
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical investigation of carbon dioxide adsorption on MgH2 with a cobalt catalyst†
Sara
Rozas
 a,
Fabiana C.
Gennari
b,
Mert
Atilhan
c,
Alfredo
Bol
de and
Santiago
Aparicio
*ae
a,
Fabiana C.
Gennari
b,
Mert
Atilhan
c,
Alfredo
Bol
de and
Santiago
Aparicio
*ae
aDepartment of Chemistry, University of Burgos, 09001 Burgos, Spain. E-mail: sapar@ubu.es
bNational Scientific and Technical Research Council (CONICET), Bariloche Atomic Centre (CNEA), R8402AGP, S. C. de Bariloche, Río Negro, Argentina
cDepartment of Chemical and Paper Engineering, Western Michigan University, Kalamazoo, MI 49008-5462, USA
dDepartment of Physics, University of Burgos, 09001 Burgos, Spain
eInternational Research Center in Critical Raw Materials for Advanced Industrial Technologies (ICCRAM), University of Burgos, 09001 Burgos, Spain
First published on 26th January 2024
Abstract
This work presents a theoretical investigation of carbon dioxide (CO2) adsorption on MgH2 and its reaction (chemisorption) with cobalt doped MgH2. The focus of this study is the properties and mechanisms involved in CO2 adsorption on clean MgH2 surfaces and the role of Co in enhancing the adsorption process. Density functional theory (DFT) calculations were performed to examine different CO2 adsorption sites on the MgH2 surface along with the adsorption distances, binding energies, and geometric parameters. The results indicate that physical adsorption of CO2 occurs on MgH2 with similar adsorption energies at different adsorption sites. The coverage effect of CO2 molecules on MgH2 was also investigated, revealing an increased affinity of CO2 with higher surface coverage. However, excessive coverage led to a decrease in adsorption efficiency due to competing surface adsorption and intermolecular interactions. The orientation of adsorbed CO2 molecules shifted from parallel to quasi-perpendicular arrangements upon adsorption, with notable deformations observed at higher coverage, which gives a hint of CO2 activation. Furthermore, the study explores the CO2 adsorption capacity of MgH2 in comparison to other materials reported in the literature, showcasing its medium to strong affinity for CO2. Additionally, the effectiveness of a single Co atom and Co clusters as catalysts for CO2 adsorption on MgH2 was examined. Overall, this theoretical investigation provides insights into the CO2 adsorption properties of MgH2 and highlights the potential of Co catalysts to enhance the efficiency of the methanation process.
Keywords: DFT; CO2 conversion; Cobalt catalyst; Charge transfer.
1 Introduction
Atmospheric carbon dioxide concentration reached 424.81 ppm (global mean) in June 2023,1 which far exceeds the natural atmospheric carbon dioxide concentrations (280 ppm)2 before the Industrial Revolution took place. This has led to severe consequences, including climate change and global warming. Carbon dioxide capture and storage, as well as carbon dioxide capture and utilisation technologies, i.e., CCSs and CCUs, respectively, have been considered a suitable approach to solve this pivotal problem.3 Among the main CO2 capture procedures (pre-combustion, post-combustion, and oxyfuel combustion4–6), the post-combustion process is the most engineered technology as it can be retrofitted to existing power plants.7 However, challenges remain, such as the requirement for high-performance materials or the demand for energy for the high sorbent regeneration process.8 In this process, once CO2 gas is captured by the sorption material from industrial flue gases, it can be stored in suitable geological formations9 in the case of CCS or reused in different industrial processes in the case of CCUs. This may create extra operational needs (CCUs with conversion), such as fuels or polymeric materials,10 or be ready to use (CCUs without conversion), such as heat transfer fluid.11A possible route for CCU is methane production (methanation) from captured CO2 through hydrogenation. CO2 methanation is one of the most relevant applications for recycling carbon dioxide, owing to its high gravimetric heat (55.5 MJ kg−1).12 Different means for CO2 methanation have been proposed, including photocatalytic,13 thermal catalytic,14 or metal-catalyzed processes,15 among others. Recent techno-economical analyses of the most mature technologies showed several drawbacks, with the main need being the development of more efficient sorbent/catalysts to allow scaling up to the industrial level.16 A possible route for CO2 methanation stands on the Sabatier reaction,17i.e., the hydrogenation reaction of CO2 to CH4, which is thermodynamically favourable  18 but must be catalyzed due to strong kinetics limitations. Likewise, the hydrogen required for the methanation may be provided to the chemical reactor19 or it could be produced in situ. This last option could allows capturing and converting CO2 simultaneously as well as ensuring the green origin of the hydrogen used.
18 but must be catalyzed due to strong kinetics limitations. Likewise, the hydrogen required for the methanation may be provided to the chemical reactor19 or it could be produced in situ. This last option could allows capturing and converting CO2 simultaneously as well as ensuring the green origin of the hydrogen used.
Among the materials which can potentially be used as hydrogen sources, MgH2 has shown a large hydrogen storage capacity.20–22 Nevertheless, high hydrogenation/dehydrogenation temperatures remain technologically and economically unaffordable for large-scale application of MgH2. However, Ni- and Co-additives have been shown to positively alter hydrogen absorption/desorption kinetics.23,24 Cui et al.25 confirmed that transition metal nanoparticles (NPs) of Ti, Nb, V, Co, Mo, and Ni enhance dehydrogenation performance due to a decrease in electronegativity. Dehydrogenation co-catalyzed with MgH2 was reported by Gennari et al. showing hydrogen absorption/desorption temperatures dropping from 350 to 250 °C (ref. 26) and activation energies from 143 to 110 kJ mol−1. Liu et al. reported improved hydrogen absorption/desorption performance in terms of H2 temperature release (198.9 °C) and activation energies (76.66 kJ mol−1) using Co/Pd@CNTs catalyst.27 Therefore, taking advantage of MgH2 material as a hydrogen source, CO2 hydrogenation and conversion into valuable products has been assessed from an experimental point of view using MgH2 as substrate.28 Therefore, MgH2 has been considered a substrate for H2 supply in the CO2 methanation process due to its natural capacity to release H2 above 350 °C.18 Theoretical studies have already been performed in this context; Manggada et al. employed an MgH2 substrate for the CO2 hydrogenation using a Mo-phosphonic acid catalyst to report a great reduction in the interaction energy barriers in the presence of the catalyst.29 Cu/ZrO2 system has also been evaluated in hydrogenation of CO2 to methanol showing potential active reaction sites.30 Moreover, Chen et al. reported DFT calculations revealing an enhanced weakening of the H–Mg surface bonds by introducing Mg vacancies, thus facilitating CO2 molecule adsorption on the MgH2 substrate.31 Experimentally, Co-based catalysts for this reaction have been studied widely.32,33 Computational studies based on theoretical models are in demand to accurately assess and evaluate active sites in materials.34
CO2 methanation assays with MgH2 substrate as a portable hydrogen medium under thermal conditions were performed in previous work on cobalt catalysts. Results showed the mechanisms for direct CO2 reduction to CH4 in non-catalyzed systems, however, global mechanisms of catalyzed conversion are based on the Sabatier process that is enhanced by a Co catalyst.35 In this regard, the aim of the present work is to analyze for the first time CO2–MgH2 interactions and adsorption mechanisms with and without adsorbed atoms of Co. We shed light on the driving forces and electronic properties in this reaction using a theoretical approach based on the density functional theory (DFT).
