
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of polymer nanoparticle-based complex coacervate hydrogels using polymerisation-induced self-assembly derived nanogels†
Ruiling
Du
 ab and
Lee A.
Fielding
*ab
ab and
Lee A.
Fielding
*ab
aDepartment of Materials, School of Natural Sciences, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: lee.fielding@manchester.ac.uk
bHenry Royce Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 23rd February 2023
Abstract
This paper reports a generic method to prepare polymer nanoparticle-based complex coacervate (PNCC) hydrogels by employing rationally designed nanogels synthesised by reversible addition-fragmentation chain-transfer (RAFT)-mediated polymerisation-induced self-assembly (PISA). Specifically, a poly(potassium 3-sulfopropyl methacrylate) (PKSPMA) macromolecular chain-transfer agent (macro-CTA) was synthesised via RAFT solution polymerisation followed by chain-extension with a statistical copolymer of benzyl methacrylate (BzMA) and methacrylic acid (MAA) at pH 2. Thus, pH-responsive nanoparticles (NPs) comprising a hydrophobic polyacid core-forming block and a sulfonate-functional stabiliser block were formed. With the introduction of methacrylic acid into the core of the NPs, they become swollen with increasing pH, as judged by dynamic light scattering (DLS), indicating nanogel-type behaviour. PNCC hydrogels were prepared by simply mixing the PISA-derived nanogels and cationic branched polyethyleneimine (bPEI) at 20% w/w. In the absence of MAA in the core of the NPs, gel formation was not observed. The mass ratio between the nanogels and bPEI affected resulting hydrogel strength and a mixture of bPEI and PKSPMA68-P(BzMA0.6-stat-MAA0.4)300 NPs with a mass ratio of 0.14 at pH ∼7 resulted in a hydrogel with a storage modulus of approximately 2000 Pa, as determined by oscillatory rheology. This PNCC hydrogel was shear-thinning and injectable, with recovery of gel strength occurring rapidly after the removal of shear.
Introduction
Hydrogels are widely used in everyday and specialist applications such as contact lenses,1 adsorbents for water purification,2 soft tissue engineering,3 drug delivery,4 and energy storage5 owing to their high water content and highly tuneable mechanical properties. An important class of hydrogels are ‘polymeric complex coacervates’.6 These gels are typically formed from two oppositely charged macromolecules via reversible ionic bonds.7,8 Complex coacervate gels have been studied for decades to yield an interesting range of soft materials, and have been widely used as a strategy for encapsulation in cosmetic, pharmaceutical industry and food science industries.9,10 Complex coacervate-based underwater adhesives inspired by marine organisms, such as sandcastle worms which secrete coacervated proteins, have also been reported.11,12Another interesting class of soft hydrogels are particle-based gels. Polymer–nanoparticle interactions have been employed to allow the formation of tuneable and self-healing hydrogels.13 For instance, Appel et al.14 synthesized self-assembled hydrogels by mixing hydroxypropylmethylcellulose derivatives and PEG-poly(lactic acid) nanoparticles. Similarly, collagen-gold hybrid hydrogels with adjustable mechanical properties were prepared using a biomineralization-inspired trigger.15 Highly swellable microgels synthesized via seed-feed emulsion polymerisation have been investigated as injectable materials that can form macroscopic hydrogels.16 Substantial swelling of such microgels (in excess of 200 times by volume) facilitates their pH-triggered physical gelation, which is subsequently followed by re-enforcement by chemical crosslinking.17
Recently, there has been an interest in preparing nanoparticle-based complex coacervate gels. Wu et al.18 prepared polymer/microgel complex coacervate gels by mixing pre-formed polyacid microgel nanoparticles (NPs) and cationic branched poly(ethylene imine) (bPEI) followed by thermal annealing under mild conditions. This new family of gels was shapeable, super-stretchable, self-healing, adhesive, had low cytotoxicity and could be toughened using Ca2+. More recently, graphene nanoplatelets were introduced to these hydrogels, conferring electrical conductivity as well as tensile and compressive strain-sensing capability.19 However, such microgels were prepared by seed-feed emulsion polymerisation. This has the benefit of producing particles of tuneable size with adjustable swelling ratios but it is relatively limited in allowing the formation of NPs with precise molecular weight, functionality distribution and morphological control.
Polymerisation-induced self-assembly (PISA) via reversible addition-fragmentation chain-transfer (RAFT) polymerisation is a popular method for preparing self-assembled block copolymer NPs in recent years.20–23 It is a robust and efficient route to generate amphiphilic copolymer NPs of controllable size, morphology, and surface chemistry. Typically, a solvophilic homopolymer, also known as a macromolecular chain-transfer agent (macro-CTA) or stabiliser, is prepared and extended with monomer(s) in a selective solvent for the second block, resulting in NP formation during polymerisation. Depending on the initial solubility of the monomers used, PISA can proceed by either emulsion polymerisation (insoluble monomer), and dispersion polymerisation (soluble monomer).22,24 PISA can be used to prepare block copolymer NPs at high solids content (up to 50% w/w) and is suitable for large-scale industrial production.24,25 Furthermore, the surface chemistry of PISA-derived NPs can be readily modified by selecting macro-CTA(s) with the desired distribution of functional groups, which enables the precise design of NPs exhibiting e.g., non-ionic,26,27 anionic28–30 or cationic coronas.31,32 For example, Wen et al.28 prepared poly(potassium 3-sulfopropyl methacrylate)-poly(benzyl methacrylate) (PKSPMA-PBzMA) spherical diblock copolymer NPs with tuneable diameters (20–200 nm) in alcohol/water mixtures by varying the copolymer composition, and/or by altering the co-solvent composition. Douverne and co-workers30 explored the effect of varying the anionic charge density in the steric stabilizer block on the occlusion efficiency of the nanoparticles within host crystals via statistical copolymerisation of anionic and non-ionic monomers. In addition, Fielding and co-workers33 pre-mixed poly(methacrylic acid) (PMAA) and poly(glycerol monomethacrylate) macro-CTAs prior to the extension of 2-hydroxypropyl methacrylate via RAFT aqueous dispersion polymerization in order to prepare NPs with tuneable morphologies and adjustable anionic character.
