
Achieving high-capacity aqueous supercapacitors via anion-doped construction of dual redox centers in NixCo1−xSeO3†
Tao
Li
,
Jinyue
Song
,
Hongguang
Fan
,
Yanpeng
Wang
,
Yusheng
Luo
,
Chenchen
Shao
,
Qingping
Li
and
Wei
Liu
 *
*
School of Materials Science and Engineering, Ocean University of China, Qingdao 266100, P. R. China. E-mail: weiliu@ouc.edu.cn
First published on 2nd May 2024
Abstract
In asymmetric supercapacitors, transition metal selenates are promising electrodes, but their capacity are limited by a single redox center. To further enhance the performance of transition metal selenates, NixCo1−xSeO3 (NCSeO) doped with N and Cl was prepared on nickel-plated carbon cloth (NCSeO–NCl–NiCC). During electrochemical reactions, NCSeO can be converted to M(OH)2 (M = Ni/Co) and OH− is replaced by N and Cl. Two redox centers, M(OH)2/MOOH and M(OH)xN2−x/NO3−, are formed during charging and discharging, which is attributed to the increased capacity of the NCSeO–NCl–NiCC electrode. On NCSeO, the substitution of Cl facilitates the regulation of the electronic structure and enhances the stability of N-doping. The optimised electrode exhibits a high capacity of 417 mA h g−1 at 1 A g−1 and an impressive rate capability of 235 mA h g−1 at 50 A g−1. Asymmetric supercapacitors with this design have an ultra-high energy density of 73.6 W h kg−1, as well as an excellent rate and cycling performance with a capacitance retention of 97.8% after 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at a current density of 20 A g−1.
000 cycles at a current density of 20 A g−1.
Introduction
As a result of their high power density and excellent cycling performance, supercapacitors are becoming increasingly popular in plug-in vehicles, machinery and equipment, and backup power sources. However, the low energy density limits their widespread use.1 Therefore, current research on supercapacitors has focused on improving their energy density while maintaining their high power density. Currently, microstructure design, dropwise addition of redox electrolytes and widening of the voltage window are used to improve the energy density of materials, but new strategies are urgently required to improve competitiveness in practical applications.2–4 A proven strategy for increasing the energy density of SCs is the use of battery-based electrode materials. Among them, transition metal oxides have been widely used due to their high theoretical specific capacitance and excellent redox reversibility. Han et al. constructed a 3D NiCo-LDH nanosheet network with high-quality loading on activated carbon cloth by electrodeposition.5 When the mass-loading is 16 mg cm−2 and the current density is 10 mA cm−2, the areal capacitance of the NiCo(NA)-LDH@ACC electrode is 27.3 F cm−2 and the specific capacitance is 1709 F g−1 at 0.62 A g−1. Wu et al. prepared Ni(OH)2/HGO nanocomposites by in situ growth of Ni(OH)2 nanosheets on highly oxidized graphene oxide HGO.6 The composite Ni(OH)2/HGO electrode demonstrated a high capacity of 1430.9 F g−1 at 5 A g−1 and 850 F g−1 at an ultra-high current density of 60 A g−1, enabling excellent fast charging. Research on battery-based electrode materials, however, lacks fine regulation of the charging and discharging processes, limiting further advancement.The theoretical capacity of an electrode material is determined by the number of electrons transferred from each redox center. It is possible to overcome the single-electron transfer limitation through multi-electron transfer processes, but it is more difficult to realize due to the fact that multi-electron transfer processes tend to be more complex thermodynamically and kinetically.7,8 Theoretically Ni(OH)2 undergoes two oxidative redox processes during charging and discharging, Ni2+ → Ni3+ and Ni3+ → Ni4+ processes. The first oxidation process transforms layered Ni(OH)2 into NiOOH, which is more structurally stable and prevents the loss of another protonated hydrogen, making the second oxidation process more difficult.9 Currently, researchers use heteroatom doping to adjust the electronic structure and increase the carrier activity to enhance the reactivity of the electrode.10 Wang et al. prepared in situ-grown Mn-doped Ni(OH)2 on nickel foam, which exhibited high capacity, excellent multiplicity performance, and excellent cycling performance due to the presence of Mn4+ ions altering the electronic configuration, inducing a localized contraction of the metal–O/metal bond lengths, and introducing a special stacking layer disorder between the layers.11 However, compared with metal cation doping, anion doping avoids the disorganization of metal active sites due to doping of transition metals.12 Chang et al. synthesized Ni(OH)2 in saturated NaCl solution using a co-precipitation method in which OH− was partially replaced by Cl−. Cl ion doping significantly increased the interlayer H2O content by altering the polarity of the hydroxide layer to facilitate ultrafast ion transport.13 Cl ion doping also changed the electronic configuration of both Ni and Co, enhancing the electrical conductivity and redox activity. Hao et al. prepared a series of Ni(OH)2 electrode materials doped with halogen elements by electrodeposition, demonstrating that the strong electron-withdrawing property of F, the most electronegative element, causes electrons to escape from the surroundings of O and Ni and create a defect structure that enhances the reactivity and accelerates the reaction kinetics.14 It is evident that the doping of F− and Cl− plays a crucial role in improving the reactivity of nickel-based active materials.
To increase capacity, the electronic structure needs to be modulated to stimulate site activation, and redox pairs need to be added for a greater electron transfer rate. Han et al. studied the electrochemical behavior of nickel oxide (NiO) electrodes in 2 M KOH and 2 M KOH containing different concentrations of the Na2S2O8 electrolyte.15 The reaction mechanism of NiO electrodes in the mixed Na2S2O8 electrolyte is oxidative reduction of Ni2+ to Ni3+ through two different channels, OH− and S2O8−. The NiO electrode has excellent discharge capability in the redox electrolyte, with a specific capacitance as high as 6317.5 F g−1 at 0.5 A g−1. The capacity is enhanced by two redox reactions of the electrode and the electrolyte by adding a redox electrolyte in the electrolyte, but it has poor cycling stability. Therefore, there is an urgent need to find new atoms or groups to further improve the capacity of nickel-based materials.16,17 Compared with oxygen, selenium has stronger metallic properties and lower electronegativity, thereby transition metal selenides and selenites have higher electrical conductivity.18–22 In this work, we synthesized nickel-coated carbon cloth (NiCC) by electrodeposition and grew N ion and Cl ion co-doped NixCo1−xSeO3 (NCSeO) on its surface (NCSeO–NCl–NiCC) by secondary electrodeposition. Introducing SeO32− facilitates the modulation of the electronic structure, increases the exposure of the active sites, facilitates the bonding of the N and Cl ions to the metal active sites, and promotes the diffusion of the electrolyte.19 After a period of electrochemical activation, N and Cl ions replace OH− in M(OH)2 (M = Ni/Co). The experimental results show that during the charging and discharging process, the dual redox centers are constructed through hydroxide ions and N ions will be converted to hydroxyl oxides and NO3−, thus obtaining a higher capacity. In NCSeO, Cl ions can change the electronic structure in favor of N ion attachment, thus improving electrical conductivity and redox activity. Thus, the NCSeO–NCl–NiCC electrodes have an ultra-high capacity of 417 mA h g−1 in an electrolyte of 2 M KOH. Assembled hybrid supercapacitors exhibit excellent energy/power density as well as a cycle life that is comparable to most advanced hybrid/asymmetric supercapacitors.
Experimental
Material synthesis
Firstly, surface impurities were removed by ultrasonication using a 2 cm × 1 cm carbon cloth (CC), which was washed with dilute hydrochloric acid, ethanol and deionised water. The mixture was obtained by dissolving 0.5 M NiSO4·6H2O and 1 M H3BO3 in 200 mL of deionised water (DI) with continuous stirring for 2 h. The mixture was then used as the working electrode. Then, in a three-electrode system, CC was used as the working electrode, a platinum sheet was used as the counter electrode, and a saturated calomel electrode (SCE) was used as the reference electrode. NiCC was obtained by electrodeposition at a constant potential of −1.5 V for 5 min. The working electrode was removed, rinsed repeatedly with deionised water and anhydrous ethanol and dried.0.03 M Ni(NO3)2·6H2O, 0.01 M Co(NO3)2·6H2O, 0.02 M Na2SeO3 and 0.1 M LiCl were dissolved in 200 mL of DI and the pH was adjusted to 4 by HCl. NiCC was used as the working electrode, Pt foil as the counter electrode, and a SCE as the reference electrode, and electrodeposition was carried out through 20 cycles of voltammetry at −1.2 V to 0.2 V and a scanning rate of 5 mV s−1, and the sample obtained was named NCSeO–NCl–NiCC. It was rinsed several times with DI and ethanol and finally dried under vacuum at 70 °C. The sample obtained by using NiCl2·6H2O and CoCl2·6H2O as the electrodeposition solution was NCSeO–Cl–NiCC. NCSeO–N–NiCC is a sample obtained by using Ni(NO3)2·6H2O, Co(NO3)2·6H2O, Na2SeO3, LiNO3 and HNO3 as the electrodeposition solution.
