
Recent advancement in heterogeneous CO2 reduction processes in aqueous electrolyte
Shuvojit
Mandal
 ,
Dibyendu
Ghosh
and
Praveen
Kumar
*
,
Dibyendu
Ghosh
and
Praveen
Kumar
*
School of Materials Science, Indian Association for the Cultivation of Science, Kolkata-700032, India. E-mail: praveen.kumar@iacs.res.in
First published on 12th September 2022
Abstract
Energy is essential in the human race toward prosperity and it always remains in peak demand. Conventional fossil fuels may fulfil this energy demand for now but at the cost of severe long-term adverse effects on the environment. Therefore, many research groups are working to develop efficient and affordable processes for green energy harvesting to ensure true prosperity and a healthy environment for future generations. In this context, the CO2 reduction reaction (CO2RR) is emerging as one of the most essential and potent research fields for sustainable zero-emission green energy harvesting. Although there are a lot of reviews on the CO2RR, there is a lack of systematic comparisons of various common electro or non-electro catalytic processes, discussion of progress over time, and in-depth analysis with respect to materials. This review briefly introduces numerous heterogeneous catalytic CO2RR processes, categorized based on fundamental differences, and provides an up-to-date overview of catalyst materials and improvements in catalysis setup. In this review, electrochemical (EC), photoelectrochemical (PEC), and photovoltaic cell-assisted EC/PEC (PV-EC/PEC) processes with several modifications have been emphasized, including a detailed overview of the futuristic membrane electrode assembly (MEA) method, and they are comprehensively discussed following a timeline format. Along with the underlying comparison of various processes, strategies for the further utilization of these processes/materials and challenges for future work have also been summarized.
 Shuvojit Mandal | Shuvojit Mandal is currently doing a Ph.D. at the School of Materials Sciences, Indian Association for the Cultivation of Science (IACS), Kolkata under the supervision of Prof. Praveen Kumar. After graduating in Chemistry from Scottish Church College affiliated (Calcutta University), he obtained his master’s degree in 2019 from the Indian Institute of Technology (Indian School of Mines) Dhanbad (IIT (ISM) Dhanbad), Jharkhand. His research interest is on 2D emerging materials as photo/electrocatalysts for efficient CO2 reduction application. |
 Dibyendu Ghosh | Dibyendu Ghosh obtained his Ph.D. in Physics from Jadavpur University, India in 2015. He did his 1st postdoc at the Indian Institute of Science Education and Research (IISER) Kolkata, during 2015-2018. He is currently working, from mid-2018, as a research associate at the Indian Association for the Cultivation of Science, Kolkata, India in Prof. Kumar’s research group. His Ph.D. work was on III-V semiconductor thin films for device applications. Currently, his main research focus is on 2D materials for photoelectrochemical water splitting and CO2 reduction. |
 Praveen Kumar | Dr Praveen is working as an Assistant Professor at the Indian Association for the Cultivation of Sciences (IACS), Kolkata. He is an Editorial Board Member of Materials Letters (Elsevier) and Nanotechnology (IOP) Journals. He is a recipient of several recognized awards and fellowships, including a Fulbright Nehru Professional Excellence Fellowship, MRSI Medal, MCAA Societal Impact Award, DAE Young Achiever Award, Marie Curie Postdoctoral Fellowship, etc. His research contributions cover the materials engineering of III–V/Si semiconductors, 2D-materials/MXenes, carbon nanostructures for water-splitting, the CO2RR, photodetectors, and supercapacitors. He has authored 89 publications, 4 patents (filed), and 9 books/chapters, and delivered ∼65 invited talks around the globe. |
1. Introduction
From the very beginning of civilization, energy requirements have been increasing rapidly. To meet that demand, until recent decades we have relied solely on fossil fuels. This practice increased the CO2 level in the atmospheric air day by day.1 Moreover, increased population and industrial development caused inevitable deforestation. Overall the consumption of CO2 by plants in photosynthesis was reduced, which shifted the proportion of CO2 in the atmosphere towards a higher level. The atmospheric CO2 level increased from a pre-industrial 285 ppm in 1850 to 415 ppm by 2020.2 This resulted in the global average surface temperature rising by 1.2 °C over the same period via the greenhouse effect. However, the goal of carbon neutrality is to limit the temperature increment up to 2100 by only 1.5–2 °C from the pre-industrial level after somehow reaching carbon neutrality by 2050. Also, the existing higher CO2 level will keep increasing the global surface temperature.3 This global warming has cast a huge shadow over our livelihoods and the list of fatal consequences is even larger, like flood, drought, climate change, wildfires, species extinction, glacial retreat, the melting of Arctic and Antarctic ice, and sea-level rise. Sea-level rise alone could directly affect 100 million people or more living in coastal regions because a temperature rise of 10 °C can increase sea level by 200 m.4 According to a report from the IPCC, sea level will rise by 0.24 m from the pre-industrial period with an average 1.2 °C rise in temperature, and could go up to 0.3–1.5 m by 2100.5 Therefore, beyond carbon neutrality by stopping the use of fossil fuel, we have to think of an efficient way to decrease the already high CO2 level. After four major industrial revolutions brought about by the steam engine, electricity, computers and the internet, we need a fifth one in the form of environment-friendly renewable green energy harvesting.6 However, developing a technology to use CO2 as an energy storage medium by reducing it to fuel, will solve the energy storage problem and help to decrease the atmospheric CO2 level sustainably and effectively. That is why CO2RR has been considered one of the most relevant strategies. Furthermore, if somehow green energy sources can be utilized, directly or indirectly, to provide this endothermic reduction energy, then we will reach net-zero carbon emission more easily. In electrocatalytic (EC) CO2RR, electrical energy may be supplied from green sources like photovoltaic cells, and hydro, wind, geo-thermal or nuclear power. However, solar energy can be directly utilized in photoelectrocatalytic (PEC) and photocatalytic (PC) CO2RR processes. These catalytic processes can be accomplished in both homogeneous and heterogeneous catalyst systems. Due to its many advantages and industrial viability, the heterogeneous process gained more interest from scientists and became the most convenient and reliable process over time. Recently, one new approach has focused on metal–CO2 batteries, but it is totally in its infancy and requires an enormous amount of time to find practical applicability.Water splitting can be an easy and well-developed research topic for green energy utilization.7 Still, water-reduced product hydrogen (H2) mostly behaves like an ideal gas and cannot be compressed at room temperature; therefore, storing and transporting H2 is very expensive. Herein lies the considerable superiority of CO2RR over water splitting. Most of the products of CO2RR are liquid, and even the gaseous products can easily be compressed to a liquid, which automatically minimizes the storage problem of converted energy. However, these advantages must be realized after many research challenges like finding stable catalyst materials and suppressing water reduction over CO2RR. Table 1 lists the standard reduction potentials for different CO2 reduction pathways and related products. The required reduction potentials for various products are very close; therefore, getting a selective product is really tough thermodynamically. The next important issue is the product formation rate without altering the selectivity because the required enhanced current density may be realized at the cost of selectivity or increased hydrogen evolution reaction (HER).
| Reaction | E 0/VSHE | Product |
|---|---|---|
| 2H+(aq) + 2e− → H2(g) | 0.00 | Hydrogen |
| CO2(g) + e− → CO2˙−(aq) | −1.49 | — |
| CO2(g) + H+(aq) + 2e− → HCO2˙−(aq) | −0.08 | — |
| CO2(g) + 2H+(aq) + 2e− → CO(g) + H2O(l) | −0.12 | Carbon monoxide |
| CO2(g) + 2H+(aq) + 2e− → HCOOH(l) | −0.20 | Formic acid |
| CO2(g) + 4H+(aq) + 4e− → HCHO(l) + H2O(l) | −0.07 | Formaldehyde |
| CO2(g) + 6H+(aq) + 6e− → CH3OH(l) + H2O(l) | 0.03 | Methanol |
| CO2(g) + 8H+(aq) + 8e− → CH4(g) + 2H2O(l) | 0.17 | Methane |
| 2CO2(g) + 2H+(aq) + 2e− → H2C2O4(aq) | −0.50 | Oxalic acid |
| 2CO2(g) + 12H+(aq) + 12e− → CH2CH2(g) + 4H2O(l) | 0.06 | Ethylene |
| 2CO2(g) + 12H+(aq) + 12e− → CH3CH2OH(l) + 3H2O(l) | 0.08 | Ethanol |
| 2CO2(g) + 14H+(aq) + 14e− → CH3CH3(l) + 4H2O(l) | 0.14 | Ethane |
Chen et al.8 reported that if industrial electricity is used to produce ethanol from CO2 by electrocatalysis, the electricity cost will be $0.32 per L, which is $0.06 per L less than the average ethanol fuel price in 2020. Worldwide large-scale industrial production using CO2RR is greatly expected in the near future. In addition, various governments are promoting CO2RR by giving significant financial benefits through special projects such as the ‘Rheticus Project’ in Germany, ‘Recode’ in Italy, and ‘CO2 perate’ in Belgium.9 Haldor Topsoe,10 a Danish company, has already scaled up the high-temperature electrochemical reduction of CO2 to CO with 99.95% selectivity. This field is growing exponentially and attracting higher interest from the scientific and industrial research community.
There are several recent reviews on EC CO2RR focusing on several aspects. Wang et al.11 and Hoang et al.12 thoroughly reviewed the catalyst material and product selectivity separately. Whereas Liu et al.13 focused their review on the selectivity issue for CO2RR. Moreover, a few reviews looked at specific product selectivity, like Nguyen et al.14 on carbon monoxide selectivity, Zhao et al.15 on methane formation, and Fan et al.16 on C2 product selectivity. Some groups have covered the various catalyst materials used in EC CO2RR.17–23 Likewise, a few reviews have focused solely on a particular catalyst material. Li et al.,24 Lu et al.,25 and Huang et al.26 thoroughly reviewed single-atom catalyst (SAC) materials as electrodes. Shao et al.,27 Chen et al.,28 and Gao et al.29 gave a detailed overview of metal chalcogenide materials as electrocatalysts. Choi et al.30 reviewed defect engineering and the effect of catalyst poisoning on EC CO2RR. Liang et al.31 briefly included EC CO2RR in their review of energy conversion through CO2 reduction. However, several significant reviews are also available looking exclusively at PEC CO2RR.32–37 Li et al.38 and Feng et al.39 covered PEC CO2RR concisely along with PEC water splitting, while Zheng et al.40 did so along with the nitrogen reduction reaction (NRR). Yang et al.41 made a detailed review of both EC and PEC. He et al.42 recently covered PEC, PV-EC, and PC CO2RR. However, very few reviews cover EC and PEC in detail showing a timeline advancement in the catalyst material. There has been no review to our knowledge that gave significant attention to PV-EC/PEC. Therefore, we have made a detailed briefing of all these three most important systems: EC, PEC, and PV-EC/PEC. Moreover, we have thoroughly discussed the emerging CO2RR technique of membrane electrode assembly (MEA), with its superior current density over the conventional EC process. Again, PV cells are modified with a catalyst, and that composite has been used as a whole photoelectrode. As a result, these materials show exceptionally high photoactivity. Hence, we have drawn a clear boundary in discussing photovoltaic cell modified PEC systems and general semiconducting PEC systems. We have also included a thorough overview of a basic understanding of all the sub-topics, which will benefit the readers to understand the fundamental depth of advancement in this research field. Finally, we end the discussion with future perspectives, which will provide a clear way to think about future directions related to this very futuristic research on CO2RR. We hope this article will accelerate the development of a sustainable, carbon-neutral energy cycle and add value to the scientific community.
2. Possible processes for the CO2RR: basics, merits, and demerits
There are three basic processes based on the energy input utilized in the uphill CO2RR, as demonstrated in Fig. 1: (i) electrocatalysis uses electrical power, (ii) photocatalysis uses photon (solar) energy, and (iii) thermochemical uses heat energy. However, there are some hybrid methods where different energy sources are used simultaneously. For example, in photoelectrocatalysis, both light and electricity are required, whereas in photothermal catalysis, light and heat work together to supply the energy for CO2RR. In another method, a light-harvesting photovoltaic cell is coupled with an electrocatalytic system to provide electrical power efficiently. In the main, six different methods with broad boundaries are studied for CO2 reduction: (i) photothermal, (ii) thermochemical, (iii) photocatalysis (PC), (iv) electrocatalysis (EC), (v) photoelectrocatalysis (PEC), and (vi) photovoltaic-electrocatalysis/photoelectrocatalysis (PV-EC/PEC). We will now briefly discuss all these processes in heterogeneous catalysis one by one along with their fundamentals.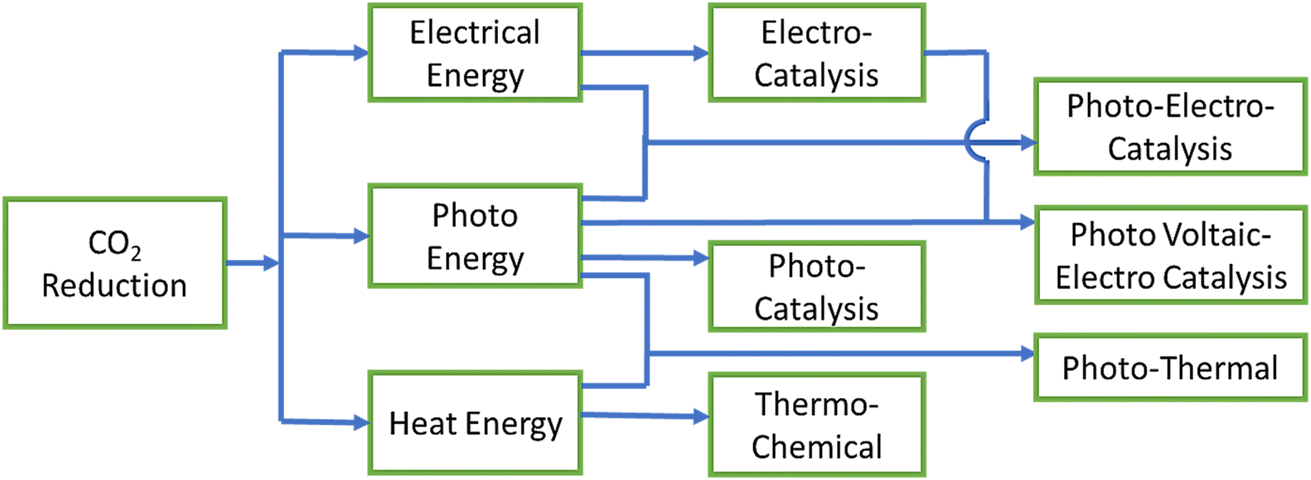 | ||
| Fig. 1 The fundamental differences between CO2RR processes with respect to energy utilization. | ||
2.1. Photothermal
The photo and thermal energy of sunlight are directly utilized in this process. In one straightforward method, a large amount of solar light is focused through a mirror/lens onto a receiver site, where the reaction occurs. Therefore, solar energy is utilized to assist the thermochemical CO2RR process.43–48 We will discuss this in more detail in the Thermochemical section (2.2). Although it requires the development of a vast infrastructure, it is highly scalable to meet industrial demand. In contrast, another way is to utilize surface plasmon resonance phenomena to use the sunlight for direct localized heating, called a plasmonic catalyst. Plasmonic catalysis relies on the resonance of the oscillations of free electrons (plasmons) upon the absorption of incident electromagnetic radiation.49 Such resonance occurs when the delocalized electrons are supplemented with an energy wave, whose frequency corresponds to the characteristic resonant frequency of the metal nanoparticles. These plasmons induce hot carriers, which have higher energy than a general carrier generated through photoexcitation.50 So, these carriers are more capable of overcoming Schottky junction barriers. Non-plasmonic phenomena can also assist this process in metallic nanoparticles via intraband and interband electronic excitation. Generally, under heated conditions, the introduction of light illumination enhances the activity in the case of a photothermal reaction.51 Depending on what reducing agent is used, there can be several options for reducing CO2. If the reducing sacrificial gas is hydrogen, then the process is called hydrogenation. Hydrogenation can be classified as reverse water gas shift (RWGS) when the product is CO, and as methanation when methane is the main product. Hydrogenation can even give rise to multicarbon products. When the sacrificial reducing agent is methane gas, the process is called a dry reformation reaction. And most importantly, it becomes comparable with PC, PEC or EC systems when the sacrificial reducing agent is water: the reaction popularized as artificial photosynthesis. Fig. 2 represents the milestones in the development of photothermal CO2RR.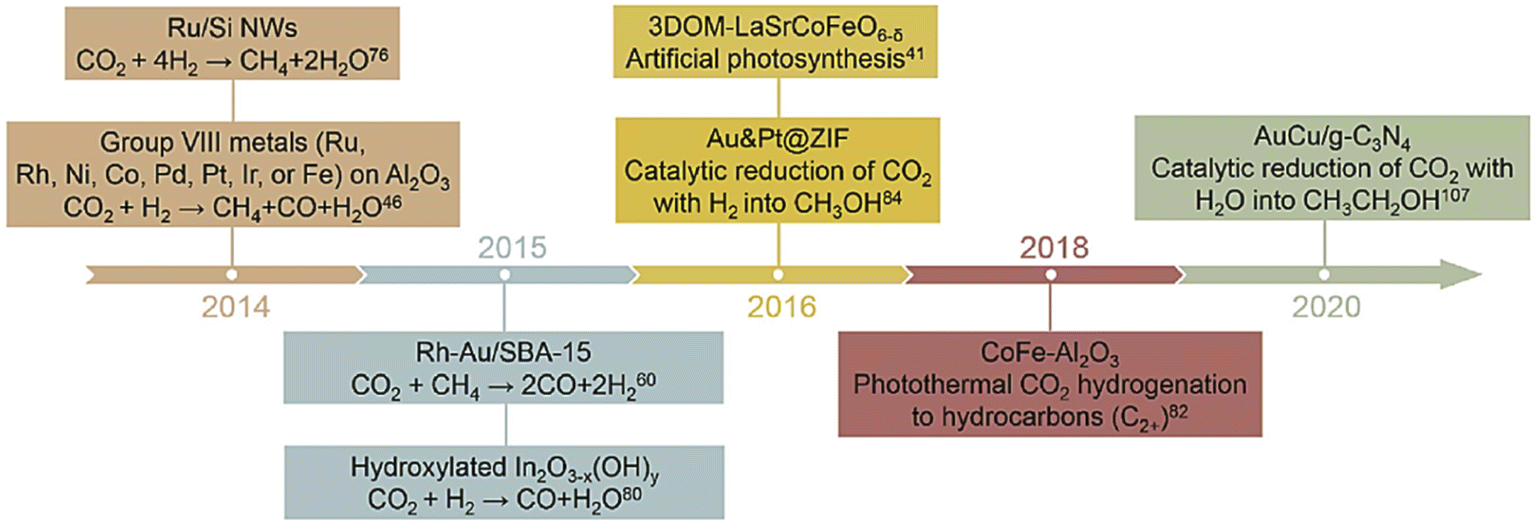 | ||
| Fig. 2 A timeline of the advances in photothermal catalytic CO2 reduction. Reprinted with permission from Zhang et al.,51 Copyright 2021, Elsevier Ltd. | ||
Photothermal artificial photoreduction is quite a new field compared to hydrogenation. In 2016, Ha et al.52 synthesized a three-dimensional microporous structure of LaSrCoFeO6−δ (LSCF), double perovskite with a poly(methyl methacrylate) template by simple in air calcination. CH4 formation with the best catalyst can reach 557.88 μmol g−1 for 8 h at 350 °C under visible light illumination, which is five times higher than with the thermochemical process (120.86 μmol g−1). Several other studies on improved photothermal artificial photosynthesis have been reported with CO and CH4 selectivity.53–56 However, recent reports even claim multicarbon products with high selectivity. In 2020, Wang et al.57 synthesized carbon-doped In2S3 nanosheets, which have been reported to produce ethylene with 50% selectivity by an artificial photosynthesis process at 150 °C under illuminated conditions. At 420 nm, the quantum efficiency was 13.3%; however, undoped In2S3 failed to produce ethylene. The reported production rate was 26.6 ± 4.1 μmol g−1 h−1, which is commendable in the case of ethylene formation. Li et al.58 made Au–Cu alloy nanoparticle (∼10 nm size) loaded ultrathin g-C3N4 nanosheets. A catalyst with 1 wt% alloy NP loading can reach a rate of ethanol formation as high as 0.89 mmol g−1 h−1 with 93.1% selectivity at 120 °C under illuminated conditions, which is 4.2 times higher than that of photocatalysis and 7.6 times higher than that of thermal catalysis alone. In 2021, Liu et al. designed a 3D spherical microfluidic evaporator loaded with Cu2O catalyst which can utilize omnidirectional solar light for photothermal CO2RR.59 From the latest reports discussed above, although artificial photosynthesis should have a very bright future in the field of photothermal CO2RR, it is still in the primary stage of development for CO2RR application.
2.2. Thermochemical
The thermochemical process is the most explored field for CO2 reduction, and the method is highly scalable. Whatever, most reports are on gas–solid interface catalysis and the liquid aqueous phase is not involved. However, recent solution-phase thermochemical CO2RR is discussed in the next paragraph after this. For the gas-phase thermochemical process, two broadly different processes considering the temperature requirement are generally reported. In one process, some catalyst is decomposed at a very high temperature (generally >1000 °C) to give rise to an O2 stream and then CO2 gas and/or water vapour are purged into the activated catalyst chamber to be reduced to CO and/H2 while re-oxidizing the catalyst. These two steps of heating to activate the catalyst and cooling by CO2 and/H2O reduction, are repeated. However, syn gas is the product in this process when CO2 and H2O both are purged, and the CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 ratio can be easily tuned. Ceria-based materials44–47,60 are the most suitable and effective for this type of thermochemical process. However, mixed oxide,43,61 ferrite-based62 and manganate-based48 perovskites have also been reported as efficient catalysts. In another process, a fixed-bed continuous flow reactor or a fluidized-bed reactor is used. In the former case, CO2 gas flows with a reagent gas over a catalyst bed, generally kept at an elevated temperature and in the latter case, gas is passed at high pressure, which can cause a few catalysts particles to fly a little bit with it, due to the high gas flow velocity. Among all the reports on CO2 hydrogenation to methanol in the past 10 years, 79% are on Cu-based material and in 75.9% of these reports ZnO is used as support. Together with these, 50% of the time Al2O3 and 32.7% time ZrO2 are used as a mixed support material.63 For example, in 2019, Wang et al. reported a Cu/ZnO/ZrO2 catalyst with a conversion efficiency of 18.2% and methanol selectivity of 80.2% at 220 °C with 3.0 MPa pressure.64 For CO2 hydrogenation to methane, transition metals, including Rh, Ru, Pd, Ni and Co, on various supports, including Al2O3, SiO2, CeO2, TiO2 and ZrO2, have been investigated to study the strong metal–support interaction in improving the catalyst activity.65 For example, Ningi et al.66 synthesized highly active Ni catalysts on a CeO2 support. At 275 °C, the plasma-decomposed Ni/CeO2 catalyst shows a CO2 conversion of 84.2% at a high space velocity of 56000 h−1, while it is only 34.7% on the regular calcined catalyst. Meanwhile, the methane formation rate of 100.3 μmol gcat−1 s−1 is achieved with a methane selectivity of 99.5%.
:H2 ratio can be easily tuned. Ceria-based materials44–47,60 are the most suitable and effective for this type of thermochemical process. However, mixed oxide,43,61 ferrite-based62 and manganate-based48 perovskites have also been reported as efficient catalysts. In another process, a fixed-bed continuous flow reactor or a fluidized-bed reactor is used. In the former case, CO2 gas flows with a reagent gas over a catalyst bed, generally kept at an elevated temperature and in the latter case, gas is passed at high pressure, which can cause a few catalysts particles to fly a little bit with it, due to the high gas flow velocity. Among all the reports on CO2 hydrogenation to methanol in the past 10 years, 79% are on Cu-based material and in 75.9% of these reports ZnO is used as support. Together with these, 50% of the time Al2O3 and 32.7% time ZrO2 are used as a mixed support material.63 For example, in 2019, Wang et al. reported a Cu/ZnO/ZrO2 catalyst with a conversion efficiency of 18.2% and methanol selectivity of 80.2% at 220 °C with 3.0 MPa pressure.64 For CO2 hydrogenation to methane, transition metals, including Rh, Ru, Pd, Ni and Co, on various supports, including Al2O3, SiO2, CeO2, TiO2 and ZrO2, have been investigated to study the strong metal–support interaction in improving the catalyst activity.65 For example, Ningi et al.66 synthesized highly active Ni catalysts on a CeO2 support. At 275 °C, the plasma-decomposed Ni/CeO2 catalyst shows a CO2 conversion of 84.2% at a high space velocity of 56000 h−1, while it is only 34.7% on the regular calcined catalyst. Meanwhile, the methane formation rate of 100.3 μmol gcat−1 s−1 is achieved with a methane selectivity of 99.5%.
We should mention another process in which catalysts are added to a solvent at a required temperature and reagent gases are purged in a closed reactor at high pressure. Recently, thermochemical CO2 hydrogenation in an aqueous solvent has been reported with high selectivity.67 For example, in 2017, Wang et al. reported methanol selectivity with a Co4N nanosheet catalyst in water solvent with a turnover frequency of 25.6 h−1. After 3 h of reaction at 150 °C, the mixed gas (CO2/H2 = 1:3) taken at 32 bar converts to 4.2 mmol of methanol with 0.5 mmol of formic acid. Under similar conditions, a long-term stability test with 16 subsequent cycles of 15 h each, without removing the catalyst (for a total of 240 h) gave rise to 211 mmol of methanol and 2 mmol of formic acid in total.68 In 2020, Ye et al. made an Ir single-atom catalyst modified In2O3 which completely tuned the methanol selectivity of In2O3 to ethanol. In an aqueous solvent at 180 °C, CO2 hydrogenation by this catalyst gave >99% ethanol with a high turnover frequency of 481 h−1.69 In 2022, Jaleel et al. reported immobilized RuCl3 on a nitrogen-doped carbon matrix with formate-only selectivity at 120 °C with mixed gas (CO2/H2 = 1:1) at 8 MPa pressure in aqueous 1 M Et3N solution. The optimized catalyst with accessible mesoporous structure had a high turnover number of 4468 in a 2 h reaction and 18212 in a 12 h reaction.70 However, thermochemical CO2 reduction with water as a sacrificial reagent is very interesting in terms of cost-effectiveness and creates a research gap to find a way other than high-temperature two-step syn-gas synthesis. So, this research field also casts a very futuristic horizon in CO2RR and can provide low-cost and efficient CO2-to-fuel conversion technology.
2.3. Photocatalysis (PC)
Photocatalysis is a straightforward way of CO2 reduction, but herein lie all the drawbacks. There is significantly less external control over product selectivity. Again, both oxidized and reduced products form on the same catalyst particle. In that case, oxidized liquid products may be re-oxidized by the photogenerated hole, thereby significantly reducing the system efficiency. However, it is more practical if the sacrificial electron donor is water (artificial photosynthesis). In that case, oxygen produced from water oxidation will mix with fuel generated from CO2 reduction and create a high potential blast risk. So, to find practical applicability, the catalyst must be very efficient so that the gas mixture can be used on the spot at the time of formation. Moreover, very few semiconductor materials can be found with a suitable band position and bandgap for CO2 reduction. There is no scope for providing external bias, as in the case of the PEC process, to overcome the required large overpotential for CO2 reduction.71 However, as the same catalyst works as anode and cathode, charge transport becomes very fast, and photogenerated electron–hole pairs get efficiently separated and diffuse to the catalyst surface due to there being a built-in electric field gradient between the two different kinds of active sites.72 PC is itself a vast field, and compiling short updates is difficult; therefore, we have kept this part out of our discussion.2.4. Photoelectrocatalysis (PEC)
Photoelectrocatalysis is the ultimate method: a stand-alone device that can fulfil both the purposes of solar light harvesting and CO2 conversion. An external bias can also be applied to optimize the selectivity with minimum activity loss. When an n-type material is dipped into an electrolyte, electrons, as majority carriers, move to the electrolyte to match the Fermi level of the redox system (VRedox) in the electrolyte. This causes band bending in the photoanode. The metal cathode (counter electrode) also comes into equilibrium with this level, but as the metal has a huge free electron density, no such kind of electronic effect was observed (Fig. 3a). Whenever the external potential is applied in forward bias, the metal Fermi level goes up, and the Fermi level of the n-type material goes down from the VRedox level. This causes enhancement in band bending (Fig. 3b). However, if the external potential is not high enough, then the quasi Fermi level splitting (QFLS) in the electrolyte-facing side will be negligible (not shown in Fig. 3b) so that the hole concentration in the valence band will be too small to oxidize water. For this reason, although metal Fermi catalysis has reached a potential where it is capable of reducing CO2, CO2RR does not occur. Now, with illumination of suitable frequency light, a steady-state QFLS occurs, enhancing both type charge-carrier concentrations. As a result, a reduction in band banding is observed and the hole concentration becomes high enough to oxidize water. Moreover, this also results in photovoltage generation (Voc), generally <QFLS, depending on the absorption efficiency and recombination of the carriers (Fig. 3c).73,74 This photovoltage also works in forward bias and, as a result, combines with the applied potential to drive the CO2RR more efficiently. In some cases, the photovoltage is sufficient to provide the total voltage required to run the full-cell reaction.75 A similar but opposite situation is observed in the case of p-type semiconductors, as shown in Fig. 3d–f.36,76 However, a narrow bandgap is very effective for maximum solar light harvesting from a broad range of light frequencies. But, in that case, these materials fail to provide the required photovoltage for CO2 reduction. A photoanode based on oxide materials has a higher bandgap; therefore, these materials, in some cases, can operate without bias or less external bias to operate the CO2RR cell. But as the photo-harvesting property is poor and higher wavelength light remains unutilized, the current density remains very low. Therefore, the most effective way is to couple the photocathode and photoanode to realize a bias-less monolithic operation with significant current density. However, this kind of coupling in a Z-scheme or, more ideally, making a tandem-type arrangement of the electrodes is challenging, and very few reports are available to date.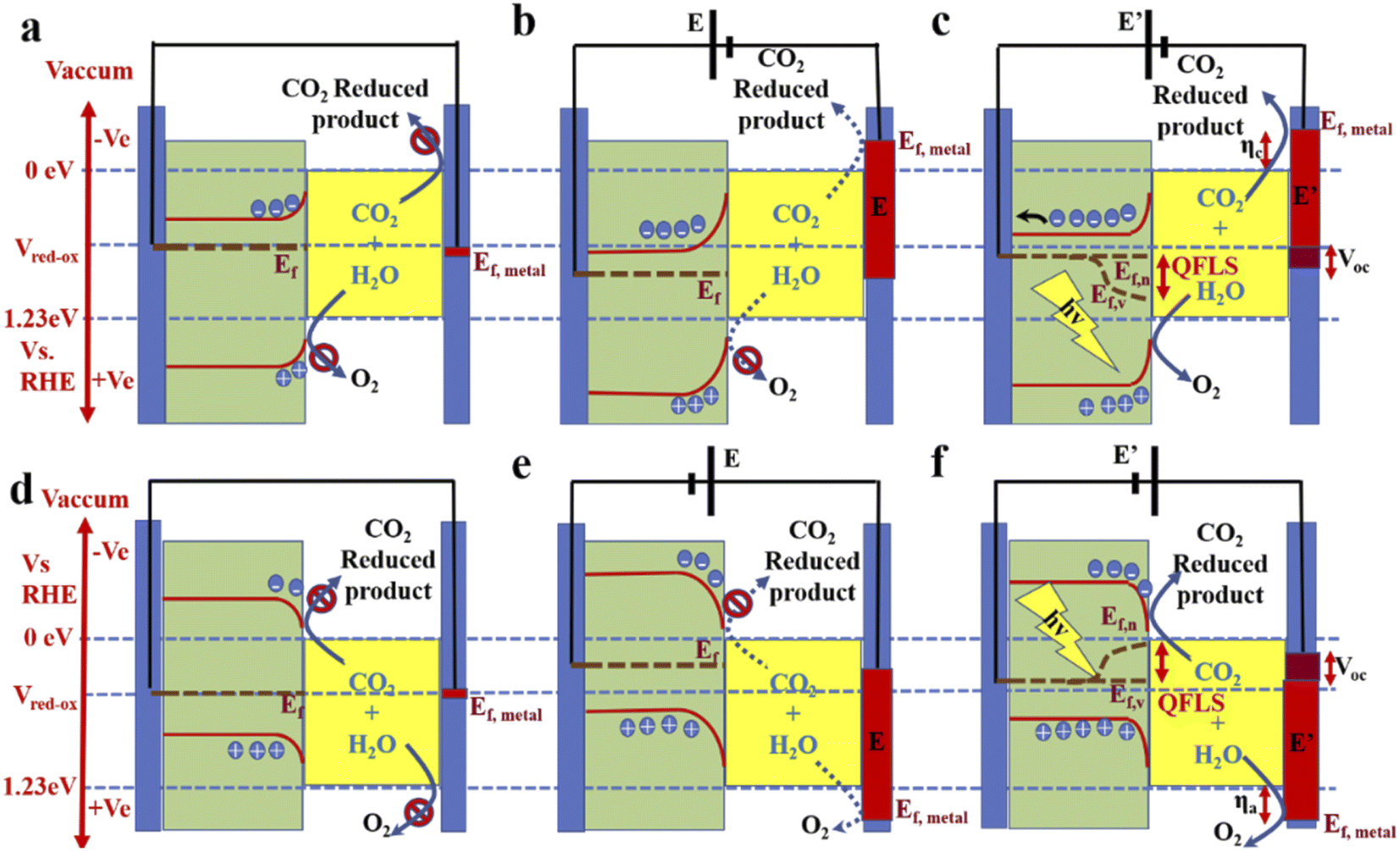 | ||
| Fig. 3 A diagram of band bending in a semiconductor electrode: (a) photoanode dipped inside the electrolyte at equilibrium, (b) with an applied external forward bias, and (c) with additional photo-illumination. (d), (e), and (f) Band diagrams for a photocathode under similar conditions to (a), (b), and (c), respectively. | ||
2.5. Electrocatalysis (EC)
In this process, CO2 is reduced with the help of an applied bias provided by an external source. However, EC is convenient, scalable, and will be viable in the future as the external potential can be provided by any green energy source. In this way, reliance on fossil fuels can be reduced. Depending upon the EC cell, many types of EC system exist. The H-type cell is the simplest, where the anode and cathode are separated by an ion exchange membrane (Fig. 4a). Here, due to the low solubility of CO2 in an aqueous electrolyte, diffusion limitation does not allow a high current density to be reached without causing enhanced water splitting. Therefore, to directly feed gas-phase CO2 to the cathode, Gas Diffusion Electrode (GDE) is explored where three-phase interfaces, made up from a solid catalyst, liquid electrolyte and CO2 gas, act as active catalytic sites (Fig. 4b). In this setup, current density improved significantly, but in the long term, GDL loses hydrophobicity and electrolyte flooding occurs to inhibit the operation. To avoid the flooding issue, an MEA setup is introduced. In MEA, an ion exchange membrane is sandwiched between the GDE and anode catalyst layer; as a result, only an anolyte is used, which reaches the catalyst surface of GDE by diffusion (Fig. 4c). This minimizes the risk of flooding; however, another problem of salt precipitation becomes significant, which blocks the pores of GDE. However, feeding humidified CO2 marginally decreases salt precipitation issue. To further improve the current density, a zero-gap MEA is constructed. A compact design, with no dedicated electrolyte chamber, significantly reduces iR loss occurring from electrolyte resistance. And the anolyte is flowed through the flow channel from external storage (Fig. 4d). In some cases, a zero-gap MEA is built where on both sides a flow channel is inserted and the cathode is fed with CO2 plus an electrolyte mixture while the anode is fed with DI-water. This is sometimes called an MEA flow cell. This setup significantly improves the salt precipitation problem (Fig. 4e). However, a simple flow cell is one where in the GDE setup, both sides of the electrolytes of the ion exchange membrane are being flowed through the electrolyte chamber from external storage. We will discuss MEA cells in a separate Section (4.2.2).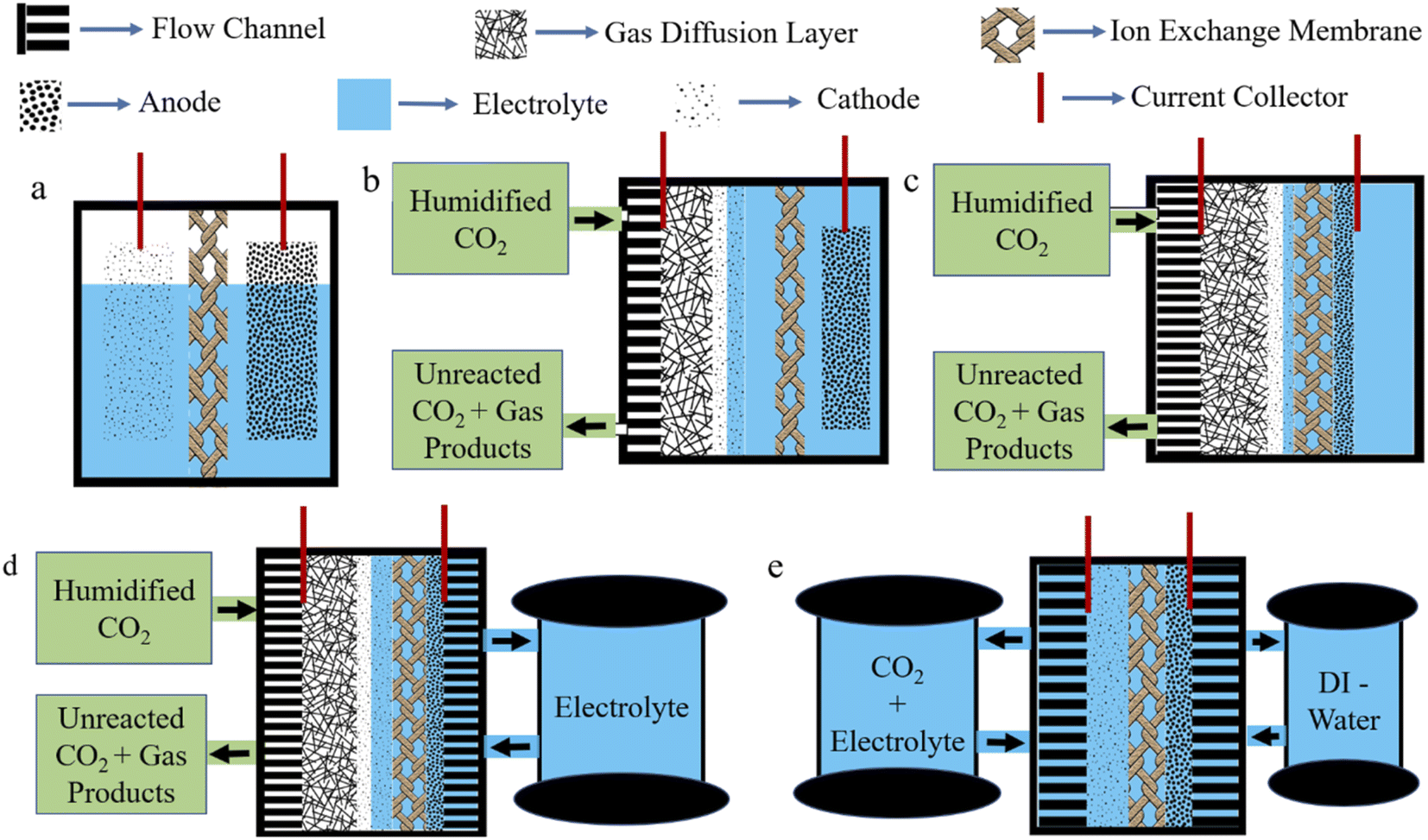 | ||
| Fig. 4 Graphical representations of (a) a conventional H-type cell, (b) a GDE cell, (c) an MEA cell, (d) a zero-gap MEA cell, and (e) an MEA flow cell. | ||
2.6. PV-electrocatalysis/photoelectrocatalysis (PV-EC/PEC)
In this system, a photovoltaic cell is coupled with an electrocatalytic cell. Photovoltaic cells are becoming more efficient and more stable in the long term day by day, and the synthesis cost is also showing a declining trend.77 Moreover, by taking stand-alone PV cells for solar light harvesting, there is no need to dip the semiconductors in the corrosive electrolyte. Hence, the chance of electrocorrosion or photocorrosion in the photo-harvesting unit is bypassed. There is another possible division called PV-PEC, where one PV cell is connected and aligned in a tandem fashion with a photoelectrode. We have discussed this kind of work in a separate paragraph in the ‘PV-EC/PEC section’ (4.3).2.7. Pros and cons of EC/PEC in aqueous electrolyte
In the above discussion, we have discussed various CO2RR processes with their superiorities and inferiorities. Now, the electrolyte can be non-aqueous, like various ionic liquids. An ionic liquid is very interesting and useful in battery and super capacitor applications due to having a broad range of stable potential windows. In EC CO2RR or water splitting, our target is to operate the electrolytic cell at a minimal voltage requirement. Moreover, ionic liquids are very costly and inflammable, which are the main constraints to industrial viability. Above all, water acts as a solvent cum sacrificial reactant which provides the hydrogen for CO2RR.3. Possible mechanism and efficiency standards for PEC/EC CO2 reduction
3.1. Possible surface electron transfer mechanism for the CO2RR
Several mechanistic pathways can even give rise to any particular product. Depending on the catalyst surface, several steps can occur in which proton transfer, electron transfer or concerted transfer of both can be possible, and water elimination, dimerization, and insertion may also occur. Possible mechanistic pathways for common CO2RR products are outlined in Fig. 5. Formate can occur via single-electron transfer forming a rate-determining CO2˙− radical anion and then a further proton/electron pair transfer.78 This pathway is most commonly reported for p-block formate-selective materials like In, Sn, Bi, Pb, and Sb. Proton coupled electron transfer (PCET) to CO2 can also occur directly from an HCOO* intermediate, which further reduces to formate.79,80 In another pathway, CO2 gets inserted into the surface-adsorbed hydroxyl group to form a bicarbonate intermediate (HCO3*), which is then reduced to an HCOO* intermediate and finally to formate.81 However, CO can be generated from an *COOH intermediate, which may be generated via PCET. For CO formation, a weak binding energy between *CO and the catalyst surface is required, depending on it, *COOH formation or CO desorption from a *CO intermediate becomes a rate-determining step (RDS).82 If decoupled electron–proton transfer gives rise to a *COOH intermediate, the radical anion formation step becomes the RDS.83–85 Whereas, formaldehyde is formed in a step-by-step PCET from a *COOH intermediate via a *CHO intermediate.86,87 Methanol is also produced from the common *CHO intermediate.88 The *CH3 intermediate, which originates from either a carbene intermediate via *CO or through a *CHO intermediate, can generate several products. It can form CH4 (ref. 89) or dimerize to ethane, or with carbon dioxide radical anion insertion90 may even produce acetic acid. In C2 product formation, *CO dimerization plays a crucial role in forming *C2O2 as the RDS. Step-by-step reduction produces a very important vinyl alcohol intermediate (CH2CHO*), which diversifies to several product-forming paths.91 Ethylene is produced from the intermediate via splitting the C–O bond.92 Ethanol is formed via an ethoxy intermediate (*OCH2CH3).93 Ethylene can also be directly formed via non-electrochemical dimerization of a carbene intermediate (*CH2).82 C2 products can also form via CO insertion in a *CHO intermediate or by dimerization of a *CHO intermediate, as shown in Fig. 5.94 For n-propanol formation, *CO undergoes several reductions to form a carbene intermediate (*CH2) and then CO insertion occurs into the carbene intermediate. Further reduction give rise to another carbene intermediate (*CHCH3) into which similarly one more CO gets inserted. Finally, reduction of that intermediate forms n-propanol. This is generally reported in such an optimal potential where both carbon monoxide and ethylene formation rates are very high. Therefore, it is thought that these are the prime precursors for n-propanol formation.95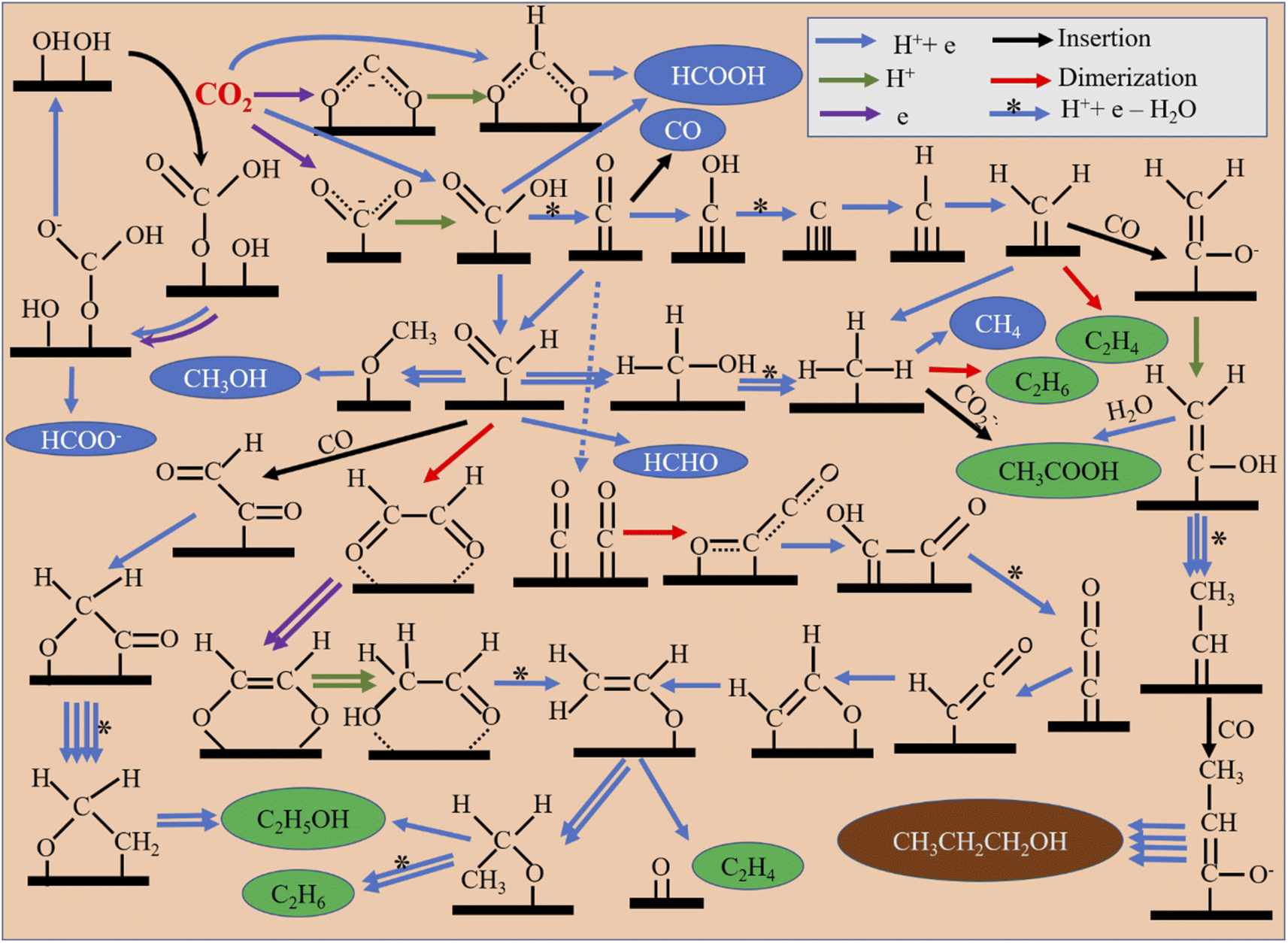 | ||
| Fig. 5 Possible mechanistic pathways for different product formation processes. | ||
3.2. Efficiency standards for the CO2RR in various catalytic processes
CO2 reduction is a complex process as it can give rise to various products depending on how many electrons are transferred during CO2 reduction. Hence, selectivity is primarily an essential parameter, as lower selectivity will cause huge product separation costs, and the catalyst cannot be industrially viable. However, selectivity does not give any idea of the activity of the catalyst. Therefore, a suitable catalyst must have optimal activity at the highest selectivity for industrial-scale production. Moreover, a suitable catalyst needs long-term stability and low synthesis cost to be industrially acceptable. Several photo-harvesting efficiency parameters denote photoactivity for photoactive catalysts. Here, we will discuss several efficiency parameters used in EC and PEC systems one by one.In EC, for a half-cell electrode study, the current density at an applied voltage is reported to denote the activity of the catalyst, which corresponds to the product formation rate. The product selectivity at the corresponding potential is reported in terms of faradaic efficiency (FE), which is defined as:96
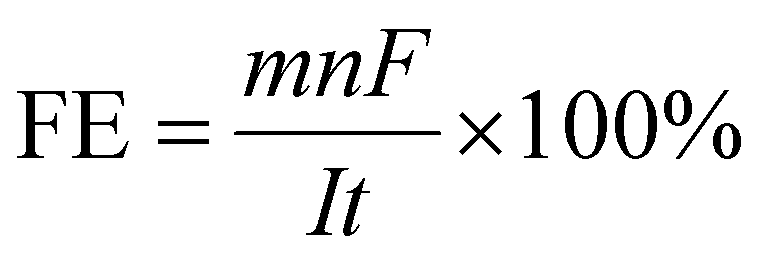 | (1) |
487C mol−1), I (A) is the reaction current, and t (s) is the reaction time. The ultimate goal is to achieve the highest FE for a particular product with a high current density at minimal overpotential. In full electrocatalytic cell operation, another parameter, electrical/energy efficiency (EE), is important, accounting for electricity loss other than through product formation to overcome overpotential, electrolyte resistance, membrane and interface potential, etc. EE can be defined as:97,98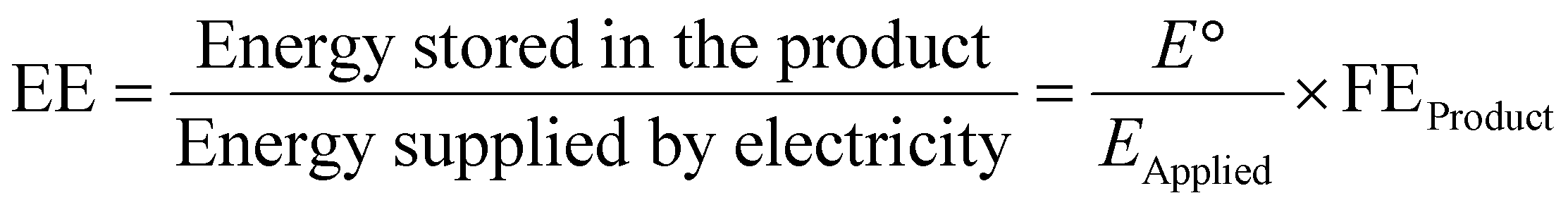 | (2) |
For PEC, in the case of an active half-cell photoelectrode study, efficiency is reported similarly to that of the EC system. In most cases, the overpotential requirement is reduced under illuminated conditions compared to dark conditions for PEC, or improved selectivity towards a product is observed. However, incident photon-to-current efficiency (IPCE) is reported to determine the light-harvesting efficiency at different wavelengths. IPCE is plotted against wavelength for representation and is defined as:99
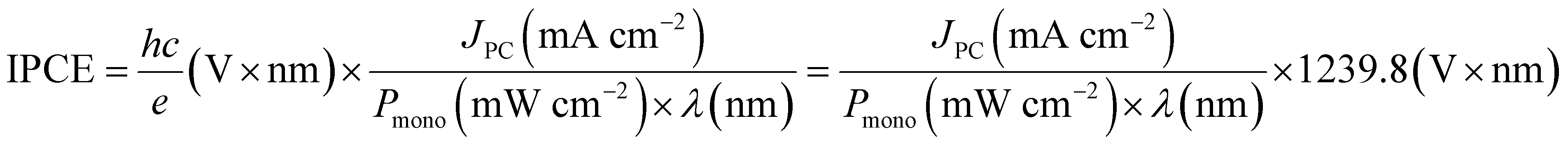 | (3) |
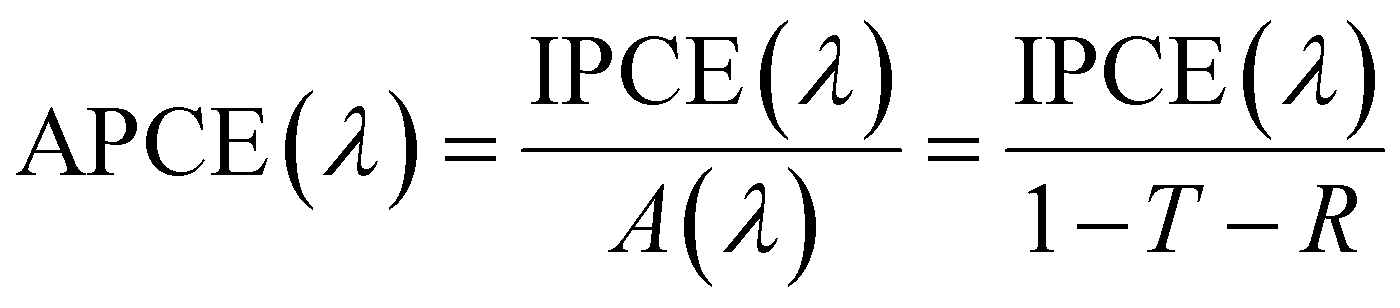 | (4) |
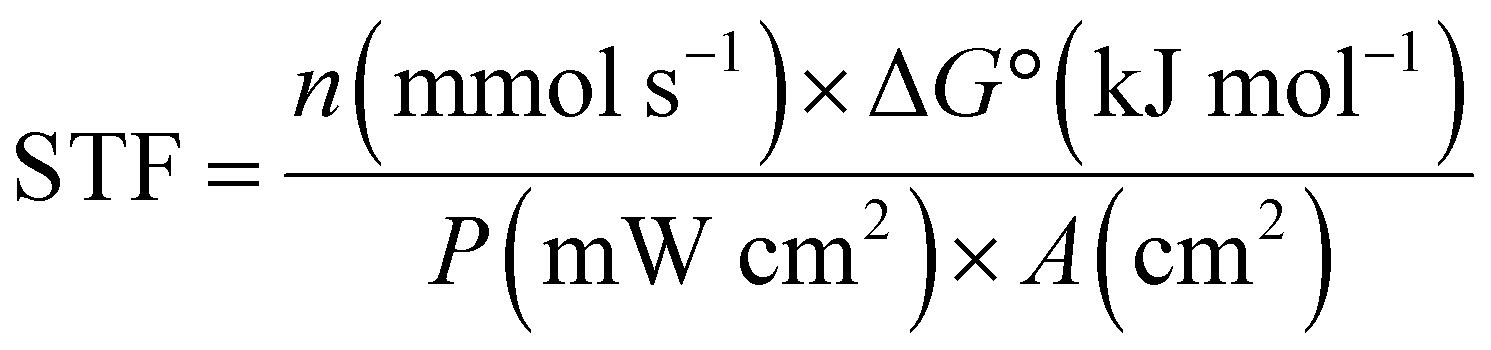 | (5) |
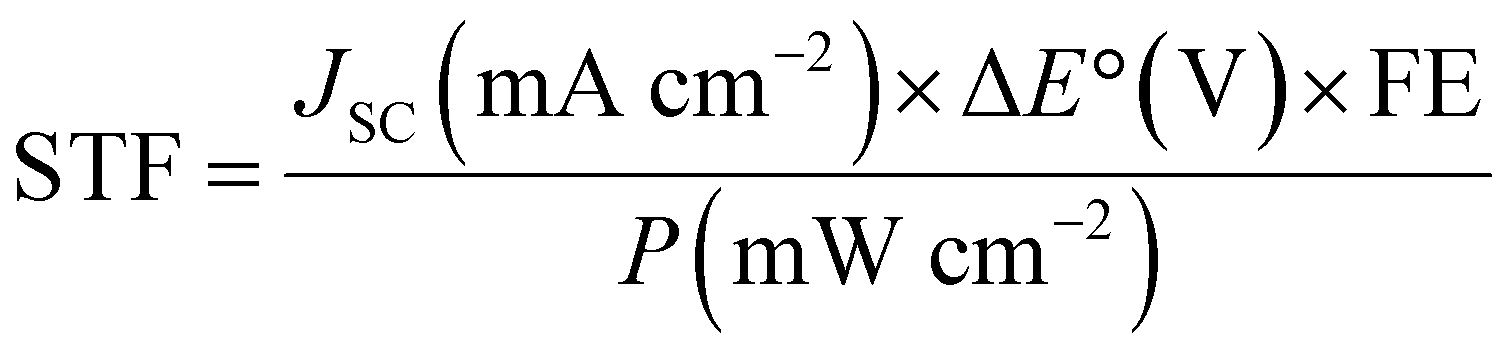 | (6) |
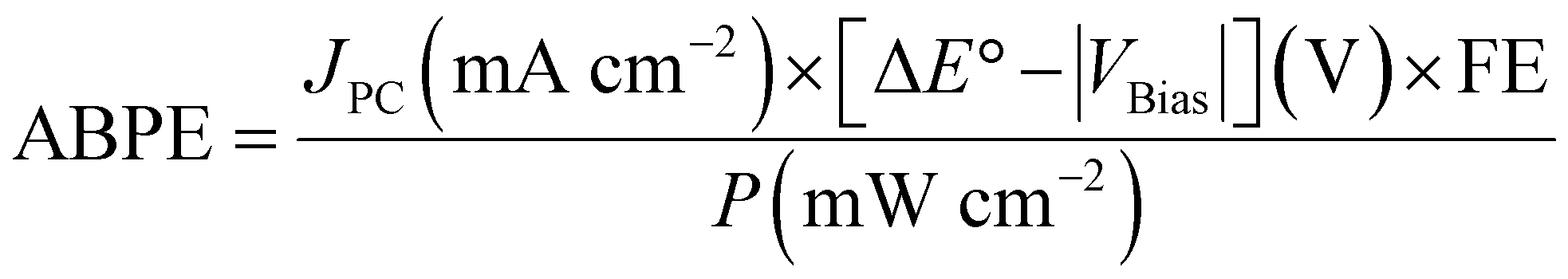 | (7) |
For PV-EC/PEC, the overall merit is reported in a complete cell operating system with cathode and anode, as the PV cell needs to be connected to both terminals for operation. However, multiple solar cells are used in series to provide the required voltage for electrocatalytic cells. Therefore, extra electrical bias is not required, and the whole device operates under light energy alone. So, reporting of STF efficiency is widespread in this kind of system. Here, activity is denoted by the operating solar current density at corresponding cell potential. Unmatched high STF is reported in this system because PV cells have become highly efficient in solar light harvesting.
4. Benchmarking of the materials and processes for the CO2RR
4.1. PEC system
There are two broad differences in selecting photoelectrode materials. In one case, an already optimized PV-cell material is integrated into the photoelectrode by modifying its surface through structural engineering or making junctions, or loading with other cocatalysts. These photoelectrodes are highly efficient and well-known photo-harvesters. Moreover, photogenerated electrons can easily migrate to the electrode surface to reduce CO2 efficiently due to good charge-carrier separation efficiency. As a result, these PV-cell modified photoelectrodes have an unmatched high current density at much lower overpotential in half-cell operating conditions. In the second case, general semiconductors, non-conventional as PV cell material, are utilized and generally have average photo-harvesting capacity. So, due to this factor, we will discuss general photoelectrode systems and PV-cell modified systems in separate sections.4.1.1.1. Photoanode with a dark cathode. In EC, the anode has no role in assisting the cathode in CO2RR except by providing another half-cell to complete the other half-reaction. But in the case of PEC, a photoactive anode can provide additional photovoltage, which will assist the dark cathode in showing CO2RR activity or in altering product selectivity. In 2015, Jin et al.102 made a cobalt carbonate (Co–Ci) tailored, thin coated, very transparent photoanode to realize anode-assisted CO2RR in the dark cathode. Fig. 5a shows a graphical illustration. They deposited WO3 on FTO, then a BiVO4 coating over this, and finally, Co–Ci was incorporated on it by a photo-electrodeposition method. Under the photoanode-illuminated conditions, they reported 46.8% CH4 selectivity at a comparatively very low anodic potential of +0.4 V vs. reversible hydrogen electrode (VRHE) with a Cu counter electrode. In 2016, Chang et al.103 reported TiO2 nanorod (NR) photocathode-assisted CO2 reduction on a Cu2O cathode. At +0.75 VRHE, the dark Cu2O cathode with an illuminated TiO2 NR photoanode as the working electrode gave a stable current density of 1.34 mA cm−2 for a 3 h test time. This corresponds to only 6.94% H2 formation with 54% CH4, 30% CO and 2.74% methanol. However, when Cu2O is used as a photocathode, it degrades rapidly. When a protective layer of TiO2 was grown by atomic layer deposition (ALD) on Cu2O nanoparticle (NP), the H2 formation increased to 41.61%, and CH4 selectivity dropped to only 22.7% due to the masking of active sites. They also found that the holes are more responsible for Cu2O degradation. Back illumination provides a short hole travelling distance to the conducting support, making it less corrosive than front illumination. They used a dark cathode to avoid photocorrosion completely. By the end of 2017, Kang and his group made a hierarchical (040)-facet exposed BiVO4 photoanode.104 They found higher positive anodic potential is (photoanode used as working electrode) shifting selectivity towards the higher reduced product. At 0.75 VRHE, they got 2e-reduced formate product with 65% selectivity, and at a small increased potential of 0.9 VRHE, the major product was 4e-reduced formaldehyde with 85% faradaic efficiency (FE). Whereas, at 1.35 VRHE, 6e-reduced product methanol and 12e-reduced C2 product ethanol were formed with 5% and 4% FE, respectively. They also found by in situ XAFS that N2-purged solution with a simulated Cu–Cu distance of 0.21 nm matched the experimental values. In the CO2-purged electrolyte, at 0.9 VRHE, when formaldehyde was the major product, the main peak shifted to 0.195 nm from 0.21 nm. At a much higher potential of 1.35 VRHE, two separate peaks at 0.18 nm and 0.21 nm corresponded to the simulated values of Cu–C and Cu–Cu bonds, respectively (Fig. 4b; the order potential increases from bottom to top). They claimed that the increase in the concentration of Cu–C bonds facilitates the formation of higher reduced methanol and ethanol. Xiaoxia et al.105 air-oxidized a Cu plate, then made a surface coating of Cu NPs of varying deposited thickness by a homemade e-beam vaporization technique. The catalyst with optimal thickness gave 53.6% methanol selectivity as a dark cathode with a current density of 1.31 mA cm−2 with an illuminated TiO2 NR photoanode. In a bare Cu2O dark cathode, more selectivity towards CH4 (∼53%) with CO (∼30%) formation was observed instead of methanol selectivity at the same current density. In 2019, Kang and his group reported another detailed study on a (040)-facet exposed BiVO4 photoanode assisted CO2RR after the work of 2017.106 They found Nafion coating on a dark TiO2 cathode could alter CO2RR selectivity from ethanol to methanol. An acetonitrile–water mixed solvent was chosen to boost CO2 solubility with tetra-ethyl ammonium perchlorate (TEAP) as an electrolyte. In situ XANES peak positions showed that the TiO2 surface contained both Ti3+ and Ti4+, but when potential was applied with illuminated photoanode, only the Ti3+ ion was found on the surface. This may happen due to higher e-density being transported from photoanode (Fig. 6c and d). Corresponding EXAFS data suggests the Ti–O interatomic distance reduced from 1.87 Å to 1.77 Å in the operating conditions and for the Nafion-coated cathode it reduced from 1.65 Å to 1.59 Å (Fig. 6e and f). The in situ XAS study setup is graphically described in Fig. 6g. They also captured the carbon dioxide radical anion by N-tert-butyl-α-phenylnitrone (PBN) and probed the resultant complex through in situ EPR (Fig. 6h and i). They hypothesized that Nafion coating enhances proton concentration at the TiO2 surface, making proton transfer kinetically faster, preventing the intermediate from dimerizing. These give rise to methanol preferentially with H2, whereas bare dark TiO2 favors dimerization and ethanol becomes a significant product. In 2020, Lin and his group made a Cu NP modified flower-like reduced graphene oxide decoration on Cu foam. They used a 5% Pt-loaded TiO2 photoanode to provide photovoltaic compensation to the dark cathode. In dark conditions, the oxygen evolution reaction started at 1.65 VAg/AgCl which under illumination shifted to a much lower potential of 0.85 VAg/AgCl. Compared to their control cathode, in the best cathode, acetic acid selectivity increased from 26.7% to 31.8% and ethanol selectivity from 28.3% to 31.1%, but no other product formation was observed.107
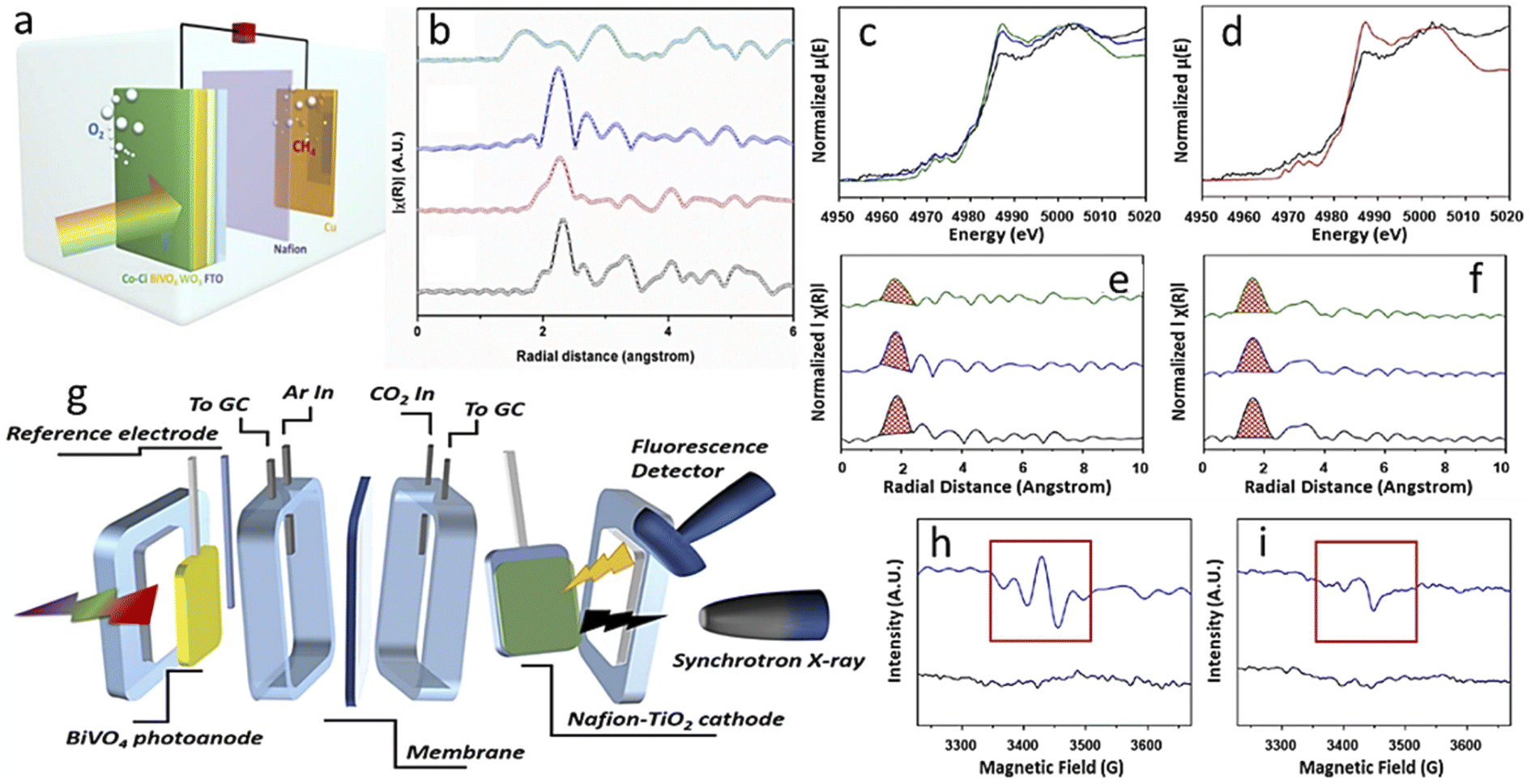 | ||
| Fig. 6 (a) Photoanode-assisted methane formation. (b) EXAFS fitting data. (c), (d) XANES spectra and (e), (f) the corresponding EXAFS fitting of basic and Nafion-modified electrodes, respectively. (g) A graphical representation of the in situ XAS setup. (h), (i) EPR data (a) is reprinted with permission from Kim et al.,102 Copyright 2015, Elsevier Ltd. (b) is from Kim et al.,104 Copyright 2018, American Chemical Society; (c)–(i) from Kang et al.,106 Copyright 2019, American Chemical Society. | ||
4.1.1.2. Photocathode with a dark anode.
A. Copper-based materials. In PEC CO2RR, copper-based materials have a monopoly over several other types of semiconductors. These materials are cheap, earth-abundant, and can significantly reduce the high overpotential requirement for CO2RR. Even some materials with Cu2O were reported to show CO2RR activity at underpotential conditions (below the required thermodynamic potential). However, these are not the only reasons for this monopoly. Copper-based materials can give rise to various higher reduced products like methanol, methane, and C2 products like ethanol, ethylene, acetate, oxalate, and C2+ products. However, the biggest drawback is that its shallow stability as a semiconducting active photo-harvester gets reduced under cathodic potential in the presence of an electrolyte. Therefore, numerous approaches have been initiated to stabilize the material either by passivating the catalyst surface with a different protective coating or utilizing a binary salt material like CuFeO2, CuBi2O4, CuInS2, etc. These modifications also additionally improve the selectivity with operational stability. Here, we will summarize the progress of primary Cu-based materials as photoelectrodes.
Cu2O . Between the two oxides of copper, Cu2O has endured because of its favorable bandgap for absorbing a more extensive range of wavelengths from solar light. Theoretically, Cu2O can reach a very high photocurrent density of 14.7 mA cm−2.108,109 Moreover, its conduction band minimum (CBM) is also located at a very negative potential, making it a superior choice as a photocathode material for CO2RR. Many studies have reported this material to produce higher reduced liquid products like methanol, ethanol, and acetone at very low cathodic overpotential, sometimes even at underpotential, which is rarely reported with other materials. But one thing that stops it being a universal photoelectrode is its fragile stability. It is highly prone to photocorrosion and electrochemical corrosion and loses its activity many fold over a brief period. However, keeping in mind its superior potential as a photocathode, many attempts have also been made to increase its stability and current density.
By 2013, Rajeswer and his group claimed the first use of copper-based oxide for CO2RR.110 On thermally synthesized CuO nanowire, they electrodeposited Cu2O and reported surprisingly high methanol selectivity of 95% at −0.2 VSHE (150 mV underpotential compared to the thermodynamic reduction potential) for 2 h of operation. They did not use any homogenous catalyst-assisting chemicals like pyridine, which are known to take an electron from the electrode and do the PCET CO2 reduction step by activating CO2. They did not undertake detailed stability tests at this time, but with simple XRD after 2 h of operation, no Cu0 formation was reported. However, after some months, they published another report with much-improved catalyst morphology and a detailed stability study.111 This one achieved an enhanced stable current of 1.05 mA for a 3 cm2 photoelectrode for 2 h of operation at 70 mW cm−2 light intensity with the same electrolyte (0.1 M Na2SO4) and potential with unaltered selectivity towards methanol. They reported the self-healing property of the photoelectrode, as a decreasing current for the first 30 min starts to rise back to the initial value after those 30 min during the 2 h test time. XRD peak intensities corresponding to Cu0 increased at 43.2° and 74.1° after 30 min of electrolysis and decreased after 2 h, supporting this claim of a self-healing property. In 2014, Li et al.112 electrodeposited double-layered Cu2O on volcano-shaped Fe2O3 nanotubes. They also found 93% methanol selectivity after a comparatively long PEC study (6 h), but with low current density. In parallel, Won et al.113 studied a mixed copper oxide with the surface deposition of several metal particles like Ag, Au, Cd, Cu, Sn, and Pb. They achieved the best activity and selectivity with Pb with an improved current density of 0.8 mA cm−2 at the cost of compromised low and mixed product selectivity, which decreased in 1 h of test time. Ba et al.114 electrodeposited p-type and n-type Cu2O and undertook electrocatalysis at −2 VAg/AgCl to get nearly 30% FE towards C2H4 with a p-type belt-like Cu2O morphology. They got almost a 3% selectivity increment with 10 mW cm−2 blue LED light emission, which was quite interesting. In 2015, after the first pioneering work in 2012, Rajeswer and his group reported another study on Cu2O for product selectivity re-distribution with applied potential, reaction time, and pH variation.115 For the PEC test at 0.2 VAg/AgCl, they found only methanol formation after 30 minutes, but after 120 minutes, the C2 product started to increase. They also noticed that methanol was the main product at 0.2 VAg/AgCl, but acetone became the main product at an increased cathodic potential of −0.2 VAg/AgCl. In contrast, at −0.4 VAg/AgCl, acetone became the only product (Fig. 7a). They also found that with a change in pH from 9 to 11, the selectivity shifted from methanol to acetone (Fig. 7b). According to the authors, at higher pH, the concentration of bicarbonate ions decreased, and carbonate ions dominated; as a result, the activity of the hydroxyl radical significantly increased with respect to the hydrogen radical. This factor insists on oxidating coupling preferentially over the simple PCET reduction. One thing that became clear is that copper oxide gets highly photocorroded and electrocorroded. The actual catalysis may not come from Cu2O, whereas in situ prepared Cu NPs on the surface of Cu2O may be responsible for an effective electrocatalytic reduction process. Therefore, at the start of 2018, Lee et al.116 tried to passivate electrodeposited Cu2O with crystalline TiO2 by dip-coating followed by annealing. They also modified the surface with Cu+ ions for improved catalytic activity. At 0 VRHE, the bare sample current drops from 2.8 mA cm−2 to only 5.3% in 30 min; however, the current density of the passivated sample became stabilized by dropping to 27.6%. At 0.3 VRHE, they reported 55% methanol selectivity in 2 h of operation under illumination, which claims further optimization of stability of passivated Cu2O. Then, Kang et al.117 published another study with TiO2 passivation with an entirely new approach. They made a coating of Cu precursor on a Cu-layer deposited FTO by electrospinning, followed by two-step calcination at 500 °C. The first calcination is done in air to oxidize and remove all the carbon precursors, and the second in 10 μtorr pressure to get pure-phase Cu2O nanofiber over CuO selectively. Finally, TiO2 overcoating with an optimal thickness of 5 nm via ALD gave reported 90% methanol with ∼0.8 mA cm−2 current density at 0.4 VRHE (0.2 V underpotential) under illuminated conditions. In 10 min, they found no mentionable activity loss. Refer to Fig. 7c and d to compare the current density and stability with previous TiO2 passivation work. Until then, none of the copper-oxide-based photocathodes could have reached at least the 1 mA cm−2 benchmark, although the theoretically predicted value was much higher.
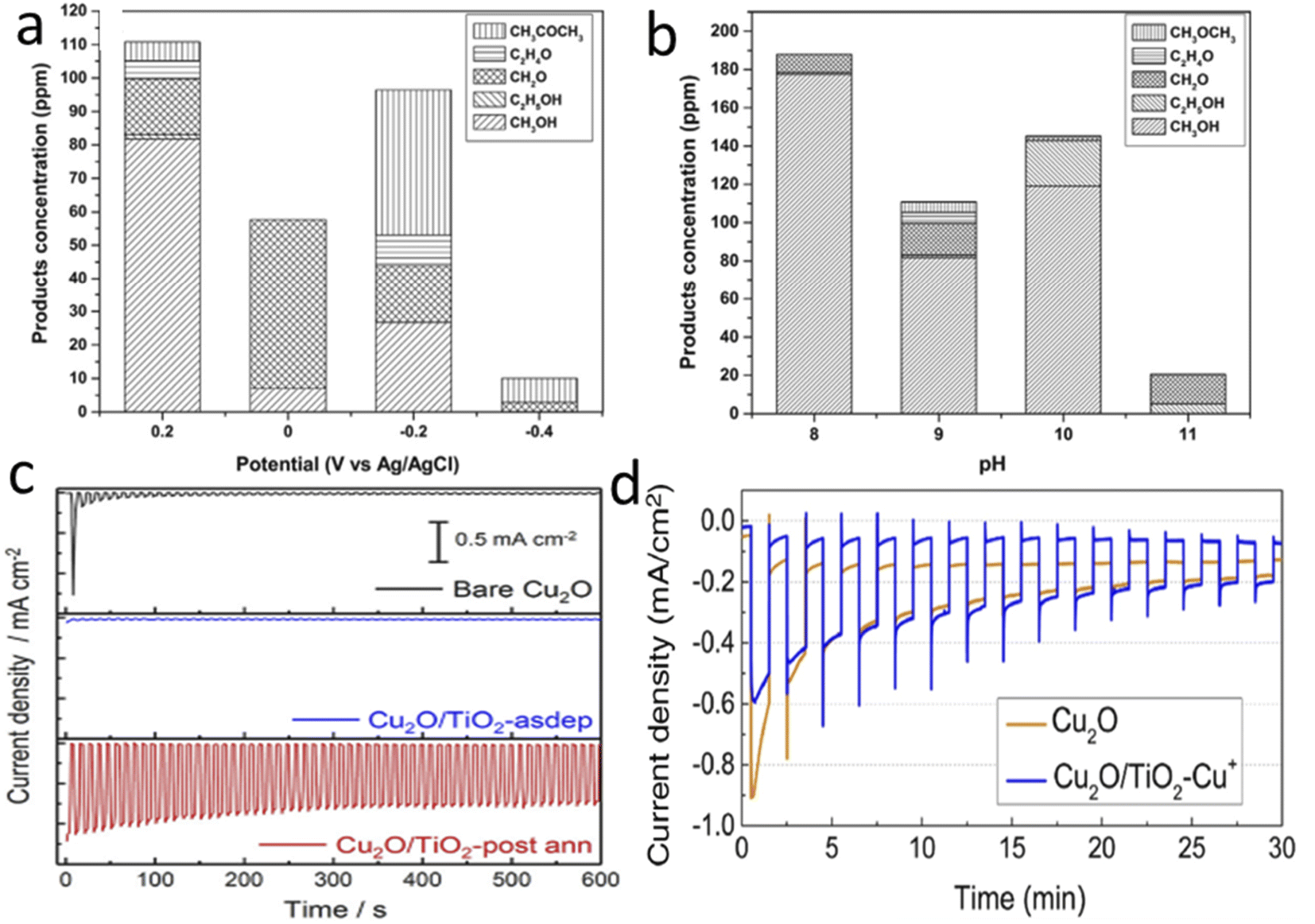 | ||
| Fig. 7 The product distributions after 2 h of the PEC CO2RR (a) at different potentials at pH 9 and (b) at different pH levels at +0.2 VAg/AgCl. Current density vs. time plots clearly showing that the data from the annealed TiO2 coated photoelectrode in the 3rd row of (c) are much more stable than those shown in (d), (a) and (b) are reproduced with permission from de Brito et al.,115 Copyright 2015, Elsevier Ltd. (c) Kang et al.,117 Copyright 2018, American Chemical Society; (d) is from Lee et al.,116 Copyright 2018, Elsevier Ltd. | ||
By the end of 2018, Li et al.118 made a strategic heterostructure with n-type TiO2 to improve the current density of Cu2O material. They suggested the Z-scheme pathway over the type-II recombination, inspired by another previous study on water splitting.119 They sandwiched Au particles between a TiO2 underlayer and an electrodeposited Cu2O surface layer, which acted as the Z-scheme favoring agent and achieved a current density as high as 1.82 mA cm−2 for PEC CO2RR at a cathodic potential of 0.11 VRHE. They also provided supporting data for their claim by preferential chemical redox deposition on the specially made exposed TiO2 surface and Cu2O exposed electrode surface by SEM element mapping data. However, they did not provide any data on product selectivity. In 2019, Zeng et al.120 reported Ti3C2 quantum dot modified Cu2O nanowire. They hydrothermally broke the Mxene sheet to form quantum dots (QD) and treated them with PEI to make a positively charged surface on the QDs. They treated Cu2O NW with poly(sodium-4-styrene sulfonate) (PSS), thereby undergoing electrostatic self-assembly with the QDs to get a negative surface charge. Finally, calcination removed the organic moieties and formed Mxene QD decorated Cu2O NRs (Fig. 8a and b for SEM images). TEM data in Fig. 8d and e confirms the presence of the QDs on Cu2O NR. This composite photocathode had a lower bandgap of 2.02 eV than the bare Cu2O NW with 2.2 eV, indicating a higher photon absorption range. Under 1 sun with optimal QD-loaded photocathode, they reported a steady current density of nearly 5 mA cm−2 (Fig. 8c) for 30 minutes of operation. They got methanol as the only detectable product, and the methanol formation was 8.25 times higher than with the unmodified Cu2O NWs. Moreover, after 6 cycles of repetition, 89% of the material activity was conserved (Fig. 8f). In 2020, many studies on passivating Cu2O were reported. Szaniawska et al.121 made a coating with tungsten carbide and a carbide-derived carbon mixture, and after a considerable loss of activity over 1.5 h, the photocathode stabilized to 0.25 mA cm−2 current density in a 4 h long operation at 0.1 VRHE. They reported methanol as the main product with a small amount of ethanol without any quantitative data.
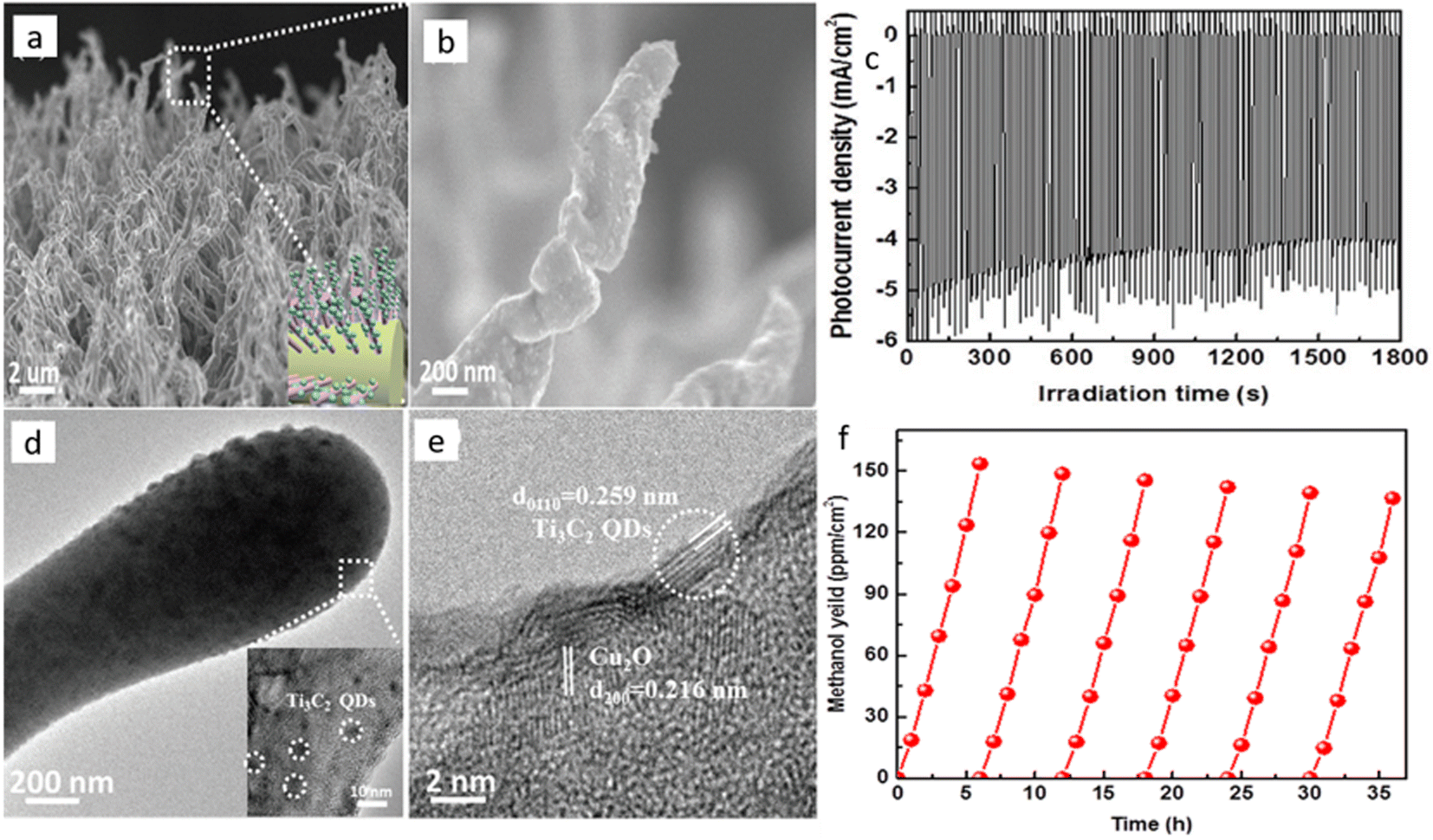 | ||
| Fig. 8 (a), (b) SEM images of Ti3C2 QD loaded Cu2O NWs. (c) The photocurrent vs. time curve. (d), (e) TEM images of the same composite. (f) Re-cyclability data over 6 cycles. Reproduced with permission from Zeng et al.,120 Copyright 2018, John Wiley and Sons. | ||
Galante et al.122 deposited aminopolysiloxane as an overlayer on Cu2O to protect the underlayer, and the free amine group of the polymeric chain acts as a CO2 activating center. In situ FT-IR confirmed that a higher applied potential enhances CO2 adsorption in the electrode surface. After 2 h of operation under illumination, 61% FE towards formate was reported at a high underpotential condition of −0.3 VAg/AgCl (0.31 VRHE). After their premium work on Cu2O passivation in 2018, Joo and his group came up with another report on Cu2O passivation in 2020.123 This time, they also synthesized the Cu2O phase by the previous calcination method, but at a comparatively higher partial pressure of O2 (in millitorr instead of μtorr pressure) with an increased calcination temperature at 650 °C instead of 500 °C. They smartly overcoated Cu2O with CuFeO2 by simply spraying an iron precursor on the Cu2O layer, followed by controlled calcination. They reported a comparatively high current density of 1.1 mA cm−2 at an external potential of 0.35 VRHE. They also got 68.6% FE towards acetate, which is rarely reported for copper oxide-based materials with 21% formate. However, they claimed higher stability of their photocathode due to sustaining 10 LSV scans with a minimal activity loss of 8% compared to the bare electrode, which lost 50% in just two LSV scans. By 2022, Zhang et al.124 reported one photocathode with pearl-like Cu2O nanowires by anodizing Cu foam and corresponding high-temperature annealing in N2 followed by electrodeposition of SnOx. With 50 min deposition time for SnOx, they got the best photocurrent density of 11.61 mA cm−2 at −0.55 VRHE with a CO formation rate of 141.79 μmol cm−2 h−1 with a formate production rate of 19.57 μmol cm−2 h−1. Again, in a 12 h stability test at −0.35 VRHE under 1 sun, CO selectivity remained at 90.32%.
Copper–iron oxide.
Jing Gu and his group utilized Mg2+-doped CuFeO2 in 2013 by a thermal process for CO2RR with very weak formate selectivity of 10% at −0.9 VSCE.125 Mg2+ doping of 0.05% could replace one trivalent cation or substitute three monovalent cations to create a hole that will increase the material's conductivity and induce a p-type character. However, after 8 h of operation, XRD data showed slight Cu0 formation, which was not even detectable in SEM. This also supports the idea that the catalysis does not originate from well-known Cu0 particles. However, after 24 h of operation, both XPS and XRD denoted the formation of Cu0. EDX also showed that the Cu:Fe ratio increased from 1:1 to 1.3:1 after 24 h. By 2015, Kang and his group electrodeposited CuFeO2/CuO heterostructure on an FTO plate with a thickness of a few micrometers.126 They reported the highest formate formation rate at a significantly low external bias of 0.15 VRHE. The photocathode with a Pt anode can operate even without a bias under illumination. With this significant achievement, they reported 1% STF efficiency, which stabilized to 0.7% after 5 h and was sustained for 7 days of operation with a formate selectivity of 90% and an expectedly low current density. ICP-MS data of the electrolyte solution taken after a seven-day test of this photocathode showed no Cu or Fe leaching. Still, they reported the partial one-electron reduction of both Cu2+ and Fe3+ in the electrode. By the end of 2016, Yang et al.127 also electrodeposited CuFeO2/CuO and found 80% FE at −0.4 VAg/AgCl towards acetate, which is very rarely reported; however, it had a very low current density. Depth-profiled XPS data confirmed that the surface layer contained Cu2+ consisting of CuO, and the underlayer contained Cu+ representing CuFeO2. They used biphosphate electrolytes to ensure that CO2 gives rise to the product, not the bicarbonate part of the electrolyte. They got a higher amount of acetate formation at the same applied potential. However, they found severe Fe3+ leaching from the electrode surface in a short time. As the Fe:Cu ratio decreased from 1.3 to 0.1, the selectivity shifted from acetate to formate, and finally, the catalyst was deactivated. By 2018, Karim and his group synthesized CuFe2O4 instead of CuFeO2 by a simple sol–gel method.128 With this photocathode, they found 68% methanol selectivity at −0.5 VNHE. In contrast, CuFeO2 is known to give mainly formate selectivity. All the above-discussed work established copper–iron oxide as a suitable catalyst for high selectivity at a minimal current density (<1 mA cm−2). Keeping this factor in mind, in 2020, Joo and his group smartly overcoated the Cu2O layer with CuFeO2 and got a 1.1 mA cm−2 current density for acetate formation at an incredibly low external potential of 0.35 VRHE.123 Yuan et al.129 decorated CuInS2 thin film with CuFeO2 NPs and got a methanol formation rate of 15.40 mol h−1 m−2 with 88% selectivity at 170 mV overpotential (−0.7 VSCE).
CuInS2. In 2013, Yuan et al.130 electrodeposited Cu–In alloys on FTO, and by annealing in the presence of sulfur, they made a CuInS2 thin film. This thin film photocathode provided 97% FE towards methanol at a very low applied overpotential of 20 mV (−0.54 VSCE). They concluded it followed a pyridine-assisted CO2RR pathway bypassing the single-electron reduced CO2 intermediate pathway, and the thin film gave a very low but stable current density up to 11 h. In 2016, they reported a single-step electrodeposition process of heterostructured graphene oxide (GO) with CuInS2.131 The reported single-step CuInS2 deposition process was similar to that of Xu et al. in 2011.132 They used DMSO as a solvent instead of H2O because the sulfur precursor, thiosulfate, is unstable in water, and a slight amount of water reduction may give rise to pinholes in the thin film. Simultaneous deposition of Cu, In, and S with GO was found to occur between −1 VSCE and −1.3 VSCE and a more negative potential resulted in the deposition of larger size particles. Surprisingly, they got the CuInS2 phase by this simple electrodeposition, without any heat treatment or calcination. However, EDS data suggested that the optimized sample deposited at −1.3 VSCE is Cu rich, and this time, they achieved a 1.39 mol h−1 m−2 methanol formation rate at a current density of 2.62 mA cm−2 with nearly the same 95% FE. A slightly higher overpotential of a total of 70 mV was observed compared to the previous work, with a stable 10 h of operation for every six cycles. By 2020, they came up with CuInS2 thin film decorated with CuFeO2 NPs and showed a methanol formation rate of 15.40 mol h−1 m−2 with 88% FE, including 10% ethanol selectivity at 170 mV overpotential (−0.7 VSCE) with up to 9 h stability in the same electrolyte solution as used previously.129
B. Zinc-based photocathode. In 2014, Lee and his group claimed the first utilization of ZnTe in PEC CO2RR.133 They synthesized ZnO nanorods (NRs) on the Zn surface, followed by coating with ZnTe using a simple hydrothermal method. They cleverly utilized a simple property of a sparingly soluble salt called the solubility product, Ksp. During hydrothermal treatment, the surface layer of ZnO NRs gets dissolved as Zn(OH)2, and at low temperatures, ZnTe has a lower Ksp. ZnTe crystallizes preferentially on ZnO NRs instead of re-crystallization of ZnO. Although it had a satisfactory current density of nearly 8 mA cm−2, this photocathode had very poor selectivity of only 23% toward CO. The next year, Lee with a different group reported a significant improvement in FE of up to 65% toward CO.134 They deposited nearly 2–3 nm of Au NPs over ZnO/ZnTe nanowire and found a nearly doubled current density of 16 mA cm−2 at the same applied potential as used the previous time of −0.7 VRHE. By 2016, the same group had published further improvement in FE towards carbon monoxide at lower overpotential.135 This time, ZnO/ZnTe NRs were overcoated with CdTe by the same step-by-step hydrothermal method, and finally, Au NPs were deposited. The photocathode provided 5 mA cm−2 current density under illumination with 80% FE at nearly zero overpotential, which is rare in a non-Cu-based material. SEM images from these three different studies are shown in Fig. 9a–f, to compare the morphological improvement in the subsequent study. In 2018, a similar dissolution and recrystallization process was utilized by Cai et al.136 to synthesize ZnSe-coated ZnO. This photocathode showed 26% of FE towards methane at 0.2 VRHE with >0.5 mA cm−2 current density, which is not usual in this material. At −0.4 VRHE, the current density increased to >1 mA cm2; however, methane selectivity reduced to 6%, and CO selectivity rose to 54%. By 2019, Wang and his group synthesized a g-C3N4/ZnTe type-II heterojunction and reported weak alcohol selectivity.137 In 2021, Wen et al.138 deposited an ultrathin protective and insulating TiO2 layer on the ZnTe QD layer by atomic layer deposition and finally decorated it with Ag3Cu co-catalyst which gave rise to a metal–insulator–semiconductor heterojunction photocathode. The open circuit voltage (OCV) was enhanced by more than 3 times in the photocathode to 0.49 V compared to only 0.16 V for ZnTe QDs, ensuring the successful formation of an effective heterojunction. This photocathode was reported to be stable for 24 h operation under 1 sun illumination at −0.2 VRHE with 86.5% CO selectivity and a comparatively high photocurrent density of 5.1 mA cm−2. In 2022, Ouyang et al.139 reported a 1D rough-surface α-Fe2O3/ZnO heterostructure nanorod array with Bi modification showing formate selectivity. The authors claimed that the sharp tip benefits from an accumulation of electrons on the active site, which enhances the catalyst activity. The photocathode under illumination reached 3.75 mA cm−2 current density at −1.2 VRHE and in a 4 h stability test at −0.65 VRHE, 61.2% formate selectivity was observed along with 13.7% methane.
 | ||
| Fig. 9 (a)–(c) SEM images of ZnO nanostructures. (d)–(f) SEM images of the respective ZnTe-overcoated nanostructures (a) and (d) are reproduced with permission from Jang et al.,133 Copyright 2014, John Wiley and Sons, (b) and (e) are from Jang et al.,134 Copyright 2015, Royal Society of Chemistry, (c) and (f) are from Jang et al.,135 Copyright 2016, American Chemical Society. | ||
C. CdSeTe. In 2014, Li et al.140 first utilized this material with anodized TiO2 nanotubes (NTs) for PEC CO2RR. A hydrothermally made nanosheet of CdSeTe was decorated on the TiO2 NT surface. They reported significantly high methanol selectivity of 87.95% at −0.8 VSCE under illuminated conditions for 9 h of operation; however, at higher potential, competitive water reduction increased. In 2016, Wei and his group reported subsequent follow-up work where they deposited CdSeTe nanoparticles by the photoelectrodeposition method on TiO2 NTs.141 The catalyst had superior electrocatalytic properties, with a weak increase in current when exposed to photo-illumination at the same applied potential. However, most surprisingly, the light irradiation made the catalyst more selective towards methanol. FE increased from 65% to 88% at the same external applied potential of −1.2 VRHE. This high methanol selectivity at a comparatively high current density of 6.95 mA cm−2 makes it a commendable photoelectrode, although it has considerable applied potential.
D. Other photocathodes. In 2008, Barton et al.142 utilized p-type GaP for photoelectrochemical CO2 conversion to methanol for the first time. In the presence of pyridinium at −0.4 VSCE, methanol FE was between 88% and 100% at pH 5.2. With a charge passing of 3C to 10C, a linear increase in methanol formation was observed, indicating good stability of the material. In 2018, Xu et al.143in situ prepared Ti3C2/g-C3N4 with different kinds of metal deposited on it. Among them, Pd-loading showed the highest selectivity for CO2 reduction. They reported 100% methanol selectivity at −0.85 V external potential, but they did not provide any data on the corresponding current density. In 2019, Ikeda et al.144 made a kesterite Cu2ZnGeS4 (CZGS) thin film on Mo-coated glass by spray pyrolysis in a sulfur atmosphere. The CO-selective material had very weak stability in the PEC environment. At −0.2 VRHE, the initial current density was less than 0.5 mA cm−2 under 1 sun, rapidly decreasing in the reported 2 h test period. In 2021, Zhou et al.145 synthesized Cu2ZnS4 (CZTS) by co-sputtering and sulfurization on Mo-coated glass. A CdS layer was grown on it by chemical bath deposition followed by annealing at 270 °C in the air or an N2 atmosphere. As calculated by DFT, annealing in the N2 atmosphere creates a sulfur vacancy in CdS which favors easy desorption of CO. Whereas, in the air atmosphere defect formation is healed by the oxygen atoms, giving a stronger CO adsorption which gives rise to C–C coupling. As a result, the average carrier lifetime was enhanced to 8.44 ns compared to the mere 0.33 ns of CZTS due to the decrease in defect states. CO TPD of CdS on the air-annealed sample had a peak at 385 °C and the N2-annealed sample had one at 356 °C, suggesting stronger CO adsorption on air-annealed CdS, supporting the DFT data. They reported a photocurrent density of 0.75 mA cm−2 with the air-annealed photocathode at −0.6 VRHE with methanol (formation rate is 1.87 μmol cm−2 h−1) and ethanol along with CO but the FE distribution was not mentioned.
4.1.1.3. Photocathode with a photoanode. PEC CO2RR with both photoactive electrodes is rarely reported because a suitable band edge alignment of both electrodes is difficult to find. And a tandem configuration already faces challenges in transmitting light of sufficient intensity to the following materials. Moreover, in an H-type cell, two compartments must be separated by an ion-exchange membrane, making the tandem configuration more complicated. If two light sources are used, it will be less attractive for practical usability. However, there are a few reports of a photoactive immobilized molecular complex with a photoanode for CO2RR. In 2016, Sahara et al.146 immobilized an Ru(II)–Re(I) metal complex on a p-type NiO photocathode. They found CO formation under photo-illumination; however, catalysis does not occur with similar Re or Ru metal complexes. They used a CoOx-deposited TaON photoanode to couple with this photocathode, and at −0.3 V vs. the photoanode, 79 nmol of CO was formed under 60 min of illuminated conditions. With the XPS study, they found that the Re/Ni ratio decreased and, consequently, the activity decreased. In 2018, Sekizawa et al.147 screened several Ru-complex-coated multilayered photocathodes. When the Ru-complex/TiO2/N,Zn codoped-Fe2O3/Cr2O3 photocathode was made in tandem with SrTiO3−x, a stable current density of 102 μA cm−2 was observed under 1 sun illumination for 3 h under bias-less conditions. In 1 h, formate FE was found to be 79% with 16% CO selectivity. Xu et al.143 used a Pd-decorated Ti3C2/g-C3N4 photocathode in tandem with a BiVO4 photoanode and reported bias-free 100% methanol formation under illumination for 20 h of test time. In 2019, Kamata et al.148 reported an Ru(II)–Re(I) metal complex on a NiO electrode anchored by methyl phosphonic acid groups, and the ligand vinyl groups of the complexes were electrochemically polymerized. This photocathode reached a CO and formate combined FE of 85%. They used CoOx-coated TaON as a photoanode with the photocathode. There are many reports with both semiconductor-only (no molecular catalyst) photoelectrodes in water splitting, but they are very rare in the case of CO2RR to date. In 2020, Kuk et al.149 made a photo-biocathode by interfacing formate dehydrogenase from Clostridium ijungdahlii (ClFDH) with a TiO2-coated CuFeO2, and CuO mixed oxide on FTO support. An FeOOH-coated BiVO4 photoanode in tandem with that photo-biocathode can generate formate with 33.5% FE at a 0.098 μmol h−1 cm−2 conversion rate under illumination in a bias-free conditions. In 2022, Reisner and his group demonstrated a hybrid photocathode for CO2 reduction by integrating BiOI as a photovoltaic cell with a co-catalyst via encapsulating the PV cell with state-of-the-art conducting graphite epoxy (GE) paste.150 A BiOI PV platelet on ITO is covered by a polycrystalline hole-transport layer of NiOx from below and electron-transport layer of ZnO on the outer side. They made 8 individual pixels each of size 0.045 cm2 on a 1.2 cm × 1.2 cm substrate. Among 16 such devices, only a total of 81 pixels were in working condition with a 0.69 ± 0.06 V open circuit voltage and −4.3 ± 0.4 mA cm−2 short circuit current density. In a PV cell stability test in an N2 atmosphere at 0 V, after an initial decay, the short circuit current remained constant for >100 h. With dendritic Cu92In8 co-catalyst loading, the BiOI|GE|Cu92In8 photocathode operated at 0 V for 1 h with CO
:H2 of 6.4. In tandem with a BiVO4 photoanode, the device operated without bias, having CO:H2 of 3.7 with an onset voltage of −0.15 V. This operation was sustained for 24 h similar to the co-catalyst at −0.3 VRHE. Table 2 highlights a few representative works on all these general photoelectrocatalytic systems discussed above.
| S. No. | Material | Electrolyte (pH) | Conditions | Activity | Selectivity/FE (%) | Study time | Year (ref.) |
|---|---|---|---|---|---|---|---|
| 1 | Cu2O/SnOx | 0.5 M NaHCO3 | 100 mW cm−2 | 11.61 mA cm−2 @ −0.55 VRHE | CO (141.79 μmol cm−2 h−1) + HCOO− (19.57 μmol cm−2 h−1) | (12 h at −0.35 VRHE) | 2022 (ref. 124) |
| 2 | 1D α-Fe2O3/ZnO/Bi | 0.1 M KHCO3 (pH = 6.8) | 100 mW cm−2 | @ −0.65 VRHE (3.75 mA cm−2 @ −1.2 VRHE) | HCOO− (61.2) + CH4 (13.7) | 4 h | 2022 (ref. 139) |
| 3 | ZnTe/TiO2/Ag3Cu | 0.1 M KHCO3 | 100 mW cm−2 | 5.1 mA cm−2 @ −0.2 VRHE | CO (86.5) | 24 h | 2021 (ref. 138) |
| 4 | Cu2ZnS4/CdS | CO2-saturated 0.1 M KHCO3 | 100 mW cm−2 | 0.75 mA cm−2 @ −0.6 VRHE | CO + CH3OH (1.87 μmol cm−2 h−1) + EtOH | — | 2021 (ref. 145) |
| 5 | CuInS2/CuFeO2 (PC) | 0.1 M acetate buffer with 10 mM pyridine at pH = 5.2 | 100 mW cm−2 CO2 flowed at 60 mL min−1 | 15.40 mol h−1 m−2 @ −0.7 VSCE | CH3OH (88) + EtOH (10) | Up to 9 h | 2020 (ref. 129) |
| 6 | TiO2/CuFeO2 and CuO/formate dehydrogenase (PC) + BiVO4/FeOOH (PA) | CO2-saturated phosphate buffer | 100 mW cm−2 | 0.098 μmol h−1 cm−2 @ bias-less condition | HCOO− (33.5) | — | 2020 (ref. 149) |
| 7 | Cu2O/CuFeO2 | 0.5 M KHCO3 | 100 mW cm−2 | 1.1 mA cm−2 @ 0.35 VRHE | Acetate (68) + formate (21) | — | 2020 (ref. 123) |
| 8 | Cu2O NW/Ti3C2 QD | 0.5 M Na2SO4 | 100 mW cm−2 | 5 mA cm−2 @ 0 VRHE | CH3OH (100) | 2019 (ref. 120) | |
| 9 | M(Pd)–Ti3C2/g-C3N4(PC) + BiVO4 (PA) | 0.1 M KHCO3 (6.8) | No data | ∼0.8 mA cm−2 @ −0.85 V external bias | CH3OH/+HCOO− (100) | 20 h | 2018 (ref. 143) |
| 10 | TiO2 NR (PA) + Cu NP/Cu2O (DC) | 0.1 M NaOH (13) (anolyte); 0.1 M KHCO3 (9.3) (catholyte) | 100 mW cm−2 160 mL CO2 re-flowed throughout experiment | 1.31 mA cm−2 @ 0.75 VRHE (−0.7 VRHE for dark cathode as a working electrode) | CH3OH (53.6) | 3 h | 2018 (ref. 105) |
| 11 | ZnO/ZnSe (PC) | 0.5 M NaHCO3 (7.5) | 100 mW cm−2 | ∼0.5 mA cm−2 @ 0.2 VRHE | CH4 (26) | 2018 (ref. 136) | |
| ∼1 mA cm−2 @ −0.4 VRHE | CO (53) | ||||||
| 12 | BiVO4 (PA) + Cu (DC) | 0.5 N NaCl | 100 mW cm−2 CO2 bubbling at 100 sccm | 0.1 mA cm−2 @ 0.75 VRHE | HCOOH (65.4) 1.22 μmol h−1 | 11 h | 2018 (ref. 104) |
| <0.4 mA cm−2 @ 0.9 VRHE | HCHO (85) 2.68 μmol h−1 | ||||||
| 13 | Cu2O/TiO2 | 0.5 M KHCO3 (8.8) | 100 mW cm−2 CO2 bubbling | 0.8 mA cm−2 @ 0.4 VRHE | CH3OH (90) | 10 min (stable) | 2018 (ref. 117) |
| 14 | CuFeO2/CuO | 0.1 M NaHCO3 | 100 mW cm−2 CO2 bubbling | <1 mA cm−2 @ −0.4 VAg/AgCl | Acetate (80) | — | 2017 (ref. 127) |
| 15 | CdSeTe NPs/TiO2 NTs (PC) + Pt wire | 0.1 M KHCO3 | 100 mW cm−2 CO2 flowed at 40 mL min−1 | 6.97 mA cm−2 @ −1.2 VRHE | CH3OH (88) | 300 min | 2016 (ref. 141) |
| 16 | ZnO/ZnTe/CdT/Au (PC) | 0.5 M KHCO3 (7.5) | 100 mW cm−2 | 5 mA cm−2 @ −0.11 VRHE | CO (∼80) | — | 2016 (ref. 135) |
| 17 | CuInS2/GO (PC) | 0.1 M acetate buffer with 10 mM pyridine at pH = 5.2 | 100 mW cm−2 CO2 saturated solution | 1.39 mol h−1 m−2 @ −0.59 VSCE | CH3OH (97) | 10 h for 6 times | 2016 (ref. 131) |
| 18 | ZnO/ZnTe/Au (PC) | 0.5 M KHCO3 (7.5) | 100 mW cm−2 CO2 saturated solution | 16 mA cm−2 @ −0.7 VRHE | CO (65) | 3 h | 2015 (ref. 134) |
| 19 | Co–Ci/BiVO4/WO3 (PA) + Cu (DC) | 0.5 M KHCO3 (7.5) | 100 mW cm−2 CO2 purging | ∼0.25 mA cm−2 @ 0.4 VRHE | CH4 (46.8) | 2 h | 2015 (ref. 102) |
| 20 | CuFeO2/CuO (PC) | 0.1 M KHCO3 (6.5) | 100 mW cm−2 CO2 saturated solution | <1 mA cm−2 @ bias-less condition STF = 1% | HCOO− (90) | 7 days | 2015 (ref. 126) |
| 21 | ZnO/ZnTe (PC) | 0.5 M KHCO3 (7.5) | 100 mW cm−2 79 mL CO2 circulated | 8.2 mA cm−2 @ −0.7 VRHE | CO (23) | 3 h | 2014 (ref. 133) |
| 22 | Hybrid CuO–Cu2O (PC) | 0.1 M Na2SO4 | 70 mW cm−2 CO2 bubbling | ∼0.34 mA cm−2 (calculated) @ −0.2 VSHE (150 mV under potential) | CH3OH (94–96) | 2 h | 2013 (ref. 111) |
A. Studies with Si solar cells. In 2016, Wang et al.151 monolithically integrated highly n-type doped GaN NRs on an Si solar cell. Further loading of Cu co-catalyst achieved 19% FE towards CH4 after a 100 min test under a 300 W light source at −1.4 VAg/AgCl. During this testing time, the current density stabilized at nearly 28 mA cm−2 after losing 30% from the maximum value, which is even significant for a simple H-type cell. They also showed that the Cu co-catalyst plays a crucial role in giving 30 times higher selectivity towards CH4 with CO formation. In 2018, Rao and his group made a photocathode on silicon solar cells by drop-casting porous Sn nanowire obtained by reducing SnO2.152 Under 1 sun illumination, this photoelectrode gave a partial current density of 10 mA cm−2 for formic acid formation at −0.4 VRHE with 59.2% corresponding FE. Moreover, they claimed 90% retention of current density after 3 hours of the test under illumination. To compare the effect of silicon PV cells, they also deposited the catalyst on an FTO support. Product distributions at different potentials are shown in Fig. 10a. This figure shows that photovoltage generated in solar cells partially compensates for the external potential requirement but does not alter the product distribution profile. Hu et al.153 made a robust Ag-decorated Si- photoelectrode, which could reach 90% FE towards CO formation at a cathodic potential of −0.5 VRHE with a significant current density of 8 mA cm−2 for a 4000 s run under 0.5 sun. Although, at 1 sun illumination, CO selectivity dropped to <40% at −0.8 VRHE due to mass transfer limitation, as claimed. However, they reported 12 h of stable operation with 8–9 mA cm−2 current density, but the carbon monoxide FE was reduced to 60% by the end of the experiment. In 2019, numerous studies on silicon solar cell modified PEC CO2RR were reported. Wei et al.154 synthesized Si-NW by chemical etching followed by loading with Pt, Au, Pd, and mainly Ag NPs. They found Au had the highest CO selectivity; however, in the case of Ag, with the increase in NP size, the selectivity toward CO decreased, i.e., the component ratio of syn-gas can be monitored with Ag NP size. This photoelectrode provided a stable 4 mA cm−2 current density at −1 VRHE for 10 h of the experiment under 1 sun. Inspired by the previously discussed work by Hu et al.,153 Ding and his group attempted Bi3+-assisted Si etching and robust surface growth of metallic bismuth.155 They found that a longer growth time for Bi provided higher water reduction suppression capability but suffered larger current density loss due to shielding of the photoactive Si cell underlayer. The optimized catalyst reported nearly 5 mA cm−2 current density at −0.623 VRHE with 90% formate selectivity instead of CO as obtained in Ag-modified previous work. Gong et al.156 also integrated their bismuth nanotube (NT) electrocatalyst with a hierarchical Si substrate. They attached the catalyst on the Si-nanowire of 5 μm in length and 500 nm diameter, grown on the pyramidal microstructure of the Si substrate. Fig. 10b and c are SEM images and Fig. 10d is a graphical illustration. The composite photoelectrode reached a saturated photocurrent density of 17 mA cm−2, which is very close to the theoretical maximum of the silicon substrate. Under the illumination of 0.5 sun at −0.4 VRHE, the photocathode provided a stable current density of 8 mA cm−2 for a 5 h long operation, and FE was 96% which dropped by up to 90% at the end of the study. Zhou et al.157 reported similar work to Wang et al. with three different co-catalysts (Co, Sn, Ni) NP loaded instead of Cu. Co and Ni mainly favor water reduction; however, an Sn co-catalyst is highly selective towards CO2RR and maintained the selectivity for a 10 h long test without any significant selectivity loss. They reported a high current density of 17.5 mA cm−2 at −0.53 VRHE with 76.9% selectivity towards formate under 1 sun illumination. Later, Gurudayal et al.158 reported a back-illuminated both-sides textured photocathode with n-type silicon. The illumination side was doped p+-type, and the electrolyte side was coated with TiO2 followed by silver, and finally, Cu dendrites were grown electrochemically by applying a high current density, Fig. 10e and f are SEM images and the corresponding elemental mappings. In the three-electrode study, they used IrO2 as a counter electrode (anode) and at −1 VRHE on the textured photocathode hydrocarbon selectivity was as high as 79 ± 6% with 30 mA cm−2 current density in 1 sun illumination. However, in a 10 h test at −0.4 VRHE for each of the next 10 days with replaced electrolyte, the current density was between 8 and 10 mA cm−2. After 2 days, H2 FE started to increase from 20% and on the 10th day, H2 FE reached 60%. Although FE towards CO increased, the FE of main products ethylene, ethanol and 1-propanol were reduced by a large margin. This increase in H2 production was reported to be due to Ir contamination in the cathode active sites from the anode, as Ir is well known for its water-splitting activity. Further, they physically removed the surface layer and regenerated the catalyst by re-depositing Cu dendrite. In a similar 10 h for each 10 day test, they got a better result, and this time on the 10th day, H2 FE increased to 40% alone. In 2020, Kan et al.159 loaded Ag NPs on a nanoporous Si surface and further modified this photocathode with minimal loading of 2-aminobenzenethiol (2-ABT) and compared this with a similarly modified non-porous planar silicon. An XPS study reveals that this modification prevents surface oxidation of Si to SiOx and forms an Ag+ layer over Ag NPs by coordination through the thiol group. Again, to further confirm the assistance of the proximal amine group in CO2RR, they also compared aniline (AP), thiophenol (TP) and 4-aminobenzenethiol (4-ABT) modified photoelectrodes with the 2-aminobenzenethiol (2-ABT) modified one. However, they found that 2-ABT provides higher FE towards CO formation compared to the others. However, at −1.2 VRHE, they obtained 10 mA cm−2 partial current density for CO with the optimized photoelectrode, which is not significant at this high cathodic potential. Kempler and his group obtained an ordered microwire of silicon and decorated it with Cu.160 This microstructured photoelectrode was found to have hydrocarbon selectivity with a partial current density of 4.1 ± 0.2 mA cm−2 and for ethylene alone it was 2.1 ± 0.2 mA cm−2 at −0.44 VRHE, but this was observed at very low FE. Moreover, this photocathode provided a stable current density of ∼25 mA cm−2 at only −0.58 VRHE under 1 sun for 48 h of experiment, unlike the other control photocathode made with planar silicon which lost significant activity after 20 min due to delamination of the Cu catalyst. However, in this long-term test, ethylene selectivity started to decrease after 4 h and for methane after 8 h, and CO selectivity increased throughout. So, microstructuring was very efficient for photo-harvesting, but the authors reported the morphological loss of Cu co-catalyst after the experiment. In 2022, Roh et al.161 made an Si nanowire interface with 7 nm of Cu NPs. The photocathode under illumination operated at >10 mA cm−2 current density at −0.5 VRHE with a significantly high ethylene partial current density of 2.5 mA cm−2. In a 50 h long test, up to 30 h ethylene FE remained ∼25% and the methane FE gradually rose >10%; however, the ethylene current density remained >1 mA cm−2 throughout the test. XPS data suggests in situ modification while PEC exposes the Si nanowire which degrades the photocathode. Dong et al.162 decorated AgX on a GaN nanowire/n+–p Si heterojunction. Although the partially reduced Ag/AgX phase acted as the active co-catalyst. Under 1 sun illumination, CO FE remained >80% at −0.4 VRHE and the current density reached 21.6 mA cm−2 at −0.6 VRHE. They reported that the saturated photocurrent density reached as high as 92 mA cm−2 under 3 sun illumination in a flow-cell configuration.
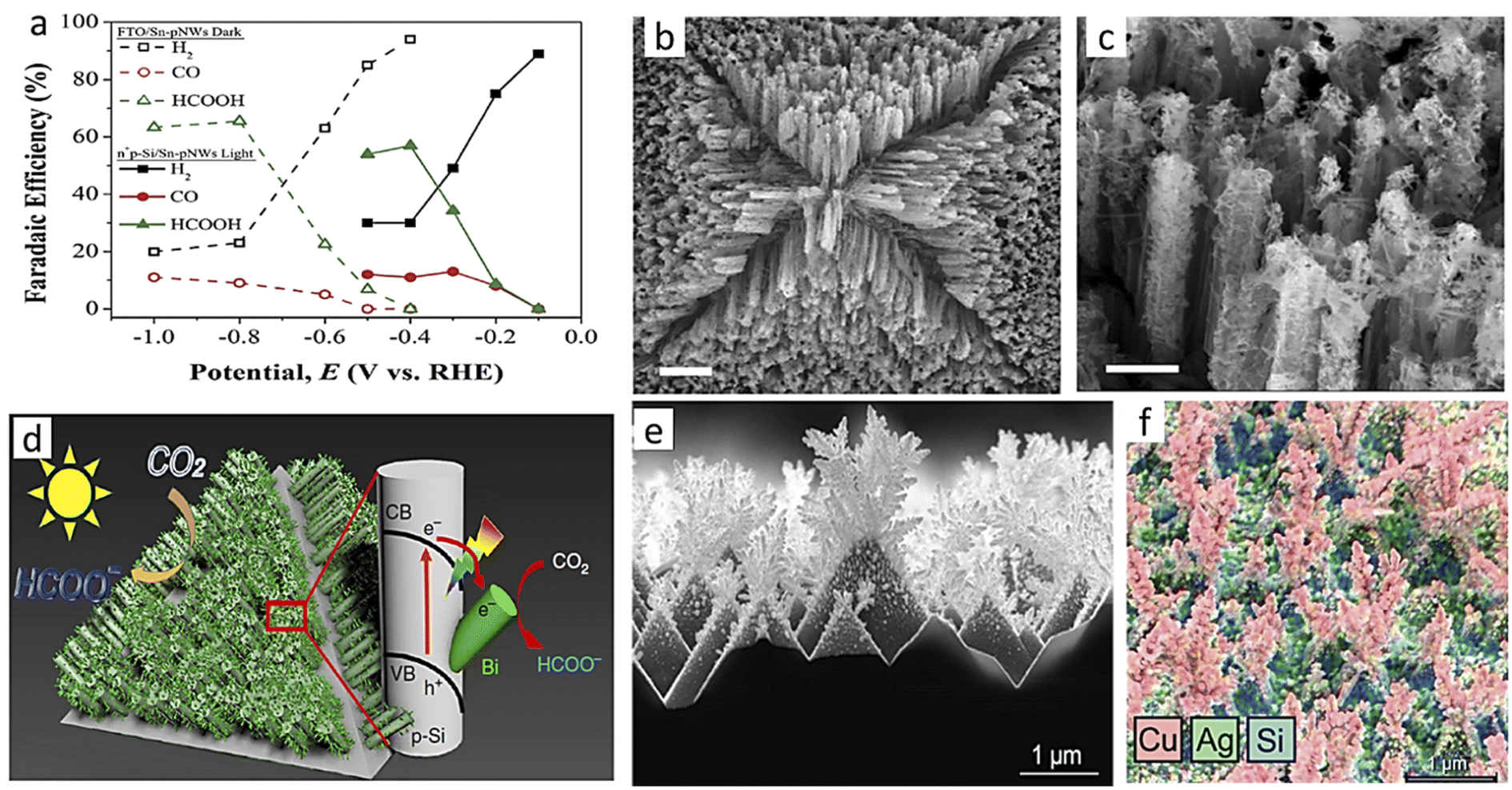 | ||
| Fig. 10 (a) FE vs. potential curve with and without a photovoltaic cell. (b) and (c) SEM images of Bi-loaded Si- nanowires. (d) A graphical representation of the photocathode. (e) An SEM image of Cu dendrites. (f) EDX mapping of the photoelectrode, (a) is reproduced with permission from Rao et al.,152 Copyright 2018, Royal Society of Chemistry; (b)–(d) from Gong et al.,156 Copyright 2019, Springer Nature Limited; (e) and (f) from Gurudayal et al.,158 Copyright 2019, Royal Society of Chemistry. | ||
B. Studies with III–V tandem solar cells. In 2016, Zhou et al. made a photoanode by making a TiO2 protective coating on a GaAs/InGaP III–V tandem solar cell and a thin Ni catalyst coating on the surface.163 All the electrodes and membranes were used with a small surface area of 0.03 cm2 to avoid pinholes. A detailed study was done on the membrane potential and product crossover with Nafion and bi-polar membrane (BPM) and also on the changes in overpotential requirement at different operating electrolyte pH values. While operating at 8.5 mA cm−2 current density with BPM separating the anolyte 1 M KOH and catholyte 2.8 M KHCO3, the photoanode needed ∼320 mV overpotential for OER, and Pd/C loaded on titanium mesh cathode required <100 mV overpotential for formate production and the membrane potential was 480 mV. Under illumination, the small-area photoanode gave stable and near-unity OER for >100 h. Moreover, in 3 h of operation of the full cell under 1 sun illumination without any bias, the FEs for formate production were ∼100, 98, 95, and 94% after 30 min, 1 h, 2 h and 3 h, respectively. The corresponding STF efficiency was also reduced from 10.5 to 9.9% at the end of the 3 h test.
C. Studies with perovskite solar cells. In 2019, Chen et al.164 integrated their alloy electrocatalyst over a lead halide perovskite PV cell layer for the first time. Their optimal catalyst, In0.4Bi0.6 eutectic alloy, had a lower melting point than the maximum sustainable temperature of 100 °C for the PV cell. So, a catalyst layer with 200 μm particle size was coated, avoiding any harm to the PV cell. Under 1 sun illumination, this integrated photocathode reached nearly 100% FE towards formate at −0.52 VRHE, whereas in dark condition, the cathode used to take 680 mV higher potential to achieve the highest faradaic efficiency. In a 1.5 h long operation under 1 sun at −0.6 VRHE, the photocathode could sustain a stable 5.5 mA cm−2 current density. Several recent studies with an embedded perovskite cell photocathode have been reported in tandem with a photoanode; therefore, we have included that kind of work in the PV-EC/PEC section (4.3). Table 3 lists a few superior reports on PV cell modified photoelectrocatalytic systems.
| S. No. | Material | Electrolyte (pH) | Conditions | Activity | Selectivity/FE (%) | Study time | Year (ref.) |
|---|---|---|---|---|---|---|---|
| 1 | p–n+ Si/GaN/AgX | 0.1 M KHCO3 | 300 mW cm−2 | Saturated current density 92 mA cm−2 | CO (−) | (12 h) | 2022 (ref. 162) |
| 2 | Si NW/7 nm Cu NP | 0.1 M KHCO3 | 100 mW cm−2 | >10 mA cm−2 @ −0.5 VRHE | C2H4 (25) + CH4 (10) | 50 h | 2022 (ref. 161) |
| 3 | Si micro wire/Cu NP | 0.1 M KHCO3 (6.8) | 100 mW cm−2 | 25 mA cm−2 @ −0.58 VRHE | CO + CH4 + C2H4 (low) | 48 h | 2020 (ref. 160) |
| 4 | Halide perovskite photovoltaic/In0.4Bi0.6 | KHCO3 | 100 mW cm−2 | 5.5 mA cm−2 @ −0.6 VRHE | HCOOH (100) | 1.5 h | 2019 (ref. 164) |
| 5 | p+–n–n− Si/TiO2/Ag coated Cu dendrite | 0.5 M CsHCO3 | 100 mW cm−2 CO2 saturated solution | 10 mA cm−2 @ −0.4 VRHE | Hydrocarbon (∼40–75) | 10 h for each 10 Days | 2019 (ref. 158) |
| 6 | n+Si (100)/Sn porous NW | 0.1 M KHCO3 (6.8) | 100 mW cm−2 CO2 bubbling at 10 sccm | 10 mA cm−2 (partial) @ −0.4 VRHE | HCOOH (59.2) | 2018 (ref. 152) | |
| CO (11.4) | |||||||
| 7 | p+–n–n− Si/GaN nano wire/Cu | 0.5 M KHCO3 (8) | 8 sun CO2 saturated solution | >30 mA cm−2 @ −1.4 VAg/AgCl [44.9 mA cm−2 @ −1.5 VAg/AgCl] | CH4 (19) | 100 min | 2016 (ref. 151) |
| 8 | GaAs/InGaP/TiO2/Ni (PA) + Pd/C/Ti mesh | 2.8 M KHCO3 (8) (catholyte) | 100 mW cm−2 CO2 bubbling | 8.7 ± 0.5 mA cm−2 @ bias-less condition (∼2.04 V) STF = 10% | HCOO− (94–100) | >100 h (PA) | 2016 (ref. 163) |
| 1 M KOH (13.7) (anolyte) |
4.2. Electrocatalytic (EC) systems
Electrocatalytic systems will be discussed in two parts considering the electrode configuration and arrangement in the operating cell. Cathode and anode assembled systems will be found in a separate section (4.2.2) on membrane electrode assembly (MEA); however, the GDE cell setup will be discussed in the general EC part (4.2.1).| S. No. | Material | Electrolyte (pH) | Conditions | Activity | Selectivity/FE (%) | Stability | Year (ref.) |
|---|---|---|---|---|---|---|---|
| Electrocatalytic systems with a GDE setup | |||||||
| 1 | Ag NP/C-paper | Ultrapure water as anolyte (1 M CsOH for activation of cathode) | CO2 flowed at 160–320 sccm | 200 mA cm−2 (partial) @ <2.8 V | CO (>90) | — | 2022 (ref. 179) |
| 2 | 2D Cu-THQ MOF | 1 M choline chloride + 1 M KOH | — | 173 mA cm−2 @ −0.43 VRHE | CO(91) | 24 h | 2021 (ref. 196) |
| 3 | Carbon cloth/Sn dense tip | Anolyte: 1 M H2SO4 | Catholyte flow rate 4 mL min−1 and CO2 flowed at 50 sccm | 18.7 mA cm−2 @ −0.76 VRHE [30 mA cm−2 @ −0.84 VRHE] | HCOO− (62.5) | 72 h | 2020 (ref. 176) |
| Catholite: 1 M KHCO3 | |||||||
| 4 | Immobilised single-atom Co | 1 M KHCO3 without GDE: 0.1 M KHCO3 | CO2 flowed without GDE: CO2 saturated solution | 211 mA cm−2 @ −0.9 VRHE without GDE: 67 mA cm−2 @ −0.9 VRHE | CO (92) without GDE: CO (91) | 50 h | 2020 (ref. 208) |
| 5 | Solid Ni–N–C | 1 M KHCO3 (7.7) without GDE: CO2 saturated 0.1 M KHCO3 (6.8) | Electrolyte flow rate 100 mL min−1, CO2 flowed at 50 mL min−1 | 200 mA cm−2 @ −1 VRHE without GDE: 10 mA cm−2 @ −0.7 VRHE | CO (∼90) | 20 h (stability test) | 2019 (ref. 206) |
| 6 | Fe3+–N–C | Catholyte: 0.5 M KHCO3 | Electrolyte flow rate of 10 mL min−1 and CO2 flowed at 40 sccm | (1.75 mmol h−1 cm−2) 94 mA cm−2 @ −0.45 VRHE without GDE: 20 mA cm−2 @ −0.47 VRHE | CO (90) | 28 h (stability test) | 2019 (ref. 205) |
| Anolyte: 1 M KOH without GDE: CO2 saturated 0.5 M KHCO3 | |||||||
| 7 | Cu2+–N–C | 0.1 M CsHCO3 | CO2 flowed at 2.5 mL min−1 | 16.2 mA cm−2 @ −1.2 VRHE | CH3OH (43) | 1 h | 2019 (ref. 207) |
| 8 | Carbon paper/Ag NWs | Anode: 2 M KOH (14.3) | 16 mL min−1 humidified CO2 flow | 130 mA cm−2 @ −1.78 VAg/AgCl, without GDE: 16 mA cm−2 @ −1.73 VAg/AgCl | CO (70) without GDE: CO (95) | 1 h | 2019 (ref. 174) |
| Without GDE: 30 mL 0.5 M KHCO3 | |||||||
| 9 | Graphite/carbon NPs/Cu/PTFE | 10 M KOH | Electrolyte flow rate of 10 mL min−1 and CO2 rate of 50 mL min−1 | 75–100 mA cm−2 @ −0.54 VRHE | C2H4 (70) | 150 h (stability test) | 2018 (ref. 171) |
| Cu/C-GDE | 275 mA cm−2 @ −0.54 VRHE | Hydrocarbon (86) including C2H4 (66) | <1 h | ||||
|
|||||||
| Electrocatalytic systems with a H-setup | |||||||
| 1 | Ni SA | 0.1 M KHCO3 | CO2 saturated | >45 mA cm−2 @ −1.5 VRHE | CO (>92) | 48 h | 2022 (ref. 209) |
| 2 | Brass mesh/Cu/S | 0.5 M KHCO3 (7.2) | CO2 saturated | 75 mA cm−2 @ −0.7 VRHE | HCOO− (∼70) | 20 × 1.5 h | 2021 (ref. 192) |
| 3 | Bi NS | 0.5 M KHCO3 (7.2) | CO2 saturated | 55 mA cm−2 @ −0.58 VRHE | HCOO− (99) | 75 h | 2020 (ref. 215) |
| 4 | Cu3N derived NW/Cu-foam | 0.1 M KHCO3 | CO2 saturated solution | >56 mA cm−2 @ −1 VRHE | Hydrocarbon (86) C2H4 (66) | 28 h (stability test) | 2019 (ref. 187) |
| 5 | Cu3N NC | 0.1 M KHCO3 (6.8) | CO2 flowed at 30 mL min−1 | 30 mA cm−2 @ −1.6 VRHE | C2H4 (60) | 20 h (selectivity drops 7%) | 2019 (ref. 188) |
| 6 | Core–shell porous-3D Cu@Sn | 0.5 M KHCO3 (7.3) | CO2 saturated solution | 16.52 mA cm−2 @ −0.93 VRHE (29.8 mmol m−2 min−1) [55 mA cm−2 @ −1.33 VRHE] |
HCOO− (100) | Until 15 h no selectivity loss | 2019 (ref. 173) |
| 7 | mp-Bi nanosheets | 0.5 M NaHCO3 (7.4) | CO2 bubbling at 20 sccm | 18 mA cm−2 @ −1.1 VRHE | HCOOH (100) | 12 h | 2018 (ref. 213) |
| 8 | Single-atom C–Zn1Ni4 ZIF-8 | 1 M KHCO3 | CO2 bubbling at 20 mL min−1 | 31.4 mA cm−2 @ −0.63 VRHE [71.5 ± 2.9 mA cm−2 @ −1.03 VRHE] | CO (97.8) | 12 h | 2018 (ref. 204) |
| 9 | Single-atom Co–N2 | 20 mL of 0.5 M KHCO3 (7.2) | CO2 bubbling at 5 mL min−1 | TOF: 18200 h−1 18.1 mA cm−2 @ −0.63 VRHE |
CO (94) | 60 h | 2018 (ref. 200) |
| 10 | Single-atom Co–N5 | 0.2 M NaHCO3 | CO2 flowed at 20 sccm | 4.5 mA cm−2 @ −0.73 VRHE | CO (>99) | 10 h | 2018 (ref. 203) |
| 11 | Cu-SA/CeO2 | 0.1 M KHCO3 | — | ∼58 mA cm−2 @ −1.8 VRHE | CH4 (58) | 9000 s | 2018 (ref. 201) |
| 12 | Immobilized single-atom Ni | 0.5 M KHCO3 | CO2 saturated solution | 28.6 mA cm−2 @ −0.81 VRHE [36.2 mA cm−2 @ −0.91 VRHE] | CO (99) | — | 2017 (ref. 199) |
| 13 | Sn–Pb alloy/carbon cloth | Catholyte: 0.5 M KHCO3 | CO2 bubbling at 30 mL min−1 | 45.7 mA cm−2 @ −2 VAg/AgCl | HCOO− (79.8) | 2 h | 2016 (ref. 168) |
| Anolyte: 0.5 M KOH | |||||||
| 14 | Electrodeposited Sn | 0.1 M KHCO3 (6.8) | CO2 bubbling | <2 mA cm−2 @ −1.4 VSCE [∼15 mA cm−2 @ −1.8 VSCE] | HCOO− (91) | 1 h | 2016 (ref. 167) |
| 15 | mp-SnO2 nanosheet/carbon cloth | 0.5 M NaHCO3 (7.2) | CO2 saturated | 45–50 mA cm−2 @ −1.6 VAg/AgCl (0.88 V overpotential) | HCOO− (87 ± 2) | 24 h | 2016 (ref. 183) |
| 16 | Partially oxidized 4-layer Co | 0.1 M Na2SO4 (6) | CO2 saturated solution | 10.59 mA cm−2 @ −0.85 VSCE (0.24 V overpotential) | HCOO− (90.1) | 40 h | 2016 (ref. 169) |
| 17 | Sn dendrite | 0.1 M KHCO3 | CO2 bubbling at 19 mL min−1 | 17.1 mA cm−2 @ −1.36 VRHE (228.6 μmol h−1 cm−2) |
HCOO− (71.6) | 18 h | 2015 (ref. 166) |
| 18 | SnO2 NP/graphene | 0.1 M NaHCO3 | CO2 bubbling | 10.2 mA cm−2 @ −1.8 VSCE | HCOO− (93.6) | 18 h | 2014 (ref. 180) |
A. Metal-based materials. At the beginning of electrocatalytic CO2RR, most of the catalyst was metallic, sometimes with nanostructures. Later on, bimetallic electrocatalysts also became popular for their selectivity-tuning application as well as to improve them further, and several hierarchical surface structures were also studied. In 2015, Xiaofeng et al.165 reported a dedicated study on grain boundaries in nanoparticles. They showed a linear dependency between the partial current density of CO formation and grain boundary surface density on Au nanoparticles. Won et al.166 made an Sn dendrite with 17.1 mA cm−2 current density at a high potential of −1.36 VRHE with 71.6% formate FE. In 2016, Zhao et al.167 electrodeposited Sn and reported improved formate selectivity of 91%. Choi et al.168 reported a carbon-cloth-supported Sn–Pd alloy with a significant current density of 45.7 mA cm−2 with 79.8% formate FE but at a high potential of −2 VAg/AgCl. Gao et al.169 showed that partially oxidized four-layer Co exhibited good formate FE of 90.1% at a low potential of −0.85 VSCE with a 40 h stable operation. In 2018, Urbain et al.170 made an Ag dendrite on Cu foam with a 27.3 mA cm−2 current density in a flow cell at −1 VRHE. The cathode was stable for the 15 h test, and CO FE peaked at 95.7%. Sergent and his group increased their catalyst stability 300-fold by sandwiching the Cu NPs between PTFE and C-NP (Fig. 11a and b).171 This approach also showed a 5% improvement in FE, resulting in 70% ethylene with a decreased but significant current density of 75–100 mA cm−2. At high pH, CO2 was readily converted to carbonate ions. Hence, high-pH electrolyte is not practical in an H-type cell. But in the case of the GDE setup, CO2 diffuses to the proximity of the catalyst surface with a very high local concentration and reacts on the interface before getting converted to carbonate ions. So, they used 10 M KOH as an electrolyte and got a very high current density of 275 mA cm−2 with effective suppression of H2 evolution. They also mixed KI with the electrolyte as iodine helps to accelerate the hydrogenation process of adsorbed CO intermediate and increases the CO2 reduction activity.171,172 In 2019, Hou et al.173 reported a core–shell-type porous 3D structure of Cu on Sn to realize 100% formate FE along with 15 h long stability. Chen et al.164 screened out In0.6Bi0.2Sn0.2 and In0.4Bi0.6 alloys among the In–Bi–Sn alloy family for formate selectivity. They also found that Bi does not form any alloy with Sn. These two materials were reported to give formate FE of 95–98% at an applied potential range of −1.2 VRHE to −1.3 VRHE. EIS study shows the smallest radius semicircle for In0.4Bi0.6 suggesting lower charge transfer resistance. In the case of a carbon-made gas diffusion layer (GDL), the GDL use to lose its hydrophobicity over time while being used in applied potential conditions that cause flooding of the electrolyte into the pore. This reduces both the activity and selectivity drastically after a certain operating time. Vázquez et al.174 synthesized Ag nanowires on carbon paper and reported 16 mA cm−2 current density at −1.73 VAg/AgCl with 95% CO FE. In the GDE setup, the current density rose to 130 mA cm−2 at −1.78 VAg/AgCl, but CO FE decreased to 70%. In 2020, Wen-Hui et al.175 adopted an intelligent strategy to maintain the GDE in a wetted condition without getting flooded to enhance the operational lifetime. They kept the Ag catalyst coated Ni-foam GDE surface towards the gas-flow side, i.e., facing the opposite side of the electrolyte, calling it ‘reverse-assembled’ GDE. Both the conventional and reverse-assembled GDE had similar activities, but for the conventional setup, the selectivity dropped rapidly in 2 h. In contrast, this strategically modified GDE was sustained for 150 h in the stability test. Lim et al.176 synthesized dense-tip Sn on GDE (see Fig. 11c and d for the SEM images) and concluded that local pH increases at the tip, which helps preferred selectivity towards CO formation. Gregorio et al.177 studied facet-dependent product selectivity on tailored Cu NPs. The particle sizes of spherical (CuSph), cubic (CuCub), and octahedral (CuOh) Cu NPs were 6 nm, 44 nm, and 150 nm, respectively. The XRD data in Fig. 11e shows the preferential presence of 100 facets in CuCub, 111 facets in CuOh, and both in CuSph with a peak ratio similar to bulk, and Fig. 11f–h are the TEM images. In the GDE flow cell, at −0.79 VRHE with 200 mA cm−2 current density, ethylene FE peaked at 31% for CuSph. However, for CuCub, at a potential of −0.75 VRHE, current density reached 300 mA cm−2 with 57% ethylene FE, and at −0.91 VRHE CuOh gave rise to 53% methane FE with 100 mA cm−2 current density which rose to 300 mA cm−2 at 0.96 VRHE, but methane FE dropped to 40% although ethylene FE was below 10%. Therefore, the methane to ethylene ratio was ∼5
:1 much lower than 1:20 in the case of CuCub. Fig. 11i provides the product distributions at different potentials for separate catalysts. CuOh was not highly stable, but CuCub was sustained in a 6 h test until the GDE got flooded. Later, Arquer et al.178 made a catalyst-ionomer planar heterojunction (CIPH) by spray coating of perfluorinated sulfonic acid (PTFA) solution, dissolved in polar solvent on a hydrophilic metal catalyst deposited on a porous polytetrafluoroethylene (PTFE) substrate. SEM images are shown in Fig. 11j, k, m and in Fig. 11k the ionomer layer is clearly visible. Fig. 11l is a graphical illustration and Fig. 11n, o are TEM images of a cryo-microtomed CIBH and the corresponding elemental mapping. PFSA ionomers such as Nafion contain both hydrophilic (–SO3− group) and hydrophobic (–CF2 group) parts and the hydrophobic domain formed by their self-assembly assists extended gas diffusion and the hydrated hydrophilic domain helps in water uptake and ion transport. They tested Ag–CIPH and Cu–CIPH and got a similar result: for example, GDE with Cu–CIPH reached 800 mA cm−2 with 60% ethylene FE whereas for GDE with a bare Cu catalyst, the current density was merely 50 mA cm−2. Both catalysts were found to have a similar electrochemical surface area (ECSA), resistance and hydrophobicity. Moreover, when the electrodes were tested in an H-type cell where dissolved CO2 was fed for CO2RR, both electrodes resulted in similar current density. Therefore, they concluded that the huge current enhancement is due to extended gas diffusion through the ionomers. However, with increased catalyst loading >3.33 mg cm−2, an astonishingly high current density of 1.32 A cm−2 was observed with a C2+ partial current density of 1.21 A cm−2 along with a significant cathodic energy efficiency (EE) of nearly 45%. In 2022, Boutin et al.179 made a GDE by spray coating Ag NPs and sustainion solution on carbon paper with IrOx as an anode. The zero-gap GDE flow cell (basically an MEA) is operated with ultrapure water as anolyte. However, for in situ activation of the cathode, 1 M CsOH in 1:3 isopropanol and water solution is injected through the CO2 feed track to provide the necessary alkali cations to the cathode. Finally, gas pressure takes the solution out. This treatment enhanced the current density by approximately 10 fold. The partial current density for CO formation increased from 75 mA cm−2 to 150 mA cm−2 or to 225 mA cm−2 when temperature was ramped from 30 °C to 40 °C or to 60 °C. This is due to improved kinetics and also to reduced series resistance, as concluded from an EIS study. The cell operated with a PV cell under 289 sun and ultrapure anolyte was used to take the extra heat from the PV cell to improve the efficiency of the overall system. After activation, the cell could reach 2 A to 10 A (25 cm2 electrode) when the voltage increased from 2.5 V to 2.8 V. In 2.5 h of operation with a cooling bath temperature set to 15 °C, average 87% CO
selectivity was obtained and after 1.5 h cathode reactivation was undertaken to maintain the operation.
 | ||
| Fig. 11 (a) A graphical representation and (b) corresponding SEM image of Cu NPs sandwiched between PTFE and C-NPs. (c) and (d) SEM images of Sn DT. (e) XRD data. (f)–(h) TEM images of spherical, cubic, and octahedral Cu NPs, respectively. (i) Product distribution profiles. (j), (k), (m) SEM images of Cu–CIPH. (l) A graphical illustration and (n) TEM image of cryo-microtomed CIBH and (o) elemental mapping data. (a) and (b) Reproduced with permission from Dinh et al.,171 Copyright 2018, American Association for the Advancement of Science. (c), (d) from Lim et al.,176 Copyright 2020, Royal Society of Chemistry, (e)–(i) from Gregorio et al.177 Copyright 2020, American Chemical Society, (j)–(o) from Arquer et al.,178 Copyright 2020, American Association for the Advancement of Science. | ||
B. Oxide/oxide-derived materials. In 2014, Meyer et al.180 synthesized SnO2 nanocrystals by a simple hydrothermal method which could reduce CO2 to formate. However, they reported an SnO2 reduction peak at −1 VSCE, which disappeared after 50 LSV cycles from −1 VSCE to −1.8 VSCE, and the active catalyst was utilized for the electrolysis study. With a 30% catalyst loading of 5 nm particle size with graphene and carbon black, the carbon-black-containing electrode had reached only 5.4 mA cm−2 at −1.8 VSCE, whereas the graphene-containing cathode could operate at a stable 10.2 mA cm−2 of current density with 93.6% FE towards formate in an 18 h long operation. In 2016, Xie et al.181 reported Sn quantum sheets sandwiched within graphene. They hydrothermally deposited amorphous carbon on SnO2 nanosheets and further calcined them in an argon atmosphere, which resulted in the reduction of the SnO2 nanosheet into Sn quantum sheet while carbon was converted into graphene, making a protective coating. The synthetic pathways are graphically shown in Fig. 12a. The robust electrode exhibited formate selectivity with a FE of minimum 85% throughout the 50 h operation (Fig. 12b) with a current density of 21.1 mA cm−2 at −1.8 VSCE. Due to the lack of long-range order in 3D, XRD failed to probe the material, XPS also failed to probe the Sn quantum sheet, whereas Raman data confirmed the formation of the graphene layer. However, the Sn sheet was probed by HRTEM (Fig. 12c) and AFM (Fig. 12d) data and XAFS data confirm Sn–Sn bonding. From the XAFS data it was also concluded that the coordination number of Sn–Sn reduced from 2 and 4 to 1.4 and 2.7. The authors claimed that this unsaturated coordination number enhances the activity, supported by the 83 mV dec−1 Tafel slope compared to 176 mV dec−1 for bulk Sn. The Nyquist plot also confirmed the lowest charge transfer resistance among other control electrodes. TGA data gives the most reliable information on the protection ability of the graphene layer. The protected Sn material showed rapid weight loss from 390 °C to 570 °C due to oxidative loss of graphene and then weight gain due to Sn oxidation. However, an unprotected control material gained weight due to Sn oxidation from as early as 200 °C (Fig. 12e). In 2017, Spurgeon and his group showed that electrochemically reduced SnO2 porous nanowires with highly dense grain boundaries obtained via a scalable solvo-plasma technique, could be much more effective in providing additional active sites for the selective reduction of CO2 to formate.182 To conclude, the enhanced activity results from a high grain boundary instead of increased active surface area due to the introduction of porosity compared to the non-porous control nanowire material (6 m2 g−1). They also made a comparison with commercially available high-surface-area (55 m2 g−1) SnO2 nanoparticles. Porous SnO2 nanowire (35 m2 g−1) with a high grain boundary was far superior, although it had a lower active surface area. At −1.0 VRHE, a current density of 10 mA cm−2 was obtained with 78% FE towards formate. In a 15 h test at −0.8 VRHE, the catalyst could reach a stable 4.8 mA cm−2 of partial current density for formate production with 80% FE. Li et al.183 reported another similar study on a hydrothermally synthesized hierarchical structure of mesoporous SnO2 on carbon cloth, which gave a robust mesoporous SnO2 catalyst by calcination in the open air. At −1.6 VAg/AgCl, the electrode could give 50 mA cm−2 of stable current density for 24 h of operation with 87 ± 2% FE towards formate production (Fig. 12f–h). Such a high current density is comparable to a GDE-type cell, and they also tested the electrode by twisting it 10 times before the test, but no activity loss was observed. Then, Schreier et al.184 tuned the mixed product selectivity of a CuO cathode by atomic layer deposition (ALD) of SnO2 on the surface. With 5 ALD cycles, they obtained the optimized peak current with intact CO selectivity, but a higher number of cycles enhanced the formate selectivity, which is known for an SnO2 catalyst. At −0.6 VRHE, the FE towards CO was 97% with a low current density, and they also claimed at higher potential, but no C2 or C3 products were obtained. After the test, the nanowire morphology was intact, but metallic Cu was found with an Sn oxidation state of 2+. They also found Cu1+; therefore, commenting on the nature of the active site needs further study. In 2019, Li et al.185 synthesized a self-templated mesoporous SnO2 nanosheet via calcination of solvothermally produced SnS2 particles. The electrode reached peak FE for formate of ∼83% at a potential of −0.9 VRHE (overpotential = 710 mV), which is at least 150 mV lower than that reported in other similar work. The electrode could give a stable 20 mA cm−2 of current density in a 10 h stability test at −0.82 VRHE with an average formate selectivity of >75%. Gong and his group synthesized defect-rich Bi nanotubes from β-B2O3 double-walled nanotubes.156 In a normal H-type cell, they reported 64 mA cm−2 current density at −1 VRHE, and at −0.7 VRHE, FE towards formate reached 93%, and it was greater than 93% up to −1 VRHE. While, in 48 h long-term operation at −0.82 VRHE, formate FE was between 98 and 100% with a stable current density of 36 mA cm−2. They further tested the material in the GDE setup to improve the current density and reported 140 mA cm−2 current density in 1 M KHCO3 electrolyte at −0.85 VRHE. In contrast, in 1 M KOH electrolyte, 210 mA cm−2 current density was observed at a much lower potential of −0.58 VRHE with >97% FE for 11–13 h of operation until the GDE electrode was flooded with electrolyte. Guo and his group deposited Ag nanoparticles on Cu2O nanowire by a galvanic replacement reaction.93 The nanowire substrate was dipped in nitric acid and silver nitrate solution for variable growth times of 1, 3, 5, and 8 min. With 5 min growth, 100–200 nm Ag nanoparticles completely covered the surface. They electrochemically reduced the material to obtain a Cu/Ag bimetallically active catalyst as a final step. At −1.05 VRHE, they reported a 18.1 mA cm−2 partial current density for ethylene formation with a 52% corresponding FE, and the overall C2 FE was 76%. In a 12 h operation, FE dropped from 50% to 44%. They suggested a ‘spillover of intermediates' from Ag to the Cu site for efficient selectivity, and operando Raman was done to support their claim. The intense C–H vibration peak at low potential on Ag/Cu compared to the control Cu material, suggested a high CO* intermediate dimerization rate on the Ag/Cu surface, whereas the Cu alone lacked this. At ≤1 VRHE, all peaks other than the CO* intermediate disappeared, and they related this to the speedy conversion kinetics at a higher potential, which uses a high local concentration of CO* intermediate.
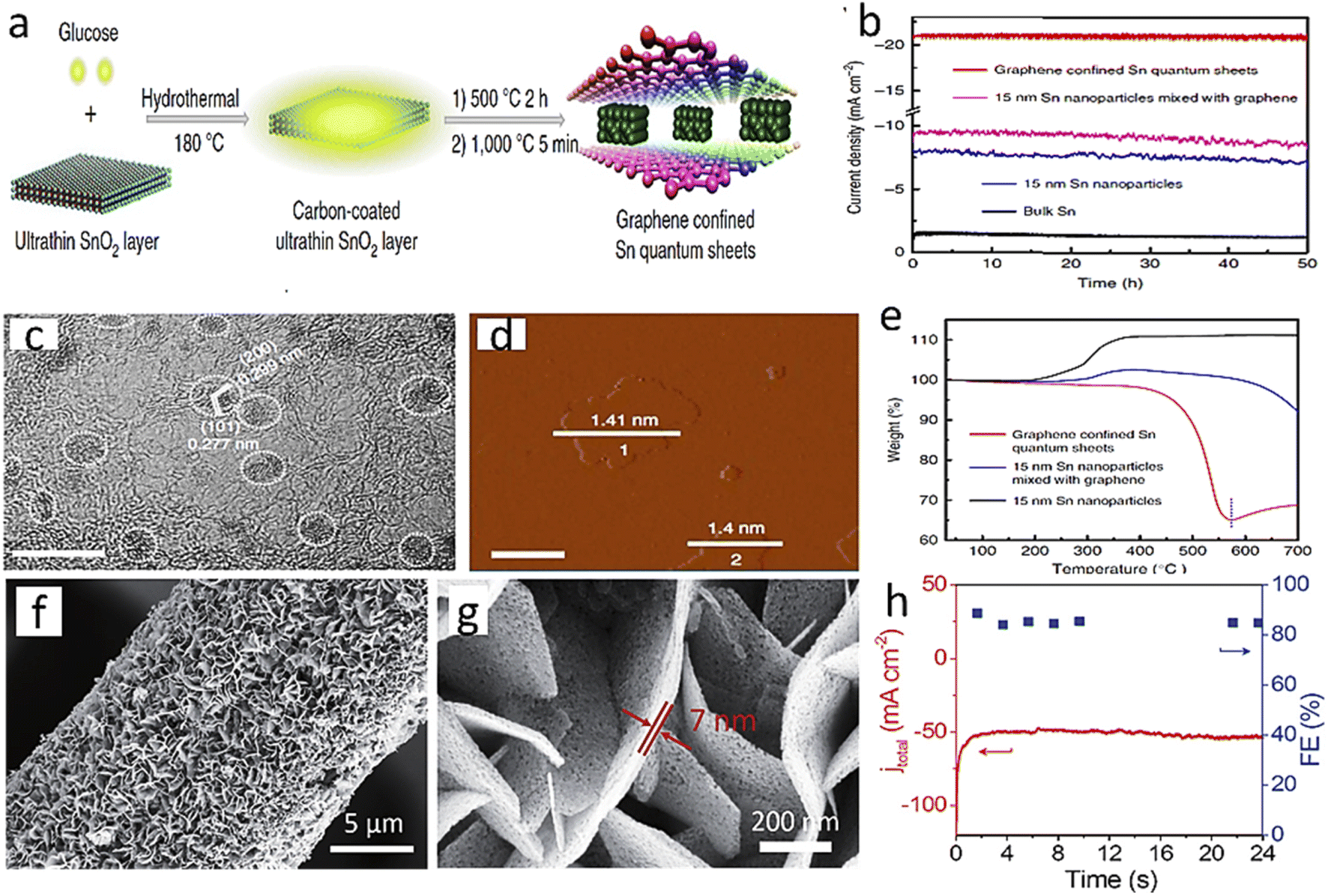 | ||
| Fig. 12 (a) A schematic diagram of a synthetic procedure. (b) 50 h chronoamperometry data at −1.8 VSCE. (c) A HRTEM image (10 nm scale). (d) An AFM image. (e) TGA data. (f), (g) SEM images of mesoporous SnO2 nanosheets. (h) The chronoamperometry curve over long-term testing, (a)–(e) are reproduced with permission from Lei et al.,181 Copyright 2016, Springer Nature Limited, and (f)–(h) from Li et al.,183 Copyright 2016, John Wiley and Sons. | ||
C. Metal nitrides. In 2018, Hou et al.186 reported an Ni-based nitride with a carbon support by calcining nickel nitrate and carbon mixture under flowing ammonia conditions at 973 K for 3 h. In 0.5 M NaCl, the partial current density towards CO was 8.7 mA cm−2 at −1 VRHE. Whereas, in a 6 h test at −0.9 VRHE, the FE was more than 84% with a partial current density of 6.3 mA cm−2, and after the test, the XRD data showed only an Ni3N phase, but no deformation peak. Surprisingly, they reported approx. 20% drops in CO selectivity in a commonly used NaHCO3 electrolyte. In 2019, Mi et al.187 synthesized Cu3N nanowire by nitridation of Cu(OH)2 nanowire directly grown on Cu foam in an ammonia environment (Fig. 13a and b). However, an active catalyst was obtained by electrochemically reducing the Cu3N to Cu nanowire at −1.2 VRHE. This material showed commendable C2 product selectivity of 86% in a non-alkaline medium, including 66% for ethylene, 12% for ethane, and 8% for ethanol with 4% C1 selectivity. Moreover, in the 28 h long operation at −1 VRHE, the material supplied a large and stable current density of 56.8 mA cm−2 (this data is obtained from Fig. 3d). Cu3N nanowire can also reduce CO2 to acetylene, but then FE towards ethylene drops to only 38%. They denoted a high-density grain boundary as the reason for the enhanced activity observed in Cu nanowire only after the electrochemical reduction of Cu3N. Yin and his group also claimed activity enhancement due to a denser grain boundary.188 Inspired by an old report (Fig. 13c and d) from 2011 on alkaline fuel cells by Wu et al.,189 they synthesized Cu3N nanocubes (Fig. 13e and f) of different particle sizes by controlling the synthesis temperature and achieved a 30 mA cm−2 current density with optimal 25 nm Cu3N nanocubes at −1.6 VRHE. In the 20 h operation, 60% ethylene selectivity dropped after 1 h and stabilized at 53%. After electrolysis, TEM data revealed small aggregations of particles which they reported as the cause of this 7% selectivity drop.
 | ||
| Fig. 13 (a) An SEM image of a Ni3N-derived Cu nanowire. (b) A TEM image showing grain boundaries. (c) and (d) TEM images of Cu3N NCs used in a fuel cell. (e) and (f) TEM images of Cu3N NCs used in an electrocatalysis study. (a) and (b) Reproduced with permission from Mi et al.,187 Copyright 2019, John Wiley and Sons, (c) and (d) from Wu et al.,189 Copyright 2011, American Chemical Society and (e), (f) from Yin et al.,188 Copyright 2019, American Chemical Society. | ||
D. Metal sulfides. In 2017, Zhao et al.190 synthesized CuS nanosheets on Ni foam support. The material reached 7.35 mA cm−2 current density at −1.1 VRHE, which is a pretty average report; however, they got a significant FE of 73 ± 5% towards CH4, which is much rare, with >15% CO FE. The material was highly robust, and after 60 h of operation, no current drop or selectivity drop was observed. In contrast, the control material with nanowire morphology with 49 ± 3% FE, rapidly lost its activity. By 2018, Shinagawa et al.191 reported a sulfur-modified Cu catalyst. They synthesized carbon-supported CuS with varying precursor amounts by 5% loading of different particle sizes. They found that a larger particle size gave higher FE towards formate. However, after electrolysis, the peak for particle size distribution shifted to a drastically lower region due to reductive reconstruction. They further synthesized Cu–S in the solvothermal process with different sulfur contents to study the effect on electrocatalysis. For their optimized catalyst, 80% formate FE was observed with 780 mV overpotential and sustained for 12 h of operation at −0.8 VRHE. Only a 5% drop in FE with the appearance of Cu2S and Cu phases was observed. In 2021, Dou and his group bath-deposited CuS nanosheets on brass mesh, which on electrocatalysis was rapidly reconstructed to a nanowire network.192 A DFT study suggested that the Cu(111) plane and residual sulfur (Raman data confirmed the presence of S residue) weakened the adsorption of HCOO* and *COOH intermediates; therefore, formate was formed without further reaction proceeding to CO. The electrocatalyst could provide a commendable high current density of 75 mA cm−2 at −0.7 VRHE with 67.8 ± 1% FE towards formate, while CO and CH4 FE were less than 1%. However, surface-modifier PTFE treatment enhanced the formate FE to 70.2 ± 1% by converting the surface from aerophobic to aerophilic. 20 cycles of 1.5 h each resulted in an average of 71.4 ± 1% FE towards formate, which proved that the evolved catalyst after initial rapid reconstruction was highly stable.
E. Metal phosphides. In 2018, Calvinho and his group reported transition metal phosphides with high C3 and C4 product selectivity in CO2RR for the first time.193 They synthesized five compounds with varying P content – Ni3P, Ni2P, Ni12P5, Ni5P4 and NiP2. Due to being conductive in nature, no conducting carbon support was used for electrode fabrication. The highest P-containing NiP2 reached 71% FE towards 2,3-furandiol at 0 VRHE, whereas at −0.1 VRHE, FE towards methylglyoxal was 84%. However, these materials favor HER at higher overpotential, so that the operating current density was very low (Fig. 14a). They also reported a different hydride transfer mechanism to O-bound intermediates along with a self-coupling-type condensation reaction instead of radical coupling of CO* intermediate for C–C bond formation, as shown in Fig. 14b. During 3 h of chronoamperometry, in the first 30 minutes, a rapid fall in current density was observed before reaching a stable current. The authors referred to this rapid fall due to surface phosphoxide reduction at the initial stage and the formation of a pH gradient inside the pores. They also reported an energy efficiency of 8% for Ni3P and 99% for NiP2. In 2021, Choi et al.194 synthesized CuP2, and introduced crystallinity via calcination at 450 °C. They obtained FE of 3.87 ± 0.66% towards 1-butanol, which was claimed to be 70 times higher than in current reports, along with >30% FE towards formate and >22% FE towards acetate at −0.6 VRHE in a 2 h test with a small current density of ∼2.5 mA cm−2. While for electrocatalysis, no peak for CO intermediates in the range 2060–2100 cm−1 was detected in in situ surface-enhanced infrared absorption (SEIRA) spectroscopy (Fig. 14c). Hence, they also supported the self-condensation-type mechanism of formaldehyde for C–C bond formation in 1-butanol instead of radical coupling of the CO* intermediate. They conducted blank electrocatalysis with 50 mM formate and 50 mM acetaldehyde in 0.5 M potassium bicarbonate and found that formaldehyde is converted to acetaldehyde with acetaldehyde forming 1-butanol. This data further supports their predicted mechanism. Therefore, phosphide material is interesting due to its unusual multicarbon product selectivity and deserves more research.
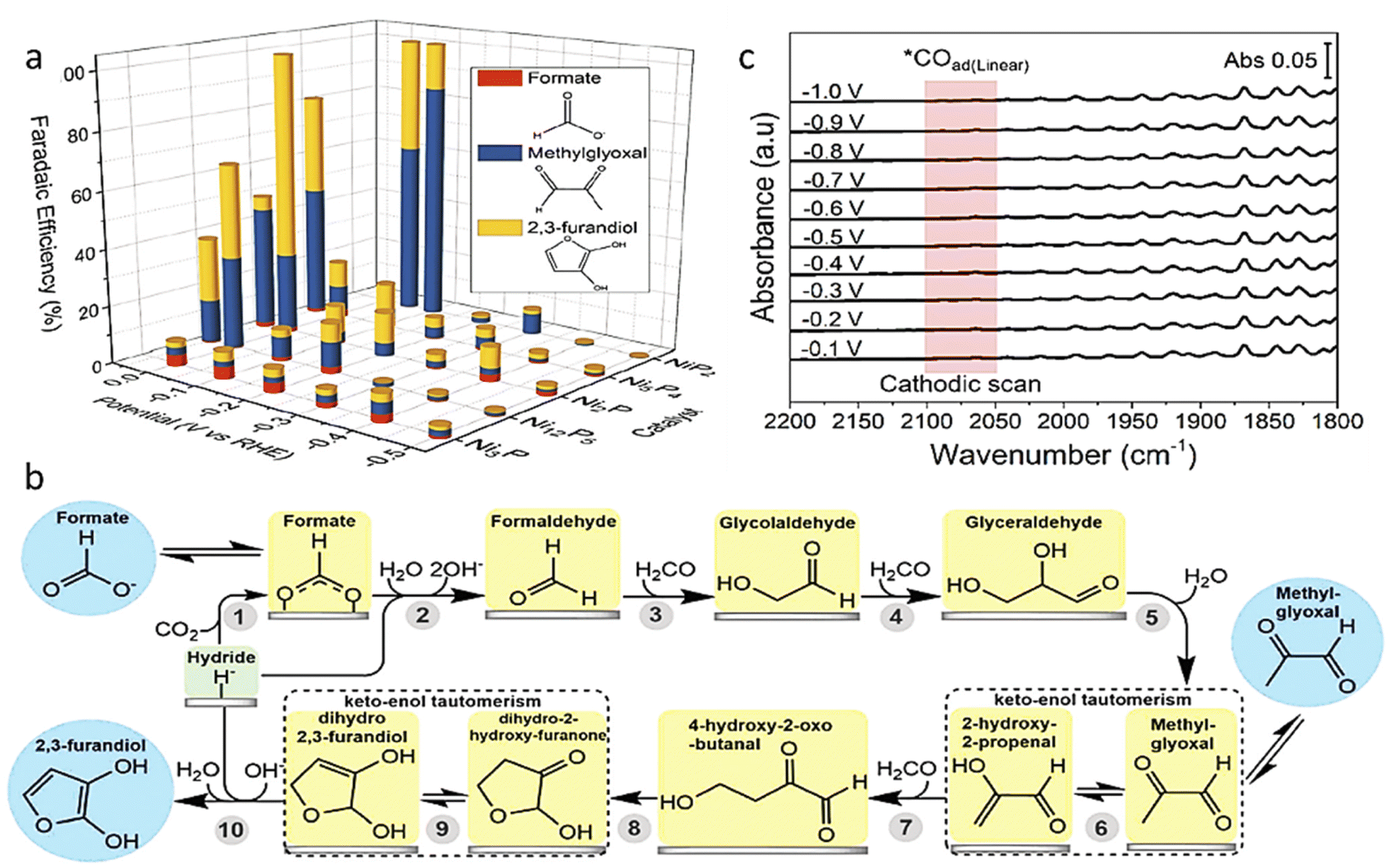 | ||
| Fig. 14 (a) FEs at different potentials for different phosphide materials. (b) A predicted mechanism where products are given with blue backgrounds, and O-bound intermediates are presented with yellow backgrounds. (c) An in situ SEIRS study at different potentials. (a), (b) Reproduced with permission from Calvinho et al.,193 Copyright 2018, Royal Society of Chemistry; and (c) from Choi et al.,194 Copyright 2021, American Chemical Society. | ||
F. Metal–organic frameworks. In 2018, Nam et al.195 reported the formation of distorted-symmetry MOFs derived in situ under coordinated Cu clusters in electrocatalytic conditions for CO2RR. A different degree of asymmetry was introduced in HKUST-1 by calcination at 250 °C. The calcination temperature was optimized from TGA analysis and subsequent EPR, XRD, and XPS data analysis. After 3 h of calcination, the XRD peak for the (111) plane disappeared due to structural distortion and ordering loss as this plane consists of the paddlewheel of Cu-dimers. XPS data suggested the appearance of the Cu1+ state and the EPR signal got sharper for a longer period of calcination. In situ XAS data showed Cu cluster formation, while the electrolytic study, which reversed back to the initial form at the end of the catalysis, suggested a small-sized cluster was formed. The 3 h calcined sample with the most substantial EPS peak gave rise to the Cu cluster with the lowest coordination number of 9.5 ± 0.9. As-prepared and 1 h calcined MOF materials were used and had 10% and 35% FE, respectively, towards ethylene. However, for a 3 h calcined sample, they reported a superior current density of 262 mA cm−2 with 45% ethylene FE at −1.07 VRHE in a flow-cell system with 1 M KOH electrolyte. Such a high current density with significant selectivity was reported for the first time on a MOF-based material. In 2021, Majidi et al.196 synthesized 2D copper tetrahydroxyquinone (Cu-THQ) nanoflakes (NFs) by liquid-phase exfoliation of a bulk Cu-THQ conducting metal–organic framework (MOF). The optimal catalyst with a lateral size of 140 nm (from a DLS study) and average thickness of 10.1 ± 6.7 nm provided 173 mA cm−2 of current density at −0.45 VRHE in a GDE setup. The corresponding FE towards CO was 91% in a hybrid electrolyte consisting of 1 M choline chloride and 1 M potassium hydroxide. After a long-term EC study at −0.43 VRHE, the XPS peak for Cu2+ shifted to lower binding energy and the authors concluded this was due to the change in the Cu2+ environment, not from its reduction. However, an operando XAS study suggested the reversible formation of Cu and Cu+.
G. Single-atom catalysts (SACs). The first work on a single-atom immobilized carbon matrix was reported by Strasser and his group in 2015.197 Since then, many groups have been working in this field. Depending on the coordination number of the anchoring site, there can be various types of sites like graphitic, pyridinic, pyrrolic, etc., in a carbon matrix (Fig. 15a). In 2017, Jiang et al.198 synthesized graphene shell (GS) encapsulated metallic Ni by electrospinning. GS-encapsulated 20 nm Ni NPs reached 4 mA cm−2 current density (20 mA mg−1) at a 700 mV overpotential with 93.2% carbon monoxide FE. However, nitrogen-doped GS assisted the thin wrapping and bonding of more Ni atoms (NiN site) with GS. And more NiN-GS loading on a high-surface-area carbon fiber paper substrate enhanced the geometrical current density to 20 mA cm−2, keeping the FE intact. This Ni atom shows a unique oxidation state with respect to encapsulated Ni NPs in an X-ray photoelectron spectroscopy (XPS) study. Three-dimensional atom probe tomography (ATP) gave the 2D atomic map of the NiN-GS catalyst shown in Fig. 15b. Analysis in the highlighted yellow part revealed that 83% of total Ni atoms are single atoms with no neighboring Ni atoms less than 2.2 Å distant. Fig. 15c and d are 2D-projected views of Ni atoms and contour plots of Ni atom concentration, respectively. Li et al.199 reported 36.2 mA cm−2 current density at −0.91 VRHE with 99% CO FE on an immobilized Ni single-atom catalyst. By 2018, Xiaoqian et al.200 attempted to control the coordination number of single Co atoms by varying the pyrolysis temperature. That is, more CoII–imidazolate–CoII linkages would decompose to release CN fragments at elevated temperatures, resulting in the decay of Co–N bonds. As a result, three atomically dispersed Co catalysts with different Co–N numbers can be selectively prepared at 800, 900 and 1000 °C, and designated as Co–N4, Co–N3, and Co–N2, respectively. Surprisingly, they obtained the best activity with Co–N2, and the material lost its activity when the coordination number increased to 4 via NH3 treatment at 400 °C. Wang et al.201 chose CeO2 nanorods (NRs) for a Cu single-atom (SA) support. They reported 58% FE for methane at −1.8 VRHE with their optimized 4% Cu-loaded CeO2 electrode. They theoretically found that an oxygen vacancy state near the Cu SA enhanced the stability of the material. They concluded from XPS and XAFS that Cu0 and Cu1+ states were dominant over Cu2+ and the formation of a Cu single-atom site. Although it had a nearly constant 58 mA cm−2 current density throughout their test period of 9000 s, the hydrogen evolution FE increased from 22% to 45%, and methane FE decreased from 58% to 40% in 0.1 M KHCO3 electrolyte. Zhipeng et al.202 synthesized Zn SA on an N-doped carbon matrix and got 91% carbon monoxide FE at a lower overpotential of 390 mV with a comparatively lower current density. Pan et al.203 showed high CO selectivity of 99% by a Co single atom also in a Co–N5 coordination environment at a low current density. Yan et al.204 reported 31.4 mA cm−2 current density at a comparatively low potential of −0.63 VRHE with 97.8% CO FE. At −1.03 VRHE, the current density reached 71.5 mA cm−2, which is exceptional in the case of an H-type cell. In 2019, Gu et al.205 found CO2 reduction activity at an astonishingly low overpotential of 80 mV with a high current density of 94 mA cm−2 in the GDE setup. Möller et al.206 reported a 10 mA cm−2 current density at −0.7 VRHE in an H-type cell with 90% CO FE with an Ni single atom in an N-doped carbon matrix. In the GDE setup, the current density increased to 200 mA cm−2 at −1 VRHE and was sustained for 20 h of operation. Fontecave et al.207 did an operando X-ray absorption (XAS) study of Cu SA and found restructuring of Cu–N4 sites into Cu nanoparticles during electrocatalysis. This happened reversibly, and the isolated Cu–N4 site regenerated after the electrocatalysis was over. They also found that an electrolyte consisting of a larger cation drastically impacted methanol selectivity. FE for methanol increased in the order Li+ (2%) < Na+ (5%) < K+ (16%) < Cs+ (43%). In 2020, Yang et al.208 synthesized Co SA immobilized in a free-standing, 3D porous crosslinked carbon nanofiber structure by electrospinning and subsequent pyrolysis (Fig. 15e–i). From the XANES spectra in Fig. 15j it was concluded that the oxidation state of Co was between 0 and +3, and from the EXAFS fitting (Fig. 15k) it was concluded that the coordination number of Co SA was 4. CO FE was >90% in a wide potential window of −0.4 VRHE to −0.9 VRHE and FE reached 97% at −0.6 VRHE. In the H-type cell, the partial current density towards CO was 65 mA cm−2 for 50 h of operation. Whereas in the GDE setup, the current density reached as high as 211 mA cm−2 at the same potential of −0.9 VRHE with a similar CO FE of 91%. In 2022, Xi et al.209 synthesized Ni single atoms in a nitrogen-doped carbon matrix with 80% of nitrogen coordinated with metal. They heated the precursor to 1300 °C in just 0.5 seconds by a Joule heating technique. A control sample made by normal heating at 1300 °C with a 2 °C min−1 ramp rate showed a dominant Ni–Ni peak in the XAFS study. The optimized material sustained >92% CO selectivity in a large potential window of −0.7 VRHE to −1.9 VRHE in an H-type cell. In a 48 h long stability test at −1.5 VRHE, >45 mA cm−2 current density (derived) was maintained. They reported a 600 mA cm−2 current density with 95% CO selectivity in the GDE flow-cell system.
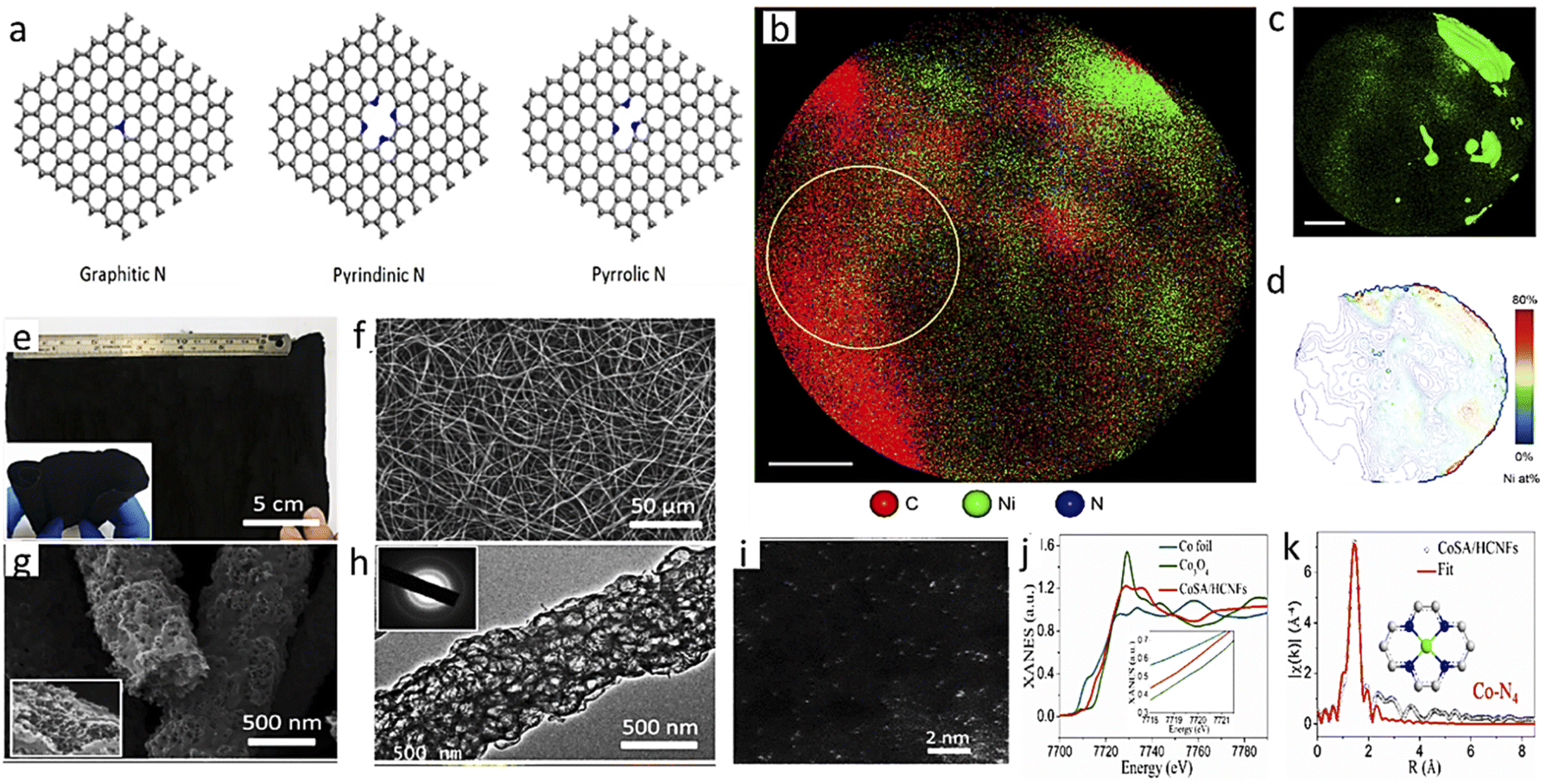 | ||
| Fig. 15 (a) Types of anchoring sites in a nitrogen-doped carbon network. (b) A 2D atom map of the NiN-GS catalyst obtained via atom probe tomography (ATP). (c) A 2D projected view of Ni atoms. Scale bar, 10 nm. (d) A contour map of the Ni concentration with an interval of 2 at%. (e) Photographs of a piece of flexible CoSA/HCNFs membrane. (f), (g) FE-SEM imaging. (h) A HR-TEM image; the inset shows the SAED pattern. (i) Aberration-corrected HAADF-STEM images of CoSA/HCNFs. (j) XANES spectra at the Co K-edge (the inset shows a magnified image) of Co foil, Co3O4, and CoSA/HCNFs. (k) Fitting for the EXAFS data of CoSA/HCNFs; the inset shows the Co–N4 structure, (a) is reproduced with permission from Pan et al.,203 Copyright 2018, American Chemical Society; (b)–(d) from Jiang et al.,198 Copyright 2017, Elsevier Ltd. (e)–(k) from Yang et al.,208 Copyright 2020, Elsevier Ltd. | ||
H. 2-D materials. 2D materials are adding a fascinating chapter nowadays to the materials chemistry field with their versatile application. The catalysis domain is also getting enriched with the advantage of high exposure of active sites in the large active surface area.
Metallic 2D bismuth has been well studied in recent years. Although Sn is mainly known for its formate selectivity, 2D bismuth secured superiority in formate selectivity. In 2018, Zhang et al.210 successfully exfoliated bulk bismuth to nanosheets with exposed (110) facets of 1.2–1.5 nm thickness by probe sonication. This material could give rise to 16.5 mA cm−2 current density at −1.1 VRHE with a formate FE of 86%. The bulk bismuth could only reach 6.8 mA cm−2 current density at the same potential with a much lower formate FE of 64.9%. In a 10 h operation, average formate FE was 92.6%, and the authors claimed no catalyst degradation by XPS, XRD or Raman analysis. Further, Su et al.211 converted Bi2S3 to bismuth oxide sulfate by heating at 500 °C in an O2 environment. Finally, ∼10 nm thick bismuth NSs with exposed (110) facets were obtained via electrochemical reduction of this intermediate. The SEM images in Fig. 16a and b show synthesized Bi NSs attached to a carbon-fiber electrode. This material managed to sustain more than 90% formate FE in the potential range of −0.9 VRHE to −1.2 VRHE. In a 5 h operation, nearly 12.5 mA cm−2 stable current density was observed at −1 VRHE with >95% formate FE. Han et al.212 hydrothermally synthesized bismuth oxyiodide nanosheets (NSs) (Fig. 16c and d), where Bi2O2 slabs were sandwiched between iodide ion layers. Electrochemical reduction of these 2D sheets gave rise to a nearly nanometer-thick bismuth NS (Fig. 16e) without any significant change in volume or crystallinity. At −1.27 VSCE, the formate FE was only 16%, rapidly increasing to >95% at −1.5 VSCE and maintaining nearly 100% up to −1.7 VSCE. The partial current density of 24 mA cm−2 was observed at −1.74 VSCE, whereas the commercial bismuth nanopowder did not even reach 6 mA cm−2. In 10 h of catalysis at −1.5 VSCE (680 mV overpotential), the electrode managed to provide a stable current density of 15–16 mA cm−2. They also demonstrated a complete cell device with an IrO2 anode, which initiated at 2.1 V and operated at 10 mA cm−2 current density with 3.2 V external voltage. In 3 h of operation in full-cell mode, the average formate FE was 95% and electricity to formate conversion efficiency was 47%, which is commendable. Later, Yang and his group synthesized a cathode with a few atomic thick (3.5–7 nm) mesoporous Bi NS with nearly 100% formate FE.213 They solvothermally prepared bismuth oxycarbonate NS in N-methyl pyrrolidine (NMP) solvent. And they further electrically reduced them to Bi NS, confirmed by XRD, with introduced mesoporosity due to volume contraction, keeping the morphology intact. At −0.9 VRHE, formate selectivity was 99%, and in 12 h long operation at −0.8 VRHE, the average FE was 94% with a current density of 5 mA cm−2. In 2020, Wang et al.214 hydrothermally grew a few nanometer thick bismuth oxyselenides on a nitrogen-doped ordered mesoporous carbon (OMC-N) support. Electrochemical reduction then converted the bismuth oxyselenide to bismuth NSs. An electrode with bismuth NSs could reach 51 mA cm−2 current density with 90% formate FE at −1 VRHE. However, the OMC-N support in the electrode induced a little bit of methanol selectivity, which peaked at −0.6 VRHE with 67% methanol FE, and at −0.87 VRHE peak formate efficiency of 72% was observed. Moreover, the support helped to sustain 35 h of operation with 620 mV overpotential with ∼80% formate FE. They also fabricated a device with Ir/C as an anode, and the device could generate a 6.43 mA cm−2 current density with a battery of 3.1 V open circuit potential. They further studied the catalyst in flow-cell mode, and at −0.465 VRHE, bismuth NSs reached 95.29% formate FE whereas the supported ones reached 86.34%. For the bismuth NS flow-cell, current density reached as high as 347 mA cm−2 at −0.5 VRHE, which is undoubtedly significant. Yang and his group obtained atomically thin bismuth NSs by chemically reducing bismuth chloride.215 They found this 0.65 nm thin NS (Fig. 16f–i) has an exposed (111) facet whereas a thicker NS has an exposed (011) facet. Theoretically, they found that the (011) facet strongly binds to the intermediate, making the thinner NS a superior catalyst. A small Tafel slope of 87.6 mV dec−1 compared to 127.6 mV dec−1 of a thicker one also supported the phenomenon. At −0.58 VRHE, the electrode with thin NS maintained 99% formate FE for 75 h of operation, but the current density was poor due to blocked active sites in the stacking layers. Therefore, they mixed the catalyst with a small amount of inactive carbon black and got a stable partial current density of 54 mA cm−2 for 75 h. This high current density in low overpotential with an H-type cell was a milestone in improving the material. After electrocatalysis, a high-resolution XPS study of bismuth 4f showed no degradation of the catalyst (Fig. 16j). The catalyst remained active even in air annealing at 400 °C, proving the robustness of the catalyst.
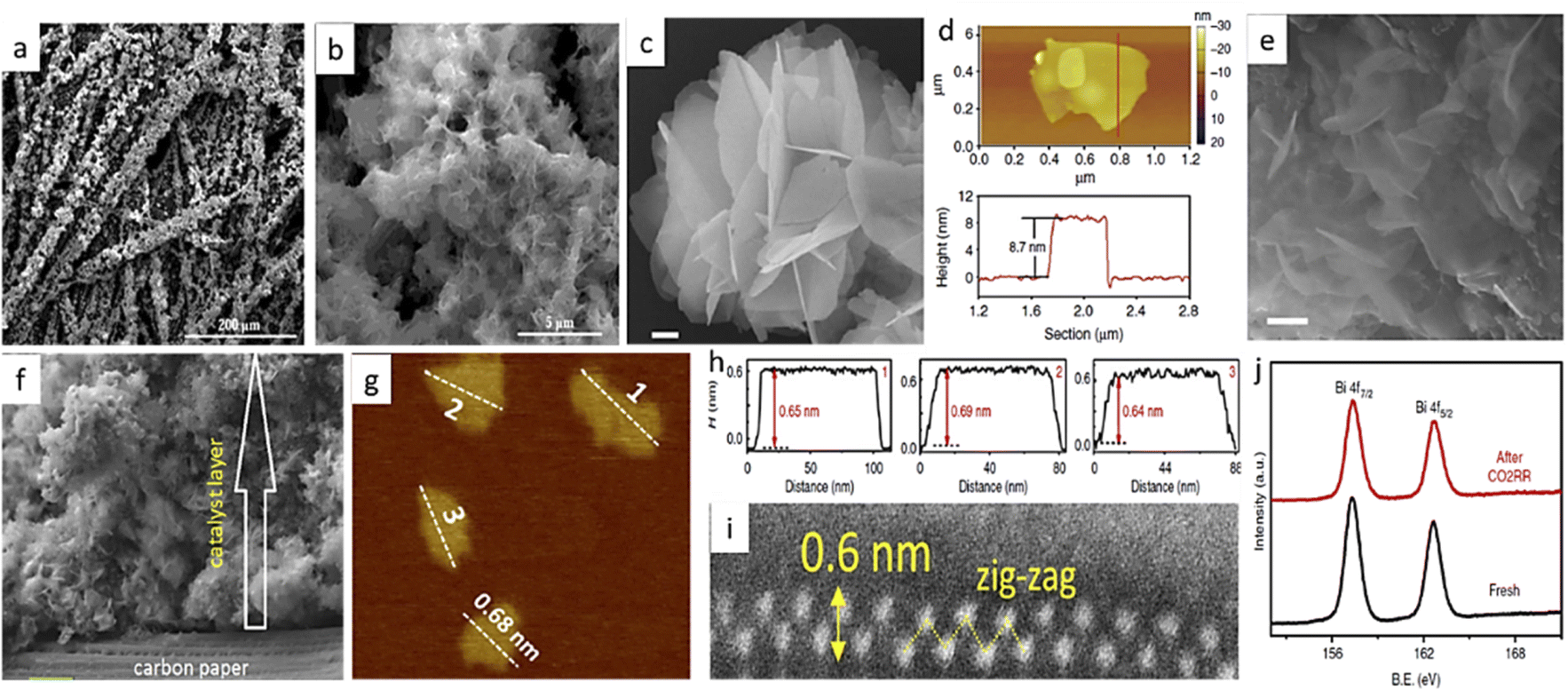 | ||
| Fig. 16 (a), (b) SEM images of Bi NSs on carbon fiber. (c) An SEM image of BiOI NSs; 200 nm scale bar. (d) An AFM image and the corresponding height profile. (e) An SEM image of topotactically reduced Bi NS; 200 nm scale bar. (f) An SEM image of a bismuthene catalyst layer on a carbon paper electrode; 1.0 μm scale bar. (g) An AFM image and (h) corresponding height profiles for the three bismuthene NSs marked in (g). (i) A lateral HAADF-STEM image of a bismuthene NS, showing a zig-zag structured layer with single-atom thickness. (j) High-resolution Bi 4f XPS spectra before and after 75 h of the CO2RR process at −0.58 VRHE, (a), (b) are reproduced with permission from Su et al.,211 Copyright 2018, John Wiley and Sons, (c)–(e) from Han et al.212 Copyright 2018, Springer Nature Limited, (f)–(j) from Yang et al.,215 Copyright 2020, Springer Nature Limited. | ||
:1 CNP and CuPC loading, they got commendable methane selectivity of 62% for the first time, with the partial current density for methane as high as 136 mA cm−2 at −4.0 V cell voltage. The activity was conserved over a more than 110 h stability test with DI-water flushing of the cathode at periodic intervals, as shown in Fig. 17e. The product selectivity dependence with different loading ratios is shown in Fig. 17f. In a report by Kim et al.,220 the effect of CO2 flow rate and partial pressure on zero-gap MEA cells was studied. Their Ni SA catalyst supported on a carbon matrix (Ni–NC) could give 100% CO FE in 1 atm of CO2 flow with −299.1 mA cm−2 current density at −3 V. When the partial pressure of CO2 was reduced from 1 atm to 0.1 atm, Ni–NC sustained CO FE at 93% even at 0.1 atm partial CO2 pressure, but for commercial Ag NPs, CO FE dropped from 94% to 40% at −3 V. They also found that CO FE on Ag NPs dropped due to low CO2 concentration, not because of the amount of CO2 reaching the GDE. Hou et al.221 reported formate production in an MEA flow cell with InN NPs. The cell was stable at a constant current density of 64.1 mA cm−2 in an 88 h long run with more than 90% formate selectivity, and the corresponding cell voltage was ∼2.96 V. In 2022, Liu et al.222 made ultrathin CuO nanoplates by anodic oxidation which self-reduced to a Cu/Cu2O heterogeneous interface during electrocatalysis. In an H-type cell, the catalyst could sustain 62.2% ethylene FE for a 50 h long test in neutral KCl electrolyte at −0.8 VRHE. The interface of the heterostructure and Cu+ content remained intact after a long-term test. In a flow cell, the partial current density for ethylene was 92.5 mA cm−2 at −0.81 VRHE and the corresponding FE and EE were 84.5% and 47.6%. While in full-cell operating mode at −3.1 V, the current density reached 75 mA cm−2 with ethylene FE 77.3% and EE 28.9 ± 1.3%. However, in 55 h long operation at constant 150 mA cm−2 current density, an average 74% ethylene FE was observed and the corresponding cell voltage was ∼4.5 V (derived data). Finally, to reach a higher current density, an MEA configuration with 0.5 M KHCO3 electrolyte was explored. Fig. 17g and h clearly show that the highest ethylene FE was observed at 100 mA cm−2 in a flow cell whereas in an MEA cell at 200 mA cm−2 the current density and cell voltage for MEA were also lower. At 200 mA cm−2 operating current density, a considerably high ethylene FE of 80.0 ± 2.2% and EE of 27.6 ± 0.8% were reported. Table 5 features recent developments in MEA cell setups.
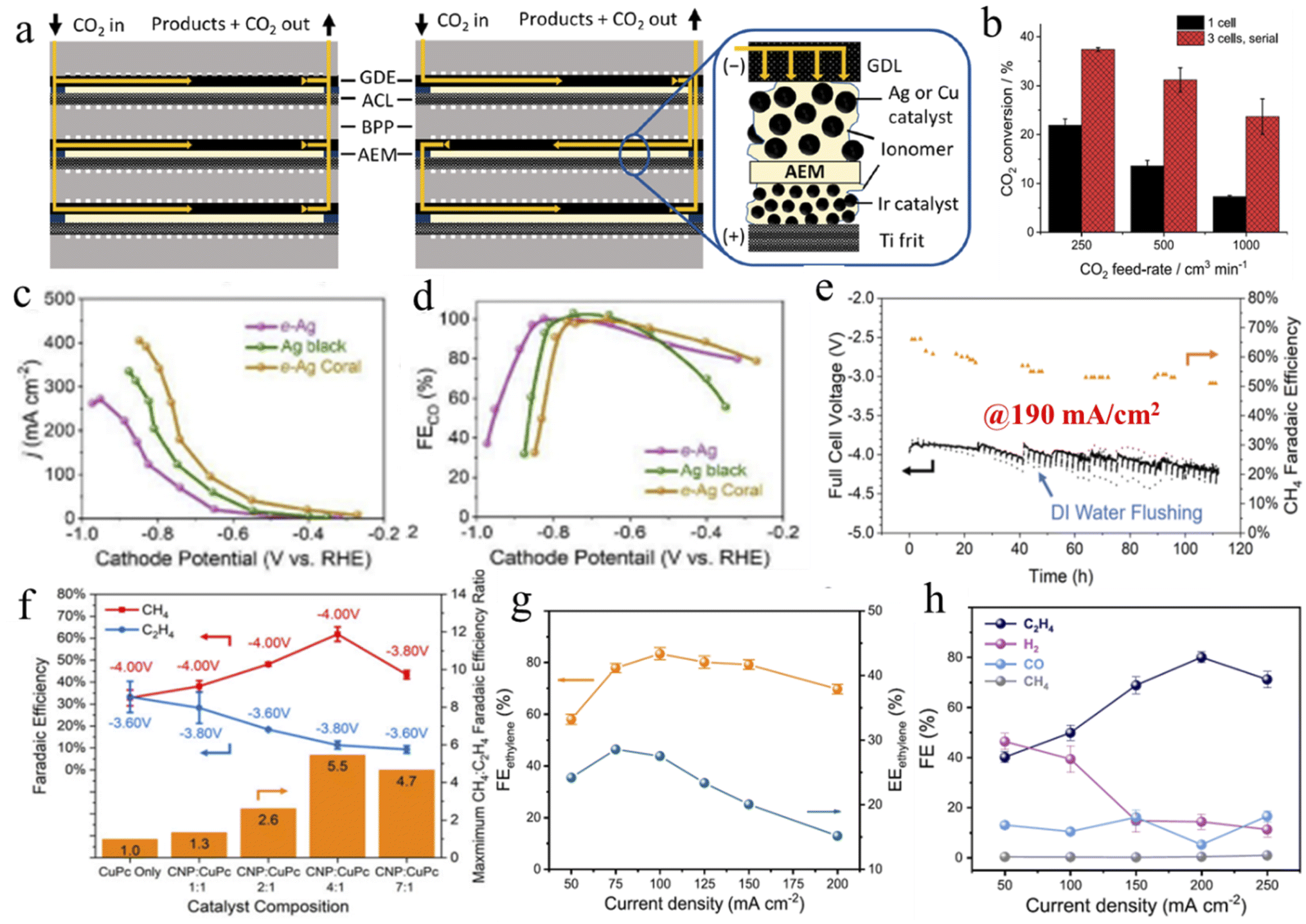 | ||
| Fig. 17 (a) Graphical representations of a layer-stacked MEA cell. (b) CO2 conversion efficiency with flow rate. (c), (d) LSV data for a cathode GDE and corresponding FE values, respectively. (e) Long-term stability test data in cell mode. (f) Selectivity dependence on catalyst loading. (g), (h) Ethylene FE changes with current density in a flow cell and MEA, respectively, (a), (b) are reproduced with permission from Endrödi et al.,217 Copyright 2019, American Chemical Society; (c), (d) are from Lee et al.,98 Copyright 2020, Elsevier Ltd; (e), (f) from Xu et al.,219 Copyright 2021, Springer Nature Limited; (g), (h) are from Liu et al.,222 Copyright 2022, Springer Nature Limited. | ||
| S. No. | Material | Electrolyte (pH) | Activity | Selectivity/FE (%) | Study time | Year (ref.) |
|---|---|---|---|---|---|---|
| 1 | Cu/Cu2O hetero-interface | Normal MEA 0.5 M KHCO3 | 200 mA cm−2 @ ∼−3.5 V (derived) | C2H4 (80.2 ± 2.2) | — | 2022 (ref. 222) |
| 2 | Carbon NP + Cu cluster | Normal MEA 0.05 M KHCO3 | 136 mA cm−2 @ ∼−4 V | CH4 (62) | 110 h | 2021 (ref. 219) |
| 3 | Ni SA/C-matrix | Zero-gap MEA 0.1 M KHCO3 | −299.1 mA cm−2 @ ∼−3 V | CO (100) | — | 2021 (ref. 220) |
| 4 | InN NPs | Flow-cell MEA 0.5 M NaHCO3 | 64.1 mA cm−2 @ ∼−2.96 V | HCOO− | 88 h | 2021 (ref. 221) |
| 5 | mp-Choral Ag/PTFE/C-paper | Zero-gap MEA 1 M KOH | 1 A by 10 cm2 electrode @ 2.75 V | CO (100) | 30 h | 2020 (ref. 98) |
| 6 | Cu NP layer/PTFE treated C-paper | Normal MEA 0.1 M KHCO3 | 120 mA cm−2 @ ∼−3.75 V | C2H4 (40) | 100 h | 2019 (ref. 218) |
4.3. Photovoltaic-electrocatalytic/photoelectrocatalysis (PV-EC/PEC) devices
Like electrocatalysis, here also, the target is a high current density at a minimally applied overpotential without losing the optimal FE towards a CO2-reduced product. This setup is basically the synchronization of two independent devices – PV and electrocatalytic cells, and both can be optimized separately. As found in eqn (6) (Section 3), STF efficiency depends only on the operating photocurrent density, and the FE of the dark cathode, i.e., the overpotential, has no role in STF efficiency calculation. Therefore, a contour plot with current density and FE shows a fixed relation with STF (Fig. 18a). Now the factor FE, which depends on the dark cathode of the 2nd device, only has a limit of 100%. Hence, to realize a very high STF, enhancement of photocurrent density of PV cells must be given more importance. However, the operating current in the final integrated device can be chosen by controlling the surface area of the PV cell. To ensure that optimal current density passes through the cathode so that FE remains a maximum, the cathode surface area is also altered accordingly, so that the same current is passed throughout the integrated device. This ensures that optimal current density of PV cells is conserved, and hence, high STF is also ensured in this kind of device. To be more precise, the optimal condition of electrocatalytic geometric current density must be very close to the optimal-zone current density of the respective solar cell (Fig. 18b includes I–V plots of common PV cells).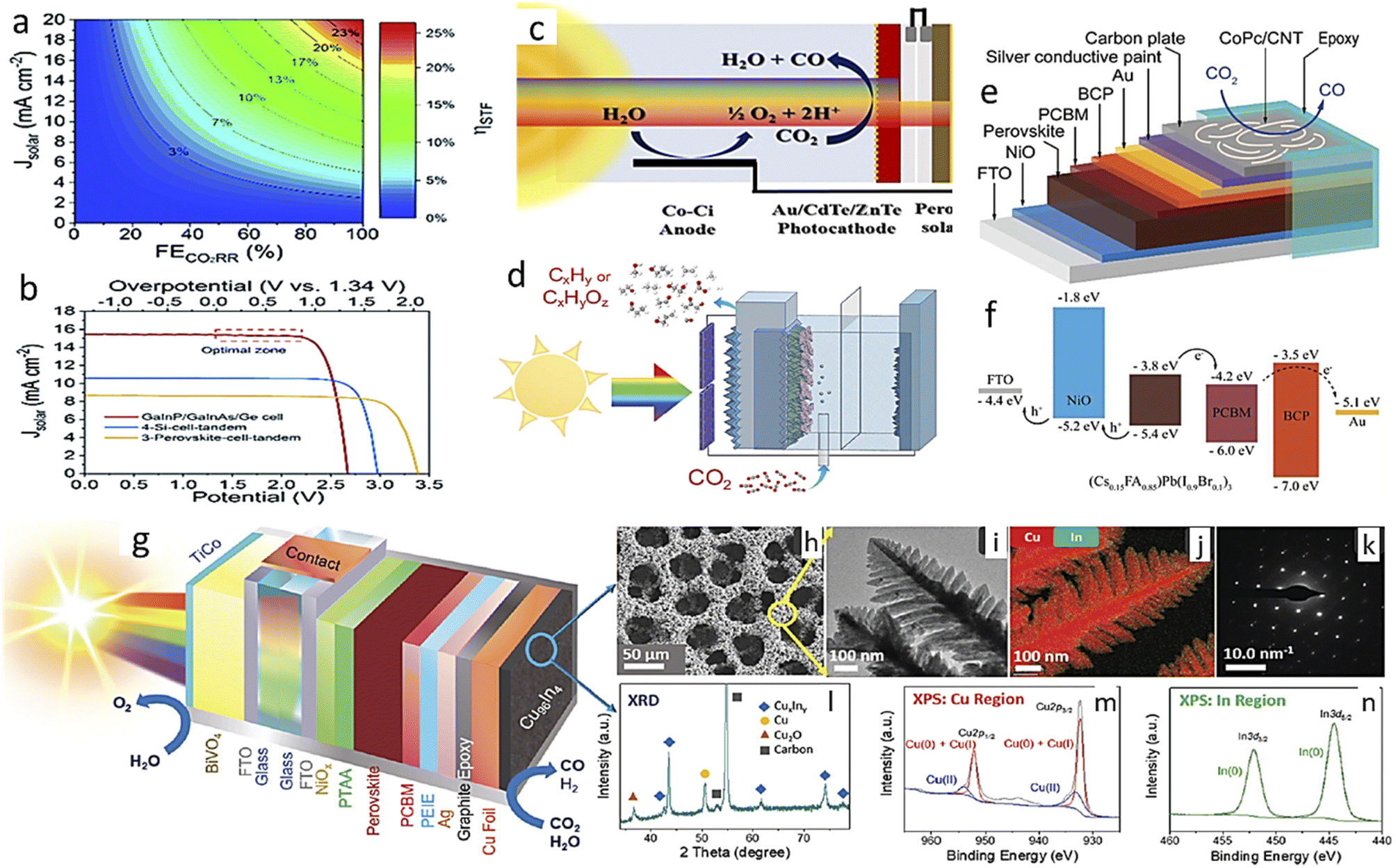 | ||
| Fig. 18 (a) Contour plot relating the operating solar current density, FE, and STF efficiency. (b) I–V characteristics of a few common solar cells. (c) and (d) Graphical representations of the device setup for PV-PE. (e) A graphical representation of a photocathode and (f) the charge separation mechanism of its corresponding layers. (g) A graphical representation of a whole cell. (h)–(n) SEM, HR-TEM, STEM mapping, SAED pattern, powder XRD, and XPS analysis in the Cu and In regions, respectively. (a), (b) Reproduced with permission from Xiao et al.,225 Copyright 2020, Royal Society of Chemistry; (c) is from Jang et al.,135 Copyright 2016, American Chemical Society; (d) is from Gurudayal et al.,158 Copyright 2019, Royal Society of Chemistry; (e), (f) from Zhang et al.226 Copyright 2020, John Wiley and Sons, and (g)–(n) from Rahaman et al.,228 Copyright 2020, Royal Society of Chemistry. | ||
In 2015, Marcel et al.223 used three halide perovskite PV cells with 0.285 cm2 surface area in series for the first time to get a 5.8 mA cm−2 operating current density, giving rise to an STF efficiency of ∼6.5%. They used 1 cm2 of anodized gold nanoparticles to get an effective porous cathode and 4.5 cm2 IrOx on the Ti anode. As a result, the actual electrode current density was 0.37 mA cm−2 for the anode and 1.65 mA cm−2 for the cathode, which was optimal for CO selectivity to remain between 80% and 90% in 18 h of operation. In 2017, Sriramagiri et al.224 scaled up the PV-EC system by connecting 5 silicon solar cells in series, reaching an operating current >1 A (1024 mA). They used a 25 cm2 cathode with Ag nanoparticles and an equal-sized anode with an Ir-coated catalyst membrane to construct a flow cell. The electrolytic cell operated at 2.85 V, but a resistive loss in the circuit of 5 cells in series with a 25 cm2 surface area each failed to deliver the optimal current. As a result, 33 cm2 surface area was suitable for optimal operation, i.e., the total photo-harvesting surface area = 5 × 33 cm2; therefore, the operating current density was reduced. Thus, with ∼75% CO FE, the STF efficiency was 6.5% which is quite average. Kun et al.198 synthesized immobilized a single Ni atom in graphene shell. In normal H-type cells with Li-treated Co3O4 as an anode, they got carbon monoxide at 2.34 V of cell potential with more than 10% STF efficiency by a GaInP2/GaAs/Ge solar cell under 50 mW cm−2 light exposure. Apart from the cathode-only analysis described in the EC Section (4.2.1), Schreier et al.184 also demonstrated a device where the material is used as both the cathode and anode. This bi-functional electrode naturally eliminates one basic problem of cathode poisoning from the deposition of another leached cation from the anode material. In 0.25 M KOH, 0.1 mA cm−2 current density was obtained at 320 mV overpotential for O2 production, but the cathode works optimally in neutral pH; therefore, they used a double-layer membrane (one anion exchange and the other cation exchange) to use different electrolytes independently without any potential build-up. With a GaInP/GaInAs/Ge multijunction solar cell with 0.5625 cm2 surface area, they got an operating current density of 11.57 mA cm−2 (the operating current was 6.51 mA) with a cell voltage of 2.38 V under 1 sun illumination. In 5 h of stable operation with 20 cm2 electrode size, the average FE towards CO was 81%, which made the STF efficiency nearly 13.4%, although the cathodic current density was as low as 0.33 mA cm−2. In 2018, Yang et al.213 made a highly formate-selective (∼100%) cathode with a Bi NS, as already discussed in the EC part (4.2.1.H); they also fabricated a device with an anode made of 20% (by weight) Ir loading in Vulcan carbon black. They used GaInP/GaInAs/Ge multijunction solar cells under 1 sun with 1 cm2 active surface area to supply the photovoltage. Under an electrocatalytic load of 2.5 V, the operating current was 3.3 mA. In a 3 h test, the operating current dropped to 1.9 mA due to the deactivation of the anode, which resulted in an average STF as low as 1.5%. Looking to high selectivity and easy hydrothermal synthesis, along with anode selection, the cathode current density also needed to be improved to reach the full potential of this cathode material. In 2019, Guo and his group demonstrated PV-EC devices with their Cu/Ag bimetallic cathode and IrO2-deposited Ti foil as an anode.93 To operate without external bias, three perovskite solar cells of 0.25 cm2 each were connected in series to provide the ∼2.8 V required for the electrocatalytic cell. For 60 min operation under 1 sun, the operating current density was 6.68 mA cm−2 at 2.76 V, which was very close to the maximum power output point of 6.63 mA cm−2 at 2.8 V of the solar cell. To sustain the 54% FE towards ethylene, the cathode surface area was made 0.085 cm2 to maintain optimal current density in the cathode. However, the ethylene STF efficiency was 4.17%, and the overall fuel STF was 7.78%. In 2020, Xiao et al.225 synthesized an additive-free GDE that can reach 18.4 mA cm−2 current density at 300 mV overpotential. They directly deposited a silver layer on a polypropylene membrane, then by anodization converted it to AgCl then further electrochemical reduction gave rise to a hierarchical porous nanostructure. The best catalyst with a 1.2 μm Ag deposition layer expanded to a 2.8 μm thick porous active catalyst layer. They also concluded that, although it had a higher number of active sites, the active layer thickness of more than 3 μm, showed lower current density due to mass transport limitation. Cheng et al.175 improved the catalytic activity tremendously. They used PV cells as well as electrodes with the same surface area of 0.31 cm2. The operating current was also as high as 4.5 mA. They claimed that this catalyst had a 50 times higher CO formation rate that other reported catalysts for PV-EC CO2RR and they intentionally used a low catalyst loading in the GDE to match the current from the PV cell. They also tested it in outdoor sunlight (<100 mW cm−2) conditions and got a result close to the laboratory experiment, with ∼18.7% of STF efficiency at 2.2 V cell voltage. In 2022, Boutin et al.179 utilized a 0.92 cm2 GaInP/GaInAs/Ge triple-junction concentrated PV cell (CPV) which could work up to 450 sun. They also found that the open circuit voltage increased from 2.33 V to 2.58 V when the light intensity increased from 132 sun to 450 sun. They used ultrapure water to synergistically cool down the CPV cell and heat up the EC cell. As the coolant was also being used as an anolyte to avoid a short circuit between the PV cell and the anode by the coolant, non-conducting ultrapure water was used. At the optimal 289 sun, the operating current was as large as 4 A and at 150 mA cm−2 a cathode current density of more than 90% CO selectivity was maintained.
Apart from the PEC study discussed in the modified solar cell part by Jang et al.,135 they also reported another approach in a PV-cell-coupled CO2RR device in 2016. They integrated their photoelectrode with PV cells and assembled them in a tandem manner to get bias-free operation with a single light source (Fig. 18c). By this approach, higher energy photons are absorbed by the photocathode, and lower energy photons can pass through it to be absorbed by the PV cell, kept in parallel. Moreover, due to the additional supply of photovoltage by the photocathode, even the lower photovoltage of single-layer perovskite was sufficient to drive the CO2RR under illuminated conditions. They reported ∼0.7 mA cm−2 of current density under bias-free conditions with a nearly similar FE of 80% towards carbon monoxide in 3 h of operation. In 2019, Gurudayal and his group also used a modified silicon solar cell photocathode in tandem with a halide perovskite solar cell (Cs0.05(MA0.17FA0.83)0.95Pb(I0.83Br0.17)3) to realize bias-free operation (Fig. 18d).158 Two series-connected solar cells of 0.5 cm2 active area each, can supply 2.1 V photovoltage and 5.8 mA of current density, and the silicon photocathode provides 0.6 V of photovoltage in addition. However, in an electrocatalytic load with 0.1 M CsHCO3, the operating current was 2.1 mA at 1.95 V cell voltage, and with 0.5 M CsHCO3, the operating current increased to 2.9 mA with a cell potential of 1.86 V. The higher electrolytic concentration also improved the hydrocarbon selectivity along with the operating current and as a result the STF efficiency for hydrocarbon increased from 0.9% to 1.5%.
In 2020, Zhang et al.226 synthesized a carbon-encapsulated cesium formamidinium lead halide [(Cs0.15FA0.85)Pb(I0.9Br0.1)3] photocathode and decorated it with cobalt phthalocyanine molecular catalyst loaded carbon nanotubes as a top layer for CO2RR. Fig. 18e and f present schematics of the layers of the photocathode and corresponding charge extraction mechanisms from the perovskite cell, respectively. The carbon-encapsulated cell had an open circuit voltage of 0.99 V and −22.85 mA cm−2 short circuit current density. The hybrid photocathode under 1 sun illumination could give −15.5 mA cm−2 current density at −0.11 VRHE, and a peak FE of 87.5% towards CO was observed at 0.17 VRHE. The photoanode of amorphous Si with IrOx loading could reach 8.18 mA cm−2 current density at 1.23 VRHE. Finally, when both electrodes were operated in tandem under 1 sun, a stable 3 mA cm−2 current density was observed for 1.5 h with 83% CO FE at the photocathode and 100% O2 formation at the photoanode. In the same year, Reisner and his group reported an integrated PV-PEC setup to realize bias-free CO2RR with OER.227 They buried the perovskite solar cell under the cathode catalyst layer and used it with a BiVO4 photoanode in a tandem arrangement. In one experiment, with an immobilized cobalt porphyrin catalyst on carbon nanotubes with triple-cation mixed halide perovskite, they reported the CO:H2 ratio in the product syn gas was 0.33 ± 0.29 under zero-bias conditions with 181 ± 20 μA cm−2 stable current density, and at 0.4 V applied voltage, the ratio reached a maximum at 1.96 ± 0.35. Moreover, the tandem cell sustained stable operation for 67 h under zero-bias conditions with a current density higher than 100 μA cm−2. Another report used Cu96In4 alloy as a CO2RR catalyst instead of a metal complex catalyst to realize a stable photocurrent density of 195 ± 20 μA cm−2 over a 10 h experiment.228Fig. 18g illustrates the cell model and Fig. 18h–n represent SEM, XRD and XPS characterizations of the alloy catalyst. A few of the interesting reports from the above discussion are highlighted in Table 6.
| S. No. | Material | PV cell | Electrolyte (pH) | Conditions | Activity | Selectivity/FE (%) | Study time | Year (ref.) |
|---|---|---|---|---|---|---|---|---|
| 1 | Ag NP/C-paper | GaInP/GaInAs/Ge At 289 sun | 1 M CsOH for activation | GDE flow cell (MEA) | 150 mA cm−2 @ solar cell potential of <2.8 V | CO (>90) | 2.5 h | 2022 (ref. 179) |
| 2 | (Cs0.15FA0.85)Pb(I0.9Br0.1)3/Co-phthalocyanine on carbon NT (PV cell embedded PC) + a-Si/IrOx (PA) | 0.5 M KHCO3 (7.2) | H-type cell with tandem configuration at 100 mW cm−2 | 3 mA cm−2 | CO (83) | 1.5 h | 2020 (ref. 226) | |
| 3 | Np-Ag/GDE/activated Ni foam | GaInP/GaInAs/Ge at 100 mW cm−2 | 1 M KOH (14) | In GDE setup: electrolyte flowed at 2 mL min−1, and CO2 flowed at 10 sccm | 14.4 mA @ solar cell potential of 2.23 V | CO (99) STF (CO) = 19.1% | 150 h (stability test) | 2020 (ref. 175) |
| 4 | np Ag/PP membrane (C) + Ni–Fe/Ni foam (A) | GaInP/GaInAs/Ge at 100 mW cm−2 | 1 M KOH with AEM membrane | In GDE setup: electrolyte flowed at 0.9 mL min−1 and CO2 flowed at 20 sccm | >15 mA @ cell potential of 2.2 V (cathode overpotential = 300 mV) | CO (100) STF (CO) = 20.1% | 28 h | 2020 (ref. 225) |
| 5 | mp-Bi nanosheets + Ni/C | GaInP/GaInAs/Ge at 100 mW cm−2 | 0.5 M NaHCO3 (7.4) | H-type cell: CO2 bubbling at 20 sccm | 3.3 mA @ solar cell potential of 2.5 V | HCOOH (100) STF (HCOOH) = 1.5% | 3 h | 2018 (ref. 213) |
| 6 | SnO2 coated CuO NW (both electrodes) | GaInP/GaInAs/Ge at 100 mW cm−2 | 0.1 M CsHCO3 (6.75) (catholyte) 0.25 M CsOH (13.3) (anolyte) in bi-polar membrane | H-type cell: CO2 flow rate at 10 mL min−1 | 11.57 mA @ cell potential of 2.38 V (cathode potential = −0.55 V) | CO (81) STF (CO) = 12.4% | 5 h | 2017 (ref. 184) |
| 7 | Np-Ag + Ir-CCM | Si-cell (5–6 cells in series) at 100 mW cm−2 | 0.5 M NaHCO3 | Flow cell: catholyte (no anolyte used) flowed at 150 mL min−1, and CO2 flowed at 25 mL min−1 | 1024 mA operating current 6.27 mA @ solar cell potential of 2.75 V | CO (75.9) STF (CO) = 6.4% | — | 2017 (ref. 224) |
| 8 | NiN-GS + Li+-tuned Co3O4 | GaInP2/GaAs/Ge at 50 mW cm−2 | 0.5 M KHCO3 (7.5) + 0.5 M K2HPO4/KH2PO4 (buffer) | H-type cell: CO2 flow rate at 50 sccm | >4.5 mA @ solar cell potential of 2.35 V | CO (86.7) STF (CO) > 10% | 10 h | 2017 (ref. 198) |
| 9 | ZnO/ZnTe/CdT/Au (PC) | CH3NH3PbI3 at 100 mW cm−2 (single light source for both absorbers) | 0.5 M KHCO3 (7.5) | H-type cell | ∼0.7 mA cm−2 @ bias less condition | CO (∼80) STF = 0.35% | 3 h | 2016 (ref. 135) |
| 10 | Au (C) + IrO2/Ti (A) | CH3NH3PbI3 perovskite (3 cells in series) at 100 mW cm−2 | 0.5 M KHCO3 (7.2) | H-type cell: CO2 flow rate at 20 mL min−1 | 5.8 mA @ solar cell potential | CO (80–90) STF (CO) = 6.5% | 18 h | 2015 (ref. 223) |
5. Summary and future prospects
Among the several processes discussed in Section 2, very few are being utilized in industry. Photothermal artificial photosynthesis via plasmonic catalysts still requires significant research to reach the commercial scale with affordability. A concentrated solar-light-assisted thermochemical process could be industrially viable because renewable solar energy will be utilized to reach a high reactor temperature. The thermochemical hydrogenation of CO2 towards various products like methanol, dimethyl ether, methane, and formate is a highly scalable process, but industrial profitability mostly depends on the cost of hydrogen and the CO2 capture process. Moreover, elevated temperature and pressure requirements decrease the convenience and energy efficiency. As the hydrogen price is higher than that of the finally obtained methanol via CO2 hydrogenation, for now, this is not economically viable.229 However, the United States has large levels of natural gas storage, so both black and green hydrogen can be used, but mainly black hydrogen with a lower unit production cost (UPC) is more viable. India and China have the potential for low-cost green hydrogen production favoring cheaper green fuel UPC via a thermochemical process.230 CRI's George Olah Renewable Methanol plant in Iceland has been operating since 2012 with industrial-scale CO2 direct hydrogenation to methanol via consuming CO2 waste from a nearby geothermal power plant using a traditional CuO/ZnO catalyst.231 And the corresponding required H2 is produced via electrocatalytic water splitting, mainly using geothermal, wind, and hydropower sources.232 The PC CO2RR is also a developing field of research and very interesting in terms of the direct utilization of solar energy. However, this field requires further long-term research to bring the product formation rate into an industrially viable range.The PEC CO2RR has been studied for the last few decades and, hence, is not an optimized field for converting CO2 into renewable fuel. Therefore, many potential semiconductors are being explored for this application. This field is growing rapidly due to its highly convenient and sustainable solution to solar light harvesting and CO2 utilization. However, it is tough to find a semiconductor with a perfect bandgap and band edge position suitable for the CO2RR. Moreover, as the electrode inside the electrolyte is exposed to light along with electrical potential, the chance of photocorrosion is enhanced along with electrocorrosion. Most catalysts, specifically oxide and sulfide materials, lose selectivity during long-term operation, and water reduction dominates over time. Therefore, it is necessary to find a protecting material that can make a light-transparent coating on the photoelectrode without altering its catalytic activity. Moreover, the improved stability of the photoelectrode will also allow PEC operation at a higher cathodic potential window to realize more reduced products along with consequent higher current densities. However, a modified solar cell photoelectrode has already shown considerable possibilities in this area. PEC also holds a very optimistic future due to its high liquid product selectivity and higher reduced product selectivity. We should look forward to improved material stability with higher current densities for commercialization, as this is the greenest approach for the CO2RR.
Due to its comparatively higher current density, EC is well ahead of other techniques in terms of industrial viability. Moreover, the electrodes are also comparatively more stable and can allow long-term operation without significant selectivity loss. With GDEs, industrially viable high current densities are observed, but most electrocatalysts with high current density are reported to be selective towards CO alone. And, CO requires further intense treatment to be used as a fuel. Recently, a few reports have improved methane and ethylene selectivity, but these catalysts also lose selectivity after long-term operation. Moreover, carbon-based GDEs lose hydrophobicity during long-term operation, and flooding hampers operation. Lots of research is required to fix these issues. Recent developments in MEA setups take the EC CO2RR to the next level in terms of both current density and energy efficiency, as electrolyte iR loss is mostly minimized in a zero-gap MEA. Apart from sophisticated cell setup and other minor drawbacks, like salt precipitation, this process is far superior in performance and electrical efficiency. Therefore, intense research may soon unleash the full potential of this EC CO2RR technique.
PV-EC is in the most advantageous position in today's race among other techniques for real-life application. The more efficient solar cells become, the more the PV-EC technique will benefit. There is only one way of increasing the STF efficiency for a selective electrode: increasing the solar current density. However, using concentrated light for PV cells can be a solution for industrial viability, as shown by Boutin et al.179 Many solar cells with low open circuit voltages can be used in series to obtain the required cell potentials. In that case, current density decreases, consequently reducing the STF efficiency. However, the Jsolar value for the CO2RR supplied by state-of-the-art solar cells still could not exceed 17.5 mA cm−2, giving a projected ηSTF of 23.4% as the highest achievable ηSTF for state-of-the-art solar cells at present.225
In conclusion, we may expect an MEA cell with minimal cell voltage to be coupled with one PV cell with decent STF efficiency, which will change the outlook of the world regarding energy harvesting and storage in the near future. However, next-generation MEAs with high current density, energy efficiency, and CO2 conversion efficiency will be perfectly suitable for industrial use. More research on earth-abundant low-cost materials with long operational life will further assist the technology to help mankind by giving a low-cost and green solution to the energy crisis and environmental degradation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
PK acknowledges financial support from the Department of Science and Technology (Grant No. DST/TMD/HFC/2k18/138).References
- J. Zhang, C. D. Sewell, H. Huang and Z. Lin, Adv. Energy Mater., 2021, 11, 1–38 CAS
.
- H. B. Iz, All Earth, 2022, 34, 16–26 CrossRef
- J. Tollefson, Nature, 2017 DOI:10.1038/nature.2017.22627
- B. U. Haq and S. R. Schutter, Science, 2008, 322, 64–68 CrossRef CAS PubMed
- V. Gray, Energy Environ., 2007, 18, 433–440 CrossRef
- J. M. Chen, Innovation, 2021, 2, 100127 CAS
- K. Roy, P. Devi and P. Kumar, Nano Energy, 2021, 87, 106119 CrossRef CAS
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS
- O. G. Sánchez, Y. Y. Birdja, M. Bulut, J. Vaes, T. Breugelmans and D. Pant, Curr. Opin. Green Sustainable Chem., 2019, 16, 47–56 CrossRef
- R. Küngas, P. Blennow, T. Heiredal-Clausen, T. Holt, J. Rass-Hansen, S. Primdahl and J. B. Hansen, ECS Trans., 2017, 78, 2879–2884 CrossRef
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC
- V. C. Hoang, V. G. Gomes and N. Kornienko, Nano Energy, 2020, 78, 105311 CrossRef CAS
- A. Liu, M. Gao, X. Ren, F. Meng, Y. Yang, L. Gao, Q. Yang and T. Ma, J. Mater. Chem. A, 2020, 8, 3541–3562 RSC
- D. L. T. Nguyen, Y. Kim, Y. J. Hwang and D. H. Won, Carbon Energy, 2020, 2, 72–98 CrossRef CAS
- R. Zhao, P. Ding, P. Wei, L. Zhang, Q. Liu, Y. Luo, T. Li, S. Lu, X. Shi, S. Gao, A. M. Asiri, Z. Wang and X. Sun, Adv. Funct. Mater., 2021, 31, 1–35 Search PubMed
- Q. Fan, M. Zhang, M. Jia, S. Liu, J. Qiu and Z. Sun, Mater. Today Energy, 2018, 10, 280–301 CrossRef
- J. J. Wang, X. P. Li, B. F. Cui, Z. Zhang, X. F. Hu, J. Ding, Y. Da Deng, X. P. Han and W. Bin Hu, Rare Met., 2021, 40, 3019–3037 CrossRef CAS
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS
- Y. Zhou and B. S. Yeo, J. Mater. Chem. A, 2020, 8, 23162–23186 RSC
- L. Hou, J. Yan, L. Takele, Y. Wang, X. Yan and Y. Gao, Inorg. Chem. Front., 2019, 6, 3363–3380 RSC
- M. M. Ayyub and C. N. R. Rao, Mater. Horiz., 2021, 8, 2420–2443 RSC
- G. M. Tomboc, S. Choi, T. Kwon, Y. J. Hwang and K. Lee, Adv. Mater., 2020, 32, 1–23 CrossRef PubMed
- A. Mustafa, B. G. Lougou, Y. Shuai, Z. Wang, S. Razzaq, E. Shagdar, J. Zhao and J. Shan, J. Mater. Chem. A, 2021, 9, 4558–4588 RSC
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 1–24 Search PubMed
- X. L. Lu, X. Rong, C. Zhang and T. B. Lu, J. Mater. Chem. A, 2020, 8, 10695–10708 RSC
- L. Huang, W. Li, M. Zeng, G. He, P. R. Shearing, I. P. Parkin and D. J. L. Brett, Composites, Part B, 2021, 220, 108986 CrossRef CAS
- X. Shao, X. Zhang, Y. Liu, J. Qiao, X. D. Zhou, N. Xu, J. L. Malcombe, J. Yi and J. Zhang, J. Mater. Chem. A, 2021, 9, 2526–2559 RSC
- Y. Chen, K. Chen, J. Fu, A. Yamaguchi, H. Li, H. Pan, J. Hu, M. Miyauchi and M. Liu, Nano Mater. Sci., 2020, 2, 235–247 CrossRef
- F. Y. Gao, Z. Z. Wu and M. R. Gao, Energy and Fuels, 2021, 35, 12869–12883 CrossRef CAS
- W. Choi, D. H. Won and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 15341–15357 RSC
- F. Liang, K. Zhang, L. Zhang, Y. Zhang, Y. Lei and X. Sun, Small, 2021, 2100323 CrossRef CAS
- P. Wang, S. Wang, H. Wang, Z. Wu and L. Wang, Part. Part. Syst. Charact., 2018, 35, 1–25 Search PubMed
- P. Ding, T. Jiang, N. Han and Y. Li, Mater. Today Nano, 2020, 10, 100077 CrossRef
- Y. Liu and L. Guo, J. Chem. Phys., 2020, 152, 100901 CrossRef CAS PubMed
- A. U. Pawar, C. W. Kim, M. T. Nguyen-Le and Y. S. Kang, ACS Sustainable Chem. Eng., 2019, 7, 7431–7455 CrossRef CAS
- H. Pang, T. Masuda and J. Ye, Chem–Asian J., 2018, 13, 127–142 CrossRef CAS
- K. Wang, Y. Ma, Y. Liu, W. Qiu, Q. Wang, X. Yang, M. Liu, X. Qiu, W. Li and J. Li, Green Chem., 2021, 23, 3207–3240 RSC
- N. Li, F. Xiang and A. Fratalocchi, Adv. Sustainable Syst., 2021, 5, 2000242 CrossRef CAS
- J. Feng, H. Huang, S. Yan, W. Luo, T. Yu, Z. Li and Z. Zou, Nano Today, 2020, 30, 100830 CrossRef CAS
- H. Zheng, S. Zhang, X. Liu and A. P. O'Mullane, Catal. Sci. Technol., 2021, 11, 768–778 RSC
- Y. Yang, S. Ajmal, X. Zheng and L. Zhang, Sustain. Energy Fuels, 2018, 2, 510–537 RSC
- J. He and C. Janáky, ACS Energy Lett., 2020, 5, 1996–2014 CrossRef CAS
- Z. Chen, Q. Jiang, H. An, J. Zhang, S. Hao, X. Li, L. Cai, W. Yu, K. You, X. Zhu and C. Li, ACS Catal., 2022, 7719–7736 CrossRef CAS
- D. Marxer, P. Furler, M. Takacs and A. Steinfeld, Energy Environ. Sci., 2017, 10, 1142–1149 RSC
- M. Hoes, C. L. Muhich, R. Jacot, G. R. Patzke and A. Steinfeld, J. Mater. Chem. A, 2017, 5, 19476–19484 RSC
- S. Abanades, A. Haeussler and A. Julbe, Appl. Phys. Lett., 2021, 119, 023902 CrossRef CAS
- A. Haeussler, S. Abanades, F. A. Costa Oliveira, M. A. Barreiros, A. P. F. Caetano, R. M. Novais and R. C. Pullar, Energy and Fuels, 2020, 34, 9037–9049 CrossRef CAS
- K. Gao, X. Liu, T. Wang, Z. Zhu, P. Li, H. Zheng, C. Song, Y. Xuan, Y. Li and Y. Ding, Sustain. Energy Fuels, 2021, 5, 4295–4310 RSC
- E. T. Kho, T. H. Tan, E. Lovell, R. J. Wong, J. Scott and R. Amal, Green Energy Environ., 2017, 2, 204–217 CrossRef
- M. Ghoussoub, M. Xia, P. N. Duchesne, D. Segal and G. Ozin, Energy Environ. Sci., 2019, 12, 1122–1142 RSC
- F. Zhang, Y.-H. Li, M.-Y. Qi, Y. M. A. Yamada, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem Catal., 2021, 1, 272–297 CrossRef
- M. N. Ha, G. Lu, Z. Liu, L. Wang and Z. Zhao, J. Mater. Chem. A, 2016, 4, 13155–13165 RSC
- K. Wang, R. Jiang, T. Peng, X. Chen, W. Dai and X. Fu, Appl. Catal., B, 2019, 256, 117780 CrossRef CAS
- J. Yan, C. Wang, H. Ma, Y. Li, Y. Liu, N. Suzuki, C. Terashima, A. Fujishima and X. Zhang, Appl. Catal., B, 2020, 268, 118401 CrossRef CAS
- L. Wang, X. Liu, Y. Dang, H. Xie, Q. Zhao and L. Ye, Solid State Sci., 2019, 89, 67–73 CrossRef CAS
- D. Liu, C. Wang, Y. Yu, B. H. Zhao, W. Wang, Y. Du and B. Zhang, Chem, 2019, 5, 376–389 CAS
- L. Wang, B. Zhao, C. Wang, M. Sun, Y. Yu and B. Zhang, J. Mater. Chem. A, 2020, 8, 10175–10179 RSC
- P. Li, L. Liu, W. An, H. Wang, H. Guo, Y. Liang and W. Cui, Appl. Catal., B, 2020, 266, 118618 CrossRef CAS
- H. Liu, H. G. Ye, M. Gao, Q. Li, Z. Liu, A. Q. Xie, L. Zhu, G. W. Ho and S. Chen, Adv. Sci., 2021, 8, 2101232 CrossRef CAS
- R. Jacot, J. M. Naik, R. Moré, R. Michalsky, A. Steinfeld and G. R. Patzke, J. Mater. Chem. A, 2018, 6, 5807–5816 RSC
- S. Zhai, J. Nam, G. Sai Gautam, K. Lim, J. Rojas, M. F. Toney, E. A. Carter, I. H. Jung, W. C. Chueh and A. Majumdar, J. Mater. Chem. A, 2022, 10, 3552–3561 RSC
- S. Zhai, J. Rojas, N. Ahlborg, K. Lim, C. H. M. Cheng, C. Xie, M. F. Toney, I. H. Jung, W. C. Chueh and A. Majumdar, Energy Environ. Sci., 2020, 13, 592–600 RSC
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed
- Y. Wang, S. Kattel, W. Gao, K. Li, P. Liu, J. G. Chen and H. Wang, Nat. Commun., 2019, 10, 1166 CrossRef PubMed
- E. C. Ra, K. Y. Kim, E. H. Kim, H. Lee, K. An and J. S. Lee, ACS Catal., 2020, 10, 11318–11345 CrossRef CAS
- N. Rui, X. Zhang, F. Zhang, Z. Liu, X. Cao, Z. Xie, R. Zou, S. D. Senanayake, Y. Yang, J. A. Rodriguez and C. J. Liu, Appl. Catal., B, 2021, 282, 119581 CrossRef CAS
- L. Lin, Y. Ge, H. Zhang, M. Wang, D. Xiao and D. Ma, JACS Au, 2021, 1, 1834–1848 CrossRef CAS
- L. Wang, W. Zhang, X. Zheng, Y. Chen, W. Wu, J. Qiu, X. Zhao, X. Zhao, Y. Dai and J. Zeng, Nat. Energy, 2017, 1–8 Search PubMed
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, J. Am. Chem. Soc., 2020, 142, 19001–19005 CrossRef CAS
- A. Jaleel, A. Haider, C. Van Nguyen, K. R. Lee, S. Choung, J. W. Han, S.-H. Baek, C.-H. Shin and K.-D. Jung, Chem. Eng. J., 2022, 433, 133571 CrossRef CAS
- Y. Subramanian, B. Mishra, S. Mandal, R. Gubendiran and Y. S. Chaudhary, Mater. Today: Proc., 2021, 35, 179–185 CAS
- J. Wei, F. L. Meng, T. Li, T. Zhang, S. Xi, W. L. Ong, X. Q. Wang, X. Zhang, M. Bosman and G. W. Ho, Adv. Funct. Mater., 2022, 32, 1–9 Search PubMed
- M. Stolterfoht, P. Caprioglio, C. M. Wolff, J. A. Márquez, J. Nordmann, S. Zhang, D. Rothhardt, U. Hörmann, Y. Amir, A. Redinger, L. Kegelmann, F. Zu, S. Albrecht, N. Koch, T. Kirchartz, M. Saliba, T. Unold and D. Neher, Energy Environ. Sci., 2019, 12, 2778–2788 RSC
- D. Guo, V. M. Caselli, E. M. Hutter and T. J. Savenije, ACS Energy Lett., 2019, 4, 855–860 CrossRef CAS
- U. Kang, S. K. Choi, D. J. Ham, S. M. Ji, W. Choi, D. S. Han, A. Abdel-Wahab and H. Park, Energy Environ. Sci., 2015, 8, 2638–2643 RSC
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC
- B. Ghosh, D. Ghosh, A. Ghosh, S. Hussain, R. Bhar and A. K. Pal, Mater. Sci. Semicond. Process., 2014, 25, 130–136 CrossRef CAS
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed
- A. Klinkova, P. De Luna, C. T. Dinh, O. Voznyy, E. M. Larin, E. Kumacheva and E. H. Sargent, ACS Catal., 2016, 6, 8115–8120 CrossRef CAS
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed
- K. H. T. Inoue, A. Fujishima and S. Konishi, Nature, 1979, 277, 637 CrossRef
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., Int. Ed., 2014, 53, 871–874 CrossRef CAS PubMed
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS
- M. Esmaeilirad, A. Kondori, B. Song, A. Ruiz Belmonte, J. Wei, K. Kucuk, S. M. Khanvilkar, E. Efimoff, W. Chen, C. U. Segre, R. Shahbazian-Yassar and M. Asadi, ACS Nano, 2020, 14, 2099–2108 CrossRef CAS PubMed
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, Green Chem., 2017, 19, 2406–2415 RSC
- S. Hanselman, M. T. M. Koper and F. Calle-Vallejo, ACS Energy Lett., 2018, 3, 1062–1067 CrossRef CAS
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS PubMed
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Grätzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20–24 CrossRef CAS PubMed
- S. K. Choi, U. Kang, S. Lee, D. J. Ham, S. M. Ji and H. Park, Adv. Energy Mater., 2014, 4, 1–7 Search PubMed
- A. Ozden, F. Li, F. P. Garcĺa De Arquer, A. Rosas-Hernández, A. Thevenon, Y. Wang, S. F. Hung, X. Wang, B. Chen, J. Li, J. Wicks, M. Luo, Z. Wang, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, ACS Energy Lett., 2020, 5, 2811–2818 CrossRef CAS
- W. H. Lee, Y. J. Ko, Y. Choi, S. Y. Lee, C. H. Choi, Y. J. Hwang, B. K. Min, P. Strasser and H. S. Oh, Nano Energy, 2020, 76, 105030 CrossRef CAS
- L. Ji, M. D. Mcdaniel, S. Wang, A. B. Posadas, X. Li, H. Huang, J. C. Lee, A. A. Demkov, A. J. Bard, J. G. Ekerdt and E. T. Yu, Nat. Nanotechnol., 2015, 10, 84–90 CrossRef CAS PubMed
- L. Zhao, R. Soc. Open Sci., 2018, 5, 181218 CrossRef CAS PubMed
- X. Zhou and C. Xiang, ACS Energy Lett., 2018, 3, 1892–1897 CrossRef CAS
- J. H. Kim, G. Magesh, H. J. Kang, M. Banu, J. H. Kim, J. Lee and J. S. Lee, Nano Energy, 2015, 15, 153–163 CrossRef CAS
- X. Chang, T. Wang, P. Zhang, Y. Wei, J. Zhao and J. Gong, Angew. Chem., 2016, 128, 8986–8991 CrossRef
- C. W. Kim, M. J. Kang, S. Ji and Y. S. Kang, ACS Catal., 2018, 8, 968–974 CrossRef CAS
- X. Chang, T. Wang, Z. J. Zhao, P. Yang, J. Greeley, R. Mu, G. Zhang, Z. Gong, Z. Luo, J. Chen, Y. Cui, G. A. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 15415–15419 CrossRef CAS PubMed
- M. J. Kang, C. W. Kim, A. U. Pawar, H. G. Cha, S. Ji, W.-B. Cai and Y. S. Kang, ACS Energy Lett., 2019, 4, 1549–1555 CrossRef CAS
- M. Zhang, X. Xuan, W. Wang, C. Ma and Z. Lin, Adv. Funct. Mater., 2020, 30, 2005983 CrossRef CAS
- J. Luo, L. Steier, M. K. Son, M. Schreier, M. T. Mayer and M. Grätzel, Nano Lett., 2016, 16, 1848–1857 CrossRef CAS PubMed
- A. Paracchino, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed
- G. Ghadimkhani, N. R. de Tacconi, W. Chanmanee, C. Janakyab and K. Rajeshwar, Chem. Commun., 2013, 49, 1297–1299 RSC
- K. Rajeshwar, N. R. De Tacconi, G. Ghadimkhani, W. Chanmanee and C. Janáky, ChemPhysChem, 2013, 14, 2251–2259 CrossRef CAS PubMed
- P. Li, H. Jing, J. Xu, C. Wu, H. Peng, J. Lu and F. Lu, Nanoscale, 2014, 6, 11380–11386 RSC
- D. H. Won, C. H. Choi, J. Chung and S. I. Woo, Appl. Catal., B, 2014, 158–159, 217–223 CrossRef CAS
- X. Ba, L. L. Yan, S. Huang, J. Yu, X. J. Xia and Y. Yu, J. Phys. Chem. C, 2014, 118, 24467–24478 CrossRef CAS
- J. F. de Brito, A. R. Araujo, K. Rajeshwar and M. V. B. Zanoni, Chem. Eng. J., 2015, 264, 302–309 CrossRef
- K. Lee, S. Lee, H. Cho, S. Jeong, W. D. Kim, S. Lee and D. C. Lee, J. Energy Chem., 2018, 27, 264–270 CrossRef
- H. Y. Kang, D. H. Nam, K. D. Yang, W. Joo, H. Kwak, H. H. Kim, S. H. Hong, K. T. Nam and Y. C. Joo, ACS Nano, 2018, 12, 8187–8196 CrossRef CAS PubMed
- J. M. Li, C. W. Tsao, M. J. Fang, C. C. Chen, C. W. Liu and Y. J. Hsu, ACS Appl. Nano Mater., 2018, 1, 6843–6853 CrossRef CAS
- J. M. Li, H. Y. Cheng, Y. H. Chiu and Y. J. Hsu, Nanoscale, 2016, 8, 15720–15729 RSC
- Z. Zeng, Y. Yan, J. Chen, P. Zan, Q. Tian and P. Chen, Adv. Funct. Mater., 2019, 29, 1–9 Search PubMed
- E. Szaniawska, I. A. Rutkowska, E. Seta, I. Tallo, E. Lust and P. J. Kulesza, Electrochim. Acta, 2020, 341, 136054 CrossRef CAS
- M. T. Galante, P. V. B. Santiago, V. Y. Yukuhiro, L. A. Silva, N. A. Dos Reis, C. T. G. V. M. T. Pires, N. G. Macedo, L. S. Costa, P. S. Fernandez and C. Longo, ChemCatChem, 2021, 13, 859–863 CrossRef CAS
- S. H. Oh, H. Y. Kang, W. H. Joo and Y. C. Joo, ChemCatChem, 2020, 12, 5185–5191 CrossRef CAS
- Y. Zhang, D. Pan, Y. Tao, H. Shang, D. Zhang, G. Li and H. Li, Adv. Funct. Mater., 2022, 32, 2109600 CrossRef CAS
- J. Gu, A. Wuttig, J. W. Krizan, Y. Hu, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Phys. Chem. C, 2013, 117, 12415–12422 CrossRef CAS
- U. Kang, S. K. Choi, D. J. Ham, S. M. Ji, W. Choi, D. S. Han, A. Abdel-Wahab and H. Park, Energy Environ. Sci., 2015, 8, 2638–2643 RSC
- X. Yang, E. A. Fugate, Y. Mueanngern and L. R. Baker, ACS Catal., 2017, 7, 177–180 CrossRef CAS
- K. M. Rezaul Karim, H. R. Ong, H. Abdullah, A. Yousuf, C. K. Cheng and M. M. Rahman Khan, Int. J. Hydrogen Energy, 2018, 43, 18185–18193 CrossRef CAS
- J. Yuan, C. Gu, W. Ding and C. Hao, Energy and Fuels, 2020, 34, 9914–9922 CrossRef CAS
- J. Yuan and C. Hao, Sol. Energy Mater. Sol. Cells, 2013, 108, 170–174 CrossRef CAS
- J. Yuan, P. Wang, C. Hao and G. Yu, Electrochim. Acta, 2016, 193, 1–6 CrossRef CAS
- X. H. Xu, F. Wang, J. J. Liu, K. C. Park and M. Fujishige, Sol. Energy Mater. Sol. Cells, 2011, 95, 791–796 CrossRef CAS
- J. W. Jang, S. Cho, G. Magesh, Y. J. Jang, J. Y. Kim, W. Y. Kim, J. K. Seo, S. Kim, K. H. Lee and J. S. Lee, Angew. Chem., Int. Ed., 2014, 53, 5852–5857 CrossRef CAS PubMed
- Y. J. Jang, J. W. Jang, J. Lee, J. H. Kim, H. Kumagai, J. Lee, T. Minegishi, J. Kubota, K. Domen and J. S. Lee, Energy Environ. Sci., 2015, 8, 3597–3604 RSC
- Y. J. Jang, I. Jeong, J. Lee, J. Lee, M. J. Ko and J. S. Lee, ACS Nano, 2016, 10, 6980–6987 CrossRef CAS PubMed
- C. Cai, Y. F. Xu, H. Y. Chen, X. D. Wang and D. Bin Kuang, Electrochim. Acta, 2018, 274, 298–305 CrossRef CAS
- Q. Wang, X. Wang, Z. Yu, X. Jiang, J. Chen, L. Tao, M. Wang and Y. Shen, Nano Energy, 2019, 60, 827–835 CrossRef CAS
- P. Wen, H. Li, X. Ma, R. Lei, X. Wang, S. M. Geyer and Y. Qiu, J. Mater. Chem. A, 2021, 9, 3589–3596 RSC
- T. Ouyang, Y. Ye, C. Tan, S. Guo, S. Huang, R. Zhao, S. Zhao and Z. Liu, J. Phys. Chem. Lett., 2022, 13, 6867–6874 CrossRef CAS PubMed
- P. Li, J. Zhang, H. Wang, H. Jing, J. Xu, X. Sui, H. Hu and H. Yin, Catal. Sci. Technol., 2014, 4, 1070–1077 RSC
- W. Wei, Z. Yang, W. Song, F. Hu, B. Luan, P. Li and H. Yin, J. Colloid Interface Sci., 2017, 496, 327–333 CrossRef CAS
- E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef CAS
- Y. Xu, S. Wang, J. Yang, B. Han, R. Nie, J. Wang, Y. Dong, X. Yu, J. Wang and H. Jing, J. Mater. Chem. A, 2018, 6, 15213–15220 RSC
- S. Ikeda, S. Fujikawa, T. Harada, T. H. Nguyen, S. Nakanishi, T. Takayama, A. Iwase and A. Kudo, ACS Appl. Energy Mater., 2019, 2, 6911–6918 CrossRef CAS
- S. Zhou, K. Sun, J. Huang, X. Lu, B. Xie, D. Zhang, J. N. Hart, C. Y. Toe, X. Hao and R. Amal, Small, 2021, 17, 2100496 CrossRef CAS
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152–14158 CrossRef CAS PubMed
- K. Sekizawa, S. Sato, T. Arai and T. Morikawa, ACS Catal., 2018, 8, 1405–1416 CrossRef CAS
- R. Kamata, H. Kumagai, Y. Yamazaki, G. Sahara and O. Ishitani, ACS Appl. Mater. Interfaces, 2019, 11, 5632–5641 CrossRef CAS
- S. K. Kuk, J. Jang, J. Kim, Y. Lee, Y. S. Kim, B. Koo, Y. W. Lee, J. W. Ko, B. Shin, J. Lee and C. B. Park, ChemSusChem, 2020, 13, 2940–2944 CrossRef CAS PubMed
- V. Andrei, R. A. Jagt, M. Rahaman, L. Lari, V. K. Lazarov, J. L. MacManus-Driscoll, R. L. Z. Hoye and E. Reisner, Nat. Mater., 2022, 21, 864–868 CrossRef CAS PubMed
- Y. Wang, S. Fan, B. AlOtaibi, Y. Wang, L. Li and Z. Mi, Chem.–Eur. J., 2016, 22, 8809–8813 CrossRef CAS PubMed
- K. R. Rao, S. Pishgar, J. Strain, B. Kumar, V. Atla, S. Kumari and J. M. Spurgeon, J. Mater. Chem. A, 2018, 6, 1736–1742 RSC
- Y. Hu, F. Chen, P. Ding, H. Yang, J. Chen, C. Zha and Y. Li, J. Mater. Chem. A, 2018, 6, 21906–21912 RSC
- L. Wei, J. Lin, S. Xie, W. Ma, Q. Zhang, Z. Shen and Y. Wang, Nanoscale, 2019, 11, 12530–12536 RSC
- P. Ding, Y. Hu, J. Deng, J. Chen, C. Zha, H. Yang, N. Han, Q. Gong, L. Li, T. Wang, X. Zhao and Y. Li, Mater. Today Chem., 2019, 11, 80–85 CrossRef CAS
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. Feng, Y. Li, W. Zhou and Y. Li, Nat. Commun., 2019, 1–10 CAS
- B. Zhou, X. Kong, S. Vanka, S. Cheng, N. Pant, S. Chu, P. Ghamari, Y. Wang, G. Botton, H. Cuo and Z. Mi, Energy Environ. Sci., 2019, 12, 2842–2848 RSC
- Gurudayal, J. W. Beeman, J. Bullock, H. Wang, J. Eichhorn, C. Towle, A. Javey, F. M. Toma, N. Mathews and J. W. Ager, Energy Environ. Sci., 2019, 12, 1068–1077 RSC
- M. Kan, Z. W. Yan, X. Wang, J. L. Hitt, L. Xiao, J. M. McNeill, Y. Wang, Y. Zhao and T. E. Mallouk, Angew. Chem., Int. Ed., 2020, 59, 11462–11469 CrossRef CAS PubMed
- P. A. Kempler, M. H. Richter, W. H. Cheng, B. S. Brunschwig and N. S. Lewis, ACS Energy Lett., 2020, 5, 2528–2534 CrossRef CAS
- I. Roh, S. Yu, C. K. Lin, S. Louisia, S. Cestellos-Blanco and P. Yang, J. Am. Chem. Soc., 2022, 2–6 Search PubMed
- W. J. Dong, P. Zhou, Y. Xiao, I. A. Navid, J. L. Lee and Z. Mi, ACS Catal., 2022, 12, 2671–2680 CrossRef CAS
- X. Zhou, R. Liu, K. Sun, Y. Chen, E. Verlage, S. A. Francis, N. S. Lewis and C. Xiang, ACS Energy Lett., 2016, 1, 764–770 CrossRef CAS
- J. Chen, J. Yin, X. Zheng, H. Ait Ahsaine, Y. Zhou, C. Dong, O. F. Mohammed, K. Takanabe and O. M. Bakr, ACS Energy Lett., 2019, 4, 1279–1286 CrossRef CAS
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4606–4609 CrossRef CAS PubMed
- D. H. Won, C. H. Choi, J. Chung, M. W. Chung, E. H. Kim and S. I. Woo, ChemSusChem, 2015, 8, 3092–3098 CrossRef CAS PubMed
- C. Zhao and J. Wang, Chem. Eng. J., 2016, 293, 161–170 CrossRef CAS
- S. Y. Choi, S. K. Jeong, H. J. Kim, I. H. Baek and K. T. Park, ACS Sustainable Chem. Eng., 2016, 4, 1311–1318 CrossRef CAS
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS
- F. Urbain, P. Tang, N. M. Carretero, T. Andreu, J. Arbiol and J. R. Morante, ACS Appl. Mater. Interfaces, 2018, 10, 43650–43660 CrossRef CAS PubMed
- C. T. Dinh, T. Burdyny, G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. Pelayo García De Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CrossRef CAS
- X. Hou, Y. Cai, D. Zhang, L. Li, X. Zhang, Z. Zhu, L. Peng, Y. Liu and J. Qiao, J. Mater. Chem. A, 2019, 7, 3197–3205 RSC
- M. De Jesús Gálvez-Vázquez, S. Alinejad, H. Hu, Y. Hou, P. Moreno-García, A. Zana, G. K. H. Wiberg, P. Broekmann and M. Arenz, Chimia, 2019, 73, 922–927 CrossRef
- W. H. Cheng, M. H. Richter, I. Sullivan, D. M. Larson, C. Xiang, B. S. Brunschwig and H. A. Atwater, ACS Energy Lett., 2020, 470–476 CrossRef CAS
- J. Lim, P. W. Kang, S. S. Jeon and H. Lee, J. Mater. Chem. A, 2020, 8, 9032–9038 RSC
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef
- E. Boutin, M. Patel, E. Kecsenovity, S. Suter, C. Janáky and S. Haussener, Adv. Energy Mater., 2022, 2200585 CrossRef CAS
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Nat. Commun., 2016, 7, 1–8 Search PubMed
- B. Kumar, V. Atla, J. P. Brian, S. Kumari, T. Q. Nguyen, M. Sunkara and J. M. Spurgeon, Angew. Chem., 2017, 129, 3699–3703 CrossRef
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 1–9 Search PubMed
- N. Han, Y. Wang, J. Deng, J. Zhou, Y. Wu, H. Yang, P. Ding and Y. Li, J. Mater. Chem. A, 2019, 7, 1267–1272 RSC
- P. Hou, X. Wang, Z. Wang and P. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 38024–38031 CrossRef CAS PubMed
- Y. Mi, S. Shen, X. Peng, H. Bao, X. Liu and J. Luo, ChemElectroChem, 2019, 6, 2393–2397 CrossRef CAS
- Z. Yin, C. Yu, Z. Zhao, X. Guo, M. Shen, N. Li, M. Muzzio, J. Li, H. Liu, H. Lin, J. Yin, G. Lu, D. Su and S. Sun, Nano Lett., 2019, 4–9 Search PubMed
- H. Wu and W. Chen, J. Am. Chem. Soc., 2011, 133, 15236–15239 CrossRef CAS
- Z. Zhao, X. Peng, X. Liu, X. Sun, J. Shi, L. Han, G. Li and J. Luo, J. Mater. Chem. A, 2017, 5, 20239–20243 RSC
- T. Shinagawa, G. O. Larrazábal, A. J. Martín, F. Krumeich and J. Pérez-Ramírez, ACS Catal., 2018, 8, 837–844 CrossRef CAS
- T. Dou, Y. Qin, F. Zhang and X. Lei, ACS Appl. Energy Mater., 2021, 4, 4376–4384 CrossRef CAS
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 RSC
- M. Choi, S. Bong, J. W. Kim and J. Lee, ACS Energy Lett., 2021, 2090–2095 CrossRef CAS
- D. H. Nam, O. S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C. T. Dinh, F. P. García De Arquer, Y. Wang, Z. Liang, A. H. Proppe, C. S. Tan, P. Todorović, O. Shekhah, C. M. Gabardo, J. W. Jo, J. Choi, M. J. Choi, S. W. Baek, J. Kim, D. Sinton, S. O. Kelley, M. Eddaoudi and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef CAS PubMed
- L. Majidi, A. Ahmadiparidari, N. Shan, S. N. Misal, K. Kumar, Z. Huang, S. Rastegar, Z. Hemmat, X. Zou, P. Zapol, J. Cabana, L. A. Curtiss and A. Salehi-Khojin, Adv. Mater., 2021, 33, 1–8 CrossRef PubMed
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS
- K. Jiang, S. Siahrostami, A. J. Akey, Y. Li, Z. Lu, J. Lattimer, Y. Hu, C. Stokes, M. Gangishetty, G. Chen, Y. Zhou, W. Hill, W. Bin Cai, D. Bell, K. Chan, J. K. Nørskov, Y. Cui and H. Wang, Chem, 2017, 3, 950–960 CAS
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS
- Z. Chen, K. Mou, S. Yao and L. Liu, ChemSusChem, 2018, 11, 2944–2952 CrossRef CAS PubMed
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC
- J. Gu, C. S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed
- H. Yang, Q. Lin, Y. Wu, G. Li, Q. Hu, X. Chai, X. Ren, Q. Zhang, J. Liu and C. He, Nano Energy, 2020, 70, 104454 CrossRef CAS
- D. Xi, J. Li, J. Low, K. Mao, R. Long, J. Li, Z. Dai, T. Shao, Y. Zhong, Y. Li, Z. Li, X. J. Loh, L. Song, E. Ye and Y. Xiong, Adv. Mater., 2021, 34, 2104090 CrossRef PubMed
- W. Zhang, Y. Hu, L. Ma, G. Zhu, P. Zhao, X. Xue, R. Chen, S. Yang, J. Ma, J. Liu and Z. Jin, Nano Energy, 2018, 53, 808–816 CrossRef CAS
- P. Su, W. Xu, Y. Qiu, T. Zhang, X. Li and H. Zhang, ChemSusChem, 2018, 11, 848–853 CrossRef CAS PubMed
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1–8 CrossRef
- H. Yang, N. Han, J. Deng, J. Wu, Y. Wang, Y. Hu, P. Ding, Y. Li, Y. Li and J. Lu, Adv. Energy Mater., 2018, 8, 1–6 Search PubMed
- X. Wang, W. J. Yin, Y. Si, X. Wang, X. Guo, W. Guo and Y. Fu, J. Mater. Chem. A, 2020, 8, 19938–19945 RSC
- F. Yang, A. O. Elnabawy, R. Schimmenti, P. Song, J. Wang, Z. Peng, S. Yao, R. Deng, S. Song, Y. Lin, M. Mavrikakis and W. Xu, Nat. Commun., 2020, 11, 1088 CrossRef CAS PubMed
- H. Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, E. S. Jeong, V. Subramanian, U. Sim and K. T. Nam, J. Mater. Chem. A, 2019, 7, 10651–10661 RSC
- B. Endrödi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Török, A. Danyi and C. Janáky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef PubMed
- C. M. Gabardo, C.
P. O'Brien, J. P. Edwards, C. McCallum, Y. Xu, C. T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS
- Y. Xu, F. Li, A. Xu, J. P. Edwards, S. F. Hung, C. M. Gabardo, C. P. O'Brien, S. Liu, X. Wang, Y. Li, J. Wicks, R. K. Miao, Y. Liu, J. Li, J. E. Huang, J. Abed, Y. Wang, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 4–10 CrossRef
- D. Kim, W. Choi, H. W. Lee, S. Y. Lee, Y. Choi, D. K. Lee, W. Kim, J. Na, U. Lee, Y. J. Hwang and D. H. Won, ACS Energy Lett., 2021, 6, 3488–3495 CrossRef CAS
- P. Hou, X. Wang and P. Kang, J. CO2 Util., 2021, 45, 33–35 Search PubMed
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed
- M. Schreier, L. Curvat, F. Giordano, L. Steier, A. Abate, S. M. Zakeeruddin, J. Luo, M. T. Mayer and M. Grätzel, Nat. Commun., 2015, 6, 1–6 Search PubMed
- G. M. Sriramagiri, N. Ahmed, W. Luc, K. D. Dobson, S. S. Hegedus and F. Jiao, ACS Sustainable Chem. Eng., 2017, 5, 10959–10966 CrossRef CAS
- Y. Xiao, Y. Qian, A. Chen, T. Qin, F. Zhang, H. Tang, Z. Qiu and B. L. Lin, J. Mater. Chem. A, 2020, 8, 18310–18317 RSC
- H. Zhang, Y. Chen, H. Wang, H. Wang, W. Ma, X. Zong and C. Li, Adv. Energy Mater., 2020, 10, 2002105 CrossRef CAS
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194 CrossRef CAS PubMed
- M. Rahaman, V. Andrei, C. Pornrungroj, D. Wright, J. J. Baumberg and E. Reisner, Energy Environ. Sci., 2020, 13, 3536–3543 RSC
- M. Liu, Y. Yi, L. Wang, H. Guo and A. Bogaerts, Catalysts, 2019, 9, 275 CrossRef
- T. N. Do, C. You and J. Kim, Energy Environ. Sci., 2022, 15, 169–184 RSC
- P. S. Murthy, W. Liang, Y. Jiang and J. Huang, Energy and Fuels, 2021, 35, 8558–8584 CrossRef CAS
- I. Ganesh, Renew. Sustain. Energy Rev., 2014, 31, 221–257 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2022 |