
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphido-borane-supported stannates†
Keith
Izod
 *,
Atheer M.
Madlool
,
Alex
Craig
and
Paul G.
Waddell
*,
Atheer M.
Madlool
,
Alex
Craig
and
Paul G.
Waddell
Main Group Chemistry Laboratories, School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: keith.izod@ncl.ac.uk
First published on 24th December 2022
Abstract
The reactions between SnCl2 and three equivalents of the alkali metal phosphido-borane complexes [R2P(BH3)]M yield the corresponding tris(phosphido-borane)stannate complexes [LnM{R2P(BH3)}3Sn] [R2 = iPr2, LnM = (THF)3Li (2Li), (Et2O)Na (2Na), (Et2O)K (2K); R2 = Ph2, LnM = (THF)Li (3Li), (THF)(Et2O)Na (3Na), (THF)(Et2O)K (3K); R2 = iPrPh, LnM = (THF)4Li (4Li)]. In each case X-ray crystallography reveals an anion consisting of a trigonal pyramidal tin centre coordinated by the P atoms of the phosphido-borane ligands. These tris(phosphido-borane)stannate anions coordinate to the alkali metal cations via their BH3 hydrogen atoms in a variety of modes to give monomers, dimers, and polymers, depending on the alkali metal and the substituents at the phosphorus centres. In contrast, reactions between SnCl2 and three equivalents of [tBu2P(BH3)]M (M = Li, Na) gave the known hydride [M{tBu2P(BH3)}2SnH], according to multinuclear NMR spectroscopy.
Introduction
Phosphido-borane anions [R2P(BH3)]− (also known as phosphanyl-borohydrides or mono(borane)phosphanides) are versatile ligands which are isoelectronic with silanide anions [R2MeSi]−. These anions are also key intermediates in the synthesis of chiral phosphines and in the catalytic dehydrocoupling of phosphine–boranes to give poly(phosphino-borane) materials.1,2 However, in spite of this, and the relatively simple synthesis of phosphido-borane ligands, they have received only limited attention.3 While the majority of reported phosphido-borane complexes are of the hard alkali or alkaline earth metals,4,5 a small number of late transition metal complexes have been isolated;6 only a very few complexes of these ligands with p-block elements are known.4a,7,8Phosphido-borane anions may bind to metal centres via their phosphorus centres and/or their borane hydrogen atoms, leading to a variety of coordination modes. In complexes with hard alkali or alkaline earth metals BH3⋯M contacts are favoured, whereas with softer late transition metals P–M contacts dominate. Interestingly, it has recently been shown that phosphido-borane anions may exhibit temperature-dependent ditopic character, such that, at low temperatures, [Ph2P(BH3)]Li reacts as a P-nucleophile, whereas at high temperatures this compound acts as a B–H reducing agent towards ketones, suggesting Li–P coordination at higher temperatures and Li⋯BH3 coordination at low temperatures.9
Crystallographically characterised phosphido-borane complexes of the p-block elements are limited to the aluminium complexes [Li(TMEDA)2][{Me2P(BH3)}4Al], [(MeOtBu)Li[{Me2P(BH3)}4Al]]∞,4a and [(Et2O)2Li{Ph2P(BH3)}Al(C6F5)3],7 and the recently reported hydridostannate complex [(Me2NH)2Li{tBu2P(BH3)}2SnH] (1) [TMEDA = N,N,N′,N′-tetramethylethylenediamine].8 Compound 1 was formed unexpectedly from the reaction between three equivalents of tBu2P(BH3)H and one equivalent each of Sn(NMe2)2 and Li(NMe2). The formation of 1 was proposed to proceed via a deprotonation/β-hydride elimination from the tris(phosphido-borane)stannate Li{tBu2P(BH3)}3Sn, with concomitant formation of an unidentified P–B bonded poly(phosphino-borane) side-product (Scheme 1).
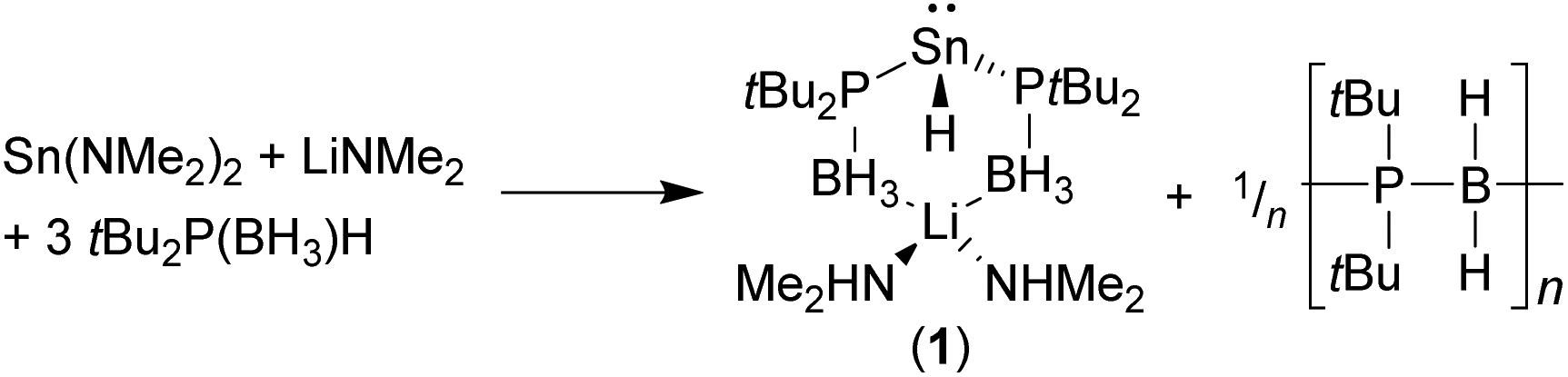 | ||
| Scheme 1 Isolation of a bis(phosphido-borane)hydridostannate (1). | ||
In an effort to elucidate the stability of such tris(phosphido-borane)stannate anions, and building on our previous work on s-block phosphido-borane complexes,4f–i,5a,d we have explored the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction between a selection of alkali metal phosphido-borane complexes and tin(II) chloride. Herein we report the results of this study and comment on the stability of the resulting tris(phosphido-borane)stannate complexes.
:1 reaction between a selection of alkali metal phosphido-borane complexes and tin(II) chloride. Herein we report the results of this study and comment on the stability of the resulting tris(phosphido-borane)stannate complexes.