Material characterization
The crystal structure of the samples was characterized by X-ray diffraction (Bruker AXS, Germany). The micro-morphology of the samples was characterized by scanning electron microscopy (SEM, Hitachi S4800) and transmission electron microscopy (TEM, JEM-2100F). X-Ray photoelectron spectroscopy (XPS) tests were performed on a Thermo ESCALAB 250 XI spectrometer using a monochromatic Al-Ka X-ray source. Raman spectroscopy was performed with a micro-Raman (Raman, Renishaw inVia). Fourier transform infrared (FTIR) spectroscopy was performed on a Nicolet 330 FT-IR spectrometer.Density functional theory (DFT) calculations
All density generalization calculations were performed using the Vienna Ab initio Simulation Program (VASP). Spin polarization was performed using the projected augmented wave (PAW) formulation. According to the literature, the GGA+U method was used with Uef (Ni) = 6.2 eV.23 During structural optimization, the total energy convergence criterion was set to 10−6 eV, and the forces on each atom were relaxed to less than 10−5 eV Å−1. All structural optimization calculations were performed using a plane wave cutoff energy of 600 eV. The Brillouin zone sampling of NCSeO–N–NiCC, NCSeO–Cl–NiCC, and NCSeO–NCl–NiCC was sampled using a 5 × 5 × 7 grid. A 20 Å vacuum layer was used to avoid interactions and a 3 × 3 × 1 k-point grid was used for calculations.24,25Electrochemical measurements
The electrode was used as the working electrode. The Pt foil electrode was the counter electrode and the Hg/HgO electrode was the reference electrode, and the tests were carried out using a CHI 6144 (Shanghai Chenhua Instrument Corp.) workstation. Cyclic voltammetry (CV), constant current charge/discharge (GCD) and electrochemical impedance spectroscopy (EIS) measurements were performed in a 2 M KOH aqueous electrolyte. CV curves were tested over a window of 0.0–0.7 V and constant current GCD over a window of 0.0–0.55 V. EIS was performed in the range of 0.01 Hz to 100 kHz. The resistivity of active materials was tested using an ST-2722 semiconductor powder resistivity tester (Suzhou Jingge Electronics). Asymmetric supercapacitors (ASCs) were used with NCSeO–NCl–NiCC electrodes as positive electrodes and commercial activated carbon (AC) as negative electrodes. A mixture of AC, conductive acetylene black and PVDF was made in the ratio of 7:2:1 and the mixture was coated on a piece of carbon cloth and tested in an electrolyte of 2 M KOH.
Results and discussion
Fabrication and characterization of the electrode
Fig. 1a shows the preparation process of the NCSeO–NCl–NiCC electrode. In order to enhance the bonding of the fluid collector and the active substance, a nickel-plated layer was added to the surface of the CC by electrodeposition. The SEM images of CC and NiCC are shown in Fig. S1 (ESI†), and it can be seen that the Ni layer covers the CC surface completely, which makes the CC surface more flat and improves the electronic conduction and mechanical bonding between the CC and the active material. Subsequently, due to the high electrical conductivity of the Ni coating, the NCSeO nanorods can grow uniformly on NiCC to generate the NCSeO–NiCC composite structure.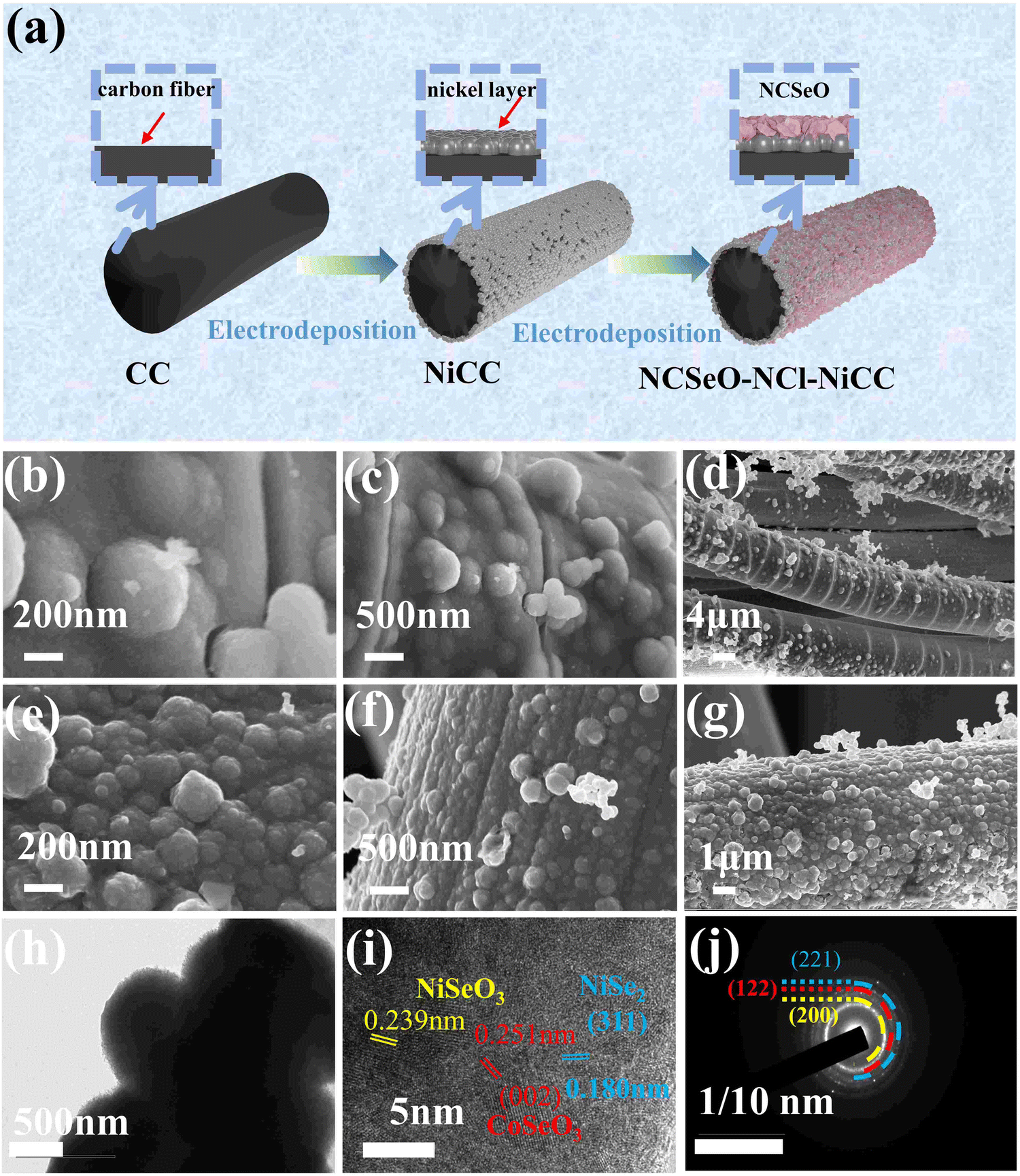 | ||
| Fig. 1 (a) Schematic of the synthesis process for NCSeO–NCl–NiCC. (b)–(d) SEM images of NCSeO–Cl–NiCC (e)–(g) SEM images of NCSeO–NCl–NiCC (h)–(j) TEM, HRTEM and the corresponding SAED images of NCSO–NCl–NiCC. | ||
Subsequently, due to the high electrical conductivity of the Ni coating, the NCSeO nanorods can grow uniformly on NiCC to generate the NCSeO–NiCC composite structure. During the electrodeposition process, NCSeO first covers the NiCC completely and gradually generates NCSeO nanoparticles, and each NCSeO particle is spherical with a uniform diameter of about 200 nm and grows side by side on the CC surface (Fig. 1b and c). The NCSeO layer cracks and forms a structure similar to bamboo joints when only Cl− is present in the electrodeposition solution, which makes it very prone to fracture and crack under stress, resulting in poor cycling performance of NCSeO–Cl–NiCC (Fig. 1d). When NO3− and Cl− were present in the electrodeposition solution, the NCSeO nanoparticles had a faster growth rate, generating small spheres with a diameter of 200 nm, which were tightly immobilized side-by-side on the surface of the carbon cloth. The surface of the NCSeO layer becomes flat and no longer shows bamboo-like cracks, which results in excellent structural stability and continuous conductivity of NCSeO–NCl–NiCC (Fig. 1e–g). From the cross-section of NCSeO–NCl–NiCC (Fig. S1c, ESI†), the CC, Ni layer, and NCSeO layer can be clearly identified, and the NCSeO layer is tightly wrapped around the Ni layer to form a relatively strong bond. The excellent interfacial bonding of the Ni layer also contributes to the extension of the collector, which in turn enhances electronic conduction in the NCSeO layer.26 A homogeneous distribution of Ni, Co, Se, O, and C in the EDS mapping images confirms the formation of NCSeO.
