
Lithium salt of a pro-mesogenic [closo-CB11H12]− derivative: anisotropic Li+ ion transport in liquid crystalline electrolytes†
Litwin
Jacob
a,
Leszek
Niedzicki
b,
Rafał
Jakubowski
 a,
Damian
Pociecha
c and
Piotr
Kaszyński
*ade
a,
Damian
Pociecha
c and
Piotr
Kaszyński
*ade
aCentre of Molecular and Macromolecular Studies, Polish Academy of Sciences, 90-363 Łódź, Poland. E-mail: piotr.kaszynski@cbmm.lodz.pl
bFaculty of Chemistry, Warsaw University of Technology, 00-664 Warsaw, Poland
cFaculty of Chemistry, University of Warsaw, 02-089 Warsaw, Poland
dFaculty of Chemistry, University of Łódź, 91-403 Łódź, Poland
eDepartment of Chemistry, Middle Tennessee State University, Murfreesboro, TN 37130, USA
First published on 24th May 2024
Abstract
Li+ ion conduction in two aligned liquid crystalline electrolytes consisting of 10 mol% Li+ salt of a pro-mesogenic anion derived from [closo-1-CB11H12]− in non-ionic hosts was investigated. Using electrochemical impedance spectroscopy (EIS), the ionic conductivity in the parallel (σ‖) and perpendicular (σ⊥) directions of the electrolyte samples was determined using two types of cells: an interdigitated gold electrode and a nylon 6-coated ITO cell. The ratio of ionic conductivities σ⊥/σ‖ in the electrolyte with a nona(ethylene oxide) spacer was about 3 in the entire SmA phase, while in the shorter homologue, the ratio monotonically increases from about 0.4 to 2.9. The liquid crystalline behavior of the hosts and the electrolytes was investigated by optical, thermal, and powder XRD methods.
Introduction
Liquid crystals1,2 (LCs) have gained considerable interest as electrolyte materials due to their ability to form self-organized structures with ion conductive channels.3–5 The inherent partial organization of LC electrolytes6 results in macroscopic ordering and, consequently, formation of 1D,7,8 2D9–12 or 3D13–15 pathways for ion transport. This property of LC electrolytes is advantageous over those of the conventional organic electrolytes,16–18 which have random molecular orientation (isotropic). Moreover, LC electrolytes can be aligned in specific directions, and studies have shown increased ionic conductivity (up to three orders of magnitude) in the aligned monodomain smectic phase of the electrolyte compared to the unaligned material.19,20 It has been demonstrated that LC electrolytes can suppress undesired processes,16 such as dissolution of electrodes and current collectors, Li metal dendrite growth, etc., which are commonly observed in the conventional liquid electrolytes during the charge–discharge cycles without the aid of external factors.21–23 LC electrolytes have also shown good electrochemical stability (up to 4.3 V vs. the Li/Li+ reference electrode) and high oxidative resistance, which are important for high voltage battery applications.4Although Li+ ion transport in LCs has been known for some decades, only recently Kato et al. demonstrated the first thermotropic LC electrolytes for applications in Li-ion batteries.3 They showed moderate ionic conductivity in the range of 10−6 to 10−5 S cm−1 at temperatures 40–100 °C, and the Li-ion battery developed using such a LC electrolyte exhibited a reversible charge–discharge for both positive and negative electrodes. They also studied the anisotropy of Li+ ion transport in an aligned photoactive LC electrolyte system, in which ionic conductivity in perpendicular and parallel directions was found to be 3.1 × 10−6 S cm−1 and 3.9 × 10−7 S cm−1, respectively, at 116 °C (σ⊥/σ‖ ≈ 8).14
Typically, LC-based electrolytes are obtained by dissolving metal ion salts, such as LiBF4, LiAsF6, LiPF6, and LiN(SO2CF3)2,24 in a LC host at concentrations up to 40 mol%. To ensure sufficient solubility of the salts, the hosts are polar LC containing oligo(ethylene oxide) chains19,25,26 or organic carbonate3,4,11,27 fragments, or ionic LC with imidazolium12,14 or ammonium13 groups. Another approach to LC electrolytes involves a single component ion pair, rather than a Li salt additive.28 Although these halogenated lithium salts are frequently used in commercial batteries, there are, however, concerns related to their chemical and electrochemical stability and impact of the decomposition products on the overall battery performance.29 Therefore, to address these shortcomings, new designs of stable and halogen-free lithium salts are being developed. Particularly interesting in this context are closo-borate anions, which are weakly coordinating, inorganic, three-dimensional, σ-aromatic boron hydrides.30–33closo-Borate anions have recently received significant attention as new solid electrolyte systems,34–43 owing to their high ionic conductivity44 and superior chemical/electrochemical stability.44–49
A combination of Li+ salts of weakly coordinating closo-borate anion derivatives as the source of Li+ ions, and anisotropic liquid crystals as a host constitutes an intriguing approach to high-performance electrolytes for Li-ion batteries. Recently, we demonstrated the first examples of such LC electrolytes containing a Li+ salt of anion S1, which is based on a 1,12-disubstituted [closo-1-CB11H12]− derivative structurally similar to the LC host 1H[n] (Fig. 1).50 The ionic conductivity of unaligned electrolyte samples was found to monotonically increase from 10−7 to 10−4 S cm−1 with increasing temperature (40–115 °C) and Li+ salt concentration (5–15 mol%). Higher conductivity is expected along the ionic planes in aligned smectic electrolyte samples. To accomplish this goal, the host and the ionic additive were redesigned. Thus, two molecules of host 1H[n] were conceptually connected into a “dimer”, in which two, three-ring mesogens are bridged with an oligo(ethylene oxide) linker in 2F[n] (with a rod–coil–rod architecture) providing solvation for the Li+ ions in the ionic interlayer (Fig. 1). The two fluorine atoms on the biphenyl core provide some degree of stabilization of the host against anodic oxidation and induce high-temperature nematic behaviour of the electrolyte, which helps to achieve alignment of the smectic phase.
 | ||
| Fig. 1 Structures of the LC host 1H[n] and ionic additive S1[Li] in the previous design (left, ref. 50) and the newly designed host 2F[n] and additive S2[Li] (right). In the boron cage of anions S1 and S2 each unsubstituted vertex (green) is a BH fragment and the dot represents a carbon atom. | ||
The redesigned salt additive S2[Li] is shown in Fig. 1. It was assumed that the negatively charged {closo-1-CB11} cage resides primarily in the ionic interlayer and the presence of the B(12)-hexyl chain in anion S1 unfavorably affects the mobility of the Li+ ions. Therefore, the alkyl chain in S1 was removed from the {closo-1-CB11} cluster and the ester group was moved deeper into the hydrophobic section of the anion S2 increasing its overall chemical stability. The overall length of the hydrophobic fragment in S2 is similar to that in the non-ionic host.
Herein we describe the synthesis and extensive characterization of three homologues 2F[n] (n = 4, 6, 9) and the ionic additive S2[Li]. The hosts were analyzed by optical, thermal, and powder XRD methods. Two of the hosts were used to formulate 10 mol% liquid crystalline electrolyte solutions 2F[n]–S2[Li], which were investigated for mesogenic behavior and by electrochemical impedance spectroscopy (EIS) for Li+ ion conductivity in aligned samples.
Results and discussion
Synthesis
The liquid crystalline hosts 2F[n] were prepared according to the procedure shown in Scheme 1. Thus, 4-bromo-2,3-difluorophenol (3) was reacted with ditosylate Ts(OCH2CH2)nOTs (4[n], n = 4, 6, 9) in the presence of t-BuOK to give dibromides 5[n] isolated in 69–90% yield after column chromatography. The subsequent reaction of 5[n] with 4-hydroxyphenylboronic acid in the presence of the Pd2(dba)3 catalyst and [HPCy3][BF4] ligand resulted in diols 6[n] obtained in 61–74% yield. Finally, the esterification of the diols 6[n] with 4-hexylbenzoic acid in the presence of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) and catalytic amounts of 4-dimethylaminopyridine (DMAP) gave the LC hosts 2F[n] in 60–78% yield. The use of DCC instead of EDC·HCl for the synthesis of 2F[4] resulted in contamination of the product with urea byproducts, which were difficult to remove. The requisite ditosylates 4[n] were prepared according to a general procedure51 and full procedures and analytical data are listed in the ESI.†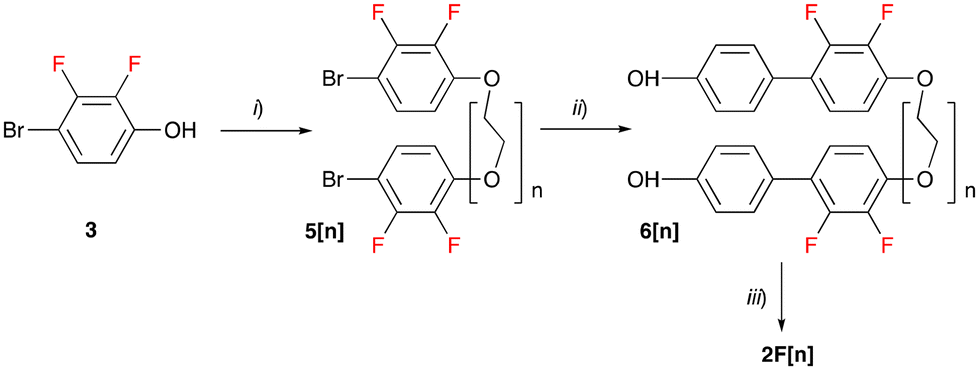 | ||
| Scheme 1 Preparation of LC hosts 2F[n]. Reagents and conditions: (i) t-BuOK, EtOH, Ts(OCH2CH2)nOTs (4[n]), reflux, 18 h, and 69–90% yield; (ii) 4-OHC6H4B(OH)2, Pd2(dba)3, [HPCy3][BF4], t-BuOK, THF/H2O, reflux, 48 h, and 61–74% yield; (iii) 4-hexylbenzoic acid, EDC·HCl, DMAP, CH2Cl2, rt, 18 h, and 60–78% yield. | ||
The pro-mesogenic ionic additive S2[Li] was prepared as shown in Scheme 2. Thus, the previously reported52 phenol 7[NMe4] was esterified with 4-(4-trans-pentylcyclohexyl)benzoyl chloride giving the ion pair S2[NMe4] in 61–69% yield. Attempts at the synthesis of S2[NMe4] through Steglich-type esterification of 7[NMe4] in the presence of DCC or EDC·HCl gave a product contaminated with urea byproducts. Salt S2[Li] was obtained by exchanging the [NMe4]+ cation in S2[NMe4] with the Li+ cation in a H2O/Et2O system.
