
Field induced slow magnetic relaxation in a linear homotrinuclear manganese heterospin coordination compound with S = 7/2 ground state and intriguing spin density distribution†
Igor N.
Shcherbakov
 *a,
Ilya I.
Krotkii
a,
Victoria I.
Kazachkova
a,
Sergey N.
Lyubchenko
a,
Nikolay N.
Efimov
b,
Arshak A.
Tsaturyan
cd and
Vladimir A.
Lazarenko
e
*a,
Ilya I.
Krotkii
a,
Victoria I.
Kazachkova
a,
Sergey N.
Lyubchenko
a,
Nikolay N.
Efimov
b,
Arshak A.
Tsaturyan
cd and
Vladimir A.
Lazarenko
e
aSouthern Federal University, Chemistry Department, Rostov-On-Don, 344006, Russian Federation. E-mail: shcherbakov@sfedu.ru
bKurnakov Institute of General and Inorganic Chemistry of the Russian Academy of Sciences, Moscow, 119991, Russian Federation
cUniversité Jean Monnet Saint-Etienne, CNRS, Institut d'Optique Graduate School, Laboratoire Hubert Curien UMR 5516, 42023, Saint-Étienne, France
dInstitute of Physical and Organic Chemistry, Southern Federal University, 194/2 Stachka Ave., Rostov-On-Don, 344090, Russia
eNational Research Center “Kurchatov Institute”, Moscow, 123182, Russian Federation
First published on 1st April 2024
Abstract
We report a first example of field-induced (HDC = 2500 Oe) slow magnetization relaxation in the homotrinuclear linear heterospin manganese coordination compound with S = 7/2 ground state, based on the bidentate 3,5-di-tert-butyl-1,2-benzoquinone-1-monooxime (HL) ligand with composition {[MnL3]Mn[MnL3]}.
Single molecule magnets (SMMs)1–3 have attracted constant interest due to their potential technological applications4–8 since the early 1990s. The first object with registered slow relaxation of magnetization was a well-known Mn12 complex.9,10 So far, in the 3d-metal series SMM behaviour has been identified for Cr(II), Mn(III), Fe(III), Fe(II), Fe(I), Co(II), and Ni(II) systems.11–13 From this point of view the Mn(II) ion as a potential SMM component is not promising. In a predominant number of compounds, it is in a high spin (HS) state with local S = 5/2, in which five 3d-electrons are distributed with close to spherical symmetry resulting in very small magnetic anisotropy. Nevertheless, a very scarce amount of mononuclear HS-Mn(II) compounds with slow relaxation of magnetization were reported recently.14–17 High stability of the isotropic d5 HS electronic state determines the fact that the local low spin (LS) state with S = 1/2 of Mn(II) is extremely rare. And even in the LS state Mn(II) is not a good SMM candidate because for S = 1/2 spin systems the magnetic anisotropy D parameter is undefined. SMM properties for Mn(II) compounds were reported earlier in the heteronuclear compounds with other metals, for example with lanthanides,18,19 mixed valence Mn(II/III/IV), and CuII/MnII.20 Homopolynuclear Mn(II) compounds with slow magnetization relaxation have not been reported to the best of our knowledge.
Reaction of 3,5-di-tert-butyl-1,2-benzoquinone-1-monooxime (HL)21–23 with Mn(ClO4)2·6H2O in a sealed ampule in isopropanol solution in the presence of triethylamine readily produced dark olive crystals with composition MnL2, which appears to be the trinuclear coordination compound {[MnL3]Mn[MnL3]} (1) (see Scheme 1).
