
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic stannylation of white phosphorus†
Marion
Till
a,
Jose
Cammarata
a,
Robert
Wolf
 *a and
Daniel J.
Scott
*b
*a and
Daniel J.
Scott
*b
aUniversity of Regensburg, Institute of Inorganic Chemistry, 93040, Regensburg, Germany. E-mail: robert.wolf@ur.de
bUniversity of Oxford, Department of Chemistry, OX1 3TA, Oxford, UK. E-mail: daniel.scott@chem.ox.ac.uk
First published on 14th July 2022
Abstract
Organophosphorus compounds (OPCs) are highly important chemicals, finding numerous applications in both academia and industry. Herein we describe a simple photocatalytic method for the stannylation of white phosphorus (P4) using a cheap, commercially-available distannane, (Bu3Sn)2, and anthraquinone as a simple photocatalyst. Subsequent ‘one pot’ transformation of the resulting stannylated monophosphine intermediate (Bu3Sn)3P provides direct, convenient and versatile access to valuable OPCs such as acylated phosphines and tetraalkylphosphonium salts.
White phosphorus (P4) – the most chemically important allotrope of this ubiquitous and abundant element – acts as the common precursor from which all commercially valuable and academically important organophosphorus compounds (OPCs) are prepared. The current methods used for the industrial synthesis of these myriad useful P1 products include the oxidation of P4 with toxic Cl2 gas to generate PCl3 which can subsequently be transformed into a variety of OPCs by reaction with nucleophiles (Scheme 1a). As an alternative route, initial acid- or base-mediated disproportionation of P4 can be used to generate highly toxic PH3 gas which is then employed for the hydrophosphination of unsaturated organic substrates.1
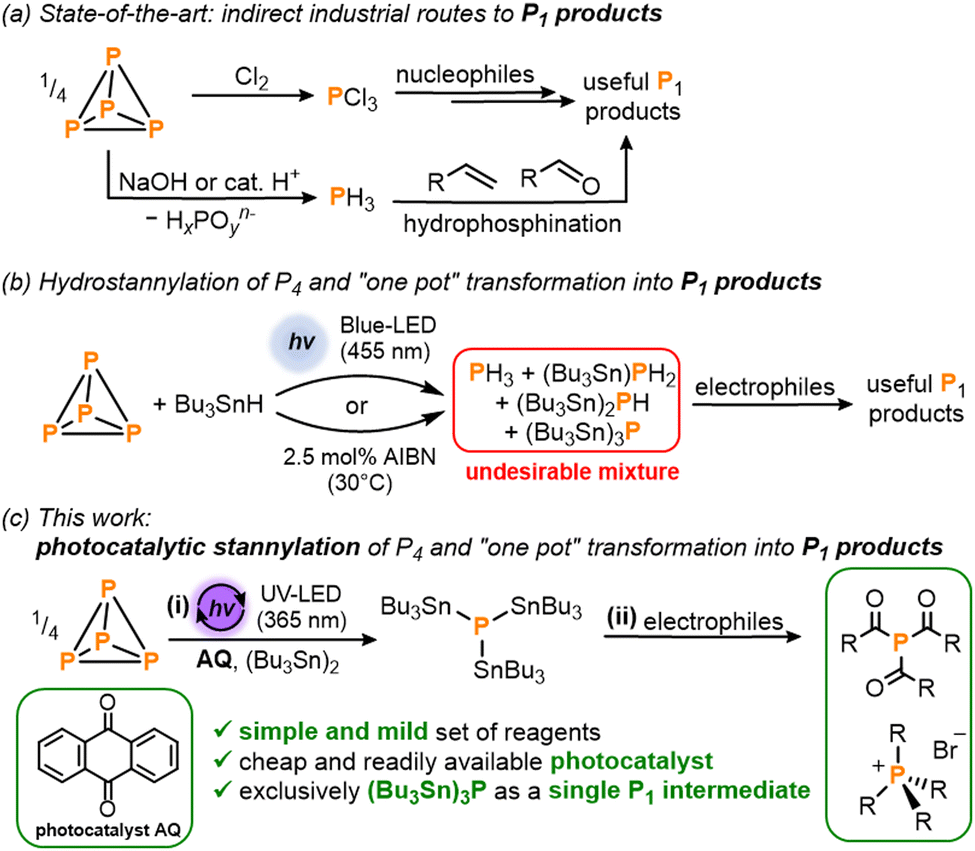 | ||
| Scheme 1 (a) Current state-of-the-art industrial methods for the synthesis of valuable P1 products.1 (b) Recently reported hydrostannylation of white phosphorus (P4) using Bu3SnH followed by reaction with electrophiles to generate useful P1 products in a ‘one-pot’ fashion.5 (c) This work: (i) photocatalytic stannylation of P4 using the photocatalyst anthraquinone (AQ) and hexabutyldistannane (Bu3Sn)2; and (ii) subsequent functionalization of the intermediate (Bu3Sn)3P with electrophiles into products such as triacylphosphines and tetraalkylphosphonium salts. | ||
Given the drawbacks of these methods, a highly prominent aim has long been to find ways of bypassing these multi-step procedures. In particular, there is a longstanding desire to develop more step-efficient direct – and, ideally, catalytic – methods to functionalize P4 and generate OPCs in a single reaction.
As a result, for several decades comprehensive efforts have been made to better understand the fundamental reactivity of P4.2 However, it is only very recently that it has finally become possible to successfully transform P4 directly into a variety of useful P1 products.3 Moreover, and despite these extensive investigations, the number of successful examples remains extremely low, and those that do exist still suffer from substantial limitations.4 As such, there remains a clear need to expand the range of strategies available for direct, productive P4 activation, with new catalytic methods being particularly desirable.4a
In one of our own contributions to this area, we recently reported a simple ‘one pot’ method in which the classical radical reagent tri-n-butyltin hydride (Bu3SnH) is used for initial hydrostannylation of P4 (Scheme 1b).5 This reductive P4 activation is mediated either by light or by a chemical radical initiator such as AIBN (azobis(isobutyronitrile)) which can initiate a radical chain reaction that breaks down the P4 tetrahedron, yielding a mixture of hydrostannylated phosphines (Bu3Sn)xPH3−x (x = 0–3). Key to this mechanism is the attack of stannyl radicals (Bu3Sn˙) on the P–P bonds of P4. The resulting (Bu3Sn)xPH3−x mixture can then be converted into a number of important and useful OPCs by reaction with electrophiles.5
Unfortunately, one significant disadvantage of this hydrostannylation strategy is the complexity of the (Bu3Sn)xPH3−x mixture, which complicates ‘downstream’ reaction development by requiring functionalization of two different types of bond (P–Sn and P–H), both of which are distributed over four distinct molecules. Moreover, the presence of gaseous PH3 as a component of this mixture has been suggested to have a limiting effect on overall yields as it can easily be lost during subsequent manipulations,4a and it is also problematic from a safety perspective.
These drawbacks would be overcome if the initial P4 reduction step could instead furnish a single species with just one functionalizable motif, but with reactivity otherwise similar to (Bu3Sn)xPH3−x. To achieve this, we describe herein a simple photocatalytic strategy for the atom-precise stannylation of P4 using the cheap, commercially-available distannane (Bu3Sn)2 and simple benzophenone derivatives as photocatalysts (Scheme 1c). This new procedure generates exclusively the stannylated monophosphine (Bu3Sn)3P and subsequent, simplified ‘one pot’ transformations with electrophiles afford valuable OPCs including acylated phosphines and alkylated phosphonium salts.
Based on the analysis above, we sought to develop a new method by which P4 could be selectively transformed into (Bu3Sn)3P as the sole product.6 It is worth noting that the closely related product (Ph3Sn)3P has previously been prepared from P4 using Ph3SnCl as the stannylating reagent, but this required use of a relatively elaborate Ti(III) reagent as a halogen atom abstractor.3f Instead, we imagined that an ideal reagent for such a reaction would be the distannane (Bu3Sn)2, which is cheap to purchase and could in principle provide the target phosphine with perfect atom economy.7 Indeed, Sn–Sn homolysis of (Bu3Sn)2 is known to furnish Bu3Sn˙ radicals, which previous work has shown are capable of adding to P4.3f,5 However, achieving this homolysis directly requires extreme temperatures or very high energy UV light irradiation that is known to lead to unselective reactivity, and is also unlikely to be compatible with P4.8–10 Fortunately, it has been reported that simple ketones can be used as photocatalysts to access Bu3Sn˙ radicals by Sn–Sn bond cleavage under much lower energy irradiation.11
The light-driven photocatalytic stannylation of P4 was therefore targeted, based on the mechanistic proposal outlined in Scheme 2.9 It was anticipated that photoirradiation of the ketone R2CO would first provide an excited state, [R2CO]*,12 capable of reacting with (Bu3Sn)2 to generate a stannylated ketyl radical and a free Bu3Sn˙ radical.11 The former could then thermally release a second Bu3Sn˙ radical to close the catalytic cycle. Once formed, these Bu3Sn˙ radicals would then add to the P–P bonds of P4, ultimately breaking it down to generate (Bu3Sn)3P as the only P1 product.13
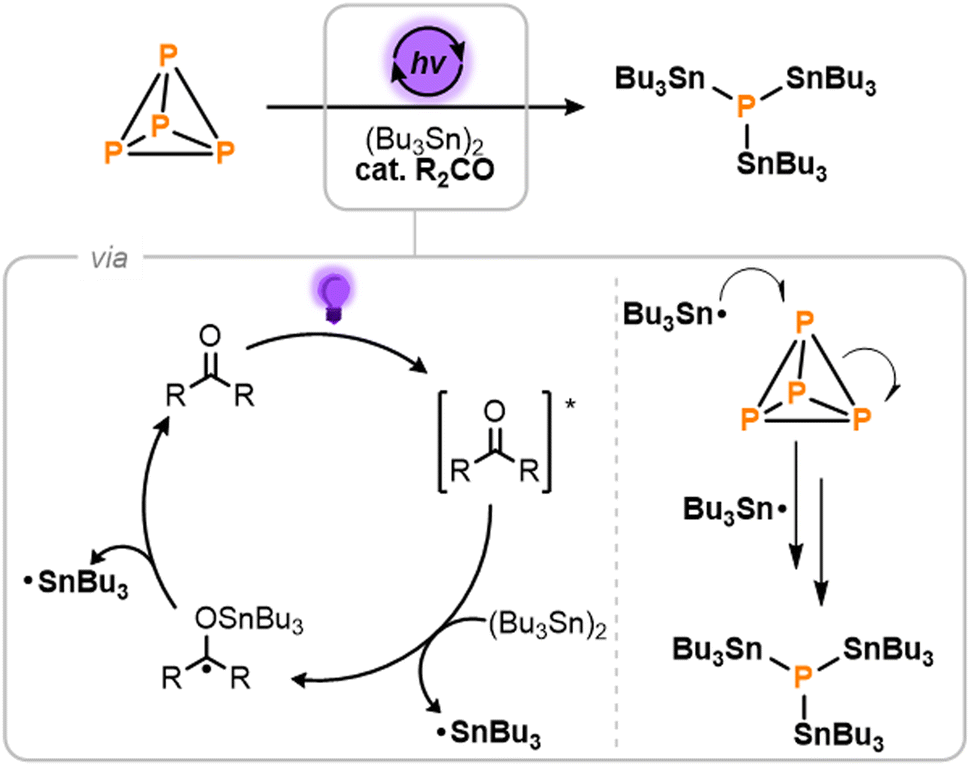 | ||
| Scheme 2 Proposed mechanism for the light-driven, photocatalytic stannylation of P4 in the presence of hexabutyldistannane, (Bu3Sn)2, and a ketone photocatalyst, R2CO. | ||
To begin, benzophenone (BP) was chosen as a proof-of-principle photocatalyst due to both its simplicity and the fact that its photoreactivity towards hexaalkyldistannanes has been studied previously.11d Gratifyingly, after an initial optimization the photocatalytic stannylation of P4 could successfully be achieved, with use of 25 mol% BP (all stoichiometries, in both equiv. and mol%, are defined per P atom) and a 3.3-fold excess (5 equiv.) of (Bu3Sn)2 providing 50% conversion to the target stannylated phosphine (Bu3Sn)3P after stirring under near UV LEDs overnight (Scheme 3; see also ESI,† S3). Control experiments confirmed that all reaction components (P4, (Bu3Sn)2, BP, irradiation) were necessary for the reaction to proceed productively (see ESI,† S3, Table S1).
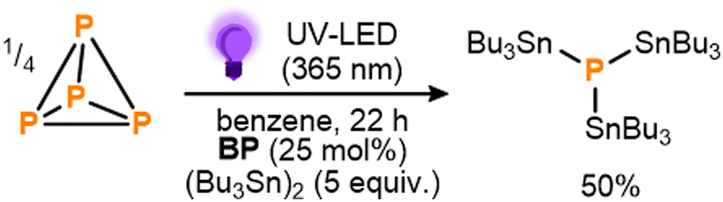 | ||
| Scheme 3 Initial conditions for the direct, photocatalytic stannylation of P4 into (Bu3Sn)3P optimized using benzophenone (BP) as photocatalyst. Stoichiometries in equiv. and mol% are defined per P atom. | ||
These initial results provided a clear proof-of-principle for the proposed mechanistic strategy. Notably, the observed conversion indicates the activation of at least three Sn–Sn bonds per available equivalent of BP,14 making this a rare example of a system where P4 activation has been achieved catalytically, using an otherwise inert substrate.5,9a,9b,9e,15 Nevertheless, in order to improve the reaction outcome further, a broader range of benzophenone derivates was subsequently screened, with several found to provide markedly improved performance (see ESI,† S5). Particularly impressive results were achieved using anthraquinone (AQ) and following brief further optimization (see ESI,† S5 and S7) 79% conversion to (Bu3Sn)3P could be achieved using significantly reduced loadings of both AQ (12.5 mol%) and (Bu3Sn)2 (3 equiv.) over the same timeframe (Scheme 4; see also ESI,† S7). Based on the catalytic cycle proposed in Scheme 2, this would correspond to a turnover number (TON) of 10.0 for AQ. Further reductions in catalyst loading to 6.3 mol% or 2.5 mol% were found to lead to even higher TONs (16.8 and 28.2, respectively), albeit at the cost of lower overall conversions (see ESI,† S7, Table S11).
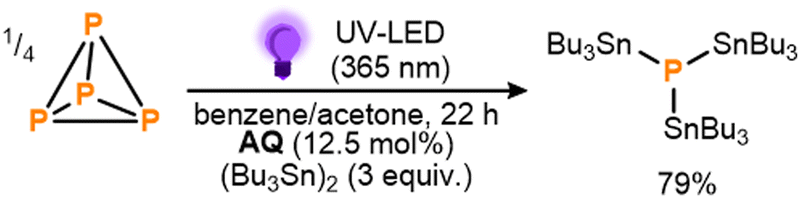 | ||
| Scheme 4 Optimized conditions for the direct, photocatalytic stannylation of P4 into (Bu3Sn)3P using anthraquinone (AQ) as photocatalyst. Stoichiometries in equiv. and mol% are defined per P atom. | ||
With the stannylation of P4 optimized, attention was then shifted to its subsequent, ‘one pot’ transformation into other useful P1 products. Having previously developed procedures for the analogous transformation of the phosphine mixture (Bu3Sn)xPH3−x, which includes (Bu3Sn)3P as a minor component, it was anticipated that addition of electrophiles to photocatalytically-generated (Bu3Sn)3P should be similarly productive,4,5 especially since neither the AQ photocatalyst nor the (Bu3Sn)2 starting material is expected to show appreciable reactivity towards such substrates. And, indeed, in situ addition of a variety of acid chlorides yielded the corresponding triacylphosphines (R(O)C)3P (R = Ph, Cy, Ad, tBu, iPr, nBu, Me) with good conversions of up to 75% (Scheme 5a(i)).5,16 Notably, and in comparison to our previously-reported hydrostannylation system, no exclusion of light and no additional base were required for this step, highlighting both the robustness and simplicity of (Bu3Sn)3P as a “P3−” synthon, relative to (Bu3Sn)xPH3−x.
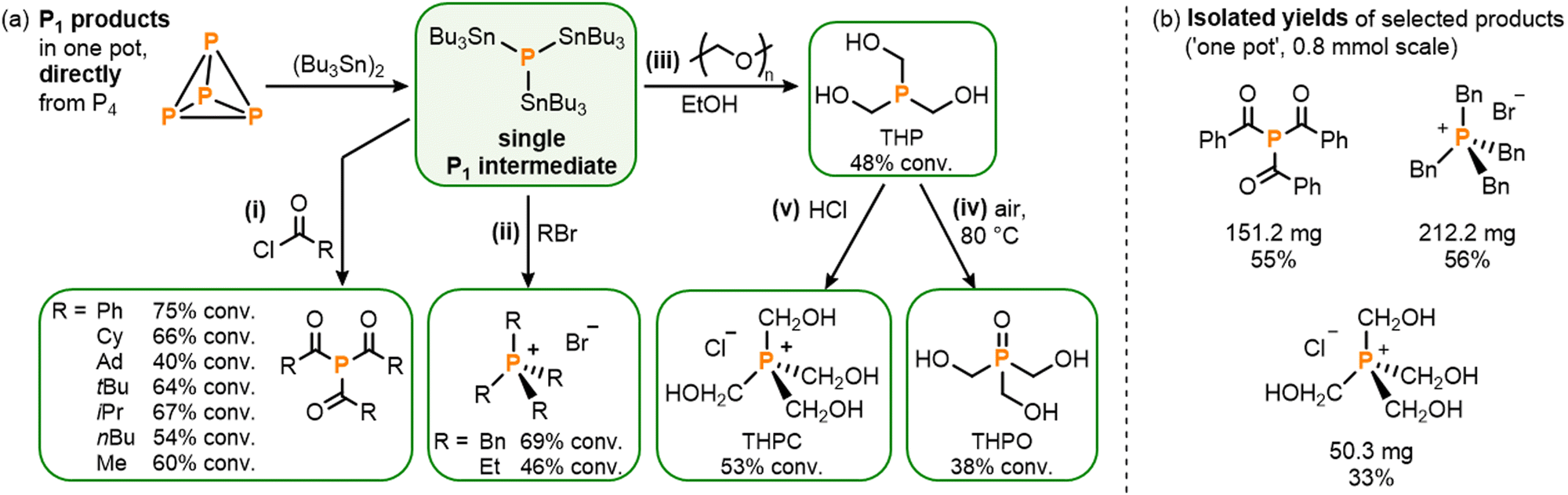 | ||
| Scheme 5 (a) One-pot synthesis directly from P4, via photocatalytically generated P1 intermediate (Bu3Sn)3P, of (i) triacylphosphines (R(O)C)3P (4 equiv. RC(O)Cl, R = tBu, Ph, Me, nBu, Cy, iPr, Ad), (ii) phosphonium salts [R4P]Br (5 equiv. RBr, R = Bn, Et, 60–80 °C), (iii) tris(hydroxymethyl)phosphine, THP (EtOH, 3 equiv. paraformaldehyde), (iv) tris(hydroxymethyl)phosphine oxide, THPO (as for (iii) then air, 80 °C), and (v) tetrakis(hydroxymethyl)phosphonium chloride, THPC (as for (iii) using 12.5 equiv. paraformaldehyde, then 10 equiv. HCl); and (b) Isolated yields for reactions on preparative scale (0.8 mmol). Stoichiometries in equiv. are defined per P atom. | ||
Similarly, reaction of (Bu3Sn)3P with alkyl bromides RBr (R = Bn, Et) under moderate heating successfully provided ‘one pot’ access to the corresponding phosphonium salts, [R4P]Br, including tetrabenzylphosphonium bromide, [Bn4P]Br, which is a known precursor for useful Wittig chemistry (Scheme 5a(ii)).17 Again, no auxiliary base was required for these reactions, in contrast to the analogous procedures via (Bu3Sn)xPH3−x where the absence of base leads to a 50% reduction in yield.5
Finally, another industrially important class of P1 products was targeted. Hydroxymethyl-substituted phosphine derivatives are used as flame-retardant materials (among a number of other applications),18 and could be accessed by reacting the stannylated monophosphine (Bu3Sn)3P with paraformaldehyde in EtOH to furnish tris(hydroxymethyl)phosphine, (HOCH2)3P (THP; Scheme 5a(iii)).18a Subsequent exposure to air then yielded the corresponding phosphine oxide, (HOCH2)3PO (THPO; Scheme 5a(iv)),18b while the phosphonium salt tetrakis(hydroxymethyl)phosphonium chloride, [(HOCH2)4P]Cl (THPC),18c,18d could be accessed by quenching the in situ generated THP with HCl, all in one pot (Scheme 5a(v)).
To demonstrate the viability of these reactions on a preparative scale the triacylphosphine (Ph(O)C)3P and the phosphonium salts [Bn4P]Br and THPC were selected as representative examples for isolation (Scheme 5b; see ESI† S9). At 0.8 mmol scale (PhC(O))3P could be isolated in 55% yield,19 which compares well with our previously-reported hydrostannylation method (51%). [Bn4P]Br could also be isolated in good 56% yield, and THPC in a more modest yield of 33%.19
For this last reaction, efforts were also made to recover the Sn-containing compounds present at the end of the reaction. We have previously shown that for the analogous synthesis of THPC via (Bu3Sn)xPH3−x recovery of the Bu3SnCl byproduct allows for convenient regeneration and recycling of the Bu3SnH starting material, thus minimizing the formation of organotin-containing waste. Bu3SnCl can also be used to regenerate (Bu3Sn)2 through a net one-electron reduction,8 meaning similar recycling should be feasible for this newer system, provided Bu3SnCl can again be cleanly recovered. Satisfyingly, Bu3SnCl could indeed be recovered during THPC workup through simple washing with diethyl ether, being isolated as part of an otherwise clean mixture with unreacted (Bu3Sn)2 in an excellent overall yield of 92% (1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, see ESI† S9).
:1 molar ratio, see ESI† S9).
In conclusion, we have developed a simple, new method for the direct transformation of P4 into a variety of commercially and academically interesting OPCs. The reaction proceeds through a photocatalytic stannylation of white phosphorus, which generates (Bu3Sn)3P with perfect atom economy as a single, convenient P1 intermediate using an inexpensive, commercially available distannane and a simple photocatalyst. This method can be used to prepare a variety of different products through inclusion of a range of different electrophilic substrates, and we have demonstrated that the Sn-containing byproducts of the reaction can in principle be recovered and recycled. These results expand the currently very limited range of strategies that are available for the direct functionalization of P4, and suggest the intriguing possibility that P4 activation might also be achievable by reaction with other weak E–E bonds under similar photocatalytic conditions.
The project received funding from the European Research Council (ERC CoG 772299). DJS is also grateful to the Alexander von Humboldt foundation, and to the EPSRC for award of an Early Career Fellowship (EP/V056069/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) W. Gleason, JOM, 2007, 59, 17–19 CrossRef CAS; (b) G. Bettermann, W. Krause, G. Riess and T. Hofmann, Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2000 Search PubMed; (c) D. E. C. Corbridge, Phosphorus: Chemistry, Biochemistry, and Technology, CRC Press, 6th edn, 2013 Search PubMed.
- (a) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed; (b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed; (c) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed; (d) C. M. Hoidn, D. J. Scott and R. Wolf, Chem. – Eur. J., 2021, 27, 1886–1902 CrossRef CAS PubMed; (e) L. Giusti, V. R. Landaeta, M. Vanni, J. A. Kelly, R. Wolf and M. Caporali, Coord. Chem. Rev., 2021, 441, 2139276–2139375 CrossRef; (f) Y. H. Budnikova, T. V. Gryaznova, V. V. Grinenko, Y. B. Dudkina and M. N. Khrizanforov, Pure Appl. Chem., 2017, 89, 311–330 CrossRef CAS.
- (a) S. Reichl, E. Mädl, F. Riedelberger, M. Piesch, G. Balázs, M. Seidl and M. Scheer, Nat. Commun., 2021, 12, 5774 CrossRef CAS PubMed; (b) M. Donath, K. Schwedtmann, T. Schneider, F. Hennersdorf, A. Bauzá, A. Frontera and J. J. Weigand, Nat. Chem., 2022, 14, 384–391 CAS; (c) Y. Mei, Z. Yan and L. L. Liu, J. Am. Chem. Soc., 2022, 144, 1517–1522 CrossRef CAS PubMed; (d) D. H. R. Barton and J. Zhu, J. Am. Chem. Soc., 1993, 115, 2071–2072 CrossRef CAS; (e) D. H. R. Barton and R. A. Vonder Embse, Tetrahedron, 1998, 54, 12475–12496 CrossRef CAS; (f) B. M. Cossairt and C. C. Cummins, New J. Chem., 2010, 34, 1533–1536 RSC; (g) S. K. Ghosh, C. C. Cummins and J. A. Gladysz, Org. Chem. Front., 2018, 5, 3421–3429 RSC.
- (a) D. J. Scott, Angew. Chem., 2022, e202205019, DOI:10.1002/anie.202205019; (b) H. Grützmacher, Nat. Chem., 2022, 14, 362–364 Search PubMed.
- D. J. Scott, J. Cammarata, M. Schimpf and R. Wolf, Nat. Chem., 2021, 13, 458–464 CrossRef CAS PubMed.
- Previous work has shown that (Bu3Sn)3P can be prepared through stannylation of (Bu3Sn)xPH3−x with Bu3SnOMe. However, this requires addition of an extra stoichiometric reagent and results in formation of MeOH as a byproduct which could lead to competing reactivity upon subsequent addition of electrophiles, see ref. 5.
- (a) A. Darwish and J. M. Chong, Synth. Commun., 2004, 34, 1885–1890 CrossRef CAS; (b) T. N. Mitchell, Encyclopedia of Reagents for Organic Synthesis – Hexabutyldistannane, Wiley, 2012 Search PubMed; (c) T. N. Mitchell and G. Walter, J. Chem. Soc., Perkin Trans. 2, 1977, 1842–1847 RSC.
- A. G. Davies, Organotin Chemistry, Wiley, 2004 Search PubMed.
- For previous examples where photochemical methods have been used to functionalize P4, see: (a) U. Lennert, P. B. Arockiam, V. Streitferdt, D. J. Scott, C. Rödl, R. M. Gschwind and R. Wolf, Nat. Catal., 2019, 2, 1101–1106 CrossRef CAS PubMed; (b) P. B. Arockiam, U. Lennert, C. Graf, R. Rothfelder, D. J. Scott, T. G. Fischer, K. Zeitler and R. Wolf, Chem. – Eur. J., 2020, 26, 16374–16382 CrossRef CAS PubMed; (c) R. Rothfelder, V. Streitferdt, U. Lennert, J. Cammarata, D. J. Scott, K. Zeitler, R. M. Gschwind and R. Wolf, Angew. Chem., Int. Ed., 2021, 60, 24650–24658 CrossRef CAS PubMed; (d) M. Till, V. Streitferdt, D. J. Scott, M. Mende, R. M. Gschwind and R. Wolf, Chem. Commun., 2022, 58, 1100–1103 RSC; (e) G. Lu, J. Chen, X. Huangfu, X. Li, M. Fang, G. Tang and Y. Zhao, Org. Chem. Front., 2019, 6, 190–194 RSC; (f) D. Tofan and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 7516–7518 CrossRef CAS PubMed; (g) L. P. Wang, D. Tofan, J. Chen, T. V. Voorhis and C. C. Cummins, RSC Adv., 2013, 3, 23166–23171 RSC.
- Cleavage of P4 under photoirradiation (256 nm) can generate highly reactive P2 (ref. 9f and g). However, this requires very high energy UV light (256 nm) and so seems unlikely to be a major, relevant pathway for the reactivity reported herein, which uses significantly lower energy UV sources (365 nm). Nevertheless, the possibility that this could act as a minor competing pathway, for example leading to minor reaction side-products, cannot yet be fully excluded.
- (a) W. P. Neumann, H. Hillgärtner, K. M. Baines, R. Dicke, K. Vorspohl, U. Kobs and U. Nussbeutel, Tetrahedron, 1989, 45, 951–960 CrossRef CAS; (b) M. Harendza, J. Junggebauer, K. Leßman, W. P. Neumann and H. Tews, Synlett, 1993, 286–288 CrossRef CAS; (c) D. J. Hart, R. Krishnamurthy, L. M. Pook and F. L. Seely, Tetrahedron Lett., 1993, 34, 7819–7822 CrossRef CAS; (d) M. J. Tomaszeski and J. Warkentin, J. Chem. Soc., Chem. Commun., 1993, 18, 1407–1408 RSC.
- (a) J. A. Dantas, J. T. M. Correia, M. W. Paixão and A. G. Corrêa, ChemPhotoChem, 2019, 3, 506–520 CrossRef CAS; (b) B. Dinda, Essentials of Pericyclic and Photochemical Reactions: Photochemistry of Carbonyl Compounds (Chapter 8), Springer, 2016 Search PubMed; (c) B. König, Chemical Photocatalysis, de Gruyter, 2013 CrossRef; (d) W. A. Green, Industrial Photoinitiators: A Technical Guide, CRC Press, 2010 CrossRef; (e) J. P. Fouassier and J. Lalevée, Photoinitiators: Structures, Reactivity and Application in Polymerization, Wiley, 2021 CrossRef; (f) J. Cervantes-González, D. A. Vosburg, S. E. Mora-Rodriguez, M. A. Vázquez, L. G. Zepeda, C. V. Gómez and S. Lagunas-Rivera, ChemCatChem, 2020, 12, 3811–3827 CrossRef.
- Alternative elementary steps such as “outer-sphere” energy transfer between [R2CO]* and (Bu3Sn)2 could also be possible, but would be expected to lead to the same overall outcome. For additional mechanistic discussion, please see ESI,† S2.
- Since conversion to (Bu3Sn)3P requires activation of 1.5 Sn–Sn bonds per P atom.
- L. Riesel, M. Kant and R. Helbing, Z. Anorg. Allg. Chem., 1990, 580, 217–223 CrossRef CAS.
- G. Becker, Z. Anorg. Allg. Chem., 1977, 430, 66–76 CrossRef CAS.
- H. Schmidbaur, U. Deschler, B. Milewski-Mahrla and B. Zimmer-Gasser, Chem. Ber., 1981, 114, 608–619 CrossRef CAS.
- (a) K. V. Katti, H. Gali, C. J. Smith and D. E. Berning, Chem. Res., 1999, 32, 9–17 CrossRef CAS; (b) M. J. Chen, C. R. Chen, Y. Tan, J. Q. Huang, X. L. Wang, L. Chen and Y. Z. Wang, Ind. Eng. Chem. Res., 2014, 53, 1160–1171 CrossRef CAS; (c) W. J. Vullo, Ind. Eng. Chem. Process Des. Dev., 1966, 5, 346–349 CAS; (d) W. J. Vullo, J. Org. Chem., 1968, 33, 3665–3667 CrossRef CAS.
- Yields of isolated materials have not been corrected for the presence of trace impurities observable by 31P{1H} NMR spectroscopy. See ESI† S9.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc03474c |
| This journal is © The Royal Society of Chemistry 2022 |