
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoresponsive properties of polyacrylamides in physiological solutions†
Kristýna
Kolouchová
 a,
Volodymyr
Lobaz
a,
Hynek
Beneš
a,
Victor R.
de la Rosa
bc,
David
Babuka
ad,
Pavel
Švec
ae,
Peter
Černoch
a,
Martin
Hrubý
a,
Richard
Hoogenboom
*b,
Petr
Štěpánek
a and
Ondřej
Groborz
*afg
a,
Volodymyr
Lobaz
a,
Hynek
Beneš
a,
Victor R.
de la Rosa
bc,
David
Babuka
ad,
Pavel
Švec
ae,
Peter
Černoch
a,
Martin
Hrubý
a,
Richard
Hoogenboom
*b,
Petr
Štěpánek
a and
Ondřej
Groborz
*afg
aInstitute of Macromolecular Chemistry, Czech Academy of Science, Heyrovsky square 2, 162 06 Prague 6, Czech Republic. E-mail: ondrej.groborz@seznam.cz
bDepartment of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281-S4, 9000 Ghent, Belgium. E-mail: richard.hoogenboom@ugent.be
cAVROXA BV, Technologiepark-Zwijnaarde 82, B-9052 Ghent, Belgium
dDepartment of Biophysics, Institute of Physics, Faculty of Mathematics and Physics, Charles University, Ke Karlovu 3, Prague 2, 121 16, Czech Republic
eDepartment of Physical and Macromolecular Chemistry, Faculty of Sciences, Charles University, Hlavova 8, Prague 2, 128 00, Czech Republic
fInstitute of Biophysics and Informatics, Charles University, First Faculty of Medicine, Salmovská 1, 120 00 Prague 2, Czech Republic
gDepartment of Organic and Medicinal Chemistry, Charles University, Faculty of Science, Hlavova 8, 128 43 Prague 2, Czech Republic
First published on 13th August 2021
Abstract
Polymer solutions with a lower critical solution temperature (LCST) undergo reversible phase separation when heated above their cloud point temperature (TCP or CPT). As such, they have been proposed for a wide range of biomedical applications, from injectable drug depots to switchable coatings for cell adhesion. However, in systematic studies, the TCP of these thermoresponsive polymers has been mostly measured in non-physiological solutions, thereby hindering the development of their medicinal applications. Here, we analysed the thermoresponsive properties of four acrylamide-based polymers with LCST, namely poly[(N-2,2-difluoroethyl)acrylamide] (pDFEA), poly[(N-isopropyl)acrylamide] (pNIPAM), poly[(N,N-diethyl)acrylamide] (pDEA), and poly[(N-acryloyl)pyrrolidine] (pAP). As shown by turbidimetry, their TCP in phosphate saline buffer (PBS) and foetal bovine serum (FBS) were consistently lower than those reported in the literature, typically assessed in pure water, even when using the same setup. In addition, these physiological solutions affected the variation of TCP as a function of polymer concentration (1.25 to 10.0 mg mL−1) and molar mass (20 to 50 kg mol−1). As shown by isothermal calorimetry, interactions between proteins in FBS and polymer aggregates were predominantly exothermic, which indicates that protein–polymer complexes are formed through enthalpically driven processes. In conclusion, the TCP of thermoresponsive polymers strongly depends on solvent composition and therefore should be measured under physiological conditions for future medicinal applications.
Introduction
Many hydrophilic polymers with a lower critical solution temperature (LCST) form homogeneous solutions below a threshold temperature (cloud point temperature, TCP or CPT), but they separate into two phases with high and low concentrations when heating above this TCP.1 Numerous applications for such “smart polymers” have been proposed, across various fields, including switchable substrates for cell/tissue culture,2–5 drug delivery systems,6,7in situ depot formation, controlled drug release (therapy/theranostics),7–11 gene delivery/therapy,12–14 tissue engeneering,4,15–17 wound dressing,18,19 biosensors,20 vaccines/immunotherapy,21 and injectable brachytherapy,22,23 among others.24,25 Therefore, developing such applications requires understanding the properties of these polymers, particularly their TCP.T CP depends on polymer molar mass (dispersity)1,26–29 and concentration,1,26,30 on solution pH9,31–33 and on the concentration and type of ions33–41 (due to the Hofmeister effect34), proteins and other macromolecules33,42–45 in solution (due to the excluded volume/molecular crowding effect,46 competition for solvent, or non-covalent protein–polymer interactions47), among other factors.1,27,48 Polymer tacticity1,49 and terminal moiety (e.g., a chain transfer agent, CTA)1,50,51 and even fairly subtle environmental changes, including changes in solution pH,32,40,52 can also affect the TCP. Several theories, especially the lattice fluid theory with hydrogen-bonding corrections (LFT-HB)53 and other (more advanced) models,45,54–57 reliably describe TCP as a function of polymer concentration, but they are rather complicated and heavily rely on empirical data.45,53–57 Unsurprisingly, many authors agree with the need to assess the thermoresponsive properties of a polymer experimentally.26,27,34,36,45,54,58
Many techniques can be used to determine the TCP of a given polymeric system, such as turbidimetry/spectrophotometry,26,59 rheology,60 refractometry,61 infrared spectroscopy,62 small-angle neutron scattering,63,64 dynamic light scattering and small-angle/-wide angle X-ray scattering (DLS, SAXS WAXS),60,65,661H or 19F NMR (nuclear magnetic resonance) spectroscopy,63,64 isothermal and differential scanning (micro)calorimetry,67,68 among others. Spectrophotometry (turbidimetry) is arguably the most common method for determining the TCP of a solution of a thermoresponsive polymer because this method is widely available, quick, sensitive, and robust, accurately providing the TCP at both high and relatively low concentrations. However, beam wavelength,59 heating and stirring rates,59,66 as well as the selected threshold absorbance,26,59 can alter the measurement sensitivity and thus shift the experimental TCP.59 Moreover, this method may be inaccurate for absorbing (coloured), non-homogeneous samples32,59 and for polymer solutions with a very low refractive index increment (dn/dc) – for such samples, DLS is a more reliable option.59 In turn, DLS may provide additional information about the variation in the size of molecular assemblies as a function of temperature (with some bias concerning larger structures),65 but this method is more time demanding and laborious than turbidimetry.65
Notwithstanding the wide range of methods for determining the TCP of thermoresponsive polymers and for their thorough characterisation, most previous systematic studies have only done so in non-physiological solutions. Furthermore, the solvent (PBS, FBS, plasma, serum, and interstitial fluid) effect on the TCPs of such polymer solutions has never been systematically assessed, with most researchers analysing the effects of molar weight or salts/proteins on TCP separately and under different conditions. Similar homopolymers prepared/measured under different conditions may display vastly different thermoresponsive properties, as shown by meta-analysis.1,26 Therefore, understanding how molar mass and salts/proteins affect TCP requires assessing all these effects with one batch of polymers in a head-to-head study. While numerous thermoresponsive homopolymers have already been reported, only poly[(N-isopropyl)acrylamide] (pNIPAM)33–37,43,44,67,69 and poly[(N-acryloyl)pyrrolidine] (pAP)37 have been extensively studied in both water and (at least partly) physiologically relevant solutions. Those studies have shown that TCP is significantly lower in those solutions than in water.
