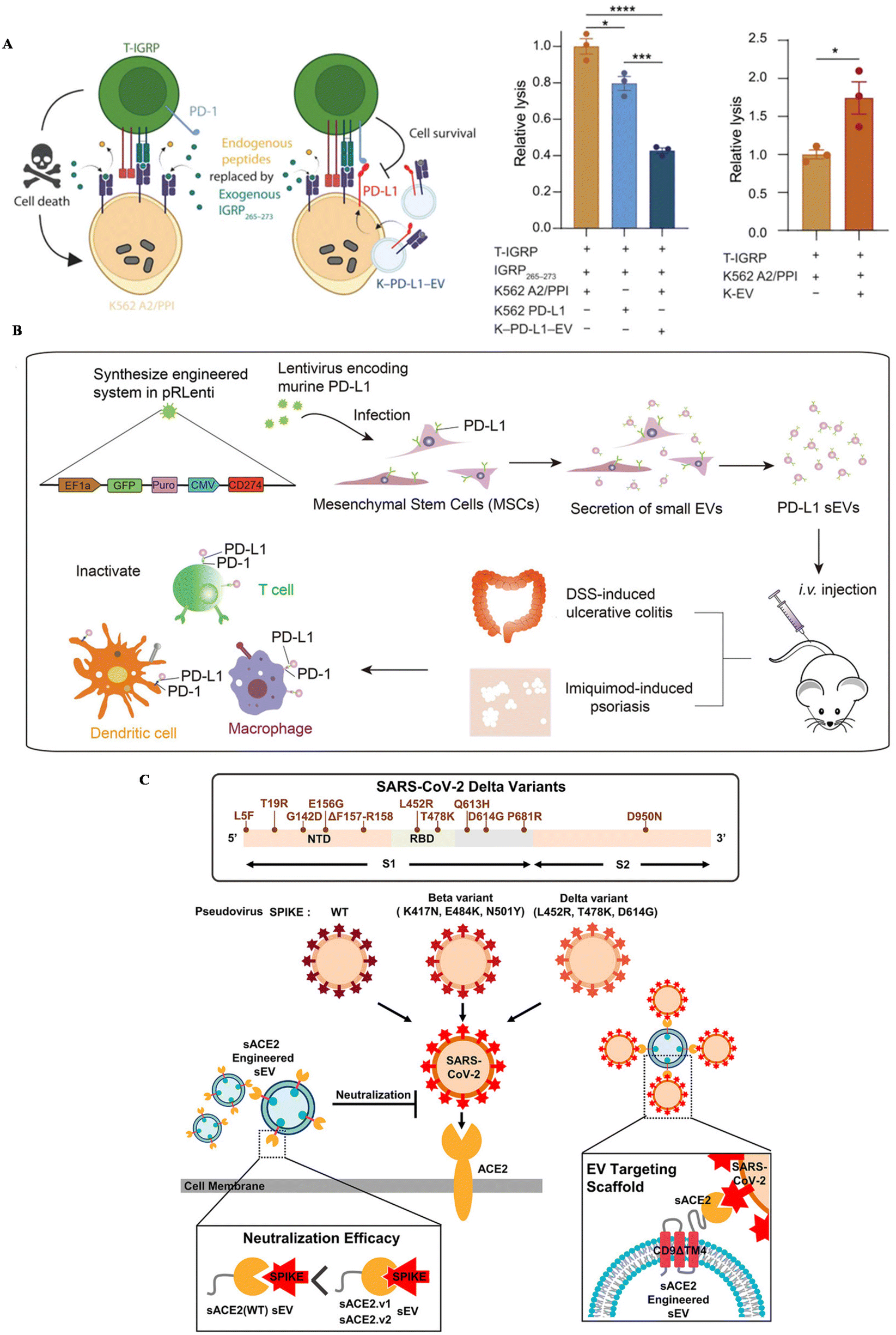 | ||
| Fig. 3 The schematic of utilizing engineered EVs for inflammatory diseases. (A) The mechanism of EV–PD–L1 to induce cell killing of T cells. Reproduced with permission.113 Copyright 2023, the American Association for the Advancement of Science. (B) The schematic illustration of the preparation of PD–L1–MSC EVs and the immune boosting mechanism. Reproduced with permission.114 Copyright 2021, Wiley. (C) The schematic of utilizing engineered EVs for infection. Reproduced with permission.56 Copyright 2022, Wiley. | ||
| Diseases | Parent cells | Surface engineering | Outcomes | Ref. |
|---|---|---|---|---|
| I diabetes | K562 | PD–L1 | Suppressing T cell activation | 113 |
| Autoimmune diseases | MSCs | PD–L1 | Reshaping the inflammatory ecosystem | 114 |
| RA | MSCs | Dextran sulfate | Reprogramming macrophages in inflammatory joints | 111 |
| COVID-19 | HEK293 | ACE-2 variants | Protecting K18-hACE-2 mice from infection | 56 |
| COVID-19 | HEK293 | ACE-2 | Protecting mice from infection | 115 |
| COVID-19 | HEK293 | COVID-19 nanobody | Neutralizing SARS-CoV-2 | 55 |
| COVID-19 | MSCs | ACE-2 | Inhibiting the infection of pseudovirus | 116 |
Our group genetically engineered MSC EVs with the overexpression of PD–L1 and treat two autoimmune mouse models with MSC-EV–PD–L1 (Fig. 3B).114In vitro, engineered EVs could bind with T cells, macrophages and dendritic cells, reverse the activation of T cells and BMDCs, and polarize macrophages towards M2 as well. In autoimmune mouse models, MSC-EV–PD–L1 could accumulate in the inflammatory colon and skin in ulcerative colitis and psoriasis, respectively. Furthermore, the excessive inflammation was relieved after the accumulation of engineered EVs, with a remarkable reduction of T cells and macrophages in the inflamed tissue, indicating the clinical potential of this strategy in the future.
Infectious diseases are usually caused by the infection of bacteria, fungi and viruses. SARS-CoV-2, a coronavirus that was first discovered in 2019, has led to millions of deaths all over the world. ACE-2, a protein that is expressed in some cells, has been regarded as a receptor for the spike protein of this virus, leading to further viral infection. Kim et al. engineered EVs to display ACE-2 variants on CD9 scaffolds (Fig. 3C).56 ACE-2-EVs could inhibit the infection of wild-type viruses and delta variants in cell lines. Additionally, these EVs are also confirmed to protect mice expressing human ACE-2 from the viral infection, with a down-regulated proinflammatory response after treatment. Besides, Wu et al. developed ACE-2-engineered extracellular vesicles, protecting the infection of pseudovirus with the S protein in various cells in vitro and suppressing the entry of pseudovirus in the mouse model.115 Xie et al. also modified ACE-2 EVs, neutralizing the pseudovirus in vitro and block the infection of pseudovirus in the hACE2 mouse model.116 Moreover, Scott embedded an anti-CoV-2 nanobody in CD63, a widely expressed protein in EVs, to neutralize virus.55 The results confirmed that EVs with the nanobody can successfully bind with the spike protein of SARS-CoV-2 and protect cells in vitro.