Whilst there have been many examples of PISA-derived NPs with functional coronas, there are relatively limited examples where the core of the nanoparticle also provides functionality. For example, Mable et al.,34 demonstrated the preparation of framboidal triblock copolymer vesicles where the core-forming blocks phase separated within the wall of the assembled structure. Additionally, schizophrenic NPs have recently been reported whereby the core and the corona invert depending on the external pH.35 There are a few reports on PISA-derived thermosensitive nanogels based on thermosensitive monomers (e.g. N-acryloyl glycinamide,36N-isopropylacrylamide,37 3-dimethyl-(methacryloyloxyethyl)ammonium propanesulfonate).38 However, there are no examples of PISA-derived particles which display pH-sensitive nanogel-type behaviour. In this work, we hypothesize that PISA can be used to prepare pH-sensitive nanogel particles by incorporating a carboxylic acid comonomer into the insoluble block. Furthermore, we expect that this new class of copolymer nanoparticles should be useful for the preparation of functional NP coacervate gels with tuneable properties.
Herein, the formation of PISA-derived copolymer NP coacervate gels was investigated (Scheme 1). Firstly, nanogels were prepared by chain extending PKSPMA68 with a statistical copolymer of BzMA and MAA at pH 2. This allowed pH-responsive spherical NPs with an anionic sulfonate corona and a hydrophobic polyacid core to be obtained. The resulting size, morphology and swellability of the NPs were characterised via dynamic light scattering (DLS) and aqueous electrophoresis. Subsequently, a series of polymer/NP complex coacervate (PNCC) gels were prepared by the addition of bPEI at various bPEI-to-NPs mass ratios (MR). The gel strength and shear thinning behaviour of these coacervate gels were characterised via strain-dependent oscillatory shear rheology.
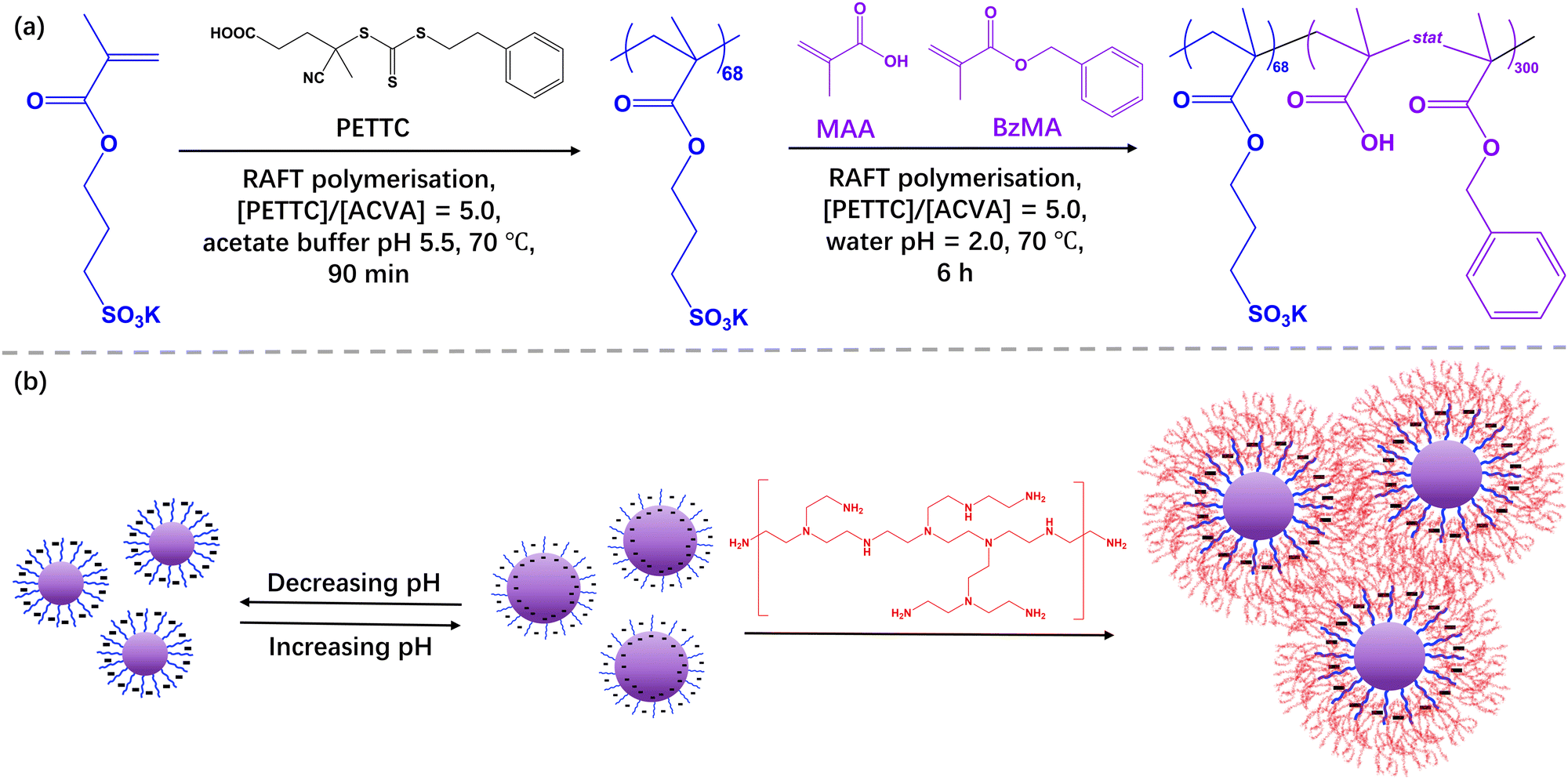 | ||
| Scheme 1 Synthesis of poly(potassium 3-sulfopropyl methacrylate) (PKSPMA) macro-CTA via RAFT solution polymerisation at 70 °C (15% w/w, pH 5.5), followed by RAFT-mediated PISA of benzyl methacrylate (BzMA) and methacrylic acid (MAA) in water at pH 2 at 70 °C (20% w/w). (b) Schematic representation of how the solution pH affects the resulting nanoparticle diameter and bPEI/NPs hydrogel preparation. | ||
Experimental
Materials
Potassium 3-sulfopropyl methacrylate (KSPMA, 98%), methacrylic acid (MAA, 99%), branched polyethyleneimine (bPEI, 10 kDa) and 4,4′-azobis(4-cyanovaleric acid) (ACVA, 98%) were purchased from Sigma-Aldrich (UK) and used as received. Benzyl methacrylate (98%) was purchased from Alfa Aesar (UK) and passed through a column of activated basic alumina to remove inhibitors and impurities before use. 4-Cyano-4-(2-phenylethane sulfanylthiocarbonyl) sulfanylpentanoic acid (PETTC) was synthesised in-house using previously reported protocols.28 1,4-dioxane was purchased from Honeywell (UK) and used as received. PKSPMA68 (denoted S68) was prepared in-house (see ESI†) following literature protocols via RAFT solution polymerisation (Mn 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1 and Mw/Mn of 1.34, Fig. S2, ESI†).28,39,40 Deuterium oxide (D2O) for NMR characterisation was purchased from Cambridge Isotope Laboratories (UK). Methanol (> 99.9%) was purchased from Fisher Scientific (UK) and used as received. Dialysis tubing (regenerated cellulose, MWCO = 3.5 kDa, diameter = 29 mm) was received from Fisher Scientific (UK). Deionised water (DI water) was used in all experiments.