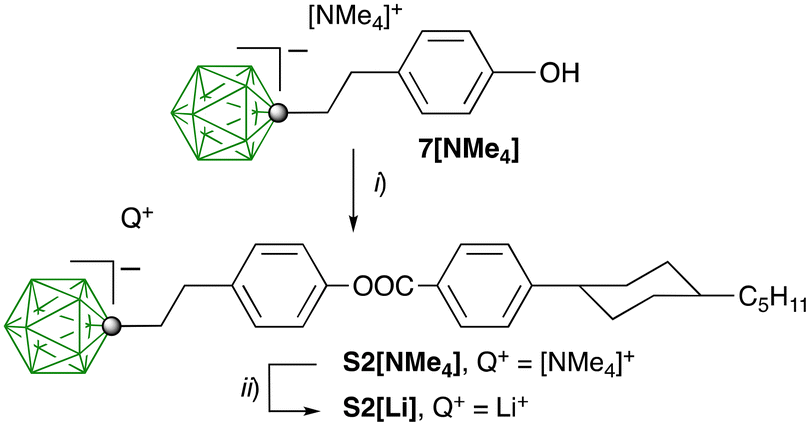 | ||
| Scheme 2 Preparation of the lithium salt S2[Li]. Reagents and conditions: (i) 4-(4-trans-pentylcyclohexyl)benzoyl chloride, NaH, THF, rt, 12 h, and 61–69% yield; (ii) (1). 20% HCl. (2). LiOH/LiCl, H2O/Et2O. | ||
Electrolyte mixtures, 2F[6]–S2[Li] and 2F[9]–S2[Li], containing 10 mol% of salt S2[Li] in host 2F[n], for impedance analysis were prepared by dissolving exact amounts of the components in CH2Cl2, followed by the evaporation of the clear solutions and drying in vacuo.
Liquid crystalline properties
The mesogenic behavior of hosts 2F[n] and mixtures 2F[n]–S2[Li] was investigated with differential scanning calorimetry (DSC) and polarized optical microscopy (POM). The results are shown in Table 1 and Fig. 2 and 3.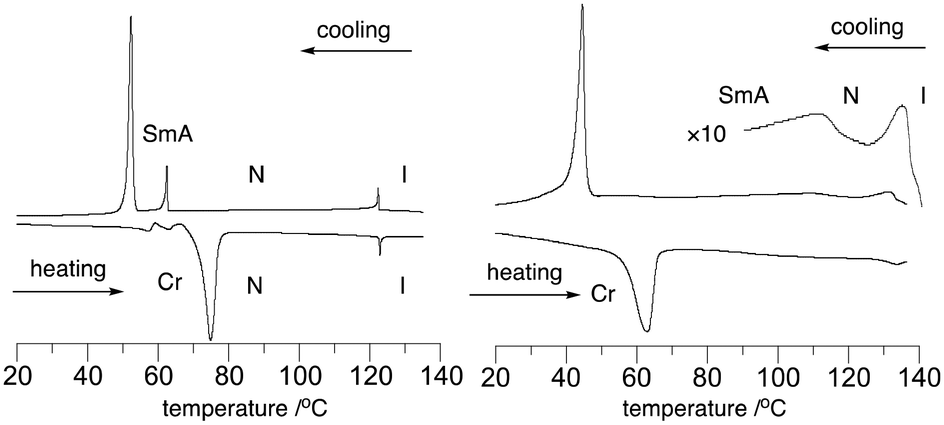 | ||
| Fig. 2 DSC trace of the pure host 2F[6] (left) and 10 mol% solution of S2[Li] in 2F[6] (right). The heating and cooling rates are 10 K min−1. | ||
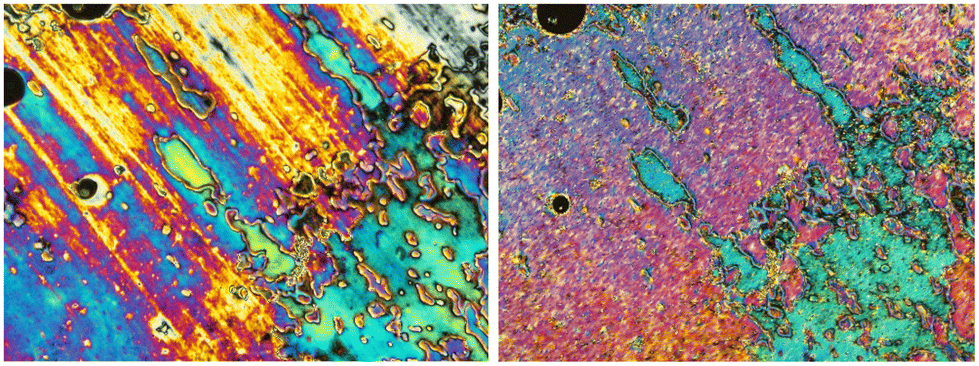 | ||
| Fig. 3 Optical textures of 2F[6] obtained on cooling observed in polarized light in the same region of the sample: nematic phase at about 90 °C (left) and SmA phase just below the N–SmA transition at 58 °C (right). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| a Peak of transition recorded on first heating at 10 K min−1; enthalpy in kJ mol−1 (in italics). Cr – crystalline, SmA – smectic A, SmX – undefined smectic phase, and I – isotropic phase. b Monotropic transition. c Recorded on cooling. d Optical observation. e 10 mol% of S2[Li] in 2F[n]. Peak of transition recorded on the second heating at a rate 10 K min−1. See the ESI† for details. | |
|---|---|
|
2F[4] |
Cr 110 (81.3) (SmA 83, 4.2)b,c N 161 (2.6) I |
| 2F[6] | Cr 75 (36.5) (SmA 62, 1.6)b,c N 123 (1.7) I |
| 2F[9] | Cr 69 (103.7) (SmA 65)b,c,d N 77 (1.8) I |
|
2F[6]–S2[Li]e |
Cr 66 SmA 110c N 134 I |
|
2F[9]–S2[Li]e |
Cr 48 SmX 52 SmA 88 N 92 I |
The analysis of hosts 2F[n] revealed the presence of enantiotropic nematic (N) and monotropic smectic (Sm) phases. Thus, the enantiotropic phase of 2F[6] (Fig. 2) exhibits a schlieren texture typical of a nematic phase (Fig. 3), while shearing the monotropic phase gave a homeotropic alignment, consistent with the behavior of a SmA phase. The data in Table 1 demonstrate that the elongation of the oligo(ethylene oxide) linker leads to rapid destabilization of the nematic phase, as is evident from the lowering of the N–I transition from 161 °C in 2F[4] to 77 °C in 2F[9]. At the same time, the Sm–N transition temperature decreases moderately from 83 °C in 2F[4] to about 65 °C in the next two homologues. Consequently, the range of the nematic phase decreases from approximately 78 K in 2F[4] to 12 K in 2F[9]. These results suggested that 2F[6] and 2F[9] are the most suitable hosts for the ionic additive S2[Li].