Results and discussion
The reaction between SnCl2 and three equivalents of the phosphido-borane complexes [M{R2P(BH3)}] (M = Li, Na, K)4i gave the corresponding tris(phosphido-borane)stannate complexes [LnM{R2P(BH3)}3Sn] as yellow or red crystals in each case [Scheme 2; R2 = iPr2, LnM = (THF)3Li (2Li), (Et2O)Na (2Na), (Et2O)K (2K); R2 = Ph2, LnM = (THF)Li (3Li), (THF)(Et2O)Na (3Na), (THF)(Et2O)K (3K); R2 = iPrPh, LnM = (THF)4Li (4Li)]. Although the yields of isolated crystalline material were low to moderate, 31P{1H}, 11B{1H} and 119Sn{1H} NMR spectra of the crude reaction solutions indicated that the reactions were essentially quantitative in each case.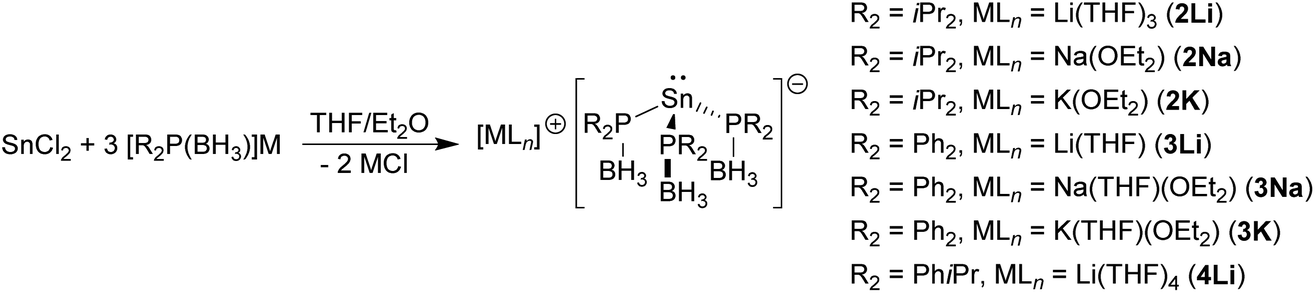 | ||
| Scheme 2 Synthesis of tris(phosphido-borane)stannate complexes 2M, 3M, and 4Li. | ||
For each of these stannate complexes the 31P{1H} NMR spectrum consists of a singlet exhibiting satellites due to coupling to 117Sn/119Sn, while the 119Sn NMR spectrum consists of a binomial quartet. This clearly indicates that the tris(phosphido-borane)stannate anion persists in solution, even in strong donor solvents such as THF. The 31P{1H} and 119Sn{1H} NMR spectra of 2Li, 2Na, and 2K were all recorded in d8-THF and are remarkably similar, with 31P and 119Sn chemical shifts ranging from 8.7 to 10.3 ppm and −171 to −181 ppm, respectively, and 31P–119Sn coupling constants of 1910–1915 Hz (Table 1). In contrast, the 31P and 119Sn chemical shifts and 31P–119Sn coupling constants of 3Li (−10.4 and −46 ppm, and 1708 Hz, respectively), which were recorded in d8-THF, are quite different from those of 3Na and 3K (−32.6/−33.2 and −113/−87 ppm, and 1428/1461 Hz, respectively), which were recorded in d8-toluene. This suggests that a similar structure, possibly a separated ion pair [M(THF)n][{R2P(BH3)}3Sn], is adopted by the stannate complexes in the strong donor solvent THF, while a different structure, possibly a contact ion pair, persists in toluene. Similar behaviour has been observed for the tris(phosphido)stannate complex [(THF)LiSn[P(Ph){CH(SiMe3)2}]3], which exists as a cage complex in which the Li cation is coordinated by the three P centres in toluene solution, but as a separated ion pair [Li(THF)4][Sn[P(Ph){CH(SiMe3)2}]3] (5) in THF.10 For all of the tris(phosphido-borane)stannate complexes isolated, except 3K, 1H NMR spectroscopy indicates that exposure of the crystalline solids to vacuum leads to partial loss of coordinated solvent.
| Solvent | 11B/ppm | 31P/ppm | 119Sn/ppm | J(31P–119Sn)/Hz | |
|---|---|---|---|---|---|
|
a 2H: iPr2P(BH3)H, 3H: Ph2P(BH3)H, 4H: iPrPhP(BH3)H.
b Data from ref. 4i.
|
|||||
|
2Ha |
CDCl3 | −40.0 | 1.0 | — | — |
| 2Li | d 8-THF | −39.9 | 8.7 | −181 | 1910 |
| 2Na | d 8-THF | −38.1 | 10.3 | −176 | 1915 |
| 2K | d 8-THF | −38.1 | 9.7 | −171 | 1910 |
|
3Ha,b |
CDCl3 | −45.3 | 27.8 | — | — |
| 3Li | d 8-THF | −32.5 | −10.4 | −46 | 1708 |
| 3Na | d 8-Toluene | −33.8 | −32.6 | −113 | 1428 |
| 3K | d 8-Toluene | −33.2 | −20.1 | −87 | 1461 |
|
4Ha,b |
CDCl3 | −42.8 | 15.2 | — | — |
| 4Li | d 8-Toluene | −36.8 | −14.0 | −113 | 1420 |
In contrast to the above, the reaction between SnCl2 and three equivalents of either [Li{tBu2P(BH3)}] or [Na{tBu2P(BH3)}] did not give the corresponding tris(phosphido-borane)stannate complexes. Instead, the 31P{1H} and 119Sn NMR spectra of the crude reaction mixtures consist of a singlet at 24.0 ppm, exhibiting 119Sn satellites (JPSn = 1700 Hz), and a 1:2:1 triplet of doublets at −287 ppm (JPSn = 1700, JHSn = 97 Hz), respectively. These are consistent with the formation of the tin(II) hydrides [M{tBu2P(BH3)}2SnH] (M = Li, Na; Scheme 3), the lithium derivative of which (1) was previously isolated by Wright and co-workers as its Me2NH complex (for comparison, the 31P and 119Sn chemical shifts of 1 are reported as 18.9 and −276 ppm, respectively, with JPSn = 1708, JHSn = 101 Hz).8 This supports Wright's proposal that the formation of 1 from the reaction between LiSn(NMe2)3 and tBu2P(BH3)H proceeds via the tris(phosphido-borane)stannate intermediate [Li{tBu2P(BH3)}3Sn], which undergoes a deprotonation/β-hydride elimination reaction to give the hydride.
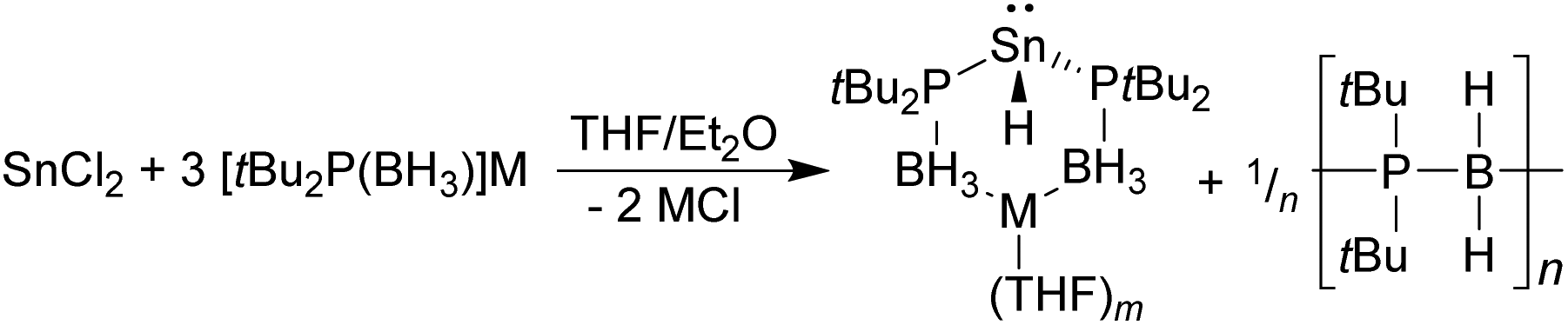 | ||
| Scheme 3 Reaction between SnCl2 and three equivalents of [M{tBu2P(BH3)}] (M = Li, Na). | ||
Similar hydride formation was not observed for any of the reactions between three equivalents of the phenyl- or isopropyl-substituted compounds [M{R2P(BH3)}] and SnCl2, even after extended periods in THF solution, suggesting that the instability of the putative stannate [Li{tBu2P(BH3)}3Sn] may be due to steric effects. All of the isolated stannate complexes decomposed on exposure to atmospheric moisture and oxygen; exposure of solutions of 2M or 3M in THF to daylight led to gradual decomposition over a period of several days, yielding elemental tin and a large number of unidentified phosphorus-containing products. However, in one case, in the absence of light and air, we observed a gradual decomposition of 3Li in THF over three days. In the 31P{1H} NMR spectrum of this solution the signal at −10.9 ppm due to 3Li was gradually replaced by a broad signal at −14.3 ppm exhibiting tin satellites (JPSn = 1555 Hz) and a sharp triplet at −35.9 ppm, also exhibiting tin satellites (JPP = 23.6 Hz, JPSn = 867, 806 Hz), in an approximately 2:1 ratio (see the ESI†). In the 119Sn{1H} NMR spectrum the sharp quartet at −48.5 ppm was gradually replaced over the same period by a sharp triplet of doublets (JPSn = 843, JP′Sn = 1615 Hz) at −24.8 ppm; the 119Sn and 119Sn{1H} NMR spectra of this solution were identical, precluding the formation of a tin hydride. However, this reaction did not proceed to completion and after 3 days decomposition ceased. Careful inspection of the 31P{1H} NMR spectrum revealed the presence of [Ph2P(BH3)]Li as a contaminant in the solution (very broad signal at −32 ppm; the literature value for the 31P chemical shift of [{Ph2P(BH3)}Li(OEt2)2]∞ is −32.6 ppm).4i The signal due to this species gradually disappeared, to be replaced by a broad signal at −6.8 ppm, consistent with the formation of the phosphido-bis(borane) complex [Ph2P(BH3)2]Li (the literature value for the 31P chemical shift of the related complex [Ph2P(BH3)2]K(18-crown-6) is −11.1 ppm).4c It therefore appears that, in this case, 3Li reacted with the excess [Ph2P(BH3)]Li present, removing one of the borane groups from 3Li to give [Li{Ph2P(BH3)}2(Ph2P)Sn] and [Ph2P(BH3)2]Li. In support of this, we have previously observed that alkali metal phosphido-borane complexes are able to abstract BH3 groups from neutral phosphine–boranes to give phosphido-bis(borane) anions.4h
X-ray crystallography reveals that each of 2M, 3M and 4Li crystallises as an alkali metal salt of a tris(phosphido-borane)stannate anion, in which the tin centre is coordinated by the phosphorus atoms of three phosphido-borane anions in a trigonal pyramidal geometry. While the core structure of the tris(phosphido-borane)stannate anion is essentially the same for all compounds, the coordination of the alkali metal cation differs significantly in each case. Compound 2Li crystallises as a contact ion pair in which the lithium cation is coordinated by three molecules of THF and one of the BH3 groups of the tris(phosphido-borane)stannate anion in a κ2-manner (Fig. 1).