100 g mol−1 and Mw/Mn of 1.34, Fig. S2, ESI†).28,39,40 Deuterium oxide (D2O) for NMR characterisation was purchased from Cambridge Isotope Laboratories (UK). Methanol (> 99.9%) was purchased from Fisher Scientific (UK) and used as received. Dialysis tubing (regenerated cellulose, MWCO = 3.5 kDa, diameter = 29 mm) was received from Fisher Scientific (UK). Deionised water (DI water) was used in all experiments.
Preparation of PKSPMA68-P(BzMAm-stat-MAA1−m)300 block copolymer nanoparticles via RAFT aqueous emulsion polymerisation
A typical protocol for the synthesis of PKSPMA68-P(BzMA0.6-stat-MAA0.4)300, denoted S68-P(B0.6-A0.4)300, is as follows. PKSPMA68 macro-CTA (0.40 g, 0.03 mmol), BzMA monomer (879 mg, 5.00 mmol), MAA monomer (286 mg, 3.33 mmol), ACVA (1.6 mg, 5.5 μmol; CTA/initiator molar ratio = 5.0) were weighed into a 25 mL round bottomed flask. Deionised water and 0.25 M HCl were added to adjust the reaction solution to pH 2.0 to prevent ionisation of the –COOH groups on MAA during the polymerisation and produce a 20% w/w aqueous solution. The solution was then purged with N2 for 30 min prior to immersion in an oil bath set at 70 °C. The heated reaction solution was stirred for 6 h before the polymerisation was quenched by cooling in an ice bath and exposure to air. Monomer conversions were determined via gravimetry by drying approximately 0.1 g of the final dispersion at 120 °C until constant weight using a Kern DBS 60-3 Moisture Analyser. Conversions were calculated based on the measured solids content and the theoretical solids content at full monomer conversion (see ESI†). In subsequent reactions, the ratio between BzMA and MAA was varied. Unfortunately, neither GPC nor NMR could be utilised to characterise S68-P(Bm-A1−m)300 nanoparticles owing to their inability to dissolve in any common solvents due to their highly amphiphilic nature.28Preparation of bPEI/PKSPMA68-P(BzMAm-stat-MAA1−m)300 mixtures
The following gives an example of the synthesis of a binary polyelectrolyte combination of S68-P(B0.6-A0.4)300 NPs and bPEI at a bPEI-to-NP mass ratio of 0.14 (denoted as bPEI/S68-P(B0.6-A0.4)300_MR0.14). The mixture was prepared by first transferring NP dispersion (2.0 mL; 20% w/w; pH∼ 2.5) to a 7 mL vial. Then bPEI (10 kDa) was diluted to 20% w/w with water (resulting solution pH ∼11.8) and 0.28 mL was injected into the NP dispersion dropwise. The mixture was mechanically stirred until it formed a smooth and uniform soft gel. Polymer/NP complex coacervates (PNCCs) with bPEI-to-NP mass ratios of 0.06 to 0.22 were subsequently prepared. The pH of the subsequent mixture was not adjusted unless otherwise noted.Characterization
Results and discussion
Preparation of pH-responsive diblock copolymer nanoparticles PKSPMA68-P(BzMAm-stat-MAA1−m)300via PISA
PKSPMA68 macro-CTA (Fig. S1 and S2, ESI†) was chain-extended via RAFT-mediated PISA of BzMA and MAA in acidic aqueous solution (pH 2) at 20% w/w to form sulfonate-functional NPs (Scheme 1). MAA was introduced as a statistical comonomer to confer carboxylic acid functionality to the core-forming block during the polymerisation and therefore introduce pH-responsive functionality to the core of these NPs. A low reaction pH was chosen to ensure the formation of a relatively hydrophobic second block during the polymerisation to ensure particle self-assembly. Specifically, a series of S68-P(Bm-stat-A1−m)300 copolymer nanoparticles were prepared while targeting core-forming block compositions with m ranging from 0.2 to 1.0. The target DP of the second block was fixed at 300 to ensure self-assembly, and high comonomer conversions (> 99%) were obtained within 6 h in all cases, as judged by gravimetry, which is consistent with previous literature on related PISA formulations.28,35The diluted BzMA-rich NP dispersions (m = 1.0, 0.8, 0.6) had diameters of approximately 50 nm at pH 2 (Table S1, ESI†). However, with decreasing BzMA content (m = 0.4, 0.2), the particles were found to be both somewhat larger (111 nm and 256 nm respectively) and more polydisperse, even though the MAA component should be in its protonated, non-ionic form at pH 2. Thus, the inclusion of relatively high amounts of MAA comonomer results in the formation of swollen, or less compact, micelles even at low pH. When the pH was increased to 10, the MAA-based NPs became swollen due to ionisation of the carboxylic acid groups in the NP cores (Fig. 1a), with the degree of swelling correlating with the MAA content.