 | ||
| Scheme 1 Synthesis of compound 1. | ||
The synthetic procedure and characterization of 1 are detailed in the ESI.† A probable reaction mechanism is stepwise formation of anionic fac-[MnL3]− units and further coordination of two [MnL3]− anions by Mn2+ with formation of trinuclear compound 1. An earlier analogous trinuclear Mn(II) compound was reported based on α-phenylazobenzaldehyde oxime as ligand.24
The structure of compound 1 was established with the single crystal X-ray diffraction (SCXRD) technique. The crystallographic parameters, structure refinement statistics and selected structural parameters for 1 are given in Tables S1 and S2, ESI.† The molecular structure of 1 is presented in Fig. 1. The formula unit corresponds to {[MnL3]Mn[MnL3]}. The compound is highly symmetric and belongs to the D3 symmetry group. Three Mn ions lie on the C3 symmetry axis (a view of the molecule along the C3 axis is presented in Fig. S2, ESI†). The middle manganese Mn2 atom is at the intersection point of the C3 axis and three perpendicular C2 axes (the angles between the C2 axes are 60°). So, six L− oximate ligands are symmetry equivalent, and both [MnL3]− units are symmetry equivalent and, hence, so are both terminal manganese ions. The three linearly arranged Mn(II) cations are triply bridged by oximate N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of the monodeprotonated ligands,21,22 and the interatomic metal–metal distance is d(Mn1⋯Mn2) = 3.6153(13) Å. The terminal Mn centres are in a 3O3N distorted octahedral donor environment of three L− ligands coordinated in a facial manner with oxygen O1 and nitroso nitrogen N1 atoms (CSM(OC-6) = 3.531, see Table S3, ESI†). The coordination bonds are rather short d(Mn1–O1) = 1.941(3) and d(Mn1–N1) = 1.912(3) Å, and noticeably differ in length (by ca. 0.03 Å).
O groups of the monodeprotonated ligands,21,22 and the interatomic metal–metal distance is d(Mn1⋯Mn2) = 3.6153(13) Å. The terminal Mn centres are in a 3O3N distorted octahedral donor environment of three L− ligands coordinated in a facial manner with oxygen O1 and nitroso nitrogen N1 atoms (CSM(OC-6) = 3.531, see Table S3, ESI†). The coordination bonds are rather short d(Mn1–O1) = 1.941(3) and d(Mn1–N1) = 1.912(3) Å, and noticeably differ in length (by ca. 0.03 Å).
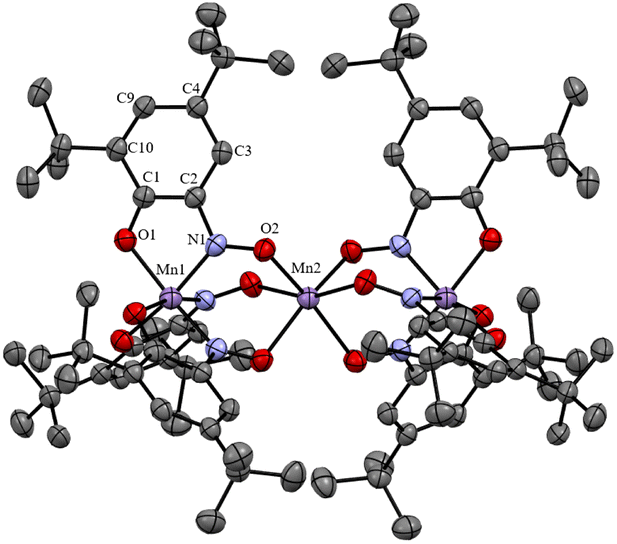 | ||
| Fig. 1 Molecular structure of {[MnL3]Mn[MnL3]} (1) according to the SCXRD data (hydrogen atoms are not shown). Only symmetry non-equivalent atoms are labelled. | ||
The central Mn2 is in a trigonally distorted octahedral 6O coordination environment (CSM(OC-6) = 1.398, see Table S3, ESI†). Mn2 forms much longer equal Mn2–O2 bonds of 2.138(3) Å, which are unambiguously falling in the range of Mn–O coordination bond lengths for Mn(II) in a local HS state. Short coordination bonds of Mn1(Mn1′) ions can arise for different reasons – firstly, due to the LS (S = 1/2) state of the terminal Mn(II) cations, and the {[MnII−LSL3]MnII−HS[MnII−LSL3]} state of 1 or, secondly, due to intramolecular oxidation of Mn(II) to Mn(IV) by reduction of the two L− ligands with formation of two dianion radical species (in analogy with close Mn coordination compounds with catecholate and semiquinonate radical ligands25,26) and the [{MnIV(L2˙−)2(L−)}MnII−HS{MnIV(L2˙−)2(L−)}] state of 1. In the latter case because of the high symmetry of molecule 1 charge (minus five) and spin density (two unpaired electrons) must be smeared over three ligands in each [MnL3]− unit. It appeared to be hard to distinguish these cases based on solely structural data. Of course, intramolecular reduction of the ligand due to metal oxidation must pronouncedly influence the structure of the ligand, namely, the distribution of the bond lengths in the benzoquinone monooximate moiety. Such an influence, for example, for N-arylamidophenoxides of 3d metal complexes, was studied in ref. 27. For coordination compounds of benzoquinone monoxime derivatives such structural studies are not available. The problem for comparison of data in ref. 27 with structural data of the ligand in 1 is that the L− anionic ligand is isoelectronic to neutral iminoquinone. One-electron reduction of L− will lead to a dianion radical (isoelectronic to iminosemiquinone anion-radical) and two-electron reduction to the trianion (isoelectronic to dianion arylamidophenoxide). So, direct comparison of structural data for L in our case is not possible, because it involves species with different formal charges. To overcome this problem, the correlation coefficients between the lengths of C–C, C–N, and C–O bonds in the benzoquinone-monooxime moiety of L in 1 and average bond lengths in metal N-arylamidophenoxides with different ligand charges were calculated (see Table S5, ESI†). Close correlation coefficients were found for structural data of iminoquinone (oxidation state 0, r = 0.93) and iminosemiqinone (oxidation state −1, r = 0.95), indicating that it is not possible to unambiguously choose between the monoanionic or dianion radical state of the ligand L in the compound 1.
The temperature dependence of molar magnetic susceptibility χ for compound 1 was measured in a static magnetic field of HDC = 5000 Oe strength (see Fig. 2, reduced magnetisation and the low temperature region are shown in Fig. S3, ESI†).
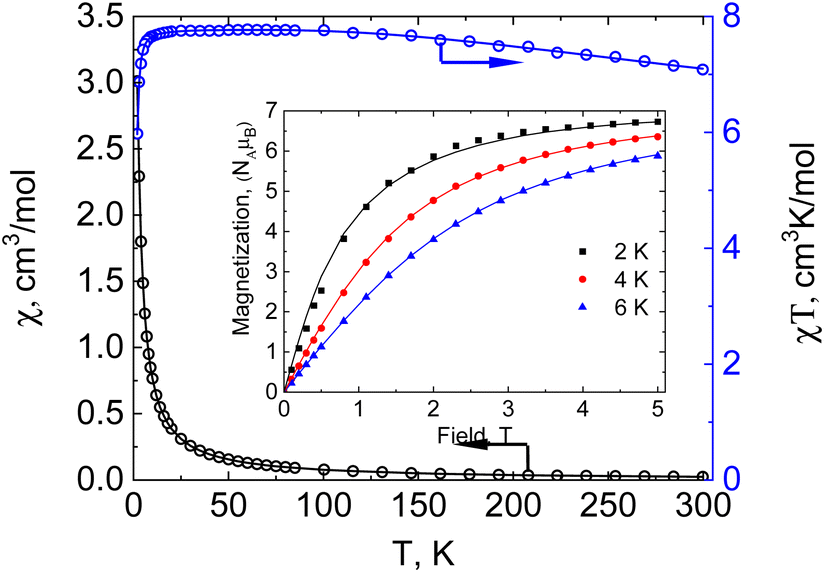 | ||
| Fig. 2 Temperature dependence of χT for 1 measured at HDC = 5000 Oe. Inset: magnetization vs. field for 1 measured at T = 2, 4 and 6 K. Theoretical curves (solid lines) are calculated with JMn1–Mn2 = +67.3(3) cm−1, giso(Mn1) = 1.960(8), giso(Mn2) = 1.998(3), DMn2 = −0.727(8) cm−1, zJ′ = −0.00321(12) cm−1. | ||