Addition of 10 mol% of Li+ salt S2[Li] to host 2F[6] raised its clearing temperature by about 11 K and increased the stability of the Sm phase by nearly 50 K in the mixture 2F[6]–S2[Li] (Table 1 and Fig. 2). Similarly, a 10 mol% mixture of 2F[9]–S2[Li] has higher N–I and Sm–N transition temperatures than the pure host by about 15 and 23 K, respectively. The observed transitions are broad and much better visible on cooling from the isotropic phase. These smectogenic and phase transition broadening effects of an added salt are typically observed in such electrolytes.19,20
Powder XRD analysis
Powder XRD measurements (PXRD) confirmed the formation of a smectic phase in 2F[n] hosts. Thus, diffractograms for 2F[6] and 2F[9] exhibit a sharp signal in the small angle region and a broad diffused halo (4.4 Å) in the wide-angle region (Fig. 4), which is a typical pattern for a lamellar structure, such as the SmA phase, with no long-range positional order within the layers. For compound 2F[6], an additional weak harmonic signal corresponding to layer periodicity is observed, evidencing non-sinusoidal modulation of electron density along the layer normal.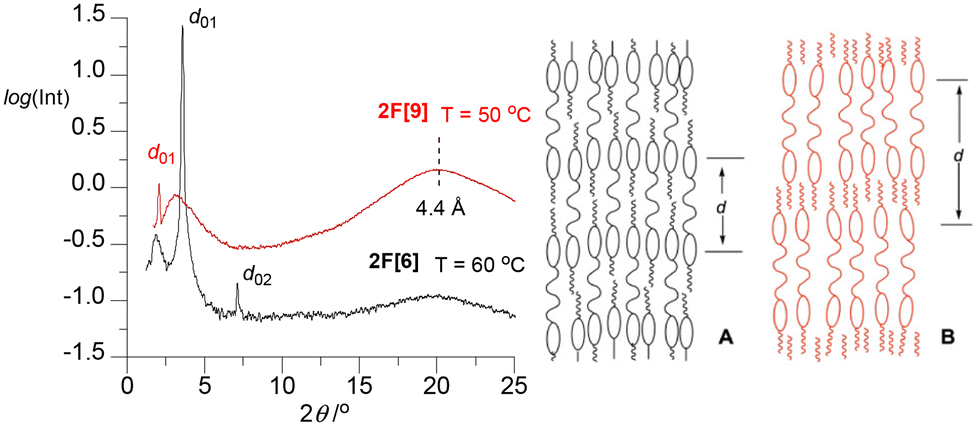 | ||
| Fig. 4 Left: powder XRD patterns for 2F[6] and 2F[9] in the smectic phase measured at 60 °C and 50 °C, respectively. Right: two extreme molecular arrangements A and B in the SmA layer of 2F[n]. | ||
Detailed analysis of data for 2F[6] demonstrates that the sharp reflection at 2θ = 3.58° labelled as d01 represents a spacing of 24.6 Å. This value is considerably smaller than the length of half of a molecule of 2F[6] in the most extended conformation (about 33 Å, DFT, Fig. 5). This suggests that the repeating units (layers) in the smectic phase of 2F[6] consist of single mesogenic cores (about half of the dimeric molecules) constituting an intercalated smectic structure. In addition, there is significant interdigitation and/or coiling up of the hexa(ethylene oxide) and terminal hexyl chains, as well as a low degree of nanosegregation of hydrophobic and polar molecular segments. This situation is schematically shown in diagram A in Fig. 4. The observed broad feature at 2θ = 1.9° (d = ∼45 Å) may indicate the formation of local, instantaneous structures (fluctuations) with smectic layer thickness defined by the entire molecule length.
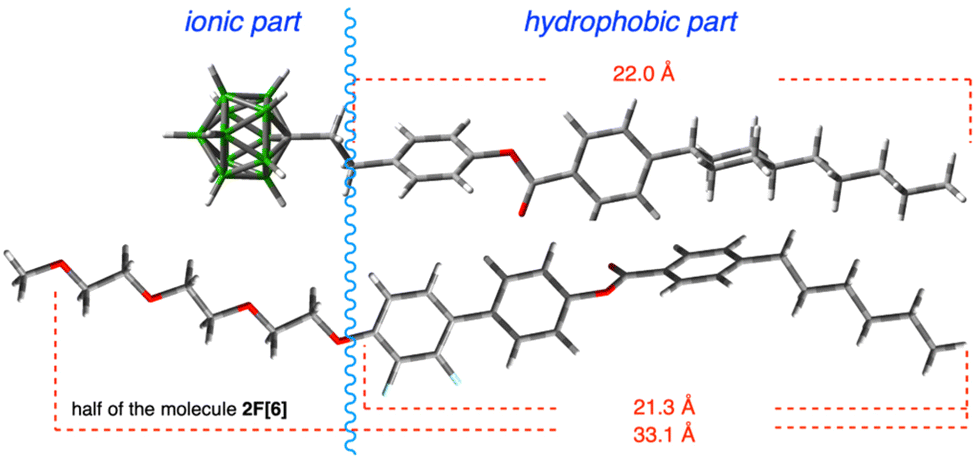 | ||
| Fig. 5 Equilibrium geometry of anion S2 (top) and half of the host 2F[6] (bottom) in most extended conformations optimized at the B3LYP/6-31(d) level of theory in vacuum. | ||
Similar analysis of results for 2F[9] demonstrates that apart from the sharp low angle XRD signal corresponding to the layer thickness, there is an additional broad reflection due to fluctuations of two competing molecular arrangement modes. However, in this case the lowest angle signal at 2θ = 2.03° (d = 42.6 Å) is sharp, while the one centered at 2θ = 3° (d = ∼27 Å) is broad. A comparison of the measured thickness of the smectic layers with the estimated molecular length of 2F[9] (75.2 Å) suggests that the layers are made of entire molecules, as depicted in diagram B (Fig. 4), which are strongly interdigitated and/or oligo(ethylene oxide) chains are coiled up. As a result, the measured periodicities are considerably shorter than those estimated from the molecular dimensions. The broad halo at 2θ = 3° corresponds to fluctuations of intercalated-type molecular packing.
Impedance spectroscopy
Li+ ion conduction in two aligned liquid crystalline electrolyte samples (10 mol% solutions of S2[Li] in 2F[6] and in 2F[9]) was investigated in the temperature range of 40–140 °C using electrochemical impedance spectroscopy (EIS). An ITO cell with a nylon-6 alignment layer and a thickness of 10 μm, and an interdigitated gold electrode with a gap size of 10 μm were used for measuring ionic conductivity in the direction perpendicular (σ⊥) and parallel (σ‖) to the smectic layer plane, respectively (Fig. 6).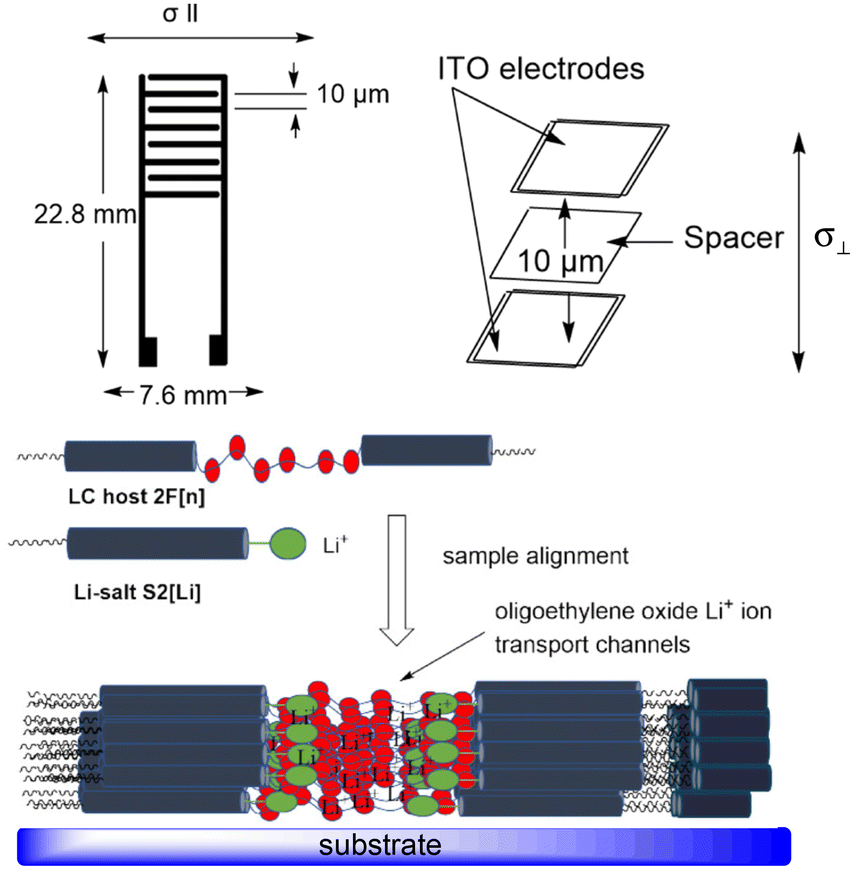 | ||
| Fig. 6 Schematic representation of the electrodes and electrolyte. Top: interdigitated gold electrode and ITO cell. Bottom: parallelly aligned electrolyte sample inside the cell with oligo(ethylene oxide) channels for Li+ ion transport. | ||
The alignment of the electrolyte smectic phase was confirmed by optical microscopy, which showed a fan-shaped multidomain texture consistent with a planar alignment in both cells (Fig. 7). This indicates that the molecules are oriented approximately parallel to the substrate and organized in smectic domains with no or weak alignment, while the ion conductive planes propagate in the perpendicular direction (Fig. 6). Therefore, the ITO cell measures conductivity along the conductive planes (perpendicular to the substrate, σ⊥), while the gold interdigitated electrode provides conductivity parallel to the substrate (σ‖) through an unaligned, multidomain smectic phase.