The nanosphere structure of NCSeO was further confirmed by TEM (Fig. 1h), where solid nanospheres were clearly seen to be closely aligned. The lattice stripes with spacings of 0.239 and 0.251 nm in the HRTEM image (Fig. 1i) of NCSeO can be attributed to the (211) and (002) facets of NiSeO3 and CoSeO3, respectively. A lattice stripe of 0.180 nm can be attributed to the (311) face of NiSe2 suggesting that a portion of NiSe2 appeared during the electrodeposition process. This mixed phase exhibits more active sites for redox reactions, which is favorable for the electrochemical performance of the NCSeO electrode. The SAED patterns clearly demonstrate the polycrystalline character of the nanorods (Fig. 1j). The (221) and (122) facets in the diffracted ring are the most important features of the nanorods. The lattice fringes of the diffraction ring further confirm the generation of NiSeO3 and CoSeO3.27
The Fig. 2a shows the XRD patterns of NCSeO–NCl–NiCC and NCSeO–Cl–NiCC between 10° and 70°. The broad peak located at 26° corresponds to CC, and the two characteristic peaks at 44.5° and 51.8° correspond to the (111) and (200) planes of Ni (JCPDS #04-0850), indicating the formation of a Ni coating on the CC surface.28 The remaining diffraction peaks correspond to NiSeO3 (JCPDS #70-0243), CoSeO3 (JCPDS #70-0242), and NiSe2 (JCPDS #74-2075), respectively, suggesting that NiSe2 can be partially generated from NCSeO–NCl–NiCC and NCSeO–Cl–NiCC at the same time as NCSeO grows on the CC, which is also consistent with Raman results (Fig. 2b).29,30 NCSeO–NCl–NiCC and NCSeO–Cl–NiCC exhibit similar XRD patterns, indicating that the addition of N does not affect the lattice of NCSeO. The valence and chemical composition of the NCSeO–NCl–NiCC and NCSeO–Cl–NiCC samples were investigated by XPS, and the full spectra of the two samples are shown in Fig. 2c, and six elements, Ni, Co, Se, O, Cl and C, are present in both samples. The high-resolution Ni 2p spectra of the NCSeO–NCl–NiCC indicate that the peaks at 855.9 eV and 873.4 eV correspond to Ni2+ and the peaks at 857.2 eV and 874.9 eV correspond to Ni3+ (Fig. 2d).31Fig. 2e shows the XPS spectrum of Co 2p, which can be similarly divided into spin orbitals of Co2+ and Co3+ and vibrational satellite peaks (sat.).32 The peaks of Ni 2p3/2 and Ni 2p1/2 in NCSeO–NCl–NiCC are shifted toward high binding energy by 0.2 and 0.17 eV and the peaks of Co 2p3/2 and Co 2p1/2 are shifted toward high binding energy by 0.19 and 0.17 eV, respectively, which implies that the electron density of Ni and Co decreases after N ion doping. N ions may extract electrons from nearby metal atoms, which leads to a decrease in the electron density around Ni and Co.33 The peaks of Se 3d at binding energies of 54.4 eV and 55.3 eV correspond to Se 3d5/2 and Se 3d3/2, respectively, and the peak of the spectrum at 59 eV corresponds to SeOx (Fig. 2f).34 The XPS spectra of N 1s in Fig. 2g show a distinct peak located at 398.2 eV, which is attributed to the Ni–N characteristic bond.35 In the XPS spectrum of Cl, two major characteristic peaks at 199.9 and 198.2 eV can be detected, which can be confirmed as Cl 2p1/2 and Cl 2p3/2 (Fig. 2h).36 This proves that both N and Cl can be successfully doped with NCSeO. Fig. S2 (ESI†) illustrates the XPS spectrum of N in NCSeO–N–NiCC, which indicates that there are Ni–N bonds present in NCSeO–N–NiCC. Based on a comparison of the atomic weights of N in NCSeO–Cl–NiCC, NCSeO–N–NiCC and NCSeO–NCl–NiCC, it can be concluded that Cl is favorable to N doping in NCSeO. In Fig. 2i, the binding energies of N after N doping have been calculated using density functional theory (DFT). The binding energy of N in NCSeO–N–NiCC is 0.26 eV, whereas it is −1.06 eV in NCSeO–NCl–NiCC, which indicates that it is more difficult for N to be doped directly on NCSeO. The Cl significantly reduces N doping binding energy and enhances efficiency and stability, which is consistent with the XPS results.37,38
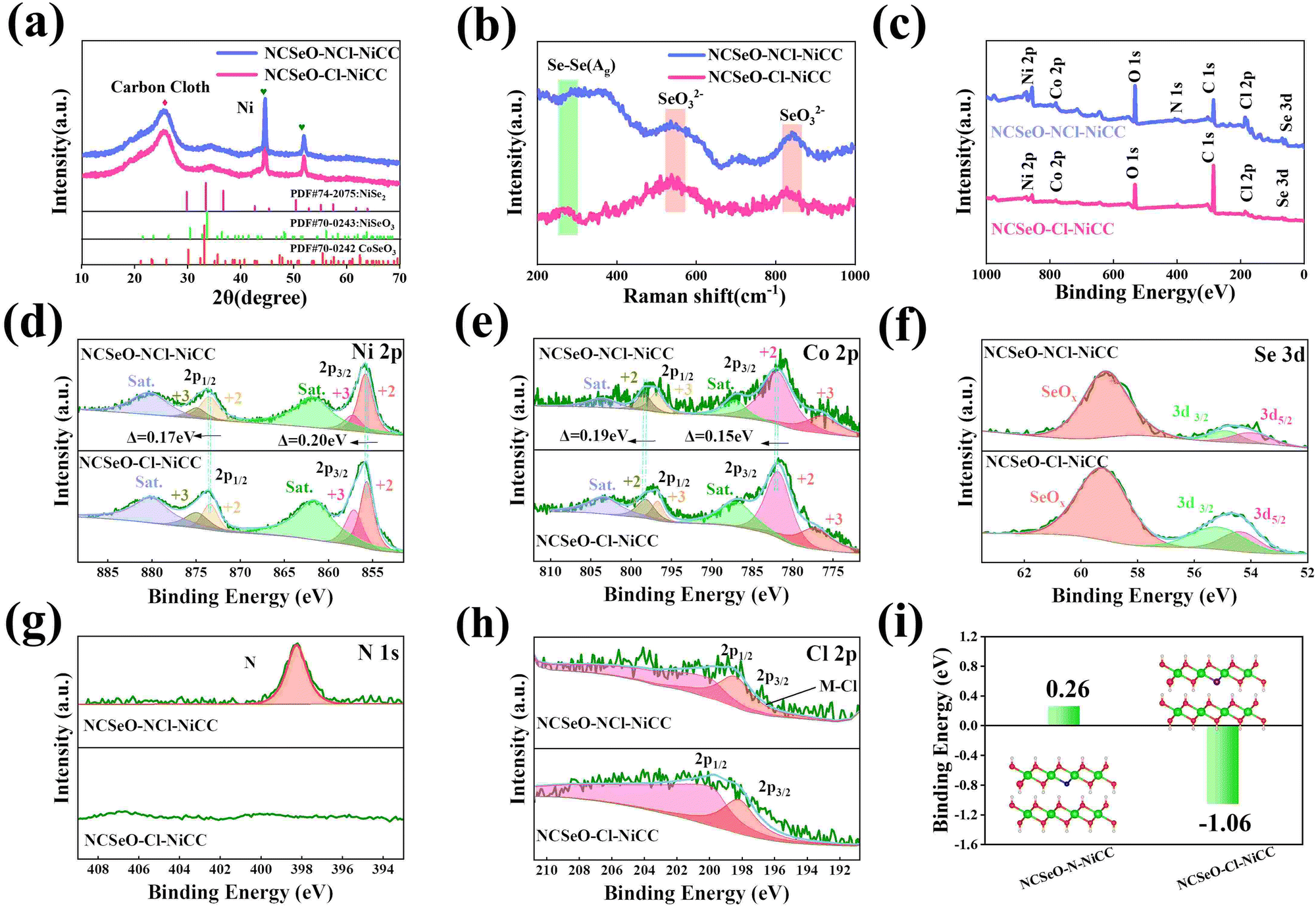 | ||
| Fig. 2 (a) XRD pattern, (b) Raman spectra, (c) the full spectrum of NCSeO–NCl–NiCC and NCSeO–Cl–NiCC. High-resolution XPS spectra of NCSeO–NCl–NiCC and NCSeO–Cl–NiCC samples: (d) Ni 2p, (e) Co 2p, (f) Se 3d, (g) N 1s, and (h) Cl 2p. (i) The binding energy of NCSeO–Cl–NiCC and NCSeO–Cl–NiCC. (The grey, red, pink, blue and purple balls are Ni, O, H, N and Cl atoms, respectively.) | ||
Electrochemical performance
The electrochemical performance of NCSeO–NCl–NiCC was evaluated using a three-electrode configuration in a 2 M KOH aqueous electrolyte. Fig. 3a shows the cyclic voltammetry (CV) curves of the samples in the potential range of 5 mV s−1 and 0.0–0.7 V. The NCSeO–NCl–NiCC electrode has the largest CV curve closure area and the highest peak current, showing better electrochemical reactivity. Fig. S3(a)–(c) (ESI†) show the CV curves of different electrodes at a scan rate of 5–50 mV s−1. At 50 mV s−1, NCSeO–NCl–NiCC shows well-maintained redox peaks, while NCSeO–Cl–NiCC and NCSeO–N–NiCC exhibits a severely distorted CV curve. From the above results, it can be concluded that the introduction of N can reduce the polarization during the electrochemical process of NCSeO and enhance the electrochemical stability. Fig. 3b shows the GCD curves of various electrodes in the range of 0–0.55 V at a current density of 1 A g−1. Compared with the other