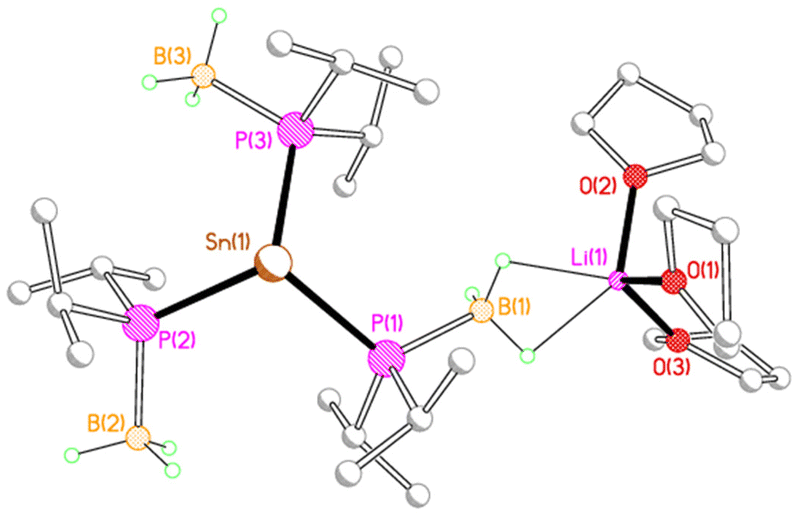 | ||
| Fig. 1 Molecular structure of 2Li with C-bound H atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Sn(1)–P(1) 2.6349(6), Sn(1)–P(2) 2.6219(6), Sn(1)–P(3) 2.6241(6), P(1)–B(1) 1.956(3), P(2)–B(2) 1.957(3), P(3)–B(3) 1.957(3), Li(1)⋯B(1) 2.381(5), Li(1)–O(1) 1.916(5), Li(1)–O(2) 1.926(5), Li(1)–O(3) 1.955(5), P(1)–Sn(1)–P(2) 103.459(19), P(1)–Sn(1)–P(3) 103.853(18), P(2)–Sn(1)–P(3) 104.400(19). | ||
In contrast, 2Na crystallises as a corrugated sheet polymer, due to multiple BH3⋯Na contacts (Fig. 2). Each sodium ion is coordinated by a molecule of diethyl ether, along with three BH3 groups from three adjacent tris(phosphido-borane)stannate anions, with one κ1 and two κ2 BH3⋯Na contacts. Thus, each stannate anion is bridged by three sodium ions to give a sheet polymer network. Compound 2K adopts a similar corrugated sheet structure, in which each stannate anion is bridged by three potassium ions to three adjacent anions (see the ESI†). However, reflecting the increased ionic radius of potassium compared with sodium, each potassium ion is coordinated by one molecule of diethyl ether, along with one κ3 and two κ2 BH3⋯K contacts.
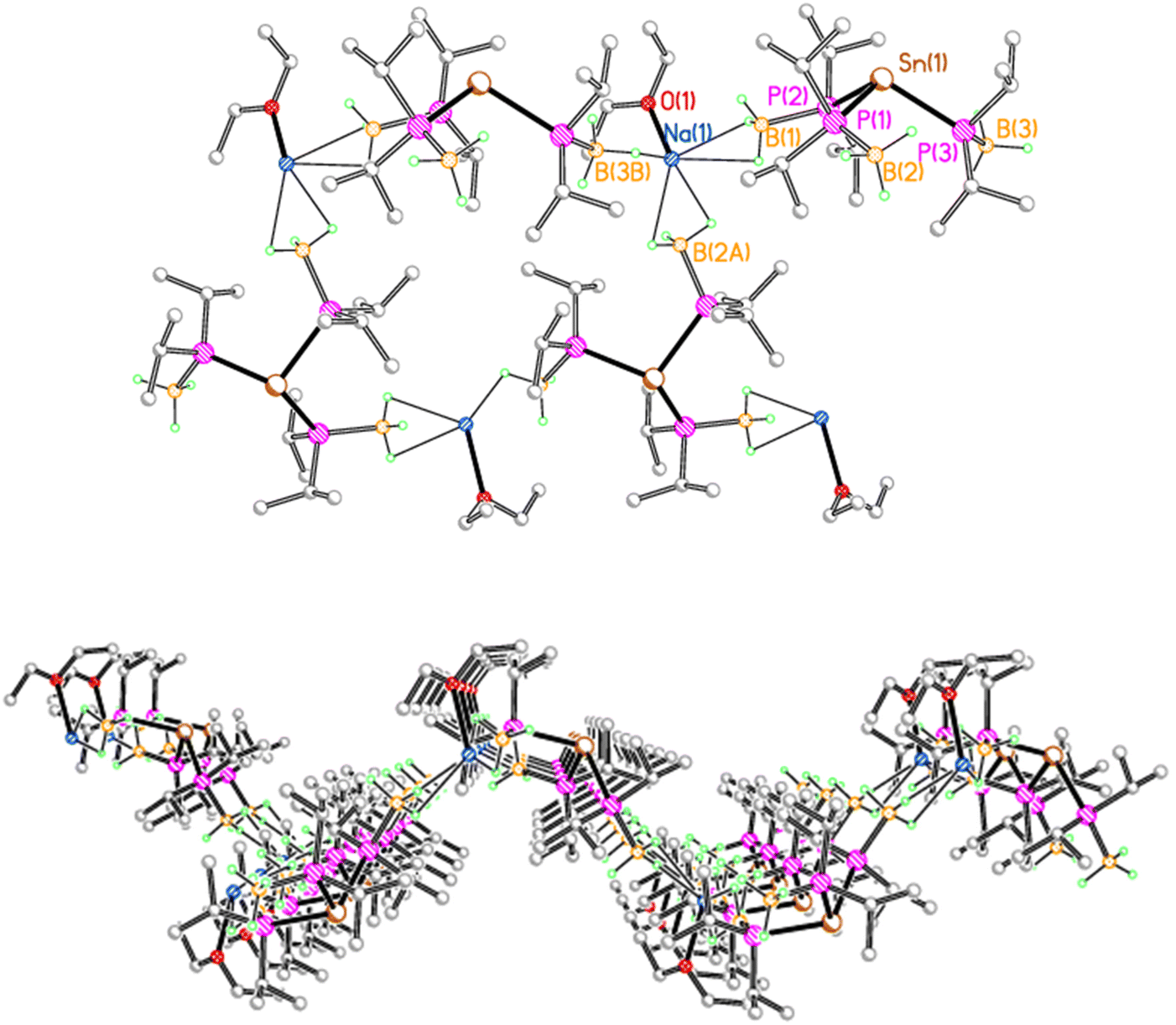 | ||
| Fig. 2 Polymeric structure of 2Na, with C-bound H atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Sn(1)–P(1) 2.6424(7), Sn(1)–P(2) 2.6325(7), Sn(1)–P(3) 2.6401(7), P(1)–B(1) 1.947(3), P(2)–B(2) 1.956(3), P(3)–B(3) 1.952(3), Na(1)⋯B(1) 2.742(4), Na(1)⋯B(2A) 2.779(3), Na(1)⋯B(3B) 2.814(3), Na(1)–O(1) 2.333(2), P(1)–Sn(1)–P(2) 101.79(2), P(1)–Sn(1)–P(3) 104.78(2), P(2)–Sn(1)–P(3) 102.46(2). | ||
The three phenyl-substituted tris(phosphido-borane)stannate complexes 3Li, 3Na, and 3K, adopt a rather different structural motif in the solid state, in which each alkali metal ion is coordinated by three BH3 groups from the same stannate anion. Compounds 3Li and 3Na crystallise as discrete monomers (Fig. 3). In 3Li the lithium ion is coordinated by a molecule of THF and by the three BH3 groups, with one κ1 and two κ2 BH3⋯Li contacts. Compound 3Na crystallises with two independent molecules in the asymmetric unit. In both molecules the sodium ions are coordinated by one molecule each of THF and diethyl ether; in molecule 1 further coordination is provided by three κ2 BH3⋯Na contacts, whereas in molecule 2 the sodium ion is further coordinated by two κ2 and one κ1 BH3⋯Na contacts.