The value of the χT product for complex 1 observed at 300 K is 7.08 cm3 K mol−1, which is much lower than the 13.125 cm3 K mol−1 value expected for three independent Mn(II) ions in a local HS state assuming g = 2. So, the terminal Mn ions are for sure not in the d5 HS state, as is supported by XRD data. If a proposition is made about the LS Mn(II) state of the terminal manganese centres, then the uncoupled value must be 5.125 cm3 K mol−1, significantly lower than the experimental value, but if ferromagnetic exchange interaction between Mn centres is considered sufficiently strong to keep a parallel arrangement of electrons at room temperature, then seven aligned electron spins give the theoretical χT value of 7.875 cm3 K mol−1, only slightly higher than the value observed at 300 K. Upon lowering of the temperature, χT increases steadily and reaches a maximum value of 7.78 cm3 K mol−1 at 60 K and then decreases to 7.70 cm3 K mol−1 at ca. 14 K and drops down to 5.98 cm3 K mol−1 at 2 K. Such behaviour supports significant ferromagnetic exchange between spin centres. The field dependence of magnetization (in the range 0–5 T) was measured at 2, 4 and 6 K (see the inset in Fig. 2). Measured at 2 K the magnetization is 6.73NAμB at 5 T, close to the saturation value of 7 NAμB, supporting the S = 7/2 spin of the ground state for compound 1. The temperature dependence of magnetic susceptibility and the field dependence of magnetization were simultaneously fitted by the PHI program,28 assuming a three spin system – two with one unpaired electron and one with five. For the two terminal Mn1 and Mn1′ ions the isotropic g-factor was considered the same due to symmetry relation and different from the g-factor of the middle Mn2. Exchange interactions between Mn2 and Mn1 (Mn1′) centres were considered as equal, JMn1–Mn1′ was arbitrarily taken to be zero. So, the spin Hamiltonian was taken as: 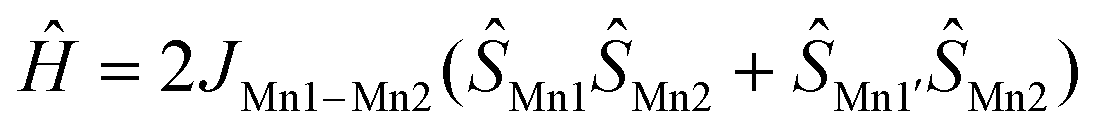 . The molecular field zJ′ was considered. Addition of axial anisotropy parameter D on the Mn2 appeared to be essential for accurate description of the field magnetization dependence, especially at 2 K. A graphical representation of experimental data and fitted dependencies is given in Fig. 2. A very nice description of the experimental data is obtained with JMn1–Mn2 = +67.3(3) cm−1, giso(Mn1) = 1.960(8), giso(Mn2) = 1.998(3), DMn2 = −0.726(8) cm−1, zJ′ = −0.00321(12) cm−1 (residual value R = 0.0054). So, very strong ferromagnetic exchange (extremely unusual for Mn–Mn exchange coupling) interaction determines the ground spin state of 7/2 in 1.
. The molecular field zJ′ was considered. Addition of axial anisotropy parameter D on the Mn2 appeared to be essential for accurate description of the field magnetization dependence, especially at 2 K. A graphical representation of experimental data and fitted dependencies is given in Fig. 2. A very nice description of the experimental data is obtained with JMn1–Mn2 = +67.3(3) cm−1, giso(Mn1) = 1.960(8), giso(Mn2) = 1.998(3), DMn2 = −0.726(8) cm−1, zJ′ = −0.00321(12) cm−1 (residual value R = 0.0054). So, very strong ferromagnetic exchange (extremely unusual for Mn–Mn exchange coupling) interaction determines the ground spin state of 7/2 in 1.
