
A plethora of isomerization processes and hydrogen scrambling in the fragmentation of the methanol dimer cation: a PEPICO study
Xiangkun
Wu
 a,
Xiaoguo
Zhou
b,
Saša
Bjelić
a,
Patrick
Hemberger
a,
Bálint
Sztáray
c and
Andras
Bodi
*a
a,
Xiaoguo
Zhou
b,
Saša
Bjelić
a,
Patrick
Hemberger
a,
Bálint
Sztáray
c and
Andras
Bodi
*a
aPaul Scherrer Institute, 5232 Villigen, Switzerland. E-mail: andras.boedi@psi.ch
bHefei National Laboratory for Physical Sciences at the Microscale, Department of Chemical Physics, University of Science and Technology of China, Hefei 230026, China
cUniversity of the Pacific, Department of Chemistry, Stockton, CA 95211, USA
First published on 21st December 2021
Abstract
The valence photoionization of light and deuterated methanol dimers was studied by imaging photoelectron photoion coincidence spectroscopy in the 10.00–10.35 eV photon energy range. Methanol clusters were generated in a rich methanol beam in nitrogen after expansion into vacuum. They generally photoionize dissociatively to protonated methanol cluster cations, (CH3OH)nH+. However, the stable dimer parent ion (CH3OH)2+ is readily detected below the dissociation threshold to yield the dominant CH3OH2+ fragment ion. In addition to protonated methanol, we could also detect the water- and methyl-loss fragment ions of the methanol dimer cation for the first time. These newly revealed fragmentation channels are slow and cannot compete with protonated methanol cation formation at higher internal energies. In fact, the water- and methyl-loss fragment ions appear together and disappear at a ca. 150 meV higher energy in the breakdown diagram. Experiments with selectively deuterated methanol samples showed H scrambling involving two hydroxyl and one methyl hydrogens prior to protonated methanol formation. These insights guided the potential energy surface exploration to rationalize the dissociative photoionization mechanism. The potential energy surface was further validated by a statistical model including isotope effects to fit the experiment for the light and the perdeuterated methanol dimers simultaneously. The (CH3OH)2+ parent ion dissociates via five parallel channels at low internal energies. The loss of both CH2OH and CH3O neutral fragments leads to protonated methanol. However, the latter, direct dissociation channel is energetically forbidden at low energies. Instead, an isomerization transition state is followed by proton transfer from a methyl group, which leads to the CH3(H)OH+⋯CH2OH ion, the precursor to the CH2OH-, H2O-, and CH3-loss fragments after further isomerization steps, in part by a roaming mechanism. Water loss yields the ethanol cation, and two paths are proposed to account for m/z 49 fragment ions after CH3 loss. The roaming pathways are quickly outcompeted by hydrogen bond breaking to yield CH3OH2+, which explains the dominance of the protonated methanol fragment ion in the mass spectrum.
1. Introduction
Proton transfer processes are important in biology,1 interstellar chemistry,2 analytical chemistry3 and atmospheric chemistry.4 Inter- and intramolecular proton transfer usually takes place along hydrogen bonds. Hydrogen atom and proton transfer is also a common gas-phase ion–molecule reaction between both polar and apolar species.5–7 The cluster phase, as an intermediate state between the gas and condensed phases, represents a simplified environment to understand the chemistry of the latter and to create bridges between the two. Weakly bound clusters, whether by van der Waals interactions or hydrogen bonds, fill the gap between condensed-phase reactive environments and isolated species interacting in the gas phase. Much remains unknown even about the simplest systems, such as the water dimer. Its photoelectron spectrum,8 exhibiting major discrepancies compared with Franck–Condon simulations9 as well as the appearance energy of H3O+ has only recently been determined.10 Vibrational studies of both small11 and large12 methanol and protonated methanol cation clusters found evidence for delocalized protons between hydroxyl groups, i.e., conventional hydrogen bonding. However, the important role of post-MP2 electron correlation in resolving the differences between experiment and harmonic predictions for the vibrational red shift of the simplest organic hydrogen bond in the methanol dimer was only revealed in 2014.13 The dissociative ionization of ethanol commences with methyl loss, after which a parallel channel opens, yielding the protonated monomer.14,15 Inter- and intramolecular methyl migrations have been proposed to account for the higher-energy processes.16 Cα–Cβ and, at higher energies, Cβ–Cγ bond cleavage has also been observed in the dissociative ionization of propanol dimers, the latter yielding a mixed, proton bound propanol/ethanol adduct.17 The dissociative ionization of glycerol, a polyalcohol, and its dimer is also governed by intramolecular proton transfers.18 This shows the rich chemistry that makes the study of weakly bound systems particularly interesting, especially if the reactivity is induced by controlled excitation or ionization. Ionization often induces intermolecular proton transfer in clusters,19 as evidenced by the detection of protonated cluster fragments in the photoionization of, e.g., water,10,20,21 ammonia22 and methanol clusters.23–25Photoionization mass spectrometry, combined with vacuum ultraviolet (VUV) synchrotron radiation, is a powerful tool to study dissociative photoionization mechanisms and energetics.26 Imaging photoelectron photoion coincidence spectroscopy (iPEPICO)27,28 offers a further analytical dimension by electron kinetic energy analysis, thereby controlling the energy balance of photoionization and allowing for photoion mass-selected threshold photoelectron spectra to be recorded.29 It has been proven to be powerful in the analysis of reactive mixtures,30,31 as a multiplexed, sensitive, selective, and universal detection technique.32 PEPICO has also unveiled dissociative photoionization mechanism of clusters,10,15,33 revealed complex dissociation mechanisms with parallel and sequential steps,34,35 and delivered accurate thermochemistry and energetics of elusive species.36–38 Ivan Powis and co-workers have used iPEPICO to investigate the photoelectron circular dichroism and the dissociative ionization processes of homochiral glycidol and butanediol clusters.39,40 This work illustrates beautifully the complex chemistry even moderately large H-bond networks can bring about in ion chemistry.
The conformational space and kinetic properties of gaseous and liquid methanol have been studied for a long time.41,42 As the simplest aliphatic alcohol, methanol can theoretically participate in conventional O–H⋯O hydrogen bonding as well as unconventional, “blue-shifted,”43 sometimes referred to as improper, intermolecular H-bonds by C–H⋯O.44 As seen in the proton transfer along an unconventional H bond in the dimethyl ether dimer cation33 as well as in the role of analogous halogen bonding to render the F3S+⋯F–SF4 adduct more stable than the classically bound F3S–SF5+ in the dissociative ionization of CF3SF5 dimers,45 such bonds often play a determining role in ion chemistry, and also affect the reactivity of (protonated) species in solution. Gas-phase methanol dimers have a relatively simple hydrogen bond structure,46 which makes them ideal subjects to study their ion chemistry.
The ionization of methanol clusters was studied by multiphoton ionization,47,48 electron impact ionization,24,49 and VUV single photon ionization.23,50,51 Booze and Baer applied threshold PEPICO to study clusters and discussed the (protonated) methanol dimer in a proof-of-principle work.52 Besides the methanol and the methanol dimer cation, only protonated methanol cluster cations were observed. Ionization may usher in barrierless proton transfer even along unconventional hydrogen bonds.33 This proton transfer, which goes hand in hand with large geometry change, is driven by the resulting stabilization of the cluster. Thus, the Franck–Condon factors at the adiabatic ionization energy are negligible and the excess energy of the cluster will be quite large already at the ionization onset, which is often sufficient for fragmentation. This mechanism has been supported by the observation of unprotonated water cluster ions, when stabilized by evaporative cooling using argon.53 By studying selectively deuterated CH3OD and CD3OH samples, proton transfer both from hydroxyl and methyl group have been detected in electron ionization experiments, e.g.:
| (CD3OH)n + hν → (CD3OH)n−1H+ + CD3O˙ + e− | (1) |
| (CD3OH)n + hν → (CD3OH)n−1D+ + ˙CD2OH + e− | (2) |
The relative ratio between proton transfer from the methyl and hydroxy groups at a given photon energy was found to depend on the cluster size. On the one hand, when larger neutral clusters were generated, overall proton transfer from the OH group, according to eqn (1), was promoted.24 On the other hand, when considering individual mass spectra, proton transfer from the methyl group was found to be more and more likely with increasing cluster size.49 This suggests dynamic competition between proton transfer processes and evaporative stabilization of larger clusters. Lee et al. also evaluated MP2/6-31G(d,p) energies at Hartree–Fock optimized geometries of dimer cation isomers to obtain the dissociation energy to form protonated methanol and CH3O or CH2OH neutral fragments.24 They reported two cation dimer structures, both with a traditional, O⋯H–O hydrogen bond, with the more stable one corresponding to a bridging hydrogen from the methyl group, in agreement with our results here (see below). Aside from peaks due to CH3O or CH2OH loss as well as monomer evaporation, Morgan et al. reported water loss from the protonated dimer, leading to protonated dimethyl ether together with dimethyl ether loss in larger protonated clusters, leading to a water subunit in the fragment ion in electron impact and 266 nm multiphoton ionization studies.48 While complex dynamics has been revealed by electron and multiphoton ionization experiments, these ionization techniques hardly allow for internal energy selection, lead to high parent ion energies and intense fragmentation. This makes it difficult to disentangle the fragmentation processes in general and to understand bonding and fragmentation in the dimer cation in particular.
Martrenchard et al. used VUV photoionization to study the proton transfer reaction in selectively deuterated (CD3OH)2+ methanol dimer ions and measured the threshold energy of methyl and hydroxyl hydrogen transfer channels as 10.1 and 10.2 eV, relative to the neutral dimer, respectively.50 They also varied the delay time between electron detection and the application of the ion extraction field in a pulsed setup, and found that deuteron transfer from the methyl group was a slower process than proton transfer from the hydroxyl group. Assuming 10.2 eV as the dissociative photoionization threshold to CH3OH2+ + CH3O˙, i.e., proton transfer from the hydroxyl group followed by fragmentation, and considering that ˙CH2OH is 0.4 eV more stable than CH3O˙, the thermodynamic threshold to proton transfer from the methyl group was established at 9.8 eV, implying a tight transition state for methyl H transfer and a reverse barrier of 0.3 eV. We will show here that the CH3OH2+ + CH3O˙ channel is inaccessible below 10.5 eV. This implies hydroxymethyl as the sole leaving neutral fragment and means that the different rate constants were probably due to kinetic isotope effects in the partially deuterated dimer ion.
Tsai et al. used a tunable vacuum ultraviolet laser to investigate the methanol dimer (CD3OH)2 in the 10.5–10.9 eV photon energy range.51 They also explored the methanol dimer cation potential energy surface (PES) at unprecedented detail at the B3LYP/6-311+G(3df,2p)//B3LYP/6-31+G(d) level of theory. Three distinct methanol dimer cation isomers were identified, including O⋯H–C, O–H⋯C, O–H⋯O type hydrogen bonds, all of which corresponded to proton-transfer structures from the neutral. Isomerization and fragmentation transition states were also reported. These results have also inspired our potential energy surface exploration efforts (see below). Furthermore, they reported that the branching ratio towards the proton-transfer products from the hydroxyl group (1) rose considerably with photon energy, as opposed to the more stable deuteron-transfer products from the methyl group (2). These results were measured more than 300 meV above threshold and above the energy range studied herein but complement our measurements and will be referred to later.
Here, we focus on the fragmentation of internal energy selected methanol dimer cations close to their ionization energy, which we measured by double imaging photoelectron photoion coincidence spectroscopy (i2PEPICO) using single photon ionization with tunable vacuum ultraviolet synchrotron radiation. This allows for internal energy selection of the parent ion to plot the breakdown diagram with up to meV accuracy. Moreover, the PES for the dissociative photoionization of methanol dimer have been recalculated using composite methods to account for the discrepancies between the PES of Tsai et al.51 and our experimental observations.
2. Experimental and computational
Double Imaging Photoelectron Photoion Coincidence (i2PEPICO) experiments were performed using the prototype CRF-PEPICO spectrometer54 at the VUV beamline55,56 of the Swiss Light Source, Paul Scherrer Institute. Synchrotron radiation from a bending magnet was collimated, dispersed by a grazing incidence monochromator with a 600 lines mm−1 laminar or a 150 lines mm−1 blazed grating, and focused onto a 200 μm exit slit in a differentially pumped rare gas filter. The gas filter was filled with a mixture of krypton, neon, and argon at a pressure of 10 mbar over an optical length of 10 cm to suppress higher-order radiation above 14.0 eV. The photon beam entered the experimental chamber with the ionization region ca. 50 cm downstream from the focus. The photon energy was calibrated using the Ar 11s′–13s′ autoionization lines in first and second orders of the grating. The photon energy resolution was 3–8 meV, depending on the grating used.57Previous experience has shown that efficient cooling in a supersonic expansion often leads to condensation and even aerosol formation with a sample prone to cluster formation, such as water,10 ethanol,15 or dimethyl ether.33 Cluster formation in the expansion is a complex non-equilibrium process,58 which often yields relatively flat cluster size distributions, implying that the overwhelming majority of the sample is found in the large clusters.59 In dimethyl ether measurements,33 we found that less efficient collisional cooling can promote dimer formation to a degree that dimer and protonated dimer signals—a shorthand for larger clusters—are comparable in intensity. To promote selective dimer formation, methanol (CH3OH, CH3OD, CD3OH and CD3OD, all from Sigma-Aldrich) was seeded in nitrogen at a flow rate of 30 sccm and expanded through a 200 μm nozzle from a stagnation pressure slightly below 100 mbar into high vacuum to form a continuous molecular beam. When switching between OH- or OD-containing samples, light or heavy water was used to saturate the inside surfaces of the sampling lines as well as the chamber walls with the corresponding hydrogen isotope to prevent isomer exchange by wall collisions. After introducing the methanol isotopologues, we waited until the isotope distribution in the mass spectra stabilized. The pressure in the source chamber was maintained at 1 × 10−5 mbar by turbomolecular and cryogenic pumps with a total pumping speed of 7500 L s−1. The beam was skimmed as it entered the ionization chamber, yielding a background pressure of 3 × 10−6 mbar, pumped by a 1500 L s−1 cryogenic pump (baseline pressure ca. 1–2 × 10−7 mbar). The photon beam intersects the molecular beam in the ionization volume, from which the produced electrons and ions are extracted in opposite directions by a 260 V cm−1 constant electric field. Electrons were detected in velocity map imaging conditions to measure their kinetic energy, whereas space focusing conditions were applied on the ion side. Both electrons and ions are detected by position-sensitive delay-line anode detectors (Roentdek, DLD40). Threshold electrons with less than 4 meV kinetic energy are projected onto the central spot of the detector together with kinetic electrons without an off-axis momentum component. The kinetic energy electron contamination of the center spot was subtracted to obtain the threshold ionization signal based on the signal in a small ring around the center, as proposed by Sztáray and Baer.60 As the electron time of flight (TOF) is negligible relative to the ion TOF, electron hits can be used as the start signal for the ion TOF analysis.61 The fractional abundance of the parent and fragment ions in the threshold photoionization mass spectra is plotted in the breakdown diagram as a function of photon energy. Due to the long ion acceleration region, metastable fragmentation processes with unimolecular rate constants in the 103 < k/s−1 < 107 range show up as asymmetrical TOF peaks broadened towards higher flight times, which can be modelled with the help of the ion optics parameters to measure and model the dissociation rate constants.62
Quantum chemical calculations were performed using Gaussian 16 A.03.63 Optimized geometries for reactants, transition states, intermediates and products were located on the ground state potential energy surface using density functional theory at the B3LYP/6-311++G(d,p) level. Tight transition states, i.e., saddle points on the PES, were found using constrained geometry scans and the stationary points were confirmed by frequency analysis. The relevant stationary points on the potential energy surface were re-calculated using the G4 composite method64 to obtain ionization, activation, and reaction energies.
3. Results and discussion
3.1. Threshold ionization mass spectra
Threshold PEPICO experiments were carried out in the 10.00–10.35 eV photon energy range with 4 meV steps. Fig. 1a shows representative threshold PEPICO mass spectra with the main ion peaks at m/z 33, 64, 65, 97, 129 and 161. They are attributed to the methanol dimer ion (CH3OH)2+ at m/z 64 and to protonated methanol (cluster) ions (CH3OH)nH+, n = 1–5, at m/z (32n + 1). At 10.1 eV, the intensity ratio of the protonated and unprotonated dimers in threshold photoionization is ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The m/z 33 peak is asymmetric close to its appearance in the spectrum, i.e., protonated methanol is formed in a slow dissociation in the acceleration region of the mass spectrometer. According to statistical rate theory,65 slow rate constants can be due to a large reactant density of states, implying numerous degrees of freedom and/or a deep potential energy well; or a tight transition state with a small number of states, i.e., low activation entropy. The methanol dimer is relatively small and not strongly bound. Therefore, an asymmetric daughter ion peak indicates a tight transition state, with a likely saddle point along the reaction coordinate. Fragmentation by quantum tunneling may also be slow at threshold, as seen in the H-loss dissociation of energy selected ethanol cations.66 In acetone dissociative ionization, H-transfer from one methyl group to the other is responsible for methane loss to form the ketene cation. It takes place entirely by H-atom quantum tunneling, and is completely suppressed by deuteration.67 Selective deuteration will also be used to identify if quantum tunneling contributes to the fragmentation of methanol dimer cations (see below). If the lowest-energy dissociative photoionization process is fast on the timescale of the experiment, the parent ion fractional abundance drops to zero at the energy where even the original zero internal energy neutrals gain enough energy to dissociate after ionization, i.e., at the 0 K appearance energy, E0.68 If the dissociation is slow compared to the time scale of mass analysis, a certain amount of excess energy is needed to speed it up enough so that it takes place before the parent ion enters the drift region of the time-of-flight mass analyzer. This excess energy is the kinetic shift, and results in a blue shift of the parent disappearance energy in the breakdown diagram. Thus, the fact that methanol dimer cations are not detected above a photon energy of ca. 10.15 eV confirms that they are not formed by dissociative photoionization of heavier clusters. Furthermore, since the falling dimer signal correlates with the rising protonated monomer signal, we conclude that dimer cations are the sole source of protonated methanol by dissociative ionization in the studied energy range.
:1. The m/z 33 peak is asymmetric close to its appearance in the spectrum, i.e., protonated methanol is formed in a slow dissociation in the acceleration region of the mass spectrometer. According to statistical rate theory,65 slow rate constants can be due to a large reactant density of states, implying numerous degrees of freedom and/or a deep potential energy well; or a tight transition state with a small number of states, i.e., low activation entropy. The methanol dimer is relatively small and not strongly bound. Therefore, an asymmetric daughter ion peak indicates a tight transition state, with a likely saddle point along the reaction coordinate. Fragmentation by quantum tunneling may also be slow at threshold, as seen in the H-loss dissociation of energy selected ethanol cations.66 In acetone dissociative ionization, H-transfer from one methyl group to the other is responsible for methane loss to form the ketene cation. It takes place entirely by H-atom quantum tunneling, and is completely suppressed by deuteration.67 Selective deuteration will also be used to identify if quantum tunneling contributes to the fragmentation of methanol dimer cations (see below). If the lowest-energy dissociative photoionization process is fast on the timescale of the experiment, the parent ion fractional abundance drops to zero at the energy where even the original zero internal energy neutrals gain enough energy to dissociate after ionization, i.e., at the 0 K appearance energy, E0.68 If the dissociation is slow compared to the time scale of mass analysis, a certain amount of excess energy is needed to speed it up enough so that it takes place before the parent ion enters the drift region of the time-of-flight mass analyzer. This excess energy is the kinetic shift, and results in a blue shift of the parent disappearance energy in the breakdown diagram. Thus, the fact that methanol dimer cations are not detected above a photon energy of ca. 10.15 eV confirms that they are not formed by dissociative photoionization of heavier clusters. Furthermore, since the falling dimer signal correlates with the rising protonated monomer signal, we conclude that dimer cations are the sole source of protonated methanol by dissociative ionization in the studied energy range.
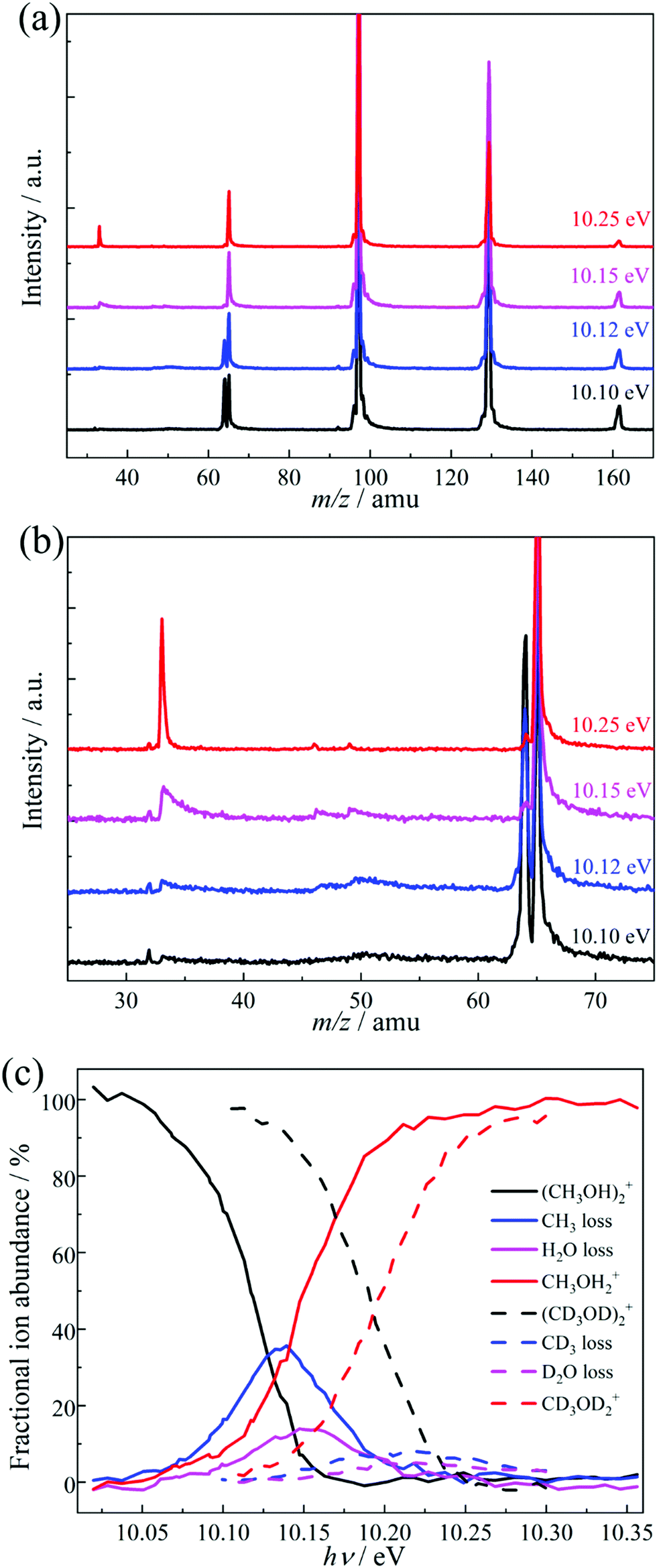 | ||
| Fig. 1 (a) Threshold photoionization mass spectra of light methanol clusters at four representative photon energies. (b) Enlarged view of the spectra in (a) from m/z 25 to 75 (c) experimental breakdown diagram of the light and perdeuterated methanol dimer. | ||
Fig. 1b shows an enlarged view of the m/z 25–75 range. Besides the asymmetric m/z 33 peak, two small asymmetric peaks are also visible at m/z 46 and 49, especially at lower photon energies. In a first approximation, all daughter ion peak shapes measure the same depletion rate of the parent ion, i.e., the sum of the dissociation rate constants. The relative fragment ion peak areas (branching ratios) correspond to the rate constant ratios. Consequently, similar metastable peak shapes rising in coincidence with the protonated methanol fragment ion of the dimer strongly suggest that the precursor of all three of these fragment ions is the same, i.e., the methanol dimer cation. Thus, the peaks at m/z 46 and 49 can be attributed to H2O and CH3 loss from (CH3OH)2+, respectively. The three fragment ions appear at a similar photon energy of ca. 10.05 eV, but the H2O- and CH3-loss fragments are only seen in a ca. 200 meV energy range and disappear completely by 10.25 eV. Methyl and water loss is quickly outcompeted by [CH3O] loss yielding CH3OH2+ at m/z 33 and are only observable in the energy range in which the parent ion is metastable. In the absence of internal energy selection and due to the experimental parameters and conditions, these fragment ions could not be observed in previous methanol dimer studies.50–52 The breakdown diagram for the light and perdeuterated methanol dimers is shown in Fig. 1c.
Methanol has two non-equivalent hydrogen atoms in the hydroxyl and methyl functional groups. As shown in the literature, selective deuteration can help reveal the hydrogen or proton scrambling mechanism, and the role of quantum tunneling.48,49,62,69Fig. 2 shows representative TOF distributions of CH3OH, CH3OD, CD3OH, and CD3OD methanol dimer samples. In CH3OD, the m/z 34, 35, 68, 101, 134 and 167 are the main peaks, which are assigned to (CH3OD)H+ and (CH3OD)nD+ (1 ≤ n ≤ 5). In CD3OH, the m/z 37, 36, 71, 106, 141 and 176 are the main products, attributed to (CD3OH)D+ and (CD3OH)nH+ (1 ≤ n ≤ 5). There is a marked difference between the protonated monomer and the protonated cluster signal. As seen in the selectively deuterated samples, proton or deuteron transfer from the methyl group dominates the protonated monomer signal (see eqn (2) for n = 2), while the protonated cluster peaks evidence primarily a proton or deuteron added from the hydroxyl group (eqn (1) for n > 2). TOF distributions of the CH3OD and CD3OH samples in the m/z 30–45 region (Fig. 2) show that proton transfer from the methyl group according to (2) is about 3 times more likely than reaction (1) in both isotopologues when forming the protonated monomer from the dimer cation. This ratio does not change markedly in the photon energy range of the breakdown diagram or with the deuteration pattern. Methanol clusters and protonated clusters have universally been found to be bound by conventional H-bonds in the gas phase and in rare gas matrices, as well.12,70 Therefore, it is surprising that the original participant in the intermolecular hydrogen bond in the neutral dimer is not the primary source of the proton in the protonated methanol fragment ion. The potential energy surface of the dimer may hold the keys to this conundrum. However, the experimental data themselves define the set of hydrogen atoms which scramble prior to dissociation. Approximately one fourth of the signal is attributed to two hydroxyl hydrogens in protonated methanol, thus, they both must participate in the exchange. If more than one methyl hydrogen were to intermingle with these, we should also detect the protonated dimer with both O-hydrogens stemming from the methyl group. While this peak is not entirely absent, its intensity is minuscule. Thus, three hydrogen atoms are expected to be mixed, two hydroxyl ones and a methyl one. Perfect scrambling would result in a HMeHOH:HOH2 ratio of 2:1 in the OH2 group of the protonated monomer, cf. the observed 3:1. This suggests somewhat hindered hydrogen exchange, but is in far better agreement with observation than any alternative set of scrambling hydrogen atoms. For comparison, if two methyl hydrogens were present in the set, the statistically expected HMe2:HMeHOH:HOH2 ratio would be 1:4:1.
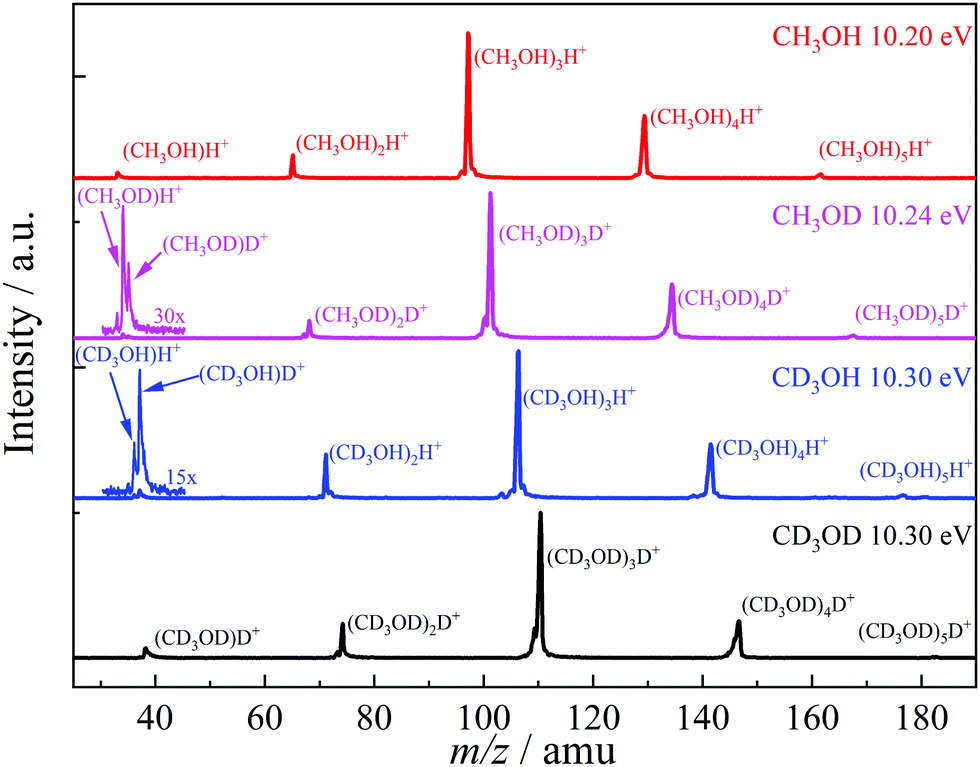 | ||
| Fig. 2 Threshold photoionization mass spectra of light, selectively deuterated and perdeuterated methanol clusters at representative photon energies. | ||
3.2. Potential energy surface of the methanol dimer cation
The potential energy surface exploration is guided by the appearance of the breakdown diagram and the limited loss of locational identity of the hydrogen atoms prior to the dissociation of the dimer cation, as discussed above. We have searched for reaction coordinates and a dissociation mechanism consistent with the observed fragmentation channels as a function of parent ion internal energy. We have explored multiple bond-breaking and isomerization pathways and only report the channels that are likely to contribute in and slightly above the energy range of the experiment.
Fig. 3a displays the pathways of the light methanol dimer cation [1] to produce the fragments of m/z 33, 46 and 49. The dimer cation [1] is formed in a barrierless proton transfer after vertical ionization of the neutral dimer. Its conceptually simplest fragmentation involves the cleavage of the hydrogen bond leading to protonated methanol [2] and a methoxy radical fragment. The dissociative photoionization energy for this fragmentation is 10.52 eV at the G4 level of theory, confirmed by the CBS-QB3 and W1U methods as 10.59 and 10.55 eV, respectively. This calculated thermochemical limit is almost 400 meV higher than the highest possible dissociative photoionization threshold to m/z 33 that can be reconciled with the breakdown diagram in Fig. 1c, and rules out direct H-bond breaking to yield the methoxy radical at low energies. This also means that the 10.2 eV threshold energy reported by Martrenchard et al.50 does not correspond to methoxy radical formation. The hydroxymethyl radical is almost 400 meV more stable than the methoxy radical (Fig. 3a). Thus, the channel leading to [2] + CH2OH is primarily responsible for the dimer cation fragmentation in the energy range of this study. Based on the disappearance energy of the parent ion, the dissociative ionization barrier is lower than ca. 10.15 eV, which is equal to the G4-computed dissociative ionization energy. This implies that there is no or only a small reverse barrier along the reaction coordinate, in contrast to the previously proposed reverse barrier of 0.4 eV.50 The structure of the immediate precursor to [2] + CH2OH is trivially [4], as suggested by Tsai et al. (structure II in Fig. 3, ref. 51), as well. However, they also reported a transition state, structurally similar to [3]‡, between the prompt dimer cation [1] and [4] at ca. 9.6 eV using density functional theory (TS3, ref. 51). This is likely a computational artifact, because of two reasons. A low-lying transition state would imply that the rate-determining step is fragmentation, which would likely be fast with a shallow, 0.8 eV potential energy well and over a loose transition state. Experimentally, however, we observe a metastable dimer parent ion. More importantly, as will be shown shortly, lower-barrier dissociative photoionization channels are open once [4] is formed. Based on the potential energy surface, if [4] could be formed at 9.6 eV, we would expect the parent ion to vanish from the breakdown diagram just above 9.8 eV, primarily by methyl and water loss. This counterfactual assumption leads to the conclusion that the transition state must lie higher, in fact almost at, when not above, the reaction energy to CH2OH formation. The lowest-lying transition state we found between [1] and [4] is [3]‡, at an energy of 10.12 eV. The intermediate [4] at 9.32 eV may then produce [2] + CH2OH at 10.15 eV without a reverse barrier via a loose transition state. Our B3LYP-calculated activation energy to [3]‡ agrees with the results of Tsai et al.,51 which, in view of the G4 results and the experimental observation, suggests that the previously reported and too low activation energy to isomerization is indeed a density functional theory artifact. The isomerization channel over [3]‡ corresponds to transferring a proton from the methyl group to the hydroxyl group of the other methanol unit. Direct dissociation from [4] would imply, e.g., exclusively (CD3OH)D+ as fragment ion of (CD3OH)2+, in contrast to the observed, ca. 1:3 abundance of (CD3OH)H+:(CD3OH)D+. Therefore, hydrogen atom scrambling must take place after proton (or deuteron) transfer from the methyl group. Indeed, the isomerization step in [4] converting the C⋯HO hydrogen bond back to an O⋯HO bond group in [6] is associated with a negligible barrier at [5]‡ and significant stabilization of the system. The minimum [6] provides us with a set of three oxygen-bound hydrogen atoms, one of which originated from a methyl group. This agrees with the previously deduced set of H atoms to be scrambled to obtain the experimentally observed protonated methanol isotopologue ratios. In Fig. 3b, we use CH3OD as an example to discuss the H-scrambling mechanism. The HOD moiety in CH2O(D)⋯H(D)OCH3+ [6] can rotate to form CH2O(D)⋯D(H)OCH3+ [6′] over the low-lying transition state [18]‡ at 9.43 eV. This only exchanges an O⋯H–O hydrogen bond to O⋯D–O. H/D exchange in [6] is achieved by rotating the OD group in the CH2OD moiety to form [6′′] after passing transition state [19]‡ at 9.75 eV. Transition states [18]‡ and [19]‡ both lie below the CH3/H2O-loss transition state [7]‡ at 9.84 eV, which allows for H/D exchange prior to fragmentation. If [6], [6′], and [6′′] were formed with equal probability, one would expect a 2:1 isotopologue ration for the protonated methanol fragment ion. The fact that [6′′] is underrepresented is easy to rationalize because of the relatively high-lying transition state [19]‡, which apparently precludes complete randomization. Similar to HOD rotation in [6], the activation energy of HOD rotation in [4] over [20]‡ is minuscule and leads to a C⋯D–O H-bond in [4′] instead of C⋯H–O in [4]. Actual H/D exchange can only take place by crossing the isomerization barrier back to [1′] or by an even higher-lying H-exchange rotational transition state [21]‡ at 10.51 eV. These processes expand the set of scrambled hydrogens and would lead to more fragment ion isotopologues and a markedly different isotopologue distribution compared to the experimentally observed one, including CH3OH2+ from (CH3OD)2+ by [4′] or [4′′]. The fact that this broader H-scrambling is not observed is due to the tight and high-energy transition state [3]‡. Once the system isomerizes to [4], dissociation is always faster and back-isomerization to [1] is suppressed.
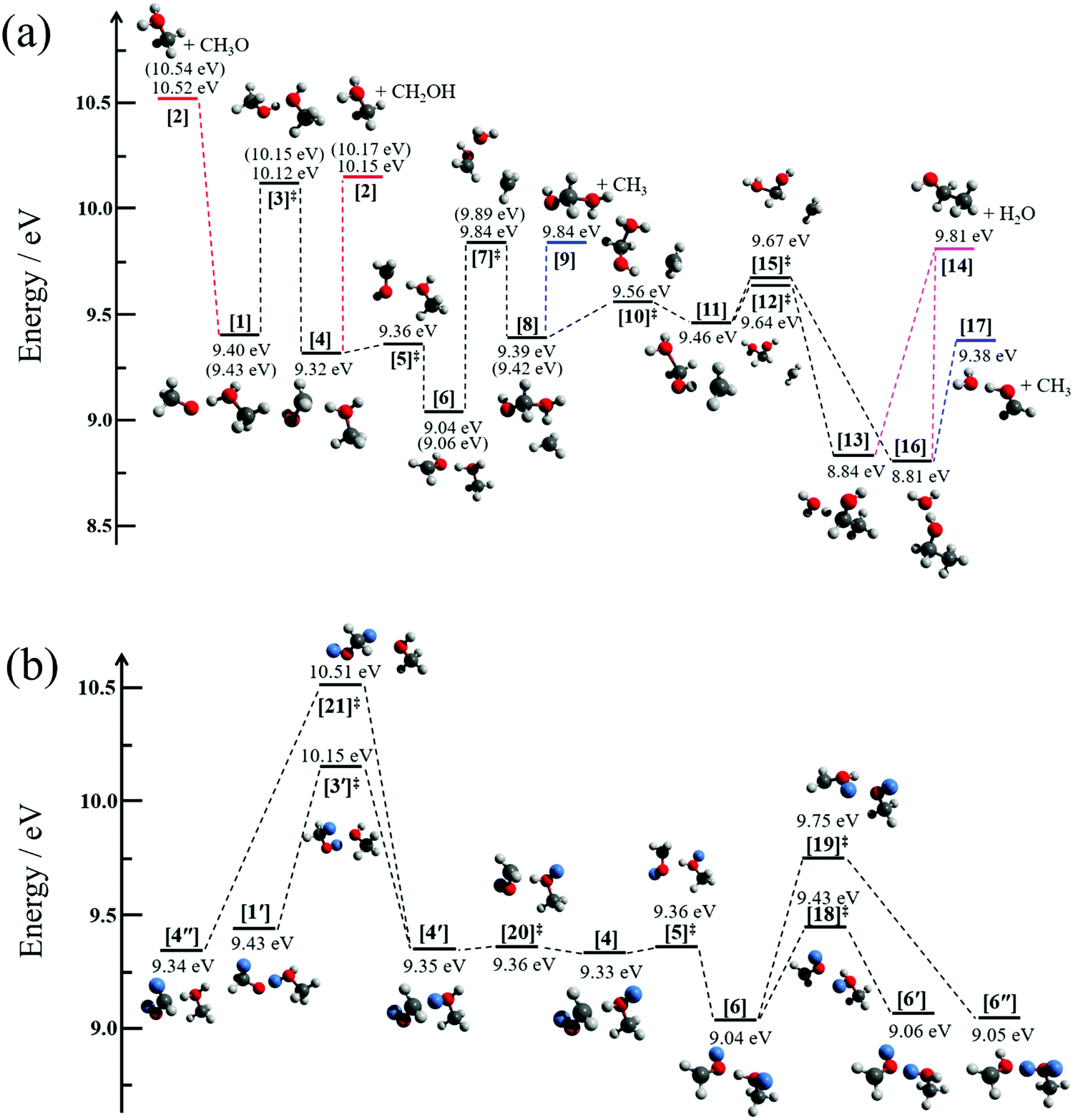 | ||
| Fig. 3 (a) Fragmentation pathways of the light methanol dimer cation to produce the fragment ions m/z 33, 46, and 49 by hydroxymethyl, water, and methyl loss, respectively. The G4-computed energies are given with respect to the neutral dimer. The energy thresholds for the perdeuterated dimer cation are given in parentheses. (b) Hydrogen scrambling mechanism in the dimer cation shown with the help of the (CH3OD)2+ dimer cation. | ||
In addition to hydroxymethyl loss, H2O and CH3 loss was also observed experimentally, and we explored the reaction coordinates leading to them, too. In the stable intermediate [6] at 9.04 eV, the methyl group can roam around the cation core and explore the potential energy well of intermediate [8] at 9.39 eV through transition state [7]‡ at 9.84 eV. Alternatively, this large-amplitude methyl motion can also lead to the methyl-loss fragment ion OHCH2OH2+, [9], without a reverse barrier and at the same, 9.84 eV threshold. Another methyl roaming transition state [10]‡ is found 0.17 eV above the minimum [8]. The reaction path bifurcates following the next intermediate minimum [11] at 9.46 eV. Depending on the direction from which the roaming methyl group attacks the central carbon to form a C–C bond, it yields intermediates [13] and [16] at 8.84 and 8.81 eV, respectively, after passing the virtually isoenergetic transition states [12]‡ and [15]‡, ca. 0.2 eV above [11]. Intermediate [13] exhibits an unusual C⋯H–O hydrogen bond, while intermediate [16] is bound by a conventional H2O⋯H–O hydrogen bond. Both [13] and [16] may lose water to yield the ethanol cation [14] at 9.81 eV, but [16] is more likely to lose CH3 to form the H-bonded water–formaldehyde complex cation [17] at 9.38 eV. This complex cation [17] is only bound by a H-bond and may lose H2O to yield the formaldehyde cation (CH2O+, m/z 30). The threshold, however, lies at 10.63 eV (G4 value), i.e., only at energies where water and methyl losses are already outcompeted by direct bond-breaking processes. In contrast to carbon–carbon bond formation and water loss to yield the ethanol cation at 9.81 eV, we could not find a reasonable path to dimethyl ether cation formation, the product energy of which would only be moderately higher at 9.90 eV. The cation core precursors to dimethyl ether cation by methyl roaming appear to be significantly higher in energy than the ethanol cation formation pathway.
Thus, the three lowest-energy fragmentation channels leading to fragment ions m/z 33, 46 and 49 share a common transition state [3]‡ at 10.12 eV. This is the highest-lying transition state in the methyl- and water-loss channels by a significant margin and is also computed to be only 30 meV lower in energy than the product energies of hydroxymethyl loss to form protonated methanol. After the system surmounts this transition state, the lowest energy fragmentation channels correspond to H2O and CH3 loss. However, these take place by a roaming mechanism and are comparably slow. The activation entropy of the higher-lying protonated methanol formation channel is larger, which is why it quickly becomes dominant after it is energetically allowed.
It is interesting to note that both [4] with an unconventional hydrogen bond and [6] are more stable minima than the prompt dimer cation structure [1]. Yet, infrared investigations of methanol cluster cations only reported conventional, mostly circular H-bound structures with methanol subunits and delocalized hydrogens shared between the hydroxyl groups.11,12 The dimer cation PES offers clues as to why this could be the case: if cluster ions have enough internal energy to isomerize over the transition state analogous to [3]‡ in the dimer, they also have enough energy to fragment afterwards. Thus, isomerization to more stable cluster ions is inherently followed by fragmentation, and the stabilized cluster ion does not survive. In the dimer cation, this means that the minimum [6] can only be a fleeting intermediate, as the barrier to forming it is higher in energy than the barrier to its fragmentation. Thus, methanol cluster cations formed by vertical ionization from neutral clusters will decay until trapped in a conventionally bound protonated cluster cation.
The PES also explains the dimer experimental observations qualitatively and resolves the discrepancies with previously suggested energetics and isomerization processes. Thus, we decided to put it to the test by constructing a statistical model to describe the dissociative ionization processes.
3.3. Statistical model
Unimolecular fragmentation processes of valence-ionized cations are generally statistical, i.e., the ergodic hypothesis holds, and the system can explore the energetically available phase space.68 In addition to isolated-state behavior,71 impulsive dissociation processes72 and roaming73 may also burst the limits of the statistical model. Indeed, the TOF distributions in Fig. 1c show a comparable and almost constant branching ratio between the methyl and water loss. This is in contrast with statistical branching on the potential energy surface, which prefers methyl loss, because both higher activation entropy and lower-energy channels are available for it. We only apply statistical theory to the steps prior to roaming according to eqn (3):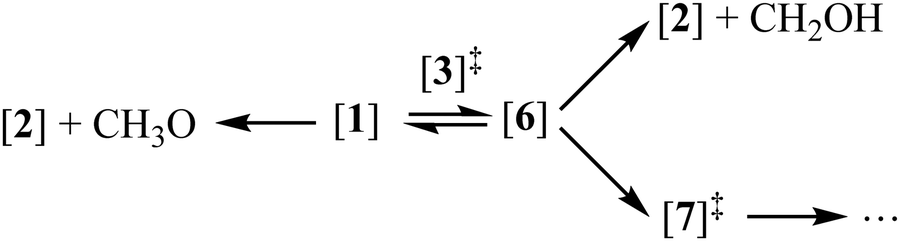 | (3) |
The isomerization of the parent ion over [3]‡ yields [6] promptly, i.e., fast interconversion is assumed for [4]–[5]‡–[6]. Branching between hydroxymethyl loss and methyl or water loss is determined by the fragmentation to [2] and the rates over transition state [7]‡. We also included the direct dissociation of [1] based on ab initio parameters, which takes place without isomerization over a loose transition state above the studied energy range and does not allow for H-scrambling. The modeling results, shown in Fig. 4a, were obtained by relaxing the transition state [3]‡ energy to 10.11 eV (vs. 10.12 eV at G4), the [2] + CH2OH threshold at 10.12 eV (vs. 10.15 eV at G4) and the CH3- and H2O-loss transition state [7]‡ at 9.83 eV (vs. 9.84 eV at G4). Aside from these threshold energies and barriers, only the transition state frequencies were scaled to reproduce the measured rate curve.68 Because of the complex mechanism, the model is overparameterized, yet the agreement with theory is excellent. When the energy is raised above the direct dissociation threshold to [2] + CH3O, direct dissociation is predicted to become the dominant channel quickly, due to its loose transition state. This explains the observation of Tsai et al. that H-scrambling is suppressed at higher energies.51 The modeling also yields a methanol dimer temperature of ca. 120 K. Thereafter, we have adapted the model to account for the dissociative ionization of the perdeuterated CD3OD sample by updating the state functions and energy thresholds based on the shifts in the harmonic frequencies upon deuteration. The goal was not to obtain the best fitting model to the perdeuterated methanol data, which would be easily attained because of the numerous model parameters, but to check whether the adapted model reproduces the shift in the breakdown diagram faithfully, i.e., if the light methanol model as implemented is physically meaningful and has predictive power. Without further optimization, the deuterated breakdown diagram is indeed reproduced quite well. Minor differences are only seen in the decay of the water and methyl loss signal above 10.2 eV, which takes place more slowly than predicted by the model. This suggests that quantum tunneling, which would be suppressed upon deuteration67 and was not included in the model, does not play a role in dissociative photoionization, and the ca. 50 meV shift in the breakdown diagram is entirely due to zero-point and classical reaction kinetics effects upon deuteration. As shown in Fig. 3a, changing zero-point energies lead to a shift of 20–30 meV in the isomerization as well as fragmentation thresholds. The blue-shift of the breakdown diagram in Fig. 4a is larger, which can be explained by the consistently lower effective fragmentation rate constants of the perdeuterated sample. Fig. 4b shows the isomerization rate constant over [3]‡, i.e., the rate-determining step at the dissociative photoionization onset. The lower dissociation rate constants are due to a lowering of the vibrational frequencies and a corresponding increase in the precursor density of states.
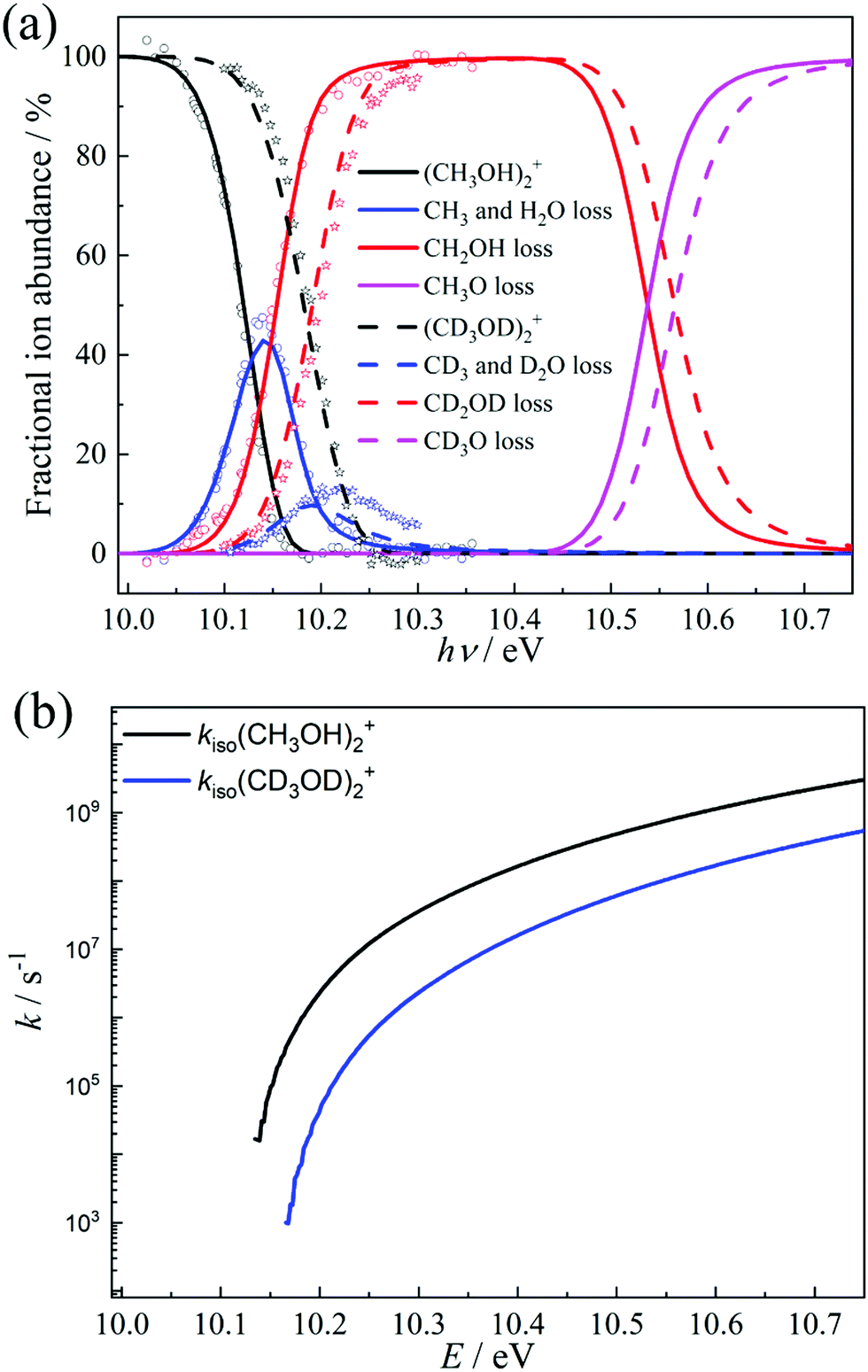 | ||
| Fig. 4 (a) Statistical model of the dissociative ionization of the methanol and the perdeuterated methanol dimer. Open circles and stars represent the experimental fractional abundances in the methanol dimer and perdeuterated methanol dimer experiments, respectively. The solid and dashed lines show the simulated methanol dimer and perdeuterated methanol dimer breakdown curves, respectively. (b) Isomerization rate constant of (CH3OH)2+ and (CD3OD)2+ over [3]‡ in the statistical model as a function of energy relative to the neutral. | ||
4. Conclusions
The unimolecular fragmentation mechanism of methanol dimer cations was studied experimentally by photoelectron photoion coincidence in the 10.00–10.35 eV photon energy range. To understand the H-scrambling mechanism and the possible role of quantum tunneling in the fragmentation of the cation, we also recorded data on selectively deuterated and perdeuterated methanol. The only intact parent ion detected below the methanol ionization energy is the dimer cation, (CH3OH)2+. The larger, protonated species (CH3OH)nH+ (n > 2) are shown not to interfere with the fragmentation products of the dimer cation by hydroxymethyl, methyl, and water loss. Isotope labeling helped show that hydroxyl hydrogens contribute most of the protons to the larger protonated clusters, but three out of four protonated monomers carry a proton from the methyl group of the other methanol unit in the dimer. Moreover, the H2O and CH3 loss fragments m/z 46 and 49 were observed in the dissociative photoionization of methanol dimer measurement for the first time. Both appear at similar energy (ca. 10.05 eV) and disappear from the mass spectra 200 meV higher. The methanol dimer, thus, has low-energy dissociative photoionization channels leading to m/z 33, 46, and 49 fragment ions, and fragmentation is slow at threshold.The dimer cation potential energy surface was explored using density functional theory and composite methods to explain the observations. Five different reaction paths were revealed from the (CH3OH)2+ methanol dimer parent ion: two to produce m/z 33 by CH2OH or CH3O loss, a path to the ethanol cation at m/z 46 by H2O loss, and two paths to form two constitutional isomers at m/z 49 by CH3 loss. Direct fragmentation of the dimer cation is energetically forbidden in the studied photon energy range. Experiments on deuterated methanol confirmed that the main pathway to protonated methanol formation is hydroxymethyl radical loss, possible after proton transfer from a methyl group in the dimer. This is accompanied by limited H-scrambling in the parent ion. Once a methyl group is deprotonated over transition state [3]‡, the other methyl group may also roam around the cation core, leading to the newly observed methyl- and water-loss fragment ions, including the ethanol cation after C–C bond formation.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Experiments were carried out at the VUV beamline of the Swiss Light Source of the Paul Scherrer Institute with support from the PSI CROSS project funding initiative. Additional financial support from the National Natural Science Foundation of China (Grant No. 21903079 and 21873089) and the Swiss Federal Office for Energy (BFE Contract No. SI/501269-01) is gratefully acknowledged. B. S. was supported by the National Science Foundation (CHE-1665464).References
- A. Douhal, S. Kim and A. Zewail, Nature, 1995, 378, 260–263 CrossRef CAS PubMed
.
- D. E. Woon, Adv. Space Res., 2004, 33, 44–48 CrossRef CAS
- L. C. Short, S.-S. Cai and J. A. Syage, J. Am. Soc. Mass Spectrom., 2007, 18, 589–599 CrossRef CAS PubMed
- H. Vehkamäki and I. Riipinen, Chem. Soc. Rev., 2012, 41, 5160–5173 RSC
- F. P. Abramson and J. H. Futrell, J. Chem. Phys., 1966, 45, 1925–1931 CrossRef CAS
- S. Gupta, E. Jones, A. G. Harrison and J. J. Myher, Can. J. Chem., 1967, 45, 3107–3117 CrossRef CAS
- C. R. Moylan and J. I. Brauman, Annu. Rev. Phys. Chem., 1983, 34, 187–215 CrossRef CAS
- S. Hartweg, G. A. Garcia and L. Nahon, J. Phys. Chem. A, 2021, 125, 4882–4887 CrossRef CAS PubMed
- E. Kamarchik, O. Kostko, J. M. Bowman, M. Ahmed and A. I. Krylov, J. Chem. Phys., 2010, 132, 194311 CrossRef PubMed
- A. Bodi, J. Csontos, M. Kállay, S. Borkar and B. Sztáray, Chem. Sci., 2014, 5, 3057–3063 RSC
- Y. Hu, H. Fu and E. Bernstein, J. Chem. Phys., 2006, 125, 154306 CrossRef CAS PubMed
- K. Tono, J. Kuo, M. Tada, K. Fukazawa, N. Fukushima, C. Kasai and K. Tsukiyama, J. Chem. Phys., 2008, 129, 084304 CrossRef PubMed
- M. Heger, M. A. Suhm and R. A. Mata, J. Chem. Phys., 2014, 141, 101105 CrossRef PubMed
- W. Li, Y. Hu, J. Guan, F. Liu, X. Shan and L. Sheng, J. Chem. Phys., 2013, 139, 024307 CrossRef PubMed
- A. Bodi, J. Chem. Phys., 2013, 139, 144306 CrossRef PubMed
- W. Xiao, Y. Hu, W. Li, J. Guan, F. Liu, X. Shan and L. Sheng, J. Chem. Phys., 2015, 142, 024306 CrossRef PubMed
- Y. Tao, Y. Hu, W. Xiao, J. Guan, F. Liu, X. Shan and L. Sheng, Phys. Chem. Chem. Phys., 2016, 18, 13554–13563 RSC
- F. Bell, Q. N. Ruan, A. Golan, P. R. Horn, M. Ahmed, S. R. Leone and M. Head-Gordon, J. Am. Chem. Soc., 2013, 135, 14229–14239 CrossRef CAS PubMed
- J. H. Litman, B. L. Yoder, B. Schläppi and R. Signorell, Phys. Chem. Chem. Phys., 2013, 15, 940–949 RSC
- O. Björneholm, F. Federmann, S. Kakar and T. Möller, J. Chem. Phys., 1999, 111, 546–550 CrossRef
- L. Belau, K. R. Wilson, S. R. Leone and M. Ahmed, J. Phys. Chem. A, 2007, 111, 10075–10083 CrossRef CAS PubMed
- W. Kamke, R. Herrmann, Z. Wang and I. Hertel, Z. Phys. D: At., Mol. Clusters, 1988, 10, 491–497 CrossRef
- K. D. Cook, G. G. Jones and J. W. Taylor, Int. J. Mass Spectrom. Ion Phys., 1980, 35, 273–292 CrossRef CAS
- S. Y. Lee, D. N. Shin, S. G. Cho, K. H. Jung and K. W. Jung, J. Mass Spectrom., 1995, 30, 969–976 CrossRef CAS
- O. Kostko, L. Belau, K. R. Wilson and M. Ahmed, J. Phys. Chem. A, 2008, 112, 9555–9562 CrossRef CAS PubMed
-
K. R. Wilson and F. Qi, Photoionization and Photoinduced Processes in Mass Spectrometry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2021, ch. 5, pp. 159–213 DOI:10.1002/9783527682201.ch5
- T. Baer and R. P. Tuckett, Phys. Chem. Chem. Phys., 2017, 19, 9698–9723 RSC
- X. Wu, X. Tang, X. Zhou and S. Liu, Chin. J. Chem. Phys., 2019, 32, 11–22 CrossRef CAS
- A. Bodi, P. Hemberger, D. L. Osborn and B. L. Sztáray, J. Phys. Chem. Lett., 2013, 4, 2948–2952 CrossRef CAS
- P. Hemberger and A. Bodi, Chimia, 2018, 72, 227–232 CrossRef CAS PubMed
- P. Hemberger, V. B. Custodis, A. Bodi, T. Gerber and J. A. van Bokhoven, Nat. Commun., 2017, 8, 15946 CrossRef PubMed
- P. Hemberger, J. A. van Bokhoven, J. Pérez-Ramírez and A. Bodi, Catal. Sci. Technol., 2020, 10, 1975–1990 RSC
- B. L. Yoder, K. B. Bravaya, A. Bodi, A. H. West, B. Sztáray and R. Signorell, J. Chem. Phys., 2015, 142, 114303 CrossRef PubMed
- X. Wu, X. Zhou, P. Hemberger and A. Bodi, Phys. Chem. Chem. Phys., 2020, 22, 2351 RSC
- X. Wu, X. Zhou, S. A. Bjelić, P. Hemberger and A. Bodi, J. Phys. Chem. A, 2021, 125, 3327–3340 CrossRef CAS PubMed
- M. Steglich, V. B. Custodis, A. J. Trevitt, G. daSilva, A. Bodi and P. Hemberger, J. Am. Chem. Soc., 2017, 139, 14348–14351 CrossRef CAS PubMed
- K. Voronova, K. M. Ervin, K. G. Torma, P. Hemberger, A. Bodi, T. Gerber, D. L. Osborn and B. L. Sztáray, J. Phys. Chem. Lett., 2018, 9, 534–539 CrossRef CAS PubMed
- X. Wu, X. Zhou, P. Hemberger and A. Bodi, Phys. Chem. Chem. Phys., 2019, 21, 22238–22247 RSC
- I. Powis, S. Daly, M. Tia, B. C. de Miranda, G. A. Garcia and L. Nahon, Phys. Chem. Chem. Phys., 2014, 16, 467–476 RSC
- S. Daly, I. Powis, G. A. Garcia, M. Tia and L. Nahon, J. Chem. Phys., 2017, 147, 013937 CrossRef PubMed
- R. W. Larsen, P. Zielke and M. A. Suhm, J. Chem. Phys., 2007, 126, 194307 CrossRef PubMed
- K. Lin, X. Zhou, Y. Luo and S. Liu, J. Phys. Chem. B, 2010, 114, 3567–3573 CrossRef CAS PubMed
- P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253–4264 CrossRef CAS PubMed
- A. J. Barnes, J. Mol. Struct., 2004, 704, 3–9 CrossRef CAS
- A. Bodi, P. Hemberger and R. P. Tuckett, J. Phys. Chem. A, 2021, 125, 2601–2611 CrossRef CAS PubMed
- I. Bakó and G. Pálinkás, THEOCHEM, 2002, 594, 179–184 CrossRef
- S. Morgan and A. Castleman Jr, J. Phys. Chem., 1989, 93, 4544–4550 CrossRef CAS
- S. Morgan, R. Keesee and A. Castleman Jr, J. Am. Chem. Soc., 1989, 111, 3841–3845 CrossRef CAS
- M. S. El-Shall, C. Marks, L. W. Sieck and M. Meot-Ner, J. Phys. Chem., 1992, 96, 2045–2051 CrossRef CAS
- S. Martrenchard, G. Grégoire, C. Dedonder-Lardeux, C. Jouvet and D. Solgadi, PhysChemComm, 1999, 2, 15–19 RSC
- S.-T. Tsai, J.-C. Jiang, Y. T. Lee, A. Kung, S. Lin and C.-K. Ni, J. Chem. Phys., 1999, 111, 3434–3440 CrossRef CAS
- J. A. Booze and T. Baer, J. Chem. Phys., 1992, 96, 5541–5543 CrossRef CAS
- R. T. Jongma, Y. Huang, S. Shi and A. M. Wodtke, J. Phys. Chem. A, 1998, 102, 8847–8854 CrossRef CAS
- B. Sztáray, K. Voronova, K. G. Torma, K. J. Covert, A. Bodi, P. Hemberger, T. Gerber and D. L. Osborn, J. Chem. Phys., 2017, 147, 013944 CrossRef PubMed
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 610, 597–603 CrossRef CAS
- A. Bodi, P. Hemberger, T. Gerber and B. Sztaray, Rev. Sci. Instrum., 2012, 83, 083105 CrossRef PubMed
-
A. Kramida, Y. Ralchenko, J. Reader and NIST ASD Team, NIST Atomic Spectra Database (version 5.9), National Institute of Standards and Technology, Gaithersburg, MD, 2021, available: https://physics.nist.gov/asd DOI:10.18434/T4W30F
- N. Gimelshein, S. Gimelshein, C. C. Pradzynski, T. Zeuch and U. Buck, J. Chem. Phys., 2015, 142, 244305 CrossRef CAS PubMed
- B. Bandyopadhyay, O. Kostko, Y. Fang and M. Ahmed, J. Phys. Chem. A, 2015, 119, 4083–4092 CrossRef CAS PubMed
- B. Sztáray and T. Baer, Rev. Sci. Instrum., 2003, 74, 3763–3768 CrossRef
- A. Bodi, B. Sztáray, T. Baer, M. Johnson and T. Gerber, Rev. Sci. Instrum., 2007, 78, 084102 CrossRef PubMed
- A. Bodi, T. Baer, N. K. Wells, D. Fakhoury, D. Klecyngier and J. P. Kercher, Phys. Chem. Chem. Phys., 2015, 17, 28505–28509 RSC
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al.Gaussian 16 Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed
-
T. Baer and W. L. Hase, Unimolecular Reaction Dynamics: Theory and Experiments, Oxford University Press, New York, 1996 Search PubMed
- A. Bodi, M. D. Brannock, B. Sztáray and T. Baer, Phys. Chem. Chem. Phys., 2012, 14, 16047–16054 RSC
- A. Bodi, T. Baer, N. K. Wells, D. Fakhoury, D. Klecyngier and J. P. Kercher, Phys. Chem. Chem. Phys., 2015, 17, 28505–28509 RSC
- B. Sztáray, A. Bodi and T. Baer, J. Mass Spectrom., 2010, 45, 1233–1245 CrossRef PubMed
- M. F. Heringa, J. G. Slowik, A. S. Prévôt, U. Baltensperger, P. Hemberger and A. Bodi, J. Phys. Chem. A, 2016, 120, 3397–3405 CrossRef CAS PubMed
- F. Kollipost, J. Andersen, D. Mahler, J. Heimdal, M. Heger, M. Suhm and R. Wugt Larsen, J. Chem. Phys., 2014, 141, 174314 CrossRef CAS PubMed
- J. Harvey, A. Bodi, R. P. Tuckett and B. Sztáray, Phys. Chem. Chem. Phys., 2012, 14, 3935–3948 RSC
- K. Voronova, K. G. Torma, J. P. Kercher, A. Bodi and B. Sztáray, Int. J. Mass Spectrom., 2019, 438, 63–71 CrossRef CAS
- K. J. Covert, K. Voronova, K. G. Torma, A. Bodi, J. Zádor and B. Sztáray, Phys. Chem. Chem. Phys., 2018, 20, 21085–21094 RSC
| This journal is © the Owner Societies 2022 |