
CO2 hydrogenation to formic acid on Pd–Cu nanoclusters: a DFT study†
D.
Chattaraj
 *a and
C.
Majumder
b
*a and
C.
Majumder
b
aProduct Development Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India. E-mail: debchem@barc.gov.in; Fax: +91 22 2550 5151; Tel: +91 22 2559 6446
bChemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
First published on 5th January 2023
Abstract
Carbon dioxide (CO2) hydrogenation to formic acid is a promising method for the conversion of CO2 to useful organic products. The interaction of CO2 with hydrogen (H2) on PdmCun (m + n = 4, 8 and 13) clusters to form formic acid (HCOOH) has been explored using density functional theoretical (DFT) calculations. Pd2Cu2, Pd4Cu4 and 13-atom Pd12Cu clusters are found to be the most stable among all of the PdmCun (m + n = 4, 8 and 13) clusters with binding energies of −1.75, −2.16 and −2.40 eV per atom, respectively. CO2 molecules get adsorbed on the Pd2Cu2, Pd4Cu4 and Pd12Cu clusters in an inverted V-shaped way with adsorption energies of −0.91, −0.96 and −0.44 eV, respectively. The hydrogenation of CO2 to form formate goes through a unidentate structure that rapidly transforms into the bidentate structure. To determine the transition state structures and minimum energy paths (MEPs) for CO2 hydrogenation to formic acid, the climbing image nudge elastic band (CI-NEB) method has been adopted. The activation barriers for the formation of formic acid from formate on Pd2Cu2 and Pd4Cu4 are calculated to be 0.79 and 0.68 eV, respectively whereas that on the Pd12Cu cluster is 1.77 eV. The enthalpy for the overall process of CO2 hydrogenation to formic acid on the Pd2Cu2, Pd4Cu4 and Pd12Cu clusters are found to be 0.83, 0.48 and 0.63 eV, respectively. Analysis of the density of states (DOS) spectra show that the 4d orbital of Pd, the 3d orbital of Cu, and the 2p orbitals of C and O atoms are involved in the bonding between CO2 molecules and the Pd2Cu2 clusters. The CO2 adsorption on the PdmCun (m + n = 4 and 8) clusters has also been explained in terms of the charge density distribution analysis.
Introduction
In recent decades, increased consumption of fossil fuels has resulted in a significant increase in the concentration level of carbon-dioxide (CO2) in the atmosphere. This has changed the global climate and caused ocean acidification.1–4 CO2 sequestration, carbon capture and storage have been suggested as different new ways to manage global CO2 levels. However, the catalytic conversion of CO2 into useful chemical products e.g. methane (CH4), methanol (CH3OH) and formic acid (HCOOH) has attracted considerable attention in recent years.5–14 CO2 is abundant in nature; it is an inexpensive feed stock for chemical processes and its removal from the atmosphere will reduce the greenhouse effect significantly. One very good example is CO2 conversion to the useful liquid hydrocarbon formic acid (HCOOH) through hydrogenation. Formic acid is an important chemical used in making animal feed, in tannery and dying leather and textiles and as a food preservative.15 Currently, formic acid synthesis is mainly performed through a two-step process: (i) carbonylation of methanol to methyl formate (HCOOCH3) using a high pressure of toxic CO as the feedstock and (2) hydrolysis of HCOOCH3 to formic acid and methanol. Formic acid production by direct hydrogenation of CO2 is a promising alternative way in terms of economy, ecology and safety. CO2 is extremely stable both thermodynamically and kinetically, exhibiting negative reaction entropies and high Gibb's energies. Hence, an effective, cheap and recyclable catalyst for this conversion of CO2 to formic acid and methanol is urgently required.A number of reports on experimental and theoretical studies of CO2 conversion to methanol and formic acid catalytically on metallic and bimetallic surfaces have been reported.16,17 Several Cu based catalysts like Pd–Cu, Ga–Cu, Ni–Cu, Co–Cu, Cu/ZnO and Cu/ZrO have been developed experimentally for enhancing the conversion of CO2 to CH3OH and HCOOH.18–21 Among these, a Pd–Cu catalyst is one of the most active bimetallic catalysts for CO2 hydrogenation. Pd has been widely used as a promising catalyst to enhance CO/CO2 hydrogenation. Melian-Cabrera et al. have found that the reduced state of Cu has been maintained via hydrogen spill over by Pd doped Cu-based catalysts.22 The reducibility of Cu sites is enhanced by Pd addition because of electron donation from Pd. The Pd–Cu catalysts exhibit a strong synergistic promotion of the methanol formation rate compared to that of pure Cu because of the combination of Cu and Pd. In the DFT study by Liu et al., a Cu(111) surface with a Pd monolayer substituted on the topmost layer has been employed as a Pd–Cu bimetallic system to explore the underlying mechanism of methanol synthesis from CO2 hydrogenation.19 In formic acid formation, CO2 hydrogenation is one of the most important steps. In this aspect, it is assumed that bimetallic Pd–Cu can act as a good catalyst for formic acid formation also. The experimental studies are not sufficient to elucidate a catalytic process over the catalytic surface and therefore, DFT calculations are essential to illustrate the reaction mechanism of CO2 hydrogenation on bifunctional catalyst.
A large number of transition metal complexes, mostly Ir, Rh and Ru-based ones, have been used for CO2 hydrogenation to formic acid. In recent times, the synthesis of formates and formic acid using homogeneous catalysis have been reviewed by several research groups.16,17,23–25 Sredojevic et al. have studied the formic acid synthesis by CO2 hydrogenation over single-atom catalysts based on Ru and Cu embedded in graphene.26 They made this synthesis possible with low activation energy by proceeding with the mechanism of H2 dissociation over the metal atom prior to CO2 adsorption. A Ni and Ni–Fe bimetallic system has also been found to be suitable for CO2 reduction to formic acid and other valuable chemicals.27,28 Most recently, Liu et al. have reported CO2 hydrogenation to formate and formic acid by a bimetallic palladium-copper hydride cluster.29 The reaction between PdCuH4− and CO2 to produce formic acid and PdCuH2− has been analysed by mass spectrometric analysis. Different isomers of PdCuH4− and PdCuCO2H4− are identified by anion photo electron spectroscopy (PES) and electronic structure calculations. Their mechanistic study confirms the metastable structure as the catalytic driving force.
Hydrogen adsorption, dissociation and diffusion on the PdCu surface has been shown in our earlier study.30 If CO2 gets adsorbed on the PdCu surface, the CO2 reduction to formic acid and methanol is possible. Aiming for this, using DFT, we have investigated CO2 adsorption on the PdCu surface with all possible orientations. But, it has been found that the PdCu surface does not adsorb CO2 in ambient conditions. In our next attempt, we have partially substituted the Pd atoms with Pt, and the Cu atoms with Ag, and Au on the PdCu surface to see whether the CO2 molecule gets adsorbed or not. It is observed that the CO2 molecule does not get adsorbed in any of these compositions of the PdCu surface. As, CO2 does not get adsorbed on the PdCu surface, the target of CO2 reduction to formic acid has been eliminated. Further, we have focused on the finite size of the PdCu clusters which have a large surface area, a high number of dangling bonds and large number of active sites. CO2 adsorption on the PdmCun (m + n = 4, 8 and 13) clusters has been studied using DFT based computational methodology. It is found that CO2 gets adsorbed on the PdmCun (m + n = 4, 8 and 13) clusters. As, the PdCu clusters also take a H2 molecule, the CO2 reduction to formic acid has been studied here on the PdmCun (m + n = 4, 8 and 13) clusters.
Although many researchers have focussed on CO2 hydrogenation for formic acid and methanol synthesis, to the best of our knowledge, formic acid formation from CO2 on nanoclusters, especially, Pd–Cu nanoclusters is rare. Nanoparticles have a high surface to volume ratio and better reactivity compared to their surface and bulk counterparts, so they can act as a better catalyst compared with those forms. Any real-life catalytic system would require the use of a support system. Here, small PdCu clusters are considered to study the basic reaction mechanism of formic acid formation by CO2 hydrogenation. By considering reactions on free clusters, which have higher degrees of freedom, it is possible to underscore the reaction mechanism at the smallest scale length. The results of these calculations are useful to study the reaction mechanism on supported clusters. Here, in this computational study, formic acid formation by CO2 hydrogenation on Pd–Cu nanoclusters has been investigated using a DFT formalism.
