
Twinned crystal Cd0.9Zn0.1S/MoO3 nanorod S-scheme heterojunctions as promising photocatalysts for efficient hydrogen evolution†
Jie
Chen
a,
Ying
Xie
 *a,
Haitao
Yu
a,
Zhenzi
Li
b and
Wei
Zhou
*b
*a,
Haitao
Yu
a,
Zhenzi
Li
b and
Wei
Zhou
*b
aKey Laboratory of Functional Inorganic Material Chemistry, Ministry of Education, School of Chemistry and Materials Science, Heilongjiang University, Harbin, 150080, PR China. E-mail: xieying@hlju.edu.cn
bShandong Provincial Key Laboratory of Molecular Engineering, School of Chemistry and Chemical Engineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan, Shandong 250353, PR China. E-mail: zwchem@hotmail.com
First published on 3rd May 2024
Abstract
Leveraging solar energy through photocatalytic hydrogen production from water stands out as one of the most promising approaches to address the energy and environmental challenges. The choice of catalyst profoundly influences the outcomes of photocatalytic reactions, and constructing heterojunctions has emerged as a widely applied strategy to overcome the limitations associated with single-phase photocatalysts. MoO3, renowned for its high chemical stability, encounters issues such as low photocatalytic efficiency and fast recombination of photogenerated electrons and holes. To tackle these challenges, the morphology of MoO3 has been controlled to form nanorods, simultaneously suppressing the aggregation of the catalyst and increasing the number of surface-active sites. Moreover, to facilitate the separation of photogenerated charge carriers, Cd0.9Zn0.1S nanoparticles with a twin crystal structure are deposited on the surface of MoO3, establishing an S-scheme heterojunction. Experimental findings demonstrate that the synergistic effects arising from the well-defined morphology and interface interactions extend the absorption range to visible light response, improve charge transfer activity, and prolong the lifetime of charge carriers. Consequently, Cd0.9Zn0.1S/MoO3 S-scheme heterojunctions exhibit outstanding photocatalytic hydrogen production performance (3909.79 μmol g−1 h−1) under visible light irradiation, surpassing that of MoO3 by nearly nine fold.
Introduction
Using solar energy photocatalytic technology to split water for hydrogen production is one of the most ideal solutions to address energy and environmental issues nowadays. Photocatalysts can sustainably convert solar energy into storable chemical energy, and water splitting for hydrogen production is a typical example.1 It is worth considering that catalysts play a decisive role in photocatalytic reactions, and one key to effectively achieving visible light photocatalytic water splitting lies in the appropriate selection of photocatalytic materials. Developing semiconductors with suitable bandgap and visible light response is of great importance.2 Currently reported catalytic systems are primarily composed of nitrides, oxides, phosphides, sulfides, graphene, perovskite materials, and so on.3–8 Furthermore, experimental research on their physical structure, morphology, chemical structure, band structure, and applications is highly interesting. In particular, developing photocatalysts with high stability and excellent catalytic efficiency will be highly desired in this field.9,10The impact of photocatalysts on photocatalytic hydrogen production primarily lies in their bandgaps. Narrow bandgap semiconductors have higher solar light absorption and utilization, while wide bandgap semiconductors possess stronger oxidation or reduction capabilities. Therefore, a single-phase semiconductor cannot simultaneously have strong redox capabilities and high visible light absorption. To solve these issues, researchers have obviously improved the catalytic activity of the semiconductors through many methods such as element doping,11 morphology control,12 defect engineering,13,14 and building heterojunctions.15,16 Among these approaches, the construction of type II heterojunctions,17 Z-scheme heterojunctions,18 and S-scheme heterojunctions19,20 has become a focal point. Type II heterojunctions enable the separation of photogenerated electrons and holes, facilitating effective spatial charge transfer. However, effective photogenerated charge carriers are concentrated at relatively low oxidation or reduction potentials, making this design less ideal.21 Z-scheme heterojunction photocatalysts can enhance the charge separation efficiency while maintaining strong oxidation–reduction capabilities. Nonetheless, the internal electric field consumes some photogenerated electrons and holes, reducing the utilization efficiency of electrons and holes in practical applications.22 In this context, Yu and others have introduced a novel concept known as the S-scheme heterojunction,23,24 which is composed of a reducing photocatalyst (RP) and an oxidizing photocatalyst (OP) with a staggered band structure. Different from traditional type II heterojunctions, the S-scheme heterojunctions can maintain highly active holes and electrons and thus exhibit much higher photocatalytic activities.25–28 More specifically, the photogenerated electrons and holes are retained in the valence band (VB) of OP and the conduction band (CB) of RP, allowing the participation of strong oxidation holes and reduction electrons in the subsequent photocatalytic reactions.29,30
According to this concept, MoO3 with a bandgap of 2.8–3.2 eV and with abundant reserves is non-toxic and has high chemical stability, and it was adopted as the OP component to achieve good UV and visible light response. However, it has drawbacks such as low photocatalytic efficiency and easy recombination of photogenerated electrons and holes. To overcome these drawbacks, one promising approach to enhance the photocatalytic performance of the catalysts is controlling their morphologies.31,32 Additionally, combining them with other materials to form heterojunctions can improve light absorption and suppress charge recombination, resulting in excellent catalytic activity.33,34 To further overcome the drawback of MoO3 and construct better S-scheme heterojunctions, sulfides are usually chosen as promising candidates. Because they have suitable band structures and oxidation–reduction potentials, as well as excellent visible light absorption capabilities, allowing them to rapidly generate electron–hole pairs and thus exhibit good photocatalytic performance.35–37 At the same time, by forming a heterojunction with the very stable MoO3, the stability of sulfides can be improved in turn owing to the strong adhesion between them, leading to a synergistic effect that simultaneously enhances the overall photocatalytic performance and cycling stability of the material.
Motivated by the above considerations, MoO3 nanorods were intentionally prepared as the basic component to grow defect-rich Cd0.9Zn0.1S nano-twin crystals. The successful construction of the S-scheme heterojunction preserves the strong reduction and oxidation capabilities of the photogenerated electrons in the conduction band (CB) of MoO3 and the photogenerated holes in the valence band (VB) of Cd0.9Zn0.1S, while the induced internal electric field at the interface facilitates the charge separation efficiency and prolongs the lifetime of the carriers. In addition, Cd0.9Zn0.1S nano-twin crystals with sulfur vacancies can effectively improve the visible light absorption, and they can be stabilized via the strong interfacial interactions, being responsible for the excellent stability of the composite. The two components of the S-scheme heterojunction collaborated intimately with each other to determine the overall photocatalytic performance.