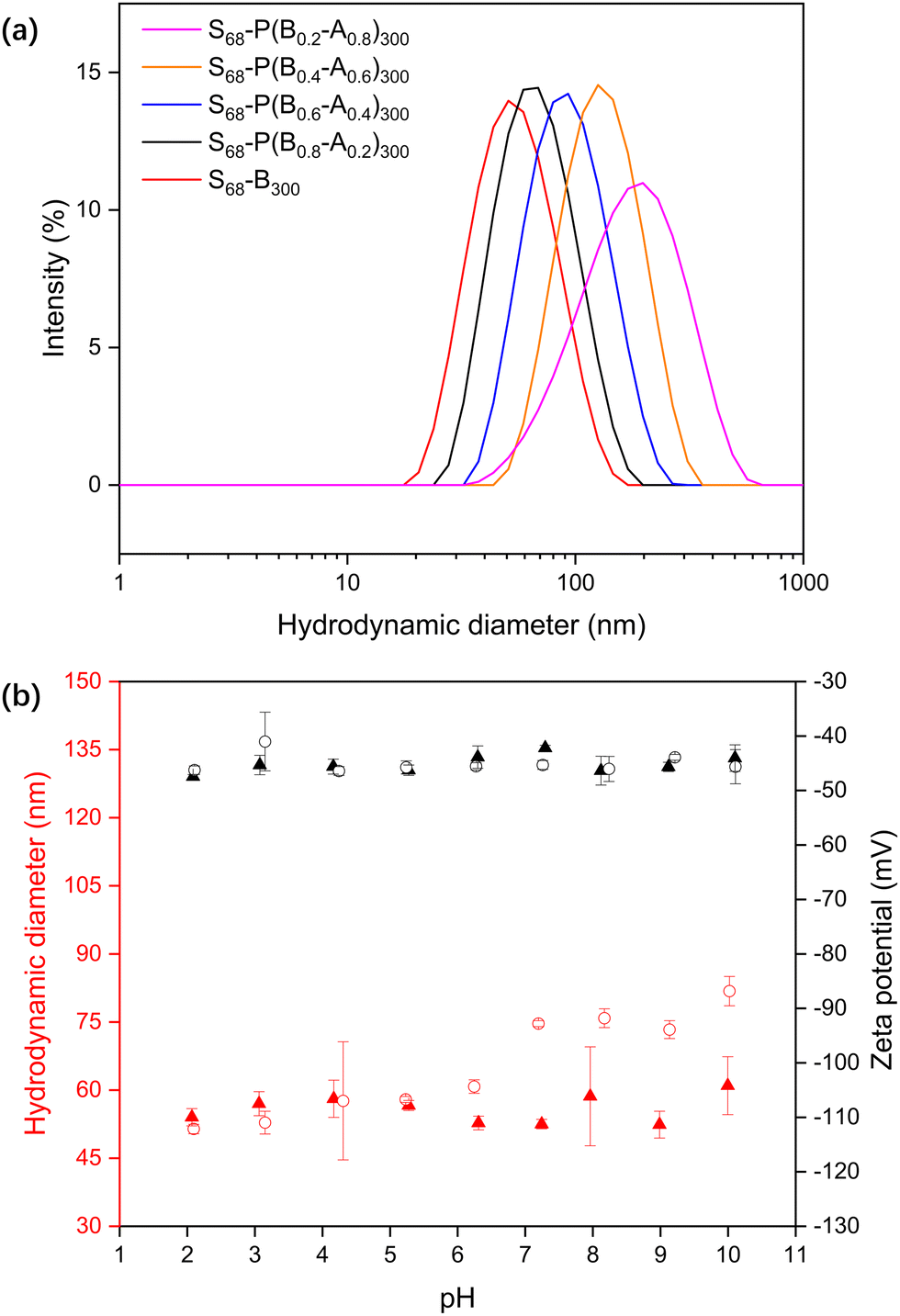 | ||
| Fig. 1 S68-P(Bm-A1−m)300 diblock copolymer nanoparticles prepared at 20% w/w solids via RAFT-mediated statistical copolymerisation of BzMA and MAA in water at pH 2. (a) DLS intensity-average size distributions of S68-P(Bm-A1−m)300 (m = 0.2, 0.4, 0.6, 0.8, 1.0) dispersions at pH 10; (b) dynamic light scattering and aqueous electrophoresis data as a function of pH obtained for S68-B300 (triangles) and S68-P(B0.6-A0.4)300 (circles) diblock copolymer nanoparticles. Measurements were conducted at a copolymer concentration of approximately 0.1% w/w in the presence of 1 mM KCl as a background electrolyte. | ||
The response of the S68-P(Bm-stat-A1−m)300 NPs as a function of pH was analysed by titrating the dispersions and monitoring their size and zeta potential (Fig. 1b). The zeta potentials of both S68-B300 and S68-P(B0.6-A0.4)300 are highly anionic and pH-independent (approximately −45 mV), even at relatively low pH. This is because the PKSPMA corona block is a strong anionic polyelectrolyte. The S68-B300 particle diameter was independent of pH, with a constant diameter of ∼50 nm. In contrast, the S68-P(B0.6-A0.4)300 particles are swollen at high pH (51 nm at pH 2 to 84 nm at pH 10). This is due to ionisation of the MAA residues in the NP core and subsequent nanogel behaviour.