Surface-engineered extracellular vesicles derived from MSCs have been tested in clinical trials for the treatment of COVID-19 (Table 4). Tel-Aviv Sourasky Medical Center evaluated the biosafety of exosomes derived from MSCs and overexpressed CD24 in patients with either moderate or severe COVID-19 infection (NCT04747574). Eli Sprecher proposed to treat patients suffering from COVID-19 with exosomes overexpressed CD24 at a dose of 1010 daily for five days (NCT04969172). Other two medical societies also registered the use of engineered MSC EVs with the overexpression of CD24 for phase II. Athens Medical Society proposed to treat SARS-CoV-2 positive patients with two doses of modified EVs, 109 and 1010, respectively (NCT04902183). OBCTCD-24 Ltd registered to evaluate the safety and therapeutic efficacy of engineered MSC EVs (EUCTR2021-004259-17-GR).
4.3. Engineered EVs for the treatment of other diseases
Cardiovascular diseases are one of the leading causes of death in the world.117 Extracellular vesicles have been utilized in the therapy of cardiovascular diseases, due to the specific RNAs contained in EVs.118 However, the targeting ability of EVs has limited their further application. Therefore, to enhance the targeting capacity of MSC EVs, Li et al. modified EVs with platelet membranes (Table 3).71 With the expression of platelet specific proteins, like CD62P, EVs could bind with monocytes in vitro and target the infracted hearts in vivo. Pei et al. also utilized a platelet membrane to improve the targeting ability of macrophage-derived EVs.119 Moreover, targeting peptides have been modified on the surface of blank EVs to improve their retention in specific tissues. For example, MSC EVs conjugated with the c(RGDyk) peptide, could cross the blood–brain barrier (BBB) and target the ischemic brain via intravenous injection.63 MSC EVs could also target the ischemic heart after fusing with another targeting peptide. Besides, Wang et al. genetically modified MSCs with CD47 and developed their EVs for myocardial infarction reperfusion injury treatment.120 With the expression of CD47, engineered EVs could accumulate 5 times in the heart, compared with unmodified EVs.| Diseases | Parent cells | Surface engineering | Outcomes | Ref. |
|---|---|---|---|---|
| Cardiac repair | MSCs | Platelet membrane | Accumulating in the infracted hearts | 71 |
| Viral myocarditis | Macrophages | Platelet membrane | Increasing the targeting capacity of EVs | 119 |
| Cerebral ischemia | MSCs | Targeting peptide | Crossing the BBB | 63 |
| Myocardial infarction | CDCs | Targeting peptide | Increasing retention of the infarcted heart | 65 |
| Myocardial infarction | MSCs | Targeting peptide | Increasing retention | 120 |
| Heart injury | CDCs | Targeting peptide | Targeting affinity to cardiomyocytes | 54 |
| Angiogenesis | K562 | RGD targeting peptide | Increasing targeting of blood vessels | 64 |
| Myocardial infarction reperfusion injury | MSCs | CD47 | Improving biodistribution in the heart | 135 |
| Central nervous system injuries | M2 microglia | DA7R; SDF-1 | Enhancing accumulation in the ischemic area | 122 |
| White matter injury | MSCs | SIRP-α | Attenuating neuroinflammation | 123 |
| Alzheimer's disease | MSCs | SHP2 | Restoring mitophagy in AD mice | 127 |
| Alzheimer's disease | MSCs | Hypoxic condition | Regulating immune responses in AD mice | 128 |
| Alzheimer's disease | MSCs | AD condition | Prolonging lift-time of AD mice | 129 |
| Alzheimer's disease | MSCs | RVG peptide | Enhancing the accumulation of EVs | 130 |
| Title | Sponsor | Status | Registered time | Reference |
|---|---|---|---|---|
| A phase I feasibility study to evaluate the safety of CD24-exosomes in patients with moderate/severe COVID-19 infection | Tel-Aviv Sourasky Medical Center | Phase I | 2020.9.25 | NCT04747574 |
| A phase II randomized, double-blind, placebo-controlled study to evaluate the safety and efficacy of exosomes overexpressing CD24 to prevent clinical deterioration in patients with moderate or severe COVID-19 infection | Eli Sprecher | Phase II | 2021.7.11 | NCT04969172 |
| A phase II randomized, single-blind dose study to evaluate the safety and efficacy of exosomes overexpressing CD24 in 109 dose versus 1010 dose, for the prevention of clinical deterioration in patients with moderate or severe COVID-19 | Athens Medical Society | Phase II | 2021.6.21 | NCT04902183 |
| A clinical trial for the safety and efficacy of exosomes that overexpress CD24 for preventing the deterioration of moderate or severe COVID-19 | OBCTCD-24 | Phase II | 2022.1.28 | EUCTR2021-004259-17-GR |
| Phase II trial of a vaccination with tumor antigen-loaded dendritic cell-derived exosomes on patients with unresectable non-small cell lung cancer responding to induction chemotherapy | Gustave Roussy, Cancer Campus, Grand Paris | Phase II | 2010.7.8 | NCT01159288 |
| A first-in-human study of CDK-002 (exoSTING) in subjects with advanced/metastatic, recurrent, injectable solid tumors | Codiak BioSciences | Phase I/II | 2020.10.18 | NCT04592484 |
| A phase 1/2a study of CDK-003 in patients with cutaneous T-cell lymphoma (CTCL) | Codiak BioSciences | Phase I/IIa | 2021.12.14 | NCT05156229 |
| A study of exoASO-STAT6 (CDK-004) in patients with advanced hepatocellular carcinoma (HCC) and patients with liver metastases from either primary gastric cancer or colorectal cancer (CRC) | Codiak BioSciences | Phase I | 2022.5.16 | NCT05375604 |
| iExosomes in treating participants with metastatic pancreatic cancer with KrasG12D mutation | M.D. Anderson Cancer Center | Phase I | 2021.1.27 | NCT03608631 |
Apart from MSC EVs, cardiosphere-derived cells (CDCs) could also be utilized in the targeting treatment of cardiovascular diseases. Vandergriff et al. engineered CDC-EVs with a cardiac homing peptide, achieving increased retention of the exosomes (Fig. 4A).65 Besides, Wang and colleagues engineered targeting EVs through a donor cell-assisted membrane modification, to increase the targeting affinity to blood cells.64 Moreover, Mentkowski also modified CDCs with a cardiomyocyte-specific peptide (CMP) and isolated CMP-EVs for cardiomyocyte targeting (Fig. 4B).54 CMP EVs could selectively bind with cardiomyocytes in vitro, with negligible binding affinity to cardiac fibroblasts and HUVECs in vitro. These EVs also showed enhanced uptake in the heart compared with blank EVs, indicating their therapeutic potential in the future.
