
Metal–organic framework-derived CuO catalysts for the efficient hydrogenolysis of hardwood lignin into phenolic monomers†
Qian
Xu
,
Qiang
Wang
,
Ling-Ping
Xiao
 *,
Xiao-Ying
Li
,
Xi
Xiao
,
Meng-Xin
Li
,
Meng-Ran
Lin
,
Yu-Man
Zhao
and
Run-Cang
Sun
*
*,
Xiao-Ying
Li
,
Xi
Xiao
,
Meng-Xin
Li
,
Meng-Ran
Lin
,
Yu-Man
Zhao
and
Run-Cang
Sun
*
Liaoning Key Lab of Lignocellulose Chemistry and BioMaterials, Liaoning Collaborative Innovation Center for Lignocellulosic Biorefinery, College of Light Industry and Chemical Engineering, Dalian Polytechnic University, Dalian 116034, China. E-mail: lpxiao@dlpu.edu.cn; rsun3@dlpu.edu.cn
First published on 24th October 2023
Abstract
The selective reductive catalytic deconstruction (RCD) of lignin into phenolic monomers provides the possibility for making the full use of lignocellulose. However, the widespread use of precious metal catalysts and the harsh reaction conditions present the challenge of poor industrial utilization in the current research. Herein, we report a metal–organic framework (MOF)-derived copper oxide catalyst (CuO/c-UiO-66), which exhibits superior catalytic properties in the RCD of hardwood lignin and affords high yields (up to 42.8 wt%) of monomeric phenols via the C–O bond scission. The mechanistic reactions using lignin model compounds reveal that phenolic compounds with propyl or propanol end chains are selectively produced during the catalytic hydrogenolysis reaction. The enhanced catalytic reactivity is attributed to the synergy of acid and base sites of the catalyst, which facilitates the C–O bond cleavage process. The new insights of this study provide guidance toward the rational design of Cu-based catalysts for RCD of lignin.
1. Introduction
Lignocellulose is a nonedible biomass feedstock, which is made up of cellulose (30–50%), hemicellulose (20–35%), and lignin (15–30%).1 In particular, lignin is a heterogeneous biomacromolecule, representing the largest and most widely renewable carbon resource.2,3 Such sustainable carbon feedstock can be used as an alternative to produce bio-fuels and bio-based materials.4,5 Currently, to relieve the fossil energy crisis, increasing attention has been put into the efficient utilization of lignocellulose materials to replace petroleum-derived chemicals with sustainable alternatives.6,7 Phenolic monomers from lignin depolymerization serve as an upstream product of aromatic chemicals.8–11 However, the natural complexity and heterogeneity of lignin restrain its efficient utilization.12 Additionally, the lignin undergoes structural rearrangement during the isolation or pulping process and forms irreversible C–C bonds while sacrificing the tenuous β-O-4′ linkages, thus resulting in the difficult transformation of industrial lignin.13 Therefore, the recalcitrant lignin in the papermaking industry is always burned as fuel to provide the energy needed for the pulp industry.14,15 In spite of extensive efforts, the efficient transformation of lignin into valuable chemicals is still challenging.15–17Many efficient strategies have been explored for lignin depolymerization. Reductive catalytic deconstruction (RCD) is an effective method to produce aromatic platform chemicals from lignin.18–22 This strategy enables the efficient hydrogenolysis of lignin to phenolic compounds without affecting the chemical carbohydrate's structural integrity for further processing.20,23 The hydrogen-donor suitable solvents, degree of reaction, and catalysts could increase the product yield in RCD.21,24,25 Lower-grade aliphatic alcohols, which have hydrogen donor characteristics, are often applied as organic solvents as they not only increase dissolution at the depolymerization interface and prevent char formation but also offer in situ hydrogen resources during the hydrogenolysis process of lignin.26 Moreover, they behaved differently as hydrogen donors during the hydrogenolysis of lignocellulose owing to their different reactivity.27 For example, Kim and co-workers28 reported that the more polar alcohols seemed to be much effective in inhibiting the repolymerization process during the catalytic hydrogenolysis of biomass.
The fundamental challenge of RCD of woody sawdust is the selective dissociation of aryl-ether linkages while preserving aromaticity, which requires tailored catalysts. In this process, many precious metal catalysts, such as Ru3,29,30 and Pd,31,32 have been reported to produce monomeric phenols with end chains by cleaving the C–O linkage from β-O-4′ lignin units. Sels' group33 used a commercial Ru/C catalyst for the hydrogenolysis of birch wood and obtained 50.5 wt% phenolic compounds, wherein guaiacol and syringol with propyl side chains were the major monomers. Huang and co-workers34 disclosed that 55 wt% mono-aromatics could be obtained from native birch lignin with small amounts of Pd/C. Hu et al.35 revealed that the bimetallic catalyst (Ni–Pd) was more active during the hydrogenolysis of birch native lignin than the parent monometallic catalysts, which was due to the ‘synergistic effects’.36–38 However, the capability of the bimetallic catalysts for hydrogenolysis of native lignin is still unclear.17 Moreover, precious metal catalysts show limitations in large-scale industrial applications due to their scarcity and high price. Therefore, a number of researchers have taken note of the development of non-precious metal catalysts. For example, MoOx/SBA-15 showed high selectivity to produce monolignols and etherized derivatives in the hydrogenolysis of woody biomass.39 Rautiainen and co-workers40 reported that the Co-based catalyst could effectively break down the β-O-4′ bonds of birch lignin, affording the yield of aromatic monomers up to 34 wt%. For lignin hydrogenolysis, catalysts based on copper have been rarely investigated. Ford and co-workers41,42 described a Cu-doped porous metal oxide-based catalyst for lignin decomposition to cyclohexyl derivatives by hydrogenolysis of phenyl ether bonds in super-critical methanol. Hensen et al.43 presented a CuMgAlOx catalyst for catalytic degradation of alkaline lignin in ethanol, which resulted in 23 wt% monomer yield. The Cu20PMO catalyst has been used for the hydrogenolysis of aryl-ether linkages, which was effective for C–O bond scission.44
Metal–organic frameworks (MOFs) have attracted great interest owing to their chemical tenability, large surface area, high catalytic activity, pore structure, and abundant metal sites.45–48 The framework can limit tiny metal nanoparticles within either holes/cages or crystalline defects, thus creating a metal–support surface for catalysis. Recently, Song's group23 reported selective production of phenolic compounds under different reaction conditions from Eucalyptus lignin with a Ni@ZIF-8 catalyst. Notably, the active metal centers and abundant metal sites of MOFs prompted our enthusiasm for developing active catalysts with a MOF-analogue structure.49 We are also concerned with recent advances in the study of MOF-derived catalysts, where the active metal center is anchored to the derivatized framework, forming catalyst structural sites that exhibit excellent catalytic performance for catalysis.50,51 Zr-based MOFs are widely used in catalysis due to their unique high specific surface area and tunable crystal structure.52–55 We conjectured that UiO-66 containing Zr atoms might regulate the selectivity control in RCD of lignocellulosic biomass. Herein, we report that CuO supported on a UiO-66 derived framework (CuO/collapse-UiO-66, abbreviated as CuO/c-UiO-66) acts as a highly efficient catalyst for the RCD of the native lignin in hardwood lignocellulosic biomass. Moreover, we have improved the performance of Cu-based catalysts in terms of stability as compared to previously reported catalysts (CuO/C) with the same CuO active site.56 In addition, this catalytic system could obtain a range of phenolic compounds in high yields by switching reaction conditions. Finally, the behavior of lignin model compounds over CuO/c-UiO-66 was also investigated to verify the reaction mechanism in this work.