The geometry, energetics and reaction mechanism are of prime importance for the synthesis of formic acid on the bimetallic clusters. Here, CO2 hydrogenation with H adsorbed on the PdmCun (m + n = 4, 8 and 13) clusters is systematically studied using a state of the art first principles method for the generation of formic acid. The reaction pathways for the conversion of CO2 to formic acid have been revealed by a nudge elastic band (NEB) method. The calculated properties for the formic acid formation provide comprehensive data which will be useful for experimental understanding.
Results and discussion
Structural optimization of PdmCun (m + n = 2, 4, 8 and 13) clusters
After establishing the properties of the PdCu dimer, the total energy and binding energy of different PdmCun (m + n = 4, 8 and 13) clusters are calculated. A large number of geometrical isomers of each composition of the PdmCun (m + n = 4, 8 and 13) clusters are shown in Tables S3–S5 (ESI†). The specific coordinates of the clusters are given in Table S9 (ESI†). The results reveal that the small PdCu clusters favor a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 composition (e.g. Pd2Cu2, Pd4Cu4) and for the 13-atom cluster it is Pd12Cu where the Cu atom is at the centre of the icosahedron. The ground state geometric configurations for each composition of the PdmCun (m + n = 2, 4, 8 and 13) clusters are shown in Fig. 1.
:1 composition (e.g. Pd2Cu2, Pd4Cu4) and for the 13-atom cluster it is Pd12Cu where the Cu atom is at the centre of the icosahedron. The ground state geometric configurations for each composition of the PdmCun (m + n = 2, 4, 8 and 13) clusters are shown in Fig. 1.
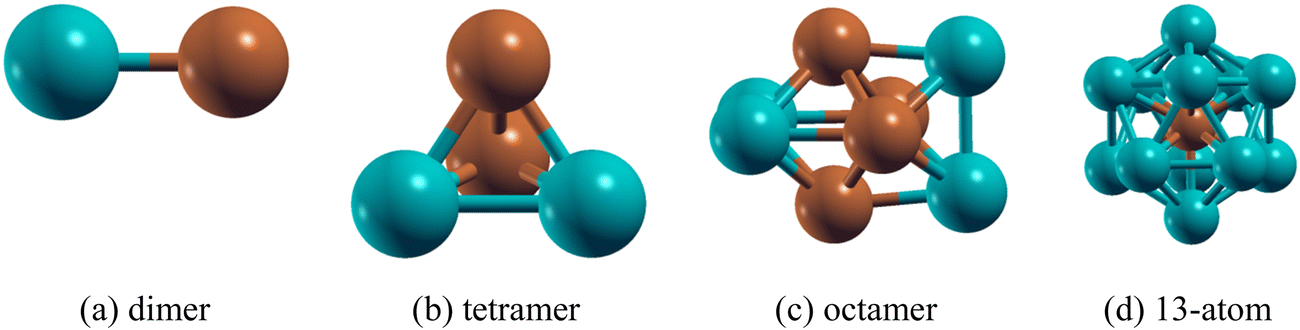 | ||
| Fig. 1 Stable isomers of thePdmCun (m + n = 2, 4, 8 and 13) clusters (a) dimer (b) tetramer (c) octamer (d) 13-atom cluster (atomic colours: green = Pd, brown = Cu). | ||
:3, 2:2 and 3:1), apart from the homoatomic clusters, are considered for calculation. Accordingly, the various possible isomeric structures of Pd4, Cu4, Pd3Cu, Pd2Cu2 and PdCu3 clusters are optimized. The average binding energy, average bond length and spin moments of these clusters are summarized in Table 2.
| Number of atom (n) | System | Average B.E. (eV per atom) | Average Pd–Cu B.L. (Å) | Spin moment (μB) |
|---|---|---|---|---|
| n = 4 | Pd4 | −1.67 | 2.60 (Pd–Pd) | 0 |
| Pd3Cu | −1.74 | 2.50 | 1 | |
| Pd2Cu2 | −1.75 | 2.47 | 0 | |
| PdCu3 | −1.62 | 2.46 | 1 | |
| Cu4 | −1.59 | 2.25 (Cu–Cu) | 0 | |
| n = 8 | Pd8 | −2.07 | 2.67 (Pd–Pd) | 2 |
| Pd7Cu | −2.10 | 2.54 | 3 | |
| Pd6Cu2 | −2.09 | 2.51 | 2 | |
| Pd5Cu3 | −2.15 | 2.50 | 1 | |
| Pd4Cu4 | −2.16 | 2.56 | 0 | |
| Pd3Cu5 | −2.09 | 2.50 | 1 | |
| Pd2Cu6 | −2.14 | 2.55 | 2 | |
| PdCu7 | −2.13 | 2.56 | 1 | |
| Cu8 | −2.14 | 2.28 (Cu–Cu) | 0 | |
| n = 13 | Pd13 | −2.32 | 2.73 (Pd–Pd) | 8 |
| Pd12Cu | −2.40 | 2.60 | 7 | |
| PdCu12 | −2.28 | 2.62 | 4 | |
| Cu13 | −2.24 | 2.53 (Cu–Cu) | 5 |
The binding energies of the PdmCun clusters are shown in Table 2. It is seen that Pd2Cu2 is the most stable structure among all of the PdmCun (m + n = 4) clusters with the average binding energy of −1.75 eV per atom. Table S3 (ESI†) shows that Pd4 forms a triangular pyramid (Tpy) structure as the lowest energy with an average Pd–Pd bond length of 2.60 Å. The substitution of a single Pd atom by a Cu atom in Pd4 resulting in Pd3Cu does not change the Tpy geometry and the average Pd–Cu bond length for that is found to be 2.50 Å. Further substitution of a Pd atom in Pd3Cu forming Pd2Cu2 does not allow a change of its Tpy geometry and here the average Pd–Cu bond length is found to be 2.47 Å. The further substitutions of the Pd atoms in Pd2Cu2 resulting in PdCu3 and Cu4 changes the geometry to planar rhombus. The Pd2Cu2 cluster can be visualized as a combination of a maximum of two PdCu dimer moieties which gives stability to Pd2Cu2 whereas in Pd3Cu and PdCu3 only one PdCu dimer moiety is present. The spin moment of both Pd4 and Cu4 clusters are calculated to be 0 μB. The substitution of Pd atoms in the Pd4 cluster and Cu atoms in the Cu4 cluster increases the spin moment except for Pd2Cu2 which is paramagnetic in nature.