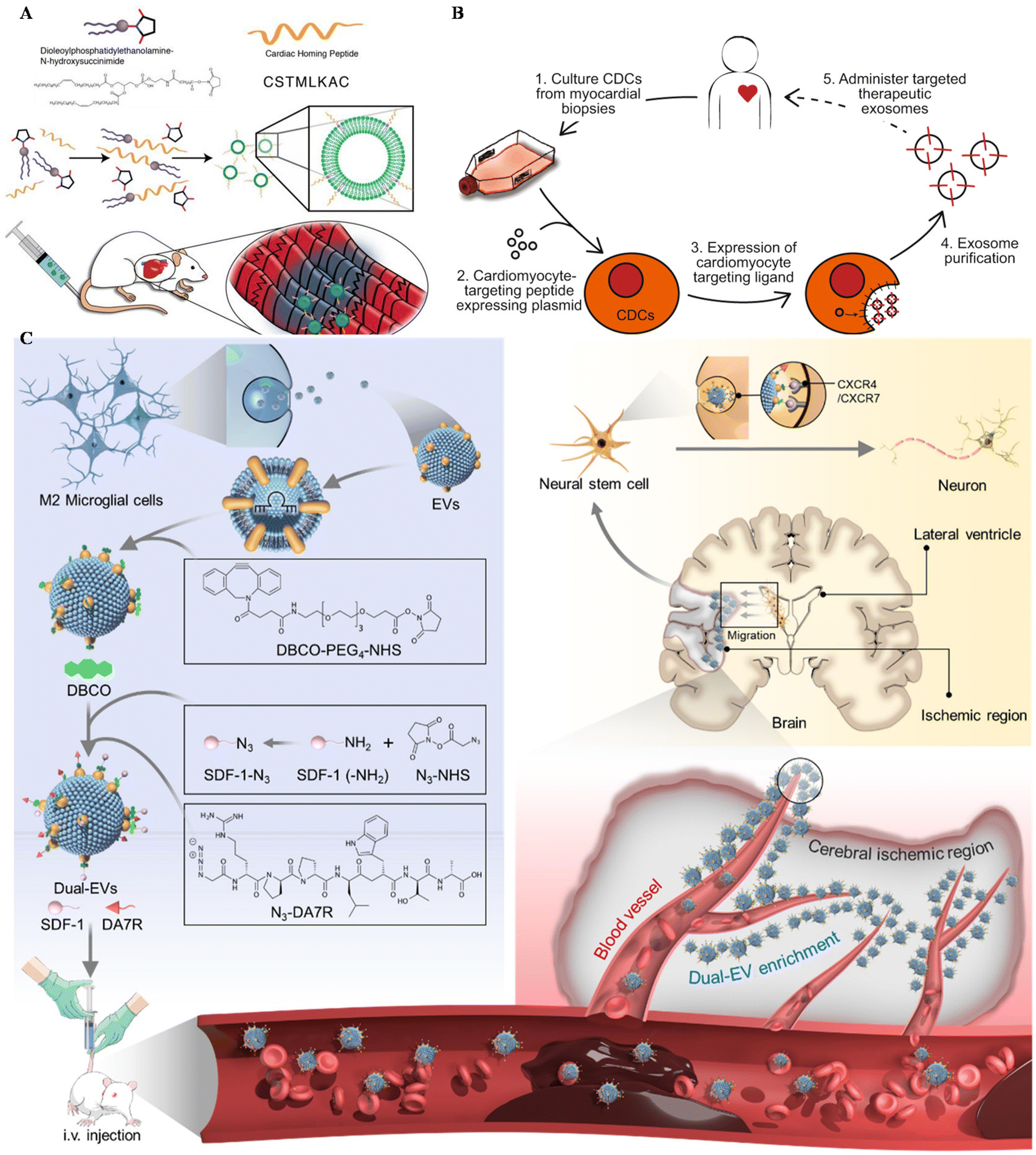 | ||
| Fig. 4 Utilizing surface-engineered EVs in other diseases. (A) The preparation of myocardium-targeting exosomes. Reproduced with permission.65 Copyright 2018, Ivyspring International. (B) Schematic diagram for the construction of CDC-derived targeted exosomes. Reproduced with permission.54 Copyright 2019, Springer Nature. (C) The schematic of utilizing engineered EVs for neurological diseases. Reproduced with permission.122 Copyright 2022, Elsevier. | ||
Central nervous system (CNS) injuries including stroke, Parkinson's disease and Alzheimer's disease (AD) are disturbing human health.121 As free drugs are difficult to accumulate in the CNS due to the blood–brain barrier (BBB), EVs seem to be a potential platform to carry drugs across the BBB, alleviating CNS injury. Ruan et al. modified EVs derived from M2 microglia with the targeting peptide and the stem cell recruiting factor to enhance the accumulation of EVs in the ischemic area of the stroke mouse model and increase neurogenesis (Fig. 4C).122 Besides, Gao engineered MSCs with SIRP-α and isolated EV–SIRP-α to calm down neuroinflammation.123 These engineered EVs could not only alleviate white matter injury, but also induce M2 polarization due to the recruitment of Treg.
AD is reported as a progressive neurodegenerative disorder with a huge prevalence globally, which could be alleviated by mitophagy.124 Tyrosine phosphatase-2 (SHP-2) is a phosphatase whose expression is upregulated during mitophagy.125 SHP-2 is a potential drug to induce mitophagy but its application is hindered by its poor BBB penetration and poor stability.126 Considering this, our group displayed SHP-2 on the surface of MSC EVs to solve this problem.127 EVs-SHP-2 had high BBB penetration ability in AD mice, because of the existence of Aβ1–42. The higher accumulation of SHP-2 led to a remarkable reduction of Aβ1–42, and improvement of cognitive decline, indicating therapeutic potential in AD treatment. Besides, researchers also engineered EVs by exposing parental MSCs within different conditions and collecting these EVs to treat AD. For instance, Cui et al. utilized EVs from hypoxia-conditioned MSCs to regulate immune responses in the brains and alleviate Aβ accumulation.128 Santamaria et al. developed EVs from MSCs exposed to AD mouse brain homogenates in vitro and these EVs could recover memory function and prolong the lifespan of AD mice.129 Additionally, Cui et al. modified EVs with the RVG peptide, to enhance the target capacity of MSC EVs. This strategy successfully increases the delivery efficacy of EVs in AD treatment and improves the memory capacity of AD mice.130
5. Conclusion and perspectives
Engineered extracellular vesicles, working as drug delivery systems, are currently a subject of extensive research and development. Their present status involves successful laboratory demonstrations of using surface-engineered EVs in the treatment of various diseases, including tumors, autoimmune diseases, infections and other diseases shown above. Early-stage clinical trials are also exploring their potential, particularly in COVID-19. Furthermore, there is growing commercial interest in the applications of EVs, leading to increased investment and collaboration in this field.Surface engineering of extracellular vesicles (EVs) encompasses various techniques aimed at enhancing their stability, targeting specificity, and therapeutic efficacy. These techniques include chemical conjugation, where molecules like lipids, peptides, antibodies, or aptamers are covalently attached to EV surfaces, ensuring stable attachment and imparting specific functionalities. Besides, genetic engineering modifies the EV surface composition during biogenesis within donor cells, allowing for precise control over surface properties. Click chemistry offers selective and efficient conjugation of bioactive molecules, enabling rapid and specific modification of EV surfaces. Moreover, comprehensive characterization ensures biological compatibility and evaluates changes in EV size, morphology, and surface properties post-modification. These techniques enable precise customization of EV surface properties, facilitating the development of tailored EV-based therapeutics and diagnostics with enhanced efficacy and specificity.