Results and discussion
The preparation scheme of the S-scheme heterojunction composite is shown in Fig. 1(a), in which the in situ solvothermal method was applied. The synthetic details are summarized in the ESI.† To determine the crystal structures of the materials, X-ray diffraction (XRD) was conducted, as shown in Fig. S1 of the ESI† and Fig. 1(b). The peaks for MoO3 at 12.78°, 23.33°, 25.69°, 27.32°, 33.76°, 38.97°, 49.26°, 59.20°, and 64.53° correspond to the (020), (111), (040), (021), (111), (060), (002), (241), and (062) crystal planes, respectively, which are in accordance with the standard card (JCPDS card number 76-1003). For Cd0.9Zn0.1S, the peaks at 24.93°, 26.66°, 28.33°, 36.82°, 43.90°, 48.12°, and 52.11° correspond to the (100), (002), (101), (102), (110), (103), and (112) crystal planes, respectively, which are similar to the hexagonal CdS lattice according to the standard card (JCPDS card number 80-0006) (Fig. S1 of the ESI†). The diffraction peaks fall between those of CdS and ZnS, suggesting that the prepared sample is not a mixture of CdS and ZnS phases but rather a single-phase structure, consistent with previous literature studies.38–40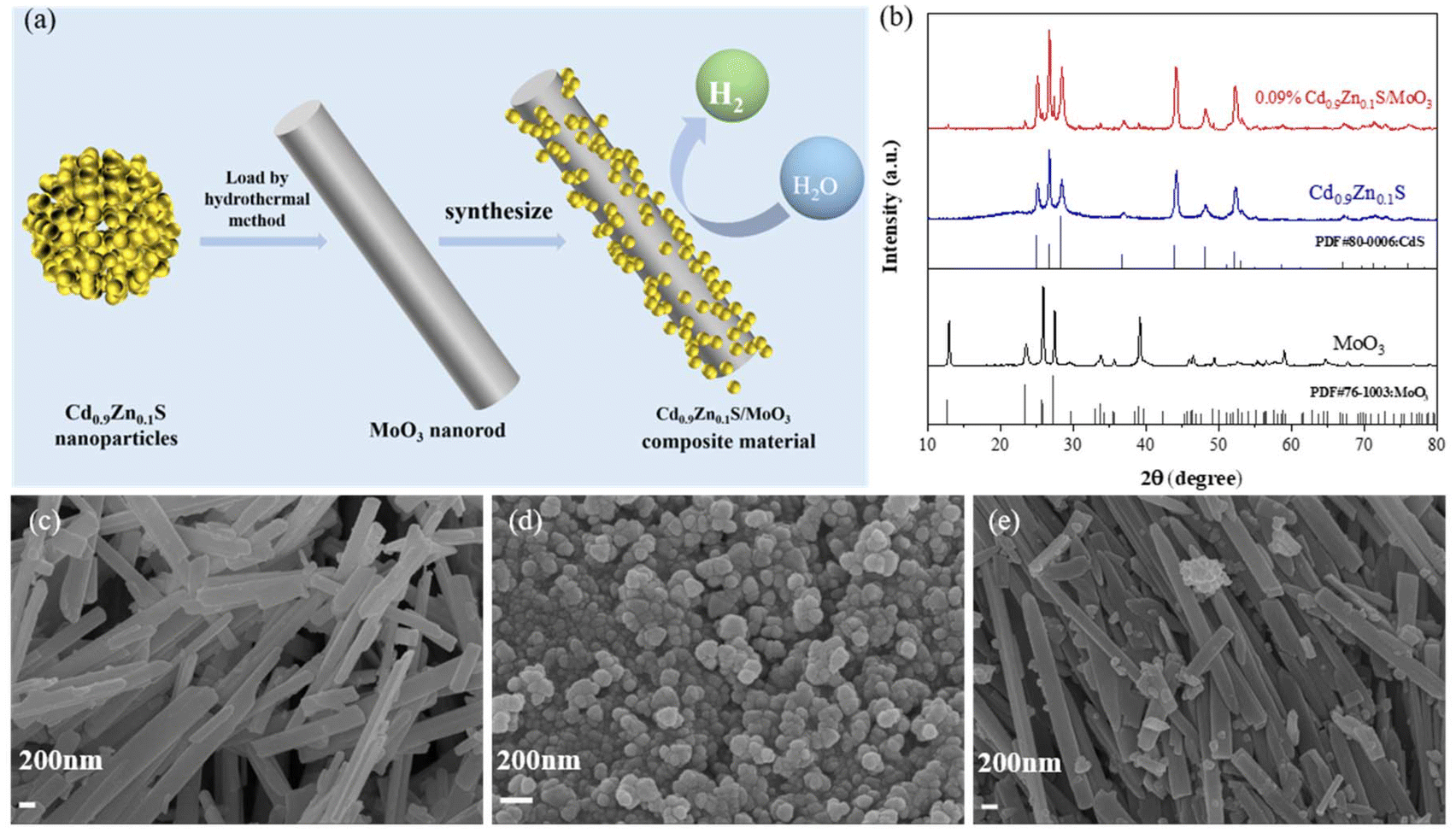 | ||
| Fig. 1 (a) Scheme for the synthesis of 0.09% Cd0.9Zn0.1S/MoO3. (b) XRD patterns of MoO3, Cd0.9Zn0.1S and 0.09% Cd0.9Zn0.1S/MoO3. SEM images of (c) MoO3, (d) Cd0.9Zn0.1S, and (e) 0.09% Cd0.9Zn0.1S/MoO3. | ||
After forming the composite, the diffraction peaks of both MoO3 and Cd0.9Zn0.1S are visible, indicating the absence of any other impurities. The intensity of the diffraction peaks also confirms the good crystallinity of the three samples. This result demonstrates the successful preparation of the composite material and provides the possibility for further research on the synergistic effects of the two components. Scanning electron microscopy (SEM) is used to characterize the microstructure and morphology of different samples. The observations revealed that MoO3 (Fig. 1(c) and Fig. S2(a)†) appears as smooth-surfaced nanorods with a width of ∼200 nm and a length of ∼4 μm, while Cd0.9Zn0.1S (Fig. 1(d)) appears as evenly distributed nanoparticles with moderate interstitial spaces. Cd0.9Zn0.1S/MoO3 (Fig. 1(e) and Fig. S2(b)†) shows a morphology with Cd0.9Zn0.1S nanoparticles distributed on the surface of MoO3 nanorods. This observation also indicates the successful preparation of the Cd0.9Zn0.1S/MoO3 composite.
Transmission electron microscopy (TEM) was used to further characterize the microstructure of the samples. Fig. 2(a)–(g) reveal that the Cd0.9Zn0.1S/MoO3 composite material consists of two phases: MoO3 and Cd0.9Zn0.1S. The (040) crystal planes of the MoO3 nanorods exhibit an interplanar spacing of approximately 0.35 nm, which is the same as the interplanar spacing of single-phase MoO3 (Fig. S3†).41 This suggests that the MoO3 in the composite still possesses its pure phase. The (101) crystal planes of the Cd0.9Zn0.1S phase have an interplanar spacing of approximately 0.31 nm, and high-resolution transmission images of Cd0.9Zn0.1S (Fig. S4†) show stacking faults at some cross-sectional areas and the twin crystal structure.42–45 High-resolution images of Cd0.9Zn0.1S/MoO3 (Fig. 2(e and f)) indicate that Cd0.9Zn0.1S in the composite material also exhibits a twin crystal structure, consistent with the analysis of pure-phase Cd0.9Zn0.1S twin crystals. Particles of Cd0.9Zn0.1S with a size of approximately 60 nm are anchored on the surface of MoO3 nanorods, forming a stable interface. This tight contact between the two components induces significant changes in the electronic structure, facilitating charge separation during the photocatalytic process, which may contribute to the enhanced photocatalytic performance. Furthermore, the EDS in Fig. S5† confirms the successful synthesis of Cd0.9Zn0.1S/MoO3, and the mapping in Fig. 2(g) further validates the uniform distribution of Mo, O, Cd, Zn, and S elements throughout the region.
 | ||
| Fig. 2 (a and b) TEM images, (c and d) HRTEM images, (e and f) 0.09% Cd0.9Zn0.1S/MoO3 twin crystal structure and (g) elemental mappings of 0.09% Cd0.9Zn0.1S/MoO3. | ||
The N2 adsorption–desorption isotherms for different samples were measured, as shown in Fig. 3(a) and Fig. S6.† All samples exhibit typical type-IV isotherms with well-defined hysteresis loops, indicating the mesoporous nature of the materials. The specific surface area of the MoO3 sample is 13.70 m2 g−1 (Table S1†), with a pore size of 22.6 nm. A smaller specific surface area and pore size would significantly reduce the number of active sites and result in poor photocatalytic performance. The addition of Cd0.9Zn0.1S with a larger specific surface area and smaller nanograins to MoO3 leads to an increase in both pore size and specific surface area in Cd0.9Zn0.1S/MoO3. This means that the number of active centers in the composite material is increased, leading to a significant enhancement in the catalytic activity of the Cd0.9Zn0.1S/MoO3 photocatalyst.
