
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecularly imprinted materials for glycan recognition and processing
Yan
Zhao

Department of Chemistry, Iowa State University, Ames, Iowa 50011-3111, USA. E-mail: zhaoy@iastate.edu; Tel: +1-515-294-5845
First published on 25th April 2022
Abstract
Carbohydrates are the most abundant organic molecules on Earth and glycosylation is the most common posttranslational modification of proteins. Glycans are involved in a plethora of biological processes including cell adhesion, bacterial and viral infection, inflammation, and cancer development. Coincidently, glycosides were some of the earliest molecules imprinted and have been instrumental in the development of covalent molecular imprinting technology. This perspective illustrates recently developed molecularly imprinted materials for glycan binding and processing. Novel imprinting techniques and postmodification led to development of synthetic glycan-binding materials capable of competing with natural lectins in affinity and artificial glycosidases for selective hydrolysis of complex glycans. These materials are expected to significantly advance glycochemistry, glycobiology, and related areas such as biomass conversion.
 Yan Zhao | Yan Zhao received his BS in chemistry from Lanzhou University in 1992 and PhD from Northwestern University in 1996. After a postdoctoral stay at the University of Illinois, he worked for the Procter & Gamble Company from 1998 to 2002 and is currently a professor of chemistry at Iowa State University. His areas of interest include design and synthesis of biomimetic molecules, polymers, and nanomaterials and using them as synthetic receptors, artificial enzymes, molecular transporters, and sensors for chemical and biological applications. |
Molecular recognition of biological glycans
As the most abundant organic molecules on Earth, carbohydrates serve diverse functions in biology including structural support (e.g., cellulose), energy storage (e.g., starch and glycogen), and mediation of biological processes.1–5 At least 50% of all proteins are glycosylated and every cell is covered with a dense layer of glycans. Not surprisingly, biomolecular recognition of glycans plays vital roles in both the physiological and pathophysiological states of cells. Glycan binding is involved in numerous biological processes including cell–cell interactions, cell trafficking, fertilization, and transmembrane signaling. Bacterial and viral pathogens frequently use glycan-binding proteins (GBPs) to recognize host glycans for attachment and infection, and eukaryotic organisms employ their own GBPs to counter these attacks by detecting and responding to the exogenous glycans on the pathogens.6The importance and ubiquity of glycans in biology make synthetic glycan-binding materials (GBMs) an extremely important class of biomaterials.7 They have a wide range of applications in analytical and functional glycomics, purification of biological and synthetic glycans, interrogation and intervention of glycan-mediated biological processes, and antiviral treatment, to name a few. Nonetheless, development of GBMs capable of binding complex glycans with biologically competitive affinities faces a number of unique challenges.
First, of the 10 common monosaccharide building blocks of biological glycans, many differ by the stereochemistry of a single chiral center—e.g., galactose (Gal) and mannose (Man) from glucose (Glu), and N-acetylglucosamine (GlcNAc) from N-acetylgalactosamine (GalNAc). Not only so, these building blocks can be connected through different hydroxyl groups, by either α or β glycosidic linkages. The subtle structural differences of the resulting constructs make their distinction a daunting task.8
Second, because carbohydrates are strongly solvated in water, their molecular recognition needs to pay a considerable desolvation penalty. Even biological GBPs tend to bind their ligands with relatively low affinities, often in the millimolar-to-micromolar range.6 In contrast, protein–protein interactions generally have dissociation constants (Kd) on the order of nanomolar or even picomolar.9
Third, glycan determinants, the functional units on glycans recognized by their GBPs, typically consist of 2–6 linear monosaccharides plus side chains.10 Sometimes, multiple determinants are present in a single glycan, as shown by 1 in Fig. 1. With the advancement of supramolecular chemistry over the last decades, an innumerable number of synthetic receptors have been prepared for all kinds of guest molecules11 and some impressive receptors for sugars have been reported.12,13 However, building a complementary receptor for complex guest molecules such as 1 or 2 remains difficult.
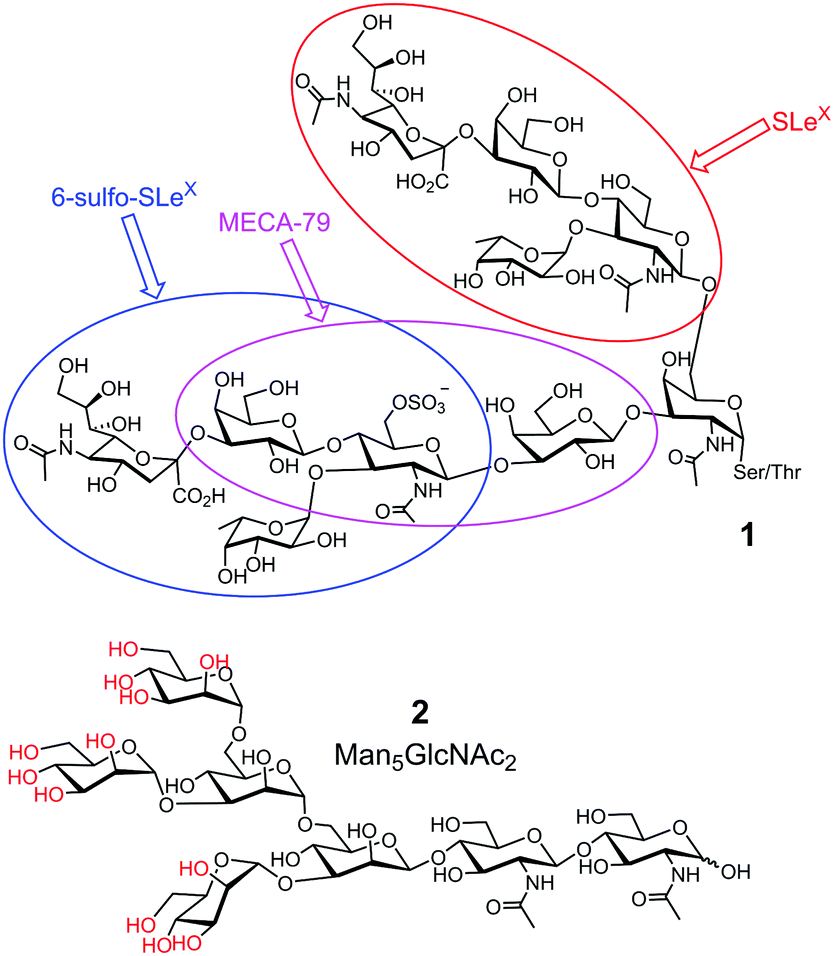 | ||
| Fig. 1 Representative biological glycans, with glycan determinants shown in different-colored ovals in 1. The hydroxyls of OVA glycan 2 that can potentially form boronate esters are colored red. | ||
Fourth, biological glycans tend to be microheterogeneous. For example, avian ovalbumin (OVA) has a single N-glycan (on Asn-292) that is characterized by its high mannose content. However, in addition to the most abundant Man5GlcNAc2 (2 in Fig. 1) and Man6GlcNAc2, a variety of other structures are present with varying numbers of Man and additional GlcNAc.14 Since many glycoproteins are glycosylated at multiple sites, microheterogeneity makes the overall structures of the glycans extremely complex. Yet, structural heterogeneity is a general rule in glycobiology, needed for precise regulation and diversification of functions.1
To bind a guest molecule strongly, a host/receptor needs to be preorganized for binding, with binding groups complementary to those on the guest.15,16 The binding interface ideally is poorly solvated prior to the guest binding. Preorganization reduces costs from loss of conformational entropy during binding and poor solvation prior to binding lowers the desolvation penalty. Although these supramolecular principles are straightforward and have been used repeatedly by chemists in the construction of all sorts of synthetic receptors,11 implementing them on subtly different, strongly solvated, complex, and microheterogeneous biological glycans is a challenge on a totally different level.
Nature, nonetheless, can teach us lessons about how to make good receptors for this unique class of molecules. Burkholderia oklahomensis EO147 agglutinin (BOA) binds high-mannose glycans such as 3α,6α-mannopentaose with Kd ≈ 50 μM in water.17 Its crystal structure shows two β-barrel-like domains with four binding sites near the flexible loops and turns connecting the β-strands of the barrels (Fig. 2a). To bind a ligand that has an abundance of hydroxyl groups, the lectin expectedly employs an extensive array of hydrogen bonds. Interestingly, even though some hydrogen bonds are formed directly between the guest and the host (e.g., with G132 or G19), even more are mediated by solvent (water molecules represented by isolated red spheres in Fig. 2b and c). As shown in Fig. 2c, the lectin also uses the indole side chain of W18 to interact with a nonpolar region of the glycan. Thus, despite the overall hydrophilic nature of the glycan guest, hydrophobic interactions may be employed to supplement hydrogen-bonding interactions. Not only deliberate hydrophobic–hydrophobic contact can be useful, but also release of water molecules from the polyamphiphilic surface of both the receptor and its glycan ligand. A polyamphiphilic surface (i.e., a molecular surface made up of many small amphiphilic parts) is known to perturb the water molecules in its solvent shell and increase their free energy.18 Although binding between two complementary hydrophilic surfaces19 is not considered a hydrophobic effect,20–22 release of high energy water molecules is a common feature and provides important driving forces in both cases.
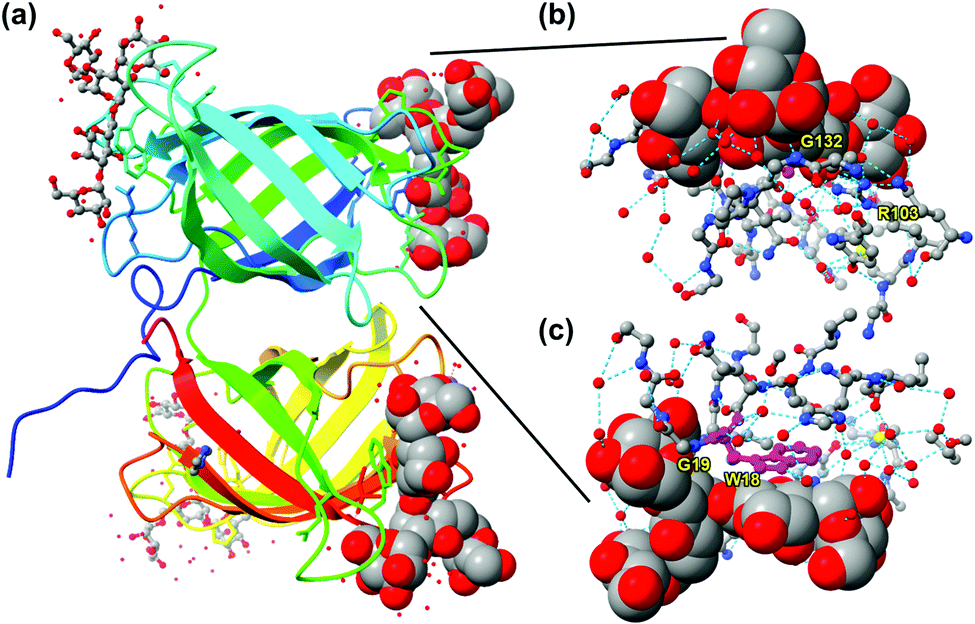 | ||
| Fig. 2 (a) Crystal structure of BOA (PDB ID: 4gk9), with the peptide chain colored from blue (N-terminus) through the rainbow spectrum to red (C-terminus). Four molecules of 3α,6α-mannopentaose are bound by the lectin and the glycans bound close to the N- and C-terminus are highlighted with sphere models. (b and c) The glycan bound near the N-terminus viewed from two different angles, with the residues at the binding interface shown. Molecular graphics was created using UCSF Chimera. Hydrogen bonds are shown by dotted cyan lines. Tryptophan 18 (W18) is colored in magenta. | ||
The main challenge in the molecular recognition of glycans is thus in the construction of a preorganized, complementary binding interface for these polyhydroxylated compounds, even if the interface is relatively hydrophilic. In traditional supramolecular chemistry, such a host is generally built through step-by-step total synthesis using conformationally constrained systems such as macrocycles.11 However, doing so for complex biological glycans such as those shown in Fig. 1 is quite unimaginable, as the host is generally more complex and larger than the guest and needs to be made water-soluble.
Macroporous sugar-binding MIPs by Wulff
Molecular imprinting provides a completely different approach toward the construction of receptors. Instead of building the host and then fit the guest into it, it simply builds the binding interface around the guest using the latter as a template (T).23–25 Wulff pioneered the concept of covalent imprinting and, interestingly, (aryl) glycosides were some of his earliest template molecules used to demonstrate the concept.26 In a series of papers published in the late 1970s, he described molecular imprinting of 4-nitrophenyl α-D-mannopyranoside (3).27–29 As shown in Fig. 3, 4-vinylphenylboronic acid (4) as the functional monomer (FM) forms boronate ester 5, which undergoes free radical polymerization with a large amount of a cross-linker. An inert solvent is also present as the porogen to form a macroporous polymer network with a high internal surface. The resulting molecularly imprinted polymer (MIP) has the template molecules embedded in the network. Removal of the templates vacates the imprinted binding sites, with the boronic acids turned into binding groups that can interact with the appropriate diols through fast and reversible boronate bond formation.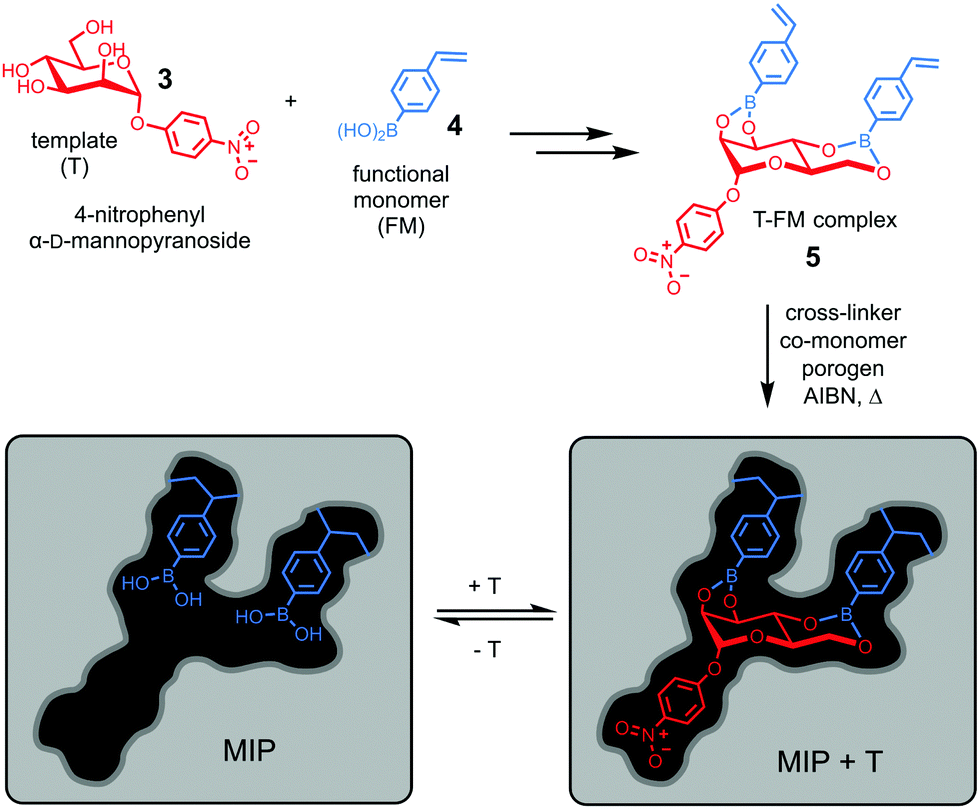 | ||
| Fig. 3 Covalent imprinting of 4-nitrophenyl α-D-mannopyranoside using 4-vinylphenylboronic acid as the FM to afford a molecularly imprinted polymer (MIP) with an imprinted binding site depicted schematically. | ||
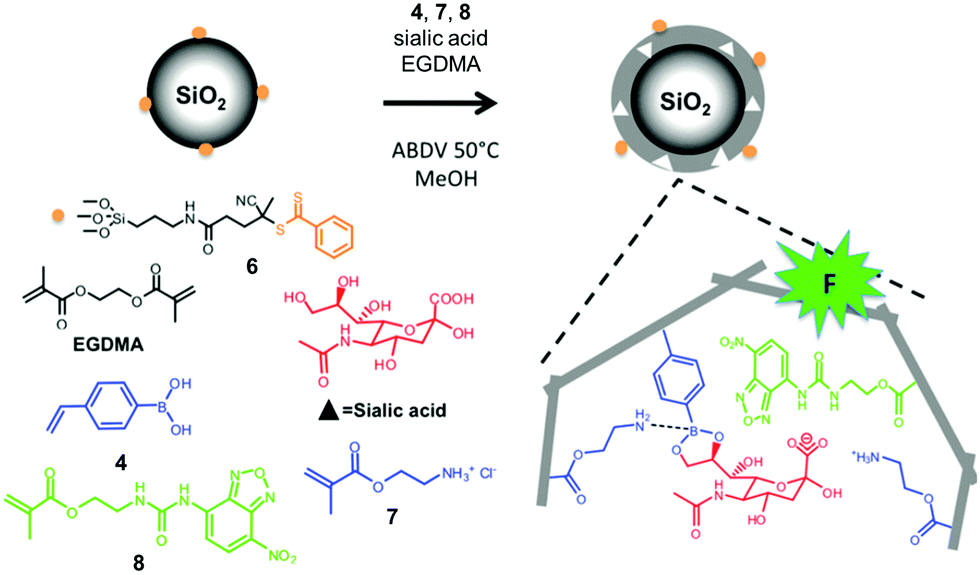 | ||
| Fig. 4 Preparation of an SA-imprinted shell on silica core particles. (Adapted with permission from ref. 34. Copyright 2015, the American Chemical Society.) | ||
The technique is remarkable from a supramolecular perspective. As demonstrated in BOA, nature achieves preorganization (of the host) and complementarity (to the guest) by the folding of a functionalized peptide chain, through her unparalleled abilities of conformational control. In contrast, preorganization of the binding site and guest-complementarity in molecular imprinting are achieved “automatically” through covalent capture of the T–FM complex by polymerization/cross-linking. Although the guest (4-nitrophenyl α-D-mannopyranoside) is far simpler than 3α,6α-mannopentaose of BOA and the binding sites are scattered within an insoluble polymer network, both the distance and the orientation of the boronic acid binding groups are optimized in a single step for the rebinding of the original template.
In these and other follow-up studies,27–31 Wulff and colleagues studied multiple parameters important to the imprinting process, including the types of cross-linker and FM, amount of cross-linker that dictates the swellability of MIP and thus the integrity of the imprinted sites, and porogen which strongly influences the morphology of MIP and accessibility of the imprinted sites. Even though only simple glycosides27–31 and small sugars32 were used, these studies not only laid much of the foundation for molecular imprinting but also greatly influenced the field of carbohydrate recognition in the decades to come.
Biocompatible carbohydrate-binding MIPs
Conventional MIPs are insoluble thermosets and their hydrophobicity makes them unsuitable for aqueous-based biological applications. Because the imprinted sites are distributed within a highly cross-linked polymer, their accessibility to guests is another issue. To improve the performance of glycan-binding MIPs, researchers developed novel methods of imprinting to overcome these and other challenges associated with traditional MIPs.One way to increase the accessibility of binding sites is to perform the imprinting on a surface. Shinkai and co-workers imprinted a boronic-acid-appended polylysine on a gold surface.33 CD spectroscopy confirmed that the template molecules (glucose or fructose) induced conformational changes of the polymer. One interesting feature of their GBM is that cross-linking of polylysine (and thus the molecular imprinting) was accomplished through multiple thiolate–gold bonds formed on the metal surface. Meanwhile, the overall low cross-linking density afforded a relatively low imprinting factor (IF < 2) and low selectivity between the sugars studied.
Sialic acid (SA) or N-acetylneuraminic acid (NANA), frequently found at the nonreducing ends of cell surface glycans, is known to be overexpressed on cancer cells. Sellergren and co-workers developed a multipronged strategy to prepare GBMs capable of detecting SA expression levels of cells (Fig. 4).34,35 To ensure good accessibility of the binding sites, they used surface-anchored RAFT initiators to form a 10–20 nm thick MIP shell on silica particles ∼200 nm in size. The SA template was bound by a combination of amine-stabilized boronate bonds and electrostatic interactions between SA and FM 7. A urea-derived co-monomer 8 was also included in the formulation, not only as a functional monomer to hydrogen-bond with the template, but also as a fluorescent probe for imaging the glycans on the cell surface. By including two FMs for cooperative interactions with the template, the researchers achieved a submillimolar binding affinity toward SA (Kd ≈ 170 μM in water with 2% methanol). In addition, the GBM had multiple SA-binding sites in the shell, to allow it to bind multiple glycans on the cell surface simultaneously. This strategy is biomimetic, since multivalency is a common feature in glycan-mediated biomolecular recognition to boost the effectiveness of (relatively weak) individual glycan–GBP interactions.6
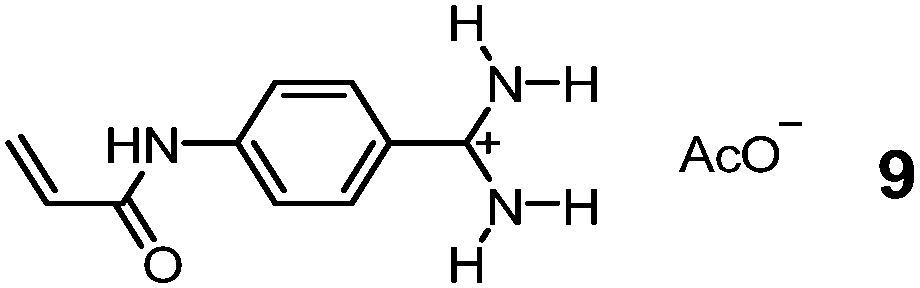
Although chemists cannot compete with nature in the synthesis and manipulation of complex molecular structures, they have the flexibility of using nonbiological materials for binding and can engineer additional functions to their GBMs for different purposes. Bui, Haupt, and colleagues took advantage of the tunable luminescence of quantum dots (QDs) and turned them into multiplexing probes for cell targeting and imaging by coating them with a MIP layer imprinted against glucuronic acid (GlcA) or SA.36 A polymerizable benzamidine FM 9 was key to the imprinting, as it bound the carboxylate of the template in water by a strong, hydrogen-bond-enforced salt bridge. The QD/MIP core–shell particles bound GlcA and SA with a Kd of 196 and 65 μM in water, respectively.37 Given only noncovalent interactions were involved in the guest binding, the affinity is impressive, on par with those of natural lectins for monosaccharides.6 These core–shell MIP particles were ∼125 nm in size, small enough to target both intracellular and pericellular terminal glycans. Even higher precision can be achieved using carbon nanodots (∼3.2 nm) as the core.38
One drawback of MIP is the heterogeneity of its imprinted sites. Especially when noncovalent binding interactions are involved, both strong- and weak-binding sites are present in the final materials and often only a small fraction of the binding sites have high binding affinities. Piletsky and co-workers came up with an ingenious method to immobilize the template molecules on the surface of glass beads to enable solid-phase synthesis of MIP nanoparticles.39,40 This allows removal of low affinity nanoparticles by simple elution, with the functionalized glass beads as the stationary phase. Moreover, the surface-anchored templates can be reused and the final MIPs have remarkable binding properties—e.g., nanomolar affinities for small molecule guests such as vancomycin.
Hyaluronic acid (HA) is a key biomarker of certain cancers but their non-immunogenicity makes it difficult to raise antibodies for them. The Kaupt group used solid-phase synthesis to prepare fluorescent MIP nanoparticles ∼70 nm in size.41 Azide-functionalized D-glucuronic acid (GlcA) 10 was clicked onto alkyne-terminated glass beads (Fig. 5a). The polymerization mixture comprised N-isopropylacrylamide (NIPAM) as a hydrogen-bonding monomer and N,N′-ethylenebis(acrylamide) (EbAm) as the cross-linker, together with the strongly binding benzamidine FM 9. The resulting MIP nanoparticles displayed excellent binding properties in water for GlcA (Ka ≈ 0.8 μM) while nonimprinted polymers (NIPs) showed negligible binding. The MIPs also had excellent selectivities, with <1% cross-reactivity for monosaccharides such as Glu, Gal, GlcNAc, GalNAc, and SA. Confocal fluorescence microscopy showed that these MIPs could label pericellular and intracellular HAs, even within the nucleus (Fig. 5b). Labeling of intracellular HAs is particularly challenging, as they tend to be masked by other HA-binding molecules present in the cell. The staining compares well with that by the natural receptor (i.e., streptavidin–HABP/FITC biotin), as shown in Fig. 5c.
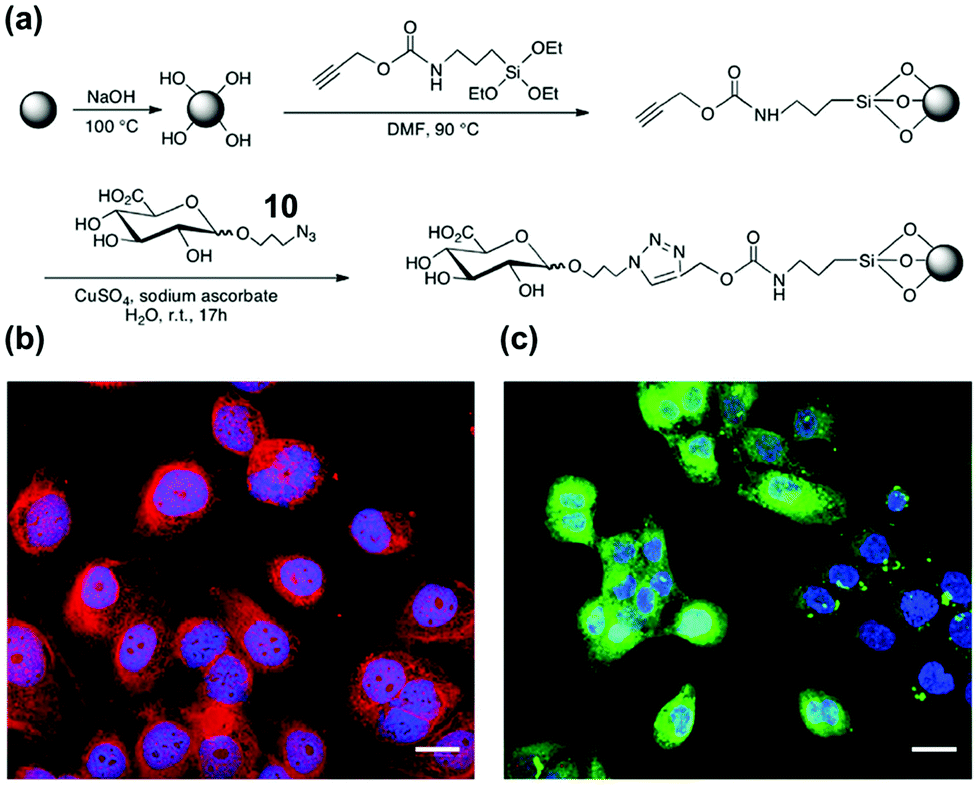 | ||
| Fig. 5 (a) Functionalization of glass beads with GlcA templates by the click reaction. (b and c) Confocal images of fixed human keratinocytes showing extracellular, intracellular, and nuclear labeling by MIPGlcA-NPs (red) and by HABP/streptavidin FITC (green). The nuclei are stained blue with Hoechst. Scale bar = 20 μm. (Adapted with permission from ref. 41. Copyright 2019, the Nature Publishing Group.) | ||
Many glycoproteins have similar (but subtly different) glycans. Thus, to achieve high selectivity in binding, a GBM not only needs to recognize the glycans but also the protein structure of the glycoprotein. The Liu group at Nanjing University developed an innovative method of oriented surface imprinting. As shown in Fig. 6a, a substrate such as a glass slide was first functionalized with boronic acid, which allowed covalent anchoring of glycoproteins on the surface.42 Instead of traditional vinyl monomers and cross-linkers, the Liu group employed dopamine and m-aminophenylboronic acid (APBA) capable of dehydration polymerization for the imprinting. The imprinted GBM afforded an outstanding Kd of 1.2 nM at pH 7.4 for horse radish peroxidase (HRP). Importantly, the binding affinity could be tuned down by nearly 100-fold, by simply adjusting the pH to 3.0, so selective capture of a glycoprotein and release was possible. The method is general, as they could also perform the imprinting on the internal pores of monolithic capillary to afford affinity columns for specific glycoproteins directly.
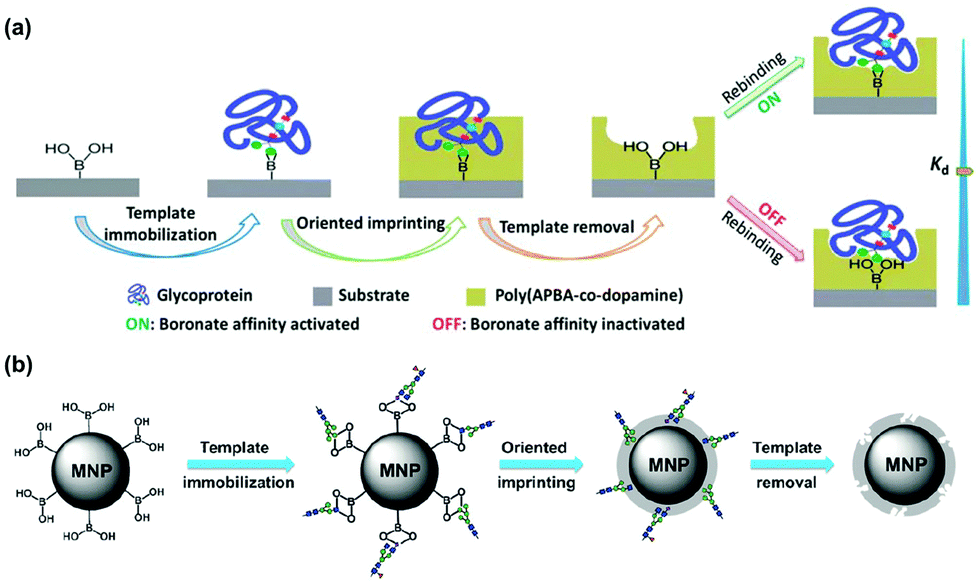 | ||
| Fig. 6 (a) Boronate affinity-based controllable oriented surface imprinting of glycoproteins. (Reproduced with permission from ref. 42. Copyright 2014, the Royal Society of Chemistry.) (b) Boronate-affinity glycan-oriented surface imprinting for producing glycan-imprinted MNPs. (Reproduced with permission from ref. 43. Copyright 2015, John Wiley & Sons, Inc.) | ||
One remarkable feature of the oriented imprinting method is the accurate control of the MIP thickness, at a rate of 3.5 ± 0.4 nm h−1 on the glass slide. This allowed the researchers to finetune the thickness of the MIP layer for protein binding (typically the thickness is kept in the range of 1/3 to 2/3 of the protein dimension). Simultaneous binding of the glycan and the protein probably was the major reason for the extraordinary binding affinity, as GBM imprinted against cleaved glycans displayed a substantially weaker binding (Kd ≈ 25 μM) toward RNaseB (Fig. 6b).43 Other factors that might have influenced the affinity include different protein targets, different materials used for the MIP layer, and different curvatures of the substrates. The authors have thoroughly optimized the imprinting method.44 The generality of the method makes these GBMs highly versatile, useful in many applications including affinity-based separation, disease diagnosis, targeting and imaging of cancer cells, and single-cell analysis.45–49
Further miniaturization of glycan-binding MIPs
The dimension of a nanomaterial is a key parameter to influence its property. Kaupt and co-workers found the size of their glycan-binding nanoparticles critical to their performance. Whereas 70 nm MIP particles stained both extra- and intracellular targets, larger (400 nm) nanoparticles prepared through precipitation polymerization displayed lower affinity for HA, targeted only extracellular glycans, and agglomerated easily in aqueous solution.41Micelles are dynamic assemblies of surfactants typically formed in aqueous solution. Ionic surfactants with a single hydrocarbon chain tend to form small spherical micelles several nanometers in size.50 By performing molecular imprinting in surfactant micelles, the Zhao group was able to reduce the size of MIPs dramatically. The so-called MINPs (molecularly imprinted nanoparticles) average ∼5 nm in diameter and are soluble in water.51 Their nanodimension, water-solubility, hydrophilic exterior, and hydrophobic interior make them great mimics of water-soluble proteins and enzymes for a variety of guests including drug molecules and peptides.52–55
Fig. 7a shows the general method of micellar imprinting. The template molecules are first solubilized by mixed micelles of an alkyne-functionalized cross-linkable surfactant such as 11a, together with divinylbenzene (DVB) and 2,2-dimethoxy-2-phenylacetophenone (DMPA, a photoinitiator). The micellar surface has a layer of terminal alkynes, enabling the micelles to be readily cross-linked by diazide 12via the highly efficient Cu(I)-catalyzed click reaction. A second round of click reaction installs a layer of hydrophilic ligands (13) on the micelle surface. UV irradiation initiates free-radical polymerization/cross-linking in the core, among DVB and the methacrylate of the surfactants around the template molecule. The templates are removed by precipitation of the MINPs from acetone and washing with organic solvents.51 Because each cross-linked micelle contains approximately 50 surfactant molecules, a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 surfactant/template ratio gives MINPs with an average of one imprinted site per particle and 25:1 gives two imprinted sites.51 MINPs are cationic if prepared from cross-linkable surfactants 11a–c but can be made anionic56 or zwitterionic54 if other surfactants such as 11d or 11e are used instead.
:1 surfactant/template ratio gives MINPs with an average of one imprinted site per particle and 25:1 gives two imprinted sites.51 MINPs are cationic if prepared from cross-linkable surfactants 11a–c but can be made anionic56 or zwitterionic54 if other surfactants such as 11d or 11e are used instead.
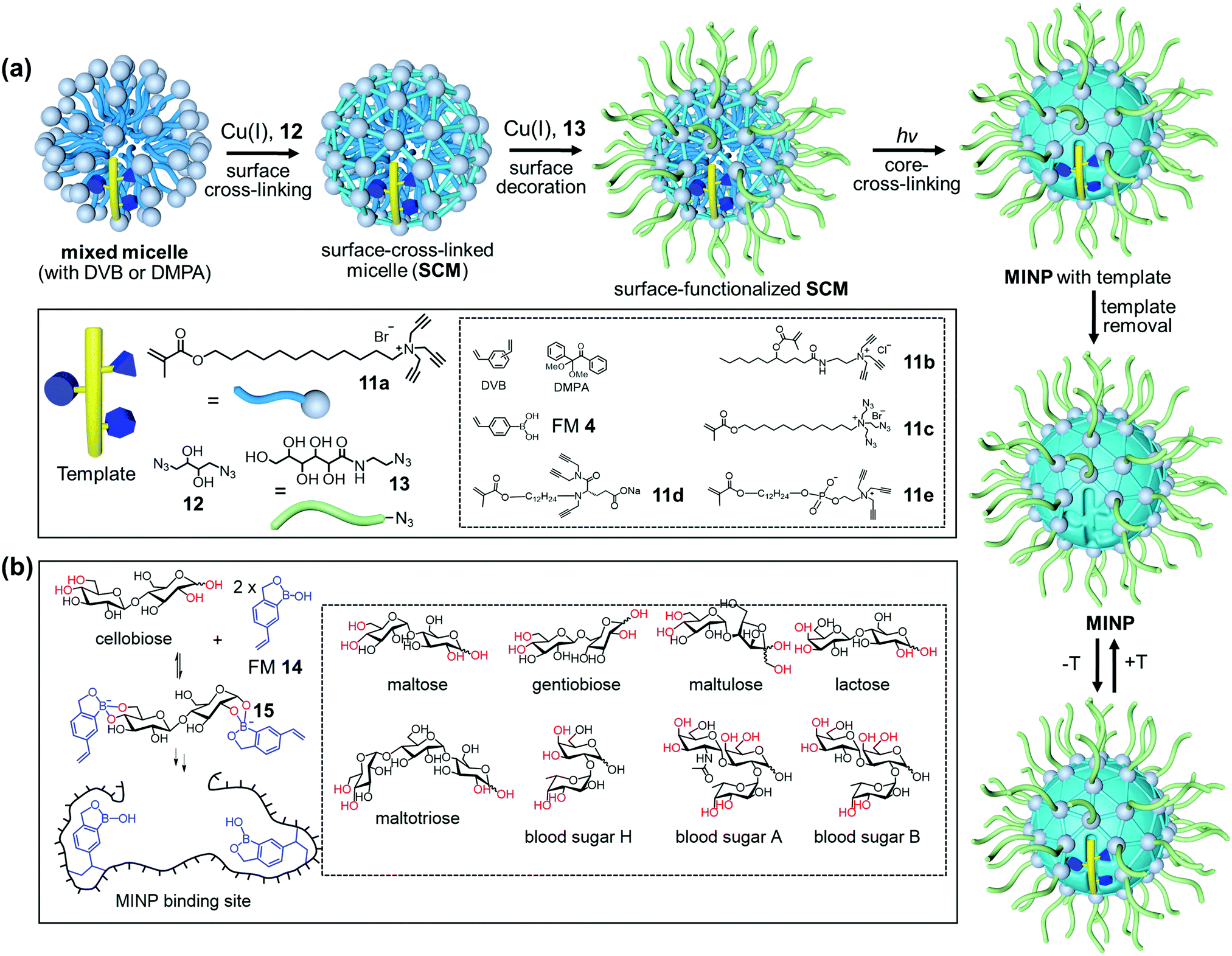 | ||
| Fig. 7 (a) Micellar imprinting. (b) MINP with boroxole binding group in the imprinted site, with the boronate-forming hydroxyls colored red. (Adapted with permission from ref. 55. Copyright 2021, the American Chemical Society.) | ||
Both covalent and noncovalent interactions may serve to bind glycans in micellar imprinting. For covalent imprinting, a boronate ester is first formed from a monosaccharide and FM 4 (4-vinylphenylboronic acid). Molecular imprinting fixes the positions of the boronic acid binding groups in the imprinted site. MINPs prepared with glucose, mannose, and galactose bind their templates with millimolar affinity in water. The inversion of a single hydroxyl in these sugars is distinguished by over >100:1 selectivity.57 The interactions of boronic acid with diols tend to be sensitive to solution pH,58,59 but the hydrophobic pocket of MINP dampens the effect, with the binding constant (Ka) changing by ∼2-fold over pH 6.5–8.5. If the sugar has a hydrophobic aglycon, the binding is stronger due to the added hydrophobic interactions.
Oligosaccharides have poor solubilities in common organic solvents, making it difficult to synthesize their corresponding boronate esters from FM 4. Vinylbenzoboroxole 14, on the other hand, reacts with mono- and oligosaccharides in situ under the micellar imprinting conditions, to form amphiphilic, anionic boronate ester such as 15 (Fig. 7b), stabilized by the cationic micelle. The amide-containing 11b works particularly well with oligosaccharides, because the amide group can hydrogen bond with the T–FM complex 15. Combination of boronate binding and hydrogen bonds makes it possible for the MINPs to bind mono- and oligosaccharides with tens of micromolar affinities.60 Oligosaccharides are differentiated on the basis of their monosaccharide building blocks, glycosidic linkages, and chain length, which has not been possible previously with synthetic GBMs. Cellobiose and lactose differ by the stereochemistry of a single hydroxyl among eight. Yet, lactose shows a cross-reactivity ratio (CRR) of 0.04 toward MINP imprinted against cellobiose. The extremely similar blood sugars A and B are differentiated by a 2:1 to 3:1 ratio in the binding.
Benefits of the boroxole-functionalized MINPs include convenient in situ imprinting (i.e., without a separate preparation of the sugar–FM complex) and their predictable selectivity for binding, through the cis-1,2-diol, cis-3,4-diol, and trans-4,6-diol (colored red in Fig. 7b and in Fig. 1 for Man5GlcNAc2). As a result, both the binding locations and the potential binding selectivities can be inferred from the sugar structure directly.
Micellar imprinting allows direct imprinting of cleaved glycan mixtures from a glycoprotein, overcoming the microheterogeneity of biological glycans. Man5GlcNAc2 (2) and Man6GlcNAc2, the two major glycans from OVA,14 can interact with 6–8 boroxole FMs, respectively (Fig. 1). The fact that the binding constant peaks at an FM/template ratio of 8:1 suggests that boronate formation is highly efficient under the micellar conditions. The binding constant reaches 16.3 × 104 M−1 in 10 mM HEPES buffer (pH 7.4), more than half of that (Ka ≈ 30 × 104 M−1) by concanavalin A (Con A),61 a natural lectin for the same glycans. The biologically competitive binding affinity allows the MINPs to shield the OVA glycans from a broad-specificity α1-2,3,6 mannosidase,62 essentially as supramolecular “protective groups” in enzymatic reactions.55,63
Boronic acids or boroxoles thus can help MINP achieve biological affinities for complex glycans. Can hydrogen-bonds do the same? After all, boronate bonds are only possible for certain type of hydroxyls (colored red in Fig. 7b) and hydrogen bonds are the primary interactions involved in natural GBP–glycan bindings.
Hydrogen bonds are ineffective for molecular recognition in water directly, due to strong competition from the solvent. Nature solves the problem by keeping them in a relatively hydrophobic microenvironment and then they become effective for protein–protein interactions64 or glycan binding.19 Since hydrogen bonds are strengthened inside a micelle65 and at the surfactant/water interface,66 they can be very powerful in micellar imprinting.
One strategy to utilize hydrogen bonds for glycan binding is to prepare MINPs from the amide-containing surfactant 11b.60 An even simpler method is to include a commercially available amide-cross-linker, N,N′-methylenebisacrylamide (MBAm) in the MINP preparation. The radical initiator (DMPA) used in micellar imprinting is hydrophobic and insoluble in water, and thus has to reside within the micelle. Once the initiating radical reacts with the methacrylate of the cross-linkable surfactant, the propagating radical is confined in the micelle. Even though MBAm is completely soluble in water, it cannot polymerize until it reaches the micelle by diffusion. Its polymerization/cross-linking produces a belt of amide groups near the surfactant/water interface, hydrogen-bonded with the glycan guest (Fig. 8). These hydrogen bonds have been shown to enhance the binding of 4-nitrophenyl-α-D-glucopyranoside 16 by 180-fold, from Ka ≈ 600 M−1 by the DVB-only MINP to 1.1 × 105 M−1 by the DVB/MBAm-MINP.67 The corresponding manno- and galactosides are bound by the glucoside-imprinted MINP less strongly, by 3–5-fold. Thus, the MBAm-functionalized MINP has a lower selectivity in binding than the boroxole-functionalized one (∼25:1 selectivity for similar changes in guest structure),60 at least when the boronate-forming hydroxyl is inverted in the guest.
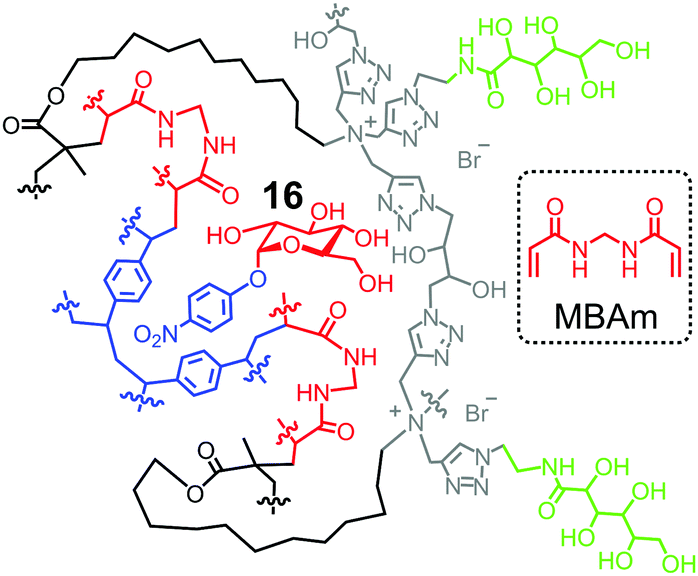 | ||
| Fig. 8 Schematic representation of MBAm-functionalized MINP to bind 4-nitrophenyl-α-D-glucopyranoside 16. | ||
Covalent and noncovalent interactions can be combined to strengthen the guest binding. When MBAm and boroxole FM 14 are both included in the formulation, MINP prepared using cleaved OVA glycans as templates binds the glycans with Ka = 64.2 × 104 M−1, thus twice as strongly as the natural lectin Con A.61 The MINP exhibits great selectivity for OVA, able to selectively extract the glycoprotein from a mixture of glycosylated and nonglycosylated proteins.67
Installation of acidic functionality for catalytic hydrolysis of glycans
One important feature of micellar imprinting is the strong imprinting effect, caused by the nanoconfinement of the imprinting process in the surface-cross-linked micelle.68 Extraordinary imprinting factors (IF) are obtained, up to 1700 for carbohydrates60,62,69 and 10000 for peptides.70 Not only so, the addition,68 removal,68 and shift71 of a single methyl (or methylene) group in the guest can be easily distinguished.
Templates 17a–c consist of a glycan and an aglycon containing a hydrolyzable imine bond. Micellar imprinting of 17a with boroxole FM 14 affords MINP(G1), whose imine is hydrolyzed in 6 M HCl at 95 °C. The aldehyde group in the imprinted site is then derivatized through reductive amination with 18a–h in DMF.72 As shown in Fig. 9a, MINP(G1) has an active site able to bind the terminal glucose of a glucose-containing oligo- or polysaccharide. Location of the active site near the micellar surface enables the unbound glucose in the substrate to reside in water, solvated properly. Most importantly, an acid group through the postmodification is placed right next to the glycosidic bond to be cleaved, with its position and acidity tuned systematically through the different amino acids used in the reductive amination. If the template contains a maltosyl or maltotriosyl, the resulting MINP(G2) and MINP(G3) will then have an active site for maltose (G2) and maltotriose (G3) instead of glucose (G1).
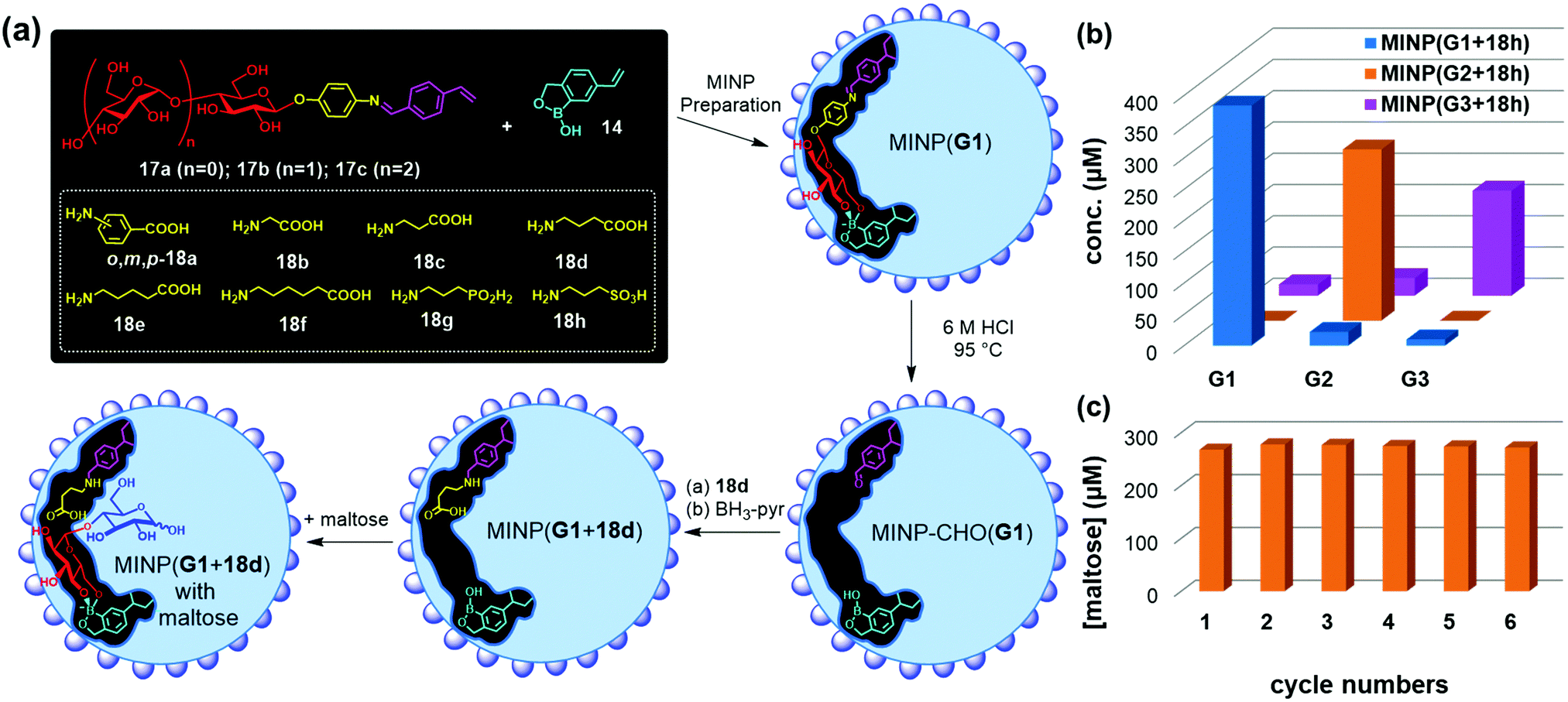 | ||
| Fig. 9 (a) Preparation of artificial glycosidase MINP(p-G1 + 18d) and its binding of maltose. (b) Product distribution (G1, G2, and G3) in the hydrolysis of amylose by the MINP catalysts after 24 h at 60 °C in H2O, with the reaction mixture (1.0 mL) dialyzed against 40 mL of deionized water using a membrane (MWCO = 500). [Amylose] = 1 mg mL−1, [MINP] = 20 μM. (c) Recyclability of MINP(G2 + 18h) in maltohexaose hydrolysis. [Maltohexaose] = 100 μM. [MINP] = 20 μM. (Adapted with permission from ref. 73. Copyright 2021, the Royal Society of Chemistry.) | ||
These MINPs indeed hydrolyze amylose with the designed selectivity in simply 60 °C water, without any buffer or other additives (Fig. 9b).73 Natural glucan 1,4-alpha-glucosidase removes one glucose residual at a time from the nonreducing end of amylose,74 and beta-amylase two glucose residues (i.e., maltose) at a time.75 The MINP-based glycosidases not only have duplicated the selectivities of these enzymes but also possess selectivity not available from natural biocatalysts—i.e., selective formation of maltotriose from a glucose-containing polymer. Because the active site of MINP(G1 + 18h)—i.e., the MINP prepared with 17a as the template and postmodified with 18h in the reductive amination—is designed to bind a terminal glucose, it hydrolyses glucose-terminated oligosaccharides such as maltose, cellobiose selectively over lactose and xylobiose.
MINP(G1–3 + 18h) binds the hydrolyzed products (glucose, maltose, and maltotriose, respectively) strongly and thus displays significant product inhibition (which is also common in natural glycosidases).76,77 Fortunately, because both MINP and the staring material (maltohexaose or amylose) are larger than the sugar products formed, product inhibition can be overcome simply by performing the hydrolysis inside a dialysis membrane permeable to the product(s) but impermeable to the starting material and the catalyst. Not only so, the product is isolated in situ, greatly simplifying the purification of the products and facile recycling of the catalyst. As highly cross-linked polymeric nanoparticles, these artificial glycosidases can be reused many times without any concerns for the loss of activity (Fig. 9c).
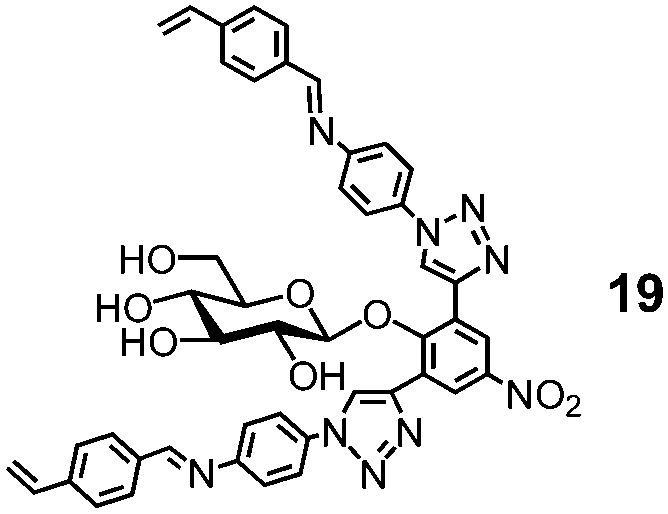
Natural glycosidase generally uses a pair of carboxylic acids to hydrolyze glycans, instead of a single sulfonic acid in MINP(G1–3 + 18h). Template 19 has two imine bonds sandwiching the glycosidic oxygen. The same MINP preparation and postmodification yield a MINP with a glucose-binding active site with a biomimetic dicarboxylic acid motif. The distance between the two acids in the natural enzyme is highly important to the cooperative catalysis, ∼5 Å for retaining glycosidases and 7–11 Å for inverting glycosidases.78 Reductive amination makes it possible to tune this distance accurately in the MINP, with the optimal results obtained in MINP(19 + 18f).79
Cellulose makes up 30–50% of the lignocellulosic biomass but needs to be depolymerized into soluble sugars before it can be converted into fuels or chemicals.80–82 Although natural enzymes (i.e., cellulases) exist for this process,77 their easily denatured protein structure limits the operating window and makes their recycling difficult. At 37 °C in aqueous buffer (pH 5), MINP(19 + 18f) hydrolyzes cellulose with an activity only a few times lower than cellulase from Aspergillus niger. On the other hand, while the natural cellulase loses its activity with an increase in the reaction temperature, the synthetic enzyme becomes more efficient, with its activity at 90 °C approaching 70% of that of the cellulase at 37 °C.79
A difficult challenge in cellulose hydrolysis comes from its high crystallinity. Although ionic liquids can dissolve cellulose to form a homogeneous solution,83,84 they denature enzymes easily, especially at elevated temperatures.85 As shown in Fig. 10a, MINP(19 + 18f) functions well in a mixture of 1-ethyl-3-methylimidazolium acetate ([C2mim]OAc) and DMSO even at 90 °C, with an activity several times higher than what the natural cellulase can achieve under its optimal aqueous conditions.
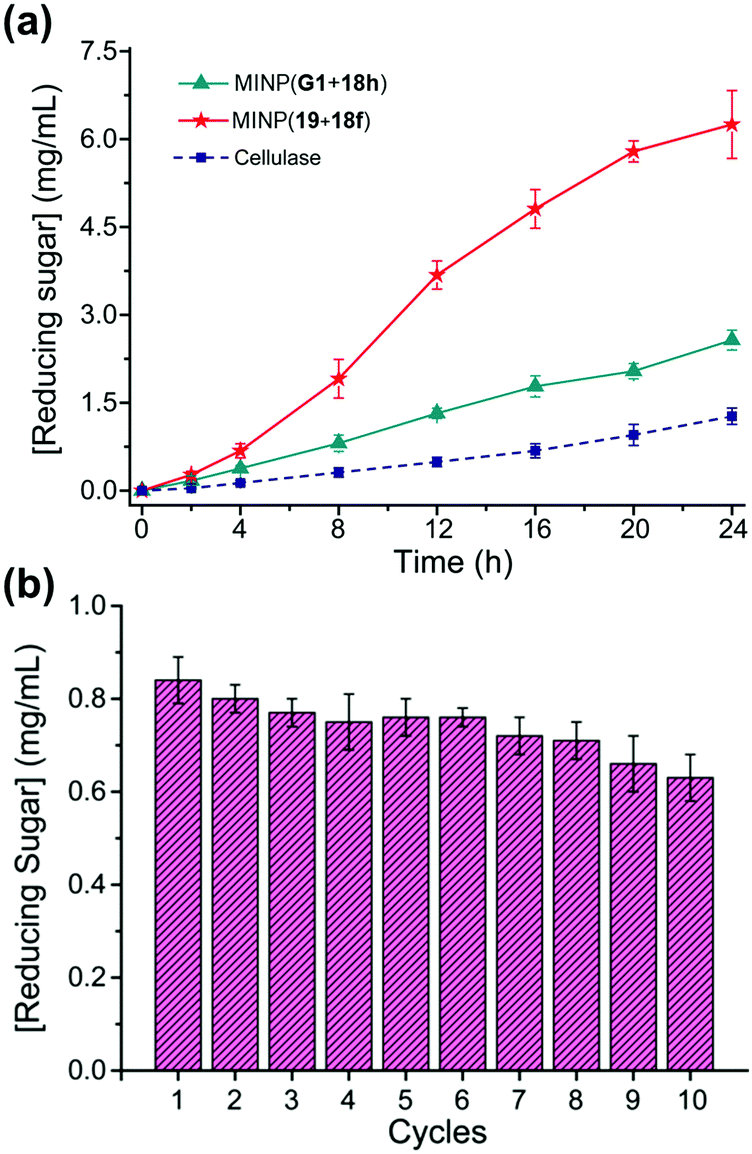 | ||
| Fig. 10 (a) Comparison of reducing sugar formed during hydrolysis of cellulose by the synthetic MINP catalysts in 2:8 [C2mim]OAc/DMSO with 5% H2O at 90 °C and natural cellulase in NaOAc buffer pH 5.0 at 37 °C. [Cellulose] = 8 mg mL−1. [Catalyst] = 2 mg mL−1. (b) Recyclability of MINP(19 + 18f) on magnetic nanoparticles for cellulose hydrolysis. (Adapted with permission from ref. 79. Copyright 2021, the American Chemical Society.) | ||
MINPs are generally decorated with monoazide 13 for enhanced hydrophilicity and facile purification (Fig. 7a). Without the surface decoration, the alkyne-containing MINPs can be “clicked” onto azide-functionalized magnetic nanoparticles to afford a reusable heterogeneous catalyst that maintains 75% of its activity after 10 cycles of hydrolysis of cellulose (Fig. 10b).79
Conclusions and outlook
Molecular recognition of subtly different, strongly solvated, complex, and microheterogeneous biological glycans had been an unreachable goal for synthetic GBMs for many years.1,7 However, decades of work from generations of researchers led to the development of molecularly imprinted materials to address this particular challenge. The largest benefit of using MIPs for glycan binding is the facile formation of preorganized, complementary imprinted sites in a single step. Not only can biologically competitive affinities be achieved nowadays, synthetic GBMs can be also endowed with functions not available in natural GBPs. These materials are expected to be useful in a wide range of applications in glycan purification, analysis, functional assay, and manipulation.1 Earlier applications of GBMs were largely proof-of-concept in nature. As materials rivaling natural GBPs in affinity and selectivity become available, glycans related to viral infection or other disease development could be targeted to address real biological problems in a practical manner. Materials development in these multidisciplinary efforts will continue to be important but needs to be tailored for the end applications. Molecular imprinting is a general method, applicable to both simple monosaccharides and complex biological glycans, using purified glycans or cleaved mixtures from glycoproteins as templates. Combined with the simplicity of the method, it gives chemists and biologists a powerful and versatile set of tools to help cracking the “sugar code”.4Wulff pioneered not only in sugar recognition but also in the creation of MIP-based artificial enzymes.23,86 Over the years, while applications of MIPs in separation, analysis, and sensing blossomed, applications in catalysis lagged behind. The high fidelity of micellar imprinting and high accessibility of imprinted sites on the surface of cross-linked micelle give an unprecedented opportunity to turn imprinted nanoparticles into synthetic enzymes. Total synthesis of carbohydrates is extremely challenging due to the prevalence of hydroxyl groups in the molecule.3 Selective, one-step hydrolysis by rationally designed synthetic glycosidase potentially can be used to produce complex glycans from precursor oligo- or polysaccharides available either naturally or prepared via enzymatic synthesis. A notable strength of the artificial glycosidases is their robustness in nonaqueous solution and high reaction temperatures—a feature extremely important in challenging conditions not possible for their natural counterparts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author thanks all the students in his group who have contributed to the papers discussed. The research in the Zhao group is supported by grants from NIGMS (R01GM138427) and the Mizutani Foundation of Glycoscience (Grant# 210051).Notes and references
-
N. R. Council, Transforming glycoscience: a roadmap for the future, National Academies Press, Washington, D.C., 2012 Search PubMed
.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS PubMed
-
J. P. Kamerling and G.-J. Boons, Comprehensive glycoscience: from chemistry to systems biology, Elsevier, Amsterdam, Boston, 1st edn, 2007 Search PubMed
- D. Solís, N. V. Bovin, A. P. Davis, J. Jiménez-Barbero, A. Romero, R. Roy, K. Smetana and H.-J. Gabius, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 186–235 CrossRef PubMed
- K. K. Palaniappan and C. R. Bertozzi, Chem. Rev., 2016, 116, 14277–14306 CrossRef CAS PubMed
- J. C. Paulson, O. Blixt and B. E. Collins, Nat. Chem. Biol., 2006, 2, 238 CrossRef CAS PubMed
-
B. Wang and G.-J. Boons, Carbohydrate recognition: biological problems, methods, and applications, Wiley, Hoboken, N.J., 2011 Search PubMed
-
A. P. Davis and T. D. James, Carbohydrate Receptors, Wiley-VCH, Weinheim, 2005 Search PubMed
- K. Van Roey, B. Uyar, R. J. Weatheritt, H. Dinkel, M. Seiler, A. Budd, T. J. Gibson and N. E. Davey, Chem. Rev., 2014, 114, 6733–6778 CrossRef CAS PubMed
- R. D. Cummings, Mol. BioSyst., 2009, 5, 1087–1104 RSC
-
J. W. Steed and P. A. Gale, Supramolecular Chemistry: From Molecules to Nanomaterials, Wiley, Weinheim, 2012 Search PubMed
- C. Ke, H. Destecroix, M. P. Crump and A. P. Davis, Nat. Chem., 2012, 4, 718–723 CrossRef CAS PubMed
- R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, Nat. Chem., 2019, 11, 52–56 CrossRef CAS PubMed
- D. J. Harvey, D. R. Wing, B. Kuster and I. B. Wilson, J. Am. Soc. Mass Spectrom., 2000, 11, 564–571 CrossRef CAS PubMed
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1986, 25, 1039–1057 CrossRef
-
H.-J. Schneider and A. K. Yatsimirsky, Principles and methods in supramolecular chemistry, Wiley, New York, 2000, pp. 137–146 Search PubMed
- M. J. Whitley, W. Furey, S. Kollipara and A. M. Gronenborn, FEBS J., 2013, 280, 2056–2067 CrossRef CAS PubMed
- U. Spohr, E. Paszkiewicz-Hnatiw, N. Morishima and R. U. Lemieux, Can. J. Chem., 1992, 70, 254–271 CrossRef CAS
- R. U. Lemieux, Acc. Chem. Res., 1996, 29, 373–380 CrossRef CAS
-
C. Tanford, The Hydrophobic Effect: Formation of Micelles and Biological Membranes, Krieger, Malabar, Fla., 2nd edn, 1991 Search PubMed
-
A. Ben-Naim, Hydrophobic interactions, Plenum Press, New York, 1980 Search PubMed
- N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS
- G. Wulff, Chem. Rev., 2002, 102, 1–28 CrossRef CAS PubMed
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef CAS
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS
- G. Wulff, W. Vesper, R. Grobe-Einsler and A. Sarhan, Makromol. Chem., 1977, 178, 2799–2816 CrossRef CAS
- G. Wulff, R. Grobe-Einsler, W. Vesper and A. Sarhan, Makromol. Chem., 1977, 178, 2817–2825 CrossRef CAS
- G. Wulff and W. Vesper, J. Chromatogr., 1978, 167, 171–186 CrossRef CAS
- G. Wulff, H.-G. Poll and M. Minárik, J. Liq. Chromatogr., 1986, 9, 385–405 CrossRef CAS
- G. Wulff, J. Vietmeier and H.-G. Poll, Makromol. Chem., 1987, 188, 731–740 CrossRef CAS
- G. Wulff and S. Schauhoff, J. Org. Chem., 1991, 56, 395–400 CrossRef CAS
- A. Friggeri, H. Kobayashi, S. Shinkai and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2001, 40, 4729–4731 CrossRef CAS PubMed
- S. Shinde, Z. El-Schich, A. Malakpour, W. Wan, N. Dizeyi, R. Mohammadi, K. Rurack, A. Gjörloff Wingren and B. Sellergren, J. Am. Chem. Soc., 2015, 137, 13908–13912 CrossRef CAS PubMed
- Z. El-Schich, M. Abdullah, S. Shinde, N. Dizeyi, A. Rosén, B. Sellergren and A. G. Wingren, Tumour Biol., 2016, 37, 13763–13768 CrossRef CAS PubMed
- M. Panagiotopoulou, Y. Salinas, S. Beyazit, S. Kunath, L. Duma, E. Prost, A. G. Mayes, M. Resmini, B. T. S. Bui and K. Haupt, Angew. Chem., Int. Ed., 2016, 55, 8244–8248 CrossRef CAS PubMed
- K. Haupt, P. X. Medina Rangel and B. T. S. Bui, Chem. Rev., 2020, 120, 9554–9582 CrossRef CAS PubMed
- B. Demir, M. M. Lemberger, M. Panagiotopoulou, P. X. Medina Rangel, S. Timur, T. Hirsch, B. Tse Sum Bui, J. Wegener and K. Haupt, ACS Appl. Mater. Interfaces, 2018, 10, 3305–3313 CrossRef CAS PubMed
- A. Poma, A. Guerreiro, M. J. Whitcombe, E. V. Piletska, A. P. F. Turner and S. A. Piletsky, Adv. Funct. Mater., 2013, 23, 2821–2827 CrossRef CAS PubMed
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443 CrossRef CAS PubMed
- P. X. Medina Rangel, S. Laclef, J. Xu, M. Panagiotopoulou, J. Kovensky, B. Tse Sum Bui and K. Haupt, Sci. Rep., 2019, 9, 3923 CrossRef PubMed
- S. S. Wang, J. Ye, Z. J. Bie and Z. Liu, Chem. Sci., 2014, 5, 1135–1140 RSC
- Z. Bie, Y. Chen, J. Ye, S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2015, 54, 10211–10215 CrossRef CAS PubMed
- R. Xing, S. Wang, Z. Bie, H. He and Z. Liu, Nat. Protoc., 2017, 12, 964 CrossRef CAS PubMed
- J. Ye, Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 10386–10389 CrossRef CAS PubMed
- S. Wang, D. Yin, W. Wang, X. Shen, J.-J. Zhu, H.-Y. Chen and Z. Liu, Sci. Rep., 2016, 6, 22757 CrossRef CAS PubMed
- Z. Liu and H. He, Acc. Chem. Res., 2017, 50, 2185–2193 CrossRef CAS PubMed
- D. Yin, X. Li, Y. Ma and Z. Liu, Chem. Commun., 2017, 53, 6716–6719 RSC
- Y. Dong, W. Li, Z. Gu, R. Xing, Y. Ma, Q. Zhang and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 10621–10625 CrossRef CAS PubMed
-
D. Myers, Surfactant Science and Technology, VCH, New York, 2nd edn, 1992, pp. 136–144 Search PubMed
- J. K. Awino and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 12552–12555 CrossRef CAS PubMed
- J. K. Awino and Y. Zhao, ACS Biomater. Sci. Eng., 2015, 1, 425–430 CrossRef CAS PubMed
- L. Duan and Y. Zhao, Bioconjugate Chem., 2018, 29, 1438–1445 CrossRef CAS PubMed
- L. Duan and Y. Zhao, J. Org. Chem., 2019, 84, 13457–13464 CrossRef CAS PubMed
- X. Li, T. M. Palhano Zanela, E. S. Underbakke and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 639–643 CrossRef CAS PubMed
- R. W. Gunasekara and Y. Zhao, Chem. Commun., 2019, 55, 4773–4776 RSC
- J. K. Awino, R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 9759–9762 CrossRef CAS PubMed
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS
- J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS
- R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 829–835 CrossRef CAS PubMed
- D. K. Mandal, N. Kishore and C. F. Brewer, Biochemistry, 1994, 33, 1149–1156 CrossRef CAS PubMed
- L. Duan, M. Zangiabadi and Y. Zhao, Chem. Commun., 2020, 56, 10199–10202 RSC
- X. Li, K. Chen and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 11092–11097 CrossRef CAS PubMed
- C. Chothia and J. Janin, Nature, 1975, 256, 705–708 CrossRef CAS PubMed
- J. S. Nowick, J. S. Chen and G. Noronha, J. Am. Chem. Soc., 1993, 115, 7636–7644 CrossRef CAS
- K. Ariga and T. Kunitake, Acc. Chem. Res., 1998, 31, 371–378 CrossRef CAS
- M. Zangiabadi and Y. Zhao, Nano Lett., 2020, 20, 5106–5110 CrossRef CAS PubMed
- K. Chen and Y. Zhao, Org. Biomol. Chem., 2019, 17, 8611–8617 RSC
- X. Li and Y. Zhao, ACS Catal., 2020, 10, 13800–13808 CrossRef CAS PubMed
- M. Zangiabadi and Y. Zhao, ACS Appl. Polym. Mater., 2020, 2, 3171–3180 CrossRef CAS
- J. K. Awino, R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 2188–2191 CrossRef CAS PubMed
- X. Xing and Y. Zhao, New J. Chem., 2018, 42, 9377–9380 RSC
- X. Li and Y. Zhao, Chem. Sci., 2021, 12, 374–383 RSC
- D. French and D. W. Knapp, J. Biol. Chem., 1950, 187, 463–471 CrossRef CAS
-
P. Bernfeld, Methods in Enzymology, Academic Press, 1955, vol. 1, pp. 149–158 Search PubMed
- Y. Yang, X. Zhang, Q. Yin, W. Fang, Z. Fang, X. Wang, X. Zhang and Y. Xiao, Sci. Rep., 2015, 5, 17296 CrossRef CAS PubMed
- C. M. Payne, B. C. Knott, H. B. Mayes, H. Hansson, M. E. Himmel, M. Sandgren, J. Ståhlberg and G. T. Beckham, Chem. Rev., 2015, 115, 1308–1448 CrossRef CAS PubMed
- D. L. Zechel and S. G. Withers, Acc. Chem. Res., 2000, 33, 11–18 CrossRef CAS PubMed
- X. Li, M. Zangiabadi and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 5172–5181 CrossRef CAS PubMed
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC
- Y. Jing, Y. Guo, Q. Xia, X. Liu and Y. Wang, Chem, 2019, 5, 2520–2546 CAS
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed
- R. M. Wahlström and A. Suurnäkki, Green Chem., 2015, 17, 694–714 RSC
- G. Wulff and J. Liu, Acc. Chem. Res., 2012, 45, 239–247 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2022 |