Formic acid formation on PdmCun (m + n = 4, 8 and 13) clusters
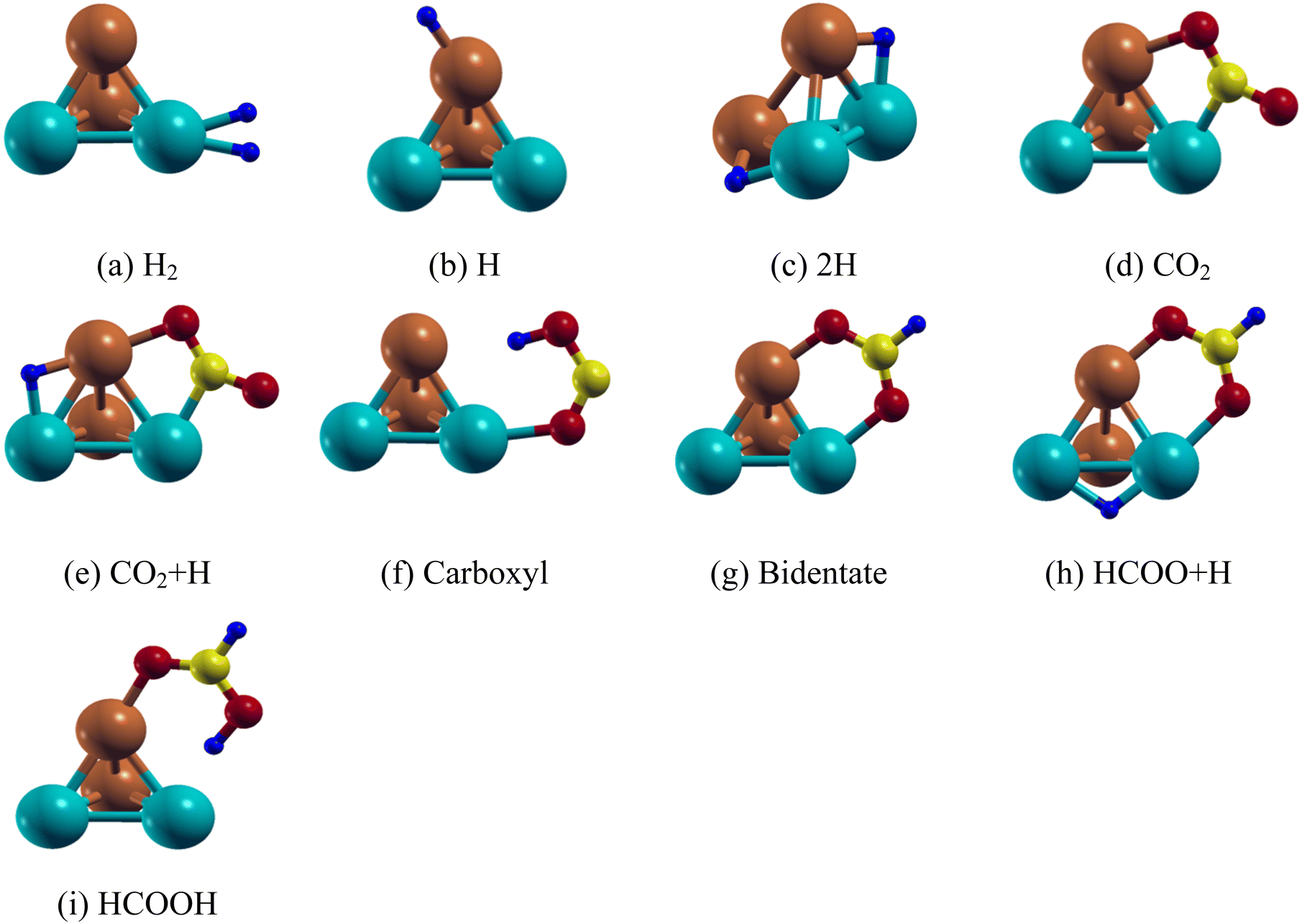 | ||
| Fig. 2 Lowest energy structure of the Pd2Cu2 clusters along with their (a) H2 (b) H (c) 2H (d) CO2 (e) CO2 + H (f) carboxyl (g) bidentate (h) HCOO + H and (i) HCOOH adsorbed species. | ||
We have first studied the CO2 adsorption on the Pd2Cu2 cluster which is the primary step for HCOOH formation. The most stable configuration of CO2 on the Pd2Cu2 shows that one of the C–O bonds is attached in such a way so that C and O atoms are attached to the Pd and Cu atoms, respectively as shown in Fig. 2(d). The binding energy of CO2 is −0.91 eV. The optimum lengths of the Cu–O and Pd–C bonds are 2.00 and 1.99 Å, respectively. As a consequence, the C–O bond is elongated from 1.16 Å (free CO2) to 1.26 Å. The O–C–O bond angle of the adsorbed CO2 is optimized to 139.8°. Another possibility of CO2 adsorption on the Pd2Cu2 cluster could be where the C and O atoms could be attached to the Cu and Pd atoms. However, this configuration is unstable. The reason behind this is the extent of electron transfer between Cu–O and Pd–C in the CO2 adsorbed Pd2Cu2 cluster. The details have been discussed in DOS analysis in the Electronic structure section.
On the basis of previous literature, it is seen that formic acid formation by hydrogenation of CO2 can take place either through formate (HCOO˙) or carboxyl (COOH) radical formation. For the Pd2Cu2 cluster, the formate route is found to be more favorable than the carboxyl route. This fact is also established here by calculating the activation energy of the corresponding processes which are discussed in later sections. Two configurations are possible for the HCOO species, carboxyl (mono-HOCO) and bidentate (bi-HCOO), distinguished by the number of O atoms that are adsorbed on the Pd2Cu2 cluster. For carboxyl, one of the two O atoms of HOCO gets attached to the Pd atom of the Pd2Cu2 cluster, respectively as shown in Fig. 2(f). The Pd–O and Cu–H bond distances are found to be 1.14 Å and 2.33 Å, respectively. The adsorption energy for carboxyl (mono-HOCO) is found to be −1.21 eV. Another possibility of carboxyl radical (mono-HOCO) adsorption on Pd2Cu2 happens where the O and H atoms of HOCO are attached to Cu and Pd atoms, respectively. However, this route is found to be not stable.
For bidentate-complex (HCOO-Pd2Cu2) formation, two O atoms are connected to the Pd–Cu edge of the Pd2Cu2 cluster as shown in Fig. 2(g) with an adsorption energy of −3.39 eV. The optimized Pd–O and Cu–O bond lengths are 2.14 and 1.94 Å, respectively. The O–C–O bond angle is 128.6° and the C–H bond length is 1.10 Å for the HCOO adsorbed on the Pd2Cu2 cluster. In this context it should be noted that here CO2 gets adsorbed on the Pd2Cu2 cluster in a V-shaped orientation, but addition of an H atom to the C of CO2 makes it possible to adsorb bidentate HCOO in an inverted V-fashion. This is because of depletion of electronic charge on the O atom which gets attached to the Pd and Cu atoms. The DOS analysis in the Electronic structure section supports this statement.
The final step is formic acid formation from bidentate formate by adding an H-atom. The adsorption of formic acid on the Pd2Cu2 cluster has two possibilities; the H atom is either attached to a Cu or a Pd atom of the cluster. It is seen that the H atom of the COOH attached to a Pd atom of Pd2Cu2 has relatively lower energy compared to that of the H attached to a Cu atom. The Pd–H configuration (Fig. 2(i)) is found to be 0.29 eV lower in energy than Cu–H configuration. The adsorption energy of the Pd–H configuration is calculated to be −1.37 eV. The Pd–H and Cu–O distances in the lower configuration are 1.96 Å and 1.95 Å, respectively. The binding energies and related bond lengths of H2, H, 2H, CO2, HCOO and HCOOH adsorbed on the Pd2Cu2 nanocluster are given in Table 3.
| Adsorbate species | Site | Bond lengths (Å) | E b (eV) |
|---|---|---|---|
| H2 | Top-Pd | Pd–H = 1.75, 1.72, H–H = 0.86 | −5.05 |
| H | Top-Cu | Cu–H = 1.51 | −2.05 |
| 2H | Cu–Pd bridge | Cu–H= 1.80, Pd–H = 1.75 | −5.19 |
| CO2 | Cu–Pd bridge | C–O = 1.26 | −0.91 |
| HCOO | Carboxyl | Cu–H = 2.33, Pd–O = 1.14, C–O = 1.23, 1.32, O–H = 1.03 | −1.21 |
| Bidentate | Pd–O = 2.14, Cu–O = 1.94, C–O = 1.28, C–H = 1.11 | −3.39 | |
| HCOOH | Pd–H | Pd–H = 1.96, O–H = 1.10, C–O = 1.31, C–O = 1.25, C–H = 1.10, Cu–O = 1.95 | −1.37 |
| Cu–H | Cu–H = 2.61, O–H = 1.05, C–O = 1.32, C–O = 1.24, C–H = 1.10, Pd–O = 2.21 | −1.08 |
The reaction between H and CO2 on PdmCun (m + n = 4, 8 and 13) leads to the formation of bidentate formate whereas that between H and formate forms formic acid. For that, co-adsorption of H and CO2 as well as H and formate HCOO on the PdmCun (m + n = 4, 8 and 13) clusters has been investigated. In the first case, H has been placed in different adsorption sites (top, bridge and hollow) of the model cluster keeping CO2 adsorbed at a fixed site. The results show that H on the Pd–Cu edge opposite to the CO2 adsorbed edge has the most stable geometry as shown in Fig. 2(e). The H adsorption energy in the pre-adsorbed Pd2Cu2–CO2 cluster is −2.36 eV as per eqn (7). On the other hand, H is found to be stable on the Pd–Pd edge of the pre-adsorbed bidentate formate (Pd2Cu2–HCOO) complex (Fig. 2(h)) with an adsorption energy of −3.13 eV as per eqn (2).