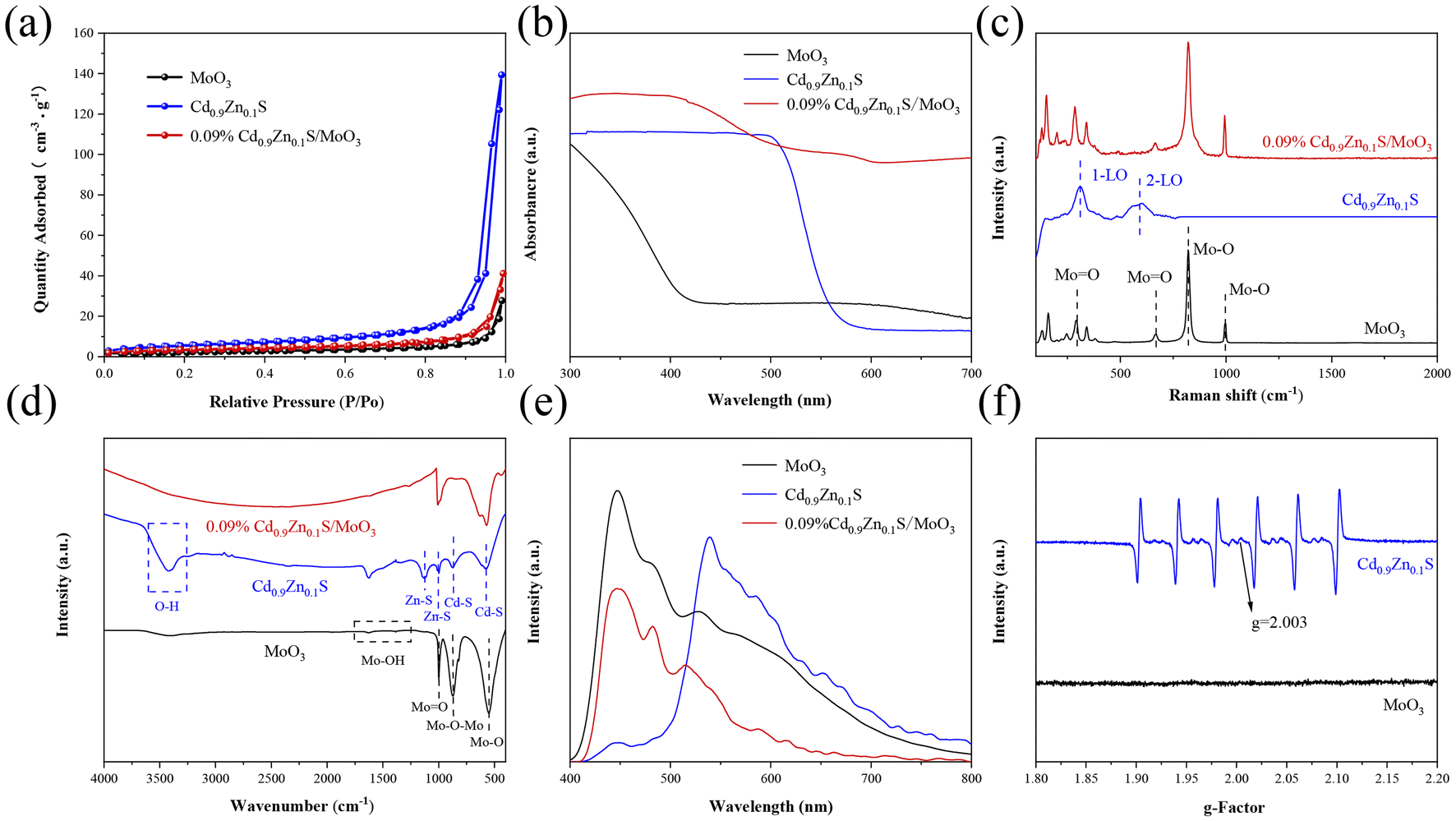 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms, (b) UV-vis DRS, (c) Raman spectra, (d) FTIR spectra, (e) PL spectra, and (f) EPR spectra of MoO3, Cd0.9Zn0.1S and 0.09% Cd0.9Zn0.1S/MoO3. | ||
Ultraviolet–visible (UV-Vis) spectroscopy is performed to analyze the light absorption range of the different samples, as shown in Fig. 3(b). The analysis reveals that the absorption edge of MoO3 is close to 440 nm. In contrast, Cd0.9Zn0.1S exhibits an absorption edge that extends into the visible light range, approximately around 567 nm. Based on the measurements, the bandgaps of MoO3 and Cd0.9Zn0.1S (Fig. S7†) are determined to be approximately 2.82 and 2.20 eV, respectively. When Cd0.9Zn0.1S is loaded onto MoO3, it significantly enhances MoO3 light absorption in the visible light range. The absorption edge of Cd0.9Zn0.1S/MoO3 experiences a redshift, expanding the light absorption range and increasing the visible light response. This improvement enhances the utilization of visible light and ultimately improves the photocatalytic performance of the heterojunction. The Raman spectra are shown in Fig. 3(c). For the original MoO3, distinct Raman peaks are observed at 126, 149, 199.5, 284, 340, 665, 821, and 992 cm−1. The Raman peaks at 284 and 665 cm−1 are associated with bending vibrations of the terminal oxygen (Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), while the peaks at 821 and 994 cm−1 are related to stretching vibrations of the coordination oxygen (Mo–O).46,47 Regarding the Cd0.9Zn0.1S twin crystal structure, its Raman spectrum displays two peaks at 307 and 599 cm−1, corresponding to the first-order and second-order longitudinal optical phonon modes (1-LO and 2-LO).48 The Cd0.9Zn0.1S/MoO3 composite material exhibits characteristic peaks of both MoO3 and Cd0.9Zn0.1S. However, the peak intensities are relatively small for Cd0.9Zn0.1S due to its lower content, and these peaks undergo some shifts, indicating a strong interaction between MoO3 and Cd0.9Zn0.1S. These results collectively confirm the successful synthesis of the Cd0.9Zn0.1S/MoO3 composite material, consistent with the XRD and TEM results mentioned above.
O), while the peaks at 821 and 994 cm−1 are related to stretching vibrations of the coordination oxygen (Mo–O).46,47 Regarding the Cd0.9Zn0.1S twin crystal structure, its Raman spectrum displays two peaks at 307 and 599 cm−1, corresponding to the first-order and second-order longitudinal optical phonon modes (1-LO and 2-LO).48 The Cd0.9Zn0.1S/MoO3 composite material exhibits characteristic peaks of both MoO3 and Cd0.9Zn0.1S. However, the peak intensities are relatively small for Cd0.9Zn0.1S due to its lower content, and these peaks undergo some shifts, indicating a strong interaction between MoO3 and Cd0.9Zn0.1S. These results collectively confirm the successful synthesis of the Cd0.9Zn0.1S/MoO3 composite material, consistent with the XRD and TEM results mentioned above.