2 Results and discussion
2.1 CO2 adsorption on clean MgH2
Different sites for CO2 molecule adsorption were considered on a clean MgH2 (0 0 1) surface (Fig. 1) in accordance with the position of the molecule along with different orientations. Gas molecules were placed on top of Mg atoms, on top of H atoms, and over the interatomic cavities. In this third configuration, CO2 molecules were parallel to the surface (XY plane) with two different orientations with the aim of studying all possible interactions (Fig. S1a, ESI†). Therefore, we have considered a slab model of 8.88 × 8.88 Å; seven different DFT calculations corresponding to seven initial structures for the adsorption of a single CO2 molecule were performed. Optimised geometries for all the considered systems are shown in Fig. 2, where CO2 bending angles and CO2–surface distances are also reported, while binding energies and further geometric parameters are presented in Table 1. Binding energies for all the considered systems are rather similar, ranging from −0.38 to −0.43 eV, except for the P4 structure, which shows the weakest interaction energy (−0.20 eV) by far. These energies are directly related to the adsorption distances, where intervals of 2.01 to 2.34 Å were observed for P1, P2, P3, P5, P6, and P7 structures and 3.11 Å for the weak P4 adsorption site. This effect may be influenced by the electronegativity difference between O–H and O–Mg atoms. The O–H electronegativity difference is smaller than that between O–Mg, leading to weaker Eads. For all the cases, CO2 intramolecular bond lengths are maintained as before the adsorption (Fig. S1c, ESI†) except for minor alterations (±0.02 Å) in P2 and P7 structures. The angle, φO1–C–O2, shows negligible bending. The bending molecule with the largest deviation from the CO2 linear isolated molecule (179.97°, Fig. S1c, ESI†) is found in the P3 structure (168.38°). After DFT geometric optimisation, the CO2 molecule on sites P1 and P6 underwent a reorientation, and it moved to the P2 adsorption site and showed adsorption energies (−0.38 and −0.40, respectively), molecule–surface distance (2.34 and 2.26 Å, respectively), and geometrical parameters similar to the P2 structure (Eads = −0.41 eV and rCO2-slab = 2.09 Å). In this adsorption position, the C atom of the CO2 is located in a small cavity located on top of a Mg atom on the second layer on the surface, and in the middle of two H atoms on the surface. The two O atoms of the CO2 molecule point to the Mg surface atoms perpendicular to the H atoms. The final geometry of CO2 at P7 is also equivalent to the P2 adsorption site, but in this case, the CO2 molecule is not fully parallel to the surface, and the binding energy is slightly larger (−0.43 eV). Therefore, P2, P3, and P5 were found to be the most active sites for CO2 adsorption. Furthermore, CO2 molecules starting at P3 and P5 evolve to a final position, where the C atoms are placed between two H atoms on the surface and the O atoms point again toward two Mg atoms on the surface; this CO2 is, however, placed over the interatomic cavity (in contrast to P1, P2, P6, and P7 structures). For the latter, binding energies, CO2–surface distances, and CO2 geometric parameters are similar to those reported in previous systems, where the shortest CO2–surface distance was found for the P3 structure (i.e., 2.00 Å). Based on the binding energy strengths, the adsorption distances, and negligible changes in CO2 geometry, physical adsorption of the CO2 molecule on the MgH2 surface can be inferred for all the proposed adsorption sites. Results agree with those reported by Chen et al. who explained the viability of CO2 hydrogenation through ionic lattice hydrogen atoms.31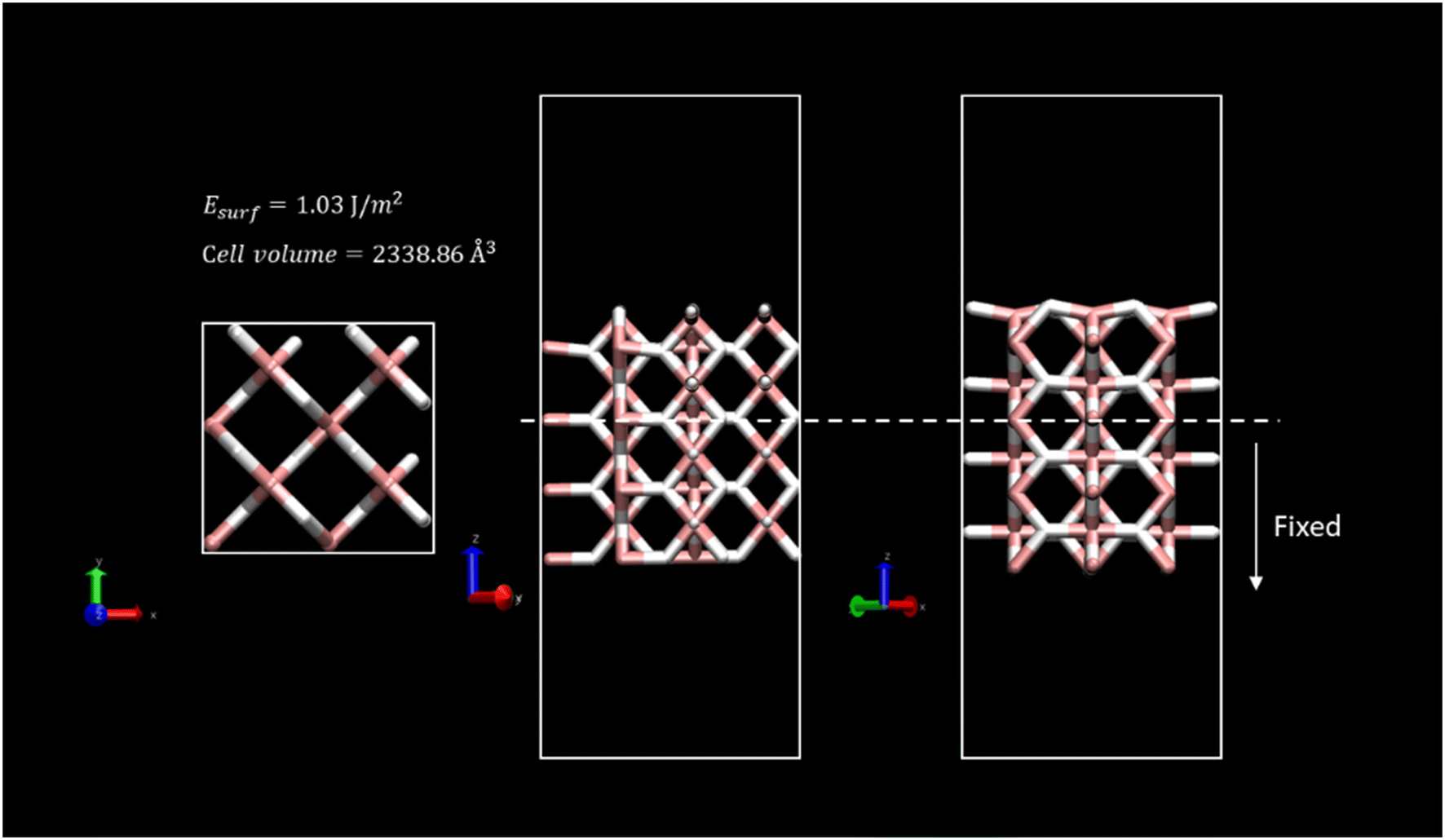 | ||
| Fig. 1 Top- and two-side-view representations of the system used in the simulations. The (0 0 1) surface energy and total volume of the optimized slab after calculation are depicted. Atom colour code: (white) hydrogen, (pink) magnesium. | ||
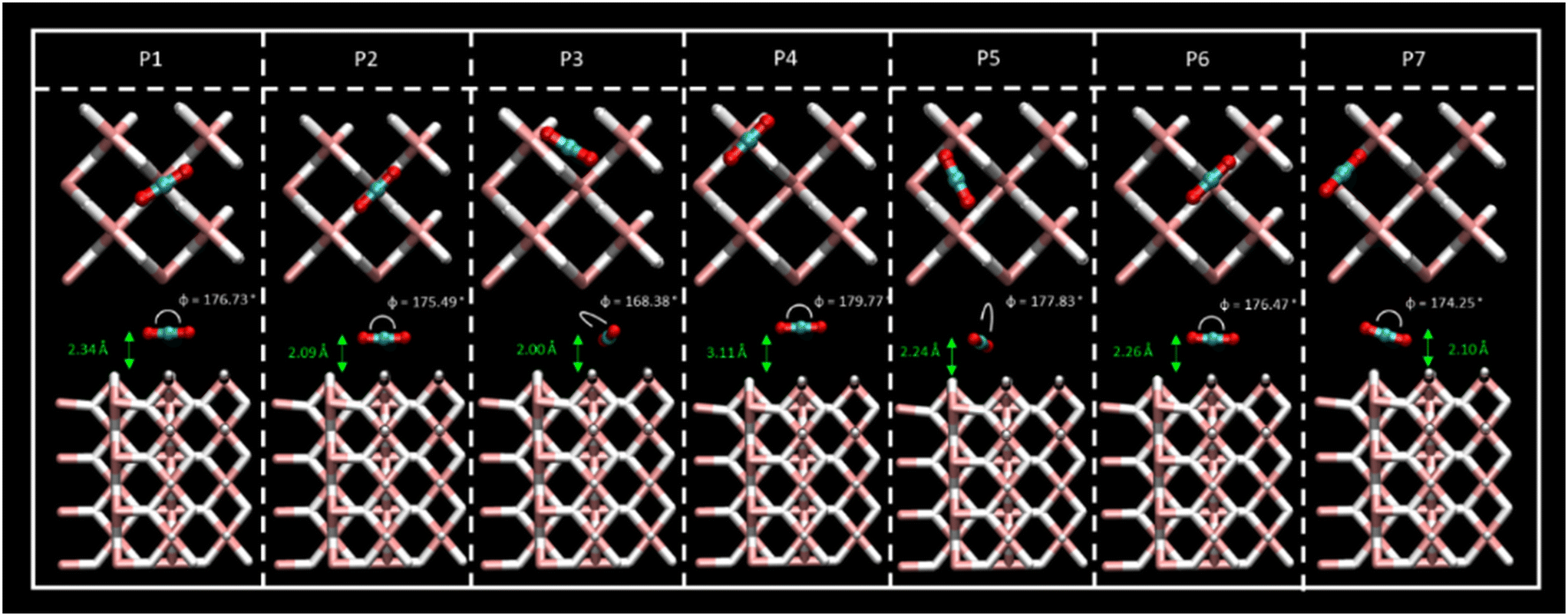 | ||
| Fig. 2 Top- and side-view of the CO2 adsorption structures for the P1–P7 sites, where CO2 bending angles and CO2–slab distances are shown. Atom colour code: (white) hydrogen, (pink) magnesium, (red) oxygen, (blue) carbon. | ||
| E ads (eV) | r C–O1 (Å) | r C–O2 (Å) | φ CO2 (deg) | r CO2-slab (Å) | |
|---|---|---|---|---|---|
| P1 | −0.38 | 1.17 | 1.17 | 176.73 | 2.34 |
| P2 | −0.41 | 1.17 | 1.17 | 175.49 | 2.09 |
| P3 | −0.38 | 1.17 | 1.19 | 168.38 | 2.01 |
| P4 | −0.20 | 1.17 | 1.17 | 179.77 | 3.11 |
| P5 | −0.39 | 1.17 | 1.17 | 177.83 | 2.24 |
| P6 | −0.40 | 1.17 | 1.17 | 176.46 | 2.26 |
| P7 | −0.43 | 1.16 | 1.17 | 174.25 | 2.10 |
To better understand the adsorption mechanism for CO2 gas, the effect of coverage of different CO2 molecules was considered. The full coverage limit (1 ML) was defined as 4 CO2 molecules (12 atoms) on top of 12 slab surface atoms (8 hydrogen + 4 magnesium atoms). Hence, an increasing number of CO2 molecules corresponding to 0.25 ML, 0.5 ML, 0.75 ML, 1 ML, and saturated 1.25 ML were added to the MgH2 surface (1, 2, 3, 4, and 5 molecules, respectively) according to Fig. S1b, ESI.† Surface saturation was used to evaluate the eventual reorientation of the CO2 molecules on the MgH2 surface. Optimized structures are shown in Fig. S2, ESI,† and the adsorption energies (per CO2 molecule) and geometric properties of each of the adsorbed CO2 are reported in Table 2. An increasing affinity of CO2 molecules to the MgH2 surface is inferred whenever enlarging surface coverage as Eads scale from −0.3 to −0.4 eV (2 to 4 adsorbed molecules). This means that each molecule adsorbs more easily with an increase in coverage. Moreover, Eads values agree with the ones reported for a single CO2 molecule (P1, P2, P3, P5, P6, and P7), leading to efficient adsorption mechanisms upon coverage. However, this behaviour is not reproduced by CO2 exceeding coverage structure (1.25 ML), where the Eads drops to −0.25 eV (per CO2 molecule). The lowering of the adsorption efficiency might be caused by the competing effects of the surface adsorption and the CO2 intermolecular interactions imposed by the structural design constraints together with the non-availability of the most suitable surface adsorption sites (P1–P3 and P5–P7), and the surplus molecules interact at a less energetically favoured site (P4). Regarding the orientation of the adsorbed molecules, despite the initial position of the CO2 being parallel to the surface, molecules tend to reorient after geometric optimisation toward quasi-perpendicular arrangements. This is more notable with the coverage effect. In contrast to adsorption structures with single CO2 molecules, CO2 bending angles and bond distances for all the structures vary upon adsorption (Table 2) and with CO2 addition. CO2 (1) molecules located on the strongest adsorption site, i.e., P2, especially deform when coverage is increased, reaching a bending angle of 123.94°, bond distances of 1.28 Å, 1.24 Å, and 1.06 Å distance to the MgH2 surface hydrogens, when n = 5 in the coverage (Fig. 3a). At this adsorption site, slab deformation is also noticeable, especially in the ω2 angle, which changes from 157.78° in the clean slab to 149.94° after the interaction (Table S1, ESI†). Indeed, it can be inferred that a weak H-bond might be present between H4 (MgH2 surface) and the C atom of CO2 (1) because of the bond distance (1.06 Å) and interacting angle (140.61°), although electronic charge transfer could also give rise to an adsorption mechanism that also explains chemical adsorption (Fig. 3b). A comparable situation is observed at the opposite side: MgH2 H5 atom with a C–H length of 2.41 Å and contact angle of 134.34°; a slab deformation of ω3 changing from 157.78° to 174.35° was observed in this case (Table S1, ESI†). Alternate orientation of CO2 molecules forming spatially perpendicular entities (non-parallel arrangement) were observed. This conformation may be adopted to avoid CO2 quadrupole–quadrupole interactions, benefiting CO2-slab interactions.36 The reported effect, i.e., a stronger Eads when coverage with n CO2 molecules increases, has already been described in previous literature for the CO2 adsorption on rutile TiO2 (1 1 0) sorbent.37 This phenomenon will be further analysed in an upcoming section using the charge transfer phenomenon.