2. Experimental section
2.1 Preparation of the catalyst
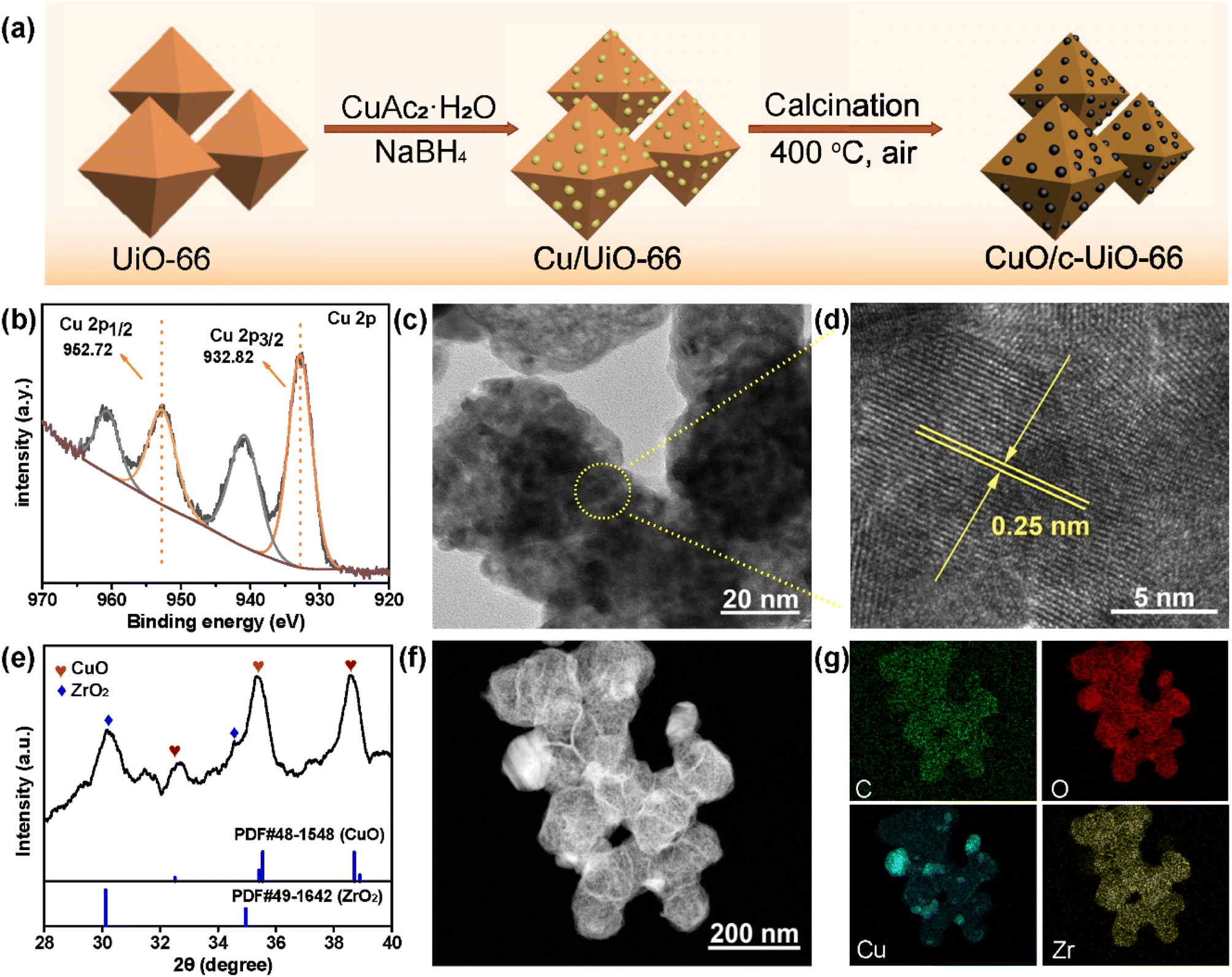 | ||
| Fig. 1 (a) Diagram of the preparation of CuO/c-UiO-66; morphology and structure characterization of CuO/c-UiO-66: (b) XRD patterns; (c) and (d) high resolution TEM images; (e) XPS spectrum of Cu 2p; and (f) and (g) HR-STEM image and corresponding EDX elemental mapping of C, O, Cu and Zr. | ||
2.2 General process for RCD of native lignin from lignocellulose
Overall, wood sawdust (50 mg), CuO/c-UiO-66 (20 mg) and a polar solvent (10 mL) were loaded into a 50 mL stainless steel batch reactor (Parr Instruments Co.), which was then sealed and purged with N2 and then pressurized with H2 (3 MPa) at room temperature. The reaction was carried out at different temperatures for a certain time. At the end of the reaction, the reactor was cooled to room temperature and carefully depressurized. The resulting mixture of reacted insoluble solids was then filtered by solid–liquid separation and washed several times with dichloromethane (DCM) to ensure maximum retention of the lignin depolymerization products. Subsequently, the solution fractions were extracted with DCM. After removal of all volatiles under vacuum, the resulting oil was quantitatively and qualitatively analyzed using gas chromatography (GC) and gas chromatography-mass spectrometry (GC-MS), respectively.2.3 Reusability of the CuO/c-UiO-66 catalyst
At the end of the hydrogenolysis process, the catalyst and carbohydrate fractions were left as insoluble fractions. In order to carry out the recycling performance test, we had to isolate the used catalyst. By blocking the carbohydrate fraction using a 200 mesh (0.074 mm) sieve, the small-sized catalyst escaped through the pores. Finally, the spent CuO/c-UiO-66 catalyst was used for the next run.2.4 General characterization
Surface areas were calculated from the adsorption data using Brunauer–Emmett–Teller (Beijing Builder Co. Ltd, China). Samples were allowed to degas under vacuum prior to measurement for 5 h at 300 °C. X-ray diffraction (XRD) analysis was performed on a Shimadzu Lab XRD-6100 diffractometer with a Cu Kα radiation source operating at 15 mA with 40 kV. X-ray photoelectron spectra (XPS) were obtained using a scanning X-ray microprobe (PHI 5000 Versa, ULAC-PHI, Inc.) with AI Kα emission and 284.80 eV C 1s peak used as an internal standard. After catalyst samples were dissolved in hydrogen fluoride (HF) solution, the Cu and Zr content was measured through inductively coupled plasma atomic emission spectroscopy (ICP-AES) using a Thermos IRIS Intrepid II XSP emission spectrometer. Transmission electron microscopy (TEM) was performed using a JEM-2100F FETEM fitted with an energy dispersive X-ray spectrometer (EDX) for analysis at 100 kV, and a high-angle annular dark-field scanning TEM (HAADF-STEM). EDX elemental mapping was carried out at 200 kV. The NH3-TPD was tested on Micromeritics AutoChem II 2920 apparatus to determine the number of acid sites. Typically, about 100 mg of the catalyst was restored in the H2 stream at 200 °C for 1 h, and before being cooled down to room temperature in the He stream. NH3-TPD was allowed to increase at temperatures from 50 °C to 650 °C at 10 °C min−1 for 30 min. CO2-TPD assays were performed on Micromeritics Autochem 2920 apparatus. A sample was restored at 300 °C, then purged for 1 h with a 10% H2/Ar stream, and chilled to 50 °C in a He flow. After being exposed to a 5% CO2/He flow (1 h; 50 °C) and purged with He (1 h; 50 °C), the sample was treated to 500 °C at 5 °C min−1 in a 30 cm per min He stream, and the dehydrogenated gas was examined by TCD.Molecular weight distributions of lignins and oily products were determined by gel permeation chromatography (GPC) utilizing a Waters 1515 isocratic pump equipped with a Dual Absorbance UV detector (Waters 2487) set at 280 nm with an Agilent Plgel 3 μm 100 Å 300 × 7.5 mm column as described previously.58 Notably, the lignin samples were firstly acetylated and then dissolved in THF (2 mg mL−1) and finally purified through a 0.45 μm syringe filter before injection.59
Gas chromatography (GC) and gas chromatography-mass spectrometry (GC-MS) analyses were performed on a Shimadzu Model 2010 plus with an HP-5 column (30 m × 0.25 mm × 0.25 mm) employing a flame ionization detector (FID) and a Shimadzu GCMS-QP2010SE with an HP-5MS (30 m × 0.25 mm × 0.25 mm) column, respectively. Injection temperature was 250 °C. The column temperature was heated from 50 °C and held for 3 min, then ramped at 8 °C min−1 to 280 °C, and held for 5 min at that temperature. The injection temperature of the FID was 200 °C. The characterization and quantification of lignin monomers within oil-based products have been performed by reference to real samples obtained from commercial sourcing or independent synthesis. The monomer yields were calculated using the formula
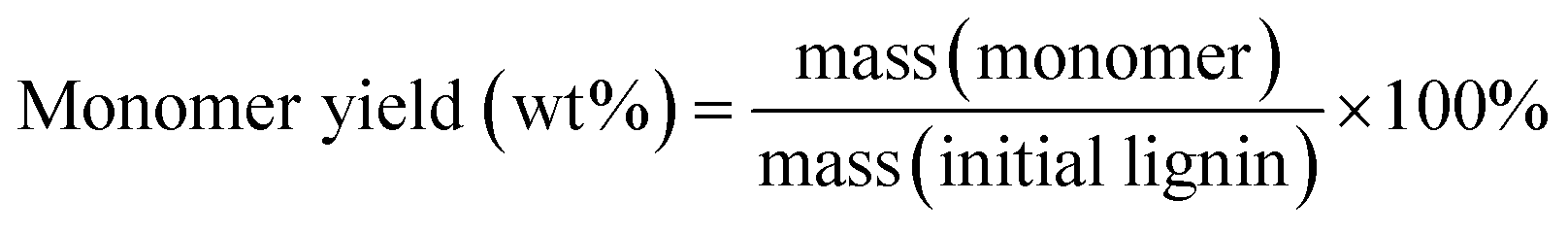 | (1) |
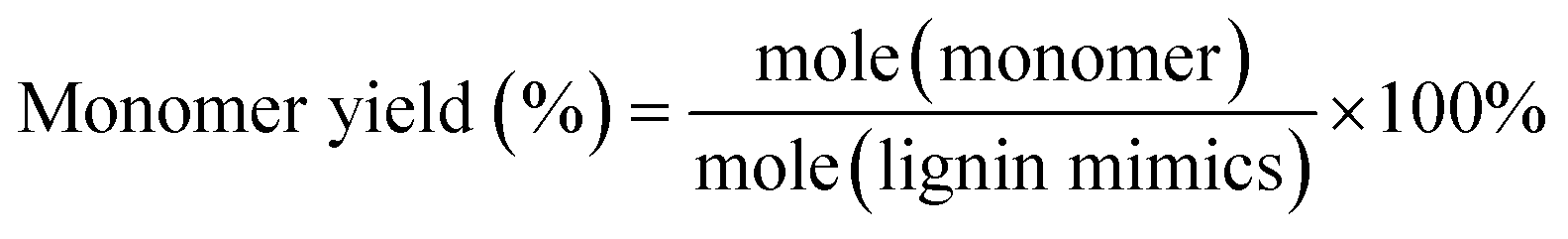 | (2) |
Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker Ascend-400 MHz spectrometer.56 Samples of lignin (∼60 mg) were added to 0.55 mL of DMSO-d6 and 0.14 mL of pyridine, and then ultrasonicated until the solids dissolved. Samples of lignin oil (∼60 mg) were solubilized in DMSO-d6. As for two-dimensional heteronuclear single-quantum correlation (2D HSQC) NMR, the solvent peak (DMSO-d6) at δC/δH 39.5/2.49 ppm was used as an internal reference.