Fourier-transform infrared (FT-IR) spectra for MoO3, Cd0.9Zn0.1S, and Cd0.9Zn0.1S/MoO3 are presented in Fig. 3(d). The analysis reveals that peaks at 553 and 880 cm−1 for MoO3 correspond to the stretching mode of three-coordinated oxygen (3Mo–O) and the stretching mode of Mo–O–Mo single bonds, respectively. The peak at 997 cm−1 is associated with the double bond vibration mode of MoO. Two vibrations are observed at around 1390 and 1632 cm−1, corresponding to the vibration modes of Mo–OH bonds and the bending vibration of adsorbed H2O, respectively.33,49 For Cd0.9Zn0.1S, the strong absorption band between 572 and 884 cm−1 is related to the stretching vibrations of Cd–S bonds, while peaks in the range of 995–1130 cm−1 are attributed to the ZnS lattice. The peak around 3400 cm−1 is related to the O–H stretching of surface-adsorbed water.50,51 The FT-IR spectrum of Cd0.9Zn0.1S/MoO3 is similar to that of MoO3 with minimal differences, suggesting that MoO3 is the major component in Cd0.9Zn0.1S/MoO3. These FT-IR results provide insights into the chemical composition and bonding characteristics of the materials and confirm the dominance of MoO3 in the Cd0.9Zn0.1S/MoO3 composite.
From Fig. 3(e), it can be observed that MoO3 exhibits strong photoluminescence (PL) emission peaks at 447, 482, and 527 nm. These peaks are the result of the rapid recombination of photogenerated electron–hole pairs and the radiative decay associated with electron capture. This indicates that the recombination of photo-generated carriers in MoO3 is severe.52 In addition, Cd0.9Zn0.1S also displays a strong PL emission peak at 539 nm, which may be attributed to charge recombination caused by surface defects, suggesting the presence of sulfur vacancies in the Cd0.9Zn0.1S twin crystal.53 In the case of the Cd0.9Zn0.1S/MoO3 composite material, the absorption peak intensity is much lower compared to those of MoO3 and Cd0.9Zn0.1S, indicating effective separation of photogenerated electrons and holes. As the heterojunction can effectively suppress the recombination of photogenerated electrons and holes, promote the utilization of photo-induced charge carriers, and enhance the charge separation efficiency, its photocatalytic performance is expected to be improved significantly.
The electron paramagnetic resonance (EPR) spectra of different samples are used to determine the defect states in the materials. As shown in Fig. 3(f), MoO3 does not exhibit any observable paramagnetic resonance absorption signal, indicating the absence of vacancies. Cd0.9Zn0.1S displays six paramagnetic resonance absorption signals, consistent with previous reports.39 There are two additional characteristic peaks observed for Cd0.9Zn0.1S at g = 2.003 and 1.995. The former can be attributed to sulfur vacancies, while the latter is related to an excess of metal ions caused by stoichiometric reduction of sulfur.39 The Cd0.9Zn0.1S/MoO3 composite material, formed by combining MoO3 with the Cd0.9Zn0.1S twin structure featuring defect states, exhibits a strong interface interaction. This interface promotes the effective separation of photogenerated charge carriers, prolonging the lifetime of electrons, and thereby significantly enhancing the photocatalytic performance.
The X-ray photoelectron spectroscopy (XPS) spectra for MoO3, Cd0.9Zn0.1S, and the Cd0.9Zn0.1S/MoO3 composite material are shown in Fig. 4(a), with the C 1s peak at a binding energy of 284.60 eV as a reference. High-resolution spectra confirm the presence of Mo, O, Cd, Zn, and S elements in the composite material. For the individual phases, in pure MoO3, the binding energy of the O 1s orbital is at 531.35 eV and 530.60 eV (Fig. 4(b)), respectively, corresponding to the surface-adsorbed OH− groups and Mo–O bonds.54,55 The binding energies of Mo 3d are 232.80 eV and 236.01 eV (Fig. 4(c)), corresponding to the Mo6+ 3d5/2 and 3d3/2 orbitals, respectively.28,56,57 In addition, the peak intensities of Mo 3d states in MoO3 are higher than those in 0.09% Cd0.9Zn0.1S/MoO3. The changes in the peak intensity may have originated from the composition change of MoO3 with respect to Cd0.9Zn0.1S in the composite. In pure Cd0.9Zn0.1S, the S 2p orbital has a binding energy of 162.15 eV (Fig. 4(d)), the Cd 3d orbitals are at 405.28 eV (3d3/2) and 412.08 eV (3d5/2) (Fig. 4(e)), and the Zn 2p orbital is at 1021.86 eV (2p3/2) and 1044.96 eV (2p5/2) (Fig. 4(f)).48 When Cd0.9Zn0.1S is introduced into MoO3, the binding energies in the composite material significantly change. The O 1s binding energy is at 531.78 eV and 530.70 eV, and the Mo 3d binding energies are at 232.83 eV and 236.03 eV. After the formation of the heterojunction with Cd0.9Zn0.1S, the binding energies of the Mo 3d orbital shift to a higher binding energy position, indicating the successful incorporation of MoO3 into Cd0.9Zn0.1S. The S 2p binding energy is at 161.93 eV, the Cd 3d binding energies are at 405.19 eV and 411.99 eV, and the Zn 2p binding energies are at 1021.50 eV and 1044.10 eV. In comparison with the literature, the values for Zn 2p states in ZnS were reported to be 1044.60 and 1021.50 eV.58,59 Although the binding energies for the Zn 2p states exhibit some differences, they may have originated from the stoichiometry change of the compound. Moreover, the formation of the heterojunction can also induce some variations. Compared with pure MoO3 and Cd0.9Zn0.1S, the signals for MoO3 in the composite shift to a higher binding energy position, indicating a decrease in the electron density. In contrast, the XPS peaks for Cd0.9Zn0.1S in the composite move to a much lower binding energy position with a significant increase in the electron density. This suggests that when MoO3 is in contact with Cd0.9Zn0.1S, strong interfacial interaction can be formed, and it is the driving force that facilitates the diffusion of electrons from MoO3 to Cd0.9Zn0.1S under X-ray radiation. As a result, the S-scheme heterojunction mechanism becomes possible.29