Binary polyelectrolyte combinations of anionic PKSPMA68-PBzMA300 NPs and cationic bPEI
Initially, aqueous solutions of PKSPMA68-PBzMA300 (20% w/w) NPs and commercially available cationic branched poly(ethylene imine) (bPEI; 10 kD; 20% w/w) were mixed at various bPEI-to-NP mass ratios (0.02 to 0.80) to give an overall polymer concentration of 20% w/w. It was initially envisaged that these amphiphilic diblock NPs would act as nanosized crosslinkers with bPEI linking adjacent S68-B300 NPs via ionic interactions between amine groups on bPEI and sulfonate groups on PKSPMA. Unfortunately, after mixing at their native pH, all samples remained as homogeneous dispersions. Zeta potential data for diluted bPEI/S68-B300 dispersions (0.1% w/w) were recorded to investigate the interaction between the cationic bPEI and anionic S68-B300 NPs (Fig. 2a). At a low mass ratio of 0.02, bPEI/S68-B300 dispersions remained highly anionic (approximately −45 mV). This is due to the large amount of sulfonate groups being in excess of the amine groups present from bPEI (bPEI has a reported a pKa of 8.518). At a relatively high mass ratio of 0.80, the behaviour of polycationic bPEI dominated the electrophoretic behaviour of the mixture, with highly cationic and pH-independent zeta potentials being observed below pH 7 (∼55 mV). However, when the pH of the dispersion was increased from 7 to 10 the zeta potential gradually decreased from 55 mV to 20 mV as the bPEI gradually became deprotonated. At intermediate MRs, charge reversal was observed. For example, at an MR of 0.10, bPEI/S68-B300 dispersions were positively charged below pH 6.5 (due to protonation of amine groups) and became negatively charged when the pH increased above 6.5 (deprotonation of amine groups). This suggested that this MR should potentially be the optimum value for investigating gel formation as a function of pH. Therefore bPEI/S68-B300 mixtures at an MR of 0.10 and total polymer concentration of 20% w/w were prepared over a range of solution pH through the addition of 1.0 M HCl during mixing.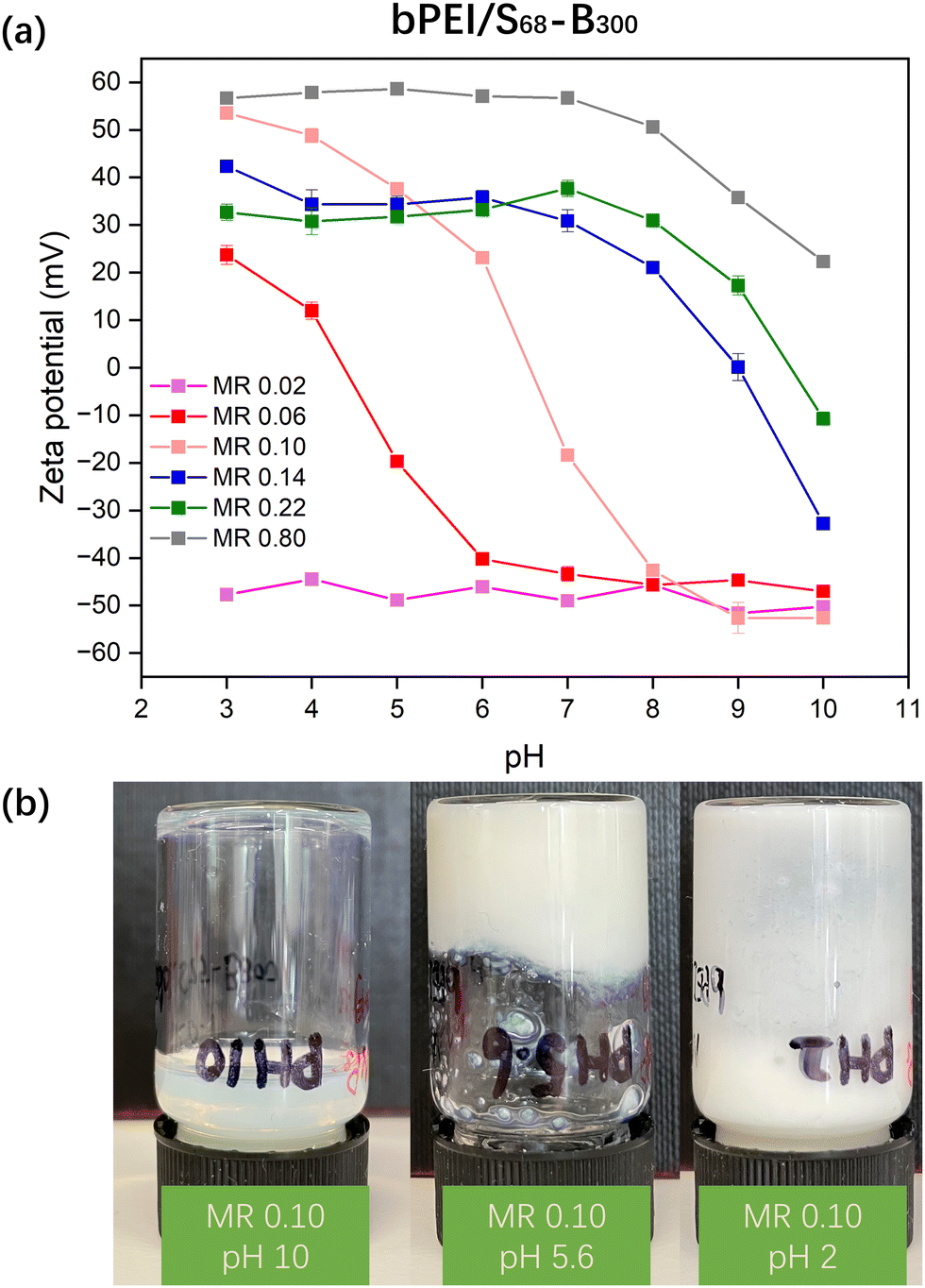 | ||
| Fig. 2 (a) Zeta potential vs. pH curves for dilute aqueous dispersions containing mixture of bPEI/S68-B300 mixtures with various bPEI-to-NPs MRs; (b) Digital image of fluids and precipitates for bPEI/S68-B300 mixtures at MR 0.10 (20% w/w) obtained at different pH values. | ||
At pH 10, it is likely that the neutral amine groups of bPEI are not efficient at forming ionic bonds with PKSPMA residues and thus did not efficiently bridge the NPs. This resulted in a homogeneous dispersion (Fig. 2b, left image). On lowering the pH of the bPEI/S68-B300 mixture (MR 0.10) the dispersion gradually became turbid. At pH 5.6, the mixture became relatively viscous (Fig. 2b, middle), which upon further inspection was not due to gelation but rather the formation of compact flocs (Fig. S3a, ESI†) at the isoelectric point (IEP) for this MR (Fig. 2a). At this pH, the protonated cationic amine groups from bPEI readily complex with the anionic S68-B300 NPs, and cause bulk flocculation of the mixture.
At pH 2.0 the mixture became free-flowing again (Fig. 2b, right) due to the NP/bPEI complexes gaining a net-cationic charge (Fig. 2a), but it remained inhomogeneous and not fully redispersed. Thus, simply mixing bPEI with charged NPs does not allow the formation of PNCCs: the use of nanogel particles is critical for the preparation of such materials.