| n CO2 | E ads (eV) | r C–O1 (Å) | r C–O2 (Å) | φ CO2 (deg) | r CO2–H (Å) | |
|---|---|---|---|---|---|---|
| 2 × CO2 | 1 | −0.30 | 1.21 | 1.19 | 155.75 | 1.97 |
| 2 | 1.16 | 1.17 | 179.18 | |||
| 3 × CO2 | 1 | −0.36 | 1.28 | 1.23 | 131.50 | 1.18 |
| 2 | 1.16 | 1.17 | 177.39 | |||
| 3 | 1.19 | 1.16 | 177.54 | |||
| 4 × CO 2 | 1 | −0.40 | 1.26 | 1.23 | 130.91 | 1.17 |
| 2 | 1.16 | 1.18 | 174.02 | |||
| 3 | 1.18 | 1.17 | 171.8 | |||
| 4 | 1.16 | 1.17 | 178.15 | |||
| 5 × CO2 | 1 | −0.25 | 1.28 | 1.25 | 123.94 | 1.06 |
| 2 | 1.17 | 1.18 | 174.76 | |||
| 3 | 1.20 | 1.17 | 175.40 | |||
| 4 | 1.18 | 1.17 | 173.54 | |||
| 5 | 1.17 | 1.17 | 173.20 |
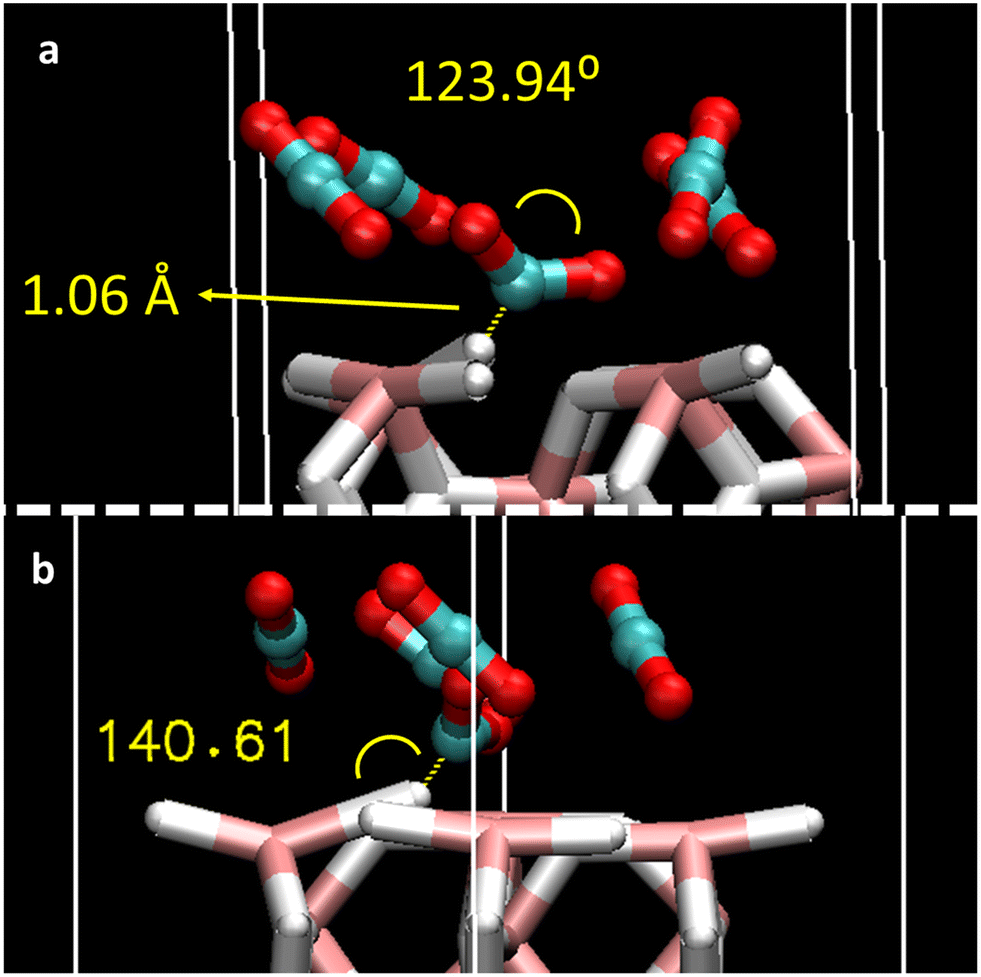 | ||
| Fig. 3 Details of the geometric parameters of (a) adsorbed CO2 bending angle and C–H4 distance and (b) Mg2–H4–C contact angle related to the CO2 (n = 1) molecule behaviour in the structure of 5 adsorbed CO2 molecules. | ||
In summary, although the P7 position is the most stable adsorption site, adsorption energies ranging from −0.38 to −0.43 eV (except for the P4 position) indicate that the MgH2 causes suitable physical adsorption of CO2 molecules. The coverage effect enhances CO2–MgH2 surface interactions until full coverage is achieved. CO2 molecular structure distortion and slab deformation upon CO2 adsorption unveil possible chemical interaction between the CO2 gas molecules and the MgH2 sorbent. Hence, activation of the CO2 molecule is demonstrated by the bending angle and bond distances of molecules and the CO2–MgH2 slab adsorption distances.
The CO2 adsorption capacity of MgH2 has been compared with that of diverse sorbent materials in the literature (Table 3). Clean MgH2 slab surface shows medium to strong affinity for CO2 molecules, and therefore, noteworthy CO2 adsorption capacity. However, the adsorption capacities of MgH2 are far from those provided by other kinds of materials, such as decorated metal–organic frameworks38 (MOFs), doped monolayers,39 γ-Al2O3 (1 0 0),40 or CuO2 (110),41 as depicted in Table 3. Binding energies for the adsorption of a single CO2 molecule on different materials reported in the literature, along with the DFT functional employed here, are given in Table 3.