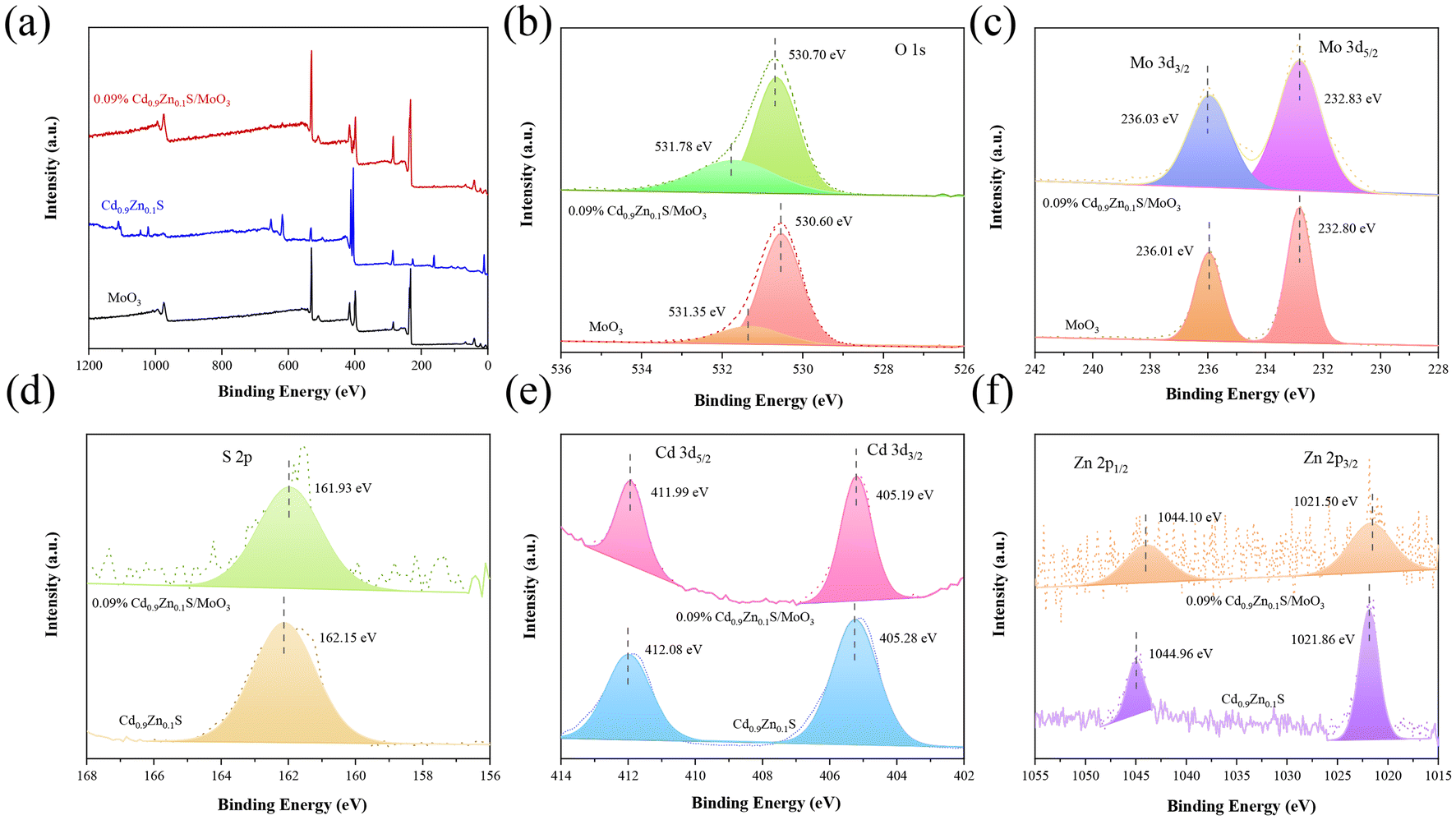 | ||
| Fig. 4 (a) XPS spectra. High-resolution XPS spectra for (b) O 1s, (c) Mo 3d, (d) S 2p, (e) Cd 3d, and (f) Zn 2p orbitals in MoO3, Cd0.9Zn0.1S and 0.09% Cd0.9Zn0.1S/MoO3. | ||
To further investigate the photocatalytic hydrogen production performance of different samples, tests were conducted in a solution composed of 90 mL of pure water and 10 mL of lactic acid under visible light irradiation. It should be noted that the applied sacrificial agent will significantly affect the photocatalytic performance. According to our tests in Fig. S8 of the ESI,† it can be found that when using triethanolamine as the sacrificial agent, the hydrogen production rate of MoO3 is only slightly lower than the value obtained by using lactic acid as the sacrificial agent. However, for Cd0.9Zn0.1S, the hydrogen production rate obtained by using the two sacrificial agents exhibits a very large difference. Generally, using sacrificial agents with poor effect may cause some difficulty in distinguishing the performance differences as it would easily induce a larger error. In this context, lactic acid was used as the standard agent to perform the subsequent photocatalytic tests. The data are shown in Fig. 5(a). The results indicate that within four hours, hydrogen production from all samples increased almost linearly. As shown in Fig. 5(b), pure MoO3 produced the least amount of hydrogen under visible light irradiation (425.19 μmol g−1 h−1). Cd0.9Zn0.1S, due to its unique twinned crystal structure, exhibited a high hydrogen production rate of 1287.09 μmol g−1 h−1. After the formation of heterojunction, it further improved the photocatalytic hydrogen production performance. In comparison with pure MoO3 and Cd0.9Zn0.1S, the performances of all composites with different MoO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cd0.9Zn0.1S ratios are better. The hydrogen production rate of 0.09% Cd0.9Zn0.1S/MoO3 is the best with a hydrogen production rate of 3909.79 μmol g−1 h−1, which is nearly 9 times higher than that of MoO3 and 3 times higher than that of Cd0.9Zn0.1S. This result demonstrates that the interface interaction between MoO3 and Cd0.9Zn0.1S has a positive impact on the photocatalytic performance. Furthermore, stability tests for MoO3 and Cd0.9Zn0.1S/MoO3 (Fig. 5(c)) indicate that the decrease in hydrogen production can be negligible, demonstrating the high stability. The XRD (Fig. S9†) and SEM (Fig. S10†) results for Cd0.9Zn0.1S/MoO3 after cycling further confirm that the structure of the catalyst remained almost unchanged. This indicates that the heterojunction formed by MoO3 and Cd0.9Zn0.1S not only effectively enhances the photocatalytic activity but also exhibits high photostability and good resistance to photo-corrosion, demonstrating the stabilization effect of the interfacial interactions on the heterojunction. To reveal the underlying catalytic mechanism, transient photocurrent (TPC) measurements, linear sweep voltammetry (LSV), electrochemical impedance spectroscopy (EIS), and Mott–Schottky analysis were performed in an electrochemical system under visible light irradiation. The results in Fig. 5(d) show that the photocurrent of Cd0.9Zn0.1S/MoO3 is much higher than that of MoO3 and Cd0.9Zn0.1S. This suggests that the Cd0.9Zn0.1S/MoO3 composite material has a relatively better ability to generate and separate photogenerated charge carriers, which is an advantage for photocatalysis.60
:Cd0.9Zn0.1S ratios are better. The hydrogen production rate of 0.09% Cd0.9Zn0.1S/MoO3 is the best with a hydrogen production rate of 3909.79 μmol g−1 h−1, which is nearly 9 times higher than that of MoO3 and 3 times higher than that of Cd0.9Zn0.1S. This result demonstrates that the interface interaction between MoO3 and Cd0.9Zn0.1S has a positive impact on the photocatalytic performance. Furthermore, stability tests for MoO3 and Cd0.9Zn0.1S/MoO3 (Fig. 5(c)) indicate that the decrease in hydrogen production can be negligible, demonstrating the high stability. The XRD (Fig. S9†) and SEM (Fig. S10†) results for Cd0.9Zn0.1S/MoO3 after cycling further confirm that the structure of the catalyst remained almost unchanged. This indicates that the heterojunction formed by MoO3 and Cd0.9Zn0.1S not only effectively enhances the photocatalytic activity but also exhibits high photostability and good resistance to photo-corrosion, demonstrating the stabilization effect of the interfacial interactions on the heterojunction. To reveal the underlying catalytic mechanism, transient photocurrent (TPC) measurements, linear sweep voltammetry (LSV), electrochemical impedance spectroscopy (EIS), and Mott–Schottky analysis were performed in an electrochemical system under visible light irradiation. The results in Fig. 5(d) show that the photocurrent of Cd0.9Zn0.1S/MoO3 is much higher than that of MoO3 and Cd0.9Zn0.1S. This suggests that the Cd0.9Zn0.1S/MoO3 composite material has a relatively better ability to generate and separate photogenerated charge carriers, which is an advantage for photocatalysis.60