Five different modification methods are utilized in the surface engineering of EVs, namely chemical engineering, genetic engineering, physical modification, metabolic engineering and lipid-based modification, each of them having distinct advantages and disadvantages. Chemical modification is mostly preferred for its high speed and flexibility, as it allows quick engineering within a short time using various molecules, including peptides and aptamers across different cells or their extracellular vesicles. However, it lacks the precision and control of genetic engineering and may introduce unintended effects on the stability and biological activity of EVs. Moreover, certain chemicals utilized in chemical engineering may be toxic to extracellular vesicles.
On the other hand, genetic modification provides a high degree of specificity and control, allowing precise adjustments to EV composition without affecting their biological activity. Genetically engineered EVs tend to exhibit long-term stability enabling target gene overexpression. Nevertheless, genetic engineering is more time-consuming and labor-intensive, requiring specialized skills and equipment and is not universally applicable to all cell types.
The advantages of physical modification include simplicity, lower costs, and minimal impact on cells. However, its drawbacks include potential unpredictable effects on vesicle stability and function, as well as challenges in achieving precise control. In contrast, metabolic engineering could have precise control over vesicle composition and yield, thus enhancing their potential for drug delivery or therapeutic efficacy. However, metabolic engineering requires a deep understanding of cellular metabolic pathways and may involve complex genetic engineering techniques, resulting in higher costs and technical hurdles. Lastly, lipid-based surface engineering utilizes the properties of lipids to modulate the surface characteristics of vesicles. Advantages include the ability to modify the external surface properties of vesicles without affecting their internal contents, thus enhancing their potential applications in drug delivery and biological imaging. However, this method requires in-depth research into lipid chemistry and biology and may face challenges related to lipid stability and vesicle structure. Researchers should select the approach that best aligns with their needs based on specific research objectives, available resources, and experimental conditions.
However, achieving the clinical use of surface-engineered extracellular vesicles is full of challenges. As extracellular vesicles are heterogeneous, it is urgent to employ various quality control (QC) measurements to ensure the stability and biosafety of engineered EVs. Moreover, the loading ratio of displayed peptides should be measured to ensure lot-to-lot consistency. Additionally, the yield of extracellular vesicles from cultured cells should also be increased with some physical methods to guarantee large scale EV production.
Looking forward, future development of surface-engineered EVs should focus on standardization and quality control, advanced large-scale production techniques, and the initiation of more clinical trials to validate the efficacy and safety of surface-engineered EVs in various disease treatments. Furthermore, exploring their application in multimodal delivery systems could open new avenues for innovative medical treatments. In summary, surface-engineered EVs working as drug delivery systems have significant potential in improving the diagnosis and treatment of various diseases.
Author contributions
Chenlu Yao: writing – original draft (lead). Hong Zhang: writing – review & editing. Chao Wang: writing – review & editing (lead).Conflicts of interest
There are no conflicts of interest to be declared.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFB3808100). This work was partly funded by the Collaborative Innovation Center of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the 111 Project.References
- C. Harding, J. Heuser and P. Stahl, J. Cell Biol., 1983, 97, 329–339 CrossRef CAS PubMed.
- J. Skog, T. Würdinger, S. van Rijn, D. H. Meijer, L. Gainche, W. T. Curry, B. S. Carter, A. M. Krichevsky and X. O. Breakefield, Nat. Cell Biol., 2008, 10, 1470–1476 CrossRef CAS PubMed.
- H. Valadi, K. Ekström, A. Bossios, M. Sjöstrand, J. J. Lee and J. O. Lötvall, Nat. Cell Biol., 2007, 9, 654–659 CrossRef CAS PubMed.
- P. D. Robbins and A. E. Morelli, Nat. Rev. Immunol., 2014, 14, 195–208 CrossRef CAS PubMed.
- E. D'Asti, D. Garnier, T. H. Lee, L. Montermini, B. Meehan and J. Rak, Front. Physiol., 2012, 3, 294 Search PubMed.
- L. Cheng and A. F. Hill, Nat. Rev. Drug Discovery, 2022, 21, 379–399 CrossRef CAS PubMed.
- X. Zhang, H. Zhang, J. Gu, J. Zhang, H. Shi, H. Qian, D. Wang, W. Xu, J. Pan and H. A. Santos, Adv. Mater., 2021, 33, 2005709 CrossRef CAS PubMed.
- F. N. Faruqu, J. T.-W. Wang, L. Xu, L. McNickle, E. M.-Y. Chong, A. Walters, M. Gurney, A. Clayton, L. A. Smyth, R. Hider, J. Sosabowski and K. T. Al-Jamal, Theranostics, 2019, 9, 1666–1682 CrossRef CAS PubMed.
- T. Smyth, M. Kullberg, N. Malik, P. Smith-Jones, M. W. Graner and T. J. Anchordoquy, J. Controlled Release, 2015, 199, 145–155 CrossRef CAS PubMed.
- M. P. Zaborowski, L. Balaj, X. O. Breakefield and C. P. Lai, BioScience, 2015, 65, 783–797 CrossRef PubMed.
- F. Tian, S. Zhang, C. Liu, Z. Han, Y. Liu, J. Deng, Y. Li, X. Wu, L. Cai, L. Qin, Q. Chen, Y. Yuan, Y. Liu, Y. Cong, B. Ding, Z. Jiang and J. Sun, Nat. Commun., 2021, 12, 2536 CrossRef CAS PubMed.
- G. Qiu, G. Zheng, M. Ge, J. Wang, R. Huang, Q. Shu and J. Xu, Stem Cell Res. Ther., 2019, 10, 359 CrossRef PubMed.
- Q. Ma, J. Bai, J. Xu, H. Dai, Q. Fan, Z. Fei, J. Chu, C. Yao, H. Shi, X. Zhou, L. Bo and C. Wang, Adv. NanoBiomed Res., 2021, 1, 2100071 CrossRef CAS.
- Q. Ma, Q. Fan, X. Han, Z. Dong, J. Xu, J. Bai, W. Tao, D. Sun and C. Wang, J. Controlled Release, 2021, 329, 445–453 CrossRef CAS PubMed.
- Q. Ma, Q. Fan, J. Xu, J. Bai, X. Han, Z. Dong, X. Zhou, Z. Liu, Z. Gu and C. Wang, Matter, 2020, 3, 287–301 CrossRef PubMed.
- W. Chiangjong, P. Netsirisawan, S. Hongeng and S. Chutipongtanate, Front. Med., 2021, 8, 761362 CrossRef PubMed.
- W. M. Usman, T. C. Pham, Y. Y. Kwok, L. T. Vu, V. Ma, B. Peng, Y. S. Chan, L. Wei, S. M. Chin, A. Azad, A. B.-L. He, A. Y. H. Leung, M. Yang, N. Shyh-Chang, W. C. Cho, J. Shi and M. T. N. Le, Nat. Commun., 2018, 9, 2359 CrossRef PubMed.