Preparation of polymer/NPs complex coacervate (PNCC) gels via mixing PKSMA68-P(BzMA0.6-stat-MAA0.4)300 and cationic bPEI
Swellable S68-P(B0.6-A0.4)300 NPs were chosen to investigate the preparation of PNCCs because they have good colloidal stability, and a low PDI at both low and high pH (Table S1, ESI†). Aqueous dispersions of S68-P(B0.6-A0.4)300 (20% w/w) and bPEI (10 kD; 20% w/w) were mixed at different bPEI-to-NPs mass ratio (0.06 to 0.22) to investigate the formation of PNCCs with these PISA-derived nanogels. On the addition of bPEI to S68-P(B0.6-A0.4)300 dispersions, the mixtures gradually became viscous. Gelation occurred, as judged by tube inversion (Fig. 3a), for samples with a MR above 0.10. Soft gels were formed at a MR of 0.12, 0.14 and 0.16 (Fig. 3a). The prepared gels remain homogeneous at room temperature under standard conditions but when subjected to centrifugation liquid–liquid phase separation occurs (Fig. S4, ESI†), indicating a complex coacervation gelation mechanism. When excess bPEI was added (≥ MR 0.18), the mixture became a low-viscosity fluid again. These observations are likely a combination of both MR and pH: as the quantity of bPEI added increases so does the pH of the mixture. Thus, gelation occurs between approximately pH 6 and 8, with a MR between 0.12 and 0.16 facilitating this. This behaviour correlates with Fig. 2b but, in contrast to the S68-B300 mixtures described above, all dispersions/gels prepared using S68-P(B0.6-A0.4)300 were homogeneous and the presence of flocs was not evident (Fig. S3, ESI†). This supports the hypothesis that the formation of PNCC gels requires NPs with (nano)gel behaviour. In this case the bPEI interacts and bridges the NPs through the anionic corona and the swollen NP cores provide the necessary gel-like behaviour.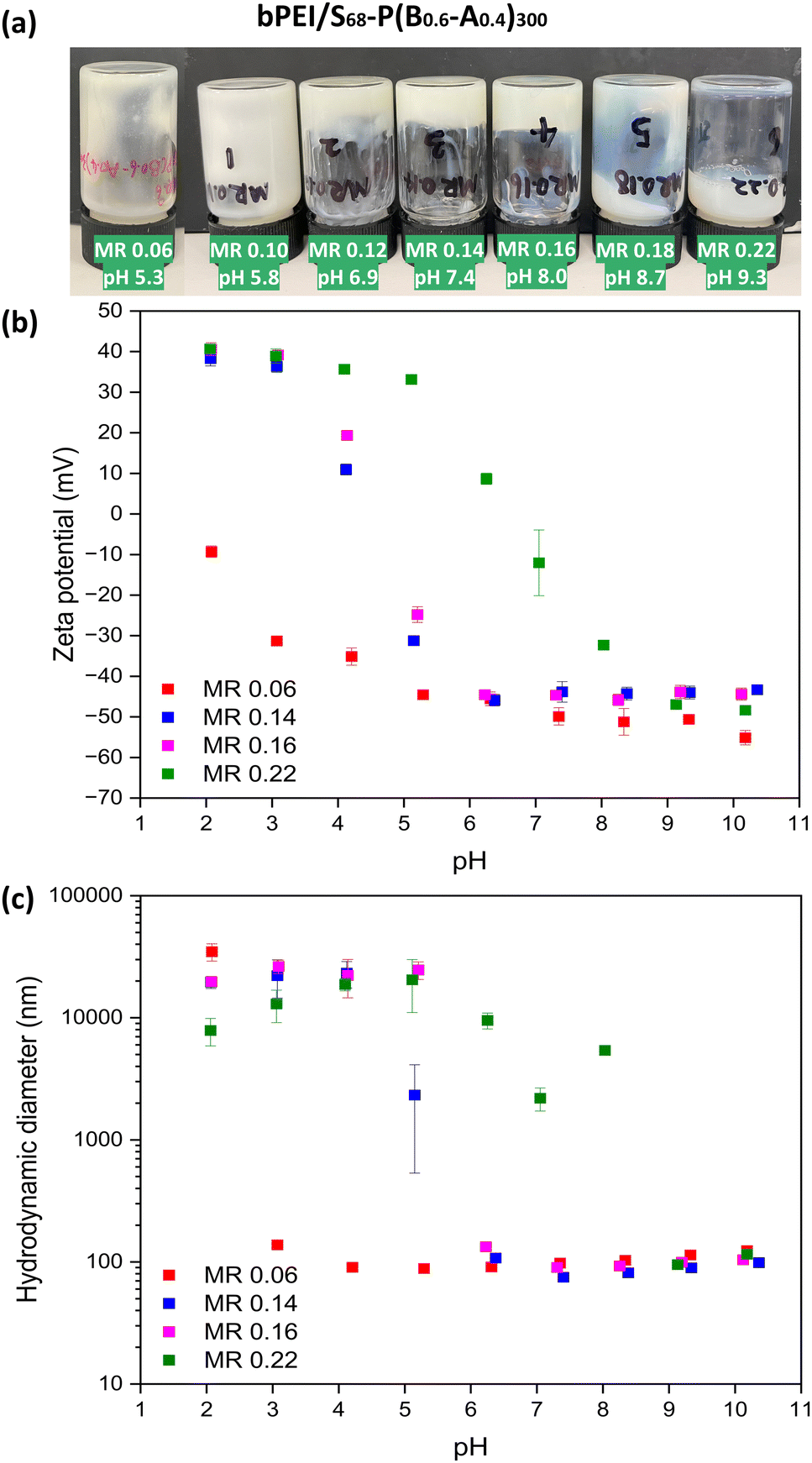 | ||
| Fig. 3 (a) Digital image of fluids and gels for bPEI/S68-P(B0.6-A0.4)300 mixtures (20% w/w) at different bPEI-to-NPs mass ratios (MR = 0.06, 0.10, 0.12, 0.16, 0.18, 0.22); (b) mean hydrodynamic diameters and (c) aqueous electrophoresis data as a function of pH obtained for dilute aqueous dispersions containing mixtures of bPEI/S68-P(B0.6-A0.4)300 at various bPEI-to-NPs mass ratios (MR = 0.06, 0.14, 0.16, 0.22). Measurements were conducted at a copolymer concentration of approximately 0.1% w/w in the presence of 1 mM KCl as a background electrolyte. | ||
Dilute aqueous dispersions of bPEI/S68-P(B0.6-A0.4)300 PNCC gels formed at various MRs were analysed by DLS and aqueous electrophoresis as a function of pH (Fig. 3b and c). In most cases, relatively small (∼100 nm) anionic NPs were observed above pH 6.0 with zeta potentials of approximately −45 mV. On lowering the pH, the zeta potential became more positive for samples with an MR ≥ 0.12, with isoelectric points between pH 4 and 8. The size of these anionic NPs slightly decreased from pH 10 to 7 followed by flocculation at pH values corresponding to observed increases in zeta potential. When the MR was 0.06 (i.e. a low bPEI concentration), the behaviour of the NPs predominated and the zeta potential and size remained relatively consistent until very low pH, whereupon aggregation occurred due to the increase in zeta potential. These observations suggest that a bPEI to NP MR which balances the number of ionic species results in optimal conditions for PNCC gel formation.