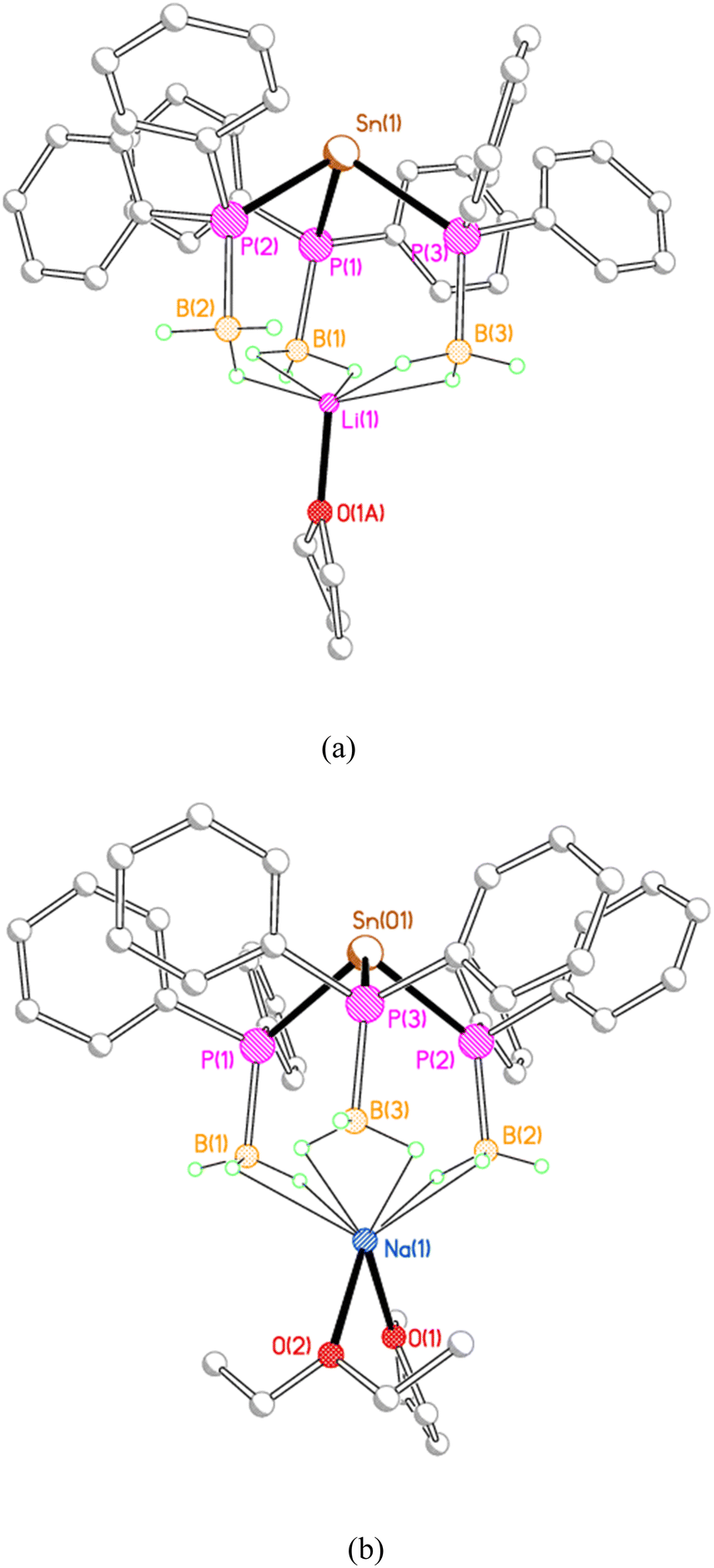 | ||
| Fig. 3 Molecular structures of (a) 3Li and (b) one of the two independent molecules of 3Na, with H atoms and minor disorder components omitted for clarity. Selected bond lengths (Å) and angles (°) [values for the second independent molecule of 3Na in square brackets]: 3Li Sn(1)–P(1) 2.6097(14), Sn(1)–P(2) 2.6444(13), Sn(1)–P(3) 2.6288(13), P(1)–B(1) 1.940(7), P(2)–B(2) 1.963(5), P(3)–B(3) 1.954(6), Li(1)⋯B(1) 2.470(13), Li(1)⋯B(2) 2.798(14), Li(1)⋯B(3) 2.477(14), Li(1)–O(1) 1.908(15), P(1)–Sn(1)–P(2) 93.60(4), P(1)–Sn(1)–P(3) 95.56(5), P(2)–Sn(1)–P(3) 95.17(4). 3Na Sn(01)–P(1) 2.6301(6), Sn(01)–P(2) 2.6072(7), Sn(01)–P(3) 2.6237(7) [Sn(02)–P(4) 2.6457(7), Sn(02)–P(5) 2.6536(7), Sn(02)–P(6) 2.6343(7)], P(1)–B(1) 1.951(3), P(2)–B(2) 1.938(4), P(3)–B(3) 1.945(3) [P(4)–B(4) 1.940(3), P(5)–B(5) 1.949(3), P(6)–B(6) 1.952(3)], Na(1)⋯B(1) 2.912(4), Na(1)⋯B(2) 2.931(4), Na(1)⋯B(3) 2.959(4), Na(1)–O(1) 2.358(2), Na(1)–O(2) 2.386(2) [Na(2)⋯B(4) 2.882(3), Na(2)⋯B(5) 2.835(4), Na(2)⋯B(6) 3.346(4), Na(2)–O(3) 2.414(15), Na(2)–O(4) 2.328(3)], P(1)–Sn(01)–P(2) 94.52(2), P(1)–Sn(01)–P(3) 96.03(2), P(2)–Sn(01)–P(3) 95.93(2) [P(4)–Sn(02)–P(5) 97.53(2), P(4)–Sn(02)–P(6) 94.11(2), P(55)–Sn(02)–P(6) 95.99(2)]. | ||
In contrast, 3K crystallises as a centrosymmetric dimer (Fig. 4). Each potassium ion is coordinated by one molecule each of THF and diethyl ether and by three κ2 BH3 groups from one stannate anion, along with a κ1 BH3 group from the second stannate anion in the dimer. Thus, one borane group from each stannate anion bridges the two potassium ions in a μ–κ2:κ1 fashion.