Unprecedented Mn⋯Mn ferromagnetic coupling and uncertainty in explanation of the structural data led us to the idea to perform DFT calculations (PBE0/6-311G(d) level of theory) of 1 and the corresponding [MnL3]− anionic unit (see computational details in the ESI†). For the ground S = 7/2 state of 1 a very interesting spin density (SD) distribution was found (see Fig. 3, left). SD of spin-up (α) electrons is located on the metal centres while there is significant spin-down (β) SD on the bridging NO moieties (values of calculated Mulliken SD and charge are shown in Table 1). Such SD distribution in the trinuclear compound is inherited from the isolated [MnL3]− unit in the S = 1/2 state (see Fig. 3, right), in which a spin-polarized electronic distribution is observed, not accompanied by charge polarization.
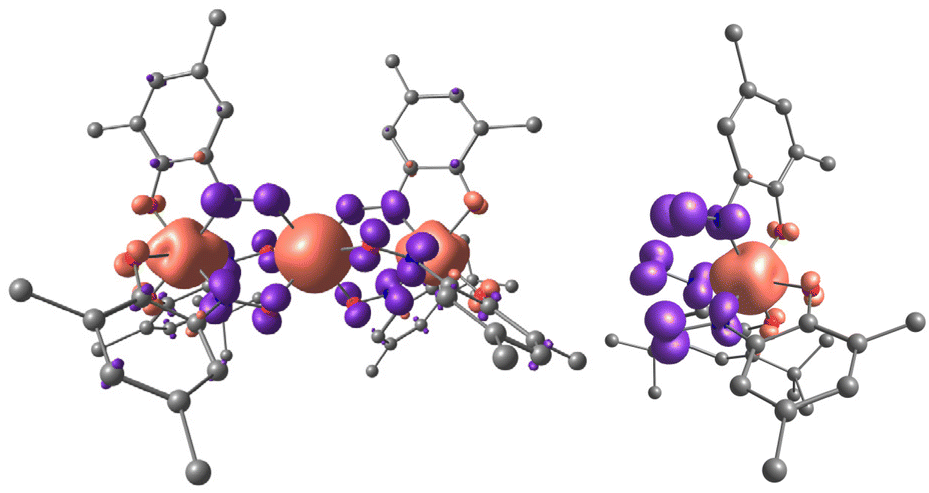 | ||
| Fig. 3 Spin density (SD) distribution in compound 1 (left) and isolated anionic fragment [MnL3]− (right). Methyl groups and hydrogen atoms are omitted for clarity. Red coloured surface – positive SD, blue – negative, contour value 0.01 e Å3 (PBE0/6-311G(d) level of theory). | ||
![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif)
![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif)
![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif)
![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif)
In compound 1 on the central Mn2 (which is undoubtedly in the +2 oxidation state) the Mulliken charge is +1.520, while the charge on the Mn1 and Mn1′ ions is +1.465, even less positive than for the Mn2 ions. Each ligand gains only 0.414 of β-SD which is predominantly located on the NO bridges (0.387 of β-SD). The total Mulliken charge on each ligand is only −0.742e. Thus, the calculated Mulliken charge and SD distribution for 1 is very far from described by the mesomeric formula [{MnIV(L2−)2(L−)}MnII{MnIV(L2−)2(L−)}] with a formal +4 Mn oxidation state of the terminal Mn ions and dianion radical structure for two of the three ligands in [ML3]− (with formal minus five charge and two unpaired electrons distributed evenly over three ligands). Also the {[MnII−LSL3]MnII−HS[MnII−LSL3]} mesomeric structure is not fully supported. The spin-polarized LS state of the [MnL3]− with SD on Mn1 (Mn1′) +2.363 and total opposite sign SDs on the three ligands of −1.242 is observed. So, the actual electronic distribution is in between these two polar mesomers, closer to the latter.
For the [MnL3]− unit the relative energies of states with total spin S = 1/2, 3/2 and 5/2 were calculated to be 0.0, 6277 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 271 cm−1, respectively, proving its local LS ground state.
271 cm−1, respectively, proving its local LS ground state.
The unusual highly spin-polarized state of [MnL3]− is obviously the reason for strong ferromagnetic spin alignment of the metal spin centres. Formally it can be described as mediated by antiferromagnetic interaction of the ligand β-SD (located on NO bridging groups) with the Mn1 and Mn2 metal centres.