 | ||
| Fig. 7 Photomicrographs of the planarly aligned SmA phase of 2F[6]–S2[Li] in the interdigitated gold electrode (left) and the ITO cell (right) at 100 °C and 90 °C, respectively. The vertical stripes are gold electrodes with 10 μm gaps between them. | ||
The results shown in Fig. 8 demonstrate that Li+ ion conductivity σ increases with increasing temperature in SmA and nematic phases for both electrolytes and in both parallel and perpendicular directions reaching the value of 0.1 mS cm−1 above 90 °C, which is in a range of interest for practical applications.29 The observed trend is consistent with decreasing organization of the phase and, consequently, its viscosity with increasing temperature. The overall conductivity is in a range of 10−6 to 10−4 S cm−1 and comparable to that observed for amorphous poly(ethylene oxide) materials and liquid crystals containing oligo(ethylene oxide) chains.19
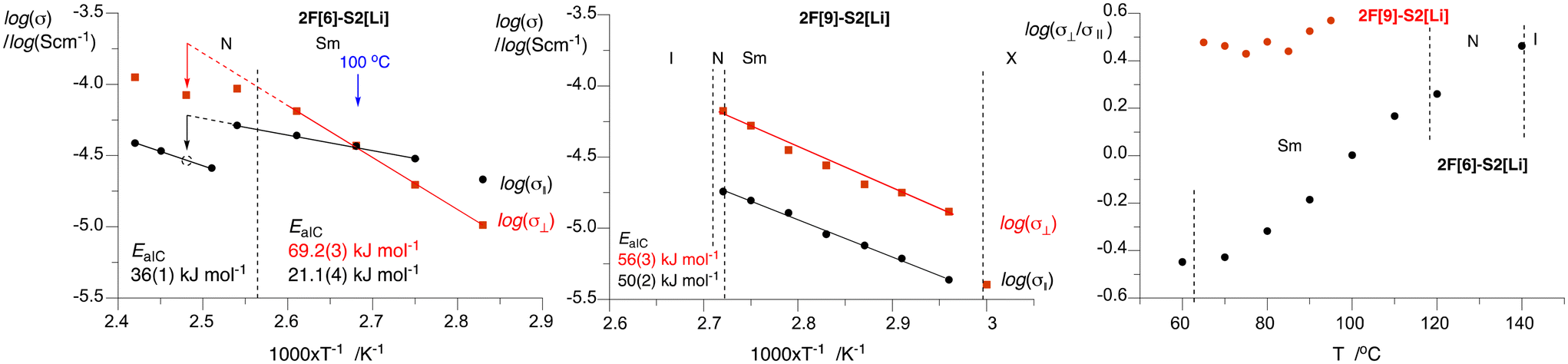 | ||
| Fig. 8 Arrhenius analysis and activation energies EaIC for the anisotropic conductivity of 10 mol% solutions of 2F[6]–S2[Li] (left) and 2F[9]–S2[Li] (middle) in the perpendicular (⊥, red squares) and parallel (‖, black dots) directions to the smectic layer plane. The dotted lines represent extrapolation from the SmA phase and the dotted black circle is interpolated data point from three σ‖ values in the nematic phase. Right: A plot of log(σ⊥/σ‖) vs. temperature. Details are provided in the ESI.† | ||
Detailed analysis of the conductivity data demonstrates that the character of the Arrhenius logσ(T−1) plots for the parallel and perpendicular directions is significantly different for the two electrolyte systems (Fig. 8). For the 2F[9]–S2[Li] electrolyte the conductivity in the perpendicular direction (σ⊥) is higher than that in the parallel direction (σ‖) by an approximately constant factor of 3 in the entire SmA phase (65–90 °C). Arrhenius analysis of the logσ(T−1) plots indicates similar activation energies (EaIC) of the conductivity process in both directions: 56(3) kJ mol−1 in the perpendicular direction and 50(2) kJ mol−1 in the parallel direction. These values are lower than those reported earlier for unaligned samples of 1H[n]–S1[Li].50
The results for the 2F[6]–S2[Li] electrolyte showed more complex behavior: at temperatures below 100 °C the conductivity in the parallel direction is higher than that in the perpendicular direction (σ⊥/σ‖ < 1), while above 100 °C the trend reverses and the ratio σ⊥/σ‖ reaches the value of 2.9 at 140 °C (nematic–isotropic transition temperature). The observed behavior is a consequence of a significantly higher activation energy for conductivity in the perpendicular (EaIC = 69.2(3) kJ mol−1) than that in the parallel direction (EaIC = 21.1(4) kJ mol−1). The former is similar to that observed before for an unaligned electrolyte smectic phase of 1H[n]–S1[Li]50 and typical of ionic liquid and liquid polymer electrolytes,53,54 while the latter is about half of that observed for conductivity in the higher homologue. The observed different behaviors in the conductivity of the two electrolytes and the large difference in the EaIC values in the shorter homologue are likely related to the length of the linking oligo(ethylene oxide) chain and hence differences in the structures of their ionic sublayers. It is plausible that the short hexa(ethylene oxide) chain in 2F[6]–S2[Li] does not provide sufficient solvation for the Li+ ions and the width of the flexible ionic layer for facile ion transport, which may lead to incomplete nanosegregation of the ionic and hydrophobic molecular components. Such a situation is hinted by XRD measurements of the host (vide supra). In contrast, the three unit longer nona(ethylene oxide) chain in 2F[9]–S2[Li] provides adequate solvation, flexibility, and nanosegregation of the ionic sublayer needed for “normal” ion transport in the material. Such effects of the length of the oligo(ethylene oxide) chain on Li+ ion transport were reported recently.55,56
The observed high EaIC value for the conduction in the perpendicular direction in the 2F[6]–S2[Li] system is responsible for the observed steep, by nearly two orders of magnitude, increase of conductivity σ⊥ in a 60 K range (60 °C to 120 °C).
Upon transition of the planar SmA phase to the planar nematic phase in 2F[6]–S2[Li] at about 110 °C, the trend in conductivity in both directions changes: they become lower with apparently different slopes (EaIC), although there are too few data points for quantitative analysis. The change in the parallel conductivity, σ‖, is abrupt, while σ⊥ changes gradually over some 20 K. This is consistent with the transformation of the planar SmA to a planar nematic phase, in which the molecules maintain an orientation parallel to the substrate still allowing for facile transport of ions solvated by the oligo(ethylene oxide) fragments in the direction perpendicular to the substrate. In contrast, the unaligned smectic domains collapse into less organized nematic domains, resulting in a drop of ionic conductivity and an apparent increase of EaIC.
Arrhenius analysis of high temperature σ‖ data in the nematic phase suggests EaIC = 36(1) kJ mol−1 for ionic conduction in the parallel direction, which is nearly twice higher than that in the SmA phase. A comparison of conductivity at 130 °C, extrapolated from the experimental logσ‖ in the SmA phase and interpolated from logσ⊥ in the nematic phase, shows that both values are lower in the nematic phase by a factor of 2.2.
Conclusions
Three liquid crystalline hosts 2F[n] with varied lengths of the linking oligo(ethylene oxide) chain and lithium salt S2[Li] containing an anionic derivative of [closo-1-CH11H12]−, structurally similar to the host, were synthesized and characterized. Thermal analysis of the hosts revealed an enantiotropic nematic phase and a monotropic SmA phase with decreasing transition temperatures with increasing length of the linker (increasing n). Addition of 10 mol% of salt S2[Li] has favorable effects on the liquid crystalline properties: it significantly enhances the stability of the SmA phase at the expense of the nematic phase and increases the TNI. The narrow range nematic phase present in electrolytes 2F[6]–S2[Li] (24 K) and 2F[9]–S2[Li] (4 K) helps to achieve a planar alignment of their SmA phase.Impedance spectroscopy revealed ionic conductivity between 0.004 and 0.1 mS cm−1 in the temperature range 60–140 °C for the two electrolyte systems, albeit with different trends. In the lower homologue 2F[6]–S2[Li] the activation energy EaIC for Li+ ion conductivity in the perpendicular direction, σ⊥, along the ionic layers, was three times higher than that in the parallel direction, σ‖, across the ionic layers, which led to inversion of the σ⊥/σ‖ ratio at 100 °C. In the higher homologue, 2F[9]–S2[Li], the σ⊥/σ‖ ratio was nearly constant and about 3 in the entire SmA region, which was consistent with expectations. These differences in the behavior of the two electrolytes were ascribed to different structures of the ionic sublayer. The obtained results indicate that systems, such as 2F[9]–S2[Li], are promising for further development of new electrolytes.
Experimental
General
Reagents and solvents were obtained commercially. The derivative 7[NMe4] was obtained according to a literature procedure.52 NMR spectra were obtained at 400 MHz (1H), 101 MHz (13C), 128 MHz (11B), and 376 MHz (19F), in CDCl3 and referenced to the solvent (δ = 7.26 ppm for 1H and δ = 77.16 ppm for 13C) or in CD3CN and referenced to the solvent (δ = 1.94 ppm for 1H and δ = 118.26 ppm for 13C) or in DMSO-d6 and referenced to the solvent (δ = 2.50 ppm for 1H and δ = 39.52 ppm for 13C)57 or to an external sample of neat BF3·Et2O in CDCl3 and CD3CN (11B NMR, δ = 0.0 ppm) or to an external sample of neat CF3COOH in CDCl3 and DMSO-d6 (19F NMR δ = 76.55 ppm). 11B and 19F NMR chemical shifts were taken from the H-decoupled spectra.A polarized optical microscope with a hot stage was used to identify the liquid crystalline phase. A TA Discovery DSC 2500 instrument was used for thermal analysis of the samples. Samples of about 1.0 mg under a flow of nitrogen gas were used for the thermal analysis and the heating and cooling rates were 10 K min−1. Melting points were determined on a Melt-Temp II apparatus in capillaries. High-resolution mass spectrometry was conducted with the TOF-MS ES method. Broad angle X-ray diffraction studies were performed for unaligned samples using a Bruker D8 GADDS instrument (Cu Kα radiation, λ = 1.54 Å, incident beam formed by a Göbel mirror monochromator and a point collimator, 0.5 mm, two-dimensional detector Vantec 2000).