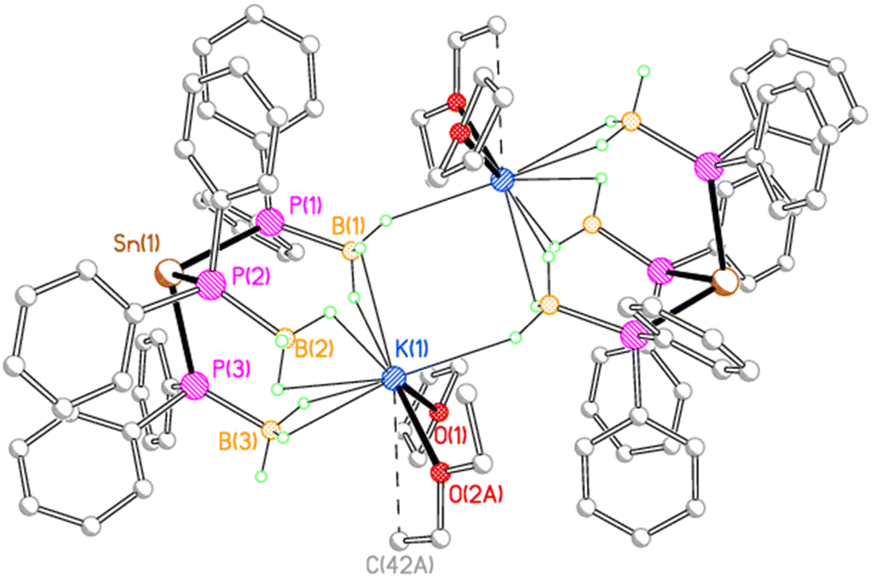 | ||
| Fig. 4 Molecular structure of 3K with C-bound H atoms and minor disorder components omitted for clarity. Selected bond lengths (Å) and angles (°): Sn(1)–P(1) 2.6386(7), Sn(1)–P(2) 2.6427(7), Sn(1)–P(3) 2.6455(8), P(1)–B(1) 1.936(4), P(2)–B(2) 1.950(4), P(3)–B(3) 1.941(4), K(1)⋯B(1) 3.213(4), K(1)⋯B(2) 3.343(4), K(1)⋯B(3) 3.241(4), K(1)⋯B(1A) 3.686(3), K(1)–O(1) 2.704(2), K(1)–O(2A) 2.719(6), P(1)–Sn(1)–P(2) 95.28(2), P(1)–Sn(1)–P(3) 97.54(2), P(2)–Sn(1)–P(3) 96.94(2). | ||
Given the two different structural motifs adopted by the isopropyl- and phenyl-substituted compounds 2M and 3M, it seemed of interest to investigate the structure of a compound possessing both phenyl and isopropyl substituents at phosphorus (4Li). Unexpectedly, this compound crystallised as a separated ion pair, with no short contacts between the tris(phosphido-borane)stannate anion and the [Li(THF)4]+ cation (Fig. 5).
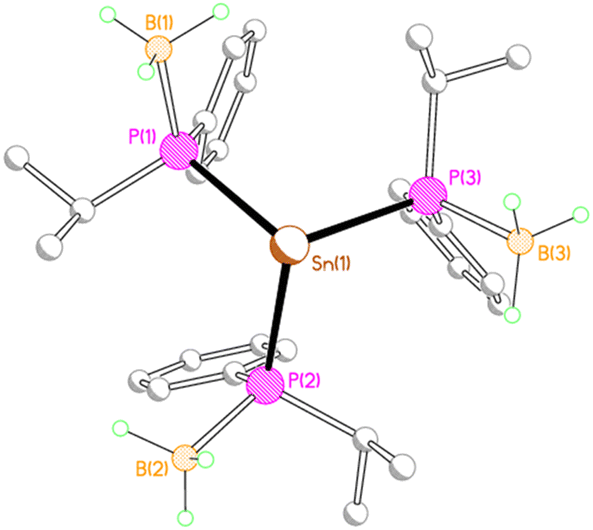 | ||
| Fig. 5 Solid-state structure of the anion of 4Li with C-bound H atoms and minor disorder components omitted for clarity. Selected bond lengths (Å) and angles (°): Sn(1)–P(1) 2.6244(7), Sn(1)–P(2) 2.6323(6), Sn(1)–P(3) 2.6324(7), P(1)–B(1) 1.945(4), P(2)–B(2) 1.947(3), P(3)–B(3) 1.951(3), Li(1)–O(1) 1.905(5), Li(1)–O(2A) 1.895(7), Li(1)–O(3A) 1.929(8), Li(1)–O(4A) 1.940(7), P(1)–Sn(1)–P(2) 100.47(2), P(1)–Sn(1)–P(3) 101.73(2), P(2)–Sn(1)–P(3) 100.13(2). | ||
Across all of the compounds studied there is little variation in the Sn–P and P–B distances, which fall in the ranges 2.6072(7)–2.6455(8) and 1.936(4)–1.963(5) Å, respectively. The Sn–P distances are similar to other Sn(II)–P distances;11 for example, the Sn–P distances in [Li(THF)4][Sn[P(Ph){CH(SiMe3)2}]3] (5) are 2.649(2) Å,10 whereas the Sn–P(terminal) distances in Sn[P{CH(SiMe3)2}(C6H4-2-CH2NMe2)]2 are 2.5906(9) and 2.6061(9) Å for the two independent molecules in the asymmetric unit.12
The degree of pyramidalisation at the tin centre within the set of compounds 2Li, 2Na and 2K is also rather similar [sum of P–Sn–P angles = 311.71, 309.03 and 309.65°, respectively], but this differs significantly from the degree of pyramidalisation of tin in 3Li, 3Na, and 3K [sum of P–Sn–P angles = 284.33, 286.48 and 289.76°, respectively]. This clearly reflects the different binding modes between the stannate anions and alkali metal cations within these two sets of compounds. In 2M the alkali metals are bound at the periphery of the stannate anions and so the P–Sn–P angles are close to those expected for a trigonal pyramid. Whereas, in 3M the stannate anion acts as a pseudo-tridentate ligand to the alkali metal centres; this necessarily leads to a reduction in the P–Sn–P angles in 3M compared with 2M. Notably, the sum of P–Sn–P angles in 3M increases as the size of the alkali metal ion increases from Li to Na and K, as expected, but is essentially identical for 2Li, 2Na and 2K. In the separated ion pair compound 4Li the pyramidalisation of the tin centre [sum of angles at tin = 302.33°] appears to be intermediate between the pyramidalisation of the tin centres in 2M and 3M. This is in marked contrast to the sum of P–Sn–P angles in 5, which crystallises with a discrete tris(phosphido)stannate anion (sum of P–Sn–P angles in 5 = 274.23°).10
Conclusions
A series of tris(phosphido-borane)stannate complexes [LnM{R2P(BH3)}3Sn] (M = Li, Na; R = Ph, iPr; L = THF, Et2O) is readily accessible by the simple treatment of SnCl2 with three equivalents of the corresponding alkali metal phosphido-borane salt. However, similar reactions between three equivalents of the bulky phosphido-borane complexes [M{tBu2P(BH3)}] (M = Li, Na) and SnCl2 led to decomposition to the corresponding hydridostannate [M{tBu2P(BH3)}2SnH].Although all of the isolated compounds crystallise as tris(phosphido-borane)stannate salts containing a trigonal pyramidal Sn(II) centre, they adopt three distinct binding modes, depending on the substituents at phosphorus. The isopropyl-substituted complexes [LnM{iPr2P(BH3)}3Sn] (2M) crystallise as monomers or polymers in which the alkali metal cation is coordinated by BH3 groups at the periphery of one or more stannate anions. In contrast, the phenyl-substituted compounds [LnM{Ph2P(BH3)}3Sn] (3M) adopt molecular structures in which the alkali metal cation is coordinated by all three BH3 groups from a single stannate anion. The mixed substituent complex 4Li crystallises as a separated ion pair, with no short contacts between the cation and the stannate anion. The preference for one structural motif over another is likely due to the steric bulk of the substituents: in 3M the phenyl rings are able to rotate in order to minimise steric repulsion and so all three borane groups are able to position themselves close to the alkali metal centre; this appears to be disfavoured for the more bulky isopropyl groups in 2M and so an alternative structural motif is adopted.