Considering the above, we used pNIPAM as a benchmark to directly compare its properties, particularly TCP, to those of other polyacrylamides which have not been characterised yet in both physiological and non-physiological solvents. For this purpose, we synthesised four acrylamide-based homopolymers by RAFT polymerization in three molar-mass-categories 20 to 25 kg mol−1, 30 to 35 kg mol−1 and 40 to 50 kg mol−1, namely poly[(N-2,2-difluoroethyl)acrylamide] (pDFEA), pNIPAM, poly[(N,N-diethyl)acrylamide] (pDEA), and pAP. Subsequently, we compared the TCPs of their solutions in water to those of their solutions in phosphate saline buffer (PBS) and foetal bovine serum (FBS), as models of physiologically relevant conditions.70–72 After determining the size of polymer aggregates as function of temperature, we compared the results to those from turbidimetry measurements, also measuring their TCPs as a function of their molar mass and concentration (1.25 to 10.0 mg mL−1). Lastly, we assessed the effect of different physiological solvents (PBS and FBS) on polymer aggregation by isothermal calorimetry.
Results and discussion
Polymer selection
The polymers were selected for this study because they are non-ionic homopolymers of acrylamide N-derivatives with one or two alkyl moieties, and their aqueous solutions display LCST thermoresponsiveness with various TCPs at similar polymer concentrations (pAP ≫ pNIPAM ≈ pDEA > pDFEA; Fig. 1).1,73 While pAP, pNIPAM, pDEA have been studied extensively,1,26pDFEA is a relatively new and atypical thermoresponsive acrylamide with fluorine atoms.73 As polyacrylamides, all four polymers act as hydrogen bond acceptors, but only pDFEA and pNIPAM can also act as hydrogen bond donors because they contain secondary amide moieties. In addition, pDFEA contains –CF2H moieties, i.e., lipophilic hydrogen donors74 (see Scheme S2† and chapter S10.2). All these polymers are non-toxic and biocompatible as well. Accordingly, their different properties (e.g., hydrophilicity or TCP) may be used to tailor the final materials for biomedical applications.1,73 Nevertheless, we avoided comparing copolymers because their TCP depends on the content of their individual monomers and on their architecture, thus adding other unknown variables to the equation.75 | ||
| Fig. 1 Structures of the study polymers: poly[(N-2,2-difluoroethyl)acrylamide] (pDFEA), poly[(N-isopropyl)acrylamide] (pNIPAM), poly[(N,N-diethyl)acrylamide] (pDEA), and poly[(N-acryloyl)pyrrolidine] (pAP). | ||
We targeted polymers with properties suitable for biomedical applications. For this reason, pDFEA, pNIPAM, pDEA, and pAP had a narrow molecular weight distribution (ĐM ≤ 1.11) and molar masses in three different ranges (≈![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 to 25 kg mol−1, ≈30 to 35 kg mol−1 and ≈40 to 50 kg mol−1) but all lower than the renal excretion limit77,78 so that they would not accumulate in the body.78 The end groups of these polymers contained a methyl ester (from methyl acrylate) on one end and a carboxyl group (initiator residue) on the other, which can be conveniently used to introduce tracers, dyes, and other moieties.
20 to 25 kg mol−1, ≈30 to 35 kg mol−1 and ≈40 to 50 kg mol−1) but all lower than the renal excretion limit77,78 so that they would not accumulate in the body.78 The end groups of these polymers contained a methyl ester (from methyl acrylate) on one end and a carboxyl group (initiator residue) on the other, which can be conveniently used to introduce tracers, dyes, and other moieties.
Polymer synthesis, purification, and characterisation
We prepared these four polymers via controlled reversible addition–fragmentation chain-transfer (RAFT) radical polymerisation76 (Fig. 2) using 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid as the CTA and 4,4′-azobis(4-cyanovaleric acid) (ACVA) as the initiator (see Table S1† for the initial quantities of the reagents). Subsequently, we purified and mixed the polymers first with propylamine (aminolysis of the terminal CTA)76 and then with an excess of methyl acrylate (to mask the reactive thiol moiety by Michael addition),76 as shown in Fig. 2 and Table S2.† Lastly, we purified and characterised these polymers by size exclusion chromatography and NMR spectroscopy to determine their purity and to confirm CTA removal (see chapter S10.3†). The properties of these polymers are outlined in Table 1.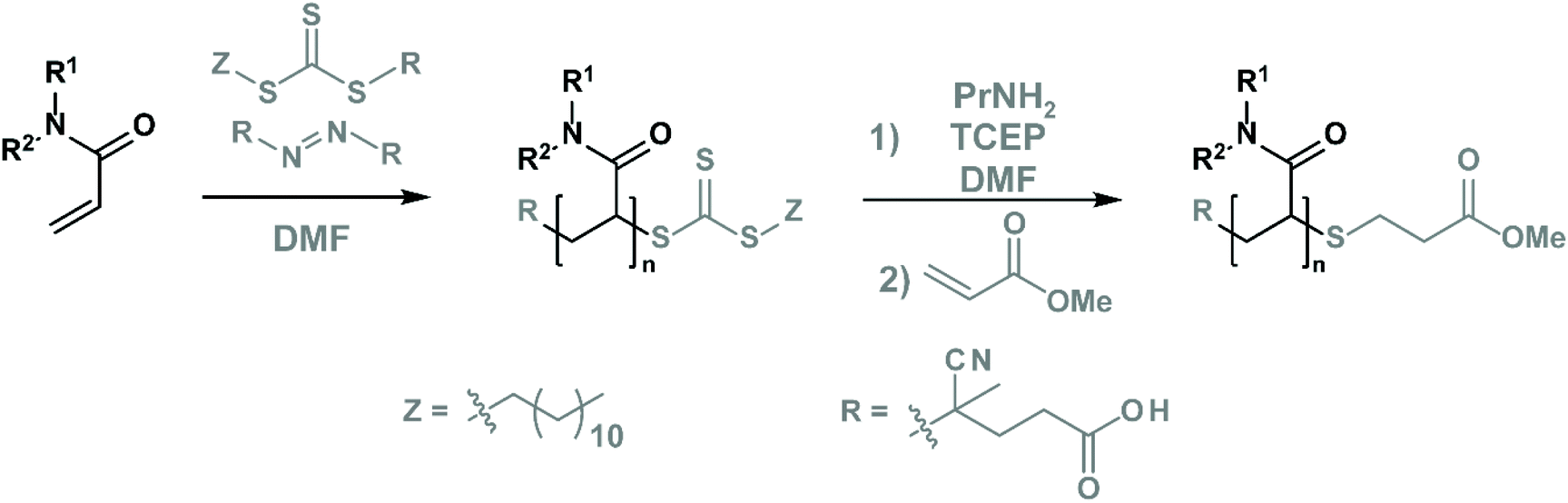 | ||
| Fig. 2 Polymer synthesis and subsequent modifications. | ||
| Polymer |
M
wa (kg mol−1) |
M
na (kg mol−1) |
Đ M | Yieldb (%) | dn/dcc (mL g−1) | |
|---|---|---|---|---|---|---|
| a Determined by SEC. b Polymerisation yield after the purification procedure based on monomer weight. c Determined by differential refractometry in PBS at 29 °C and 620 nm in PBS. | ||||||
| pDFEA | F1 | 26.2 | 24.2 | 1.08 | 68.3 | 0.088 ± 0.003 |
| F2 | 36.2 | 35.1 | 1.03 | 79.7 | 0.092 ± 0.005 | |
| F3 | 49.6 | 46.9 | 1.06 | 79.5 | 0.095 ± 0.004 | |
| pNIPAM | I1 | 20.2 | 19.6 | 1.03 | 50.6 | 0.167 ± 0.018 |
| I2 | 31.6 | 30.8 | 1.03 | 58.1 | 0.151 ± 0.012 | |
| I3 | 48.4 | 45.2 | 1.07 | 55.6 | 0.143 ± 0.013 | |
| pDEA | E1 | 22.3 | 21.2 | 1.06 | 60.5 | 0.171 ± 0.004 |
| E2 | 34.7 | 31.7 | 1.09 | 61.3 | 0.176 ± 0.011 | |
| E3 | 41.3 | 37.5 | 1.10 | 57.5 | 0.145 ± 0.010 | |
| pAP | P1 | 19.6 | 17.6 | 1.11 | 86.0 | 0.192 ± 0.007 |
| P2 | 36.0 | 32.9 | 1.09 | 77.7 | 0.180 ± 0.015 | |
| P3 | 51.2 | 46.3 | 1.11 | 79.1 | 0.164 ± 0.007 | |
PBS and FBS affect the TCP of thermoresponsive polyacrylamide solutions
The TCPs of our thermoresponsive polymers are either known (for pNIPAM33–37,43,44,67,69 and pAP37) or expected (for pDFEA and pDEA) to be lower in physiologically relevant solutions (FBS and PBS) than in pure water because these buffered solutions have a higher pH and ion concentration (as discussed in the Introduction). However, these polymers also have terminal carboxylic moieties (pKa ≈ 4.8), and while they are almost exclusively dissociated at neutral pH, unbuffered water dissolves atmospheric carbon dioxide,52 lowering the pH to values near the pKa of carboxylic acid (Table S7†). As a result, the carboxylic groups will no longer be fully deprotonated, decreasing the hydrophilicity32,52 and TCP of these polymers.52 In contrast, both PBS and FBS (140 mM; pH = 7.4) reliably maintain the pH and osmotic pressure at physiological values.70,72
Considering these differences, we have compared the effect of different solvents (water, Dulbecco's PBS70 and FBS) on TCP (c = 10.0 mg mL−1; Fig. 3). In line with previous studies, most TCP values of our polymers were higher in pure water than in PBS33–37,40,43,44,67 and FBS33,43 because the ions and proteins of buffered solutions lower the TCP of these polymers. Since differences in TCPs between solvents may be significant for many applications, polymers should be tested in an environment as similar as possible to that of the intended application.
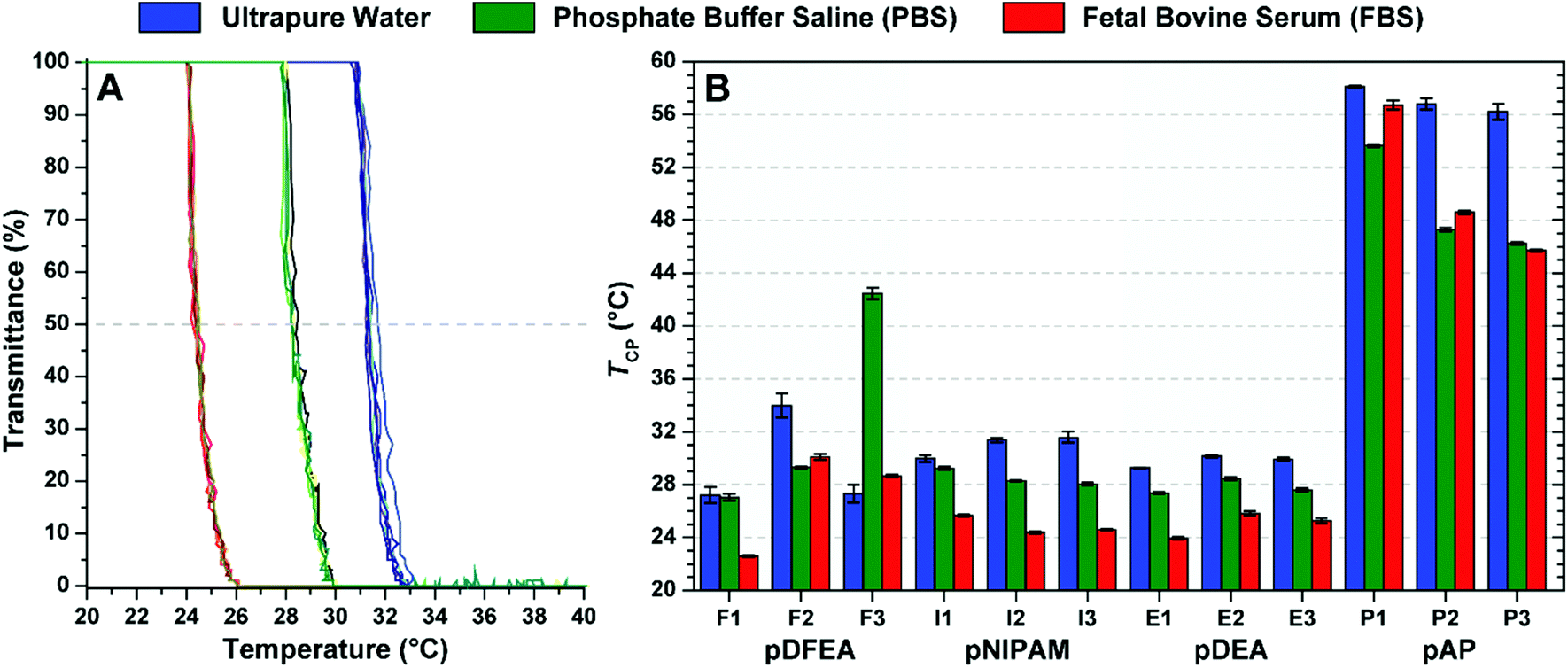 | ||
| Fig. 3 (A) Comparison of 6 turbidimetric measurements of I2 (pNIPAM) in water, PBS and FBS (c = 10.0 mg mL−1); (B) TCP of all polymers in water, PBS and FBS (c = 10.0 mg mL−1) expressed as a mean of 6 measurements ± standard deviation. The results indicate a shift in TCP. | ||
Thermoresponsive polymers may require DLS for accurate TCP determination
As shown in Fig. 3B, the TCP of pDFEA with the highest molar mass (F3) was much higher in PBS than in water. To understand the unexpected TCP of this polymer, as measured by turbidimetry, we assessed polymer aggregation as a function of temperature (10 to 50 °C) by DLS, in PBS, at a concentration of 10.0 mg mL−1 (Fig. S45–S56†). Unlike most samples, pDFEA polymers showed two major changes in population size with the increase in temperature (Fig. S45–S47†). The first change (at 26 to 30 °C) can be ascribed to unimer aggregation (radius 20 to 100 nm), and the second (from 40 to 50 °C) to aggregate coalescence into even larger polymer assemblies (radius 1000 nm or larger). Thus, turbidimetry detects the first thermal change in most samples, but only the second in F3.Long pDFEA (co)polymers, such as F3, may form non-typical nanogel-like aggregates with low polymer concentrations, as shown in our previous studies.63,64,79 In these particles, the aggregation causes only a minor local increase in polymer concentration. When combined with the low dn/dc of the solute (Table 1), this increase in polymer concentration accounts for the small difference in refractive indices between the phase-separated polymer and the bulk solution, which may prevent an accurate determination of TCP by turbidimetry.59 Upon further heating, these nanogel-like particles aggregate/coalesce, increasing the turbidity. Therefore, discrepancies in turbidimetric measurements may be explained by differences in the architecture of polymer aggregates and by the low dn/dc of pDFEA.
The effect of FBS proteins on TCP varies with polymer concentration
Proteins (in FBS) can affect the TCP of polymers indirectly (‘non-specifically’, i.e., by competing with polymers for its solvation as well as by excluded volume/crowding effect34,46) or directly (‘specifically’, i.e., by forming complexes with the polymers34,47).34 In turn, inorganic salts (in PBS and FBS) can also indirectly interfere with polymers by interacting with and destabilizing their solvation shell, thus decreasing their solubility and TCP (Hoffmeister effect), regardless of polymer concentration.33,40 Unlike inorganic salts, however, proteins affect TCP as a function of polymer concentration, as shown by our results (Fig. 4).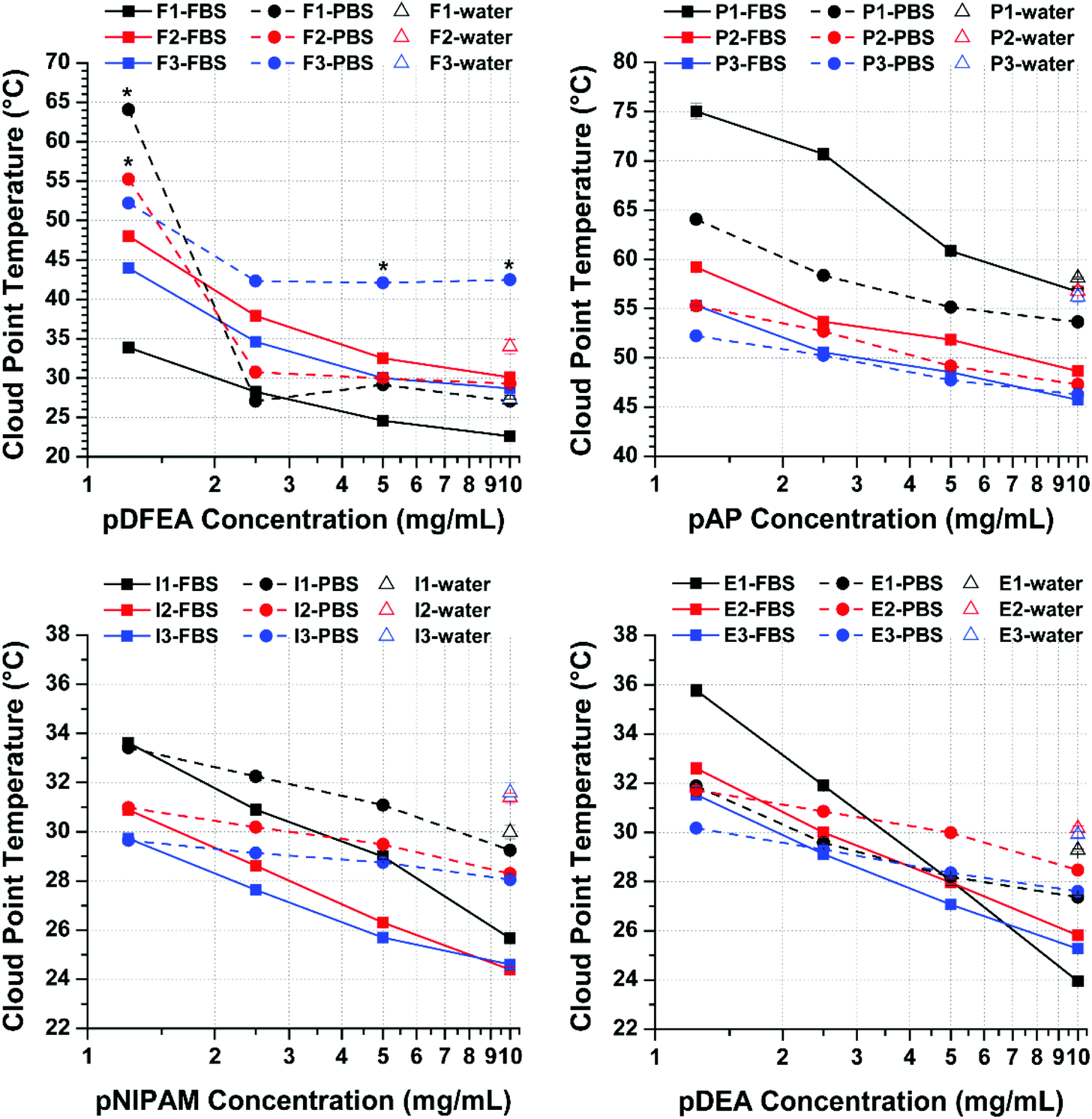 | ||
| Fig. 4 Plot of TCPs of the polymers pDFEA (top-left), pAP (top-right), pNIPAM (bottom-left) and pDEA (bottom-right) in FBS and PBS as a function of polymer concentrations. All TCPs are expressed as the mean of 6 measurement cycles ± standard deviation. Asterisks (*) indicate potential outliers. | ||
At low polymer concentrations (1.25 to 2.50 mg mL−1), polymers form soluble protein–polymer complexes (protein binding) in FBS, which prevent them from aggregating, thereby increasing their TCP above that of PBS solutions, as observed in pDEA and pNIPAM (at both low and high pAP concentrations). Because the polymer-binding capacity of proteins is limited (albeit very high for pAP), this effect is only detected at low polymer concentrations and decreases with the increase in polymer concentration (until being offset by ‘non-specific’ effects, which decrease the TCP).
At high polymer concentrations (5.00 to 10.0 mg mL−1), in contrast, proteins predominantly have a ‘non-specific’ effect by competing with polymer chains for solvation with the polymer33,34,40,41,80 (similarly to inorganic ions), thereby facilitating aggregation and lowering the TCPs. Proteins may also stabilize polymer aggregates via hydrophobic interactions, further lowering the TCPs. Under such conditions, most polymers in FBS have the lowest TCPs of all three media tested in this study (except for F3 in PBS, as discussed above).
For the purpose of this analysis (Fig. 4), we disregarded the transition temperatures of the highest polymer concentrations (20.0 and 40.0 mg mL−1) because they differed considerably between independent measurements (low reproducibility), not only in FBS but also in PBS. Nevertheless, the complete dataset is provided in Tables S3–S6.† Furthermore, in the range used in this analysis (1.25 to 10.0 mg mL−1), except for a few outliers, all three molar masses of each polymer showed similar TCP trends, that is, TCP decreased with the increase in polymer concentration, in line with previous studies.1,34–37
PBS and FBS differentially affect LCST polymer aggregation
LCST polymer aggregation is an entropy-driven endothermic process1,26,59 affected by surrounding ions/proteins. Adding these ions/proteins can induce conformational changes in the polymer (aggregation) or affect its solvation shell, both of which can be detected as heat effects.1,82–84 A decrease in enthalpy after adding ions/proteins indicates an enthalpic effect, i.e., polymers interact with ions/proteins (or new strong polymer–polymer interactions are formed). Conversely, an increase in enthalpy after adding these ions/proteins indicates an entropic effect, i.e., the loss of specific interactions, thereby increasing the entropy, e.g., due to the loss of the solvation shell.84Considering the above, we assessed the enthalpy of interaction of pDFEA, pNIPAM, pDEA, and pAP with PBS and FBS by isothermal titration calorimetry (ITC), which revealed a complex titration isotherm81 (Fig. 5B), with three phases: I, II and III (Fig. 5A). In phase I, the heat flux can be ascribed to the neutralization of the terminal carboxylic acid because adding 3 to 5 μL of PBS (pH = 7.41) or FBS (pH = 7.46) to the solution of benzoic acid had a similar effect on the heat flux (Fig. S41†). For this reason, the corresponding data points were excluded from the titration isotherms. Subsequent adding of titrant induced exo- or endothermic processes, which strongly depended on the titrant concentration (phase II on the titration isotherms). After the critical concentration of titrant (Fig. 5B), a non-zero heat flux, weakly dependent on the titrant, was still detected (phase III). Based on these results, we focused on the heat flux from phase II to analyse the solvent–polymer interactions.
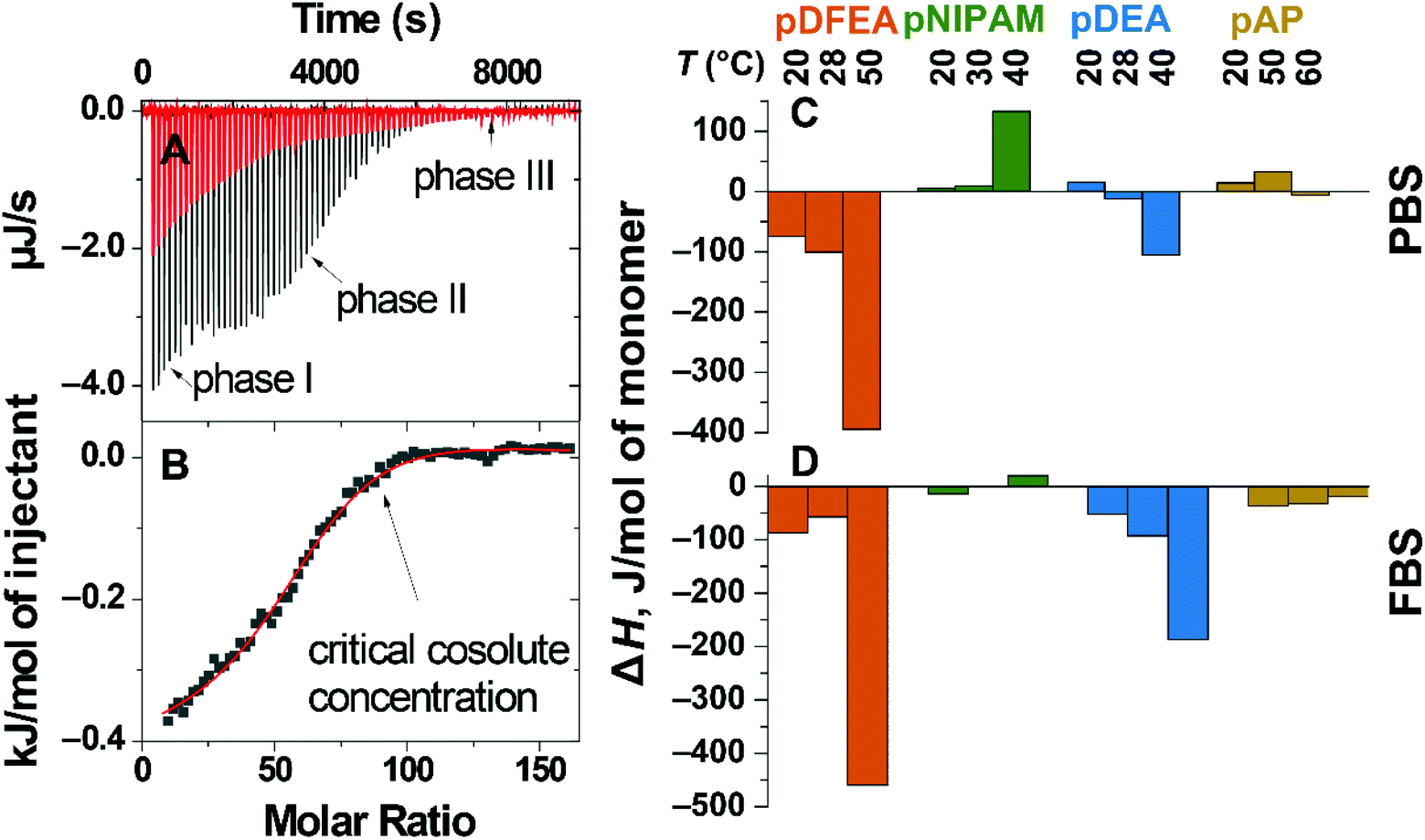 | ||
| Fig. 5 ITC of 10.0 mg mL−1F3 (pDFEA solution in ultrapure water with PBS: heat flux as a function of time, titration of PBS to water (blank, red line), titration of PBS to polymer (black line) (A); integrated heat, normalised to 1 mol of F3 injectant (scatter), fit to model (line) (B). Enthalpies of the titration of 10.0 mg mL−1 polymer solutions with PBS (C) and FBS (D), normalised to 1 mol of monomer units at three different temperatures. The values of enthalpies are outlined in Table S7.† | ||
PBS promotes aggregation in thermoresponsive polymers by decreasing their solvation shell
In non-aggregated polymers (at low temperatures), adding PBS to the solution had a positive enthalpic effect on pNIPAM, pAP and pDEA, as expected based on the Hoffmeister effect, but surprisingly had a significantly negative enthalpic effect on pDFEA. On the one hand, PBS decreases the solvation shell of all polymers (positive enthalpic effect). On the other hand, PBS increases polymer–polymer bonds in the aggregates (negative enthalpic effect). Since PBS had a net negative enthalpic effect on pDFEA, pDFEA must have strong intramolecular interactions (possibly due to hydrogen bonding between CF2H moieties74 – further investigated in chapter S7.4†).In aggregated polymers (at high temperatures), PBS had a negative enthalpic effect on pDFEA, pDEA and pAP, indicating that this buffer promotes the formation of enthalpically favourable polymer–polymer. However, adding PBS to pNIPAM had a positive enthalpic effect, suggesting further dehydration resulting from salting out. Overall, PBS disrupts the solvation shell of thermoresponsive polymers, thereby promoting aggregation.
FBS proteins stabilize LCST polymer aggregates
Adding FBS to both non-aggregated and aggregated polymers (at both low and high temperatures) had a more negative enthalpic effect than adding PBS due to the additional strong polymers–proteins interactions (Fig. 5). The amphiphilic proteins in FBS may bind to polymers and polymeric aggregates5,33via hydrophobic interactions3,5 and thus stabilize them. In all polymers, adding FBS to polymer aggregates had a stronger negative enthalpic effect than adding FBS to non-aggregating polymers, possibly because polymer aggregates are more prone to interact with proteins through hydrophobic interactions. Consequently, FBS stabilizes thermoresponsive polymer aggregates more strongly than PBS.Conclusions
The cloud point temperature (TCP) of thermoresponsive polyacrylamides is considerably lower in physiologically relevant solvents (PBS and FBS) than in water. In particular, FBS proteins stabilize LCST polymer aggregates, but the effect of FBS proteins on TCP depends on polymer concentration. At high polymer concentrations, proteins decrease the TCP by competing for solvation. At low polymer concentrations, by contrast, proteins form complexes with the polymers, thus increasing aggregation. However, proteins have limited polymer-binding capacities, so their effect decreases with the increase in concentration. In turn, PBS promotes aggregation in thermoresponsive polymers by decreasing their solvation shell. Overall, our results suggest that thermoresponsive polymers with a high potential for biomedical applications should be characterised (i) by DLS for accurate TCP determination and (ii) in physiologically relevant solutions rather than in pure water because they may be otherwise discarded merely for their unsuitable LCST behaviour in water. Moreover, our findings may enable us to better predict the biological properties of thermoresponsive polymers in vivo based on their TCP. Therefore, these results may be used to optimise polymeric drug delivery systems for in vivo applications through in vitro studies.Conflicts of interest
The authors have no conflicts of interests to declare.Acknowledgements
The authors thank Dr Carlos V. Melo for editing the manuscript and reviewing the content. The authors gratefully acknowledge the funding from the Grant Agency of Charles University, GA UK (project no. 379321). The authors gratefully acknowledge financial support from the Czech Science Foundation (GAČR; grant # 19-01602S) and from the Technology Agency of the Czech Republic (TAČR; grant # KAPPA TO01000074 ELECTTRA). R. H. acknowledges continuous support from FWO and Ghent University. Collaboration of M. H. and R. H. was supported by the bilateral CAS plus FWO project (grant # FWO-19-03).References
- V. Aseyev, H. Tenhu and F. M. Winnik, in Self Organized Nanostructures of Amphiphilic Block Copolymers II, ed. A. H. E. Müller and O. Borisov, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, vol. 242, pp. 29–89 Search PubMed.
- L. Yang, X. Fan, J. Zhang and J. Ju, Polymers, 2020, 12, 389 CrossRef CAS PubMed.
- N. Yamada, T. Okano, H. Sakai, F. Karikusa, Y. Sawasaki and Y. Sakurai, Makromol. Chem., Rapid Commun., 1990, 11, 571–576 CrossRef CAS.
- B. L. Ekerdt, C. M. Fuentes, Y. Lei, M. M. Adil, A. Ramasubramanian, R. A. Segalman and D. V. Schaffer, Adv. Healthcare Mater., 2018, 7, 1800225 CrossRef PubMed.
- T. Okano, N. Yamada, M. Okuhara, H. Sakai and Y. Sakurai, Biomaterials, 1995, 16, 7 CrossRef.
- K. Kolouchova, O. Groborz, P. Svec, Z. Starcuk, M. Slouf and M. Hruby, Biomacromolecules, 2021, 22, 2325–2337 CrossRef CAS PubMed.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS.
- A. Hatefi and B. Amsden, J. Controlled Release, 2002, 80, 9–28 CrossRef CAS.
- K. Kolouchova, D. Jirak, O. Groborz, O. Sedlacek, N. Ziolkowska, M. Vit, E. Sticova, A. Galisova, P. Svec, J. Trousil, M. Hajek and M. Hruby, J. Controlled Release, 2020, 327, 50–60 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS.
- E. Ruel-Gariépy and J.-C. Leroux, Eur. J. Pharm. Biopharm., 2004, 58, 409–426 CrossRef.
- A. M. Alhoranta, J. K. Lehtinen, A. O. Urtti, S. J. Butcher, V. O. Aseyev and H. J. Tenhu, Biomacromolecules, 2011, 12, 3213–3222 CrossRef CAS PubMed.
- M. D. Lavigne, S. S. Pennadam, J. Ellis, L. L. Yates, C. Alexander and D. C. Górecki, J. Gene Med., 2007, 9, 44–54 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- F. Doberenz, K. Zeng, C. Willems, K. Zhang and T. Groth, J. Mater. Chem. B, 2020, 8, 607–628 RSC.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS.
- D.-Q. Wu, J. Zhu, H. Han, J.-Z. Zhang, F.-F. Wu, X.-H. Qin and J.-Y. Yu, Acta Biomater., 2018, 65, 305–316 CrossRef CAS PubMed.
- I. Elloumi-Hannachi, M. Yamato and T. Okano, J. Intern. Med., 2010, 267, 54–70 CrossRef CAS PubMed.
- Y. Wei, Q. Zeng, Q. Hu, M. Wang, J. Tao and L. Wang, Biosens. Bioelectron., 2018, 99, 136–141 CrossRef CAS PubMed.
- N. Pacifici, A. Bolandparvaz and J. S. Lewis, Adv. Ther., 2020, 3, 2000129 CrossRef PubMed.
- O. Sedláček, P. Černoch, J. Kučka, R. Konefal, P. Štěpánek, M. Vetrík, T. P. Lodge and M. Hrubý, Langmuir, 2016, 32, 6115–6122 CrossRef.
- M. Hrubý, J. Kucka, O. Lebeda, H. Mackova, M. Babič, Č. Koňák, M. Studenovský, A. Síkora, J. Kozempel and K. Ulbrich, J. Controlled Release, 2007, 119, 25–33 CrossRef PubMed.
- E. Ayano and H. Kanazawa, J. Sep. Sci., 2006, 29, 738–749 CrossRef CAS.
- D. E. Bergbreiter, Chem. Rev., 2002, 102, 3345–3384 CrossRef CAS PubMed.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794–799 RSC.
- D. G. Lessard, M. Ousalem and X. X. Zhu, Can. J. Chem., 2001, 79, 1870–1874 CrossRef CAS.
- Q. Li, A. P. Constantinou and T. K. Georgiou, J. Polym. Sci., 2021, 59, 230–239 CrossRef CAS.
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
- Y. Pei, J. Chen, L. Yang, L. Shi, Q. Tao, B. Hui and J. Li, J. Biomater. Sci., Polym. Ed., 2004, 15, 585–594 CrossRef CAS PubMed.
- C. Pietsch, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 5653–5656 CrossRef CAS PubMed.
- Y. Hiruta, Y. Nagumo, Y. Suzuki, T. Funatsu, Y. Ishikawa and H. Kanazawa, Colloids Surf., B, 2015, 132, 299–304 CrossRef CAS PubMed.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS.
- R. Freitag and F. Garret-Flaudy, Langmuir, 2002, 18, 3434–3440 CrossRef CAS.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS PubMed.
- K. Skrabania, J. Kristen, A. Laschewsky, O. Akdemir, A. Hoth and J.-F. Lutz, Langmuir, 2007, 23, 84–93 CrossRef CAS.
- H. L. Judah, P. Liu, A. Zarbakhsh and M. Resmini, Polymers, 2020, 12, 2590 CrossRef CAS.
- Ł. Otulakowski, M. Kasprów, A. Strzelecka, A. Dworak and B. Trzebicka, Polymers, 2021, 13, 90 CrossRef PubMed.
- M. M. Bloksma, D. J. Bakker, C. Weber, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 724–728 CrossRef CAS PubMed.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS PubMed.
- X. Zhu, X. Gu, L. Zhang and X.-Z. Kong, Nanoscale Res. Lett., 2012, 7, 519 CrossRef PubMed.
- T. Trongsatitkul and B. M. Budhlall, Colloids Surf., B, 2013, 103, 244–252 CrossRef CAS PubMed.
- K. Bebis, M. W. Jones, D. M. Haddleton and M. I. Gibson, Polym. Chem., 2011, 2, 975–982 RSC.
- Y. Okada and F. Tanaka, Macromolecules, 2005, 38, 4465–4471 CrossRef CAS.
- R. J. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS.
- S. Suzuki, T. Sawada, T. Ishizone and T. Serizawa, Chem. Commun., 2016, 52, 5670–5673 RSC.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- B. Ray, Y. Okamoto, M. Kamigaito, M. Sawamoto, K. Seno, S. Kanaoka and S. Aoshima, Polym. J., 2005, 37, 234–237 CrossRef CAS.
- M. Hrubý, V. Šubr, J. Kučka, J. Kozempel, O. Lebeda and A. Sikora, Appl. Radiat. Isot., 2005, 63, 423–431 CrossRef.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- G. Cao, G. Li, Q. Yang, Z. Liu, Z. Liu and J. Jiang, Macromol. Rapid Commun., 2018, 39, 1700684 CrossRef PubMed.
- C. Panayiotou and I. C. Sanchez, J. Phys. Chem., 1991, 95, 10090–10097 CrossRef CAS.
- F. Afroze, E. Nies and H. Berghmans, J. Mol. Struct., 2000, 554, 55–68 CrossRef CAS.
- L. P. N. Rebelo, Z. P. Visak, H. C. de Sousa, J. Szydlowski, R. Gomes de Azevedo, A. M. Ramos, V. Najdanovic-Visak, M. Nunes da Ponte and J. Klein, Macromolecules, 2002, 35, 1887–1895 CrossRef CAS.
- C. Zhao, Z. Ma and X. X. Zhu, Prog. Polym. Sci., 2019, 90, 269–291 CrossRef CAS.
- J. Heyda and J. Dzubiella, J. Phys. Chem. B, 2014, 118, 10979–10988 CrossRef CAS PubMed.
- B. H. Chang and Y. C. Bae, Polymer, 1998, 39, 6449–6454 CrossRef CAS.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- D. G. Lessard, M. Ousalem, X. X. Zhu, A. Eisenberg and P. J. Carreau, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 1627–1637 CrossRef CAS.
- M. Philipp, R. Aleksandrova, U. Müller, M. Ostermeyer, R. Sanctuary, P. Müller-Buschbaum and J. K. Krüger, Soft Matter, 2014, 10, 7297–7305 RSC.
- F. Meersman, J. Wang, Y. Wu and K. Heremans, Macromolecules, 2005, 38, 8923–8928 CrossRef CAS.
- D. Babuka, K. Kolouchová, O. Groborz, Z. Tošner, A. Zhigunov, P. Štěpánek and M. Hrubý, Nanomaterials, 2020, 10, 2231 CrossRef CAS PubMed.
- D. Babuka, K. Kolouchova, M. Hruby, O. Groborz, Z. Tosner, A. Zhigunov and P. Stepanek, Eur. Polym. J., 2019, 121, 109306 CrossRef CAS.
- P. Štěpánek, in Dynamic Light Scattering, ed. W. Brown, Clarendon Press, Oxford, 1993, p. 760 Search PubMed.
- C. Boutris, E. G. Chatzi and C. Kiparissides, Polymer, 1997, 38, 2567–2570 CrossRef CAS.
- H. G. Schild and D. A. Tirrell, J. Phys. Chem., 1990, 94, 4352–4356 CrossRef CAS.
- K. Van Durme, G. Van Assche and B. Van Mele, Macromolecules, 2004, 37, 9596–9605 CrossRef CAS.
- K. Otake, H. Inomata, M. Konno and S. Saito, Macromolecules, 1990, 23, 283–289 CrossRef CAS.
- R. Dulbecco and M. Vogt, J. Exp. Med., 1954, 99, 167–182 CrossRef CAS.
- T. Yao and Y. Asayama, Reprod. Med. Biol., 2017, 16, 99–117 CrossRef PubMed.
- J. van der Valk, K. Bieback, C. Buta, B. Cochrane, W. G. Dirks, J. Fu, J. J. Hickman, C. Hohensee, R. Kolar, M. Liebsch, F. Pistollato, M. Schulz, D. Thieme, T. Weber, J. Wiest, S. Winkler and G. Gstraunthaler, ALTEX – Altern. Anim. Exp., 2018, 35, 99–118 Search PubMed.
- J. M. Bak and H. Lee, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1976–1982 CrossRef CAS.
- Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef CAS PubMed.
- B. D. Monnery and R. Hoogenboom, Polym. Chem., 2019, 10, 3480–3487 RSC.
- C. Zhou, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2011, 44, 1635–1641 CrossRef CAS.
- N. Bertrand, J. G. Fleischer, K. M. Wasan and J.-C. Leroux, Biomaterials, 2009, 30, 2598–2605 CrossRef CAS.
- L. Wyffels, T. Verbrugghen, B. D. Monnery, M. Glassner, S. Stroobants, R. Hoogenboom and S. Staelens, J. Controlled Release, 2016, 235, 63–71 CrossRef CAS PubMed.
- K. Kolouchova, O. Sedlacek, D. Jirak, D. Babuka, J. Blahut, J. Kotek, M. Vit, J. Trousil, R. Konefał, O. Janouskova, B. Podhorska, M. Slouf and M. Hruby, Biomacromolecules, 2018, 19, 3515–3524 CrossRef PubMed.
- H. I. Okur, J. Hladílková, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2017, 121, 1997–2014 CrossRef CAS PubMed.
- T. Wiseman, S. Williston, J. F. Brandts and L.-N. Lin, Anal. Biochem., 1989, 179, 131–137 CrossRef CAS.
- P. D. Ross and M. V. Rekharsky, Biophys. J., 1996, 71, 2144–2154 CrossRef CAS PubMed.
- J. M. Pestman, J. Kevelam, M. J. Blandamer, H. A. van Doren, R. M. Kellogg and J. B. F. N. Engberts, Langmuir, 1999, 15, 2009–2014 CrossRef CAS.
- G. Némethy, Angew. Chem., Int. Ed. Engl., 1967, 6, 195–206 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Material section, experimental & instrumental section (detailed description of monomer and polymer syntheses); processed NMR spectra (1H, 13C, 19F, 1H–13C HSQC), size exclusion chromatograms (SEC traces) of the study polymers; raw and processed turbidimetric data, raw and processed isothermal calorimetry data, dynamic light scattering data, refractive index increment (dn/dc), additional information, list of abbreviations and symbols used in this article, and author contributions (CRediT). See DOI: 10.1039/d1py00843a |
| This journal is © The Royal Society of Chemistry 2021 |