| Methodology | Material | E ads (eV) | Ref. |
|---|---|---|---|
| DFT-D3, PBE | MgH 2 | −0.41 | This work |
| DFT, PBE | MgH2 | −0.09 | 20 |
| DFT-D3, PBE | Calcite (1 0 4) | −0.38 | 42 |
| DFT, B3LYP | AlN monolayer | −0.15 | 43 |
| DFT, PBE | TiO2 Brookite (2 1 0) | −0.16 | 44 |
| DFT, PBE | TiO2 Anatase (1 0 1) | −0.20 | 45 |
| DFT, PBE | γ-Al2O3 (1 1 0) | −0.43 | 46 |
| DFT, PBE | γ-Al2O3 (1 0 0) | −0.80 | 32 |
| DFT-D2, PBE | Ni (1 1 0) | −0.29 | 47 |
| DFT-TS, PBE | TiO2 anatase (1 0 1) | −0.42 | 48 |
| DFT, PBE | CeO2 (1 1 0) | −0.24 | 49 |
| DFT, PBE | CuO2 (1 1 0) | −1.22 | 50 |
| DFT-vdW-DF2, PBE | V-MOF-74s | −0.55 | 30 |
| DFT-D, PBE | MoS2@GeSe monolayer | −0.96 | 31 |
| DFT-D, PBE | TiO2 rutile (1 1 0) | −0.09 | 29 |
| DFT-D, PBE | BC3 monolayer | −0.11 | 51 |
| DFT-D, PBE | Cu-MOF | −0.82 | 52 |
2.2 Co-doped MgH2
To improve CO2 affinity to MgH2 sorbent, the Co single-atom catalyst (SAC) and Co cluster have been proposed as catalysts for CO2 adsorption.33 Adsorption energies for the different interacting sites of SAC (Fig. S1, ESI†) and Co–surface distances are reported in Table 4. For the 5 possible adsorption sites considered along the MgH2 surface, C1 and C2 structures showed larger Eads values (−6.47 and −7.93 eV, respectively) than C3, C4, and C5 (−3.55 to −4.49 eV) (Table 4). Regarding geometry, the initial Co SAC position on C1 converged to the C2 position of Co as inferred through geometric parameters reported in Fig. 4 and Table 4. Thus, C1 and C2 sites can be considered equivalent, although the greater stability of the C2 SAC structure should be noted, which reflects the Co atom embracement by H1 and H2 atoms of the MgH2 surface (Fig. 4). Despite the C1 Co–surface distances being very similar to those in C2 (Table 4), ω2 and ω3 surface angles deformation (Table S1, ESI†) from 157.78 to 132.37° and 133.91°, respectively, manifest the insertion of the Co atom in the MgH2 surface structure. In contrast, for the C1 structure, ω2 and ω3 surface angles present negligible deformation (from 157.78 to 158.00° and 160.82° respectively) (Table S1, ESI†). For this reason, the C2 Co SAC structure was considered in the following section for CO2 adsorption studies.| E ads (eV) | r Co–H1 (Å) | r Co–H2 (Å) | |
|---|---|---|---|
| C1 | −6.47 | 1.52 | 1.50 |
| C2 | −7.93 | 1.57 | 1.57 |
| C3 | −4.49 | 2.34 | 2.17 |
| C4 | −3.55 | 1.56 | 1.56 |
| C5 | −4.49 | 1.56 | 1.56 |
 | ||
| Fig. 4 Top- and side-view of the Co SAC structures for the C1–C5 sites and Co–Mg distances. Atom colour code: (white) hydrogen, (pink) magnesium, (green) cobalt. | ||
When considering multiple Co atom catalysts, stronger adsorption energies were observed on MgH2 compared to SAC. Eads (per Co atom) range from −3.09 to −10.55 eV and from −7.18 to −9.43 eV for one-by-one Co atom addition and 1/4 to full surface coverage, respectively (Tables S2 and S3, ESI†). Interatomic Co distances, which vary from 2.16 to 4.15 Å, for the one-by-one clusterization and full coverage systems (Tables S4 and S5, ESI,† respectively), agree with Co clusterization atom distances reported in the literature.53 Catalyst adsorption on the MgH2 surface causes geometric disruption of the first and second layers of MgH2, and the Co embracement effect observed for the SAC structures is also reproduced for the structures with multiple Co atoms (Fig. S3 and S4, ESI†). A structure with seven Co atoms is the most energetically stable system (−10.55 eV) for n Co clusterization on catalyzed MgH2. The geometric parameters (Table S1, ESI†) of the converged structure indicate that H4 and H5 atoms were displaced along the x-direction (Fig. S3, ESI†) and closely (1.64 and 1.62 Å) interacted with the central Co atom (Fig. S5, ESI†), while Mg2–H4 and Mg3–H5 distances extended from 1.82 Å to 2.05 and 2.51 Å, respectively (Table S1, ESI†). For the structure with full coverage of Co (4/4, Fig. S4, ESI†), the Eads were slightly weaker (−9.43 eV) than that for the cluster with 7 Co atoms (Table S3, ESI†). However, the geometric disorder of the first MgH2 layer is larger, exhibiting considerable displacement of atoms and disruption of the H–Mg–H angles (Table S1, ESI†), triggering initial structural decomposition and Co atoms integration on the MgH2 solid surface.
For all the reported Co catalyzing structures, large adsorption energies and non-negligible changes in surface structure were found. Therefore, the Co catalyst is intended to be capable of weakening substrate bond forces, and thus, the mechanisms of interaction between CO2 molecule and MgH2 surface are expected to be enhanced.
2.3 CO2 adsorption on Co/MgH2
In this section, CO2 and CO2 + 2/4 H2 adsorption on Co/MgH2 systems are discussed. First, one CO2 molecule adsorption on Co SAC/MgH2 was analyzed for all the SAC structures reported in the previous section. In the case of C1 and C2 structures, only the C2 SAC position was considered as Co SAC spatial location is equivalent, and the energetic stability is stronger. Both the dispositions of the CO2 molecule considered initially, i.e., parallel and perpendicular to the surface on top of the SAC atom, manifested large CO2Eads in the range of −6.61 to −12.43 eV (Table 5). In the case of the C2 adsorption site, CO2 adsorption energy for the parallel disposition became smaller compared to the perpendicular disposition. However, for C3, C4, and C5 structures, parallel CO2 disposition showed larger interacting energies with respect to the surface. In general, larger adsorption energies (more negative values) rely on stronger interactions and greater disruption of CO2 geometric parameters (Table 5). Whereas in CO2 perpendicular disposition, no changes were noticed in the geometry of the gas molecules, while significant structural changes were found for the parallel disposition molecule, except for CO2 adsorption on the C2 structure. CO2–Co SAC distances are similar for all the structures (1.81 to 1.98 Å). The C3, C4, and C5 structures featured intense disruption of the CO2 bending angles (134.94 to 140.19°) (Table 5). Therefore, despite detecting C1 and C2 as the most stable SAC structures, CO2 adsorption turned out to be biased for C3, C4, and C5 (in that order), as depicted through reported Eads (Table 5). Adsorption on the C3 structure provides stronger interaction energy (−12.43 eV), but also the most disordered MgH2 first layer.| E ads (eV) | r C–O1 (Å) | r C–O2 (Å) | φ CO2 (deg) | r C–Co (Å) | r O1–Co (Å) | |
|---|---|---|---|---|---|---|
| C2 CO2 ∥ | −6.61 | 1.20 | 1.20 | 180 | 1.98 | |
| C2 CO2 ⊥ | −9.71 | 1.18 | 1.19 | 179.66 | 1.93 | |
| C3 CO 2 ∥ | −12.43 | 1.27 | 1.27 | 136.99 | 1.82 | |
| C3 CO2 ⊥ | −8.86 | 1.19 | 1.18 | 178.23 | 1.85 | |
| C4 CO2 ∥ | −10.17 | 1.23 | 1.29 | 140.19 | 1.81 | |
| C4 CO2 ⊥ | −9.38 | 1.18 | 1.19 | 179.37 | 1.94 | |
| C5 CO2 ∥ | −8.42 | 1.28 | 1.24 | 134.94 | 1.98 | |
| C5 CO2 ⊥ | −7.98 | 1.17 | 1.18 | 179.85 | 3.24 |
Two adsorption mechanisms are inferred from this analysis: the first one and the most likely to occur (stronger interacting energy) is adsorption through the C atom of the CO2 molecules. The second one, which is energetically weaker, is adsorption via the O atom with a molecular disposition perpendicular to the surface (Fig. S6, ESI†). From the former mechanism, chemical adsorption manifestation is proposed to occur based on the CO2 and MgH2 surface structure perturbation (Table S1, ESI†), whilst physisorption is inferred for the latter adsorption mechanism. Hence, CO2 chemical activation through carbon atoms is observed as an effect that should be attributed to the Co catalyst.
CO2 adsorption on a catalyst with two Co atoms on the MgH2 surface presented much weaker adsorption energies for both parallel and perpendicular disposition of the gas molecule (−2.71 and −2.69 eV, respectively), which ended up converging in the same geometrical position (Fig. S7, ESI†). Although the distortion in the CO2 molecular structure in the two-Co structure is slightly stronger (Table 6), the adsorption mechanisms of both the SAC and two-Co catalyst seem to behave similarly. Moderate surface deformation was observed (Table S1, ESI†).
| E ads (eV) | r C–O1 (Å) | r C–O2 (Å) | φ CO2 (deg) | r C–Co1 (Å) | r C–Co2 (Å) | r Co1–Co2 (Å) | |
|---|---|---|---|---|---|---|---|
| Co SAC/C3 | |||||||
| CO 2 ∥ | −12.43 | 1.27 | 1.27 | 136.99 | 1.82 | ||
| CO2 ⊥ | −8.86 | 1.19 | 1.18 | 178.23 | 1.85 | ||
| 2 Co | |||||||
|---|---|---|---|---|---|---|---|
| CO 2 ∥ | −2.71 | 1.24 | 1.34 | 129.37 | 1.84 | 2.24 | 2.71 |
| CO2 ⊥ | −2.69 | 1.25 | 1.33 | 130.59 | 1.83 | 2.2 | 2.81 |
Further investigation was carried out on the CO2 adsorption mechanism on MgH2 by taking into consideration H2 molecules that mimic H2 release from a distinct hydrogen storage substrate. Thus, 4 and 2 H2 molecules, according to different experimentally reported methanation mechanisms (Sabatier process and direct CO2 reduction),26 were considered, along with one CO2 molecule for the C2 SAC and two Co atoms catalyzing MgH2 structures. Geometrically optimized systems displayed in Fig. S8 (ESI†) yield adsorption energies in the range of −2.61 to −3.43 eV with similar CO2 molecule distortion geometries (Table 7). CO2 molecule deformation is found to be in line with the previously reported adsorbed molecules, whereas H2 molecules do not interact within the system as no structural changes occur on the molecules themselves and there are no significant interactions that could be observed within the MgH2 surface or the CO2 molecule either (Fig. S8, ESI†). It might be highlighted that for the C2 SAC structure interaction with 1 CO2 and 4 H2 molecules, the CO2 molecule is turned parallel to the surface leading to CO2–MgH2 full contact, in contrast to the remaining structures (Fig. S8, ESI†). Unremarkable slab surface deformation was observed for these structures (Table S1, ESI†).