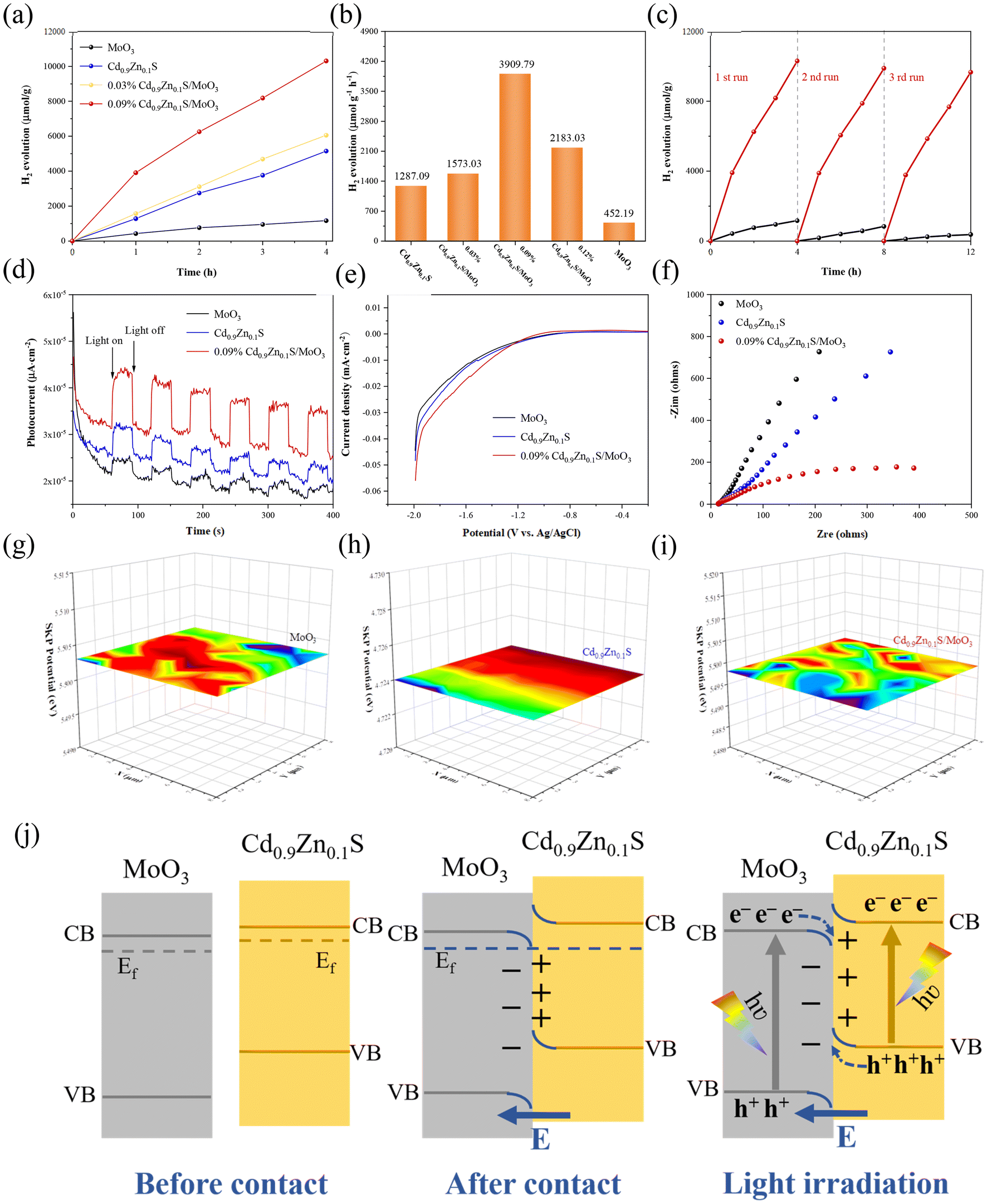 | ||
| Fig. 5 (a) Photocatalytic hydrogen evolution rates of MoO3, Cd0.9Zn0.1S, 0.03% Cd0.9Zn0.1S/MoO3, and 0.09% Cd0.9Zn0.1S/MoO3 under visible light irradiation (λ > 400 nm). (b) Photocatalytic hydrogen evolution rates of MoO3, Cd0.9Zn0.1S, 0.03% Cd0.9Zn0.1S/MoO3, and 0.09% Cd0.9Zn0.1S/MoO3 under visible light irradiation (λ > 400 nm). (c) Cyclic tests of MoO3 and 0.09% Cd0.9Zn0.1S/MoO3. (d) TPC plots, (e) LSV plot statistics over six times, (f) EIS Nyquist plots of MoO3, Cd0.9Zn0.1S and 0.09% Cd0.9Zn0.1S/MoO3. SKP maps of (g) MoO3, (h) Cd0.9Zn0.1S, and (i) 0.09% Cd0.9Zn0.1S/MoO3, respectively. (j) Photocatalytic mechanism maps of 0.09% Cd0.9Zn0.1S/MoO3 S-scheme heterojunction composites. | ||
Furthermore, as confirmed by the LSV curves in Fig. 5(e), Cd0.9Zn0.1S/MoO3 exhibits the lowest overpotential, indicating less resistance when electrons transfer from its surface to adsorbed intermediates, thus facilitating the hydrogen evolution reaction.60 The EIS results which can be used to evaluate the charge carrier conductivity for different samples are shown in Fig. 5(f). Among the three samples, the Cd0.9Zn0.1S/MoO3 composite material has the smallest semicircle, indicating the lowest charge transfer resistance and the highest electron–hole pair separation efficiency.60 In addition, the positive slope in the Mott–Schottky plots (Fig. S11(a and b)†) indicates that the materials have n-type semiconductor properties. The estimated flat-band potentials (VFB) for MoO3 and Cd0.9Zn0.1S are −0.79 and −0.83 V, respectively, according to which the band positions of MoO3 and Cd0.9Zn0.1S can be determined. Moreover, the carrier densities of photogenerated electrons were calculated, and the values for MoO3, Cd0.9Zn0.1S, and Cd0.9Zn0.1S/MoO3 are ∼1.74 × 1029, 1.64 × 1029 and 5.11 × 1029 cm−3 (Fig. S12†), respectively. A lower slope corresponds to a higher carrier concentration, stronger charge transfer activity, and better photocatalytic performance.61
As shown in Fig. 5(g–i), scanning kelvin probe microscopy (SKP) was used as a surface potential measurement technique to measure the work function or surface potential of materials. Work function (WF) can be calculated using the formula (5) in the ESI†, and the work functions of MoO3, Cd0.9Zn0.1S, and Cd0.9Zn0.1S/MoO3 are found to be 5.503 eV, 4.724 eV, and 5.497 eV, respectively. As the work functions and Fermi levels of MoO3 and Cd0.9Zn0.1S are different, their contact thus results in a bending of surface energy bands and the formation of a built-in electric field. Cd0.9Zn0.1S has a smaller work function, and its Fermi level is higher than that of MoO3, indicating the electron flow from Cd0.9Zn0.1S to MoO3.42–45
Based on the provided information, the photocatalytic mechanism of the Cd0.9Zn0.1S/MoO3 composite material can be summarized in Fig. 5(j) and Fig. S13.† MoO3 is an oxidative photocatalyst with a relatively high work function and low Fermi level, which gives it oxidative properties. Cd0.9Zn0.1S, on the other hand, is a reductive photocatalyst with a lower work function and a higher Fermi level, exhibiting reductive properties. When MoO3 and Cd0.9Zn0.1S come into close contact, electrons spontaneously transfer from Cd0.9Zn0.1S to MoO3 until their Fermi levels are equal. This results in the electron loss of Cd0.9Zn0.1S with a positive charge at the interface, while MoO3 gains electrons and carries a negative charge at the interface. The charge state near the interface creates an internal electric field. Simultaneously, due to the loss of electrons, the band edge of Cd0.9Zn0.1S is bent upward, while the band edge of MoO3 is bent downward due to electron accumulation. Under visible light irradiation, electrons in both MoO3 and Cd0.9Zn0.1S can be excited from the valence band (VB) to the conduction band (CB), leaving behind holes in the valence band. Thanks to the formation of the S-scheme heterojunction, the electrons in the CB of MoO3 will transfer to Cd0.9Zn0.1S and recombine with the holes in its VB maximum, as confirmed by the XPS test under X-ray radiation. Therefore, the photogenerated electrons in the CB of Cd0.9Zn0.1S and the holes in the VB of MoO3 can be preserved, and the participation of these highly active carriers in subsequent photocatalytic reactions thus leads to an optimal photocatalytic performance. This unique S-scheme heterojunction promotes the effective separation of photogenerated electrons and holes, prolongs the lifetime of photogenerated charge carriers, and enhances the redox capacity, making Cd0.9Zn0.1S/MoO3 exhibit excellent photocatalytic performance.