The effect of varying the bPEI-to-NPs MR on PNCC gel strength was investigated via strain-dependent oscillatory rheology (Fig. 4 and Fig. S5, ESI†). For MRs between 0.10 and 0.18, G′ was higher than G′′ at low strain, which is consistent with the tube inversion tests for hydrogel formation in Fig. 3a. The weaker gels (MRs of 0.10 and 0.18) showed moderate strain hardening until the yield point between strains of approximately 1 to 10%. More robust gels were formed with MRs between 0.12 and 0.16 (Fig. 4b), with linear viscoelastic behaviour from 0.03% to ∼5% strain. bPEI/S68-P(B0.6-A0.4)300_MR 0.14 had the highest gel strength in the linear viscoelastic region (G′ = 2 kPa). As noted above, this empirical observation is most likely a result of both the MR and pH being optimal for this NG. For example, the NG size and composition, PEI molecular architecture, solution pH and NG-to-PEI stoichiometry will influence the observed gel properties and are the subject of ongoing studies. Angular frequency sweep data at 0.2% strain for bPEI/S68-P(B0.6-A0.4)300_MR 0.14 (Fig. S6, ESI†) indicated that this mixture remains a gel with increasing angular frequency with highly frequency dependent moduli observed (G′ ∼ 6.5 kPa at 100 rad s−1).
 | ||
| Fig. 4 Rheological characterisation of PNCC hydrogels containing bPEI and PISA derived nanoparticles; (a) strain-dependent (ω = 10 rad s−1, 25 °C) oscillatory shear rheology of bPEI/S68-P(B0.6-A0.4)300_MR0.14 hydrogel (20% w/w); (b) gel strength of bPEI/S68-P(B0.6-A0.4)300 mixtures (20% w/w) at different bPEI-to-NPs mass ratios (MR = 0.10, 0.12, 0.14, 0.16, 0.18); (c) high (500%) and low (0.5%) strain steps for bPEI/S68-P(B0.6-A0.4)300_MR0.14 hydrogel (20% w/w) over three cycles (ω = 10 rad s−1, 25 °C). | ||
Shear-thinning behaviour occurred at ε > 1.2% and, as expected, at relatively high strains (12.7%) G′′ became larger than G′ indicating network breakdown. The recovery of the gels after the application of high shear was investigated by the cyclical application of high and low strain (Fig. 4c). At high strain (500%), G′ and G′′ are both dramatically reduced and cross-over, indicating liquid like behaviour. When high strain was removed (0.5% strain), the gel recovered rapidly back to its original strength. Over repeated cycles the gel strength increased slightly which can be attributed to water loss over the course of this measurement, rather than an intrinsic stiffening effect. The relatively low strength, rapid recovery and shear-thinning behaviour enables these PNCC gels to be readily injectable (see Fig. S7, ESI†) which may allow them to be used in applications such as gel printing or as an injectable/mouldable material.
Compared to the microgel-based complex coacervate gels reported by Wu et. al. (before heat or salt treatment, known as pre-gels),18 the PNCC gels prepared herein are relatively weak. This is likely due to the relatively low swelling ratio of the PISA-derived S68-P(B0.6-A0.4)300 NPs in relation to the microgels used by Wu et al. Additionally, the differences in copolymer particle morphology, such as the use of a strongly anionic sulfonate corona, may explain the differences in gel strength. Nevertheless, this demonstration that PISA-derived NGs can be synthesised and used to prepare injectable PNCCs provides a platform for development of future systems with improved strengths, tuneable functionalities, and responsive behaviour.41,42
Conclusions
Sulfonate functional nanogels containing P(MAA-stat-BzMA) nanoparticle cores were prepared by RAFT mediated PISA. These NPs can be conveniently prepared in water at pH 2 and swell with increasing pH. Interestingly, these particles do not require the use of a cross-linking monomer to maintain their structure at high pH.PKSPMA68-P(BzMA0.6-stat-MAA0.4)300 NPs were subsequently used to prepare polymer-NPs hybrid coacervate gels by mixing them with oppositely charged bPEI. It was demonstrated that the incorporation of MAA within the core-forming block of the NPs is very important for gel formation, since NPs with fully hydrophobic PBzMA cores did not form gels upon the addition of bPEI.
The mixture of bPEI and PKSPMA68-P(BzMA0.6-stat-MAA0.4)300 NPs resulted in either a low-viscosity liquid or mouldable coacervate hydrogel depending on the bPEI-to-NPs MR. At an MR of 0.14 at pH ∼7, the hybrid coacervate gel had a storage modulus of approximately 2000 Pa and exhibited shear-thinning behaviour, allowing these gels to be injectable.
This PISA-derived complex coacervate gel formation strategy is versatile and can be modified e.g., by changing the NPs to other swellable PISA-derived NPs. This may ultimately allow them to be readily tailored for applications in areas such as injectable or structural biomaterials, scaffolds for biomineralisation, soft robotics and additive manufacturing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1, and EP/P025498/1 and the Sustainable Materials Innovation Hub, funded through the European Regional Development Fund OC15R19P.Notes and references
- C. Maldonado-Codina and N. Efron, Ophthalmic Physiol. Opt., 2004, 24, 551–561 CrossRef PubMed
.