| E ads (eV) | r C–O1 (Å) | r C–O2 (Å) | φ CO2 (deg) | r C–Co1 (Å) | r C–Co2 (Å) | r Co1–Co2 (Å) | r H1–H2 (Å) | |
|---|---|---|---|---|---|---|---|---|
| Co SAC/C2 | ||||||||
| 1 CO2 + 2 H2 | −2.61 | 1.26 | 1.26 | 136.72 | 1.83 | — | — | 0.75 |
| 1 CO 2 + 4 H 2 | −3.43 | 1.32 | 1.25 | 129.73 | 1.80 | — | — | 0.75 |
| 2 Co | ||||||||
|---|---|---|---|---|---|---|---|---|
| 1 CO2 + 2 H2 | −3.10 | 1.28 | 1.28 | 136.36 | 1.90 | 1.90 | 2.66 | 0.75 |
| 1 CO 2 + 4 H 2 | −3.43 | 1.28 | 1.29 | 136.36 | 1.90 | 1.89 | 2.66 | 0.75 |
2.4 Insights into the charge transfer analysis and adsorption mechanisms
Charge density difference – or spatial charge redistribution – maps for the most stable aforementioned systems are shown in Fig. 5. Panel (a) represents the charge density for the clean slab; panels (b) to (d) depict the charge density difference of carbon dioxide on MgH2; panels (e) to (f) show the charge density difference of Co atoms on MgH2; and panels (h) to (k) show the charge density difference of CO2 and H2 on Co/MgH2. Blue and yellow surfaces represent charge density depletion and accumulation, respectively. The difference plot slices prepared for complementary charge density in relevant regions of the system aid in comprehending the charge density transfer effect (Fig. 6), where red and blue colours represent maximum and minimum charge density accumulation, respectively. In panels (b) to (d) of Fig. 5, charge accumulation around oxygen and surface hydrogen atoms is outlined, whereas atomic charge depletion is observed for central carbon atoms, justifying the physisorption effect. Moderate charge transfer between CO2 molecules and MgH2 is shown, except for the CO2 (2) molecules in panel (d), Fig. 5. Contrarily, large charge density transfer can be observed along with Co adsorption on the MgH2 surface. The chemisorption of Co SAC on the MgH2 surface is confirmed through strong charge interactions. An analogous effect is observed for the subsequent addition of Co atoms (Fig. 5f and g). More complex charge transfer patterns are observed in panels h to k. Combining panels (h) to (k) of Fig. 5 and 6, we can observe a double charge transfer effect: first, from the Co atoms adsorbed to the H atoms on the MgH2 surface, and second, from the carbon of the CO2 molecule adsorbed to the Co catalyst atoms. Hydrogen molecules do not seem to interchange charge density either with catalyzing surface or CO2 molecules as no surfaces are associated with H2 molecules.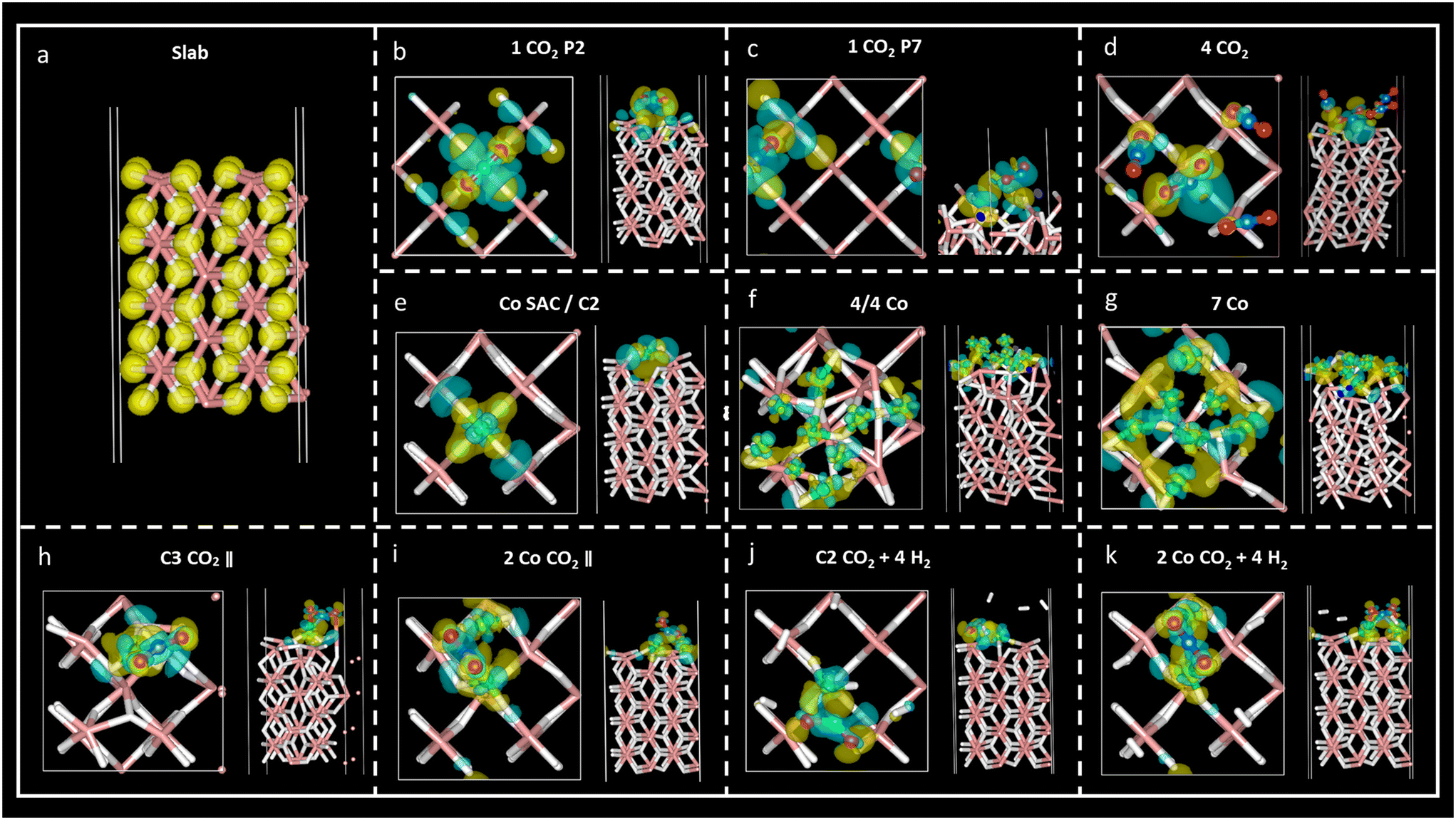 | ||
| Fig. 5 Charge density difference maps (ρsystem − ρslab − ρCo − ρgas) for the most stable structures (a–k) with the isosurface value of 0.005e. The yellow region represents charge density accumulation, while the blue region represents charge depletion. | ||
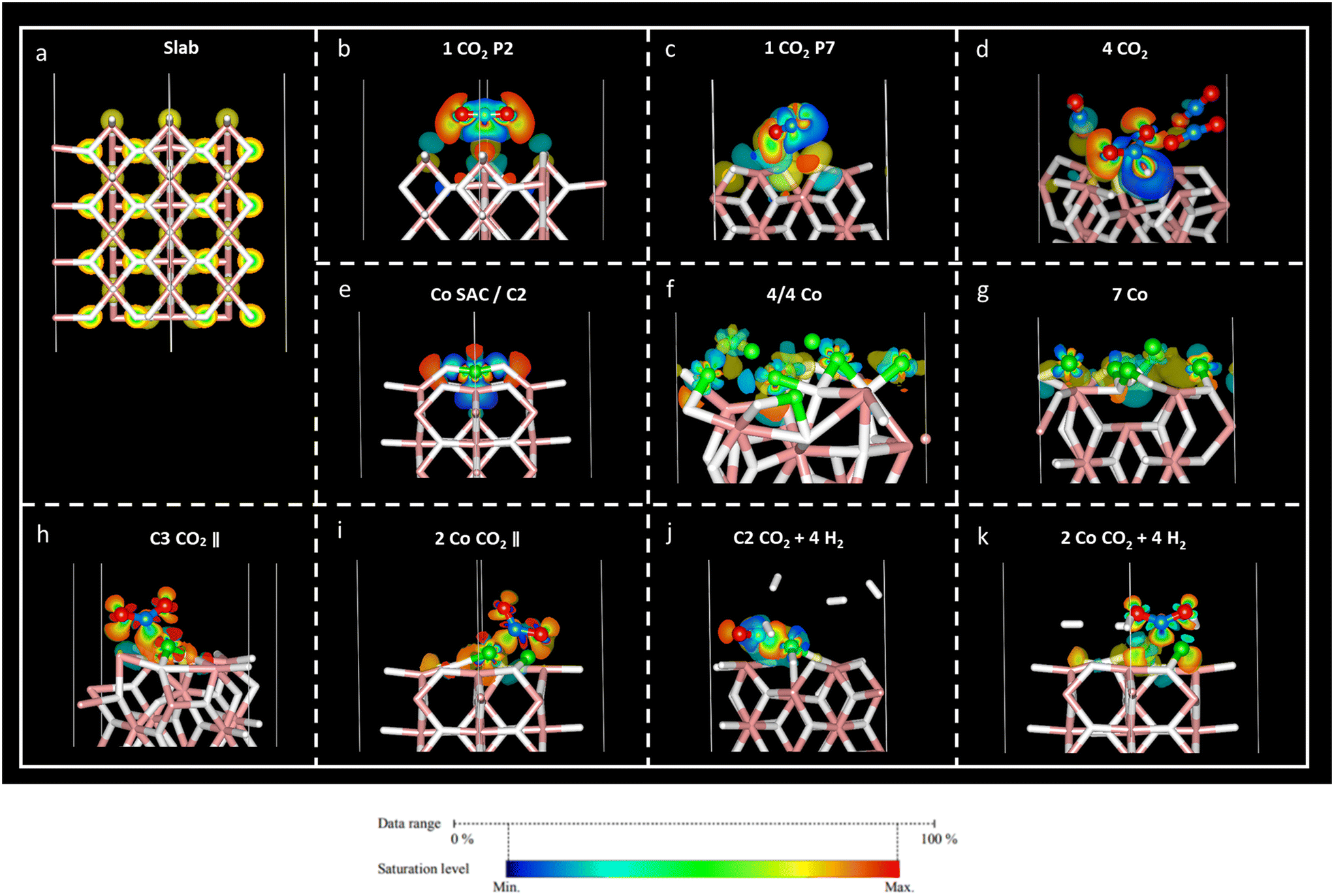 | ||
| Fig. 6 Charge density difference slice for the most stable structures (a–k) considering an isosurface value of 0.005e. Red-like colours represent charge density accumulation, while the region leaning to blue shades represents charge depletion. | ||
Lastly, to gain insight into the CO2 adsorption mechanisms, charge density transfer was investigated via valence Bader charges analysis. Valence electronic charges for CO2 and H2 molecules, first and second layers of MgH2 and Co atoms were considered before and after the adsorption of the corresponding atoms/molecules (Tables S6–S8, ESI†). Next, charge transfer was calculated as the difference between the isolated surface/CO2 molecule/Co atom and the valence Bader charges for the overall systems of the CO2 molecule adsorbed on a clean MgH2 surface, Co atom adsorbed on a clean MgH2 surface, and CO2/H2 adsorbed on the catalysing systems (Tables S8–S10, ESI,† respectively). Positive and negative charge differences indicate charge uptake and charge loss, respectively. This means that an atom with a positive charge difference has electronic charge accumulation (negative ionic charge). Given the reported results, minor charge differences between CO2 atoms and MgH2 surface atoms confirm the physisorption mechanism at P2 and P7 sites (Table S9, ESI†). However, a significant charge difference was found within the CO2 atoms: uptake of electronic charge by O atoms (+0.76 to +0.85) from the central C atom (which losses −1.51 and −1.67, respectively), favouring weak van der Waals interactions with the hydrogens on the MgH2 surface (Table 1).