So, we tested an alternative description of the magnetochemistry data considering a five spin-centre system: SMn1 = SMn1′ = 3/2, SGL = SGL′ = 1 (fictious centres GL and GL′ describe spin density on the group molecular orbitals of the L3 units involving NO moieties with two unpaired electrons) and SMn2 = 5/2 with the following spin Hamiltonian: 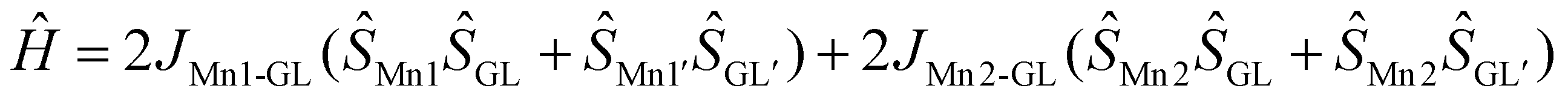 . For all spin centres an isotropic g factor was considered, for GL (GL′) centres g was fixed at 2.0 to avoid overparameterization. D was varied for the terminal Mn centres. The set of the best fit parameters was found to be the following: gMn1 = gMn1′ = 1.9797(10); gMn2 = 2.0061(13); DMn1 = DMn1′ = −1.561(10) cm−1; zJ′ = −0.00326(10) cm−1; JMn1–GL = −240.5(7) cm−1; JMn2–GL = −219(3) cm−1 (residual sum is R = 0.0052). The quality of the fit is the same as for the three spin centres description, and the fitted g-factors for the Mn1 and Mn2 centres and zJ′ values are rather close. The extremely strong ferromagnetic exchange interaction with JMn1–Mn2 = +67.3(3) cm−1 in the case of the three-spin system is replaced by two strong antiferromagnetic exchange interactions of fictious ligand spin centres GL with Mn1 (JMn1–GL = −240.5(7) cm−1) and Mn2 (JMn2–GL = −219(3) cm−1) metal centres, resulting in the same S = 7/2 ground spin state.
. For all spin centres an isotropic g factor was considered, for GL (GL′) centres g was fixed at 2.0 to avoid overparameterization. D was varied for the terminal Mn centres. The set of the best fit parameters was found to be the following: gMn1 = gMn1′ = 1.9797(10); gMn2 = 2.0061(13); DMn1 = DMn1′ = −1.561(10) cm−1; zJ′ = −0.00326(10) cm−1; JMn1–GL = −240.5(7) cm−1; JMn2–GL = −219(3) cm−1 (residual sum is R = 0.0052). The quality of the fit is the same as for the three spin centres description, and the fitted g-factors for the Mn1 and Mn2 centres and zJ′ values are rather close. The extremely strong ferromagnetic exchange interaction with JMn1–Mn2 = +67.3(3) cm−1 in the case of the three-spin system is replaced by two strong antiferromagnetic exchange interactions of fictious ligand spin centres GL with Mn1 (JMn1–GL = −240.5(7) cm−1) and Mn2 (JMn2–GL = −219(3) cm−1) metal centres, resulting in the same S = 7/2 ground spin state.
ac magnetic measurements were initially performed at 2 K at zero and applied external magnetic field. At zero field no out-of-phase signal is registered for 1, application of the field leads to development of the out-of-phase signal (Fig. S4, ESI†). The optimal value of dc field was HDC = 2500 Oe and ac susceptibility measurements were performed at this field. The frequency dependence of the imaginary part of ac susceptibility is shown in Fig. 4, and both components of the dynamic magnetic susceptibility are presented in Fig. S5, ESI.† Relaxation times were plotted against the inverse of temperature and fitted with the theoretical expressions for different relaxation mechanisms and their combination (Fig. 5).