- Y. Chen, L. Chen, H. Bai and L. Li, J. Mater. Chem. A, 2013, 1, 1992–2001 RSC
- I. M. El-Sherbiny and M. H. Yacoub, Global Cardiol. Sci. Pract., 2013, 2013, 38 CrossRef PubMed
- Z. Sun, C. Song, C. Wang, Y. Hu and J. Wu, Mol. Pharmaceutics, 2019, 17, 373–391 Search PubMed
- Y. Shi, L. Pan, B. Liu, Y. Wang, Y. Cui, Z. Bao and G. Yu, J. Mater. Chem. A, 2014, 2, 6086–6091 RSC
- A. M. Rumyantsev, N. E. Jackson and J. J. De Pablo, Annu. Rev. Condens. Matter Phys., 2021, 12, 155–176 CrossRef
- H. Bixler and A. Michaels, Encycl. Polym. Sci. Technol., 1969, 10, 765–780 CAS
- A. S. Michaels and R. G. Miekka, J. Phys. Chem., 1961, 65, 1765–1773 CrossRef CAS
- I. M. Martins, M. F. Barreiro, M. Coelho and A. E. Rodrigues, Chem. Eng. J., 2014, 245, 191–200 CrossRef CAS
- T. Moschakis and C. G. Biliaderis, Curr. Opin. Colloid Interface Sci., 2017, 28, 96–109 CrossRef CAS
- H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729–733 CrossRef CAS PubMed
- M. Dompé, F. J. Cedano-Serrano, O. Heckert, N. van den Heuvel, J. van der Gucht, Y. Tran, D. Hourdet, C. Creton and M. Kamperman, Adv. Mater., 2019, 31, 1808179 CrossRef PubMed
- E. Larrañeta, S. Stewart, M. Ervine, R. Al-Kasasbeh and R. F. Donnelly, J. Funct. Biomater., 2018, 9, 13 CrossRef PubMed
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 1–9 Search PubMed
- R. Xing, K. Liu, T. Jiao, N. Zhang, K. Ma, R. Zhang, Q. Zou, G. Ma and X. Yan, Adv. Mater., 2016, 28, 3669–3676 CrossRef CAS PubMed
- B. Rodriguez, M. Wolfe and M. Fryd, Macromolecules, 1994, 27, 6642–6647 CrossRef CAS
- Z. Cui, W. Wang, M. Obeng, M. Chen, S. Wu, I. Kinloch and B. R. Saunders, Soft Matter, 2016, 12, 6985–6994 RSC
- S. Wu, M. Zhu, D. Lu, A. H. Milani, Q. Lian, L. A. Fielding, B. R. Saunders, M. J. Derry, S. P. Armes and D. Adlam, Chem. Sci., 2019, 10, 8832–8839 RSC
- N. T. Nguyen, J. Jennings, A. H. Milani, C. D. Martino, L. T. Nguyen, S. Wu, M. Z. Mokhtar, J. M. Saunders, J. E. Gautrot and S. P. Armes, Biomacromolecules, 2022, 23, 1423–1432 CrossRef CAS PubMed
- B. Charleux, G. Delaittre, J. Rieger and F. D’Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed
- N. J. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS PubMed
- X. Zhang, S. Boisse, W. Zhang, P. Beaunier, F. D’Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC
- Y. Li and S. P. Armes, Angew. Chem., 2010, 122, 4136–4140 CrossRef
- Q. Yue, S.-P. Wen and L. A. Fielding, Soft Matter, 2022, 18, 2422–2433 RSC
- S.-P. Wen, J. G. Saunders and L. A. Fielding, Polym. Chem., 2020, 11, 3416–3426 RSC
- A. Hanisch, P. Yang, A. N. Kulak, L. A. Fielding, F. C. Meldrum and S. P. Armes, Macromolecules, 2016, 49, 192–204 CrossRef CAS
- M. Douverne, Y. Ning, A. Tatani, F. C. Meldrum and S. P. Armes, Angew. Chem., 2019, 131, 8784–8789 CrossRef
- K. Nieswandt, P. Georgopanos and V. Abetz, Polym. Chem., 2021, 12, 2210–2221 RSC
- S.-P. Wen and L. A. Fielding, Soft Matter, 2022, 18, 1385–1394 RSC
- L. A. Fielding, C. T. Hendley IV, E. Asenath-Smith, L. A. Estroff and S. P. Armes, Polym. Chem., 2019, 10, 5131–5141 RSC
- C. Mable, K. Thompson, M. Derry, O. Mykhaylyk, B. Binks and S. Armes, Macromolecules, 2016, 49, 7897–7907 CrossRef CAS PubMed
- S. L. Canning, T. J. Neal and S. P. Armes, Macromolecules, 2017, 50, 6108–6116 CrossRef CAS PubMed
- T. N. Tran, S. Piogé, L. Fontaine and S. Pascual, Macromol. Rapid Commun., 2020, 41, 2000203 CrossRef CAS PubMed
- S. Piogé, T. N. Tran, T. G. McKenzie, S. Pascual, M. Ashokkumar, L. Fontaine and G. Qiao, Macromolecules, 2018, 51, 8862–8869 CrossRef
- W. Fu, C. Luo, E. A. Morin, W. He, Z. Li and B. Zhao, ACS Macro Lett., 2017, 6, 127–133 CrossRef CAS PubMed
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS PubMed
- J. Ma, H. M. Andriambololona, D. Quemener and M. Semsarilar, J. Membr. Sci., 2018, 548, 42–49 CrossRef CAS
- X.-F. Xu, C.-Y. Pan, W.-J. Zhang and C.-Y. Hong, Macromolecules, 2019, 52, 1965–1975 CrossRef CAS
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra and GPC chromatogram of PKSPMA macro-CTA; digital photographs and rheology data of bPEI/NPs mixtures; summary of particle diameters at low and high pH. See DOI: https://doi.org/10.1039/d2sm01534j |
| This journal is © The Royal Society of Chemistry 2023 |