In the case of the four-CO2-molecules system, physical adsorption could be inferred for CO2 (1), CO2 (3), and CO2 (4) molecules on MgH2, but chemisorption was confirmed for the CO2 (2) molecule as significant charge transfer was observed between the H4 and C1 (2) atoms (Table S9, ESI†). This result is validated by the previously observed bending angle of the gas molecule, as well as adsorption distance (Table 2) and accumulation/depletion charge density map (Fig. 5d).
The charge difference reported for Co atoms on MgH2 confirms strong interactions between the sorbent and selected catalyst as a large charge transfer was found between the hydrogen atoms on the surface and Co atoms (Table S11, ESI†). Therefore, strong adsorption of Co atoms on the MgH2 surface was also corroborated by large adsorption energies and short interatomic distances (Tables 4, S2, and S3, ESI†). The charge difference of Co4, Co5, and Co9 (Co atoms on the top of the surface H atoms), as well as Co4, for seven Co and 4/4 systems, respectively, are particularly pronounced according to charge density maps (Fig. 5f and g).
Finally, the CO2 and H2 adsorption on the catalysis systems depicted moderate charge transfer between the MgH2 surface and Co atoms combined with CO2 adsorption on top of the Co atoms. In these structures, partial charge transfer from the C central CO2 atom developed toward the O atoms, in parallel to partial charge transfer to the Co catalyst, leading to large ionic charge accumulation on the Co atoms. Thus, strong interactive forces between the CO2 gas molecules and the catalysis surface are confirmed. Along with large Eads and short adsorption distances, these findings corroborate the chemical nature of the adsorption mechanism. Additionally, considerable charge density is distributed along the CO2 oxygen atoms, which may lead to possible interactive sites for H2 adhesion. However, despite that, low reactivity was found for H2 molecules according to the reported charge transfer and charge density plots.
Based on the reported results, two main conclusions can be reached: i) high CO2 adsorption capacity of MgH2 was confirmed with a preferential physisorption mechanism for the clean surface and a chemisorption route for the Co-catalysed surface, and ii) no interaction could be detected between the adsorbed CO2 and H2 molecules.
3 Conclusions
In this theoretical inquiry, we systematically investigated the properties and mechanisms governing carbon dioxide (CO2) adsorption on magnesium hydride (MgH2) surfaces augmented with Co atoms. The focal point of our analysis was an unaltered MgH2 surface and the impact of Co species on the CO2 adsorption process. The findings unveiled the physical manifestation of the adsorption phenomena on the MgH2 surface, whereby disparate adsorption sites are present indicating uniform adsorption energies. The influence of CO2 molecular coverage on MgH2 demonstrated increased affinity at enhanced surface coverage, reaching an optimal saturation point after which excessive coverage occurred and induced a decline in adsorption efficiency. Following adsorption, the orientational configuration of CO2 molecules transitioned from parallel to quasi-perpendicular arrangements, accompanied by discernible deformations at heightened coverage.In the context of comparative analyses with alternative materials, MgH2 emerged as a noteworthy medium with a strong affinity for CO2. Furthermore, our scrutiny of the efficacy of Co single atoms or clusters for CO2 adsorption on MgH2 indicated the superiority of Co single atoms for adsorption at specific sites that were reflected in heightened adsorption energies. This mechanism entails the activation of CO2 molecules through perturbation of molecular properties, characterisation of CO2–MgH2 slab interactions, and facilitation of electronic charge transfer.
These observations collectively contribute to a deep comprehension of the underlying properties and mechanisms of CO2 adsorption on MgH2 in the presence of Co. Importantly, they provide pivotal insights into the initial reaction step, thereby guiding the rational design of more efficient CO2 conversion technologies.
4 Computational methods
Quantum Espresso 6.5 software54 with the PW package was used for DFT spin-polarised calculations. For all the atoms involved in simulations, projector augmented wave (PAW) pseudopotentials were used,55,56 together with the revised version57 of the PBE exchange–correlation functional.50,58,59 Periodic boundary conditions (PBC) were applied, and dispersion forces were also included via the semi-empirical correction by Grimme, DFT-D3.60 The Burai graphical user interface of Quantum Espresso was used to build the systems. The DMol3 package from the BIOVIA materials studio environment was also employed for geometry pre-optimisation and initial systems equilibration (DFT-PBE).Magnesium hydride is characterized by a tetragonal crystal system with a P42/mnm space group (α-MgH2).61,62 Experimental lattice parameters are a = b = 4.512 Å and c = 3.016 Å.63 From this data, bulk lattice was relaxed with a k-point Monkhorst–Pack grid64 of 12 × 12 × 12, which was converged before considering production runs (see Table S12 and Fig. S9, ESI†) to allow the necessary energy accuracy, along with a cut-off energy of 40 Ry (544.23 eV), according to the largest value recommended in the employed pseudopotentials.
The new lattice parameters of the relaxed system were a = b = 4.503 Å and c = 3.01 Å. To mimic the bulk crystal, a 2 × 2 supercell with 4 layers of depth was generated. A surface with (0 0 1) Miller index was considered for the slab model since it is the most suitable surface regarding vacancy formation energies, and thus, the dehydrogenation process.48,65 A twenty-Angstrom vacuum layer was set above the model slab to avoid the interaction of neighbouring MgH2 cells. For all the calculations, two bottom layers of the slab were fixed to maintain crystal behaviour. Fig. 1 shows the overall simulated cell of dimensions, 8.88 × 8.88 × 30.47 Å, enclosing 96 atoms.
Surface energy, Esurf, for the clean MgH2 slab was calculated according to eqn (1) because the symmetry of the two-sided slab guarantees the double counting of the surface energy. Ebulk is the energy of the bulk MgH2, and Aslab is the surface area of a side of the calculated supercell slab model.
 | (1) |
Once the Esurf of the clean MgH2 slab and the energy ECO2 of the isolated CO2 molecule were computed, CO2 adsorption energy (Eads) on non-catalyzing systems was also calculated for different positions of the carbon dioxide molecule (P1–P7) as well as for different numbers of the CO2 molecules (n = 1–5) according to eqn (2):
| Eads = (Esurf+nCO2 − Esurf − nECO2)/n | (2) |
| Eint Co/MgH2 = (Esurf+nCo − Esurf − nECo)/n | (3) |
Accordingly, CO2 adsorption energy (Eads) on the catalyzing systems was evaluated for different dispositions of one CO2 molecule (parallel and perpendicular arrangement with respect to the MgH2 surface) and for one molecule of CO2 and 2–4 hydrogen molecules (in agreement with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:4 CO2/H2 reaction ratio) to understand interaction mechanisms. One Co SAC and two Co-catalyzed MgH2 systems were considered in this section. eqn (4) describes CO2 or CO2 + 2/4 H2 adsorption energies:
:2 and 1:4 CO2/H2 reaction ratio) to understand interaction mechanisms. One Co SAC and two Co-catalyzed MgH2 systems were considered in this section. eqn (4) describes CO2 or CO2 + 2/4 H2 adsorption energies:
| Eads = EnCo/MgH2+nCO2+nH2 − EnCo/MgH2 − nECO2 − nEH2 | (4) |
For the systems in which more than one carbon dioxide molecule or hydrogen molecule is considered, the calculated adsorption energy was divided by the number of molecules to estimate the adsorption energy per molecule. In Fig. S1, ESI,† top view of the considered positions for one CO2 molecule (P1–P7, Fig. S1a, ESI†) and 1 to 5 CO2 molecules (Fig. S1b, ESI†) over MgH2 (0 0 1) surface are shown as well as the initial bond distances and bending angle of the optimized carbon dioxide molecule (Fig. S1c, ESI†). Accordingly, SAC positions of Co (C1–C5), Co clusterization (1 Co–9 Co), and Co coverage arrangements (1/4 Co, 2/4 Co, 3/4 Co, and 4/4 Co) are shown in Fig. 1d, S1e and f, ESI,† respectively.