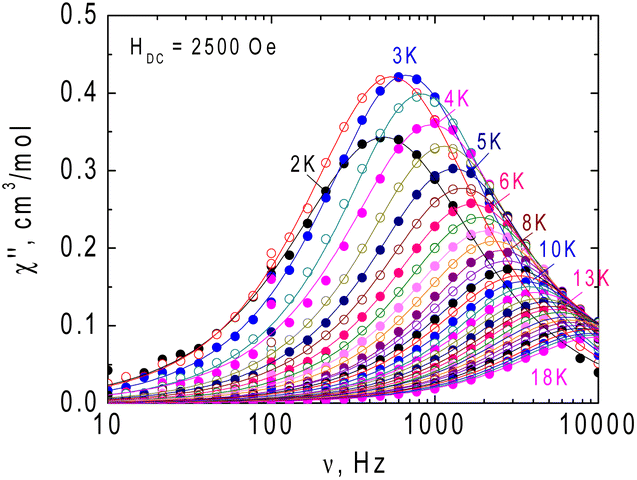 | ||
| Fig. 4 Frequency dependence of the imaginary component (χ′′) of dynamic magnetic susceptibility for 1 in the range 2–18 K at a dc supporting magnetic field of HDC = 2500 Oe. Solid lines are approximations of the experimental data using a generalized Debye model. | ||
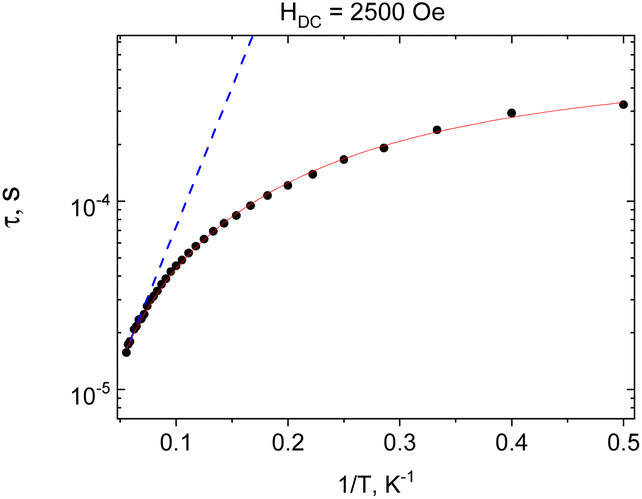 | ||
| Fig. 5 Dependence of the relaxation time and the inverse of temperature τ(1/T) of 1 (HDC = 2500 Oe). Blue dotted line – approximation of the high temperature part by the Arrhenius law. Red solid line – approximation by the sum of Raman and QTM-type contributions. Black circles – experimental data. | ||
The nonlinear course of this dependence evidences the contribution of non-Orbach relaxation mechanisms. The temperature dependence of the relaxation times vs. the inverse of temperature was nicely fitted (R2 = 0.998) with the sum of two contributions – from a Raman process τRaman−1 = CRamanTn and quantum tunnelling of magnetization (QTM, τQTM−1 = B) (red solid line in Fig. 5). The obtained best-fit parameters of the relaxation processes are CRaman = 390(40) K−n s−1, n = 1.78(5), and B = 1.74(10) × 103 s−1.
Approximation of the high-temperature part of the temperature dependence of the relaxation time with the Arrhenius law (blue dotted line in Fig. 5) gives an effective magnetization reversal barrier of Ueff/kB = 34(2) K and τ0 = 2.4(3) × 10−6 s.
In conclusion, a linear homotrinuclear manganese heterospin complex with S = 7/2 ground state showing field induced slow magnetization relaxation was synthesized. Terminal [MnL3]− units are in a local S = 1/2 state, the central Mn(II) ion is in a HS (S = 5/2) state. Spin polarization in [MnL3]− with partial β-spin density on bridging NO groups is determining the strong ferromagnetic interaction between Mn ions (JMn1–Mn2 = +67.3(3) cm−1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Russian Science Foundation (project no. 22-23-00956). N. N. E. thanks the state assignment of the IGIC RAS in the field of fundamental scientific research for magnetic measurements at the JRC PMR IGIC RAS.References
- O. Kahn, Molecular Magnetism, VCH Publishers, New York, 1993 Search PubMed.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- V. V. Lukov, V. A. Kogan, S. I. Levchenkov, I. N. Shcherbakov and L. D. Popov, Russ. J. Coord. Chem., 2015, 41, 1–15 CrossRef CAS.