Preparation of host 2F[4]
To a suspension of diphenol 6[4] (250 mg, 0.42 mmol, 1.0 eq.) and 4-hexylbenzoic acid (188 mg, 0.91 mmol, 2.2 eq.) in CH2Cl2 (10 mL) were added dicyclohexylmethanediimine (DCC, 205 mg, 0.99 mmol, 2.4 eq.) and 4-dimethylaminopyridine (DMAP, 20 mg, 0.17 mmol, 0.4 eq.). The reaction mixture was stirred overnight at room temperature. The progress in the reaction was monitored using TLC (SiO2, CH2Cl2/EtOAc, 9:1). After the reaction was complete, it was filtered, and the collected filtrate was concentrated using a rotary evaporator to obtain the crude product. The target compound was isolated by column chromatography (SiO2, CH2Cl2/EtOAc, 9:1) to give 319 mg (79% yield) of 2F[4] as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.2 Hz, 4H), 7.54 (d, J = 7.0 Hz, 4H), 7.32 (d, J = 8.1 Hz, 4H), 7.27 (d, J = 8.9 Hz, 4H), 7.10 (td, J = 8.4, 2.2 Hz, 2H), 6.86–6.80 (m, 2H), 4.25 (t, J = 4.8 Hz, 4H), 3.92 (t, J = 4.8 Hz, 4H), 3.79–3.70 (m, 8H), 2.70 (t, J = 7.7 Hz, 4H), 1.70–1.59 (m, 4H), 1.39–1.28 (m, 12H), 0.89 (t, J = 6.7 Hz, 6H). 19F{1H} NMR (376 MHz, CDCl3) δ −139.8 (d, J = 19.6 Hz, 2F), −156.1 (d, J = 19.4 Hz, 2F).13C{1H} NMR (101 MHz, CDCl3) δ 165.4, 150.7, 149.6, 148.9 (dd, JF–C = 249, 11.1 Hz), 147.8 (dd, JF–C = 7.9, 2.9 Hz), 142.0 (dd, JF–C = 248, 15.0 Hz), 132.5, 130.4, 130.0 (d, JF–C = 3.1 Hz), 128.8, 126.9, 123.8 (t, JF–C = 3.9 Hz), 122.8 (d, JF–C = 10.9 Hz), 122.1, 110.2 (d, JF–C = 3.5 Hz), 71.1, 70.8, 69.7, 69.6, 36.2, 31.8, 31.3, 29.1, 22.7, 14.2. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C58H62F4O9Na: 1001.4228, found: 1001.4188. Anal. calcd for C58H62F4O9: C, 71.15; H, 6.38. Found: C, 71.09; H, 6.22.
General procedure for the synthesis of 2F[6] and 2F[9]
To a suspension of diphenol 6[6] (200 mg, 0.29 mmol) or diphenol 6[9] (239 mg, 0.29 mmol) and 4-hexylbenzoic acid (120 mg, 0.58 mmol) in CH2Cl2 (10 mL) were added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl, 177 mg, 0.93 mmol) and 4-dimethylaminopyridine (DMAP, 35 mg, 0.29 mmol). The reaction mixture was stirred overnight at room temperature. The progress in the reaction was monitored using TLC (SiO2, CH3CN/CH2Cl2, 1:4). After the reaction was complete, water (20 mL) was added into the reaction mixture and the CH2Cl2 layer was extracted (3 × 15 mL). The combined organic layers were dried over Na2SO4 and concentrated using the rotary evaporator to obtain the crude product. The target compound was isolated by column chromatography (SiO2, CH2Cl2/CH3CN, 4:1) to obtain the pure product 2F[n]. The product was further purified by precipitation from a solution of ethyl acetate and hexane.
Preparation of host 2F[6]
A suspension of diphenol 6[6] (351 mg, 0.51 mmol, 1.0 eq.) and 4-hexylbenzoic acid (231 mg, 1.12 mmol, 2.2 eq.) in CH2Cl2 (15 mL), when reacted in the presence of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC·HCl, 311 mg, 1.63 mmol, 3.2 eq.) and 4-dimethylaminopyridine (DMAP, 62 mg, 0.51 mmol, 1 eq.), resulted in a 360 mg (64% yield) of 2F[6] as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.3 Hz, 4H), 7.55 (d, J = 7.6 Hz, 4H), 7.32 (d, J = 8.3 Hz, 4H), 7.27 (d, J = 8.6 Hz, 4H), 7.10 (td, J = 8.4, 2.2 Hz, 2H), 6.87–6.81 (m, 2H), 4.25 (t, J = 4.8 Hz, 4H), 3.90 (t, J = 4.8 Hz, 4H), 3.77–3.73 (m, 4H), 3.72–3.65 (m, 12H), 2.70 (t, J = 7.7 Hz, 4H), 1.70–1.61 (m, 4H), 1.40–1.25 (m, 12H), 0.89 (t, J = 6.4 Hz, 6H). 19F{1H} NMR (376 MHz, CDCl3) δ −139.7 (d, J = 19.9 Hz, 2F), −156.1 (d, J = 19.7 Hz, 2F). 13C{1H} NMR (101 MHz, CDCl3) δ 165.4, 150.7, 149.7, 149.0 (dd, JF–C = 249, 11.0 Hz), 147.8 (dd, JF–C = 8.2, 2.7 Hz), 142.0 (dd, JF–C = 248, 15.1 Hz), 132.6, 130.4, 130.0 (d, JF–C = 3.0 Hz), 128.8, 126.9, 123.8 (t, JF–C = 4.1 Hz), 122.8 (d, JF–C = 11.0 Hz), 122.1, 110.2 (d, JF–C = 3.5 Hz), 71.1, 70.8, 70.7, 69.7, 69.6, 36.2, 31.8, 31.3, 29.1, 22.7, 14.2. 13C {1H;19F} NMR (126 MHz, CDCl3) δ 165.4, 150.7, 149.7, 148.9, 147.8, 142.0, 132.6, 130.4, 130.0, 128.8, 127.0, 123.8, 122.8, 122.1, 110.2, 71.1, 70.8, 70.7, 69.7, 69.6, 36.2, 31.8, 31.3, 29.1, 22.7, 14.2. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C62H70F4O11Na: 1089.4752, found: 1089.4716. Anal. calcd for C62H70F4O11: C, 69.78; H, 6.61. Found: C, 69.56; H, 6.55.Preparation of host 2F[9]
A suspension of diphenol 6[9] (319 mg, 0.39 mmol, 1.0 eq.) and 4-hexylbenzoic acid (175 mg, 0.85 mmol, 2.2 eq.) in CH2Cl2 (15 mL), when reacted in the presence of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC·HCl, 178 mg, 0.93 mmol, 2.4 eq.) and 4-dimethylaminopyridine (DMAP, 20 mg, 0.16 mmol, 0.4 eq.), resulted in a 297 mg (64% yield) of 2F[9] as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.3 Hz, 4H), 7.55 (d, J = 7.0 Hz, 4H), 7.32 (d, J = 8.3 Hz, 4H), 7.28 (d, J = 8.7 Hz, 4H), 7.10 (td, J = 8.5, 2.3 Hz, 2H), 6.87–6.81 (m, 2H), 4.25 (t, J = 4.8 Hz, 4H), 3.90 (t, J = 4.8 Hz, 4H), 3.77–3.72 (m, 4H), 3.71–3.67 (m, 4H), 3.67–3.63 (m, 20H), 2.70 (t, J = 7.7 Hz, 4H), 1.70–1.61 (m, 4H), 1.40–1.24 (m, 12H), 0.89 (t, J = 6.8 Hz, 6H). 19F{1H} NMR (376 MHz, CDCl3) δ −139.7 (d, J = 19.4 Hz, 2F), −156.1 (d, J = 19.8 Hz, 2F). 13C{1H} NMR (101 MHz, CDCl3) δ 165.3, 150.7, 149.6, 149.0 (dd, JF–C = 250, 11.0 Hz), 147.7 (dd, JF–C = 8.3, 2.8 Hz), 142.0 (dd, JF–C = 248, 15.0 Hz), 132.5, 130.4, 130.0 (d, JF–C = 3.2 Hz), 128.8, 126.9, 123.7 (t, JF–C = 4.1 Hz), 122.8 (d, JF–C = 10.8 Hz), 122.0, 110.2 (d, JF–C = 3.5 Hz), 71.1, 70.74, 70.68, 69.64, 69.60, 36.2, 31.8, 31.2, 29.0, 22.7, 14.2. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C68H83F4O14: 1199.5719, found: 1199.5742. Anal. calcd for C68H82F4O14: C, 68.10; H, 6.89. Found: C, 68.03; H, 6.72.General procedure for the synthesis of dibromides 5[n]
To a solution of 4-bromo-2,3-difluorophenol (3, 100 mg, 0.48 mmol) and t-BuOK (118 mg, 1.05 mmol) in EtOH (10 mL), ditosylate 4[n] (0.24 mmol, see the ESI†) was added. The resulting solution was refluxed overnight, and EtOH was removed using the rotary evaporator. Water was added to the resulting crude mixture and extraction was performed in CH2Cl2 (3 × 10 mL). The combined organic layer was dried (Na2SO4) and the solvent was removed in a rotary evaporator to get the crude product. The target compound was isolated by column chromatography (SiO2, CH2Cl2/EtOAc, 9:1).