The formation of formic acid by the reaction of CO2 and H occurs through a bidentate formate route. These reaction pathways have been investigated using DFT and CI-NEB methods. The overall reaction pathway has been divided into two parts: (i) CO2 to bidentate formation and (ii) bidentate to formic acid formation on the PdmCun (m + n = 4, 8,13) clusters. The initial and final images of the reaction pathways obtained from the stable geometrics are shown in Fig. 2 and Fig. S1 and S2 (ESI†). The intermediate geometrics are obtained by NEB calculations. Formic acid formation from CO2 on the PdmCun (m + n = 4, 8, 13) clusters is discussed in the proceeding subsections.
NEB for CO2 to formic acid conversion on Pd2Cu2 cluster
Based on the adsorption results, the reaction pathways for CO2 to formic acid formation on the Pd2Cu2 tetramer cluster has been studied (see Fig. 3). The reaction between CO2 and H to form formate on the Pd2Cu2 cluster is shown in Fig. 3(a). From the figure, it is seen that H is stable in the bridge of the Pd–Cu edge along with a co-adsorbed CO2 molecule. The Pd–H and Cu–H bond lengths are found to be 1.71 and 1.62 Å, respectively. This H moves from its stable site to a C-atom of CO2 to form formate which gets attached to the Pd2Cu2 cluster with its two O-atoms. The intermediate images are shown in Fig. 3(a). The activation barrier for this reaction of formate formation from CO2 is found to be 0.81 eV.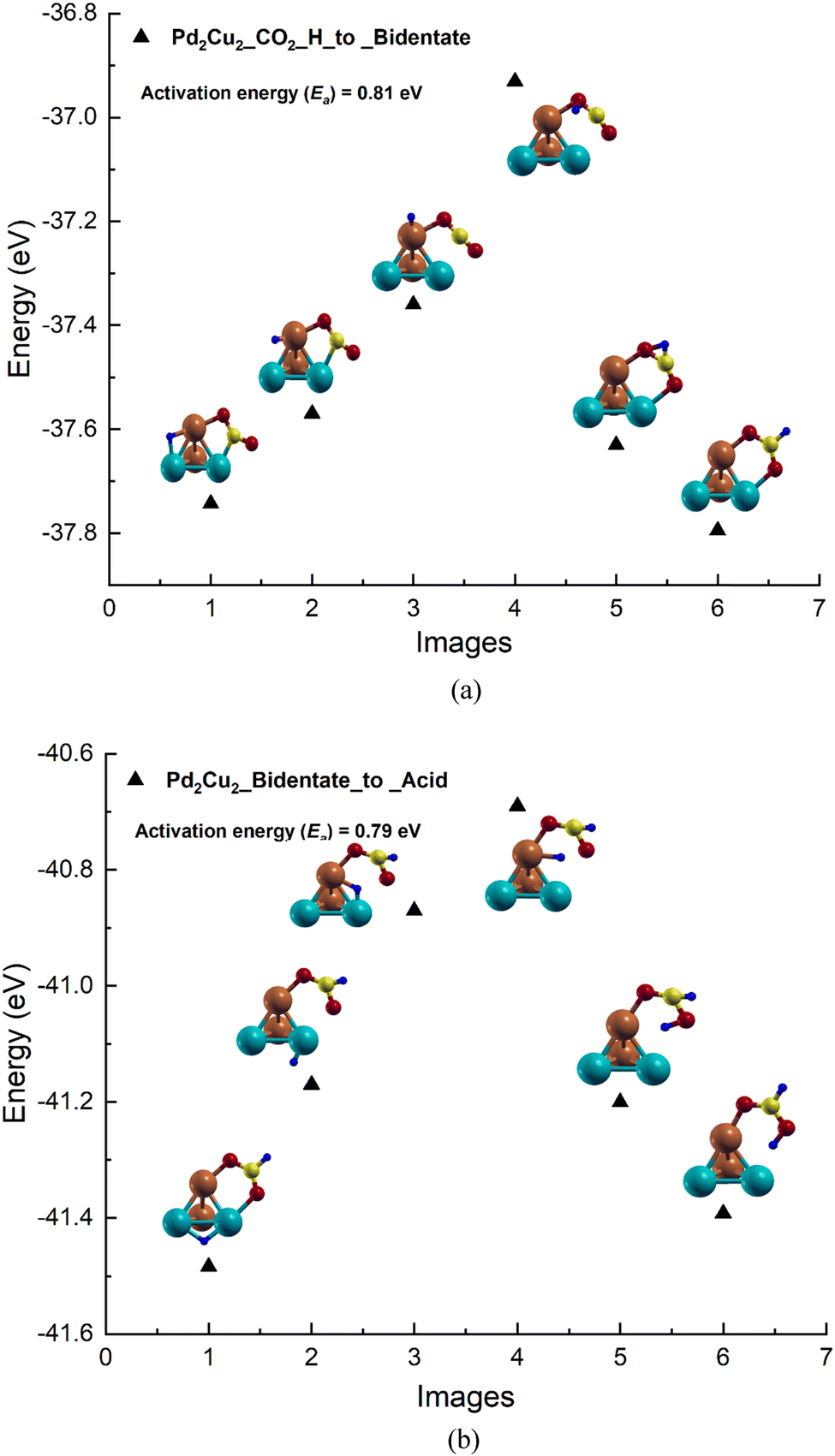 | ||
| Fig. 3 The potential energy profile for the formation of (a) bidentate formate by the reaction of H-atoms and CO2 (b) formic acid by the reaction of H-atoms and formate on the Pd2Cu2 cluster. | ||
In the next NEB calculations i.e. the formic acid formation by the reaction of formate and a H-atom is depicted and shown in Fig. 3(b). In this graph, image 1 is the initial state where H is stable on the Pd–Pd edge of Pd2Cu2 along with the adsorbed bidentate formate. From the figure, it is seen that H gets transferred to the Pd–Cu edge of Pd2Cu2 where formate is already adsorbed with its two O-atoms, which is shown in image 3. This H is further transferred to one of the O-atoms of formate closer to the Pd atom to form formic acid as shown image 6 of Fig. 3(b). The activation barrier for this reaction of formate and H to formic acid is found to be 0.79 eV which is more or less similar to the previous step of formate formation.
CO2 hydrogenation to formic acid on Pd4Cu4 octamer
In line with the study for CO2 hydrogenation on the Pd2Cu2 cluster, we have started investigating the adsorption behaviour of H2 on the Pd4Cu4 cluster. The optimization has been performed by placing the H2 molecule in on-top positions of the Pd and Cu sites along with the other bridge and hollow sites available on the cluster. The distance of the Pd/Cu to H is kept at ∼3 Å. The results suggest that H2 favours the on-top Cu site (Fig. S1(a), ESI†) with an adsorption energy of −4.91 eV. The two Cu–H bond distances are 1.68 Å and the H–H bond is elongated up to 0.83 Å. The adsorption of the H-atom on the top, bridge and hollow sites of the Pd4Cu4 cluster is also investigated. Among different configurations, the H atoms prefer the hollow site of a triangle formed by two Pd and one Cu atom (Fig. S1(b), ESI†). The adsorption energy for this is −2.87 eV with an average Pd–H distance of 1.75 Å. The stability of two H atoms on the Pd4Cu4 cluster has also been investigated. It is found that both of the H atoms prefer to bind with the Cu–Pd and Pd–Pd bridge sites as shown in (Fig. S1(c), ESI†). The binding energy for these two H atoms is calculated to be −5.87 eV with Cu–H and Pd–H bond lengths of 1.76 and 1.82 Å, respectively.Like Pd2Cu2, CO2 adsorption on this Pd4Cu4 cluster is also studied by placing the molecule at different well-defined sites. It is found that the CO2 molecule gets adsorbed in a V-shaped fashion on the Pd4Cu4 cluster as shown in Fig. S1(d) (ESI†). The adsorption energy for this configuration is −0.96 eV. The Cu–O and Pd–C bonds are 1.93 and 1.98 Å, respectively. One of the C–O bonds attached to Cu–Pd edge of the Pd4Cu4 octamer is elongated from 1.16 to 1.26 Å. The adsorption nature of the CO2 molecule in the Pd4Cu4 cluster is same as that on the Pd2Cu2 moiety and the reason lies within the nature of bonding. Like Pd2Cu2, here formic acid formation on the Pd4Cu4 cluster takes place through the formate route. The HCOO species can be adsorbed in two forms, carboxyl (mono HOCO) and bidentate (bi-HCOO) depending on the number of oxygen atoms that get attached to the Pd4Cu4 cluster. In case of carboxyl binding, O and C-atoms are attached to Pd and C atoms, respectively as shown in Fig. S1(f) (ESI†). The Pd–O and Cu–C bond distances are 1.99 Å each and the adsorption energy for monodentate carboxyl HOCO is −2.44 eV.
Like, for the Pd2Cu2 cluster, the co-adsorption of H and CO2 has been compared with H and HCOO on the Pd4Cu4 cluster.
In the case of bidentate HCOO, it binds to the Pd–Cu edge of the Pd4Cu4 cluster where Cu is in the octahedron frame and Pd atom is out of the frame (Pd–Pd edge) as shown in Fig. S1(g) (ESI†). The Pd–O and Cu–O bond distances are 2.09 and 1.91 Å, respectively. The adsorption energy is found to be −3.67 eV. The C–H bond distance and O–C–O bond angle are 1.11 Å and 104°, respectively. It is interesting to report that CO2 gets adsorbed to a Cu–Pd edge of Pd4Cu4 through a C bond and in a V-shape fashion (Fig. S1(d), ESI†) but addition of a H atom to a C-atom in CO2, resulting in bidentate formate HCOO, favors the inverted V-shaped form for the adsorption of bidentate formate (bi-HCOO). The reason is depicted in the electronic structure analysis in the Electronic structure section.
The final step is formic acid formation from bidentate formate by addition of an H-atom. Adsorption of formic acid on the Pd4Cu4 cluster has two possibilities; the H atom is either attached to a Cu or a Pd atom of the cluster. It has been found that the protonic H atom of the HCOOH is attached to the Pd atom of the Pd4Cu4 with an adsorption energy of −1.17 eV (Fig. S1(i), ESI†). However, the adsorption of H with Cu is not stable. The adsorption energy of this adsorbed formic acid configuration is −1.37 eV. The Pd–H and Cu–O distances of this adsorbed formic acid configuration are 1.94 Å and 1.93 Å, respectively.
Like, Pd2Cu2, the co-adsorption of H atoms and CO2 as well as H atoms and formate HCOO have been studied on the Pd4Cu4 cluster. Among different isomeric geometries, the H atom favors the hollow site (a triangle composed of two Pd and Cu atoms) as shown in Fig. S1(e) (ESI†). The position of the CO2 is fixed and it is attached to the Pd–Cu edge adjacent to the H atom adsorption site. The H-adsorption energy for this Pd2Cu2–CO2 moiety is found to be −2.60 eV as per equation (2). Similarly, a H atom in the same position i.e. in the mid of the triangle composed of two Pd and Cu atoms, adjacent to the adsorbed bidentate formate site, has the lowest energy structure as shown in Fig. S1(h) (ESI†). The H-adsorption energy for this Pd2Cu2–HCOO moiety is found to be −2.79 eV. The binding energies and related bond lengths of H2, H, 2H, CO2, HCOO and HCOOH adsorbed on the Pd4Cu4 nanocluster are given in Table S1 (ESI†).
NEB for CO2 to formic acid conversion on Pd4Cu4 cluster
After investigating the formic acid formation on the Pd2Cu2 cluster, we have studied the same process on the higher order cluster Pd4Cu4. Here also, the process of formic acid formation has been divided into two parts i.e. CO2 to formate and formate to formic acid formation. Fig. S3(a) (ESI†) shows the bidentate formate formation pathway by the reaction of CO2 and H-atoms on a dodecahedron Pd4Cu4 cluster. The initial image of this NEB calculation is that H is stable on the Pd–Cu edge with CO2 adsorbed on the adjacent Pd–Cu edge. The overall reaction pathway as shown in Fig. S3(a) (ESI†) shows the H atom movement from its stable position to the C atom of CO2 to form formate. The activation barrier for this reaction is found to be 0.79 eV.In the next step, the reaction pathway for formate to formic acid formation on the Pd4Cu4 octahedron is shown in Fig. S3(b) (ESI†). Here the H-atom on the Pd–Cu edge of Pd4Cu4 has a stable position along with the formate adsorbed on the adjacent Pd–Cu edge. The NEB calculation shows the H movement from its stable position to the O-atom which was attached to the Pd atom in the formate. The activation energy for this reaction is calculated to be 0.68 eV which is lower compared to the first step of formate formation on this Pd4Cu4 cluster.
CO2 hydrogenation to formic acid on Pd12Cu cluster
At first, the adsorption properties of H2 molecules on the Pd12Cu icosahedron cluster have been studied. For that, a H2 molecule was placed on the top of the Pd atom at a distance of ∼3 Å, setting the H–H bond length equal to the value of the free H2 molecule, 0.75 Å. The H2 at the top of the Pd-atom has the lowest energy structure as shown in Fig. S2(a) (ESI†). The adsorption energy of the H2 molecule adsorbed on this top Pd atom is found to be −5.06 eV. The two Pd–H bond distances are optimized to 1.70 Å, whereas the H–H bond distance is 0.88 Å.Now the adsorption of the H-atom on different sites (top, bridge and hollow) of the Pd12Cu icosahedron cluster is also investigated. Among the different structures, it has been found that the H at the mid of the triangle constituted by three Pd atoms has the stable structure as shown in Fig. S2(b) (ESI†). The adsorption energy and average Pd–H distance are −2.85 eV and 1.83 Å, respectively. The stability of two H atoms on the Pd12Cu cluster has also been investigated. It is found that both the H atoms prefer to bind with the three Pd atom hollow sites (fcc and hcp) as shown in (Fig. S2(c), ESI†). The binding energy for these two H atoms is calculated to be −5.13 eV with an average Pd–H bond length of 1.83 Å, respectively.
After studying the adsorption of H2 molecules and H-atoms, the adsorption of CO2 gas molecules on the Pd12Cu icosahedron cluster and the energetics associated with that have been investigated. It has been observed that one C–O bond, of bond length 1.27 Å, of the CO2 molecules is parallelly attached to the Pd–Pd bond of the CuPd12 cluster as shows in Fig. S2(d) (ESI†). The other O-atom of that CO2 molecule is not bonded to any atom of that cluster. Initially a linear CO2 molecule is placed parallelly to the Pd–Pd edge of the Pd12Cu cluster. In the most stable geometry the shape of the CO2 molecule became V-shaped with an O–C–O bond angle of 105°. The adsorption geometry together with the calculated binding energy of −0.44 eV implies that the CO2 molecule is bonded to the cluster.
Both by experiment and theoretical studies, it is established that HCOO is a key intermediate for CO2 hydrogenation to formic acid and methanol.45,46 There are two configurations of HCOO species, carboxyl (mono-HOCO) and bidentate (bi-HCOO), distinguished by the number of O atoms that are adsorbed on the Pd12Cu cluster. For carboxyl (mono-HOCO), the C–O bond of HOCO binds to the Pd–Pd edge of the Pd12Cu cluster as shown in Fig. S2(f) (ESI†). The O–H bond of HOCO does not attach to the cluster, it remains attached to the C-atom. The Pd–C, Pd–O and C–O bond lengths of mono-HOCO adsorbed on the Pd12Cu cluster are found to be 1.96, 2.26 and 1.25 Å, respectively. The adsorption energy for this mono-HOCO moiety is −2.50 eV. For, bidentate-HCOO, it binds to the Pd–Pd edge of the Pd12Cu cluster through two O-atoms (see Fig. S2(g), ESI†) with an adsorption energy of −3.22 eV. The two Pd–O bond lengths are identical at 2.10 Å.
The co-adsorptions of H atoms and CO2 as well as H atoms and formate HCOO on the icosahedron Pd12Cu cluster are similar to those on the PdmCun (m + n = 4, 8) clusters. Among the different isomeric geometries, the H atom is found to be most stable in the mid of the triangle composed of three Pd atoms as shown in Fig. S2(b) (ESI†). The position of the CO2 is fixed and it is attached to the Pd–Pd edge adjacent to the H atom adsorption site (Fig. S2(e), ESI†). The H-adsorption energy for this Pd12Cu-CO2 moiety is found to be −2.87 eV. Similarly, the H atom in the same position i.e. in the mid of the triangle composed of three Pd atoms, adjacent to the adsorbed bidentate formate site, has the lowest energy structure as shown in Fig. S2(h) (ESI†). The H-adsorption energy for this Pd12Cu–HCOO moiety is found to be −2.84 eV.
After formate adsorption, we turned our attention to adding an H atom to formate to produce a formic acid molecule on the Pd12Cu cluster. For the adsorption of the final product formic acid on the Pd12Cu cluster, there are two possibilities of HCOOH adsorption, where the two H-atoms are in cis and trans positions. The trans configuration (shown in Fig. S2(i), ESI†) is found to be 0.43 eV more stable than the cis one. The Pd–H and Pd–O bond distances in the trans configuration are 2.16 and 2.19 Å, respectively. The adsorption energy for that trans species is −0.90 eV. The binding energies and related bond lengths of H2, H, 2H, CO2, HCOO and HCOOH adsorbed on the Pd12Cu nanocluster are given in Table S2 (ESI†).
NEB for CO2 to formic acid conversion on Pd12Cu cluster
In this case, the process of formic acid formation from CO2 has also been divided into two parts like the previous two clusters (i) CO2 to bidentate formate (ii) formate to formic acid. The first step is the reaction of CO2 and H to form bidentate formate, which is shown in Fig. S4(a) (ESI†). Here, all of the adsorbed species i.e. CO2 and H-atoms are on the Pd–Pd edge as the outer shell of the Pd12Cu cluster is made up with Pd atoms. The H-atom moves from the Pd–Pd edge to the adjacent CO2. At first, it goes to one of the O-atoms of CO2 and is then transferred to the C atom of that CO2 to form formate. The activation barrier for the reaction is found to be 1.92 eV which is higher than the same process on the Pd2Cu2 and Pd4Cu4 clusters.For the bidentate formate to formic acid conversion the H-atom moves from the Pd–Pd edge to the apex Pd-atom with which one of the O-atoms of formate (HCOO) is attached as shown in image 3 of Fig. S4(b) (ESI†). This H-atom further gets attached to that O-atom and forms formic acid where two H-atoms are in the trans position with respect to each other. The activation barrier for the reaction is found to be 1.77 eV.
From the six NEB calculations (Fig. 3 and Fig. S3, S4, ESI†), it is seen that, formate and formic acid formation require more energy in the Pd12Cu cluster compare to that in the PdmCun (m + n = 4, 8) clusters. The reason may be the Pd–Cu and Pd–Pd edge of the clusters where CO2 and formate HCOO got adsorbed. Also, the activation energy for CO2 to formate and formate to formic acid formation decreases as the PdCu cluster size increases except for the stable Pd12Cu magic cluster.
Thermochemistry of CO2 hydrogenation to formic acid
Depending on the stability of the H2 molecule and the two H atoms on the PdmCun (m + n = 4, 8 and 13) clusters, we have extended our study to the hydrogenation of CO2 to formic acid via the formate route on these clusters. The overall reaction can be given as follows:| H2 + PdCu → H2–PdCu* | (1) |
| H2–PdCu* → 2H–PdCu* | (2) |
| 2H–PdCu* + CO2 → H–PdCu–HCOO* | (3) |
| H–PdCu–HCOO* → PdCu–HCOOH* | (4) |
| PdCu–HCOOH* → PdCu + HCOOH | (5) |
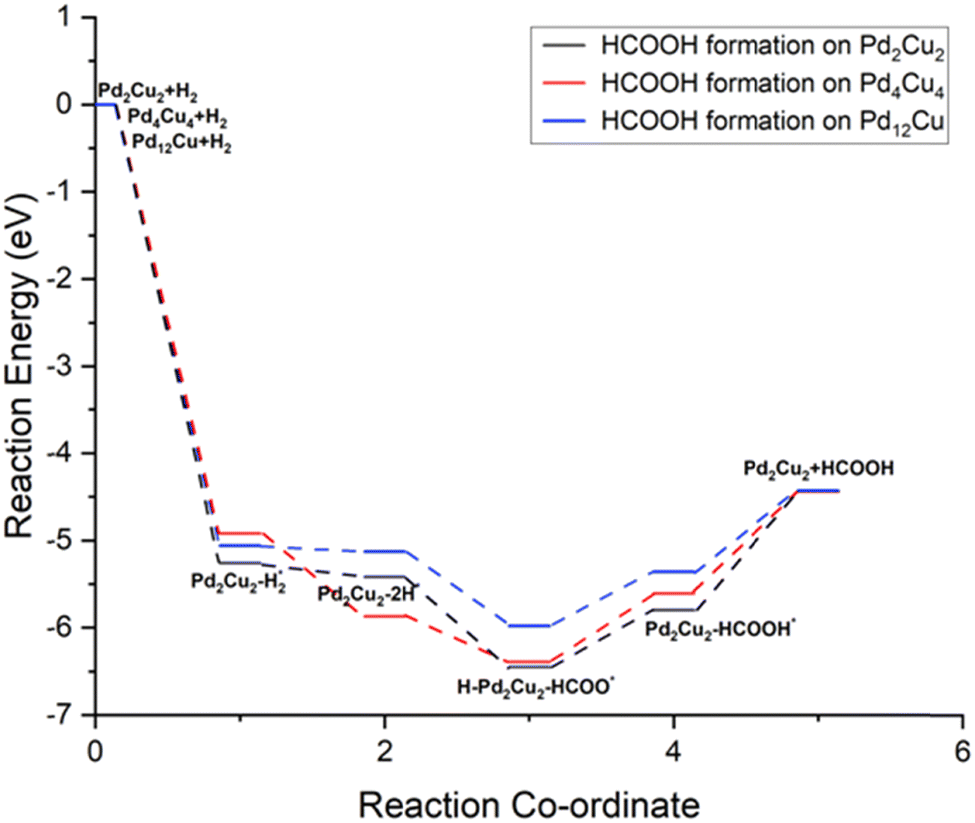 | ||
| Fig. 4 Potential energy diagram of CO2 hydrogenation to formic acid on the PdmCun (m + n = 4, 8 and 13) clusters. | ||
Electronic structure
Adsorption of CO2 on the PdmCun (m + n = 4, 8 and 12) clusters is the crucial step for the formation of formic acid and methanol either through the formate or carboxyl route. The reason behind the adsorption of CO2 on these clusters in a particular fashion needs to be explored. From the Fig. 2(d) and Fig. S1(d) and S2(d) (ESI†), it is seen that CO2 gets adsorbed either on the Pd–Cu edge (for Pd2Cu2 and Pd4Cu4 clusters) or on the Pd–Pd edge of the Pd12Cu icosahedron cluster. In the PdmCun (m + n = 4, 8 and 12) clusters, a linear CO2 molecule has been placed in various positions like on top of Pd, on top of Cu, on the Pd–Pd edge, on the Cu–Cu edge, at the mid of the Pd–Cu–Cu triangle, at the mid of the Pd–Pd–Cu triangle etc. Among those, CO2 gets adsorbed on the Pd–Cu edge in an inverted V-shaped fashion with the C-centre attached to the Pd-atom and one O-centre attached to the Cu atom of the clusters. The other O-atom of the CO2 remains unattached to any Pd or Cu atom of these clusters. A similar configuration for CO2 adsorption has been seen in the case of the Pd4Cu4 and Pd12Cu clusters also.To understand the nature of the chemical bonding present in the CO2 adsorbed Pd2Cu2 cluster, the total and orbital projected density of states for Pd2Cu2 and Pd2Cu2–CO2 have been calculated and shown in Fig. S5 and S6 (ESI†). From the Fig. S5(b) (ESI†), it is seen that the Pd-4d and Cu-3d orbitals are mainly contributing to the Fermi energy in the Pd2Cu2 cluster, whereas in the Pd2Cu2–CO2 moiety, the 2p orbital of the C and O atoms are contributing along with the Pd-4d and Cu-3d orbitals (Fig. S6(b), ESI†). The presence of finite DOS at the Fermi level indicates the metallic nature and the electron conduction behaviour of the Pd2Cu2 and Pd2Cu2–CO2clusters. In Fig. S6(b) (ESI†), the DOS peaks of the C-atom which range from 0–5 eV are overlapping with DOS of the Pd atom to which it is attached. Whereas, the DOS spectra of one of the O-atoms (1O atom) of the CO2 is overlapping with those of Cu atom to which it is attached. The DOS spectra (p-orbital DOS) of the other O-atom (2O atom) which is not attached to any Pd or Cu atoms has its position much below the Fermi level mainly ranging from −12.5 eV to −5 eV. From the charge density distribution table given in Table S6 (ESI†), it is seen that the C-atom takes charges from two O-atoms and donates that to one Pd atom of the Pd–Cu edge of the Pd2Cu2 cluster. Whereas, the Cu atom of that Pd–Cu edge donates charges to one of the O-atoms of the CO2 (1O atom) to which it is attached and becomes an electron deficient centre. This charge transfer facilitates the bond formation between the Pd–Cu edge and the CO2 molecule and helps the CO2 molecule to get adsorbed on that edge in an inverted V-shaped fashion. A similar type of bonding and charge transfer has been seen in the case of the Pd4Cu4 cluster and its CO2 adsorbed counterpart. The supporting information of the charge density distribution is given in Table S7 (ESI†).
After exploring the nature of the bonding in the Pd2Cu2 and Pd4Cu4 clusters and their CO2 adsorbed counter parts, the same has been investigated for the Pd12Cu clusters and its CO2 adsorbed species in terms charge density distributions. From the charge density distribution table given in Table S8 (ESI†), it is seen that one of the two Pd atoms of the Pd–Pd edge (1Pd atom) to which the C atom of CO2 is attached acts as an electron depletion centre as it receives electronic charges from two O atoms via a C-atom centre. Another Pd atom of that Pd–Pd edge (2Pd atom) donates electronic charges to the O-atom of the CO2 to which it is attached and it becomes an electron deficient centre. This promotes the CO2 to get adsorbed in an inverted V-shaped way like the previous cases for the Pd2Cu2 and Pd4Cu4 clusters.
Computational details
All calculations are performed within the spin-polarized density functional theory using the plane wave-pseudopotential approach as implemented in the Vienna ab initio simulation package (VASP).47–49 The electron-ion interaction and the exchange correlation energy are described under the projector-augmented wave (PAW) method and the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE), respectively.50–52 The energy cut off for the plane wave basis set has been fixed at 500 eV for assuming a good energy convergence. Finding the ground state structure of a cluster is a challenging task. For this, several methods like, Generic Algorithm, basin-hoppins, evolutionary algorithm are used to generate a large number of geometries. However, for small clusters one can use chemical intuition, which in other words is called an educated guess structure and one can then relax these structures to the possible nearest minima. Following this, we have generated a large number of initial geometries and found out the lowest energy structure for each composition.53–55 A large number of spin states have also been considered for all these structures. The ionic optimization has been carried out using the conjugate gradient scheme and the forces on each ion are minimized up to 5 meV Å−1.56,57 A suitable simulation box of size 15 × 15 × 15 Å has been considered for all of the calculations. The k-point sampling in the Brillouin zone (BZ) are treated with the Monkhorst–Pack scheme, using the gamma point.58 The total energies of each relaxed structure using the linear tetrahedron method with Blochl corrections have been subsequently calculated in order to eliminate any broadening-related uncertainty in the energies.59 In all of the DFT based calculations, the electrons are described by the scalar-relativistic approximation, in which spin–orbit coupling (SOC) is taken into account for the core states but is neglected for the valence states.60,61 However, few studies have showed that SOC plays a crucial role for heavy element systems.62–64 Due to the presence of Pd atoms, here in this work, DFT-PBE calculations with and without SOC for the valence states have been performed for the dimers. The binding energies (Eb) of the PdmCun (m + n = 2, 4, 8, and 13) clusters have been estimated from the total energy calculations using eqn (6)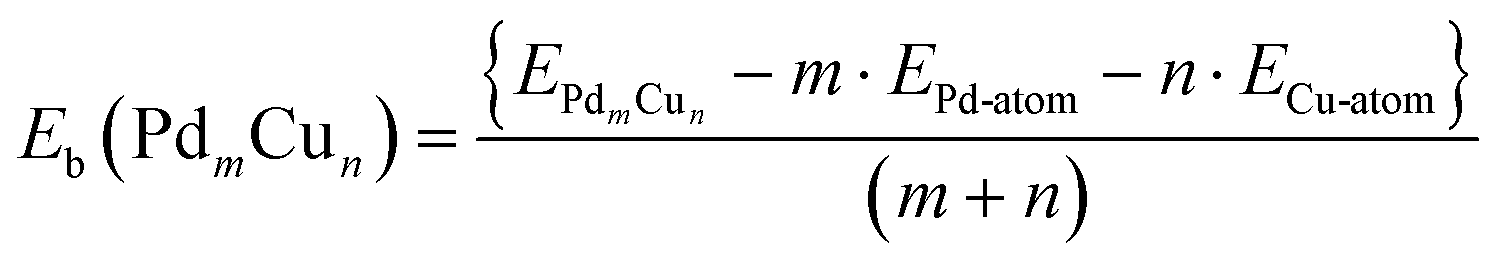 | (6) |
The adsorption energies of adsorbate on the PdmCun (m + n = 4, 8 and 13) clusters, Eads have been calculated by eqn (7)
| Eads = Eadsorbate+cluster − (Eadsorbate + Ecluster) | (7) |
Conclusion
In summary, CO2 adsorption and hydrogenation on PdmCun (m + n = 4, 8 and 13) clusters have been studied using DFT calculations. The stable adsorbed structures of the possible intermediates like adsorbed H2 molecules, one H atom, two H atoms, CO2 molecule, CO2 molecules with one H atom etc. have been evaluated and the energetics associated with those have been calculated. It has been found that CO2 got adsorbed on these clusters in an inverted V-shaped fashion and CO2 hydrogenation occurs through a formate intermediate. The first hydrogenation to formate goes through a unidentate structure which later transforms to a more stable bidentate structure easily. The second hydrogenation from formate to formic acid is energetically less difficult. An investigation of the possible pathways of forming formic acid on PdmCun (m + n = 4, 8 and 13) clusters is reported. The reaction energies for the CO2 hydrogenation to formic acid on all of the clusters have also been calculated. The nature of chemical bonding present between the adsorbate and adsorbent was investigated by electronic structure analysis. The calculated results provide a potential candidate for formic acid synthesis and the present study can be useful for CO2 conversion and utilization with Pd–Cu bimetallic catalysts.Data availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to ethical restrictions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Dr S. Kannan, Director, Radiochemistry and Isotope Group, Bhabha Atomic Research Centre (BARC) and Dr S. C. Parida, Head, Product Development Division, BARC for their interest and encouragement during progress of this work. The authors are also thankful to the members of the Computer Division, BARC for their kind cooperation during the work.References
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- Y. Yang, M. G. White and P. Liu, J. Phys. Chem. C, 2011, 116, 248–256 CrossRef.
- Y. F. Zhao, Y. Yang, C. Mims, C. H. F. Peden, J. Li and D. Mei, J. Catal., 2011, 281, 199–211 CrossRef CAS.
- Y. Yang, J. Evans, J. A. Rodriguez, M. G. White and P. Liu, Phys. Chem. Chem. Phys., 2010, 12, 9909–9917 RSC.
- W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207–2221 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- M. Zhang, M. Dou and Y. Yu, Appl. Surf. Sci., 2019, 433, 780–789 CrossRef.
- J. Yoshihara, S. Parker, A. Schafer and C. T. Campbell, Catal. Lett., 1995, 31, 313–324 CrossRef CAS.
- M. D. Porosoff and J. G. Chen, J. Catal., 2013, 301, 30–37 CrossRef CAS.
- L. Liu, H. Yao, Z. Jiang and T. Fang, Appl. Surf. Sci., 2018, 451, 333–345 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- Y. Tan, W. Nookuea and H. Li, Energy Covers Manage, 2016, 118, 204–222 CrossRef CAS.
- M. P. Fortes, A. B. Dumitriu and E. T. Zimas, Energy Procedia, 2014, 63, 7968–7975 CrossRef.
- S. T. Yong, C. W. Ooi, S. P. Chai and X. S. Wu, Int. J. Hydrogen Energy, 2013, 38, 9541–9552 CrossRef CAS.
- M. P. Fortes, J. C. Schoneberger, A. Boulamanti, G. Harrison and E. Tzimas, Int. J. Hydrogen Energy, 2016, 41(37), 16444–16462 CrossRef.
- A. Alvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef CAS.
- J. Klankermayer and W. Leitner, Philos. Trans. R. Soc., A, 2016, 374, 20150315 CrossRef PubMed.
- X. Jiang, Y. Jiao, C. Moran, X. Nie, Y. Gong, X. Guo, K. S. Walton and C. Song, Catal. Commun., 2019, 118, 10–14 CrossRef CAS.
- L. Liu, F. Fan, M. Bai, F. Xue, X. Ma, Z. Jiang and T. Fang, Mol. Catal., 2019, 466, 26–36 CrossRef CAS.
- Y. Yang, M. G. White and P. Liu, J. Chem. C, 2012, 116, 248–256 CAS.
- C. L. Chiang, K. S. Lin, H. W. Chuang and C. M. Wu, Int. J. Hydrogen Energy, 2017, 42, 23647–23663 CrossRef CAS.
- I. Melian-Cabrera, M. L. Granados and J. L. G. Fierro, Catal. Lett., 2002, 79, 165–169 CrossRef CAS.
- D. Heyl, U. Rodemerck and U. Bentrup, ACS Catal., 2016, 6, 6275–6284 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- C. Hao, S. Wang, M. Li, L. Kang and X. Ma, Catal. Today, 2011, 160, 184–190 CrossRef CAS.
- D. N. Sredojevic, Z. Sljivancanin, E. N. Brothers and M. R. Belic, ChemistrySelect, 2018, 3, 2631–2637 CrossRef CAS.
- G. Peng, S. J. Sibener, G. C. Schatz, S. T. Ceyer and M. Mavrikakis, J. Phy Chem C, 2012, 116, 3001–3006 CrossRef CAS.
- L. Gong, J. J. Chen and Y. Mu, Phys. Chem. Chem. Phys., 2017, 19, 28344–28353 RSC.
- G. Liu, P. Poths, X. Zhang, Z. Zhu, M. Marshall, M. Blankenhorn, A. N. Alexandrova and K. H. Bowen, J. Am. Chem. Soc., 2020, 142, 7930–7936 CrossRef CAS PubMed.
- N. Kumar, D. Chattaraj, P. Ghosh and C. Majumder, J. Phys. Chem. C, 2018, 122, 12920–12933 CrossRef CAS.
- I. Shim and K. Gingerich, J. Chem. Phys., 1984, 80, 5107–5119 CrossRef CAS.
- K. Huber and G. Herzberg, Molecular Spectra and Molecular Structure. IV. Constants of Diatomic Molecules, Van Nostrand Reinhold Company, New York, 1979 Search PubMed.
- M. Mors, Advances in Metal and Semiconductor Clusters, JAI Press, Greenwich, CT, 1993 Search PubMed.
- R. Ram, C. Jarman and P. Bernath, J. Mol. Spectrosc., 1992, 156, 468–486 CrossRef CAS.
- H. Kahnouji, H. Najafvandzadeh, S. Javad Hashemifar, M. Alaei and H. Akbarzadeh, Chem. Phys. Lett., 2015, 630, 101–105 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS.
- A. Mravak, S. Vajda and V. Bonacic-Kuutecky, J. Phys. C., 2022, 126, 18306–18312 CAS.
- C. Goswami, H. Saikia, B. J. Borah, M. J. Kalita, K. Tada, S. Tanaka and P. Bharali, J. Colloid Interface Sci., 2021, 587, 446–456 CrossRef CAS.
- H. Koga, A. Hayashi, Y. Ato, K. Tada, S. Tanaka and M. Okumura, Catal. Today, 2019, 331, 236–244 CrossRef.
- S. Maheswari, S. Karthikeyanm, P. Murugan, P. Sridhar and S. Pitchumani, Phys. Chem. Chem. Phys., 2012, 14, 9683–9695 RSC.
- H. Li, L. Li, A. Pederson, Y. Gao, N. Khetrapal, H. Jónsson and X. C. Zeng, Nano Lett., 2015, 15, 682–688 CrossRef CAS.
- R. Pal, A. Poddar and P. K. Chattaraj, Front. Chem., 2021, 9, 730548 CrossRef CAS PubMed.
- R. Varns and P. Strange, Phys. Status Solidi B, 2012, 249(11), 2179–2189 CrossRef CAS.
- Y. Rao, Y. Lei, X. Cui, Z. Lin and F. Chen, J. Alloys Compd., 2013, 563, 50–55 CrossRef.
- Y. Li, S. H. Chan and Q. Sun, Nanoscale, 2015, 7, 8663–8683 RSC.
- S. Kattel, P. J. Ramirez, J. G. Chen, J. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- W. Kohn and L. Sham, Phys. Rev. A, 1965, 140, 1133–1138 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- S. Bhattacharya, S. Levchenko, L. Ghiringhelli and M. Scheffler, Phys. Rev. Lett., 2013, 111(135501), 1–5 Search PubMed.
- S. Bhattacharya, S. Levchenko, L. Ghiringhelli and M. Scheffler, New J. Phys., 2014, 16, 123016 CrossRef (1–34).
- R. P. Feynman, Phys. Rev., 1939, 56, 340–343 CrossRef CAS.
- H. Hellman, Introduction to Quantum Chemistry, Deuticke, Leipzig, 1937 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef.
- D. D. Koelling and B. N. Harmon, J. Phys. C: Solid State Phys., 1977, 10, 3107–3114 CrossRef CAS.
- T. Takeda, Z. Phys. B: Condens. Matter Quanta, 1978, 32, 43–48 CrossRef CAS.
- M. J. Piotrowski, P. Piquini and J. L. F. Da Silva, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155446 CrossRef.
- O. D. Häberlen, S.-C. Chung, M. Stener and N. Rösch, J. Chem. Phys., 1997, 106, 5189–5201 CrossRef.
- M. N. Huda, M. K. Niranjan, B. R. Sahu and L. Kleinman, Phys. Rev. A: At., Mol., Opt. Phys., 2006, 73, 053201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: See supplementary Tables S1 and S2 (ESI†) for binding energies and bond lengths of different adsorbates adsorbed on Pd4Cu4 and Pd12Cu clusters, respectively; Tables S3–S5 for the different geometric isomers of PdmCun (m + n = 4, 8 and 13) clusters and Tables S6–S8 for the charge density distribution of PdmCun (m + n = 4, 8 and 13) clusters. See supplementary Fig. S1 and S2 for the lowest energy structure of Pd4Cu4 and Pd12Cu clusters along with different adsorbates on those clusters, respectively; S3 and S4 for the potential energy profile of CO2 hydrogenation to formic acid on Pd4Cu4 and Pd12Cu clusters, respectively; S5 and S6 for the density of states of Pd4Cu4 and Pd12Cu clusters along with their CO2 adsorbed counterparts, respectively. See DOI: https://doi.org/10.1039/d2cp03805f |
| This journal is © the Owner Societies 2023 |