Conclusions
An in situ growth method was applied to synthesize a special S-scheme heterojunction between the Cd0.9Zn0.1S twin crystal structure and MoO3 nanorods. This catalyst exhibited excellent photocatalytic hydrogen production performance, with a hydrogen production rate of 3909.79 μmol g−1 h−1, significantly surpassing the values of MoO3 and Cd0.9Zn0.1S. The mechanism analysis revealed that the close interfacial contact between MoO3 and Cd0.9Zn0.1S was a crucial prerequisite for the successful construction of the S-scheme heterojunction. The introduction of the Cd0.9Zn0.1S twin crystal with sulfur vacancies extended the visible light absorption and facilitated the charge separation at the interface, while the strong adhesion between the two components suppressed the photo-corrosion of the catalyst. These synergetic factors collaborated together to significantly enhance the photocatalytic hydrogen production performance of the heterojunction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support of this research by the National Natural Science Foundation of China (22279030 and 52172206), the Shandong Province Natural Science Foundation (ZR2021MB016), and the Development Plan of Youth Innovation Team in Colleges and Universities of Shandong Province.References
- H. L. Wang, W. L. Chan, D. C. Gi, L. Byoung-Hoon, J. H. Jeong, H. Park, J. Heo, M. S. Bootharaju, S. Sung-Hyuk, J. H. Kim, K. H. Ahn, D.-H. Kim and T. Hyeon, Nat. Nanotechnol., 2023, 18, 754 CrossRef PubMed.
- P. M. Rajaitha, S. Hajra, K. Mistewicz, S. Panda, M. Sahu, D. Dubal, Y. Yamauchi and H. J. Kim, J. Mater. Chem. A, 2022, 10, 15906–15931 RSC.
- J. Kröger, F. Podjaski, G. Savasci, I. Moudrakovski, A. Jiménez-Solano, M. W. Terban, S. Bette, V. Duppel, M. Joos, A. Senocrate, R. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Adv. Mater., 2022, 34, 2107061 CrossRef PubMed.
- M. Ai, L. Pan, C. Shi, Z.-F. Huang, X. Zhang, W. Mi and J.-J. Zou, Nat. Commun., 2023, 14, 4562 CrossRef CAS PubMed.
- X. Zhang, M. Gao, L. Qiu, W. Yang and Y. Yu, Chem. Eng. J., 2023, 465, 142747 CrossRef CAS.
- Q. Zhu, Q. Xu, M. Du, X. Zeng, G. Zhong, B. Qiu and J. Zhang, Adv. Mater., 2022, 34, 2202929 CrossRef CAS PubMed.
- G. Yao, S. Yang, S. Jiang, C. Sun and S. Song, Appl. Catal., B, 2022, 315, 121569 CrossRef CAS.
- K. Han, D. M. Haiber, J. Knöppel, C. Lievens, S. Cherevko, P. Crozier, G. Mul and B. Mei, ACS Catal., 2021, 11, 11049–11058 CrossRef CAS PubMed.
- S. Subudhi, S. P. Tripathy and K. Parida, Catal. Sci. Technol., 2021, 11, 392–415 RSC.
- S. Subudhi, S. P. Tripathy and K. Parida, Inorg. Chem. Front., 2021, 8, 1619–1636 RSC.
- S. Khan, M. Je, N. Nu, T. Ton, W. Lei, T. Taniike, S. Yanagida, D. Ogawa, N. Suzuki, C. Terashima, A. Fujishima, H. Choi and K.-I. Katsumata, Appl. Catal., B, 2021, 297, 120473 CrossRef CAS.
- J. Kröger, A. Jiménez-Solano, G. Savasci, V. W. H. Lau, V. Duppel, I. Moudrakovski, K. Küster, T. Scholz, A. Gouder, M.-L. Schreiber, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Adv. Funct. Mater., 2021, 31, 2102468 CrossRef.
- Y. Qin, Z. Li, Y. Duan, J. Guo, M. Zhao and Z. Tang, Matter, 2022, 5, 3260–3310 CrossRef CAS.
- H. Yang, R. Zhao, J. Wang, X. Yin, Z. Lu and L. Hou, ACS Mater. Lett., 2023, 5, 2877–2886 CrossRef CAS.
- C. M. Aitchison, S. Gonzalez-Carrero, S. Yao, M. Benkert, Z. Ding, N. P. Young, B. Willner, F. Moruzzi, Y. Lin, J. Tian, P. D. Nellist, J. R. Durrant and I. McCulloch, Adv. Mater., 2023, 2300037, DOI:10.1002/adma.202300037.
- M. Eshete, X. Li, L. Yang, X. Wang, J. Zhang, L. Xie, L. Deng, G. Zhang and J. Jiang, Small, 2023, 3, 2200041 CrossRef CAS.
- H. Zhang, X. Kong, F. Yu, Y. Wang, C. Liu, L. Yin, J. Huang and Q. Feng, ACS Appl. Energy Mater., 2021, 4, 13152–13160 CrossRef CAS.
- Y. Xing, X. Lu, Y. Li, B. Yang, Y. Huang, M. Zhang, J. Cheng, X. Min and W. Panb, J. Adv. Ceram., 2022, 11, 1789–1800 CrossRef CAS.
- S. Wang, Z. Li, G. Yang, Y. Xu, Y. Zheng, S. Zhong, Y. Zhao and S. Bai, Small, 2023, 2302717, DOI:10.1002/smll.202302717.
- L. Zhang, J. Zhang, H. Yu and J. Yu, Adv. Mater., 2022, 34, e2107668 CrossRef PubMed.
- G. Zhou, L. Meng, X. Ning, W. Yin, J. Hou, Q. Xu, J. Yi, S. Wang and X. Wang, Int. J. Hydrogen Energy, 2022, 47, 8749–8760 CrossRef CAS.