Experimental section
All manipulations were carried out using standard Schlenk techniques under an atmosphere of dry nitrogen. Diethyl ether, toluene and THF were dried prior to use by distillation under nitrogen from sodium/potassium alloy, sodium or potassium, respectively; THF was stored over activated 4 Å molecular sieves, while toluene and diethyl ether were stored over a potassium film. Deuterated toluene and THF were distilled from potassium under nitrogen and were deoxygenated by three freeze–pump–thaw cycles and stored over activated 4 Å molecular sieves. Benzylsodium,13 benzylpotassium,14 Ph2P(BH3)H (3H),15 iPr2P(BH3)H (2H),16 iPrPhP(BH3)H (4H),4i and tBu2P(BH3)H4i,17 were prepared by previously published procedures; n-butyllithium was purchased from Aldrich as a 2.5 M solution in THF and its concentration was accurately determined by titration before use. All other compounds were used as supplied by the manufacturer.1H and 13C{1H} NMR spectra were recorded on a Bruker AvanceIII 300 spectrometer operating at 300.13 and 75.48 MHz, respectively, or a Bruker AvanceIII 400 spectrometer operating at 399.78 and 100.54 MHz, respectively; chemical shifts are quoted in ppm relative to tetramethylsilane. 31P{1H}, 11B{1H}, 7Li and 119Sn{1H} NMR spectra were recorded on a Bruker AvanceIII 300 or a Bruker AvanceIII 400 spectrometer operating at 121.49 [161.83], 96.29 [128.27], 116.64 and 111.91 [149.08] MHz, respectively [values in square brackets correspond to the frequencies on the latter machine]; chemical shifts are quoted in ppm relative to external 85% H3PO4, BF3·Et2O, 0.1 M LiCl in D2O, and Me4Sn, respectively.
We were unable to obtain consistent elemental analyses for the isolated compounds due to their air- and moisture-sensitive nature and to partial loss of coordinated solvent in most cases; however, multinuclear NMR data indicate that all compounds are isolated in good purity.
[Sn{iPr2P(BH3)}3Li(THF)3] (2Li)
To a solution of iPr2PH(BH3) (0.4 g, 3 mmol) in THF (20 mL) was added nBuLi (1.2 mL, 2.45 M solution in hexane, 3 mmol) and this mixture was stirred for 30 min. This solution was added, dropwise, to a cold (−78 °C) solution of SnCl2 (0.19 g, 1 mmol) in THF (20 mL) and this mixture was stirred for 1 h. The solution was allowed to warm to room temperature and the solvent was removed in vacuo to give a sticky yellow solid. This was extracted into Et2O (10 mL) containing a few drops of THF and filtered. The filtrate was cooled to −30 °C overnight to give 2Li as yellow crystals suitable for X-ray crystallography. Yield of crystalline material 0.25 g, 18%. The coordinated THF is partially lost under vacuum; all NMR data correspond to the formula [Sn{iPr2P(BH3)}3Li(THF)0.25] (2Li′). 1H NMR (399.78 MHz, d8-THF, 25 °C): δ 0.54 (br, 9H, BH3), 1.16 (m, 18H, MeMeCH), 1.23 (m, 18H, MeMeCH), 1.78 (m, 1H, THF), 2.41 (m, 6H, MeMeCH), 3.61 (m, 1H, THF). 13C{1H} NMR (100.54 MHz, d8-THF, 25 °C): δ 19.2 (JSnC = 28.0 Hz, MeMeCH), 21.8 (JSnH = 26.5 Hz, MeMeCH), 24.9 (m, MeMeCH), 26.4 (THF), 68.3 (THF). 11B{1H} NMR (128.27 MHz, d8-THF, 25 °C): δ −39.9 (br). 13P{1H} NMR (161.83 MHz, d8-THF, 25 °C): δ 8.7 (JPSn = 1910 Hz). 7Li{1H} NMR (116.64 MHz, d8-THF, 25 °C): δ −0.6 (s). 119Sn{1H} NMR (149.08 MHz, d8-THF, 25 °C): δ −181 (q, JPSn = 1910 Hz).[Sn{iPr2P(BH3)}3Na(THF)]∞ (2Na)
To a solution of iPr2PH(BH3) (0.4 g, 3 mmol) in THF (20 mL) was added a solution of PhCH2Na (0.35 g, 3 mmol) in THF (20 mL), and this mixture was stirred for 30 min. This solution was added, dropwise, to a cold (−78 °C) solution of SnCl2 (0.19 g, 1 mmol) in THF (20 mL), and this mixture was stirred for 1 h. The mixture was allowed to warm to room temperature and the solvent was removed in vacuo to give a yellow solid. This was extracted into THF/Et2O (20 mL per 20 mL), filtered, and solvent was removed from the filtrate in vacuo. The resulting solid was crystallised from THF (5 mL) layered with Et2O (20 mL) to give yellow crystals of 2Na suitable for X-ray crystallography. The coordinated THF is partially lost under vacuum: all NMR data correspond to the formula [Sn{iPr2P(BH3)}3Na(THF)0.5] (2Na′). Yield of 2Na′ 0.21 g, 39%. 1H{11B} NMR (300.13 MHz, d8-THF, 25 °C): δ 0.54 (br, 9H, BH3), 1.15–1.29 (m, 36H, MeMeCH), 1.77 (m, 2H, THF), 2.13–2.42 (m, 6H, MeMeCH), 3.61 (m, 2H, THF). 13C{1H} NMR (75.48 MHz, d8-THF, 25 °C): δ 19.2 (JSnC = 28.3 Hz, MeMeCH), 21.8 (JSnC = 26.3 Hz, MeMeCH), 24.9 (MeMeCH), 26.4 (THF), 68.3 (THF). 11B{1H} NMR (96.29 MHz, d8-THF, 25 °C): δ −38.1 (br). 31P{1H} NMR (121.49 MHz, d8-THF, 25 °C): δ 10.3 (s, JPSn = 1915 Hz). 119Sn{1H} NMR (111.91 MHz, d8-THF, 25 °C): δ −176 (q, JPSn = 1915 Hz).[Sn{iPr2P(BH3)}3K(Et2O)]∞ (2K)
A solution of PhCH2K (0.39 g, 3 mmol) in THF (20 mL) was added to a solution of iPr2PH(BH3) (0.4 g, 3 mmol) in THF (20 mL) and this mixture was stirred for 30 min. This solution was added, dropwise, to a cold (−78 °C) solution of SnCl2 (0.19 g, 1 mmol) in THF (20 mL) and this mixture was stirred for 1 h. The reaction was allowed to warm to room temperature and the solvent was removed in vacuo. The resulting solid was extracted into THF/Et2O (20 mL per 20 mL), filtered and solvent was removed in vacuo from the filtrate. The resulting solid was crystallised from THF (5 mL) layered with Et2O (10 mL) to give 2K as yellow crystals suitable for X-ray crystallography. Yield of crystalline material 0.16 g, 25%. The coordinated diethyl ether is partially lost under vacuum; all NMR data correspond to the formula [Sn{iPr2P(BH3)}3K(Et2O)0.5] (2K′). 1H NMR (300.13 MHz, d8-THF, 25 °C): δ 0.44–0.46 (br, 9H, BH3), 1.10 (t, 3H, Et2O), 1.17–1.24 (m, 36H, MeMeCH), 2.41 (m, 6H, MeMeCH), 3.38 (q, 2H, Et2O). 13C{1H} NMR (75.48 MHz, d8-THF, 25 °C): δ 15.8 (Et2O), 19.3 (JSnH = 28.3 Hz, MeMeCH), 21.8 (JSnH = 26.1 Hz, MeMeCH), 25.0 (d, JPC = 6.0 Hz, MeMeCH), 66.4 (Et2O). 11B{1H} NMR (96.29 MHz, d8-THF, 25 °C): δ −38.1 (br). 31P{1H} NMR (121.49 MHz, d8-THF, 25 °C): δ 9.7 (s, JPSn = 1910 Hz). 119Sn{1H} NMR (111.91 MHz, d8-THF, 25 °C): δ −171 (q, JPSn = 1910 Hz).[Sn{Ph2P(BH3)}3Li(THF)] (3Li)