3. Results and discussion
3.1 Catalyst characterization
The preparation of CuO/c-UiO-66 is schematically illustrated in Fig. 1a. Clearly, the XRD pattern of crystalline phases of CuO/c-UiO-66 indicated that the CuO was dispersed on the framework of UiO-66 (Fig. 1b). The fabricated CuO/c-UiO-66 catalyst displayed typical peaks at 2θ of 32.5°, 35.5° and 38.7° indexed to (110), (11−1) and (111), respectively, which are attributed to CuO (PDF #48-1548). The pattern also showed the (111) and (200) diffractions of ZrO2 at 30.1° and 35.0°, respectively (PDF #49-1642). This revealed that the active species in the catalyst was CuO, which was in agreement with the +2 state of oxidation of Cu in CuO with the following XPS results. Notably, the Cu 2p spectrum in Fig. 1e revealed two signals at 932.82 eV and 952.72 eV, which are allotted to Cu 2p3/2 and Cu 2p1/2 corresponding to the data of CuO,60 respectively. In addition, the TEM images indicated a lattice spacing identified as 0.25 nm, which conformed to the (11−1) plane of CuO nanocrystallites (Fig. 1c and d). This result was consistent with the XRD pattern. The HAADF-STEM of CuO/c-UiO-66 suggested that CuO nanoparticles were dispersed on the Zr-based carrier framework (Fig. 1f).Catalysts with both acid–base and metal sites exhibit excellent catalytic activity for the RCD of lignin. Generally, NH3-TPD of CuO/c-UiO-66 showed two peaks, suggesting the presence of weak and medium acidic sites (Fig. 2a). The desorption peak at ∼275 °C was due to the result of NH3 adsorbed on Lewis acid sites.61 The acid site of CuO/c-UiO-66 promoted lignin depolymerization without significantly causing the recombination of lignin reaction intermediates. The surface alkalinity of CuO/c-UiO-66 was investigated by CO2-TPD (Fig. 2b), and peaks situated at low temperature ranging from 120 to 200 °C are ascribed to carbon species adsorbed to weakly basic sites, which are due to hydroxyl groups at the metal oxide surface (CuO).18 The desorption of CO2 resulted in strongly basic sites at high temperatures, which are attributed to coordinatively unsaturated O2− ions.62 The metal site has the capacity to break the ether bond and the base site has the ability to assist in hydrogenation. Moreover, it was observed that the specific surface area (69.1 m2 g−1) of CuO/c-UiO-66 decreased after the immobilization of copper on UiO-66 (294.7 m2 g−1). UiO-66 displayed a significantly higher specific surface area, which might benefit active site dispersion. On the side, the porous structure of UiO-66 confined the metal pellet effectively, forming smaller CuO particles and thus offering more active sites for RCD of lignin. The percentage by weight of elemental Cu in CuO/c-UiO-66 was determined to be 39.9% according to ICP-AES (Table S1†).
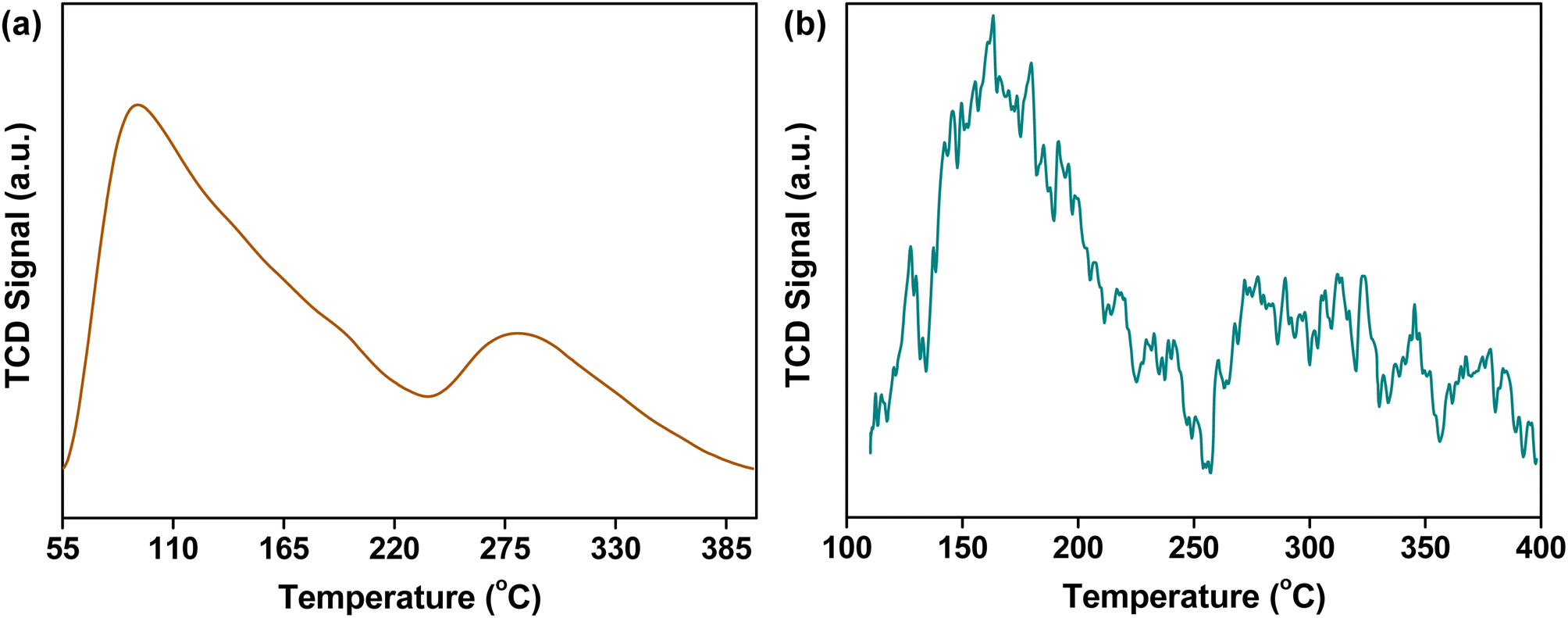 | ||
| Fig. 2 (a) NH3-TPD profile of CuO/c-UiO-66; (b) CO2-TPD profile of CuO/c-UiO-66. | ||
3.2 Catalytic hydrogenolysis of poplar sawdust
Poplar sawdust was chosen for probing the potential of CuO/c-UiO-66 for the catalytic hydrogenolysis in the lignin-first system. Firstly, we analyzed the chemical composition of poplar wood. It was found that this hardwood consisted of 24.9 wt% lignin (22.4 wt% of Klason lignin and 2.5 wt% acid-soluble lignin), 42.7 wt% cellulose, and 19.7 wt% hemicellulose (Table S5†).Then, the treatment of poplar sawdust with 20 mg CuO/c-UiO-66 under 3 MPa H2 at 240 °C in MeOH for 2 h afforded a soluble fraction with lignin oily products and an insoluble fraction with carbohydrate slurry as well as the catalyst. To identify and quantify the lignin-derived products, the lignin oily product was subjected to comparative analysis with GC by comparison with respective real samples. Clearly, it was found that 4-n-propanolsyringol (S1, 14.7 wt%), 4-n-propylsyringol (S2, 9.9 wt%), 4-n-propanolguaiacol (G1, 5.6 wt%) and 4-n-propylguaiacol (G2, 3.0 wt%) were the main monophenols, which are dependent on the total amount of lignin in the feedstock (Fig. 3a). As a contrast, a control experiment without the catalyst at 240 °C involving heating the poplar sawdust for 2 h was given, which resulted in a dark lignin oil with only 6.7 wt% total monomers (Table S7†). The average molecular weight (Mw) of the lignin oil (475 g mol−1) was sharply decreased as compared to that of DEL isolated from poplar (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 850 g mol−1) as determined by GPC (Fig. 3b). With the purpose of proving the CuO importance in the catalytic hydrogenolysis system, we used d-UiO-66 (the framework would decompose when the UiO-66 was calcined alone at 400 °C, so we denoted it as decompose-UiO-66, abbreviated as d-UiO-66) instead of CuO/c-UiO-66 at 240 °C for 2 h with 3 MPa H2 in MeOH (Fig. S9†). The obtained high content of dimethyl terephthalate indicated that the UiO-66 derived framework was broken down under the hydrogenolysis reaction conditions. This result suggested that CuO was indispensable in the hydrogenolysis reaction. Additionally, we used the CuO catalyst and ZrO2 catalyst in the reaction (Fig. S9†). It was found that the CuO catalyst alone was active in the hydrogenolysis reaction of poplar lignin, but was less selective for the propanol group and the propanol side chain products. The ZrO2 catalyst was inactive in this reaction. We speculated that CuO was the main active substance in this reaction system, and the MOF-derived framework as a support might assist in modulating the selectivity of copper oxide and further improve the catalytic activity.