- K. Yin, Z. Yan, N. Fang, W. Yu, Y. Chu, S. Shu and M. Xu, Sep. Purif. Technol., 2023, 325, 124636 CrossRef CAS.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- S. Dash, S. P. Tripathy, S. Subudhi, P. Behera, B. P. Mishra, J. Panda and K. Parida, Langmuir, 2024, 40, 4514–4530 CrossRef CAS PubMed.
- S. P. Tripathy, S. Subudhi, A. Ray, P. Behera, J. Panda, S. Dash and K. Parida, J. Colloid Interface Sci., 2023, 629, 705–718 CrossRef CAS PubMed.
- S. P. Tripathy, S. Subudhi, S. Das, M. K. Ghosh, M. Das, R. Acharya, R. Acharya and K. Parida, J. Colloid Interface Sci., 2022, 606, 353–366 CrossRef PubMed.
- S. Subudhi, G. Swain, S. P. Tripathy and K. Parida, Inorg. Chem., 2020, 59, 9824–9837 CrossRef CAS PubMed.
- T. Li, N. Tsubaki and Z. Jin, J. Mater. Sci. Technol., 2023, 169, 82–104 CrossRef.
- L. Zhang, X. Jiang, Z. Jin and N. Tsubaki, J. Mater. Chem. A, 2022, 10, 10715 RSC.
- Y. Nie, J. Liu, B. Ning, Y. Wang, Q. Cheng, S. He, Q. Guo, R. Zhao and F. Pan, Fuel Process. Technol., 2023, 250, 107871 CrossRef CAS.
- S. K. Fanourakis, J. Peña-Bahamonde and D. F. Rodrigues, Environ. Sci.: Nano, 2020, 7, 3794 RSC.
- G. A. Ashraf, R. T. Rasool, R. U. Rasool, M. F. Saleem, J. Ali, D. Ghernaout, M. Hassan, A. M. Aljuwayid, M. A. Habila and H. Guo, J. Water Process Eng., 2023, 51, 103435 CrossRef.
- O. Chong, X. Quan, C. Zhang, Y. Pan, X. Li, Z. Hong and M. Zhi, Chem. Eng. J., 2021, 424, 130510 CrossRef.
- C. Li, X. Liu, P. Huo, Y. Yan, G. Liao, G. Ding and C. Liu, Small, 2021, 17, 2102539 CrossRef CAS PubMed.
- Q. Chen, X. Chen, Q. Jiang, Z. Zheng, Z. Song, Z. Zhao, Z. Xie and Q. Kuang, Appl. Catal., B, 2021, 297, 120394 CrossRef CAS.
- X. Meng, S. Wang, C. Zhang, C. Dong, R. Li, B. Li, Q. Wang and Y. Ding, ACS Catal., 2022, 12, 10115–10126 CrossRef CAS.
- X. Zhang, Z. Zhao, W. Zhang, G. Zhang, D. Qu, X. Miao, S. Sun and Z. Sun, Small, 2016, 12, 793–801 CrossRef CAS PubMed.
- H.-B. Huang, Z.-B. Fang, K. Yu, J. Lü and R. Cao, J. Mater. Chem. A, 2020, 8, 3882–3891 RSC.
- K. Yu, H.-B. Huang, X.-Y. Zeng, J.-Y. Xu, X.-T. Yu, H.-X. Liu, H.-L. Cao, J. Lü and R. Cao, Chem. Commun., 2020, 56, 7765–7768 RSC.
- Y. Li, H. Sun, X. Cheng, Y. Zhang and K. Zhao, Nano Energy, 2016, 27, 95–102 CrossRef CAS.
- M. Liu, D. Jing, Z. Zhou and L. Guo, Nat. Commun., 2013, 4, 2278 CrossRef PubMed.
- M. Liu, L. Wang, G. Lu, X. Yao and L. Guo, Energy Environ. Sci., 2011, 4, 1372–1378 RSC.
- J. Song, H. Zhao, R. Sun, X. Li and D. Sun, Energy Environ. Sci., 2017, 10, 225–235 RSC.
- S. Lee and S. C. Glotzer, Nat. Commun., 2022, 13, 7362 CrossRef CAS PubMed.
- Y. F. Zhou, K. Bi, L. Wan, X. Ji, C. Wen, K. Huang, C. Liang, Z. B. Sun, D. Y. Fan, H. J. Yang, Y. G. Wang and M. Lei, Mater. Lett., 2015, 154, 132–135 CrossRef CAS.
- V. O. Odhiambo, T. L. Ba, Z. Kónya, C. Cserháti, Z. Erdélyi, M. C. Naomi and I. M. S. Szilágyi, Mater. Sci. Semicond. Process., 2022, 147, 106699 CrossRef CAS.
- B. Sun, J. Wu, W. Wang, H. Wang, Y. Li, Z. Guo, Y. Geng, H. Lin and L. Wang, Appl. Surf. Sci., 2021, 542, 148542 CrossRef CAS.
- A. Khan, M. Danish, U. Alam, S. Zafar and M. Muneer, ACS Omega, 2020, 5, 8188–8199 CrossRef CAS PubMed.
- H. Zheng, Z. Chen, C. Huang, L. Gao, T. Dong and J. Hu, J. Colloid Interface Sci., 2022, 606, 1882–1889 CrossRef CAS PubMed.
- T. Mahvelati-Shamsabadi and E. K. Goharshadi, Ultrason. Sonochem., 2017, 34, 78–89 CrossRef CAS PubMed.
- Q. Xi, J. Liua, Z. Wu, H. Bi, Z. Li, K. Zhu, J. Zhuang, J. Chen, S. Lu, Y. Huang and G. Qian, Appl. Surf. Sci., 2019, 480, 427–437 CrossRef CAS.
- J. Shi, H. n. Cui, Z. Liang, X. Lu, Y. Tong, C. Su and H. Liu, Energy Environ. Sci., 2011, 4, 466–470 RSC.
- P. Behera, A. Ray, S. P. Tripathy, S. Subudhi, L. Acharya and K. Parida, ACS Appl. Eng. Mater., 2023, 1, 2876–2891 CrossRef CAS.
- S. P. Tripathy, S. Subudhi, A. Ray, P. Behera, G. Swain, M. Chakraborty and K. Parida, Langmuir, 2023, 39, 7294–7306 CrossRef CAS PubMed.
- L. Liu, J. Huang, H. Yu, J. Wan, L. Liu, K. Yi, W. Zhang and C. Zhang, Chemosphere, 2021, 282, 131049 CrossRef CAS PubMed.
- S. Wang, X. Zhao, H. M. A. Sharif, Z. Chen, Y. Chen, B. Zhou, K. Xiao, B. Yang and Q. Duan, Chem. Eng. J., 2021, 406, 126849 CrossRef CAS.
- A. Ray, S. Subudhi, S. P. Tripathy, L. Acharya and K. Parida, Adv. Mater. Interfaces, 2022, 9, 2201440 CrossRef CAS.
- A. Ray, S. Sultana, S. P. Tripathy and K. Parida, ACS Sustainable Chem. Eng., 2021, 9, 6305–6317 CrossRef CAS.
- C. Wu, W. Huang, H. Liu, K. Lv and Q. Li, Appl. Catal., B, 2023, 330, 122653 CrossRef CAS.
- Y. Peng, M. Geng, J. Yu, Y. Zhang, F. Tian, Y. n. Guo, D. Zhang, X. Yang, Z. Li, Z. Li and S. Zhang, Appl. Catal., B, 2021, 298, 120570 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt00585f |
| This journal is © The Royal Society of Chemistry 2024 |