To a solution of Ph2PH(BH3) (0.6 g, 3 mmol) in THF (20 mL) was added nBuLi (1.2 mL, 2.45 M solution in hexane, 3 mmol) and this mixture was stirred for 30 min. This solution was added, dropwise, to a cold (−78 °C) solution of SnCl2 (0.19 g, 1 mmol) in THF (20 mL), and this mixture was stirred for 1 h. The reaction was allowed to attain room temperature and the solvent was removed in vacuo. The solid was extracted into toluene (20 mL) and filtered. The filtrate was concentrated to 10 mL and cooled (−25 °C) to give 3Li as pale yellow crystals suitable for X-ray crystallography. Yield of crystalline material 0.3 g, 37%. The coordinated THF is rapidly lost under vacuum; all NMR data correspond to the formula [Sn{Ph2P(BH3)}3Li(THF)0.5] (3Li′). 1H{11B} NMR (300.13 MHz, d8-THF, 25 °C): δ 1.18 (br, 9H, BH3), 1.78 (m, 2H, THF), 3.63 (m, 2H, THF), 6.98–7.11 (m, 24H, Ph), 7.45 (m, 6H, Ph). 13C{1H} NMR (75.48 MHz, d8-THF, 25 °C): δ 26.4 (THF), 68.4 (THF), 128.1, 128.4, 135.1, 137.7 (m, Ph). 11B{1H} NMR (96.29 MHz, d8-THF, 25 °C): δ −32.5 (br). 31P{1H} NMR (121.49 MHz, d8-THF, 25 °C): δ −10.4 (J(31P–119Sn) = 1708, J(31P–117Sn) = 1633 Hz). 7Li{1H} NMR (116.64 MHz, d8-THF, 25 °C): δ −0.6 (s). 119Sn{1H} NMR (111.91 MHz, d8-THF, 25 °C): δ −46 (q, JPSn = 1708 Hz).[Sn{Ph2P(BH3)}3Na(THF)(Et2O)] (3Na)
To a solution of Ph2PH(BH3) (0.60 g, 3 mmol) in THF (20 mL) was added a solution of PhCH2Na (0.35 g, 3 mmol) in THF (20 mL), and this mixture was stirred for 30 min. This solution was added, dropwise, to a cold (−78 °C) solution of SnCl2 (0.19 g, 1 mmol) in THF (20 mL) and this mixture was stirred for 1 h. The solution was allowed to warm to room temperature and the solvent was removed in vacuo. The resulting solid was extracted into THF/Et2O (20 mL per 20 mL), filtered and solvent was removed from the filtrate in vacuo. The resulting solid was crystallised from cold (−25 °C) Et2O (15 mL) containing a few drops of THF to give 3Na as orange crystals suitable for X-ray crystallography. Yield of crystalline material 0.2 g, 22%. The coordinated diethyl ether is partially lost under vacuum; all NMR data correspond to the formula [Sn{Ph2P(BH3)}3Na(THF)(Et2O)0.5] (3Na′). 1H{11B} NMR (300.13 MHz, d8-toluene, 25 °C): δ 1.14 (t, 3H, Et2O), 1.40 (m, 4H, THF), 1.91 (d, JPH = 10.5 Hz, 9H, BH3), 3.26 (q, 2H, Et2O), 3.58 (m, 4H, THF), 6.72–6.80 (m, 18H, Ph), 7.61 (m, 12H, Ph). 13C{1H} NMR (75.48 MHz, d8-toluene, 25 °C): δ 14.5 (Et2O), 25.6 (THF), 66.0 (Et2O), 68.1 (THF), 128.3, 128.4, 128.9 (Ph), 133.4 (d, JPC = 8.1 Hz, Ph). 11B{1H} NMR (96.29 MHz, d8-toluene, 25 °C): δ −33.8 (br), 31P{1H} NMR (121.49 MHz, d8-toluene, 25 °C): δ −32.6 (s, JPSn = 1428 Hz), 119Sn{1H} NMR (111.91 MHz, d8-toluene, 25 °C): δ −113 (q, JPSn = 1428 Hz).[Sn{Ph2P(BH3)}3K(THF)2] (3K)
A solution of PhCH2K (0.39 g, 3 mmol) in THF (20 mL) was added to a solution of Ph2PH(BH3) (0.60 g, 3 mmol) in THF (20 mL), and this mixture was stirred for 30 min. This solution was added, dropwise, to a cold (−78 °C) solution of SnCl2 (0.19 g, 1 mmol) in THF (20 mL) and this mixture was stirred for 1 h. The solution was allowed to warm to room temperature and the solvent was removed in vacuo. The solid was extracted into THF/Et2O (20 mL per 20 mL), filtered and solvent was removed from the filtrate in vacuo. The resulting solid was crystallised from a mixture of cold (−25 °C) Et2O (10 mL) and THF (5 mL) to give red crystals of 3K suitable for X-ray crystallography. Yield of crystalline material 0.4 g, 44%. 1H{11B} NMR (300.13 MHz, d8-toluene, 25 °C): δ 1.47 (m, 8H, THF), 1.91 (d, JPH = 9.9 Hz, 9H, BH3), 3.55 (m, 8H, THF), 6.80 (m, 18H, Ph), 7.65 (m, 12H, Ph). 13C{1H} NMR (75.48 MHz, d8-toluene, 25 °C): δ 25.8 (THF), 67.8 (THF), 128.1 (d, JPC = 7.6 Hz, Ph), 129.0 (d, JPC = 10.3 Hz, Ph), 133.7 (d, JPC = 6.2 Hz, Ph), (ipso carbon not observed). 11B{1H} NMR (96.29 MHz, d8-toluene, 25 °C): δ −33.2 (br). 31P{1H} NMR (121.49 MHz, d8-toluene, 25 °C): δ −20.1 (J(31P–119Sn) = 1461, J(31P–117Sn) = 1388 Hz). 119Sn{1H} NMR (111.91 MHz, d8-toluene, 25 °C): δ −87 (q, JPSn = 1461 Hz).[Sn{iPrPhP(BH3)}3][Li(THF)4] (4Li)
To a solution of iPrPhPH(BH3) (0.7 g, 3 mmol) in THF (20 mL) was added nBuLi (1.2 mL, 2.45 M solution in hexane, 3 mmol), and this mixture was stirred for 30 min. This solution was added, dropwise, to a cold (−78 °C) solution of SnCl2 (0.19 g, 1 mmol) in THF (20 mL) and this mixture was stirred for 1 h. The solution was allowed to attain room temperature and the solvent was removed in vacuo to give a yellow solid. This was extracted into THF/Et2O (20 mL per 20 mL), filtered and solvent was removed in vacuo from the filtrate. The resulting solid was crystallised from a cold (−25 °C) mixture of Et2O (10 mL) and THF (5 mL) to give 4Li as yellow crystals suitable for X-ray crystallography. Yield of crystalline material 0.3 g, 33%. The coordinated THF is partially lost under vacuum; all NMR data correspond to the formula [Sn{iPrPhP(BH3)}3Li(THF)2] (4Li′). 1H{11B} NMR (300.13 MHz, d8-toluene, 25 °C): δ 0.71 (dd, JPH = 13.8, JHH = 7.2 Hz, 9H, MeMeCH), 0.78 (dd, JPH = 15.9, JHH = 6.9 Hz, 9H, MeMeCH), 1.44 (m, 8H, THF), 1.56 (m, 3H, MeMeCH), 1.70 (d, JPH = 9.6 Hz, 9H, BH3), 3.65 (m, 8H, THF), 7.04–7.15 (m, 9H, Ph), 8.05 (m, 6H, Ph). 13C{1H} NMR (75.48 MHz, d8-toluene, 25 °C): δ 18.2 (Me2CH), 25.7 (THF), 27.1 (m, Me2CH), 68.1 (THF), 128.3, 129.5 (Ph), 135.1 (d, JPC = 6.0 Hz, Ph). 11B{1H} NMR (96.29 MHz, d8-toluene, 25 °C): δ −36.8 (s). 7Li{1H} NMR (116.64 MHz, d8-toluene, 25 °C): 0.4 (s). 31P{1H} NMR (121.49 MHz, d8-toluene, 25 °C): δ −14.0 (JPSn = 1420 Hz). 119Sn{1H} NMR (111.91 MHz, d8-toluene, 25 °C): −113 (q, JPSn = 1420 Hz).Crystal structure determinations of 2Li, 2Na, 2K, 3Li, 3Na, 3K, and 4Li
Measurements were made at 150 K on an Oxford Diffraction (Agilent Technologies) Gemini A Ultra diffractometer, using CuKα radiation (λ = 1.54184 Å) for 2Li and MoKα radiation (λ = 0.71073 Å) otherwise. Cell parameters were refined from the observed positions of all strong reflections. For 2K and 3Li an analytical numeric absorption correction was applied using a multifaceted crystal model based on expressions derived by R. C. Clark and J. S. Reid.18 For all other compounds intensities were corrected for absorption empirically using spherical harmonics. The structures were solved by direct or dual-space methods and refined on F2 values for all unique data; Table S1 in the ESI† gives further details. All non-hydrogen atoms were refined anisotropically, and C-bound H atoms were modelled with idealised geometry, while B-bound H atoms were located using the Fourier difference map; Uiso for all hydrogen atoms was constrained to be an appropriate multiple of the Ueq value of the parent C atom. Programs were Oxford Diffraction CrysAlisPro for data collection and processing, and programs of the SHELX family for structure solution, refinement, and molecular graphics.19Conflicts of interest
There are no conflicts of interest to declare.References
- For a recent review see: A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef PubMed.