- Y. Ito, S. Nakamura, N. Sugimoto, T. Shigemori, Y. Kato, M. Ohno, S. Sakuma, K. Ito, H. Kumon, H. Hirose, H. Okamoto, M. Nogawa, M. Iwasaki, S. Kihara, K. Fujio, T. Matsumoto, N. Higashi, K. Hashimoto, A. Sawaguchi, K.-I. Harimoto, M. Nakagawa, T. Yamamoto, M. Handa, N. Watanabe, E. Nishi, F. Arai, S. Nishimura and K. Eto, Cell, 2018, 174, 636–648 CrossRef CAS PubMed.
- S.-C. Tao, S.-C. Guo and C.-Q. Zhang, Int. J. Biol. Sci., 2017, 13, 828–834 CrossRef CAS PubMed.
- F. Puhm, E. Boilard and K. R. Machlus, Arterioscler., Thromb., Vasc. Biol., 2020, 41, 87–96 Search PubMed.
- Y. Wang, M. Zhao, S. Liu, J. Guo, Y. Lu, J. Cheng and J. Liu, Cell Death Dis., 2020, 11, 924 CrossRef CAS PubMed.
- C. Cianciaruso, T. Beltraminelli, F. Duval, S. Nassiri, R. Hamelin, A. Mozes, H. Gallart-Ayala, G. Ceada Torres, B. Torchia, C. H. Ries, J. Ivanisevic and M. De Palma, Cell Rep., 2019, 27, 3062–3080 CrossRef CAS PubMed.
- Y. Zhao, Y. Zheng, Y. Zhu, H. Li, H. Zhu and T. Liu, J. Nanobiotechnol., 2022, 20, 359 CrossRef CAS PubMed.
- A. Matsumoto, M. Asuka, Y. Takahashi and Y. Takakura, Biomaterials, 2020, 252, 120112 CrossRef CAS PubMed.
- O. Markov, A. Oshchepkova and N. Mironova, Front. Pharmacol., 2019, 10, 1152 CrossRef CAS PubMed.
- J. Chen, F. Huang, Y. Hou, X. Lin, R. Liang, X. Hu, J. Zhao, J. Wang, N. Olsen and S. G. Zheng, Cell. Mol. Immunol., 2021, 18, 2516–2529 CrossRef CAS PubMed.
- S. Shin, I. Jung, D. Jung, C. S. Kim, S.-M. Kang, S. Ryu, S.-J. Choi, S. Noh, J. Jeong, B. Y. Lee, J.-K. Park, J. Shin, H. Cho, J.-I. Heo, Y. Jeong, S. H. Choi, S. Y. Lee, M.-C. Baek and K. Yea, Biomaterials, 2022, 289, 121765 CrossRef CAS PubMed.
- I. S. Okoye, S. M. Coomes, V. S. Pelly, S. Czieso, V. Papayannopoulos, T. Tolmachova, M. C. Seabra and M. S. Wilson, Immunity, 2014, 41, 89–103 CrossRef CAS PubMed.
- M. F. Gutknecht, N. E. Holodick and T. L. Rothstein, iScience, 2023, 26, 107526 CrossRef CAS PubMed.
- F. Zhang, R. Li, Y. Yang, C. Shi, Y. Shen, C. Lu, Y. Chen, W. Zhou, A. Lin, L. Yu, W. Zhang, Z. Xue, J. Wang and Z. Cai, Immunity, 2019, 50, 738–750 CrossRef CAS PubMed.
- F. Wu, M. Xie, M. Hun, Z. She, C. Li, S. Luo, X. Chen, W. Wan, C. Wen and J. Tian, Front. Immunol., 2021, 12, 658698 CrossRef CAS PubMed.
- C. H. Wu, J. Li, L. Li, J. Sun, M. Fabbri, A. S. Wayne, R. C. Seeger and A. Y. Jong, J. Extracell. Vesicles, 2019, 8, 1588538 CrossRef CAS PubMed.
- J. Kowal and M. Tkach, in International Review of Cell and Molecular Biology, ed. C. Lhuillier and L. Galluzzi, Academic Press, 2019, vol. 349, pp. 213–249 Search PubMed.
- A. Barone, N. d’Avanzo, M. C. Cristiano, D. Paolino and M. Fresta, Biomedicines, 2022, 10, 252 CrossRef PubMed.
- F. Lyu, K. Wu, S.-Y. Wu, R. P. Deshpande, A. Tyagi, I. Ruiz, S. Yalavarthi and K. Watabe, Oncogene, 2024, 43, 319–327 CrossRef CAS PubMed.
- Y. Jiao, Y. Tang, Y. Li, C. Liu, J. He, L.-K. Zhang and Y.-Q. Guan, J. Controlled Release, 2022, 349, 606–616 CrossRef CAS PubMed.
- C. Marar, B. Starich and D. Wirtz, Nat. Immunol., 2021, 22, 560–570 CrossRef CAS PubMed.
- S. Keshtkar, N. Azarpira and M. H. Ghahremani, Stem Cell Res. Ther., 2018, 9, 63 CrossRef CAS PubMed.
- S. Rani, A. E. Ryan, M. D. Griffin and T. Ritter, Mol. Ther., 2015, 23, 812–823 CrossRef CAS PubMed.
- M. Riazifar, E. J. Pone, J. Lötvall and W. Zhao, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 125–154 CrossRef CAS PubMed.
- S. Varderidou-Minasian and M. J. Lorenowicz, Theranostics, 2020, 10, 5979–5997 CrossRef CAS PubMed.
- A. Krishnan, S. Muthusamy, F. B. Fernandez and N. Kasoju, Tissue Eng. Regener. Med., 2022, 19, 659–673 CrossRef CAS PubMed.
- M. G. Sartorio, E. J. Pardue, M. F. Feldman and M. F. Haurat, Annu. Rev. Microbiol., 2021, 75, 609–630 CrossRef CAS PubMed.
- M. Kaparakis-Liaskos and R. L. Ferrero, Nat. Rev. Immunol., 2015, 15, 375–387 CrossRef CAS PubMed.
- K. E. Bonnington and M. J. Kuehn, Biochim. Biophys. Acta, Mol. Cell Res., 2014, 1843, 1612–1619 CrossRef CAS PubMed.
- S. Qing, C. Lyu, L. Zhu, C. Pan, S. Wang, F. Li, J. Wang, H. Yue, X. Gao, R. Jia, W. Wei and G. Ma, Adv. Mater., 2020, 32, 2002085 CrossRef CAS PubMed.
- S. Wang, J. Guo, Y. Bai, C. Sun, Y. Wu, Z. Liu, X. Liu, Y. Wang, Z. Wang, Y. Zhang and H. Hao, Front. Immunol., 2022, 13, 987419 CrossRef CAS PubMed.
- Y. Cui, J. Gao, Y. He and L. Jiang, Protoplasma, 2020, 257, 3–12 CrossRef CAS PubMed.
- S. Rome, Food Funct., 2019, 10, 529–538 RSC.
- L. Yu, Z. Deng, L. Liu, W. Zhang and C. Wang, Front. Bioeng. Biotechnol., 2020, 8, 584391 CrossRef PubMed.
- N. P. Ly, H. S. Han, M. Kim, J. H. Park and K. Y. Choi, Bioact. Mater., 2023, 22, 365–383 CAS.