Preparation of dibromide 5[4]
The reaction of Ts(OCH2CH2)4OTs (4[4], 3.98 g, 7.93 mmol, 1.0 eq., see the ESI†) with 4-bromo-2,3-difluorophenol (3, 3.47 g, 16.64 mmol, 2.1 eq.) in EtOH (35 mL) in the presence of t-BuOK (1.96 g, 17.44 mmol, 2.2 eq.) gave 4.18 g (92% yield) of 5[4] as a white solid: mp 73–75 °C. 1H NMR (400 MHz, CDCl3) δ 7.19 (ddd, J = 9.4, 7.1, 2.5 Hz, 2H), 6.69 (ddd, J = 9.4, 7.6, 2.1 Hz, 2H), 4.18 (t, J = 4.7 Hz, 4H), 3.87 (t, J = 4.7 Hz, 4H), 3.74–3.65 (m, 8H). 19F{1H} NMR (376 MHz, CDCl3): δ −127.8 (d, J = 20.2 Hz, 2F), −153.0 (d, J = 20.5 Hz, 2F). 13C{1H} NMR (101 MHz, CDCl3) δ 148.8 (dd, JF–C = 248, 12.3 Hz), 148.0 (dd, JF–C = 7.5, 2.5 Hz), 142.2 (dd, JF–C = 252, 14.9 Hz), 126.5 (d, JF–C = 4.5 Hz), 111.0 (d, JF–C = 3.5 Hz), 101.2 (d, JF–C = 18.3 Hz), 71.1, 70.8, 69.8, 69.6. HRMS (AP-TOF) m/z: [M + H]+ calcd for C20H21Br2F4O5: 574.9692, found: 574.9713. Anal. calcd for C20H20Br2F4O5: C, 41.69; H, 3.50. Found: C, 41.70; H, 3.45.Preparation of dibromide 5[6]
The reaction of Ts(OCH2CH2)6OTs (4[6], 4.40 g, 7.45 mmol, 1.0 eq., see the ESI†) with 4-bromo-2,3-difluorophenol (3, 3.27 g, 15.65 mmol, 2.1 eq.) in EtOH (40 mL) in the presence of t-BuOK (1.84 g, 16.40 mmol, 2.2 eq.) resulted in 3.53 g (73% yield) of 5[6] as a colorless oily liquid. 1H NMR (400 MHz, CDCl3) δ 7.19 (ddd, J = 9.4, 7.1, 2.5 Hz, 2H), 6.70 (ddd, J = 9.5, 7.6, 2.1 Hz, 2H), 4.18 (t, J = 4.8 Hz, 4H), 3.86 (t, J = 4.8 Hz, 4H), 3.73–3.69 (m, 4H), 3.68–3.63 (m, 12H). 19F{1H} NMR (376 MHz, CDCl3) δ −127.8 (d, J = 20.1 Hz, 2F), −152.9 (d, J = 20.1 Hz, 2F). 13C{1H} NMR (126 MHz, CDCl3) δ 148.8 (dd, JF–C = 247, 12.1 Hz), 148.0 (dd, JF–C = 8.4, 2.5 Hz), 142.2 (dd, JF–C = 252, 15.0 Hz), 126.5 (d, JF–C = 4.6 Hz), 111.0 (d, JF–C = 3.3 Hz), 101.1 (d, JF–C = 18.2 Hz), 71.1, 70.72, 70.69, 69.8, 69.6. 13C {1H;19F} NMR (126 MHz, CDCl3) δ 148.8, 148.0, 142.2, 126.5, 111.0, 101.1, 71.1, 70.73, 70.69, 69.8, 69.6. HRMS (AP-TOF) m/z: [M + H]+ calcd for C24H29Br2F4O7: 663.0216, found: 663.0223. Anal. calcd for C24H28Br2F4O7: C, 43.39; H, 4.29. Found: C, 43.60; H, 4.15.Preparation of dibromide 5[9]
The reaction of Ts(OCH2CH2)9OTs (4[9], 3.03 g, 4.19 mmol, 1.0 eq., see the ESI†) and 4-bromo-2,3-difluorophenol (3, 1.93 g, 9.22 mmol, 2.2 eq.) in EtOH (20 mL) in the presence of t-BuOK (1.22 g, 10.89 mmol, 2.6 eq.) gave 2.21 g (67% yield) of 5[9] as a colorless oily liquid. The typical yield of the reaction is in the range 64–67%. 1H NMR (400 MHz, CDCl3) δ 7.19 (ddd, J = 9.4, 7.1, 2.5 Hz, 2H), 6.70 (ddd, J = 9.4, 7.7, 2.1 Hz, 2H), 4.18 (t, J = 4.8 Hz, 4H), 3.86 (t, J = 4.8 Hz, 4H), 3.73–3.68 (m, 4H), 3.68–3.60 (m, 24H). 19F{1H} NMR (376 MHz, CDCl3) δ −127.1 (d, J = 20.0 Hz, 2F), −152.3 (d, J = 20.1 Hz, 2F). 13C NMR (101 MHz, CDCl3) δ 148.8 (dd, JF–C = 248, 12.2 Hz), 148.0 (dd, JF–C = 8.3, 2.7 Hz), 142.2 (dd, JF–C = 252, 14.9 Hz), 126.5 (d, JF–C = 4.6 Hz), 111.0 (d, JF–C = 3.5 Hz), 101.1 (d, JF–C = 18.3 Hz), 71.1, 70.74, 70.69, 69.8, 69.6. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C30H41Br2F4O10: 795.1003, found: 795.0989. Anal. calcd for C30H40Br2F4O10: C, 45.24; H, 5.06. Found: C, 45.20; H, 5.01.General procedure for the synthesis of diphenols 6[n]
To a mixture of 5[n] (0.25 mmol), Pd2(dba)3 (4 mol%), [HPCy3][BF4] (8 mol%), t-BuOK (8 mol%), and (4-hydroxyphenyl)boronic acid (69 mg, 0.50 mmol) in THF (15 mL), a solution of K2CO3 (73 mg, 0.53 mmol) in H2O (10 mL) was added at room temperature. After refluxing the reaction mixture for 42 h, 10% HCl was added at room temperature until the solution became acidic. All the organic volatiles were removed in vacuo and the resulting aqueous layer was extracted in CH2Cl2 (3 × 15 mL). The extracts were combined, dried (Na2SO4) and the solvent was removed. The target diphenol 6[n] was isolated by column chromatography (SiO2, CH2Cl2/EtOAc, 3:2).
Preparation of diphenol 6[4]
The reaction of 5[4] (2.00 g, 3.47 mmol, 1.0 eq.) with 4-hydroxyphenylboronic acid (957 mg, 6.94 mmol, 2.0 eq.) in THF (25 mL) in the presence of Pd2(dba)3 (127 mg, 0.14 mmol, 4 mol%), [HPCy3][BF4] (102 mg, 0.28 mmol, 8 mol%), t-BuOK (32 mg, 0.28 mmol, 8 mol%), and K2CO3 (1.01 g, 7.29 mmol, 2.1 eq.) dissolved in H2O (25 mL) gave 1.51 g (72% yield) of 6[4] as a white solid: mp 132–135 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 2H), 7.31 (dd, J = 6.8, 1.5 Hz, 4H), 7.16 (td, J = 8.7, 2.2 Hz, 2H), 7.04 (ddd, J = 9.2, 7.8, 1.8 Hz, 2H), 6.84 (d, J = 8.7 Hz, 4H), 4.21 (t, J = 4.4 Hz, 4H), 3.77 (t, J = 4.5 Hz, 4H), 3.62–3.53 (m, 8H). 19F{1H} NMR (376 MHz, DMSO-d6) δ −140.8 (d, J = 20.9 Hz, 2F), −157.1 (d, J = 20.9 Hz, 2F). 13C{1H} NMR (101 MHz, DMSO-d6) δ 157.3, 147.8 (dd, JF–C = 246, 11.0 Hz), 146.4 (dd, JF–C = 7.8, 2.5 Hz), 140.7 (d, JF–C = 245, 15.3 Hz), 129.7 (d, JF–C = 2.9 Hz), 124.7, 123.7 (t, JF–C = 4.1 Hz), 122.3 (d, JF–C = 10.7 Hz), 115.5, 110.3 (d, JF–C = 3.3 Hz), 70.0, 69.9, 68.9, 68.8. HRMS (ESI-TOF) m/z: [M − H]+ calcd for C32H29F4O7: 601.1849, found: 601.1866. Anal. calcd for C32H30F4O7: C, 63.78; H, 5.02. Found: C, 63.84; H, 4.87.Preparation of diphenol 6[6]
The reaction of 5[6] (2.38 g, 3.58 mmol, 1.0 eq.) with (4-hydroxyphenyl)boronic acid (1.04 g, 7.67 mmol, 2.0 eq.) in THF (25 mL) in the presence of Pd2(dba)3 (132 mg, 0.14 mmol, 4 mol%), [HPCy3][BF4] (106 mg, 0.28 mmol, 8 mol%), t-BuOK (32 mg, 0.28 mmol, 8 mol%), and K2CO3 (1.04 g, 7.52 mmol, 2.1 eq.) dissolved in H2O (25 mL) gave 1.71 g (69% yield) of 6[6] as a yellow-orange colored, highly viscous oil. 1H NMR (400 MHz, DMSO-d6) δ 9.69 (s, 2H), 7.32 (d, J = 8.0 Hz, 4H), 7.17 (t, J = 8.7 Hz, 2H), 7.04 (t, J = 8.5 Hz, 2H), 6.84 (d, J = 8.1 Hz, 4H), 4.21 (bs, 4H), 3.76 (bs, 4H), 3.62–3.48 (m, 16H). 19F{1H} NMR (376 MHz, DMSO-d6) δ −141.5 (d, J = 21.0 Hz, 2F), −157.8 (d, J = 20.9 Hz, 2F). 13C{1H} NMR (125 MHz, DMSO-d6) δ 157.3, 147.9 (dd, JF–C = 245, 10.5 Hz), 146.5 (dd, JF–C = 7.9, 2.2 Hz), 140.8 (dd, JF–C = 245, 15.1 Hz), 129.9 (d, JF–C = 3.1 Hz), 124.8, 123.8 (t, JF–C = 3.9 Hz), 122.4 (d, JF–C = 10.4 Hz), 115.7, 110.4 (d, JF–C = 3.2 Hz), 70.1, 69.93, 69.90, 69.0, 68.9. 13C {1H;19F} NMR (126 MHz, DMSO-d6) δ 157.3, 147.9, 146.5, 140.8, 129.9, 124.8, 123.8, 122.4, 115.7, 110.4, 70.1, 69.93, 69.90, 69.0, 68.9. HRMS (AP-TOF) m/z: [M + H]+ calcd for C36H39F4O9: 691.2530, found: 691.2540. Anal. calcd for C36H38F4O9: C, 62.90; H, 5.55. Found: C, 62.87; H, 5.28.Preparation of diphenol 6[9]