- For examples see: (a) A.-C. Gaumont, M. B. Hursthouse, S. J. Coles and J. M. Brown, Chem. Commun., 1999, 63 RSC; (b) S. Pican and A.-C. Gaumont, Chem. Commun., 2005, 2393 RSC; (c) I. Abdellah, E. Bernoud, J.-F. Lohier, C. Alayrac, L. Toupet, C. Lepetit and A.-C. Gaumont, Chem. Commun., 2012, 48, 4088 RSC.
- For examples see: (a) A. C. Jaska, A. J. Lough and I. Manners, Dalton Trans., 2005, 326 RSC; (b) K. Lee, T. J. Clark, A. J. Lough and I. Manners, Dalton Trans., 2008, 2732 RSC; (c) C. A. Jaska, H. Dorn, A. J. Lough and I. Manners, Chem. – Eur. J., 2003, 9, 271 CrossRef CAS PubMed.
- (a) G. Müller and J. Brand, Organometallics, 2003, 22, 1463–1467 CrossRef; (b) T. I. Kückmann, F. Dornhaus, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Eur. J. Inorg. Chem., 2007, 1989–2003 CrossRef; (c) F. Dornhaus, M. Bolte, H.-W. Lerner and M. Wagner, Eur. J. Inorg. Chem., 2006, 1777–1785 CrossRef CAS; (d) F. Dornhaus and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m3573–m3575 CrossRef CAS; (e) F. Dornhaus, M. Bolte, H.-W. Lerner and M. Wagner, Eur. J. Inorg. Chem., 2006, 5138–5147 CrossRef CAS; (f) K. Izod, J. M. Watson, W. Clegg and R. W. Harrington, Inorg. Chem., 2013, 52, 1466–1475 CrossRef CAS; (g) K. Izod, J. M. Watson, W. Clegg and R. W. Harrington, Eur. J. Inorg. Chem., 2012, 1696–1701 CrossRef CAS; (h) K. Izod, J. M. Watson, W. Clegg and R. W. Harrington, Dalton Trans., 2011, 40, 11712–11718 RSC; (i) K. Izod, A. M. Madlool, A. Craig and P. G. Waddell, Eur. J. Inorg. Chem., 2022, e202200123 CAS.
- (a) K. Izod, J. M. Watson, S. M. El-Hamruni, R. W. Harrington and P. G. Waddell, Organometallics, 2017, 36, 2218–2227 CrossRef CAS; (b) L. J. Morris, M. S. Hill, M. F. Mahon, I. Manners and B. O'Patrick, Organometallics, 2020, 39, 4195–4207 CrossRef CAS; (c) L. J. Morris, N. A. Rajabi, M. S. Hill, I. Manners, C. L. McMullin and M. F. Mahon, Dalton Trans., 2020, 49, 14584 RSC; (d) K. Izod, J. M. Watson, R. W. Harrington and W. Clegg, Dalton Trans., 2021, 50, 1019–1024 RSC.
- (a) I. Amor, D. Garcia-Vivo, M. E. Garcia, M. A. Ruiz, D. Sáez, H. Hamidov and J. C. Jeffery, Organometallics, 2007, 26, 466–468 CrossRef CAS; (b) W. F. McNamara, E. N. Duesier, R. T. Paine, J. V. Ortiz, P. Kölle and H. Nöth, Organometallics, 1986, 5, 380–383 CrossRef CAS; (c) T. N. Hooper, A. S. Weller, N. A. Beattie and S. A. MacGregor, Chem. Sci., 2016, 7, 2414–2426 RSC; (d) H. Dorn, R. A. Singh, J. A. Massey, J. M. Nelson, C. A. Jaska, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2000, 122, 6669–6678 CrossRef CAS; (e) F. Dornhaus, M. Bolte, H.-W. Lerner and M. Wagner, J. Organomet. Chem., 2007, 692, 2949–2955 CrossRef CAS; (f) A. Schafer, T. Jurca, J. Turner, J. R. Vance, K. Lee, V. A. Du, M. F. Haddow, G. R. Whittel and I. Manners, Angew. Chem., Int. Ed., 2015, 54, 4836–4841 CrossRef.
- A.-M. Fuller, A. J. Mountford, M. L. Scott, S. J. Coles, P. N. Horton, D. L. Hughes, M. B. Hursthouse and S. J. Lancaster, Inorg. Chem., 2009, 48, 11474–11482 CrossRef CAS.
- M. Fernández-Millán, L. K. Allen, R. Garcia-Rodríguez, A. D. Bond, M. E. G. Mosquera and D. S. Wright, Chem. Commun., 2016, 52, 5993–5996 RSC.
- G. B. Consiglio, P. Queval, A. Harrison-Marchand, A. Mordini, J.-F. Lohier, O. Delacroix, A.-C. Gaumont, H. Gérard, J. Maddaluno and H. Oulyadi, J. Am. Chem. Soc., 2011, 133, 6472 CrossRef PubMed.
- K. Izod, J. Stewart, E. R. Clark, W. Clegg and R. W. Harrington, Inorg. Chem., 2010, 49, 4698–4707 CrossRef PubMed.
- K. Izod, Coord. Chem. Rev., 2012, 256, 2972–2993 CrossRef.
- K. Izod, J. Stewart, E. R. Clark, W. McFarlane, B. Allen, W. Clegg and R. W. Harrington, Organometallics, 2009, 28, 3327–3337 CrossRef.
- S. H. Bertz, C. P. Gibson and G. Dabbagh, Organometallics, 1988, 7, 227–232 CrossRef.
- L. Lochmann and J. Trekoval, J. Organomet. Chem., 1987, 326, 1 CrossRef CAS.
- P. Wyatt, H. Eley, J. Charmant, B. J. Daniel and A. Kantacha, Eur. J. Inorg. Chem., 2003, 4126–4226 Search PubMed.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef.
- V. de la Fuente, M. Waugh, G. R. Eastman, J. A. Iggo, S. Castillón and C. Claver, Chem. – Eur. J., 2010, 16, 6919–6932 CrossRef CAS PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- (a) CrysAlisPro, Version 1.171.36, Agilent Technologies, Oxford, UK, 2011 Search PubMed; (b) CrystalClear, Rigaku Corporation, Tokyo, Japan, 2011 Search PubMed; (c) APEX3, Version 2018.7–2, Bruker AXS Inc., Madison, USA, 2018 Search PubMed; (d) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef; (e) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic refinement details; NMR spectroscopic data. CCDC 2217486–2217492. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03587a |
| This journal is © The Royal Society of Chemistry 2023 |