To elucidate the CO2 adsorption mechanism and CO2 + H2 interactions over MgH2 (0 0 1) surface catalyzed and non-catalyzed by Co, charge density differences obtained from electron (pseudo-) charge density quantum espresso plots were evaluated. The final spatial distribution of electronic charge density upon CO2/Co/H2 adsorption was calculated by subtracting the charge densities of the CO2/Co/H2 entity (ρMgH2, ρCo, ρCO2, ρH2, respectively) from the entire final system, ρCo/MgH2+CO2+H2 according to eqn (5):
| ρfinal = ρsys − ρMgH2 − ρCo − ρCO2 − ρH2 | (5) |
VESTA and VMD were used to visualize charge density differences and to evaluate charge transfer results. Bader charges of the valence electrons were also evaluated for the largest energy configurations. For this purpose, the Bader code by the Henkelman group66–69 was employed to identify single atoms inside the system and evaluate their charge density allowing the evaluation of charge transfer phenomena upon adsorption.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This investigation was funded by the CO2 absorbing Materials Project-RISE (CO2MPRISE) which received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant Agreement No. H2020-MSCA-RISE-2016-CO2MPRISE-734873. We also acknowledge SCAYLE (Supercomputación Castilla y León, Spain) for providing supercomputing facilities. The statements made herein are solely the responsibility of the authors.References
- Global Monitoring Laboratory, Earth System Research Laboratories, https://gml.noaa.gov/ccgg/trends/, (accessed July 2023).
- IPCC, Climate Change 2007. The Physical Science Basis, Cambridge Univ. Press, Cambridge, 2007 Search PubMed.
- F. Kazemifar, A review of technologies for carbon capture, sequestration, and utilization: Cost, capacity, and technology readiness, Greenhouse Gases: Sci. Technol., 2022, 12, 200–230 CrossRef CAS.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Müller, Worldwide innovations in the development of carbon capture technologies and the utilization of CO2, Energy Environ. Sci., 2012, 5, 7281–7305 RSC.
- N. S. Sifat and Y. Haseli, A critical review of CO2 capture technologies and prospects for clean power generation, Energies, 2019, 12, 4143 CrossRef CAS.
- A. Sood, A. Thakur and S. M. Ahuja, Recent advancements in ionic liquids-based carbon capture technologies, Chem. Eng. Commun., 2021, 210, 933–954 CrossRef.
- B. Dutcher, M. Fan and A. G. Russell, Amine-based CO2 capture technology development from the beginning of 2013 – A review, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- A. I. Osman, M. Hefny, M. I. A. Abdel Maksoud, A. M. Elgarahy and D. W. Rooney, Recent advances in carbon capture storage and utilization technologies: A review, Environ. Chem. Lett., 2021, 19, 797–849 CrossRef CAS.
- A. Raza, R. Gholami, R. Rezaee, V. Rasouli and M. Rabiei, Significant aspects of carbon capture and storage–A review, Petroleum, 2019, 5, 335–340 CrossRef.
- K. M. K. Yu, I. Curcic, J. Gabriel and S. E. Tsang, Recent advances in CO2 capture and utilization, ChemSusChem, 2008, 1, 893–899 CrossRef CAS PubMed.
- G. Cau, D. Cocco and V. Tola, Performance and cost assessment of Integrated Solar Combined Cycle Systems (ISCCSs) using CO2 as heat transfer fluid, Sol. Energy, 2012, 86, 2985–2985 CrossRef.
- R. C. Weast, CRC handbook of chemistry and physics, CRC Press, Boca Raton, 1973–1974 Search PubMed.
- U. Ulmer, T. Dingle, P. N. Duchesne, R. H. Morris, A. Tavasoli, T. Wood and G. A. Ozin, Fundamentals and applications of photocatalytic CO2 methanation, Nat. Commun., 2019, 10, 3169 CrossRef PubMed.
- Z. Liu, X. Gao, B. Liu, Q. Ma, T. S. Zhao and J. Zhang, Recent advances in thermal catalytic CO2 methanation on hydrotalcite-derived catalysts, Fuel, 2022, 321, 12411 Search PubMed.
- J. Ashok, S. Pati, P. Hongmanoram, Z. Tianxi and C. Junmei, A review of recent catalyst advances in CO2 methanation processes, Catal. Today, 2020, 356, 471–489 CrossRef CAS.
- Z. Lv, H. Du, S. Xu, T. Deng, J. Ruan and C. Qin, Techno-economic analysis on CO2 mitigation by integrated carbon capture and methanation, Appl. Energy, 2024, 355, 122242 CrossRef CAS.
- A. Tripodi, F. Conte and I. Rosetti, Carbon Dioxide Methanation: Design of a Fully Integrated Plant, Energy Fuels, 2020, 34, 7242–7256 CrossRef CAS.
- P. Sabatier and J. B. Senderns, Mouvelles synthese du methane, C. R. Acad. Sci., 1902, 134, 514–516 CAS.
- K. Fakumoto, W. Zhang, R. Mizoguchi, Y. Lin, C. Chai, H. Machida and K. Narinaga, CFD simulation of CO2 methanation through the Sabatier reaction in a shell-and-tube reactor incorporating phase change on the shell side, Fuel, 2023, 349, 128126 CrossRef.
- Q. Hou, X. Yang and J. Zhang, Review on Hydrogen Storage Performance of MgH2: Development and Trends, ChemistrySelect, 2021, 6, 1589–1606 CrossRef CAS.
- E. M. Dematteis, J. Barale, M. Corno, A. Sciullo, M. Baricco and P. Rizzi, Solid-state hydrogen storage systems and the relevance of a gender perspective, Energies, 2021, 14, 6158 CrossRef CAS.
- C. N. C. Hitman, M. A. A. Aziz, A. H. Ruhaimi and M. R. Taib, Magnesium-based alloys for solid-state hydrogen storage applications: A review, Int. J. Hydrogen Energy, 2021, 46, 31067–31083 CrossRef.
- O. Zavorotynska, I. Saldan, S. Hino, T. D. Humphries, S. Deledda and B. C. Hauback, Hydrogen cycling in γ–Mg(BH4)2 with cobalt-based additives, J. Mater. Chem. A, 2015, 3, 6592–6602 RSC.
- I. Saldan, S. Hino, T. D. Humphries, O. Zavorotynska, M. Chong, C. M. Jensen, S. Deledda and B. C. Hauback, Structural changes observed during the reversible hydrogenation of Mg(BH4)2 with Ni-based additives, J. Phys. Chem. C, 2014, 118, 23376–23384 CrossRef CAS.
- J. Cui, J. Liu, H. Wang, L. Ouyang, D. Sun, M. Zhu and X. Yao, Mg-TM (TM: Ti, Nb, V, Co, Mo or Ni) core-shell like nanostructures: Synthesis, hydrogen storage performance and catalytic mechanism, J. Mater. Chem. A, 2014, 2, 9645–9655 RSC.
- M. G. Verón, H. Troiani and F. C. Gennari, Synergetic effect of Co and carbon nanotubes on MgH2 sorption properties, Carbon, 2011, 49, 2413–2423 CrossRef.
- M. Liu, X. Xiao, S. Zhao, M. Chen, J. Mao, B. Luo and L. Chen, Facile synthesis of Co/Pd supported by few-walled carbon nanotubes as an efficient bidirectional catalyst for improving the low temeprature hydrogen storage properties of magnesium hydride, J. Mater. Chem. A, 2019, 7, 5277–5287 RSC.
- M. L. Grasso, L. Fernández Albanesi, S. Garroni, G. Mulas and F. C. Gennari, Methane production by mechanochemical processing of MgH2-Li2CO3 as sources of H2 and CO2 at room temperature, J. CO2 Util., 2020, 40, 101209 CrossRef CAS.
- G. W. Manggada and S. Zhou, Phosphonic acid modification to molybdenum atom catalyst for CO2 hydrogenation on magnesium hydride surface, DISTILAT: Jurnal Teknologi Separasi, 2022, 8, 315–324 CrossRef.
- S. Polierer, J. Jelic, S. Pitter and F. Studt, On the reactivity of the Cu/ZrO2 system for the hydrogenation of CO2 to methanol: A density functional theory study, J. Phys. Chem. C, 2019, 123, 26904–26911 CrossRef CAS.
- H. Chen, P. Liu, J. Li, Y. Wang, C. She, J. Liu, L. Zhang, Q. Yang, S. Zhou and X. Feng, MgH2/CuxO hydrogen storage composite with defect-rich surfaces for carbon dioxide hydrogenation, ACS Appl. Mater. Interfaces, 2019, 11, 31009–31017 CrossRef CAS PubMed.
- V. Iablokow, S. K. Beaumnt, S. Alayoglu, V. V. Pushkarev, C. Specht, J. Gao, A. P. Alivisatos, N. Kruse and G. A. Somorjai, Size-controlled model Co nanoparticle catalyst for CO2 hydrogenation: Synthesis, characterization, and catalytic reactions, Nano Lett., 2012, 12, 3091–3096 CrossRef PubMed.