- M. Y. Konoshenko, E. A. Lekchnov, A. V. Vlassov and P. P. Laktionov, BioMed Res. Int., 2018, 2018, 8545347 Search PubMed.
- Q. Zhang, D. K. Jeppesen, J. N. Higginbotham, J. L. Franklin and R. J. Coffey, Nat. Protoc., 2023, 18, 1462–1487 CrossRef CAS PubMed.
- X. Wang, Y. Chen, Z. Zhao, Q. Meng, Y. Yu, J. Sun, Z. Yang, Y. Chen, J. Li, T. Ma, H. Liu, Z. Li, J. Yang and Z. Shen, J. Am. Heart Assoc., 2018, 7, e008737 CrossRef CAS PubMed.
- T. A. Scott, A. Supramaniam, A. Idris, A. A. Cardoso, S. Shrivastava, G. Kelly, N. A. Grepo, C. Soemardy, R. M. Ray, N. A. J. McMillan and K. V. Morris, Mol. Ther.–Methods Clin. Dev., 2022, 24, 355–366 CrossRef CAS PubMed.
- H. K. Kim, J. Cho, E. Kim, J. Kim, J. S. Yang, K. C. Kim, J. Y. Lee, Y. Shin, L. F. Palomera, J. Park, S. H. Baek, H. G. Bae, Y. Cho, J. Han, J. H. Sul, J. Lee, J. H. Park, Y. W. Cho, W. Lee and D. G. Jo, J. Extracell. Vesicles, 2022, 11, e12179 CrossRef CAS PubMed.
- D. Bellavia, S. Raimondo, G. Calabrese, S. Forte, M. Cristaldi, A. Patinella, L. Memeo, M. Manno, S. Raccosta, P. Diana, G. Cirrincione, G. Giavaresi, F. Monteleone, S. Fontana, G. De Leo and R. Alessandro, Theranostics, 2017, 7, 1333–1345 CrossRef CAS PubMed.
- E. Koh, E. J. Lee, G.-H. Nam, Y. Hong, E. Cho, Y. Yang and I.-S. Kim, Biomaterials, 2017, 121, 121–129 CrossRef CAS PubMed.
- M. Piffoux, J. Volatron, K. Cherukula, K. Aubertin, C. Wilhelm, A. K. A. Silva and F. Gazeau, Adv. Drug Delivery Rev., 2021, 178, 113972 CrossRef CAS PubMed.
- H. Qin, H. Li, J. Zhu, Y. Qin, N. Li, J. Shi, G. Nie and R. Zhao, Small, 2023, 2303225 CrossRef CAS PubMed.
- W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS.
- C. D. Hein, X.-M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS PubMed.
- T. Tian, H.-X. Zhang, C.-P. He, S. Fan, Y.-L. Zhu, C. Qi, N.-P. Huang, Z.-D. Xiao, Z.-H. Lu, B. A. Tannous and J. Gao, Biomaterials, 2018, 150, 137–149 CrossRef CAS PubMed.
- J. Wang, W. Li, Z. Lu, L. Zhang, Y. Hu, Q. Li, W. Du, X. Feng, H. Jia and B.-F. Liu, Nanoscale, 2017, 9, 15598–15605 RSC.
- A. Vandergriff, K. Huang, D. Shen, S. Hu, M. T. Hensley, T. G. Caranasos, L. Qian and K. Cheng, Theranostics, 2018, 8, 1869–1878 CrossRef CAS PubMed.
- T. J. Antes, R. C. Middleton, K. M. Luther, T. Ijichi, K. A. Peck, W. J. Liu, J. Valle, A. K. Echavez and E. Marbán, J. Nanobiotechnol., 2018, 16, 61 CrossRef PubMed.
- S. Rayamajhi and S. Aryal, J. Mater. Chem. B, 2020, 8, 4552–4569 RSC.
- Y.-Y. Jhan, D. Prasca-Chamorro, G. Palou Zuniga, D. M. Moore, S. Arun Kumar, A. K. Gaharwar and C. J. Bishop, Int. J. Pharm., 2020, 573, 118802 CrossRef CAS PubMed.
- M. Richter, P. Vader and G. Fuhrmann, Adv. Drug Delivery Rev., 2021, 173, 416–426 CrossRef CAS PubMed.
- S. Rayamajhi, R. Marasini, T. D. T. Nguyen, B. L. Plattner, D. Biller and S. Aryal, Biomater. Sci., 2020, 8, 2887–2904 RSC.
- Q. Li, Z. Huang, Q. Wang, J. Gao, J. Chen, H. Tan, S. Li, Z. Wang, X. Weng, H. Yang, Z. Pang, Y. Song, J. Qian and J. Ge, Biomaterials, 2022, 284, 121529 CrossRef CAS PubMed.
- J. P. K. Armstrong, M. N. Holme and M. M. Stevens, ACS Nano, 2017, 11, 69–83 CrossRef CAS PubMed.
- C. Williams, F. Royo, O. Aizpurua-Olaizola, R. Pazos, G.-J. Boons, N.-C. Reichardt and J. M. Falcon-Perez, J. Extracell. Vesicles, 2018, 7, 1442985 CrossRef PubMed.
- G. T. Lim, D. G. You, H. S. Han, H. Lee, S. Shin, B. H. Oh, E. K. P. Kumar, W. Um, C. H. Kim, S. Han, S. Lee, S. Lim, H. Y. Yoon, K. Kim, I. C. Kwon, D. G. Jo, Y. W. Cho and J. H. Park, J. Extracell. Vesicles, 2021, 10, e1207 Search PubMed.
- A. H. Mohammadi, Z. Ghazvinian, F. Bagheri, M. Harada and K. Baghaei, BioDrugs, 2023, 37, 353–374 CrossRef CAS PubMed.
- T. Limongi, F. Susa, M. Marini, M. Allione, B. Torre, R. Pisano and E. di Fabrizio, Nanomaterials, 2021, 11, 3391 CrossRef CAS PubMed.
- W. Lin, Y. Xu, X. Chen, J. Liu, Y. Weng, Q. Zhuang, F. Lin, Z. Huang, S. Wu, J. Ding, L. Chen, X. Qiu, L. Zhang, J. Wu, D. Lin and S. Qiu, Theranostics, 2020, 10, 4871–4884 CrossRef CAS PubMed.
- S. K. Horrevorts, D. A. Stolk, R. van de Ven, B. v. H. Hof, S. Duinkerken, M. Hulst, M. H. Heineke, W. Ma, S. A. Dusoswa, R. Nieuwland, J. J. Garcia-Vallejo, A. A. van de Loosdrecht, T. D. de Gruijl, S. J. van Vliet and Y. van Kooyk, Cancers, 2019, 11, 1266–1284 CrossRef CAS PubMed.
- T. Yong, Z. Wei, L. Gan and X. Yang, Adv. Mater., 2022, 34, 2201054 CrossRef CAS PubMed.
- S. Walker, S. Busatto, A. Pham, M. Tian, A. Suh, K. Carson, A. Quintero, M. Lafrence, H. Malik, M. X. Santana and J. Wolfram, Theranostics, 2019, 9, 8001–8017 CrossRef CAS PubMed.