The reaction of 5[9] (1.44 g, 1.99 mmol, 1 eq.) with 4-hydroxyphenylboronic acid (715 mg, 5.17 mmol, 2.6 eq.) in THF (25 mL) in the presence of Pd2(dba)3 (182 mg, 0.20 mmol, 10 mol%), [HPCy3][BF4] (117 mg, 0.32 mmol, 16 mol%), t-BuOK (35 mg, 0.32 mmol, 8 mol%) and K2CO3 (715 mg, 5.17 mmol, 2.6 eq.) dissolved in H2O (25 mL) gave 1.09 g (67% yield) of 6[9] as a yellow-orange colored highly viscous oil. 1H NMR (400 MHz, DMSO-d6) δ 9.66 (s, 2H), 7.32 (d, J = 6.8 Hz, 4H), 7.16 (td, J = 8.7, 2.2 Hz, 2H), 7.08–7.01 (m, 2H), 6.86 (d, J = 8.6 Hz, 4H), 4.24–4.18 (m, 4H), 3.80–3.74 (m, 4H), 3.62–3.57 (m, 4H), 3.55–3.45 (m, 24H). 19F{1H} NMR (376 MHz, CDCl3) δ −140.3 (d, J = 19.8 Hz, 2F), −156.6 (d, J = 19.8 Hz, 2F). 13C{1H} NMR (101 MHz, CDCl3) δ 156.2, 148.7 (dd, J = 248, 11.1 Hz), 147.0 (dd, JF–C = 8.4, 2.5 Hz) 141.9 (dd, JF–C = 247, 15.3 Hz), 129.9 (d, JF–C = 3.0 Hz), 126.6, 123.3 (t, JF–C = 3.8 Hz), 123.2, 115.8, 110.1 (d, JF–C = 3.6 Hz), 70.9, 70.7, 70.6, 70.55, 70.50, 69.7, 69.4. HRMS (ESI-TOF) m/z: [M − H]+ calcd for C42H49F4O12: 821.3160, found: 821.3154. Anal. calcd for C42H50F4O12: C, 61.31; H, 6.13. Found: C, 61.36; H, 6.08.Preparation of the ion pair S2[NMe4]
To a solution of phenol 7[NMe4] (100 mg, 0.29 mmol, 1 eq.) and NaH (60% dispersion in mineral oil, 17 mg, 0.44 mmol, 1.5 eq.) in THF (5 mL), was added freshly prepared 4-(4-pentylcyclohexyl)benzoyl chloride (104 mg, 0.35 mmol, 1 eq.). 4-(4-Pentylcyclohexyl)benzoyl chloride was obtained by treating 4-(4-pentylcyclohexyl)benzoic acid (98 mg, 0.36 mmol) in dry CH2Cl2 (5 mL) under argon with oxalyl chloride (40 μl, 0.46 mmol, 1.3 eq.) in the presence of two drops of DMF for 6 hours, followed by the removal of the solvent in the rotary evaporator. The reaction mixture was stirred overnight and THF was removed using the rotary evaporator. Water was added (5 mL) to the crude mixture and the white precipitate formed was collected by filtration. This was recrystallized from MeCN at ambient temperature giving 98 mg (56% yield) of pure S2[NMe4] as a white microcrystalline powder: mp 285–290 °C (dec). 1H NMR (400 MHz, CD3CN) δ 8.05 (d, J = 8.3 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.5 Hz, 2H), 7.07 (d, J = 8.5 Hz, 2H), 3.06 (s, 12H), 2.66–2.56 (m, 3H), 2.50–0.80 (bm, 11H), 2.11–2.04 (m, 2H), 1.88 (d, J = 11.6 Hz, 4H), 1.57–1.44 (m, 2H), 1.40–1.18 (m, 9H), 1.14–1.02 (m, 2H), 0.90 (t, J = 6.9 Hz, 3H). 11B NMR (128 MHz, CD3CN): δ −9.9 (d, J = 111 Hz, 1B), −13.4 (d, J = 140 Hz, 10B). 13C{1H} NMR (101 MHz, CD3CN) δ 166.1, 155.3, 150.1, 140.8, 130.9, 130.14, 130.10, 128.22, 128.18, 122.6, 118.3, 70.9 (bs), 56.17, 56.13, 56.09, 45.4, 42.2, 38.0, 37.9, 36.7, 34.7, 34.1, 32.9, 27.3, 23.4, 14.4. HRMS (ESI-TOF) m/z: [M + H]− calcd for C27H45B11O2: 522.4443, found: 522.4420. Anal. calcd for C31H56B11NO2: C, 62.71; H, 9.51; N, 2.36. Found: C, 62.15; H, 9.78; N, 2.47.Preparation of lithium salt S2[Li]
A precipitate of S2[NMe4] (85 mg, 0.14 mmol, 1 eq.) in water was acidified with 10% HCl (5 mL). The acidic form of the ester was extracted with Et2O (3 × 15 mL) and a solution of LiOH (4 mg, 0.14 mmol, 1.0 eq.) and LiCl (6 mg, 0.14 mmol, 1.0 eq.) in H2O (10 mL) was added to the combined organic layer. All organic volatiles were removed using a rotary evaporator. The aqueous mixture obtained was extracted with EtOAc (3 × 15 mL) and the combined organic layer was concentrated in a rotary evaporator. The resulting oily material was dissolved in a minimum amount of EtOAc and petroleum ether was carefully layered. This was allowed to stand for two days and the white precipitate formed was collected by filtration. The precipitate obtained was dried giving 64 mg (85% yield) S2[Li] as a solvate with H2O and AcOEt in the form of a white powder: mp 272–295 °C (dec). 1H NMR (400 MHz, CD3CN) δ 8.05 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 7.07 (d, J = 8.5 Hz, 2H), 2.65–2.57 (m, 3H), 2.50–0.80 (bm, 11H), 2.27–2.20 (m, 2H), 2.13–2.04 (m, 2H), 1.88 (d, J = 12.0 Hz, 2H), 1.51 (q, J = 12.3 Hz, 2H), 1.39–1.24 (m, 9H), 1.08 (q, J = 12.2 Hz, 2H), 0.90 (t, J = 6.9 Hz, 3H); EtOAc in the sample at δ 4.05 (q, J = Hz, 2H), 2.10 (s, 3H) and 1.20 (t, J = Hz, 3H). 11B NMR (128 MHz, CD3CN) δ −9.5 (bs, 1B), −13.4 (d, J = 140 Hz, 10B). 13C{1H} NMR (101 MHz, CD3CN) δ 166.1, 155.2, 150.0, 140.8, 130.8, 130.0, 128.1, 122.5, 70.8 (bs), 45.3, 42.1, 38.0, 37.8, 36.6, 34.6, 34.0, 32.8, 27.2, 23.3, 14.3. 7Li NMR (156 MHz, CDCl3) δ −2.6 (s, 1Li). Anal. calcd for C27H44B11O2Li: C, 61.60; H, 8.42; calcd for C27H44B11O2Li·3H2O·2AcOEt: C, 55.55; H, 8.79. Found: C, 55.85; H, 9.02.For the mixture formulation the salt S2[Li] was dried in vacuo making it highly hygroscopic.
Preparation of electrolyte solutions
Electrolyte mixtures were prepared by dissolving 10 mol% of Li+ salt S2[Li] in an appropriate LC host 2F[n]. Thus, a solution of the liquid crystalline host 2F[n] and 10 mol% of Li+ salt S2[Li] in CH2Cl2 was evaporated to dryness in a rotary evaporator and dried under high vacuum to obtain the electrolyte samples. The electrolyte sample 2F[6]–S2[Li] (150 mg) was prepared by dissolving 8 mg of S2[Li] in 142 mg of 2F[6] and 2F[9]–S2[Li] (150 mg) was prepared by dissolving 7 mg of S2[Li] in 143 mg of 2F[9].Impedance spectroscopy
Electrochemical impedance spectroscopy measurements were performed in the range of 60 to 140 °C using two types of cells for measuring the ionic conductivity in the parallel and perpendicular directions relative to the smectic layer (interdigitated gold electrode covered with a plane glass slide and ITO cell, respectively). A computer-interfaced multichannel potentiostat with a frequency range of 500 kHz to 1 Hz and a 5 mV AC signal amplitude was used for the electrochemical impedance spectroscopy measurements. Two types of cells were used for measuring the ionic conductivity in the parallel and perpendicular directions (interdigitated gold electrode and ITO cell, respectively). The cells were filled with the electrolytes in their respective isotropic phases. At each temperature point the cells were thermostated for a minimum of one hour using a cryostat–thermostat system (Haake K75 with a DC50 temperature controller), before taking a series of measurements, which were averaged.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this project was provided by the European Space Agency (NPI 542) grant. Dr Eva Oton Martinez of Miliary University of Technology is thanked for providing electrooptical nylon-6 coated ITO cells for impedance measurements and Dr Jannick Guschlbauer for technical assistance.References
- J. W. Goodby, in Handbook of Liquid Crystals, ed. J. W. Goodby, C. Tschierske, P. Raynes, H. Gleeson, T. Kato and P. J. Collings, Wiley-VCH, Weinheim, 2014, vol. 4, pp. 43–68 Search PubMed.