- D. Gatteschi, Adv. Mater., 1994, 6, 635–645 CrossRef CAS.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- F. Troiani and M. Affronte, Chem. Soc. Rev., 2011, 40, 3119–3129 RSC.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. Novak, Nature, 1993, 365, 141 CrossRef CAS.
- A. Caneschi, D. Gatteschi, R. Sessoli, A. L. Barra, L. C. Brunel and M. Guillot, J. Am. Chem. Soc., 1991, 113, 5873–5874 CrossRef CAS.
- G. A. Craig and M. Murrie, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC.
- S. Gómez-Coca, D. Aravena, R. Morales and E. Ruiz, Coord. Chem. Rev., 2015, 289–290, 379–392 CrossRef.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- R. Mičová, C. Rajnák, J. Titiš, E. Samoľová, M. Zalibera, A. Bieńko and R. Boča, Chem. Commun., 2023, 59, 2612–2615 RSC.
- T. T. da Cunha, V. M. M. Barbosa, W. X. C. Oliveira, E. F. Pedroso, D. M. A. García, W. C. Nunes and C. L. M. Pereira, Inorg. Chem., 2020, 59, 12983–12987 CrossRef CAS PubMed.
- K. Uchida, G. Cosquer, K. Sugisaki, H. Matsuoka, K. Sato, B. K. Breedlove and M. Yamashita, Dalton Trans., 2019, 48, 12023–12030 RSC.
- C. Rajnák, J. Titiš, J. Moncol, R. Mičová and R. Boča, Inorg. Chem., 2019, 58, 991–994 CrossRef PubMed.
- X.-L. Li, F.-Y. Min, C. Wang, S.-Y. Lin, Z. Liu and J. Tang, Dalton Trans., 2015, 44, 3430–3438 RSC.
- I. Oyarzabal, A. Zabala-Lekuona, A. J. Mota, M. A. Palacios, A. Rodríguez-Diéguez, G. Lorusso, M. Evangelisti, C. Rodríguez-Esteban, E. K. Brechin, J. M. Seco and E. Colacio, Dalton Trans., 2022, 51, 12954–12967 RSC.
- E. Pilichos, P. Bhunia, M. Font-Bardia, A. Ghosh, J. Mayans and A. Escuer, Dalton Trans., 2022, 51, 1779–1783 RSC.
- A. M. Ionov, S. N. Lyubchenko, V. A. Kogan and A. Y. Tsivadze, Russ. J. Coord. Chem., 2008, 34, 195–197 CrossRef CAS.
- S. N. Lyubchenko, A. M. Ionov, I. N. Shcherbakov, G. G. Aleksandrov, V. A. Kogan and A. Y. Tsivadze, Russ. J. Coord. Chem., 2006, 32, 539–544 CrossRef CAS.
- I. I. Krotkii, E. Y. Shcherbakova, S. N. Lyubchenko, N. N. Efimov, M. A. Kiskin, V. A. Lazarenko, P. A. Knyazev, S. P. Kubrin, G. S. Borodkin and I. N. Shcherbakov, Polyhedron, 2024, 251, 116876 CrossRef CAS.
- S. Chattopadhyay, P. Basu, S. Pal and A. Chakravorty, J. Chem. Soc., Dalton Trans., 1990, 3829–3833 RSC.
- D. N. Hendrickson and C. G. Pierpont, Valence Tautomeric Transition Metal Complexes in Spin Crossover in Transition Metal Compounds II, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 63–95, DOI:10.1007/b95413.
- M. W. Lynch, D. N. Hendrickson, B. J. Fitzgerald and C. G. Pierpont, J. Am. Chem. Soc., 1981, 103, 3961–3963 CrossRef CAS.
- S. N. Brown, Inorg. Chem., 2012, 51, 1251–1260 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2284760. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04123a |
| This journal is © The Royal Society of Chemistry 2024 |