850 g mol−1) as determined by GPC (Fig. 3b). With the purpose of proving the CuO importance in the catalytic hydrogenolysis system, we used d-UiO-66 (the framework would decompose when the UiO-66 was calcined alone at 400 °C, so we denoted it as decompose-UiO-66, abbreviated as d-UiO-66) instead of CuO/c-UiO-66 at 240 °C for 2 h with 3 MPa H2 in MeOH (Fig. S9†). The obtained high content of dimethyl terephthalate indicated that the UiO-66 derived framework was broken down under the hydrogenolysis reaction conditions. This result suggested that CuO was indispensable in the hydrogenolysis reaction. Additionally, we used the CuO catalyst and ZrO2 catalyst in the reaction (Fig. S9†). It was found that the CuO catalyst alone was active in the hydrogenolysis reaction of poplar lignin, but was less selective for the propanol group and the propanol side chain products. The ZrO2 catalyst was inactive in this reaction. We speculated that CuO was the main active substance in this reaction system, and the MOF-derived framework as a support might assist in modulating the selectivity of copper oxide and further improve the catalytic activity.
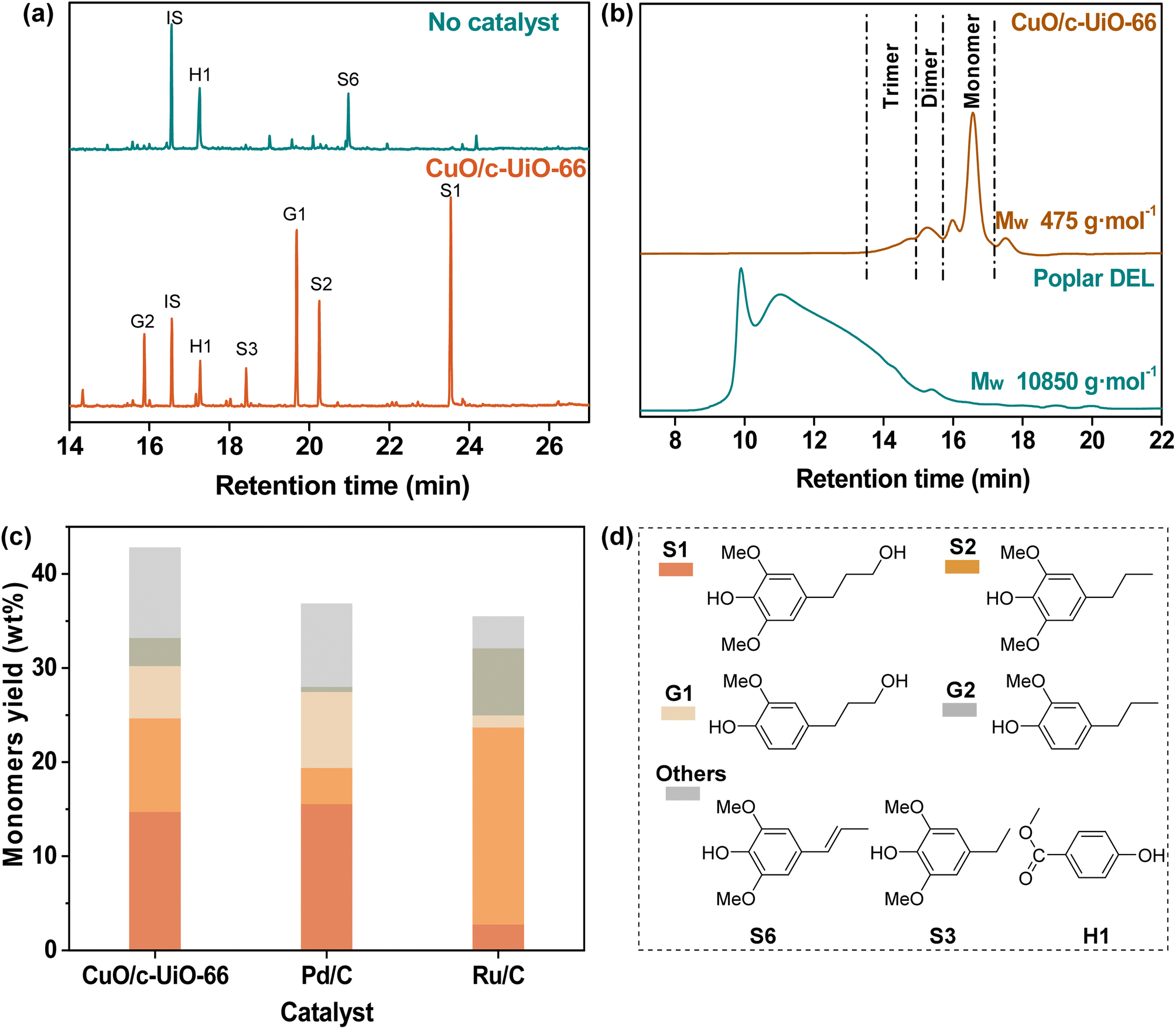 | ||
| Fig. 3 (a) GC of lignin oil obtained from the catalytic hydrogenolysis of poplar sawdust with or without the CuO/c-UiO-66 catalyst; (b) GPC analysis of double enzymatic lignin (DEL) and lignin oil obtained by RCD of poplar sawdust over the CuO/c-UiO-66 catalyst. (c) Comparison of the monomeric yields obtained from RCD of poplar sawdust over the CuO/c-UiO-66 with those of commercial catalysts of Pd/C and Ru/C. (d) Structures of monomers. Reaction conditions: poplar sawdust (50 mg), catalyst (20 mg), MeOH (10 mL), H2 (3 MPa), 240 °C, and 2 h. | ||
In general, the precious metals Pd and Ru have excellent effects on catalytic hydrogen hydrolysis. So, a control experiment with commercially available catalysts, Pd/C and Ru/C, was performed for comparison under the conditions of 240 °C at MeOH for 2 h (Fig. 3c), which afforded relatively lower monomer yields than CuO/c-UiO-66 of 36.9 wt% and 35.4 wt%, respectively (Table S9†). Notably, the fabricated CuO/c-UiO-66 exhibits comparable hydrogenolysis activity toward RCD of lignin into monophenols with those of state-of-the-art catalysts (Table S16†).39,40,63–67
These results suggested that the fabricated CuO/c-UiO-66 nanomaterial was an efficient catalyst toward the RCD of lignin into monophenols in high yields. Moreover, it was observed that monophenols with less preservation of γ-OH over the Ru/C catalyst were obtained, which could be ascribed that less hydrogenation epimer of Ru/C than CuO/c-UiO-66 gave a chance for priority hydrogenolysis.68
The aromatic and side chain regions of poplar DEL and lignin oily product of 2D HSQC NMR spectra are illustrated in Fig. 4, demonstrating the sufficient scission of lignin C–O bonds and product distributions after the RCD process. Notably, in the aromatic area, a significant reduction in signals was observed for G2, G5,6, and S2,6 (Fig. 4b). Moreover, in the side chain region, the detected cross-peaks of 4-propylphenols S/G1 are identified at δC/δH 31.7/2.50, 34.8/1.70 and 61.0/3.43 ppm (labeled in orange), and the signals at δC/δH 37.9/2.44, 24.8/1.55, and 14.1/0.87 ppm are ascribed to the propanol chain ascribed to S/G2 (labeled in blue) (Fig. 4b). Furthermore, no signal was detected for β-O-4′, demonstrating the active dissociation of lignin over the CuO/c-UiO-66 catalyst. This result revealed that nearly all the aryl-ether linkages in poplar lignin have been decomposed by C–O bond cleavage under the hydrogenolysis reaction conditions.