- J. Uchida, B. Soberats, M. Gupta and T. Kato, Adv. Mater., 2022, 2109063 CrossRef CAS PubMed.
- J. Sakuda, E. Hosono, M. Yoshio, T. Ichikawa, T. Matsumoto, H. Ohno, H. Zhou and T. Kato, Adv. Funct. Mater., 2015, 25, 1206–1212 CrossRef CAS.
- A. Kuwabara, M. Enomoto, E. Hosono, K. Hamaguchi, T. Onuma, S. Kajiyama and T. Kato, Chem. Sci., 2020, 11, 10631–10637 RSC.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- K. Salikolimi, A. A. Sudhakar and Y. Ishida, Langmuir, 2020, 36, 11702–11731 CrossRef CAS PubMed.
- H. Shimura, M. Yoshio, A. Hamasaki, T. Mukai, H. Ohno and T. Kato, Adv. Mater., 2009, 21, 1591–1594 CrossRef CAS.
- M. Yoshio, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2004, 126, 994–995 CrossRef CAS PubMed.
- K. Kishimoto, T. Suzawa, T. Yokota, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2005, 127, 15618–15623 CrossRef CAS PubMed.
- Y. Iinuma, K. Kishimoto, Y. Sagara, M. Yoshio, T. Mukai, I. Kobayashi, H. Ohno and T. Kato, Macromolecules, 2007, 40, 4874–4878 CrossRef CAS.
- A. Eisele, K. Kyriakos, R. Bhandary, M. Schönhoff, C. M. Papadakis and B. Rieger, J. Mater. Chem. A, 2015, 3, 2942–2953 RSC.
- J. H. Lee, K. S. Han, J. S. Lee, A. S. Lee, S. K. Park, S. Y. Hong, J.-C. Lee, K. T. Mueller, S. M. Hong and C. M. Koo, Adv. Mater., 2016, 28, 9301–9307 CrossRef CAS PubMed.
- T. Ichikawa, M. Yoshio, A. Hamasaki, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2011, 133, 2163–2169 CrossRef CAS PubMed.
- B. Soberats, M. Yoshio, T. Ichikawa, H. Ohno and T. Kato, J. Mater. Chem. A, 2015, 3, 11232–11238 RSC.
- T. Kobayashi, T. Ichikawa, T. Kato and H. Ohno, Adv. Mater., 2017, 29, 1604429 CrossRef PubMed.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef CAS PubMed.
- E. Quartarone and P. Mustarelli, J. Electrochem. Soc., 2020, 167, 050508 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- T. Ohtake, M. Ogasawara, K. Ito-Akita, N. Nishina, S. Ujiie, H. Ohno and T. Kato, Chem. Mater., 2000, 12, 782–789 CrossRef CAS.
- K. Kishimoto, M. Yoshio, T. Mukai, M. Yoshizawa, H. Ohno and T. Kato, J. Am. Chem. Soc., 2003, 125, 3196–3197 CrossRef CAS PubMed.
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li, N. Tavajohi and B. Li, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS.
- Z. Ahmad, Z. Hong and V. Viswanathan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26672–26680 CrossRef CAS PubMed.
- D. Gopalakrishnan, S. Alkatie, A. Cannon, S. Rajendran, N. K. Thangavel, N. Bhagirath, E. M. Ryan and L. M. R. Arava, Sustainable Energy Fuels, 2021, 5, 1488–1497 RSC.
- M. Marcinek, J. Syzdek, M. Marczewski, M. Piszcz, L. Niedzicki, M. Kalita, A. Plewa-Marczewska, A. Bitner, P. Wieczorek, T. Trzeciak, M. Kasprzyk, P. Łężak, Z. Zukowska, A. Zalewska and W. Wieczorek, Solid State Ionics, 2015, 276, 107–126 CrossRef CAS.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- M. Yoshizawa, T. Mukai, T. Ohtake, K. Kanie, T. Kato and H. Ohno, Solid State Ionics, 2002, 154–155, 779–787 CrossRef CAS.
- T. Onuma, E. Hosono, M. Takenouchi, J. Sakuda, S. Kajiyama, M. Yoshio and T. Kato, ACS Omega, 2018, 3, 159–166 CrossRef CAS PubMed.
- D. Bresser, M. Leclere, L. Bernard, P. Rannou, H. Mendil-Jakani, G.-T. Kim, T. Zinkevich, S. Indris, G. Gebel, S. Lyonnard and L. Picard, ChemSusChem, 2021, 14, 655–661 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. Poater, C. Viñas, I. Bennour, S. Escayola, M. Solà and F. Teixidor, J. Am. Chem. Soc., 2020, 142, 9396–9407 CrossRef CAS PubMed.
- J. Aihara, J. Am. Chem. Soc., 1978, 100, 3339–3342 CrossRef CAS.
- R. B. King, Chem. Rev., 2001, 101, 1119–1152 CrossRef CAS PubMed.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- J. W. Johnson and J. F. Brody, J. Electrochem. Soc., 1982, 129, 2213–2219 CrossRef CAS.
- J. W. Johnson and M. S. Whittingham, J. Electrochem. Soc., 1980, 127, 1653–1654 CrossRef CAS.
- S. G. McArthur, R. Jay, L. Geng, J. Guo and V. Lavallo, Chem. Commun., 2017, 53, 4453–4456 RSC.
- T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizuno, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., Int. Ed., 2014, 53, 3173–3177 CrossRef CAS PubMed.
- S. Li, P. Qiu, J.-X. Kang, Z. Shi, Y. Zhang, Y. Ma and X. Chen, Mater. Chem. Front., 2021, 5, 8037–8046 RSC.
- R. Asakura, L. Duchêne, R.-S. Kühnel, A. Remhof, H. Hagemann and C. Battaglia, ACS Appl. Energy Mater., 2019, 2, 6924–6930 CrossRef CAS.
- M. Green, K. Kaydanik, M. Orozco, L. Hanna, M. A. T. Marple, K. A. S. Fessler, W. B. Jones, V. Stavila, P. A. Ward and J. A. Teprovich Jr., Adv. Sci., 2022, 9, 2106032 CrossRef CAS PubMed.
- S. Kim, K. Kisu, S. Takagi, H. Oguchi and S.-I. Orimo, ACS Appl. Energy Mater., 2020, 3, 4831–4839 CrossRef CAS.
- A. W. Tomich, J. Chen, V. Carta, J. Guo and V. Lavallo, ACS Cent. Sci., 2024, 10, 264–271 CrossRef CAS PubMed.
- H. Braun, R. Asakura, A. Remhof and C. Battaglia, ACS Energy Lett., 2024, 9, 707–714 CrossRef CAS.
- W. S. Tang, M. Matsuo, H. Wu, V. Stavila, W. Zhou, A. A. Talin, A. V. Soloninin, R. V. Skoryunov, O. A. Babanova, A. V. Skripov, A. Unemoto, S.-I. Orimo and T. J. Udovic, Adv. Energy Mater., 2016, 6, 1502237 CrossRef.
- T. J. Udovic, M. Matsuo, W. S. Tang, H. Wu, V. Stavila, A. V. Soloninin, R. V. Skoryunov, O. A. Babanova, A. V. Skripov, J. J. Rush, A. Unemoto, H. Takamura and S.-I. Orimo, Adv. Mater., 2014, 26, 7622–7626 CrossRef CAS PubMed.
- W. S. Tang, A. Unemoto, W. Zhou, V. Stavila, M. Matsuo, H. Wu, S.-I. Orimo and T. J. Udovic, Energy Environ. Sci., 2015, 8, 3637–3645 RSC.
- O. Tutusaus, R. Mohtadi, N. Singh, T. S. Arthur and F. Mizuno, ACS Energy Lett., 2017, 2, 224–229 CrossRef CAS.
- A. Wahab, C. Douvris, J. Klíma, F. Šembera, J. Ugolotti, J. Kaleta, J. Ludvík and J. Michl, Inorg. Chem., 2017, 56, 269–276 CrossRef CAS PubMed.
- N. T. Hahn, T. J. Seguin, K.-C. Lau, C. Liao, B. J. Ingram, K. A. Persson and K. R. Zavadil, J. Am. Chem. Soc., 2018, 140, 11076–11084 CrossRef CAS PubMed.
- J. Guschlbauer, L. Niedzicki, L. Jacob, E. Rzeszotarska, D. Pociecha and P. Kaszyński, J. Mol. Liq., 2023, 377, 121525 CrossRef CAS.
- C. Wang, H. Wang, C. Zheng, B. Li, Z. Liu, L. Zhang, L. Yuan and P. Xu, ACS Med. Chem. Lett., 2023, 14, 92–102 CrossRef CAS PubMed.
- R. Jakubowski, A. Pietrzak, A. C. Friedli and P. Kaszyński, Chem. – Eur. J., 2020, 26, 17481–17494 CrossRef CAS PubMed.
- X. Wang, Y. Chi and T. Mu, J. Mol. Liq., 2014, 193, 262–266 CrossRef CAS.
- F. Zhang, Y. Sun, Z. Wang, D. Fu, J. Li, J. Hu, J. Xu and X. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 23774–23780 CrossRef CAS PubMed.
- M. Broszkiewicz, A. Zalewska, E. Karpierz-Marczewska and L. Niedzicki, J. Mol. Liq., 2024, 400, 124550 CrossRef CAS.
- M. Broszkiewicz, A. Zalewska and L. Niedzicki, Ionics, 2019, 25, 3651–3660 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic details, NMR spectra, DSC traces, texture photomicrographs, EIS and computational details. See DOI: https://doi.org/10.1039/d4dt01246a |
| This journal is © The Royal Society of Chemistry 2024 |