electrodes, the NCSeO–NCl–NiCC electrode can detect a longer discharge time and exhibit a stronger charge storage capacity. NCSeO–NCl–NiCC can achieve a high specific capacity of 417 mA h g−1 at 1 A g−1 (Fig. 3c), which is much higher than the NCSeO–Cl–NiCC electrode (338 mA h g−1) and the NCSeO–N–NiCC electrode (284 mA h g−1). Three different electrode materials at a current density of 1 mA cm−2 as shown in Fig. S4 (ESI†), and it can be concluded that NCSeO–NCl–NiCC has an excellent area capacitance of 630 mA h cm−2, which is higher than that of 590.4 mA h cm−2 of NCSeO–Cl–NiCC and 482.8 mA h cm−2 of NCSeO–N–NiCC. In addition, in terms of rate performance, the NCSeO–NCl–NiCC electrode also has impressive performance, with a capacity of 235 mA h g−1 remaining at 50 A g−1. Fig. 3d demonstrates that the NCSeO–NCl–NiCC electrode with the NCSeO–Cl–NiCC electrodes have a smaller voltage drop compared to the NCSeO–Cl–NiCC electrodes, which further verifies that more doping of N and Cl is favorable for the formation of a stable NCSeO structure with excellent electronic conductivity and proton conduction with ultrafast kinetics. In order to further explore the electrochemical behaviour of various electrodes, further electrochemical analyses were carried out. The conductivity values of NCSeO–NCl–NiCC, NCSeO–Cl–NiCC and NCSeO–NCl–NiCC were 2.88 S cm−1, 2.35 S cm−1 and 1.41 S cm−1, respectively, proving that the NCSeO–NCl–NiCC electrode has better conductivity. It can be concluded that the NCSeO–NCl–NiCC electrode has higher electrical conductivity, which proves that N and Cl double doping can improve the electrical conductivity of electrode materials. Fig. 3e demonstrates that the NCSeO–NCl–NiCC electrode has an anodic peak with a b-value of 0.74 and a cathodic peak with a b-value of 0.6, suggesting that it has a stronger surface-controlled process.39Fig. 3f demonstrates the Nyquist plot of the different electrodes. In the high-frequency region, NCSeO–NCl–NiCC has a smaller semicircle, corresponding to a smaller charge transfer resistance (Rct). In the low-frequency region, the NCSeO–NCl–NiCC electrode exhibits the largest slope, indicating a lower ion diffusion resistance and a faster diffusion rate. As shown in Fig. 3g, the capacitance contribution of the NCSeO–NCl–NiCC electrode grows from 27% to 67% when the scan rate increases from 0.2 mV s−1 to 5 mV s−1, which is larger than that of the NCSeO–Cl–NiCC electrode (Fig. S3d, ESI†). Combining the above results, the NCSeO–NCl–NiCC electrode exhibits a faster reaction kinetics. The NCSeO–NCl–NiCC electrode showed excellent cycling performance with a capacity retention of 93.5% after 8000 cycles at 20 A g−1 (Fig. 3h), which can be comparable or superior to the reported NiCo-oxides and NiCo-selenides (Table S1, ESI†).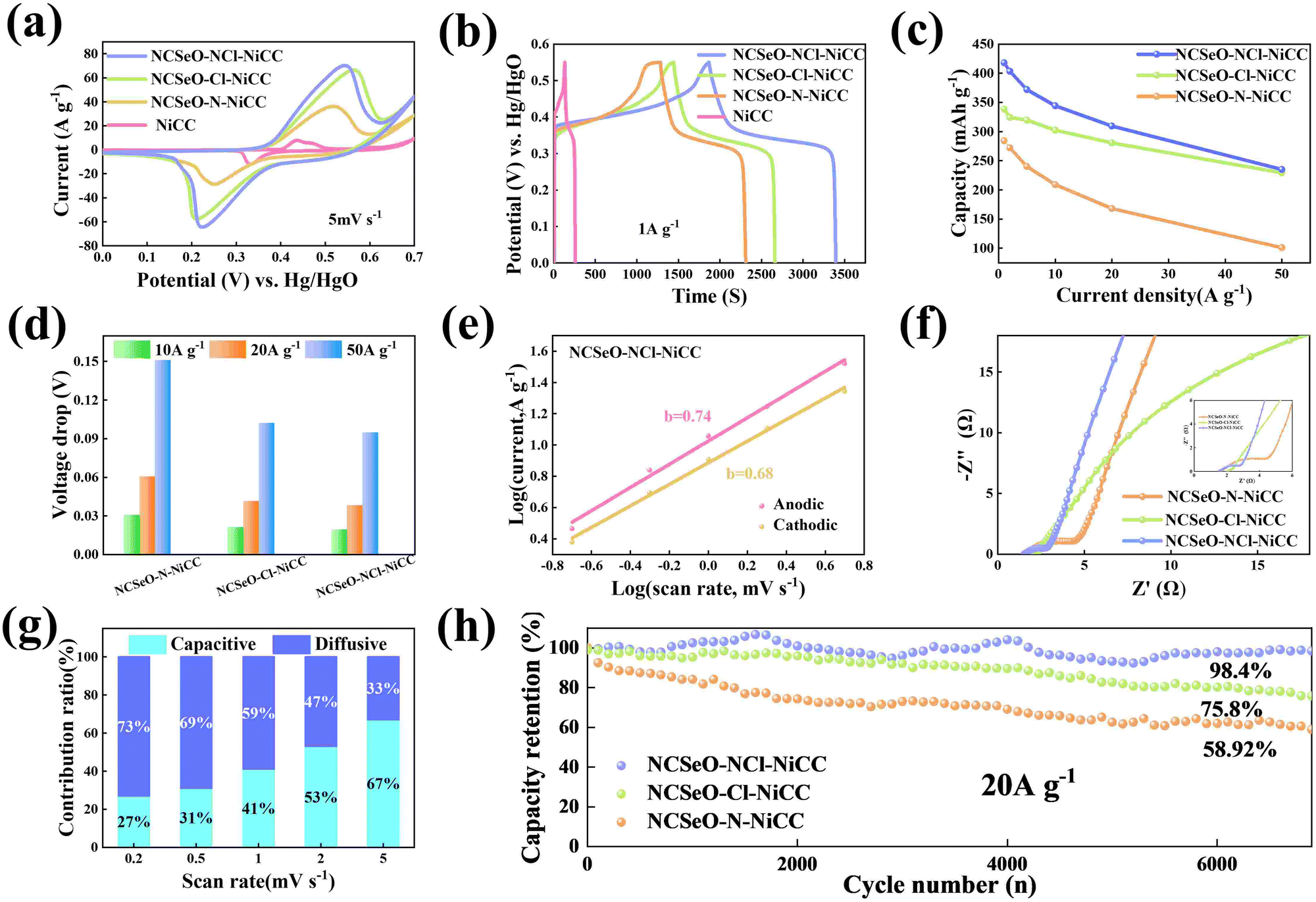 | ||
| Fig. 3 (a) CV curves of various electrodes at a scan rate of 5 mV s−1. (b) GCD curves of various electrodes at a current density of 1 A g−1. (c) Specific capacity at different current densities of various electrodes. (d) The voltage drops at high current densities of different electrodes. (e) The b-values of NCSeO–NCl–NiCC anode–cathode according to the relationship between the peak current and the scan rate. (f) EIS plots of various electrodes. (g) The percentage of the capacitive/diffusion-controlled contribution to the charge storage of the NCSeO–NCl–NiCC electrode at different scan rates. (h) The cycling performance of NCSeO–NCl–NiCC, NCSeO–Cl–NiCC and NCSeO–NCl–NiCC at a current density of 20 A g−1. | ||
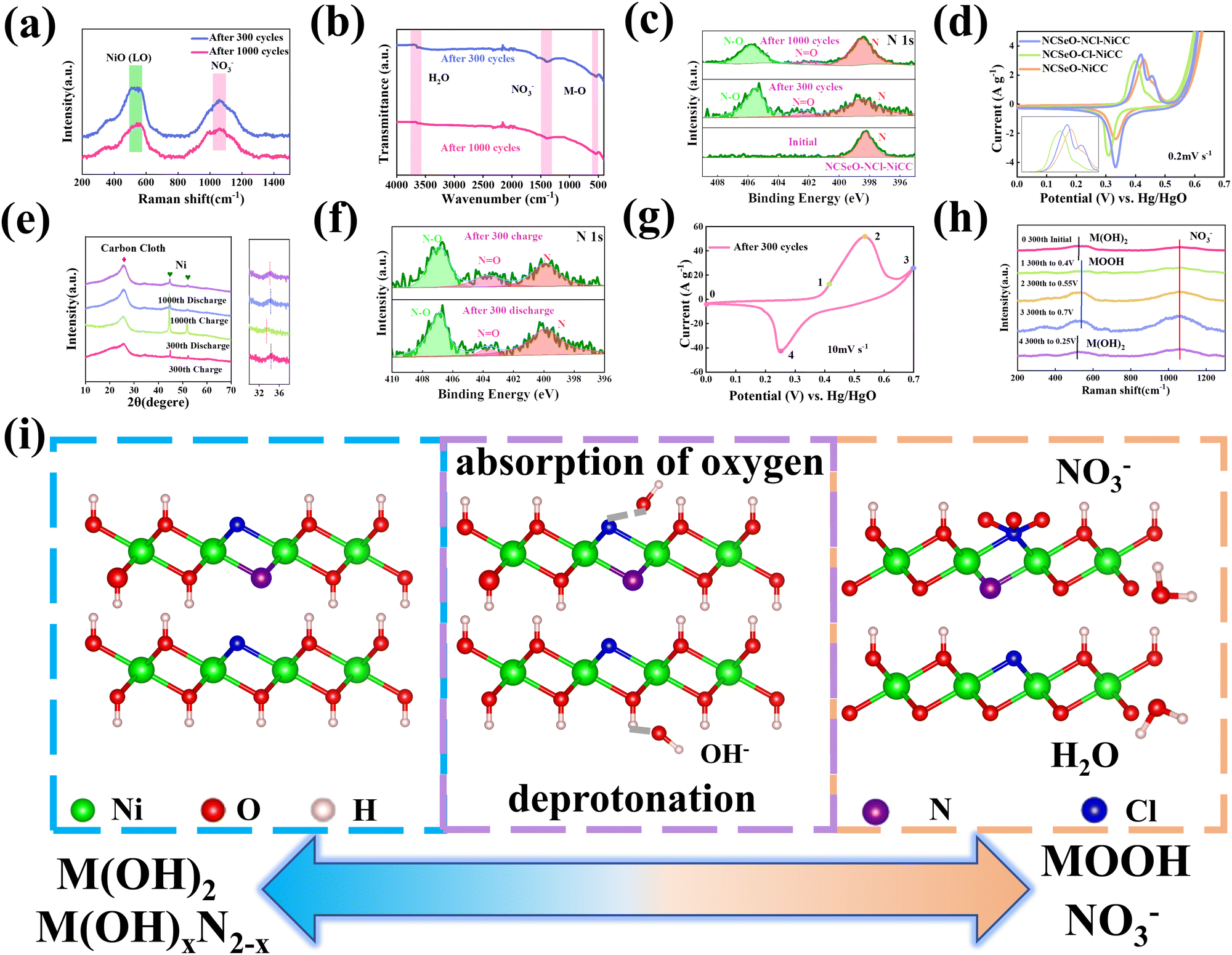
According to a previous study,40 NiSeO3 generates Ni(OH)2 in an alkaline electrolyte. Subsequently, the obtained Ni(OH)2 was used as the active species to participate in the electrochemical reaction. Fig. 4a confirms the transformation of NCSeO into M(OH)2 after 300 cycles.41 SeO32− undergoes irreversible transformation to produce SeO42− (Fig. 4b). XPS spectra of N 1s and Cl 2p indicate that N and Cl are mainly in the form of Ni/Co–N and Ni/Co–Cl bonds indicating that N and Cl replace OH− in the hydroxide layer (Fig. 4c and d).42,43 In NCSeO–NCl–NiCC and NCSeO–Cl–NiCC after the electrochemical reaction, N and Cl originally doped in NCSeO are also in situ doped into M(OH)2 and replace OH− with the transformation of NCSeO. The N doped in M(OH)2 becomes a new redox pair, which achieves the transformation of N and NO3− during charging and discharging. The crystal structure of NCSeO–NCl–NiCC is shown in Fig. S5 (ESI†). NCSeO–NCl–NiCC-1 and NCSeO–NCl–NiCC-2 are two different positions where N and Cl ions may exist with binding energies of −1.38 eV and −1.67 eV, respectively (Fig. 4e). It can be concluded that the binding of metal atoms in the hydroxide layer is more stable when N, Cl ions are located on both sides above and below the Ni atom. As shown in Fig. 4f, the density of states (DOS) of NCSeO–Cl–NiCC and NCSeO–NCl–NiCC exhibit non-zero peaks at the Fermi energy level, which implies that these two materials have some metallicity.44 A comparison of the magnified plot of DOS near the Fermi energy level shows that NCSeO–NCl–NiCC displays a larger DOS area than NCSeO–Cl–NiCC, indicating that the doping of N ions increases the number of electrons in the whole system.45 Fig. S6 (ESI†) demonstrates the SEM images of NCSeO–Cl–NiCC and NCSeO–NCl–NiCC after 300 cycles, where deepening of the cracks on the surface of NCSeO–Cl–NiCC partially produces detachment. The surface cracks of NCSeO–NCl–NiCC are significantly reduced, indicating that the doping of N atoms can alleviate the cracks caused by the structural phase transition of NCSeO during its transformation into M(OH)2, with better cycling stability.
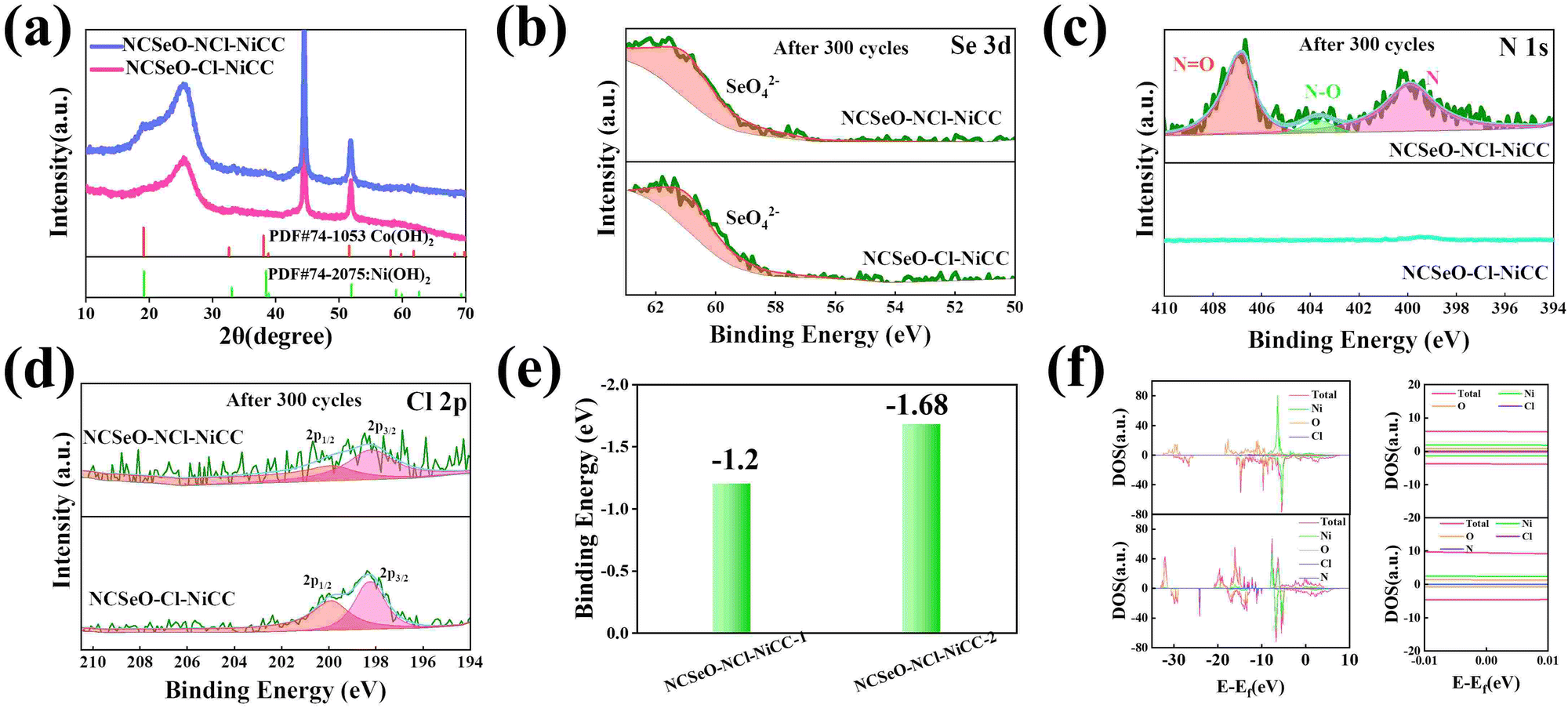 | ||
| Fig. 4 (a) XRD patterns of NCSeO–NCl–NiCC and NCSeO–Cl–NiCC after 300 cycles. High-resolution XPS spectra of NCSeO–NCl–NiCC and NCSeO–Cl–NiCC samples after 300 cycles: (b) Se 3d, (c) N 1s, and (d) Cl 2p. (e) The binding energy of NCSeO–NCl–NiCC-1 and NCSeO–NCl–NiCC-2 and (f) DOS of NCSeO–NCl–NiCC and its enlarged version near the Fermi level. | ||
In order to investigate the transformation of N and NO3− in NCSeO–NCl–NiCC during long-term cycling, it was further tested. Fig. 5a shows the Raman image of NCSeO–NCl–NiCC after 300 and 1000 cycles, and it can be seen that after long-term cycling, the Raman image of NCSeO in NCSeO–NCl–NiCC after 300 and 1000 cycles, and it can be seen that after long-term cycling, NCSeO was completely transformed into M(OH)2, and the N transformed into NO3−,46,47 and the FTIR spectrum proves a similar conclusion (Fig. 5b).48Fig. 5c shows the XPS spectrum of N of the NCSeO–NCl–NiCC after initial, 300 and 1000 cycles. After long-term cycling, part of the N remains doped in M(OH)2 as N, while the other part is converted to NO3−. After long-term cycling, part of the N remains doped in M(OH)2 as N, while the other part is converted to NO3−. Fig. 5d shows the CV curves of different samples at a scan rate of 0.2 mV s−1. There is a large oxidation peak in the CV curves of NCSeO–N–NiCC and NCSeO–Cl–NiCC, which corresponds to the reaction of M(OH)2 → MOOH. There are two distinct oxidation peaks in NCSeO–NCl–NiCC, which corresponds to the reactions of M(OH)2 → MOOH and M(OH)xN2−x → NO3− transitions, in which two types of redox reactions occur, resulting in a high specific capacity and excellent multiplicative cycling performance of the NCSeO–NCl–NiCC electrode. Fig. 5e shows the charging and discharging state ex situ XRD curves of NCSeO–NCl–NiCC after undergoing 300 and 1000 cycles. It can be seen that in the charging state the curve is shifted to the right, which corresponds to an increase in the M(OH)2 layer spacing, indicating a shift from N ions to NO3−. The discharged state corresponds to the transition of NO3− to N ions. Fig. 5f shows the N 1s high resolution XPS pattern of NCSeO–NCl–NiCC in the charged and discharged states after 300 cycles. It can be clearly seen that the peak areas of N–O and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O decrease significantly in the discharged state and increase again in the charged state, which indicates that there is a transition between N ions and NO3− during charging and discharging. Fig. 5g shows the CV curve of NCSeO–NCl–NiCC after 300 cycles. The phase transition of NCSeO–NCl–NiCC during charging and discharging after 300 cycles was further investigated using ex situ Raman spectroscopy (Fig. 5h). After undergoing 300 cycles, all the peaks of selenite and selenide disappeared due to the redox reaction between the reactive materials and OH− groups. When charged to 0.7 V, the electrode material exhibited a broad band at 551 cm−1, corresponding to MOOH.49 During the subsequent discharge, two bands at 529 cm−1 in the Raman spectrum of the electrode discharged to 0 V matched well with M(OH)2.50 The spectral band at 1048 cm−1 of the electrode material corresponds well with NO3−,46 indicating that NO3− has been present in M(OH)2 after 300 cycles. During the charging process, the Raman peak of NO3− increased with an increase in voltage, and the highest NO3− peak was observed when charged to 0.7 V, indicating the conversion of more N ions to NO3−. During the discharge process, the peak of NO3− gradually weakened, indicating the transformation of NO3− into N ions. Based on the above results, it is evident that the electrochemical mechanism of the NCSeO–NCl–NiCC electrode should be attributed to the transition between M(OH)2/MOOH and the N ion/NO3−. Fig. S7 (ESI†) shows the comparison of Cl ions in different electrodes during electrochemical charging and discharging processes, and it can be concluded that Cl ions are more stable and do not detach during the electrochemical process. Cl ion substitution facilitates the binding of N ions to MOOH by altering the polarity of the M(OH)2 layer. In addition, Cl ion substitution also changes the electronic configuration of Ni and Co, which enhances the electrical conductivity and redox activity.43 According to the above results, the reactive formula of the NCSeO–NCl–NiCC electrode can be speculated as follows:40
O decrease significantly in the discharged state and increase again in the charged state, which indicates that there is a transition between N ions and NO3− during charging and discharging. Fig. 5g shows the CV curve of NCSeO–NCl–NiCC after 300 cycles. The phase transition of NCSeO–NCl–NiCC during charging and discharging after 300 cycles was further investigated using ex situ Raman spectroscopy (Fig. 5h). After undergoing 300 cycles, all the peaks of selenite and selenide disappeared due to the redox reaction between the reactive materials and OH− groups. When charged to 0.7 V, the electrode material exhibited a broad band at 551 cm−1, corresponding to MOOH.49 During the subsequent discharge, two bands at 529 cm−1 in the Raman spectrum of the electrode discharged to 0 V matched well with M(OH)2.50 The spectral band at 1048 cm−1 of the electrode material corresponds well with NO3−,46 indicating that NO3− has been present in M(OH)2 after 300 cycles. During the charging process, the Raman peak of NO3− increased with an increase in voltage, and the highest NO3− peak was observed when charged to 0.7 V, indicating the conversion of more N ions to NO3−. During the discharge process, the peak of NO3− gradually weakened, indicating the transformation of NO3− into N ions. Based on the above results, it is evident that the electrochemical mechanism of the NCSeO–NCl–NiCC electrode should be attributed to the transition between M(OH)2/MOOH and the N ion/NO3−. Fig. S7 (ESI†) shows the comparison of Cl ions in different electrodes during electrochemical charging and discharging processes, and it can be concluded that Cl ions are more stable and do not detach during the electrochemical process. Cl ion substitution facilitates the binding of N ions to MOOH by altering the polarity of the M(OH)2 layer. In addition, Cl ion substitution also changes the electronic configuration of Ni and Co, which enhances the electrical conductivity and redox activity.43 According to the above results, the reactive formula of the NCSeO–NCl–NiCC electrode can be speculated as follows:40
| M(SeO3) + 2OH− ⇒ M(OH)2 + SeO32− | (1a) |
| M(SeO3) + 4OH− ⇒ M(OH)2 + SeO42− + H2O + 4e− | (1b) |
 | ||
| Fig. 5 (a) Raman spectra and (b) FTIR of NCSeO–NCl–NiCC after 300 cycles. (c) N 1s XPS spectra of NCSeO–NCl–NiCC for different cycles. (d) CV curves of various electrodes at a scan rate of 0.2 mV s−1. (e) Ex situ XRD patterns of NCSeO–NCl–NiCC after various cycles. (f) N 1s XPS spectra of NCSeO–NCl–NiCC at different states. (g) CV curves at a scan rate of 10 mV s−1 of NCSeO–NCl–NiCC after 300 cycles. (h) Ex situ Raman spectra of NCSeO–NCl–NiCC in an electrochemical process. (i) Schematic illustration of the energy storage mechanism of M(OH)2 and M(OH)xN2−x. | ||
Subsequently, the obtained M(OH)2 and M(OH)xN2−x participate in electrochemical reactions as reactive species:
| M(OH)2 + OH− ⇔ MOOH + H2O + e− | (2a) |
| M(OH)xN2−x + (15 − 7x)OH− ⇔ MOOH + (2 − x)NO3− + (7 − 3x)H2O + (13 − 6x)e− | (2b) |
Based on the above results, a mechanism to enhance charge storage through anionic construction of dual redox centers can be proposed, as shown in Fig. 5i. NCSeO–NCl–NiCC is able to provide more active sites. During the initial electrochemical activation, NCSeO–NCl was converted into M(OH)2, in which N and Cl ions replaced part of the O–H, while SeO32− was oxidized to SeO42− and adsorbed to the electrode surface. During the charging process, two redox reactions occur in NCSeO: on the one hand, M(OH)2 is converted to MOOH by deprotonation, a process in which Ni and Co ions, which are electron donors, are shifted to a higher valence state to balance the charge because Cl ions have higher electronegativity than OH− and can attract more electrons.51 On the other hand, as the valence states of Ni and Co increase, the N ions attached to them will likewise undergo charge transfer and react with OH− to form NO3−.52,53 Therefore, the dual redox transformations of M(OH)2/MOOH and M(OH)xN2−x/NO3− play a crucial role in the charge storage process, which is the key reason for the better performance of NCSeO–NCl–NiCC.
To further evaluate the practical application of the NCSeO–NCl–NiCC electrode, the ASC device was assembled as shown in Fig. 6a, employing carbon cloth-coated activated carbon as the negative electrode and 2 M KOH as the electrolyte (denoted as NCSeO–NCl–NiCC//AC). Fig. 6b shows the CV curves of the NCSeO–NCl–NiCC//AC device at different scan rates of 5–50 mV s−1, demonstrating a distorted rectangular shape with a pair of redox humps. From CV curves, no significant distortion is observed with an increase in the scan rate, which indicates the good multiplicative performance of the ASC device. The corresponding GCD curves at different current densities are nonlinear, which is consistent with the CV results (Fig. 6c). The specific capacity of the ASC device was calculated to be 93.97 mA h g−1 at a current density of 1 A g−1. Fig. 6d shows the Ragone plot of the NCSeO–NCl–NiCC//AC ASC device compared to other different devices. The NCSeO–NCl–NiCC//AC ASC device achieves an energy density of 73.6 W h kg−1 at a power density of 786.5 W kg−1, and at a higher power density of 12250 W kg−1, the energy density is still maintained at 19 W h kg−1, which is better than that of the recently reported supercapacitors.54–60 More importantly, the NCSeO–NCl–NiCC//AC ASC exhibits excellent high-rate cycling performance. After 20000 cycles, at a current density of 10 A g−1, the capacity of the NCSeO–NCl–NiCC//AC ASC device remained at 98.2% of the initial capacity (Fig. 6e). After 20000 cycles, a capacity retention of 97.8% can be achieved at a higher rate of 20 A g−1 (Fig. 6f). To further evaluate the actual performance, a number of visual methods were used to verify the practical application potential of the assembled device, a small electric fan that can be operated on the drive of a capacitor battery (Video S1, ESI†).
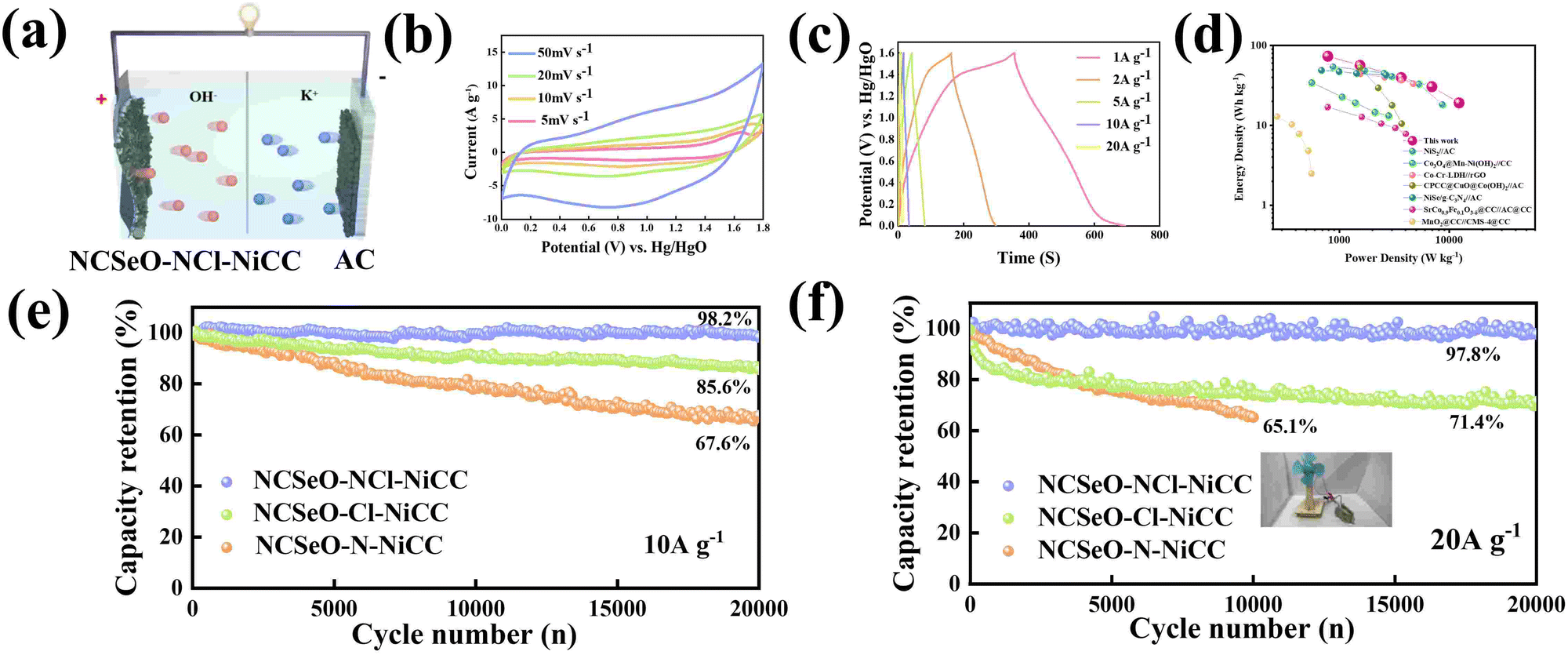 | ||
| Fig. 6 (a) Structure schematic of the NCSeO–NCl–NiCC//AC ASC device. (b) CV curves of the ASC device of NCSeO–NCl–NiCC//AC at different scan rates. (c) GCD curves of the ASC device of NCSeO–NCl–NiCC//AC at different current densities. (d) Ragone plots of NCSeO–NCl–NiCC//AC compared with different electrical energy-storage technologies. Cycling stability of the NCSeO–NCl–NiCC//AC ASC device at (e) 10 A g−1 and (f) 20 A g−1. | ||
Conclusions
In conclusion, a novel NCSeO–NCl–NiCC composite electrode was prepared by a facile electrodeposition method. The charge storage mechanism of NCSeO–NCl–NiCC is attributed to the redox reaction between M(OH)2/MOOH and M(OH)xN2−x/NO3−. The N and Cl ions replace part of the OH− in M(OH)2, and when M(OH)2 is charged, the OH− in M(OH)2 deprotonates hydrogen to produce MOOH and H2O, and the N ions adsorb OH−, which is oxidized to NO3−, resulting in two redox centers. The presence of Cl ions is conducive to the enhancement of the stability of N ion doping, and the presence of Cl ions also changes the electronic configuration of Ni and Co, which enhances the electrical conductivity and redox activity, and the NCSeO–NCl–NiCC electrode can provide an ultra-high capacity of more than 417 mA h g−1. The NCSeO–NCl–NiCC//AC ASC device exhibits excellent energy/power density and cycle life comparable to state-of-the-art hybrid/asymmetric supercapacitors with organic electrolytes. Thus, these results suggest new directions for the design and preparation of high-capacity electrode materials for ASCs with higher energy densities while maintaining their intrinsic properties of greenness, safety, and low cost.Author contributions
Tao Li: conceptualization, formal analysis, investigation, data curation, writing original draft, and writing – review and editing. Jinyue Song: DFT simulations and formal analysis. Hongguang Fan: formal analysis. Yanpeng Wang: formal analysis and investigation. Yuesheng Luo: formal analysis and investigation. Chenchen Shao: methodology and investigation. Qingping Li: methodology and investigation. Wei Liu: methodology, conceptualization, resources, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Major Basic Research Projects of the Shandong Natural Science Foundation (ZR2020ZD07) and the Key Scientific and Technological Innovation Project of Shandong (No. 2020CXGC010401).Notes and references
- Y. Qiao, J. He, Y. Zhou, S. Wu, X. Li, G. Jiang, G. Jiang, M. Demir and P. Ma, ACS Appl. Mater. Interfaces, 2023, 15, 52381–52391 CrossRef CAS PubMed.
- D. Y. Jiang, Y. Q. Wang, C. Du, M. J. Xie, J. Chen, Y. Zhang and L. Wan, J. Alloys Compd., 2024, 970, 11 Search PubMed.
- Y. Liu, N. Fu, G. Zhang, M. Xu, W. Lu, L. Zhou and H. Huang, Adv. Funct. Mater., 2017, 27, 1605307 CrossRef.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2020, 5, 5–19 CrossRef.
- X. Han, J. Li, J. Lu, S. Luo, J. Wan, B. Li, C. Hu and X. Cheng, Nano Energy, 2021, 86, 106079 CrossRef CAS.
- X. Wu, F. Zeng, X. Song, X. Sha, H. Zhou, X. Zhang, Z. Liu, M. Yu and C. Jiang, Chem. Eng. J., 2023, 456, 140947 CrossRef CAS.
- D. N. Yang, R. M. Wang, M. S. He, J. Zhang and Z. F. Liu, J. Phys. Chem. B, 2005, 109, 7654–7658 CrossRef CAS PubMed.
- D. Zhang, X. Guo, X. Tong, Y. Chen, M. Duan, J. Shi, C. Jiang, L. Hu, Q. Kong and J. Zhang, J. Alloys Compd., 2020, 837, 155529 CrossRef CAS.
- J. Kang, Y. Xue, J. Yang, Q. Hu, Q. Zhang, L. Gu, A. Selloni, L.-M. Liu and L. Guo, J. Am. Chem. Soc., 2022, 144, 8969–8976 CrossRef CAS PubMed.
- C. Li, W. H. Kan, H. Xie, Y. Jiang, Z. Zhao, C. Zhu, Y. Xia, J. Zhang, K. Xu, D. Mu and F. Wu, Adv. Sci., 2019, 6, 1801406 CrossRef PubMed.
- Z. Zhang, H. Huo, L. Wang, S. Lou, L. Xiang, B. Xie, Q. Wang, C. Du, J. Wang and G. Yin, Chem. Eng. J., 2021, 412, 128617 CrossRef CAS.
- W. He, L. Han, Q. Hao, X. Zheng, Y. Li, J. Zhang, C. Liu, H. Liu and H. L. Xin, ACS Energy Lett., 2019, 4, 2905–2912 CrossRef CAS.
- J. Chang, S. Zhang, M. Shi, J. Feng, Z. Liu, T. Wei and Z. Fan, Adv. Funct. Mater., 2022, 32, 2109225 CrossRef CAS.
- J. Hao, L. Yan, X. Zou, Y. Bai, Y. Han, C. Zhu, Y. Zhou and B. Xiang, Small, 2023, 19, 2300467 CrossRef CAS PubMed.
- J. Zou, D. Xie, J. Xu, X. Song, X. Zeng, H. Wang and F. Zhao, Appl. Surf. Sci., 2022, 571, 151322 CrossRef CAS.
- R. Y. Hu, L. Y. Liu, J. H. He, Y. Zhou, S. B. Wu, M. X. Zheng, M. Demir and P. P. Ma, J. Energy Storage, 2023, 72, 108656 CrossRef.
- Z. Jiao, Y. Chen, M. Du, M. Demir, F. Yan, Y. Zhang, C. Wang, M. Gu, X. Zhang and J. Zou, J. Alloys Compd., 2023, 958, 170489 CrossRef CAS.
- J. Xin, H. Tan, Z. Liu, L. Zhao, J. Xie, Y. Sang, W. Zhou, A. Wang, H. Liu and J. J. Wang, Nanoscale, 2019, 11, 3268–3274 RSC.
- B. Wang, W.-X. Lu, Z.-Q. Huang, D.-S. Pan, L.-L. Zhou, Z.-H. Guo and J.-L. Song, Chem. Eng. J., 2020, 399, 125799 CrossRef CAS.
- Y. Jiang, Y. Song, Z. Pan, Y. Meng, L. Jiang, Z. Wu, P. Yang, Q. Gu, D. Sun and L. Hu, ACS Nano, 2018, 12, 5011–5020 CrossRef CAS PubMed.
- G. D. Park, J. H. Hong, J. H. Choi, J. H. Lee, Y. S. Kim and Y. C. Kang, Small, 2019, 15, e1901320 CrossRef PubMed.
- M. Wang, A. Peng, J. Jiang, M. Zeng, Z. Yang, J. Chen, B. Guo, Z. Ma, B. Yu, Y. Zhang and X. Li, Chem. Eng. J., 2022, 433, 134567 CrossRef CAS.
- T. Kavinkumar, S. Seenivasan, H. Jung, J. W. Han and D.-H. Kim, J. Mater. Chem. A, 2021, 9, 21132–21141 RSC.
- B. Zhao, L. Zhang, Q. Zhang, D. Chen, Y. Cheng, X. Deng, Y. Chen, R. Murphy, X. Xiong, B. Song, C.-P. Wong, M.-S. Wang and M. Liu, Adv. Energy Mater., 2018, 8, 1702247 CrossRef.
- J. Li, Z. Liu, Q. Zhang, Y. Cheng, B. Zhao, S. Dai, H.-H. Wu, K. Zhang, D. Ding, Y. Wu, M. Liu and M.-S. Wang, Nano Energy, 2019, 57, 22–33 CrossRef CAS.
- Y. Bai, C. Liu, T. Chen, W. Li, S. Zheng, Y. Pi, Y. Luo and H. Pang, Angew. Chem., Int. Ed., 2021, 60, 25318–25322 CrossRef CAS PubMed.
- J. Xin, H. Tan, Z. Liu, L. Zhao, J. Xie, Y. Sang, W. Zhou, A. Wang, H. Liu and J.-J. Wang, Nanoscale., 2019, 11, 3268–3274 RSC.
- H. Qian, B. Wu, Z. Nie, T. Liu, P. Liu, H. He, J. Wu, Z. Chen and S. Chen, Chem. Eng. J., 2021, 420, 134567 Search PubMed.
- J. Chu, Q. Yu, K. Han, L. Xing, Y. Bao and W. Wang, Carbon, 2020, 161, 834–841 CrossRef CAS.
- R. L. Frost and E. C. Keeffe, J. Raman Spectrosc., 2009, 40, 509–512 CrossRef CAS.
- J. Li, S. Luo, G. Liu, J. Wan, J. Lu, B. Li, X. Han and C. Hu, Mater. Today Chem., 2021, 22, 100620 CrossRef CAS.
- T. Liu, Y. Zheng, W. Zhao, L. Cui and J. Liu, J. Colloid Interface Sci., 2019, 556, 743–752 CrossRef CAS PubMed.
- W. He, H. Liu, J. Cheng, Y. Li, C. Liu, C. Chen, J. Zhao and H. L. Xin, ACS Appl. Mater. Interfaces, 2022, 14, 6869–6875 CrossRef CAS PubMed.
- Y. Chen, Z. Ren, H. Fu, X. Zhang, G. Tian and H. Fu, Small, 2018, 14, 1800763 CrossRef PubMed.
- J. Yin, Q. H. Fan, Y. X. Li, F. Y. Cheng, P. P. Zhou, P. X. Xi and S. H. Sun, J. Am. Chem. Soc., 2016, 138, 14546–14549 CrossRef CAS PubMed.
- L. Zhou, M. Shao, C. Zhang, J. Zhao, S. He, D. Rao, M. Wei, D. G. Evans and X. Duan, Adv. Mater., 2017, 29, 1604080 CrossRef PubMed.
- G. Wang, Z. Yan, N. Wang, M. Xiang, Z. Xu and H. Zhu, J. Mater. Res., 2022, 37, 1714–1726 CrossRef CAS.
- G. L. Zhou, X. L. Gao, S. Z. Wen, X. L. Wu, L. L. Zhang, T. S. Wang, P. S. Zhao, J. Z. Yin and W. S. Zhu, J. Colloid Interface Sci., 2022, 612, 772–781 CrossRef CAS PubMed.
- Y. G. Wang, Y. F. Song and Y. Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- J. Y. Song, H. G. Fan, L. C. Bai, Y. P. Wang, Y. C. Jin, S. Liu, X. H. Xie, W. S. Zheng and W. Liu, Small Methods, 2023, 7, 12 Search PubMed.
- B. Li, H. Nam, J. Zhao, J. Chang, N. Lingappan, F. Yao, T. H. Lee and Y. H. Lee, Adv. Mater., 2017, 29, 5 Search PubMed.
- L. Zhao, S. J. Lei, Q. Y. Tu, L. H. Rao, W. H. Zen, Y. H. Xiao and B. C. Cheng, J. Energy Storage, 2021, 43, 9 Search PubMed.
- M. V. Konishcheva, D. A. Svintsitskiy, D. I. Potemkin, V. N. Rogozhnikov, V. A. Sobyanin and P. V. Snytnikov, ChemistrySelect, 2020, 5, 1228–1234 CrossRef CAS.
- X. X. Zhao, J. R. Feng, J. Liu, W. Shi, G. M. Yang, G. C. Wang and P. Cheng, Angew. Chem., Int. Ed., 2018, 57, 9790–9794 CrossRef CAS PubMed.
- T. Kavinkumar, S. Seenivasan, H. H. Lee, H. Jung, J. W. Han and D. H. Kim, Nano Energy, 2021, 81, 11 CrossRef.
- L. J. Bonales, N. Rodríguez-Villagra, I. Sánchez-García and O. R. Montoro, Prog. Nucl. Energy, 2022, 145, 12 CrossRef.
- R. Hao, S. S. Fang, L. Tian, R. L. Xia, Q. X. Guan, L. F. Jiao, Y. P. Liu and W. Li, Chem. Eng. J., 2023, 467, 11 CrossRef.
- R. Hao, L. Tian, C. Wang, L. Wang, Y. P. Liu, G. C. Wang, W. Li and G. A. Ozin, Chem Catal., 2022, 2, 622–638 CrossRef CAS.
- X. Bo, R. K. Hocking, S. Zhou, Y. B. Li, X. J. Chen, J. C. Zhuang, Y. Du and C. Zhao, Energy Environ. Sci., 2020, 13, 4225–4237 RSC.
- A. Y. Faid, A. O. Barnett, F. Seland and S. Sunde, Electrochim. Acta, 2020, 361, 11 CrossRef.
- J. W. Hao, J. Mou, J. W. Zhang, L. B. Dong, W. B. Liu, C. J. Xu and F. Y. Kang, Electrochim. Acta, 2018, 259, 170–178 CrossRef CAS.
- S. D. Liu, Y. Yin, Y. Shen, K. S. Hui, Y. T. Chun, J. M. Kim, K. N. Hui, L. P. Zhang and S. C. Jun, Small, 2020, 16, 14 Search PubMed.
- Y. F. Zhao, Q. Wang, T. Bian, H. J. Yu, H. Fan, C. Zhou, L. Z. Wu, C. H. Tung, D. O'Hare and T. R. Zhang, Nanoscale, 2015, 7, 7168–7173 RSC.
- G. S. Wang, Y. J. Ding, Z. H. Xu, G. M. Wang, Z. K. Li and Z. X. Yan, Chem. Eng. J., 2023, 469, 12 Search PubMed.
- S. V. Sadavar, N. S. Padalkar, R. B. Shinde, A. S. Patil, U. M. Patil, V. V. Magdum, Y. M. Chitare, S. P. Kulkarni, S. B. Kale, R. N. Bulakhe, D. S. Bhange, S. T. Kochuveedu and J. L. Gunjakar, Energy Storage Mater., 2022, 48, 101–113 CrossRef.
- S. G. Gong, Y. F. Li, Y. Su, B. Li, G. D. Yang, X. L. Wu, J. P. Zhang, H. Z. Sun and Y. F. Li, Small, 2023, 19, 10 Search PubMed.
- S. R. Khaladkar, O. Maurya, G. Gund, B. Sinha, D. Dubal, R. R. Deshmukh and A. Kalekar, J. Energy Chem., 2023, 87, 304–313 CrossRef CAS.
- M. S. U. Shah, X. Q. Zuo, A. Shah, S. I. Al-Saeedi, M. Z. U. Shah, E. A. Alabbad, H. Y. Hou, S. A. Ahmad, M. Arif, M. Sajjad and T. Ul Haq, J. Energy Storage, 2023, 65, 11 Search PubMed.
- L. Liu, G. Liu, S. Wu, J. He, Y. Zhou, M. Demir, R. Huang, Z. Ruan, G. Jiang and P. Ma, Ceram. Int., 2024, 50, 1970–1980 CrossRef CAS.
- R. H. Xu, P. P. Ma, G. F. Liu, Y. Qiao, R. Y. Hu, L. Y. Liu, M. Demir and G. H. Jiang, Energy Fuels, 2023, 37, 6158–6167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00454j |
| This journal is © the Owner Societies 2024 |