- A. Taghikhani, F. Farzaneh, F. Sharifzad, S. Mardpour, M. Ebrahimi and Z. M. Hassan, Front. Immunol., 2020, 11, 221 CrossRef CAS PubMed.
- K. Y. Choi, H. Y. Yoon, J.-H. Kim, S. M. Bae, R.-W. Park, Y. M. Kang, I.-S. Kim, I. C. Kwon, K. Choi, S. Y. Jeong, K. Kim and J. H. Park, ACS Nano, 2011, 5, 8591–8599 CrossRef CAS PubMed.
- S. Sajeesh, A. Camardo, S. Dahal and A. Ramamurthi, Mol. Pharm., 2023, 20, 2801–2813 CrossRef PubMed.
- C.-L. Liu, J. Guo, X. Zhang, G. K. Sukhova, P. Libby and G.-P. Shi, Nat. Rev. Cardiol., 2018, 15, 351–370 CrossRef PubMed.
- Y. Tian, S. Li, J. Song, T. Ji, M. Zhu, G. J. Anderson, J. Wei and G. Nie, Biomaterials, 2014, 35, 2383–2390 CrossRef CAS PubMed.
- S. Loibl and L. Gianni, Lancet, 2017, 389, 2415–2429 CrossRef CAS PubMed.
- A. Longatti, C. Schindler, A. Collinson, L. Jenkinson, C. Matthews, L. Fitzpatrick, M. Blundy, R. Minter, T. Vaughan, M. Shaw and N. Tigue, Nanoscale, 2018, 10, 14230–14244 RSC.
- S. A. A. Kooijmans, C. G. Aleza, S. R. Roffler, W. W. van Solinge, P. Vader and R. M. Schiffelers, J. Extracell. Vesicles, 2016, 5, 31053 CrossRef PubMed.
- R. Ito, N. Oue, K. Yoshida, K. Kunimitsu, H. Nakayama, K. Nakachi and W. Yasui, Virchows Arch., 2005, 447, 717–722 CrossRef CAS PubMed.
- P. Xia, H. Yuan, M. Tian, T. Zhong, R. Hou, X. Xu, J. Ma, H. Wang, Z. Li, D. Huang, C. Qu, L. Dai, C. Xu, C. Yang, H. Jiang, Y. He, F. Rückert, Z. Li, Y. Yuan and J. Wang, Adv. Funct. Mater., 2022, 33, 2209393 CrossRef.
- K. Moon, J. Hur, K. P. Kim, K. Lee and J. Y. Kang, Adv. Mater. Interfaces, 2023, 10, 2300220 CrossRef CAS.
- M. Binnewies, E. W. Roberts, K. Kersten, V. Chan, D. F. Fearon, M. Merad, L. M. Coussens, D. I. Gabrilovich, S. Ostrand-Rosenberg, C. C. Hedrick, R. H. Vonderheide, M. J. Pittet, R. K. Jain, W. Zou, T. K. Howcroft, E. C. Woodhouse, R. A. Weinberg and M. F. Krummel, Nat. Med., 2018, 24, 541–550 CrossRef CAS PubMed.
- B. Lv, Y. Wang, D. Ma, W. Cheng, J. Liu, T. Yong, H. Chen and C. Wang, Front. Immunol., 2022, 13, 844142 CrossRef PubMed.
- L. Xu, C. Zou, S. Zhang, T. S. M. Chu, Y. Zhang, W. Chen, C. Zhao, L. Yang, Z. Xu, S. Dong, H. Yu, B. Li, X. Guan, Y. Hou and F.-M. Kong, J. Hematol. Oncol., 2022, 15, 87–117 CrossRef PubMed.
- H. Phuengkham, L. Ren, I. W. Shin and Y. T. Lim, Adv. Mater., 2019, 31, 1803322 CrossRef PubMed.
- Y. Tie, F. Tang, Y.-Q. Wei and X.-W. Wei, J. Hematol. Oncol., 2022, 15, 61–94 CrossRef CAS PubMed.
- S. G. Kalathil and Y. Thanavala, Cancer Immunol., Immunother., 2016, 65, 813–819 CrossRef CAS PubMed.
- Q. F. Meng, Y. Zhao, C. Dong, L. Liu, Y. Pan, J. Lai, Z. Liu, G. T. Yu, X. Chen and L. Rao, Angew. Chem., Int. Ed., 2021, 60, 26320–26326 CrossRef CAS PubMed.
- B. Li, T. Fang, Y. Li, T. Xue, Z. Zhang, L. Li, F. Meng, J. Wang, L. Hou, X. Liang, X. Zhang and Z. Gu, Nano Today, 2022, 46, 101606 CrossRef CAS.
- L. Li, Q. Miao, F. Meng, B. Li, T. Xue, T. Fang, Z. Zhang, J. Zhang, X. Ye, Y. Kang, X. Zhang, Q. Chen, X. Liang, H. Chen and X. Zhang, Theranostics, 2021, 11, 6033–6043 CrossRef CAS PubMed.
- X. Shi, Q. Cheng, T. Hou, M. Han, G. Smbatyan, J. E. Lang, A. L. Epstein, H. J. Lenz and Y. Zhang, Mol. Ther., 2020, 28, 536–547 CrossRef CAS PubMed.
- P. Guermonprez, J. Valladeau, L. Zitvogel, C. Théry and S. Amigorena, Annu. Rev. Immunol., 2002, 20, 621–667 CrossRef CAS PubMed.
- X. Dai, Z. Wang, M. Fan, H. Liu, X. Yang, X. Wang, X. Zhou, Y. Dai, J. Zhang and Z. Li, Extracell. Vesicles, 2022, 1, 100012 CrossRef.
- F. Xu, D. Jiang, J. Xu, H. Dai, Q. Fan, Z. Fei, B. Wang, Y. Zhang, Q. Ma, Q. Yang, Y. Chen, E. A. Ogunnaike, J. Chu and C. Wang, Cell Rep., 2023, 42, 113138 CrossRef CAS PubMed.
- M. Li, H. Zhou, C. Yang, Y. Wu, X. Zhou, H. Liu and Y. Wang, J. Controlled Release, 2020, 323, 253–268 CrossRef CAS PubMed.
- K. Cheng, R. Zhao, Y. Li, Y. Qi, Y. Wang, Y. Zhang, H. Qin, Y. Qin, L. Chen, C. Li, J. Liang, Y. Li, J. Xu, X. Han, G. J. Anderson, J. Shi, L. Ren, X. Zhao and G. Nie, Nat. Commun., 2021, 12, 2041 CrossRef CAS PubMed.
- Y. Yue, J. Xu, Y. Li, K. Cheng, Q. Feng, X. Ma, N. Ma, T. Zhang, X. Wang, X. Zhao and G. Nie, Nat. Biomed. Eng., 2022, 6, 898–909 CrossRef CAS PubMed.
- W.-R. Zhuang, Y. Wang, W. Nie, Y. Lei, C. Liang, J. He, L. Zuo, L.-L. Huang and H.-Y. Xie, Nat. Commun., 2023, 14, 1675–1690 CrossRef CAS PubMed.