- G. Zhou, T. Wu, H. Xie and X. Zheng, Effect of structure on the carbon dioxide methanation performance of Co-based catalyst, Int. J. Hydrogen Energy, 2013, 38, 10012–10018 CrossRef CAS.
- A. Morales-García, F. Viñes, J. R. B. Gomes and F. Illas, Concepts, models, and methods in computational heterogeneous catalysis illustrated through CO2 conversion, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11(4), e1530 Search PubMed.
- M. L. Grasso, S. Rozas, P. A. Larochette, S. Aparicio and F. Gennari, Thermochemical transformation of CO2 for CH4 production using metal hydride and carbonates as portable sources, Int. J. Hydrogen Energy, 2023, 48, 21835–24849 CrossRef CAS.
- A. J. Stone, The theory of intermolecular forces, Oxford University Press, New York, 1997 Search PubMed.
- C. Sorescu, J. Lee, W. A. Al-Saidi and K. D. Jordan, CO2 adsorption on TiO2(110) rutile: Insight from dispersion-corrected density functional theory calculations and scanning tunneling microscopy experiments, J. Chem. Phys., 2011, 134, 104707 CrossRef PubMed.
- K. Lee, J. D. Howe, L.-C. Lin, B. Smith and J. B. Neaton, Small-molecule adsorption in open-site metal–organic frameworks: A systematic density functional theory study for rational design, Chem. Mater., 2015, 27, 668–678 CrossRef CAS.
- H. Yang, C. He, L. Fu, J. Huo, C. Zhao, X. Li and Y. Song, Capture and separation of CO2 on BC3 nanosheets: A DFT study, Chin. Chem. Lett., 2021, 32, 3202–3206 CrossRef CAS.
- Y. Pan, C.-J. Liu and Q. Ge, Adsorption and protonation of CO2 on partially hydroxylated γ-Al2O3 surfaces: A density functional theory study, Langmuir, 2008, 24, 12410–12419 CrossRef CAS PubMed.
- A. K. Mishra, A. Roldan and N. H. Leeuw, A density functional theory study of the adsorption behavior of CO2 on CuO2 surfaces, J. Chem. Phys., 2016, 145, 4958804 CrossRef PubMed.
- G. Carchini, I. Hussein, M. J. Al-Marri, R. Shawabkeh, M. Mahamoud and S. Aparicio, A theoretical study of gas adsorption on calcite for CO2 enhanced natural gas recovery, Appl. Surf. Sci., 2020, 504, 144575 CrossRef CAS.
- Y. Jiao, A. Du, Z. Zhu, V. Rudolph and S. C. Smith, Adsorption of carbon dioxide and nitrogen on single-layer aluminum nitride nanostructures studied by density functional theory, J. Phys. Chem. C, 2010, 114, 7846–7849 CrossRef CAS.
- M. M. Rodriguez, X. Peng, L. Liu, Y. Li and J. M. Andino, A density functional theory and experimental study of CO2 interaction with brookite TiO2, J. Phys. Chem. C, 2012, 116, 19755–19764 CrossRef CAS.
- D. C. Sorescu, W. A. Al-Saidi and K. D. Jordan, CO2 adsorption on TiO2 (1 0 1) anatase: A dispersion-corrected density functional theory study, J. Chem. Phys., 2011, 135, 124701 CrossRef PubMed.
- Y. Pan, C.-J. Liu and Q. Ge, Adsorption and protonation of CO2 on partially hydroxylated γ-Al2O3 surfaces: A density functional theory study, Langmuir, 2008, 24, 12410–12419 CrossRef CAS PubMed.
- O. Mohan, Q. T. Trinh, A. Benerjee and S. H. Mushrif, Predicting CO2 adsorption and reactivity on transition metals surfaces using popular density functional theory methods, J. Exp. Nanosci., 2019, 45, 1163–1172 CAS.
- V. Chihaia, D. A. Neacsu, V. Alexiev and D. Angelescu, Performances of the bond order interacting potentials on calculating the surface energies of MgH2 facets, Available at SSRN: https://ssrn.com/abstract=4282784.
- N. Kumari, N. Sinha, M. A. Haider and S. Basu, CO2 reduction to methanol on CeO2 (1 1 0) surface: A density functional theory study, Electrochim. Acta, 2015, 170, 21–29 CrossRef.
- J. P. Perdew, K. Bruke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. Yang, C. He, L. Fu, J. Huo, C. Zhao, X. Li and Y. Song, Capture and separation of CO2 on BC3 nanosheets: A DFT study, Chin. Chem. Lett., 2021, 32, 3202–3206 CrossRef CAS.
- Q. Cui, G. Qin, W. Wang, K. R. Geethalakshmi, A. Du and Q. Sun, Novel two-dimensional MOF as promising single-atom electrocatalyst for CO2 reduction: A theoretical study, Appl. Surf. Sci., 2020, 500, 143993 CrossRef CAS.
- B. Farkas and N. H. Leeuw, Towards a morphology of cobalt nanoparticles: Size and strain effects, Nanotechnology, 2020, 31, 195711 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. E. Blöchl, Generalized separable potentials for electronic-structure calculations, Phys. Rev. B, 1990, 41, 5414–5416 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B, 1990, 59, 1758–1775 CrossRef.
- Y. Zhang and W. Yang, Comment on “Generalized Gradient Approximation Made Simple”, Phys. Rev. Lett., 1998, 80, 890–890 CrossRef CAS.
- G. Prandini, A. Marrazzo, I. E. Castelli, N. Mounet and N. Marzari, Precision and efficiency in solid-state pseudopotential calculations, npj Comput. Mater., 2018, 4, 72 CrossRef.
- K. Lejaeghere, G. Bihlmayer, T. Björkman, P. Blaha, S. Blügel, V. Blum, D. Caliste, I. E. Castelli, S. J. Clark, A. Dal Corso, S. de Gironcoli, T. Deutsch, J. K. Dewhurst, I. Di Marco, C. Draxl, M. Dułak, O. Eriksson, J. A. Flores-Livas, K. F. Garrity, L. Genovese, P. Giannozzi, M. Giantomassi, S. Goedecker, X. Gonze, O. Grånäs, E. K. U. Gross, A. Gulans, F. Gygi, D. R. Hamann, P. J. Hasnip, N. A. W. Holzwarth, D. Iuşan, D. B. Jochym, F. Jollet, D. Jones, G. Kresse, K. Koepernik, E. Küçükbenli, Y. O. Kvashnin, I. L. M. Locht, S. Lubeck, M. Marsman, N. Marzari, U. Nitzsche, L. Nordström, T. Ozaki, L. Paulatto, C. J. Pickard, W. Poelmans, M. I. J. Probert, K. Refson, M. Richter, G.-M. Rignanese, S. Saha, M. Scheffler, M. Schlipf, K. Schwarz, S. Sharma, F. Tavazza, P. Thunström, A. Tkatchenko, M. Torrent, D. Vanderbilt, M. J. van Setten, V. Van Speybroeck, J. M. Wills, J. R. Yates, G.-X. Zhang and S. Cottenier, Reproducibility in density functional theory calculations of solids, Science, 2016, 351, aad3000 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional disersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 15104 CrossRef PubMed.
- J.-P. Bastide, B. Bonnetot, J.-M. Létoffé and P. Claudy, Polymorphisme de l'hydrure de magnesium sous haute pression, Mater. Res. Bull., 1980, 15, 1215–1224 CrossRef CAS.
- H. S. AlMatouk, V. Chihaia and V. Alexiev, Density functional study of the thermodynamic properties and phase diagram of the magnesium hydride, CALPHAD: Comput. Coupling Phase Diagrams Thermochem., 2018, 60, 7–15 CrossRef.
- M. Bortz, B. Bertheville, G. Bötter and K. Yvon, Structure of the high pressure phase γ-MgH2 by neutron powder diffraction, J. Alloys Compd., 1999, 287, L4 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- W.-Y. Chen, J.-J. Tang, Z.-W. Lu, M.-X. Huang, L. Liu, C.-C. He and Y.-J. Zhao, Theoretical investigation of the surface orientation impact on the hydrogen vacancy formation of MgH2, Surf. Sci., 2021, 710, 121850 CrossRef CAS.
- M. Yu and D. R. Trinkle, Accurate and efficient algorithm for Bader charge integration, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
- W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved grid-based algorithm for Bader charge allocation, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 (initial dispositions of all the considered systems); Fig. S2 (optimized geometries of systems considering nCO2 molecules); Table S1 (MgH2 surface angle and radius deformation); Table S2 (adsorption energies for Co-catalyzed systems); Table S3 (adsorption energies for Co-catalyzed systems); Table S4 (interatomic distances between the Co atoms for the 1 to 9 cluster formation); Table S5 (interatomic distances between cobalt atoms for surface coverage); Fig. S3 (optimised geometries of Co clusters systems); Fig. S4 (optimized geometries of Co clusters systems); Fig. S5 (details of the geometric parameters of the 7Co/MgH2 structure); Fig. S6 (optimized geometries of CO2 on Co SAC systems); Fig. S7 (optimized geometries of CO2 on nCo-catalyzed systems); Fig. S8 (optimized geometries of CO2 + H2 on Co-catalyzed systems); Table S6 (Bader ionic charges of CO2 adsorption on clean MgH2); Table S7 (Bader ionic charges of Co adsorption on clean MgH2); Table S8 (Bader ionic charges of CO2 and H2 adsorption on Co/MgH2); Table S9 (valence Bader charge difference for CO2/MgH2 systems); Table S10 (valence Bader charge difference for Co/MgH2 systems); Table S11 (valence Bader charge difference for CO2/Co–MgH2 systems); Table S12 (MgH2 surface energies for the k-point testing set); Fig. S9 (MgH2 surface energies as a function of the selected k-points). See DOI: https://doi.org/10.1039/d3im00096f |
| This journal is © Institute of Process Engineering of CAS 2024 |