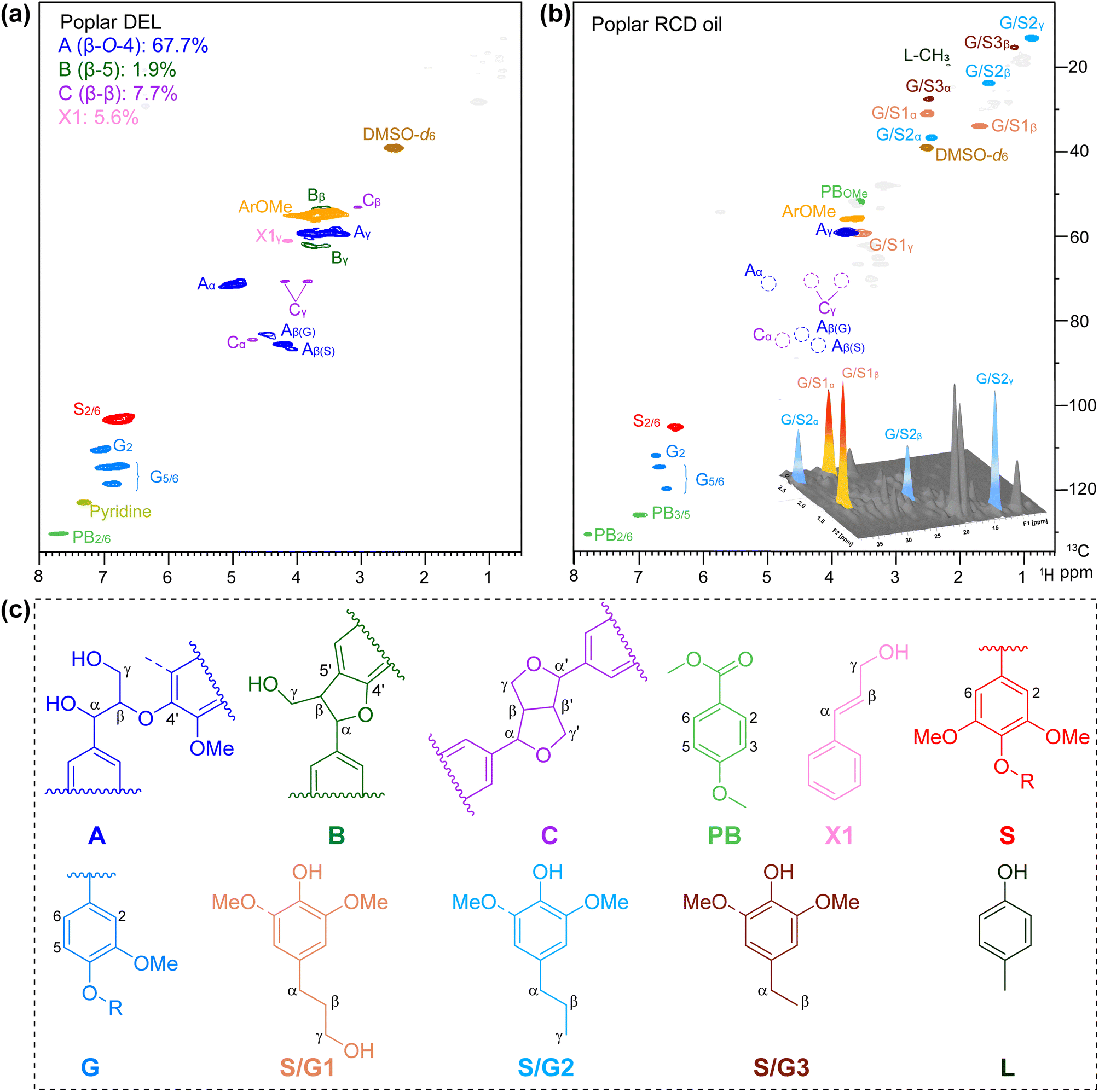 | ||
| Fig. 4 2D HSQC NMR spectrum of (a) DEL isolated from poplar and (b) lignin oily products from RCD of poplar over the CuO/c-UiO-66 catalyst. (c) Primary structure exhibited in the 2D HSQC NMR spectra: (A) β-O-4′ alkyl–aryl ether, (B) β-5′ phenylcoumaran, (C) β-β′ resinol, (PB) p-hydroxybenzoates, (X1) cinnamyl alcohol end-groups, (S) syringyl units, (G) guaiacyl units, (S1) 4-n-propanolsyringol, (S2) 4-n-propylsyringol, (G1) 4-n-propanolguaiacol, (G2) 4-n-propylguaiacol, (S3) 2,6-dimethoxy-4-ethylphenol, (G3) 4-ethylguaiacol, and (L) 4-methyl phenol. Reaction conditions: poplar sawdust (50 mg), CuO/c-UiO-66 catalyst (20 mg, 39.9 wt%), MeOH (10 mL), H2 (3 MPa), 240 °C for 4 h. | ||
3.3 Investigation of reaction conditions
In order to further understand the behavior of CuO/c-UiO-66, a series of experiments were undertaken, which aimed to further explore the optimal reaction conditions. Hydrogen might assist CuO/c-UiO-66 to resist the tendency of lignin recondensation reactions,63 so we kept the condition of 3 MPa H2 unchanged. First of all, we have designed catalysts with different Cu doping percentages for the hydrogenolysis of lignin, and the catalytic effect was relatively best when the molar ratio of Cu/Zr was 1.2 (Table S7†). Then, we explored the influence of different catalyst dosages (Fig. 5a), and the highest monomeric yield was obtained when the catalyst dosage was 20 mg. Different solvents were selected in the RCD of poplar sawdust over CuO/c-UiO-66, such as MeOH, EtOH, iPrOH, and dioxane (Fig. 5b). The CuO/c-UiO-66 catalyst showed maximum activity in MeOH toward the hydrogenolysis of poplar lignin under 240 °C for 4 h. The soluble fraction extracted by CH2Cl2 gave the oil-like product with an overall aromatic monomer yield of 42.9 wt%, from which S1 and S2 were in the highest yield. GPC analysis was applied to further examine the allocation of oligomers, dimers, and monomers in the lignin oil. A sharp decrease in Mw was observed from the 10850 g mol−1 of DEL as compared to the 470 g mol−1 of oily products (Fig. S18†). Similarly, both EtOH and iPrOH as the hydrogen donating solvents in the process showed reduced overall yields in comparison with that from MeOH in obtaining the phenolic monomers, while the high selectivity for S1 remained. This was in good agreement with the prior reports that the S unit always showed high reactivity in hydrogenolysis of lignin over late-transition-metal catalysts.69,70 Moreover, an increase of Mw was observed in the hydrogenolysis solvents EtOH (620 g mol−1) and iPrOH (1060 g mol−1) as compared to that of MeOH (470 g mol−1). This could be explained that the greater the polarity of the alcohol, the greater the hydrogen supply capacity and the more favorable to the RCD of lignin. However, in the case of iPrOH, an array of monomeric phenols with unsaturated substituents were formed in 15.0 wt% yield (Table S11†). This result might be due to the poor lignin solubility in iPrOH during this depolymerization process, thus resulting in the ether linkages among lignin units not being sufficiently fragmented.71 Moreover, the catalytic properties of the CuO/c-UiO-66 for RCD of lignin in dioxane were significantly reduced with a sharp drop in total monomer yield (25.0 wt%). This result was consistent with previous reports that alcohols performed better in the hydrogenolysis of lignin due to their superior hydrogen-donating ability.72
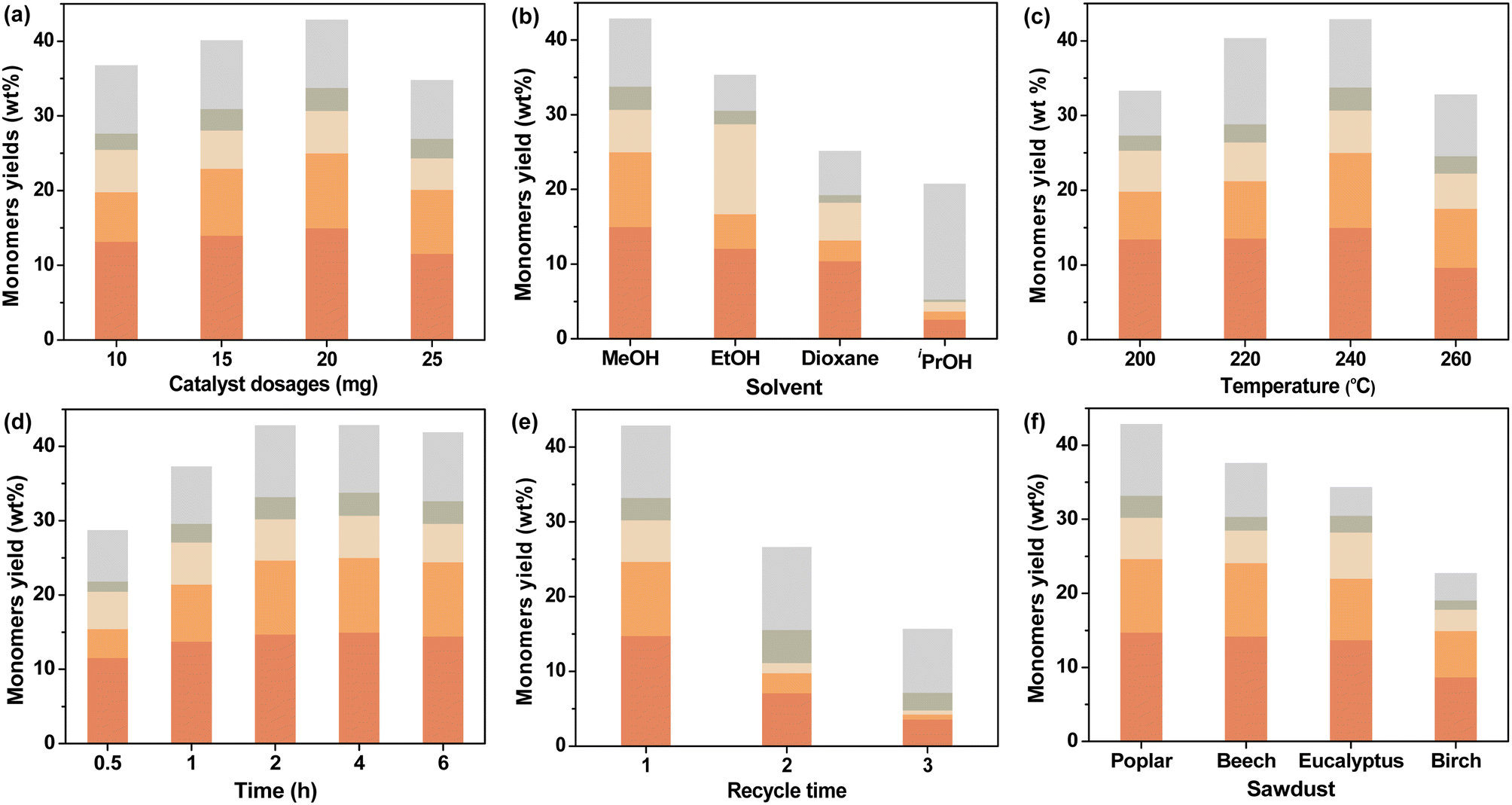 | ||
| Fig. 5 Influences of (a) catalyst dosage, (b) solvent, (c) temperature and (d) time for RCD of poplar sawdust with CuO/c-UiO-66. (e) Stability of CuO/c-UiO-66. (f) Catalytic hydrogenolysis of various hardwood sawdust. | ||
Further optimization of the reaction temperature in RCD of poplar sawdust with the CuO/c-UiO-66 catalyst has also been investigated (Fig. 5c). It was observed that raising the temperature to 260 °C, the hydrogenolysis reaction produced a lower total yield of monomers (32.9 wt%) and higher Mw values (550 g mol−1) as compared with those at 240 °C (42.9 wt%; 470 g mol−1) (Table S12 and Fig. S20†). These results are probably due to satisfactory cleavage of the majority of the C–O linkages and the retention of C–C bonds at such a temperature. Lowering the reaction temperature (∼200 and 220 °C) below 240 °C resulted in no loss of selectivity, but it led to a reduction in yield of 33.3 wt% and 40.3 wt%, respectively. The possible reason for these results was incomplete cleavage of ether bonds between lignin units due to insufficient temperature.
The influence of different reaction times on CuO/c-UiO-66-catalyzed hydrogenolysis of lignin was subsequently explored (Fig. 5d). Remarkably, 28.8 wt% monomeric phenols could be obtained at 0.5 h (Table S13†), which demonstrated the efficient catalytic activity of CuO/c-UiO-66. When prolonging the time to 1 h, a lignin oil with a monophenol yield of 37.2 wt% and Mw of 540 g mol−1 was generated. Although shorter times were also effective for the hydrogenolysis of lignin, the shortage of time might lead to insufficient exposure between the active site of CuO/c-UiO-66 and lignin, which resulted in an inadequate catalytic effect. Accordingly, the reaction times were suitably extended and the total aromatic monomer yield was stable after 2 h. That is to say, the use of a range of reaction times indicated that 2 h was sufficient for the C–O bond scissions. In summary, the favorable conditions for RCD of poplar sawdust over the CuO/c-UiO-66 catalyst in MeOH could efficiently convert lignin biopolymers to phenolic monomers with a relatively high yield (up to 42.8 wt%) and lowest Mw (475 g mol−1) with 3 MPa H2 at 240 °C for 2 h.
The ability to recover the catalyst was subsequently discussed. At the end of the hydrogenolysis process, a 200 mesh (0.074 mm) screen was used to effectively allow the small sized catalyst to escape through the pores of the screen. The separated catalyst was directly applied in the next cycle under the optimized reaction conditions. Notably, it was observed that the catalyst cycle could be performed three times with 15.7 wt% yield of monomers at the third cycle (Fig. 5e). The results of ICP analysis (Table S1†) and HADDF characterization (Fig. S4a†) of the spent catalyst showed a loss of copper species. We hypothesized that the decrease in catalytic activity could be due to the loss of the active center in the reducing hydrogen environment. Under such reaction conditions, the reactive center in the oxidation state might not be stably embedded in the MOF-derived support. Therefore, the next step is to design a Cu-based catalyst with more stable bonding between the active center and the carrier. However, to our delight, the fabricated CuO/c-UiO-66 revealed higher catalyst stability than our previously reported CuO/C.56
3.4 Hydrogenolysis of various hardwood sawdust
The hydrogenolysis of various hardwood samples, including beech, eucalyptus, and birch, was also performed over the CuO/c-UiO-66 catalyst under the optimized conditions to further evaluate the catalyst efficiency (Fig. 5f). Clearly, it was observed that all the three kinds of hardwoods afforded S1 and S2 as the main monophenols in high total yields (22.9–37.2 wt%). In a similar way to poplar sawdust, the RCD of other hardwood species underwent almost complete fragmentation of β-O-4′ linkages as illustrated by 2D HSQC NMR (Fig. S28–S30†). Suspiciously, it appeared that the copper-based catalysts were slightly less catalytically active on birch,56 and the lower β-O-4′ content in birch may be one of the reasons for this, which will be further investigated in our laboratory. Notably, the distribution of monomers and GPC results are shown in Fig. S24 and S25,† which revealed a significant decrease of Mw of lignin oils after RCD of different hardwood sawdust in comparison with those of DELs. Low molecular weight aromatic monomers could be used to produce polymers with strong thermal stability by introducing reactive groups or molecules.73–75 Furthermore, lignin monomers could be widely used in new products such as thermoplastics, coatings and resin materials.75–783.5 Mechanistic insights
In the blank control group, dimeric of β-O-4′ model compound 1 was reacted at 200 °C for 2 h (Fig. 6a). The generation of small amounts of 2-methoxy-4-(3-methoxy-1-propenyl)phenol 2 (34.7%) and 3 (32.4%) monomers was observed, as the hydrogen transfer reaction in supercritical MeOH could also achieve the cleavage of the C–O bonds, which is consistent with the blank experiments for depolymerizing poplar sawdust (Fig. 3a). For comparison, the above reaction proceeded in the presence of a combination of the CuO/c-UiO-66 catalyst and H2. It gave 4-n-propanolguaiacol G1 (9.8%), 4-n-propylguaiacol G2 (30.0%), 4-ethylguaiacol G3 (14.4%) and guaiacol 3 (55.5%) as the main products, which were derived from the hydrogenolysis of Cβ–O linkages. The acidic and basic active sites on the surface of CuO/c-UiO-66 led to the acceleration of the hydrogen transfer process, which resulted in the efficient cleavage of the C–O bonds.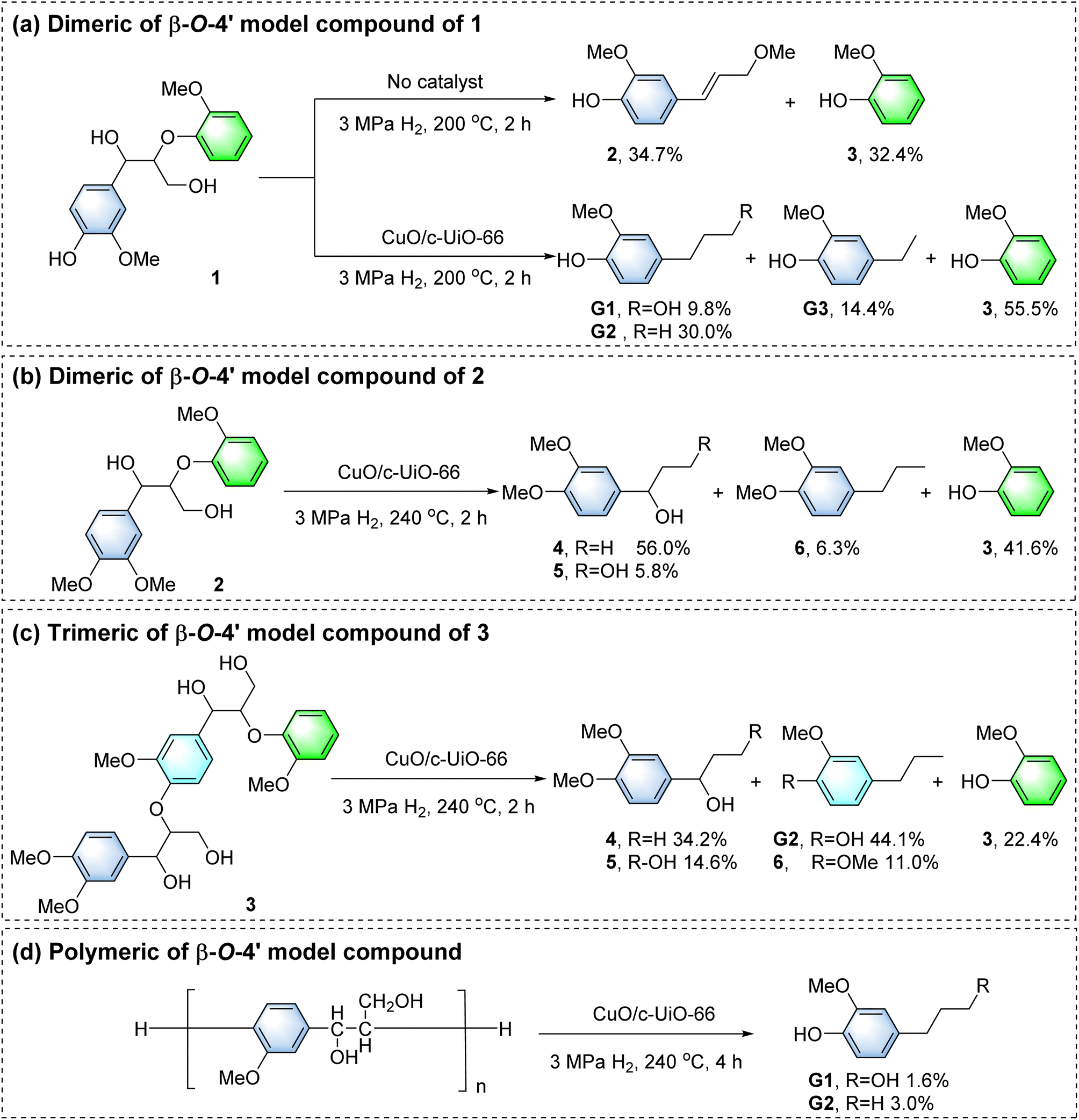 | ||
| Fig. 6 Simulated reactivity of β-O-4′ lignin model compounds over the CuO/c-UiO-66 catalyst. | ||
Compound 2 was not detected upon the addition of the catalyst as compared to the blank control group, so 2 might be an agent in the production of G1 and G2.35,79 In previous work, we confirmed that compound 2 was not generated via the reaction pathway of quinone radical products.56 Therefore, in this catalytic system, the Cα–O and Cβ–O bonds of the β-O-4′ molecule can be cleaved simultaneously. This provided important generative conditions for the production of G1 and G2. G1 almost completely reserved in the system (Fig. S37†), so the formation of G1 may be due to the cleavage of Cα–O and Cβ–O in the α-OMe β-O-4′ structure. The intermediate compound 2 undergoes a demethoxylation reaction to form G2.
The aromatic OMe groups played a key role in both oxidative polymerization and reductive depolymerization of lignin.3,80,81 In order to exclude the effect of phenolic hydroxyl groups on the products, depolymerization of nonphenolic β-O-4′ dimer 2 has been proposed (Fig. 6b). It was found that the phenolic hydroxyl group prevented the elimination of the α-OH, and the Cγ–OH bond is probably split, forming 1-(3′,4′-dimethoxyphenyl)-1-propanol 4 (56.0%). In addition, 1-(3,4-dimethoxyphenyl) propane-1,3-diol 5 (5.8%), 1,2-dimethoxy-4-n-propylbenzene 6 (6.3%) and 3 (41.6%) were also generated. This result demonstrated that there was no hydromethylation of phenolic hydroxyl groups during the β-O-4′ alcoholysis. Otherwise, the α-OH would be retained. The depolymerization of trimer 3 with CuO/c-UiO-66 also followed the same trend as the depolymerization of 2 (Fig. 6c). It should be noted that two Cβ–O bond cleavages provided 4 (34.2%), G2 (44.1%), 5 (14.6%), 3 (22.4%), and 6 (11.0%). Notably, G1 was not detected, which could also be due to γ-OH methylation of trimer 3.
To further simulate real lignin, the catalytic hydrogenation of β-O-4′ polymer was studied.82 This polymer had been broken down to G1 (1.6%) and G2 (3.0%) (Fig. 6d), which was consistent with wood biomass. GPC analysis showed a remarkably lower Mw (700 g mol−1) than β-O-4′ polymer (4190 g mol−1) (Fig. S35†), demonstrating the successful depolymerization of the macromolecule. As reported, C-2 of the G unit reacted readily with C-5 in other lignin units, leading to the formation of condensed structures83 and thus causing comparatively modest yields of the aromatic monomers.
Based on the mechanistic studies and relevant literature,39,56,63 a plausible reaction pathway for the RCD of lignin from woody biomass into phenolic monomers over the CuO/c-UiO-66 catalyst is proposed in Fig. 7. In the natural biomass, nucleophilic substitution reactions might occur between MeOH and the β-O-4′ structure, resulting in α-OMe or α,γ-OMe. Subsequently, the synergistic effects of the catalyst and hydrogen lead to the efficient scission of the C–O bonds of the methoxylated β-O-4′ intermediates, affording the target monophenols.
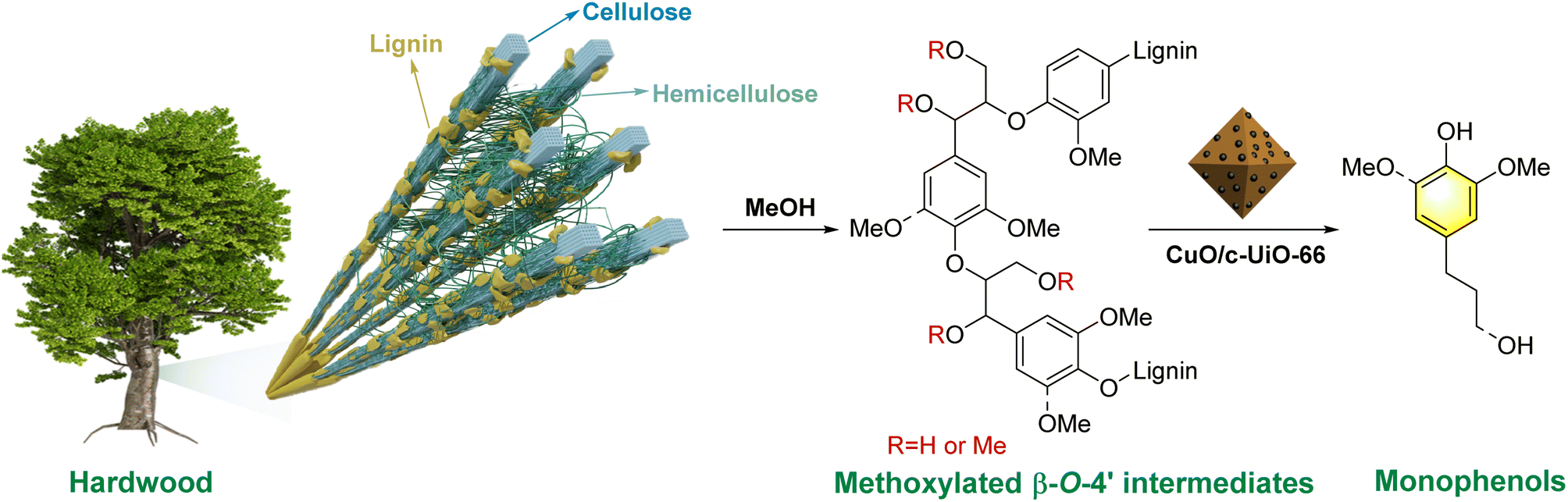 | ||
| Fig. 7 Plausible reaction pathway for the reductive catalytic deconstruction of lignin from woody biomass into phenolic monomers over the CuO/c-UiO-66 catalyst. | ||
4. Conclusions
We have reported a non-precious metal catalyst CuO/c-UiO-66 for the reductive hydrogenolysis of poplar lignin into monophenols in high yields (up to 42.8 wt%), which exhibited excellent catalytic properties. It was found that the CuO/c-UiO-66 showed satisfactory catalytic activity and good applicability in other hardwoods with high monomer yields (22.9–37.2 wt%). In view of the reactivity study of the β-O-4′ analogue, 4-propanol- and 4-propyl-substituted phenols could be formed directly over the CuO/c-UiO-66 catalyst. More specifically, it was also found that the two phenolic hydroxymethyl groups led to the retention of the α-OH group, which indicated that there was no methylation of the phenolic hydroxyl group under this catalytic system. The synergistic effect of the acid–base sites of the catalyst promoted the cleavage of the C–O bond. Of note was the optimum reactivity obtained under mild reaction conditions and the high catalytic rate of CuO/c-UiO-66. The next step is to further design a Cu-based catalyst with strong interactions between the active site and the substrate by our experimental platform for improving the cycling performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22278049 and 51961125207), the Liaoning Revitalization Talents Program (XLYC2007104), and the Scientific Research Fund Program for Liaoning Provincial Department of Education (LJKZZ20220071).References
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS PubMed.
- H. Li, A. Bunrit, N. Li and F. Wang, Chem. Soc. Rev., 2020, 49, 3748–3763 RSC.
- Z. Liu, H. Li, X. Gao, X. Guo, S. Wang, Y. Fang and G. Song, Nat. Commun., 2022, 13, 4716 CrossRef CAS PubMed.
- S. Su, L.-P. Xiao, X. Chen, S. Wang, X.-H. Chen, Y. Guo and S.-R. Zhai, ChemSusChem, 2022, 15, e202200365 CrossRef CAS PubMed.
- A. Avella, M. Ruda, C. Gioia, V. Sessini, T. Roulin, C. Carrick, J. Verendel and G. L. Re, Chem. Eng. J., 2023, 463, 142245 CrossRef CAS.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 RSC.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top Value-Added Chemicals from Biomass – Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin, United States, 2007 Search PubMed.
- A. Bohre, A. Modak, V. Chourasia, P. R. Jadhao, K. Sharma and K. K. Pant, Chem. Eng. J., 2022, 450, 138032 CrossRef CAS.
- T. Nimmanwudipong, R. C. Runnebaum, D. E. Block and B. C. Gates, Energy Fuels, 2011, 25, 3417–3427 CrossRef CAS.
- H. Luo, E. P. Weeda, M. Alherech, C. W. Anson, S. D. Karlen, Y. Cui, C. E. Foster and S. S. Stahl, J. Am. Chem. Soc., 2021, 143, 15462–15470 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191–196 RSC.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. d. Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- X. Huang, C. Atay, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Catal., 2015, 5, 7359–7370 CrossRef CAS.
- L.-P. Xiao, S. Wang, H. Li, Z. Li, Z.-J. Shi, L. Xiao, R.-C. Sun, Y. Fang and G. Song, ACS Catal., 2017, 7, 7535–7542 CrossRef CAS.
- R. C. Runnebaum, T. Nimmanwudipong, D. E. Block and B. C. Gates, Catal. Sci. Technol., 2012, 2, 113–118 RSC.
- Y. Zhai, C. Li, G. Xu, Y. Ma, X. Liu and Y. Zhang, Green Chem., 2017, 19, 1895–1903 RSC.
- Z. Sun, J. Cheng, D. Wang, T.-Q. Yuan, G. Song and K. Barta, ChemSusChem, 2020, 13, 5199–5212 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- D. Raikwar, K. Van Aelst, T. Vangeel, S. Corderi, J. Van Aelst, S. Van den Bosch, K. Servaes, K. Vanbroekhoven, K. Elst and B. F. Sels, Chem. Eng. J., 2023, 461, 141999 CrossRef CAS.
- R. M. O'Dea, P. A. Pranda, Y. Luo, A. Amitrano, E. O. Ebikade, E. R. Gottlieb, O. Ajao, M. Benali, D. G. Vlachos, M. Ierapetritou and T. H. Epps, Sci. Adv., 2022, 8, eabj7523 CrossRef PubMed.
- L. Monsigny, E. Feghali, J.-C. Berthet and T. Cantat, Green Chem., 2018, 20, 1981–1986 RSC.
- X. Liu, H. Li, L.-P. Xiao, R.-C. Sun and G. Song, Green Chem., 2019, 21, 1498–1504 RSC.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- J. Gracia-Vitoria, S. C. Gándara, E. Feghali, P. Ortiz, W. Eevers, K. S. Triantafyllidis and K. Vanbroekhoven, Curr. Opin. Green Sustainable Chem., 2023, 40, 100781 CrossRef CAS.
- K. Ye, Y. Liu, S. Wu and J. Zhuang, Ind. Crop. Prod., 2021, 172, 114008 CrossRef CAS.
- J. Zhang, Green Energy Environ., 2018, 3, 328–334 CrossRef.
- J.-Y. Kim, J. Park, H. Hwang, J. K. Kim, I. K. Song and J. W. Choi, J. Anal. Appl. Pyrol., 2015, 113, 99–106 CrossRef CAS.
- T. Ren, S. You, Z. Zhang, Y. Wang, W. Qi, R. Su and Z. He, Green Chem., 2021, 23, 1648–1657 RSC.
- F. Brienza, K. Van Aelst, F. Devred, D. Magnin, B. F. Sels, P. A. Gerin, I. Cybulska and D. P. Debecker, ACS Sustainable Chem. Eng., 2022, 10, 11130–11142 CrossRef CAS.
- K. Zhang, H. Li, L.-P. Xiao, B. Wang, R.-C. Sun and G. Song, Bioresour. Technol., 2019, 285, 121335 CrossRef PubMed.
- R. Shu, Y. Xu, L. Ma, Q. Zhang, C. Wang and Y. Chen, Chem. Eng. J., 2018, 338, 457–464 CrossRef CAS.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, M. T. Johan, K. Van Acker, B. Lagrain, D. Verboekend and F. Sels Bert, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- X. Huang, O. M. M. Gonzalez, J. Zhu, T. I. Korányi, M. D. Boot and E. J. M. Hensen, Green Chem., 2017, 19, 175–187 RSC.
- J. Hu, M. Zhao, B. Jiang, S. Wu and P. Lu, Energy Fuels, 2020, 34, 9754–9762 CrossRef CAS.
- I. Nakamula, Y. Yamanoi, T. Imaoka, K. Yamamoto and H. Nishihara, Angew. Chem., Int. Ed., 2011, 50, 5830–5833 CrossRef CAS PubMed.
- Y. Wu, S. Cai, D. Wang, W. He and Y. Li, J. Am. Chem. Soc., 2012, 134, 8975–8981 CrossRef CAS PubMed.
- R. K. Sharma, S. Yadav, S. Dutta, H. B. Kale, I. R. Warkad, R. Zbořil, R. S. Varma and M. B. Gawande, Chem. Soc. Rev., 2021, 50, 11293–11380 RSC.
- J. Sun, H. Li, L.-P. Xiao, X. Guo, Y. Fang, R.-C. Sun and G. Song, ACS Sustainable Chem. Eng., 2019, 7, 4666–4674 CrossRef CAS.
- S. Rautiainen, D. Di Francesco, S. N. Katea, G. Westin, D. N. Tungasmita and J. S. M. Samec, ChemSusChem, 2019, 12, 404–408 CrossRef CAS PubMed.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- M. Chui, G. Metzker, C. M. Bernt, A. T. Tran, A. C. B. Burtoloso and P. C. Ford, ACS Sustainable Chem. Eng., 2017, 5, 3158–3169 CrossRef CAS.
- X. Zhu, Y. Liu, Y. Xiao, L. Zhong, T. Wang, Y. Wang and W.-P. Pan, Chem. Eng. J., 2023, 467, 143402 CrossRef CAS.
- C. Guo, F. Duan, S. Zhang, L. He, M. Wang, J. Chen, J. Zhang, Q. Jia, Z. Zhang and M. Du, J. Mater. Chem. A, 2022, 10, 475–507 RSC.
- D. Yang, V. Bernales, T. Islamoglu, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2016, 138, 15189–15196 CrossRef CAS PubMed.
- Y. Xue, S. Zheng, H. Xue and H. Pang, J. Mater. Chem. A, 2019, 7, 7301–7327 RSC.
- C.-Y. Fang, S. Zhang, Y. Hu, M. Vasiliu, J. E. Perez-Aguilar, E. T. Conley, D. A. Dixon, C.-Y. Chen and B. C. Gates, ACS Catal., 2019, 9, 3311–3321 CrossRef CAS.
- Q. Wang, Y. Tan, S. Tang, W. Liu, Y. Zhang, X. Xiong and Y. Lei, ACS Nano, 2023, 17, 9565–9574 CrossRef CAS PubMed.
- Y. Wang, J. Wu, S. Tang, J. Yang, C. Ye, J. Chen, Y. Lei and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202219191 CrossRef CAS PubMed.
- C. He, J. Liang, Y.-H. Zou, J.-D. Yi, Y.-B. Huang and R. Cao, Natl. Sci. Rev., 2022, 9, nwab157 CrossRef CAS PubMed.
- J. Liang, R.-P. Chen, X.-Y. Wang, T.-T. Liu, X.-S. Wang, Y.-B. Huang and R. Cao, Chem. Sci., 2017, 8, 1570–1575 RSC.
- Y.-H. Zou, Y.-B. Huang, D.-H. Si, Q. Yin, Q.-J. Wu, Z. Weng and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 20915–20920 CrossRef CAS PubMed.
- Y.-L. Dong, Z.-Y. Jing, Q.-J. Wu, Z.-A. Chen, Y.-B. Huang and R. Cao, J. Mater. Chem. A, 2023, 11, 8739–8746 RSC.
- Q. Wang, L.-P. Xiao, Y.-H. Lv, W.-Z. Yin, C.-J. Hou and R.-C. Sun, ACS Catal., 2022, 12, 11899–11909 CrossRef CAS.
- T. Liu, X. Hong and G. Liu, ACS Catal., 2020, 10, 93–102 CrossRef CAS.
- W.-X. Li, W.-Z. Xiao, Y.-Q. Yang, Q. Wang, X. Chen, L.-P. Xiao and R.-C. Sun, Ind. Crops Prod., 2021, 170, 113692 CrossRef CAS.
- D. J. McClelland, A. H. Motagamwala, Y. Li, M. R. Rover, A. M. Wittrig, C. Wu, J. S. Buchanan, R. C. Brown, J. Ralph, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 1378–1389 RSC.
- L. Li, J. Kong, H. Zhang, S. Liu, Q. Zeng, Y. Zhang, H. Ma, H. He, J. Long and X. Li, Appl. Catal., B, 2020, 279, 119343 CrossRef CAS.
- Y. Wan, G. Yang, J. Xiang, X. Shen, D. Yang, Y. Chen, V. Rac, V. Rakic and X. Du, Dalton Trans., 2020, 49, 764–773 RSC.
- F. Arena, G. Italiano, K. Barbera, S. Bordiga, G. Bonura, L. Spadaro and F. Frusteri, Appl. Catal., A, 2008, 350, 16–23 CrossRef CAS.
- S. Van den Bosch, T. Renders, S. Kennis, S. F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- X. Gong, J. Sun, X. Xu, B. Wang, H. Li and F. Peng, Bioresour. Technol., 2021, 333, 124977 CrossRef CAS PubMed.
- S. Oh, S. Gu, J.-W. Choi, D. J. Suh, H. Lee, C. S. Kim, K. H. Kim, C.-J. Yoo, J. Choi and J.-M. Ha, J. Environ. Chem. Eng., 2022, 10, 108085 CrossRef CAS.
- M. Hou, H. Chen, Y. Li, H. Wang, L. Zhang and Y. Bi, Energy Fuels, 2022, 36, 1929–1938 CrossRef CAS.
- S. Wang, K. Zhang, H. Li, L.-P. Xiao and G. Song, Nat. Commun., 2021, 12, 416 CrossRef CAS PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- X. Ma, R. Ma, W. Hao, M. Chen, F. Yan, K. Cui, Y. Tian and Y. Li, ACS Catal., 2015, 5, 4803–4813 CrossRef CAS.
- Y. Deng, R. Gao, L. Lin, T. Liu, X.-D. Wen, S. Wang and D. Ma, J. Am. Chem. Soc., 2018, 140, 14481–14489 CrossRef CAS PubMed.
- A. L. Holmberg, J. F. Stanzione III, R. P. Wool and T. H. Epps III, ACS Sustainable Chem. Eng., 2014, 2, 569–573 CrossRef CAS.
- S. Wang, L. Shuai, B. Saha, D. G. Vlachos and T. H. Epps III, ACS Cent. Sci., 2018, 4, 701–708 CrossRef CAS PubMed.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed.
- S. Wang, A. W. Bassett, G. V. Wieber, J. F. Stanzione III and T. H. Epps III, ACS Macro Lett., 2017, 6, 802–807 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, Macromolecules, 2017, 50, 3573–3581 CrossRef CAS.
- W.-Z. Yin, L.-P. Xiao, Q. Wang, Y.-H. Lv, S.-L. Zou, Z. Wei and R.-C. Sun, Composites, Part B, 2023, 266, 111013 CrossRef CAS.
- I. Kumaniaev, E. Subbotina, J. Sävmarker, M. Larhed, M. V. Galkin and J. S. M. Samec, Green Chem., 2017, 19, 5767–5771 RSC.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- X.-J. Shen, T. Chen, H.-M. Wang, Q. Mei, F. Yue, S. Sun, J.-L. Wen, T.-Q. Yuan and R.-C. Sun, ACS Sustainable Chem. Eng., 2020, 8, 2130–2137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04927b |
| This journal is © The Royal Society of Chemistry 2023 |