- Y. Li, K. Zhang, Y. Wu, Y. Yue, K. Cheng, Q. Feng, X. Ma, J. Liang, N. Ma, G. Liu, G. Nie, L. Ren and X. Zhao, Small, 2022, 18, 2107461 CrossRef CAS PubMed.
- P. Marrack, J. Kappler and B. L. Kotzin, Nat. Med., 2001, 7, 899–905 CrossRef CAS PubMed.
- D. G. You, G. T. Lim, S. Kwon, W. Um, B. H. Oh, S. H. Song, J. Lee, D.-G. Jo, Y. W. Cho and J. H. Park, Sci. Adv., 2021, 7, eabe0083 CrossRef CAS PubMed.
- L. M. Francisco, P. T. Sage and A. H. J. I. R. Sharpe, Immunol. Rev., 2010, 236, 219–242 CrossRef CAS PubMed.
- M. W. Becker, L. D. Peters, T. Myint, D. Smurlick, A. Powell, T. M. Brusko and E. A. Phelps, Sci. Adv., 2023, 9, eadg1082 CrossRef CAS PubMed.
- F. Xu, Z. Fei, H. Dai, J. Xu, Q. Fan, S. Shen, Y. Zhang, Q. Ma, J. Chu, F. Peng, F. Zhou, Z. Liu and C. Wang, Adv. Mater., 2021, 34, 2106265 CrossRef PubMed.
- C. Wu, Q. Xu, H. Wang, B. Tu, J. Zeng, P. Zhao, M. Shi, H. Qiu and Y. Huang, Acta Pharm. Sin. B, 2022, 12, 1523–1533 CrossRef CAS PubMed.
- F. Xie, P. Su, T. Pan, X. Zhou, H. Li, H. Huang, A. Wang, F. Wang, J. Huang, H. Yan, L. Zeng, L. Zhang and F. Zhou, Adv. Mater., 2021, 33, 2103471 CrossRef CAS PubMed.
- K. Mc Namara, H. Alzubaidi and J. K. Jackson, Integr. Pharm. Res. Pract., 2019, 8, 1–11 CrossRef PubMed.
- C. M. Boulanger, X. Loyer, P.-E. Rautou and N. Amabile, Nat. Rev. Cardiol., 2017, 14, 259–272 CrossRef CAS PubMed.
- W. Pei, Y. Zhang, X. Zhu, C. Zhao, X. Li, H. Lu and K. Lv, ACS Nano, 2024, 18, 2782–2799 CrossRef CAS PubMed.
- X. Wang, Y. Chen, Z. Zhao, Q. Meng, Y. Yu, J. Sun, Z. Yang, Y. Chen, J. Li, T. Ma, H. Liu, Z. Li, J. Yang and Z. Shen, J. Am. Heart Assoc., 2018, 7, e008737 CrossRef CAS PubMed.
- C. Meisel, J. M. Schwab, K. Prass, A. Meisel and U. Dirnagl, Nat. Rev. Neurosci., 2005, 6, 775–786 CrossRef CAS PubMed.
- H. Ruan, Y. Li, C. Wang, Y. Jiang, Y. Han, Y. Li, D. Zheng, J. Ye, G. Chen, G.-Y. Yang, L. Deng, M. Guo, X. Zhang, Y. Tang and W. Cui, Acta Pharm. Sin. B, 2023, 13, 2202–2218 CrossRef CAS PubMed.
- X. Gao, H. Yang, W. Xiao, J. Su, Y. Zhang, H. Wang, W. Ni and Y. Gu, Biomater. Res., 2022, 26, 67–91 CrossRef CAS PubMed.
- D. Mantle and I. P. Hargreaves, Int. J. Mol. Sci., 2022, 23, 12603 CrossRef CAS PubMed.
- W.-Q. Qiu, W. Ai, F.-D. Zhu, Y. Zhang, M.-S. Guo, B. Y.-K. Law, J.-M. Wu, V. K.-W. Wong, Y. Tang, L. Yu, Q. Chen, C.-L. Yu, J. Liu, D.-L. Qin, X.-G. Zhou and A.-G. Wu, Free Radicals Biol. Med., 2022, 179, 76–94 CrossRef CAS PubMed.
- L. Lu, S. Zhang, C. Li, C. Zhou, D. Li, P. Liu, M. Huang and X. Shen, Cell Death Dis., 2017, 8, e2767–e2767 CrossRef CAS PubMed.
- F. Xu, Y. Wu, Q. Yang, Y. Cheng, J. Xu, Y. Zhang, H. Dai, B. Wang, Q. Ma, Y. Chen, F. Lin and C. Wang, Adv. Mater., 2022, 34, 2207107 CrossRef CAS PubMed.
- G. H. Cui, J. Wu, F. F. Mou, W. H. Xie, F. B. Wang, Q. L. Wang, J. Fang, Y. W. Xu, Y. R. Dong, J. R. Liu and H. D. Guo, FASEB J., 2018, 32, 654–668 CrossRef CAS PubMed.
- G. Santamaria, E. Brandi, P. L. Vitola, F. Grandi, G. Ferrara, F. Pischiutta, G. Vegliante, E. R. Zanier, F. Re, A. Uccelli, G. Forloni, N. K. de Rosbo and C. Balducci, Cell Death Differ., 2021, 28, 203–218 CrossRef CAS PubMed.
- G.-H. Cui, H.-D. Guo, H. Li, Y. Zhai, Z.-B. Gong, J. Wu, J.-S. Liu, Y.-R. Dong, S.-X. Hou and J.-R. Liu, Immun. Ageing, 2019, 16, 10 CrossRef PubMed.
- X. Li, T. Zhu, R. Wang, J. Chen, L. Tang, W. Huo, X. Huang and Q. Cao, Adv. Mater., 2023, 35, 2211138 CrossRef CAS PubMed.
- D. Jung, S. Shin, S. M. Kang, I. Jung, S. Ryu, S. Noh, S. J. Choi, J. Jeong, B. Y. Lee, K. S. Kim, C. S. Kim, J. H. Yoon, C. H. Lee, F. Bucher, Y. N. Kim, S. H. Im, B. J. Song, K. Yea and M. C. Baek, J. Extracell. Vesicles, 2022, 11, 12287 CrossRef CAS PubMed.
- W. Fu, C. Lei, S. Liu, Y. Cui, C. Wang, K. Qian, T. Li, Y. Shen, X. Fan, F. Lin, M. Ding, M. Pan, X. Ye, Y. Yang and S. Hu, Nat. Commun., 2019, 10, 4355 CrossRef PubMed.
- Y. Li, R. Zhao, K. Cheng, K. Zhang, Y. Wang, Y. Zhang, Y. Li, G. Liu, J. Xu, J. Xu, G. J. Anderson, J. Shi, L. Ren, X. Zhao and G. Nie, ACS Nano, 2020, 14, 16698–16711 CrossRef CAS PubMed.
- Z. Wei, Z. Chen, Y. Zhao, F. Fan, W. Xiong, S. Song, Y. Yin, J. Hu, K. Yang, L. Yang, B. Xu and J. Ge, Biomaterials, 2021, 275, 121000 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |