
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into auto-S-fatty acylation: targets, druggability, and inhibitors
Lu
Hu
*,
Zhipeng
Tao
and
Xu
Wu
 *
*
Cutaneous Biology Research Center, Massachusetts General Hospital, Harvard Medical School, 149, 13th St., Charlestown, MA 02129, USA. E-mail: LHU8@mgh.harvard.edu; XWU@CBRC2.mgh.harvard.edu
First published on 25th August 2021
Abstract
Posttranslational S-fatty acylation (or S-palmitoylation) modulates protein localization and functions, and has been implicated in neurological, metabolic, and infectious diseases, and cancers. Auto-S-fatty acylation involves reactive cysteine residues in the proteins which directly react with fatty acyl-CoA through thioester transfer reactions, and is the first step in some palmitoyl acyltransferase (PAT)-mediated catalysis reactions. In addition, many structural proteins, transcription factors and adaptor proteins might possess such “enzyme-like” activities and undergo auto-S-fatty acylation upon fatty acyl-CoA binding. Auto-S-fatty acylated proteins represent a new class of potential drug targets, which often harbor lipid-binding hydrophobic pockets and reactive cysteine residues, providing potential binding sites for covalent and non-covalent modulators. Therefore, targeting auto-S-fatty acylation could be a promising avenue to pharmacologically intervene in important cellular signaling pathways. Here, we summarize the recent progress in understanding the regulation and functions of auto-S-fatty acylation in cell signaling and diseases. We highlight the druggability of auto-S-fatty acylated proteins, including PATs and other proteins, with potential in silico and rationalized drug design approaches. We also highlight structural analysis and examples of currently known small molecules targeting auto-S-fatty acylation, to gain insights into targeting this class of proteins, and to expand the “druggable” proteome.
Post-translational protein fatty acylation covalently attaches a fatty acyl group to the amino acid residues of the protein. While several amino acid residues, such as lysine (Lys), serine (Ser), and cysteine (Cys), could be modified, S-fatty acylation on the cysteine residue is a major form of modification. The labile thioester bond enables dynamic cycling of protein acylation and de-acylation. Among the fatty acyl groups utilized in the modifications, the C16 palmitoyl group is the most common one. Therefore, S-fatty acylation is often referred to as S-palmitoylation. However, C14 myristoyl, C18 stearoyl, and other unsaturated acyl groups, such as C18:1 oleoyl, are also found in protein S-fatty acylation. Therefore, S-fatty acylation is a more accurate terminology to describe such modification and will be used in this review. Traditionally, S-fatty acylation has been known to regulate membrane association and trafficking of proteins by increasing their hydrophobicity. In addition, it has been shown that attachment of fatty acyl groups could regulate protein stability, co-factor binding and other properties. Therefore, such modifications could spatially and temporally fine-tune protein location, trafficking, stability, protein–protein interactions and activities.1–4 Such unique features of S-fatty acylation make it indispensable and ubiquitous in highly orchestrated signaling networks.5,6
S-Fatty acylation could be achieved through enzymatic and non-enzymatic processes. The evolutionarily conserved Asp–His–His–Cys (DHHC) domain containing palmitoyl acyltransferases (PATs)7 has been shown to mediate enzymatic S-fatty acylation of many membrane-bound proteins. Non-enzymatic S-fatty acylation, termed as auto-S-fatty acylation, involves a nucleophilic reaction between a Cys thiol group and a thioester moiety of fatty acyl-CoA.8 Previously, it has been suggested that the non-enzymatic S-fatty acylation is less likely to occur spontaneously under cellular physiological conditions, owing to low concentrations of free fatty acyl-CoA.9 However, such reactivity could be enhanced by increased concentration of fatty acyl-CoA, which could fluctuate due to the metabolic status of the cells. In addition, it has been shown that auto-S-fatty acylation is involved as the first step of several PAT-mediated catalysis reactions,10 in which fatty acyl-CoA reacts with the active site cysteine residue of the enzyme to form a thioester adduct via nucleophilic addition. Therefore, the unique nucleophilic properties of the cysteine residue in the protein microenvironment could also enhance auto-S-fatty acylation. Indeed, we and others have demonstrated that some proteins could undergo auto-S-fatty acylation similar to the first step of several PAT catalysis reactions upon binding to fatty acyl-CoA. Thus, enzymatic and non-enzymatic S-fatty acylation seems like two connected rather than independent processes. Auto-S-fatty acylation could regulate diverse biological functions under normal physiological conditions, coupling intracellular fatty acyl-CoA concentrations to the regulation of protein functions.
The crystal structures of ZDHHC20 and TEADs bound to palmitate offer important structural and mechanistic insights into auto-S-fatty acylation.11–13 There is usually a hydrophobic cavity in the protein to accommodate lipid chain binding. In addition, a reactive Cys residue is located in close proximity to the carbonyl group of the fatty acyl moiety, allowing facile covalent adduct formation. Therefore, auto-S-fatty acylation is not a spontaneous and non-specific reaction, but rather a process regulated by the protein structures and intracellular fatty acyl-CoA concentrations. It is reasonable to speculate that the hydrophobic cavities, which might stabilize a protein–lipid complex, are widespread among other auto-S-fatty acylated proteins.14 Such hydrophobic pockets also render auto-S-fatty acylated proteins the structural basis for small molecule binding, providing a strong foundation for their druggability.
The public database SwissPalm predicts that roughly 10% of the human proteome are putative S-fatty acylated proteins.15 Recent evidence has well documented and linked the dysfunctions of protein S-fatty acylation with various diseases, including cancers16 and neurological diseases.17,18 Therefore, it is important to understand and develop therapeutic agents to effectively modulate protein S-fatty acylation. However, the “druggability” of auto-S-fatty acylation has long been overlooked. Even for enzymatic S-palmitoylation, targeting the auto-S-fatty acylation step of several PATs would be a promising avenue for pharmacological intervention of these enzymes, which remain difficult to target with specific inhibitors. In this review, we will focus on the biological significance of auto-S-fatty acylation in human diseases and discuss the rationale of using small molecules to modulate auto-S-fatty acylation.
Auto-S-fatty acylation of non-enzymatic proteins and PATs
Some proteins without known enzymatic functions have been shown to auto-S-fatty acylate themselves (Table 1). Bet3, an evolutionarily conserved protein involved in protein trafficking, has been reported to undergo auto-S-fatty acylation. Interestingly, recombinant mouse Bet3 is covalently bound with a palmitate when expressed and purified from E. coli,19 although no PAT has been known to be expressed in bacteria. In line with this finding, both human and yeast Bet3 proteins show significant [3H]-palmitate incorporation when incubated with [3H]-palmitoyl-CoA at physiological conditions in vitro.20 A recent palmitoylated proteome study in yeast has confirmed that S-palmitoylation of Bet3 is not affected by the deficiency of any ZDHHC.21 Taken together, these results strongly support that Bet3 could undergo auto-S-fatty acylation (or auto-S-palmitoylation). Auto-S-fatty acylation has also been reported in two classes of transcriptional factors, TEADs and RFXs. Incubation of recombinant TEAD2 YBD (YAP-binding domain) with palmitoyl-CoA in vitro leads to strong S-palmitoylation,13 which was also confirmed by mass spectrometry analysis. Similarly, recombinant RFX3 protein could be auto-S-fatty acylated in vitro, and prefers the C18 stearoyl group for the modification.22 These reactions happen at physiologically relevant fatty acyl-CoA concentrations and neutral pH, supporting that auto-S-fatty acylation could happen under normal physiological conditions. Overexpression of each of the ZDHHC family of PATs failed to significantly enhance their fatty acylation levels, confirming that TEADs and RFX3 are auto-S-fatty acylated proteins. In addition, in the crystal structures of TEADs, a deep lipid-binding pocket has been identified to accommodate the fatty acyl chain, and the fatty acylated cysteine residue is located right at the entrance to the pocket, supporting its reactivity towards palmitoyl-CoA.| Proteins | Assay | Other evidence | Comments | References |
|---|---|---|---|---|
| Bet 3 | In vitro S-palmitoylation | Palmitoylated when purified from a PAT-deficiency system | Strong evidence | 19–21 |
| Cell-based ZDHHCs screening | A hydrophobic pocket bound with palmitate in crystal structure | |||
| TEADs | In vitro S-palmitoylation | Palmitoylated when purified from a PAT-deficiency system | Strong evidence | 13 and 89 |
| Cell-based ZDHHCs screening | Mass spectrometry showed TEAD2 was modified by palmitoyl group in vitro | |||
| A hydrophobic pocket bound with palmitate in crystal structure | ||||
| RFX3 | In vitro fatty acylation | N/A | Strong evidence | 22 |
| Cell-based ZDHHCs screening | ||||
| SFV p62 | In vitro S-palmitoylation | N/A | High concentration of palmitoyl-CoA | 23 |
| Lack of cell-based PATs screening | ||||
| Rhodopsin | In vitro S-palmitoylation | N/A | High concentration of palmitoyl-CoA | 24 and 25 |
| Lack of cell-based PATs screening | ||||
| Contradictory in vitro results | ||||
| Myelin proteolipid protein | In vitro S-palmitoylation | N/A | High concentration of palmitoyl-CoA | 26 |
| Myelin Po Glycoprotein | Lack of cell-based PATs screening | 27 | ||
| Gα | 28 | |||
| Erf2P | In vitro S-palmitoylation | N/A | Consistent evidence | 29 and 31 |
| Akr1p | 30 | |||
| ZDHHC2 | In vitro S-palmitoylation | Palmitoylated ZDHHC2 and 3 transfer palmitoyl group to substrates in vitro | Strong evidence | 32 |
| ZDHHC3 | ||||
| ZDHHC20 | In vitro S-palmitoylation | A hydrophobic pocket bound with 2-bromopalmitate (2-BP) in crystal structure | Strong evidence | 11 |
| Zebrafish ZDHHC15 | A hydrophobic pocket bound palmitate in modelled crystal structure |
Although there are other proteins (Table 1) being reported as auto-S-fatty acylated, the evidence supporting such conclusions is relatively weak. Almost 40 years ago, the Schmidt group described that the Semliki Forest virus (SFV) glycoprotein precursor p62 is auto-S-fatty acylated in a cell-free system.23 Unfortunately, the lack of genetic evidence made the conclusion ambiguous. The GPCR protein rhodopsin has been shown to undergo S-palmitoylation without any enzyme in vitro.24 Interestingly, it was later reported that S-palmitoylation of rhodopsin could be dramatically enhanced by partially purified PAT (10 times higher than initial rate),25 which suggests that enzyme-mediated S-palmitoylation of rhodopsin also occurs. However, it is not clear whether the auto-S-fatty acylation and enzymatic S-fatty acylation occur at the same site or not. It is possible that they are responsible for different sites or they are complementary for the same site in vivo. In addition, it is possible that such a surface reactive cysteine could have low basal activity for “auto-S-fatty acylation”, but certain PATs could significantly enhance their S-fatty acylation. Several other proteins, including the myelin proteolipid protein,26 myelin P0-glycoprotein,27 and G protein alpha subunit (Gα),28 have also been reported to be auto-S-fatty acylated in vitro. However, the use of high palmitoyl-CoA concentrations and high pH values of the reaction buffer, as well as the lack of solid cellular functional evidence, makes such claims less convincing.
In addition to these non-enzymatic proteins, several PATs have been shown to be auto-S-acylated (Table 1), based on fatty acyl adduct formation on the active site cysteine residues. Almost two decades ago, Deschenes and Davis independently reported the identification of palmitoyl acyltransferases (PATs) in yeast, Erf2p/Erf4p and Akr1p respectively. The former is responsible for S-palmitoylation of Ras2,29 whereas the latter mediates Yck2p S-palmitoylation.30 These enzymes were also observed to be S-palmitoylated, suggesting the formation of fatty acyl–enzyme adduct in the reactions. Auto-S-fatty acylation (S-palmitoylation) of Erf2 has been demonstrated by Deschenes et al. by using the [3H]-palmitoyl-CoA radioactivity assay in purified recombinant protein.31 This is the earliest observation that some PATs might initiate their catalysis through auto-S-fatty acylation. A two-step mechanism was proposed to depict the catalytic process for Erf2. In the first step, a deprotonated cysteine thiol group in the DHHC-domain attacks palmitoyl-CoA to generate a palmitoyl-enzyme intermediate, which then transfers the fatty acyl group to the substrates.
With the advance of protein purification techniques, more PATs have been shown to be auto-S-fatty acylated in biochemical assays. The Linder group32 detected significant signals of S-palmitoylation of ZDHHC2 and ZDHHC3, when purified enzymes were incubated with only palmitoyl-CoA in vitro. Palmitoylated ZDHHC2 and ZDHHC3 immediately transfer the palmitoyl moiety to the corresponding substrates after mixing with substrates, supporting a two-step ping-pong mechanism. Recently, the Banerjee group showed that treatment of human ZDHHC20 and zebrafish ZDHHC15 with palmitoyl-CoA in vitro would release free CoA, suggesting that ZDHHC20 undergoes auto-S-fatty acylation in vitro.11 Auto-S-fatty acylated intermediates were not always detected for some PATs, and different models and assays could lead to confusing and contradicting results. For example, it has been shown that auto-S-fatty acylation of ZDHHC1733 was below the detection limit in gel electrophoresis analysis. However, mass spectrometry studies suggested that ZDHHC17 is S-palmitoylated at the active site.34 Taken together, it has been confirmed biochemically that ZDHHC2, ZDHHC3, ZDHHC15, and ZDHHC20 could undergo auto-S-fatty acylation as part of their catalytic processes. It is speculated that other ZDHHC proteins could also be auto-S-fatty acylated;10 however, more evidence is still needed to confirm that auto-S-fatty acylation is a conserved mechanism involved in all PATs-mediated catalysis.
Auto-S-fatty acylation in diseases
Auto-S-fatty acylation activities of PATs in diseases
The critical role of ZDHHCs in diseases has been extensively explored in cancers16 and neural diseases17,18 (Fig. 1). In addition, metabolic disorders, infectious diseases and inflammation35 (such as viral infection and sepsis) have also been linked to the deregulation of ZDHHCs. To date, some of the PATs have been shown to have deregulated auto-S-fatty acylation levels in diseases, which will affect their PAT activities and lead to substrate dysfunction (Fig. 1).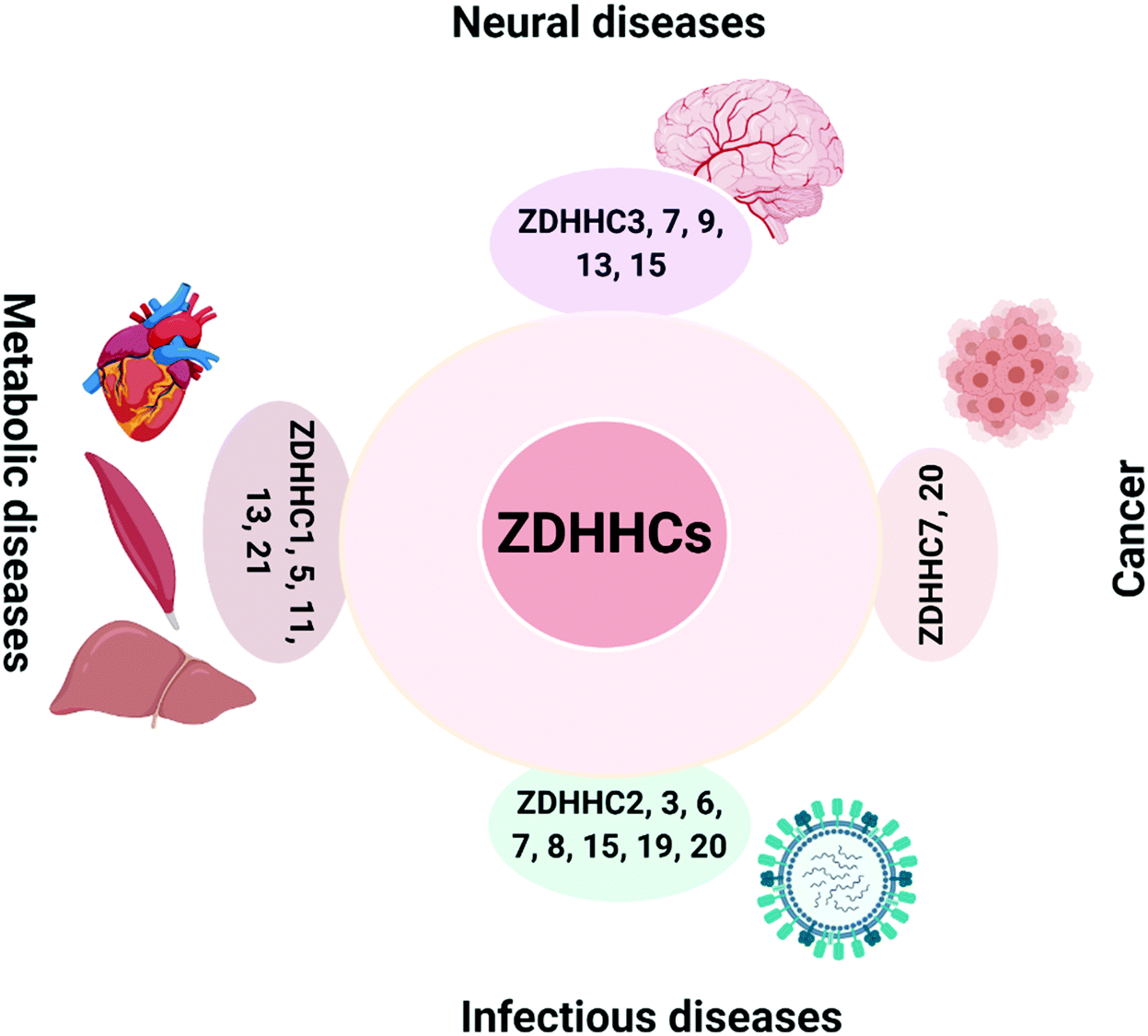 | ||
| Fig. 1 ZDHHC-family of PATs in various human diseases. PATs are involved in neurological diseases, cancers, infectious diseases and metabolic diseases. ZDHHC3,7, 9, 13 and 15 are involved in the pathogenesis of neurological diseases; ZDHHC7 and 20 are involved in cancers; ZDHHC1, 5, 11, 13 and 21 are involved in metabolic diseases; and ZDHHC2, 3, 6, 7, 8, 15, 19 and 20 are involved in infectious diseases. PATs, palmitoyl acyltransferases. The figure was created using BioRender.com. | ||
ZDHHC9 has been identified as a critical regulator of neurological functions, and ZDHHC9 deficiency in hippocampal cultures leads to shorter dendritic arbors and fewer inhibitory synapses.36,37 Indeed, ZDHHC9 could activate the ERK pathway through mediating Ras S-palmitoylation.38 Palmitoylated Ras locates to the plasma membrane and activates downstream MAPK signaling, leading to dendritic growth. On the other hand, ZDHHC9 also fine-tunes inhibitory synapse formation by palmitoylating GTPase TC10 which is required for its localization at the plasma membrane and then motivating gephyrin clustering, and inhibitory synapse formation.38 Thus, ZDHHC9 balances the excitatory and inhibitory signaling in neurological networks. Its loss-of-function mutations are implicated in neurology related diseases, such as X-linked intellectual disability (XLID) and epilepsy. These mutations impair its PAT enzyme activity by affecting its auto-S-fatty acylation levels, highlighting the importance of auto-S-fatty acylation in ZDHHC9 functions.39 Consistently, disruption of zdhhc9 in C57BL/6 mice leads to neurological impairment.40
ZDHHC13 has been shown to play a critical role in maintaining anxiety-related behaviors, motor function, and brain bioenergetics,41 which is associated with a nonsense mutation in zdhhc13. In mice, this nonsense mutation results in a premature stop codon (L203X) and a truncated ZDHHC13 without the DHQC domain (generally shown as classic DHHC in PATs).42 Consistently, DHQC deletion abolishes its auto-S-fatty acylation activity, thereby exhibiting a loss of PAT activity. Such loss-of-function mutations of ZDHHC13 lead to hypo-S-palmitoylation of many substrates, including Drp1, resulting in the dysfunction of mitochondria morphology and distribution. In addition, dysfunction of ZDHHC13 has also been linked to metabolic and degenerative diseases. For example, zdhhc13 with R425X mutation results in amyloidosis, alopecia, and osteoporosis in mice, which is probably associated with the loss of its PAT enzyme function, as it is predicted that R425X mutation leads to a truncated protein lacking active site cysteine (C456).43 Mechanistic studies showed that loss of ZDHHC13-mediated MT1-MMP S-palmitoylation was implicated in human osteoporosis.44 ZDHHC13 (R425X) mutation leads to decreased S-palmitoylation of MT1-MMP and impaired association with VEGF and osteocalcin expression in chondrocytes and osteoblasts. In mouse hepatocytes, decreased ZDHHC13 auto-S-fatty acylation leads to liver dysfunctions, lipid abnormalities, and hypermetabolism.45
ZDHHC3 and ZDHHC7 have been identified as palmitoylating enzymes for the neural cell adhesion molecule (NCAM).46S-palmitoylation of NCAM is required for its translocation to lipid rafts, where NCAM interacts with focal adhesion kinase (FAK) and fyn kinase to transiently activate MAPK/ERK signaling, thereby modulating fibroblast growth factor 2 (FGF2)-mediated neurite outgrowth, synaptogenesis, and synaptic plasticity. Interestingly, the FGF receptor (FGFR) and Src kinases phosphorylate ZDHHC3, and phosphorylation suppresses ZDHHC3 auto-S-fatty acylation and subsequent S-palmitoylation of NCAM,47 revealing a potential feedback regulation of PAT activity. Caspase-6 (CASP6) plays a critical role in neurodegenerative diseases, such as Alzheimer's and Huntington's diseases.48 Caspase-6 (CASP6) cleaves its substrates, amyloid precursor protein (APP) and the microtubule-associated protein tau, which may destabilize the microtubule and result in tangle formation. Therefore, activation of CASP6 has been implicated in neurodegeneration, while its inhibition restores neuronal functions. Palmitoyl acyltransferase ZDHHC17 (also known as HIP14, Huntingtin Interacting Protein 14) inhibits CASP6 activity through its S-palmitoylation, leading to the inhibition of CASP6 dimerization by sterically blocking the substrate-binding groove.49 Consistently, HIP14-deficiency leads to depalmitoylation and activation of CASP6, which is involved in the development of Huntington's disease (HD).50 In addition, decreased auto-S-fatty acylation levels of HIP14/ZDHHC17 are linked to its decreased PAT activity and the HD phenotype in YAC128 mice.50
Other members of ZDHHC families are undoubtedly implicated in various diseases, such as ZDHHC15 in neurodegenerative diseases,51 ZDHHC7 and ZDHHC20 in cancer,52,53 ZDHHC21 and ZDHHC1/11 in metabolic diseases,54,55 ZDHHC2/19 and ZDHHC20 in infectious diseases.56,57 However, whether deregulation of their auto-S-fatty acylation levels are linked to diseases would require further studies to elucidate, and therefore, the pathological relevance will not be discussed here. For example, a non-functional ZDHHC21 mutant (F233Δ) with phenylalanine 233 deletion is linked to tachycardic and hypotensive phenotype in mice, which was caused by ZDHHC21 mislocalization.54 More in-depth discussions of the function of all PATs in diseases could be found in many recent reviews.58–63
Taken together, ZDHHCs are promising therapeutical targets for neurological, metabolic, viral infectious diseases and cancers (Fig. 1). Designing modulators to target the auto-S-fatty acylation step of some ZDHHCs might provide feasible strategies for drug development. However, there are still significant challenges to target these proteins, and no specific modulators have been reported to inhibit any of the ZDHHCs.
Auto-S-fatty acylation of non-enzymatic proteins in diseases
Recently, several non-enzymatic auto-S-fatty acylation of proteins have been characterized, revealing novel functions in regulating normal development and diseases. These proteins are emerging as potential new therapeutic targets, expanding the current knowledge of “druggable” proteome. Transcription factors TEA/TEF (transcriptional enhancer factor)-domain (TEADs) and their co-activators YAP/TAZ regulate transcriptional activity of the Hippo pathway to modulate cell fate, organ size, and tissue regeneration.13,64 Deregulation of the TEAD–YAP has been implicated in multiple cancers,65,66 including lung, colorectal, ovarian and liver cancer. We and others discovered that auto-S-fatty acylation of TEADs occurs at evolutionarily conserved cysteine residues, mimicking palmitoylating “enzyme-like” activities.13,64 Recently, Kim and Gumbiner reported that TEADs S-palmitoylation levels decrease with high cell density, through the regulation of fatty acid synthase (FASN) and de novo fatty acyl-CoA biosynthesis. In addition, TEAD S-fatty acylation levels are also regulated through depalmitoylation by APT2. Therefore, TEAD auto-S-fatty acylation is a regulated process influenced by upstream cellular signaling cues.67 Auto-S-fatty acylation of TEADs allosterically regulates its association with the binding partner YAP/TAZ and activates downstream target gene expressions, controlling transcriptional activities of the Hippo pathway. Therefore, targeting auto-S-fatty acylation of TEADs to block TEAD–YAP transcriptional activities will be a promising strategy in cancer therapy. Indeed, several reversible and irreversible inhibitors of TEAD have been reported, which inhibit TEAD transcription activities through inhibition of its auto-S-fatty acylation.The transcription factor Regulatory Factor X 3 (RFX3) plays a critical role in the regulation of cilia formation, which are conserved organelles on the surface of most vertebrate cells. Loss of cilia has been linked to many diseases termed ciliopathies,68 including diseases of pancreas development and diabetes. We recently identified RFX3 as auto-S-fatty acylated. With the C18 stearic acid preference, RFX3 forms a dimer upon S-fatty acylation to enhance downstream transcriptional activity.22,69S-fatty acylated RFX3 upregulates the expression of ciliary genes (Dync2li1, Dnaic1, and Dnali1). Ciliogenesis and cilia elongation are essential for many signaling pathways, including the Hedgehog (Hh) pathway. Therefore, auto-S-fatty acylation of RFX3 could be related to ciliopathies and Hh signaling deregulated diseases.
In conclusion, targeting these known non-enzymatic auto-S-fatty acylated proteins and discovering more proteins with such “enzyme-like” properties would broaden the spectrum of therapeutic applications.
Targeting auto-S-fatty acylation for drug discovery
Structural information of auto-S-fatty acylated proteins could provide valuable insights into how these proteins can be targeted using small molecule modulators. In 2006, the Heinemann group reported the crystal structure of recombinant human Bet3.14 Interestingly, Bet3 harbors a hydrophobic pocket in which a palmitate is bound and forms a covalent thioester bond with Cys68 (Fig. 2a). Similar covalent modification with palmitate was also observed in the structures of TEADs. In TEAD2 and TEAD3 YBD (YAP-binding domain) structures (Fig. 2b), a palmitate is bound in a highly conserved pocket similar to Bet3. Interestingly, the carboxyl group of palmitic acid does not form a covalent bond, but points directly toward the conserved cysteine residue in TEAD2 structures, which might be resulted from the cleavage of the labile thioester bond under relatively basic conditions during purification. Refining the previously reported TEAD1–YAP complex confirmed that palmitate is covalent linked to cysteine,13 possibly preserved by YAP–peptide binding that blocks the pocket opening.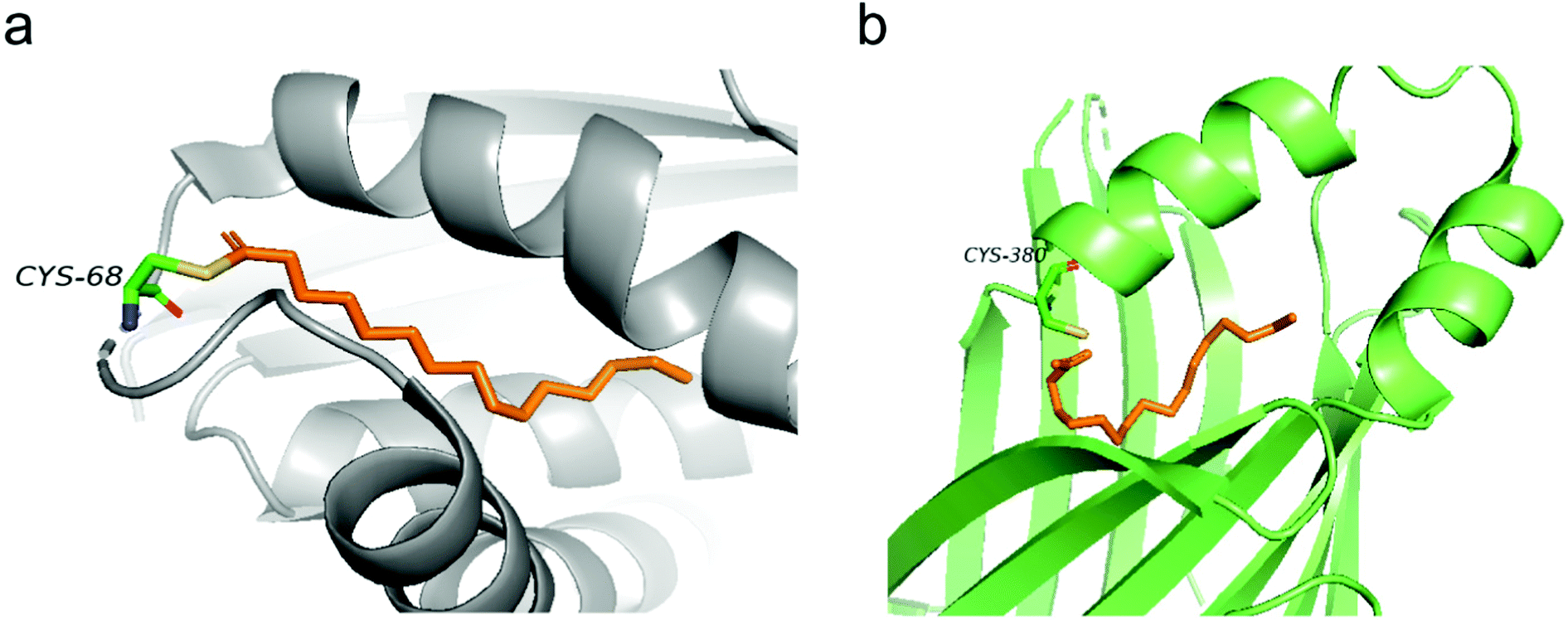 | ||
| Fig. 2 (a) Palmitate (orange) covalently attaches to C68 in the structure of human Bet3 (PDB code 1SZ7). (b) Crystal structure of human TEAD2 YBD bound with palmitate (orange) (PDB code 5HGU). | ||
Recent efforts in structural biology have revealed the high-resolution structures of human ZDHHC20 and zebrafish ZDHHS15, in which the active site cysteine was mutated to a serine.11 Both enzymes feature a teepee-like structure, harboring a hydrophobic cavity. Initially, weak electron density observed in the cavity of ZDHHC20 was attributed to a fatty acid, but it was not strong enough to be confirmed. The inhibitor 2-bromopalmitate (2-BP) was then utilized to co-purify human ZDHHC20, although it is non-specific to PATs. Structure of ZDHHC20 covalently bound to 2-BP revealed a hydrophobic cavity bound to lipid chains, as shown in the density map (Fig. 3a). In the structure of zfDHHS15, the density is strong enough to show the palmitate binding inside the cavity without using 2-BP (Fig. 3b).
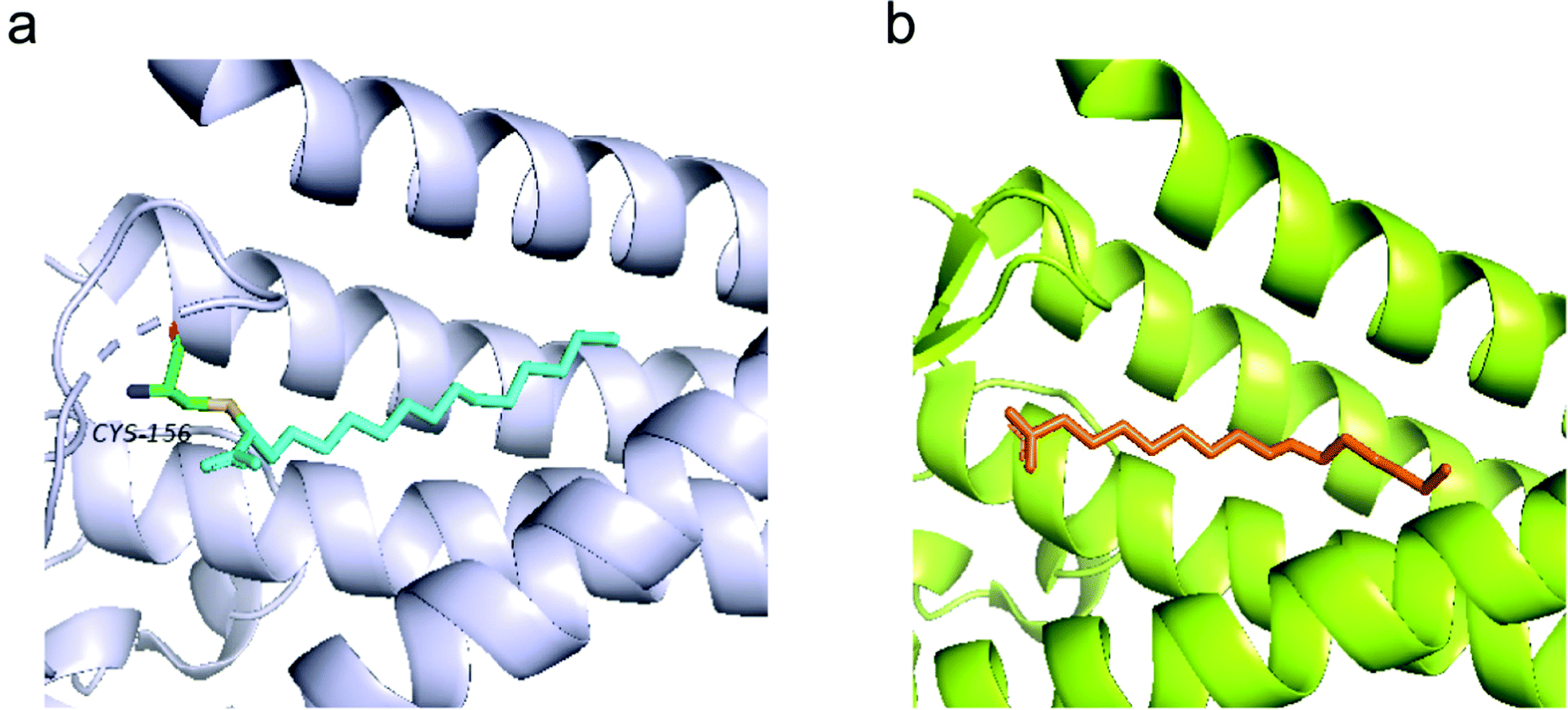 | ||
| Fig. 3 (a) Structure of human ZDHHC20 bound with 2-BP (cyan) (PDB code 6BML). (b) The modelled palmitic acid (orange) in the zfDHHS15 structure (PDB code 6BMS). | ||
In almost all of the reported structures of auto-S-fatty proteins, there is a common feature of harboring a hydrophobic cavity, potentially accommodating the lipid binding. This hydrophobic pocket anchors the lipid chain and positions the thioester bond of palmitoyl-CoA towards the active cysteine residue, facilitating the nucleophilic thioester exchange reaction. The microenvironment surrounding the active cysteine might also contribute to the reactivity by deprotonating the cysteine thiol group, and stabilizing the transition state intermediate, thereby facilitating the departure of a leaving group. Furthermore, it is speculated that such lipid-binding pocket might be conserved among other auto-S-fatty acylated proteins, providing a basis for potential small molecule targeting. A similar lipid-binding pocket also exists in Bcr–Abl, a fusion oncoprotein implicated in chronic myelogenous leukemia (CML).70N-Myristoylation and binding of myristoyl group in Abl kinase lead to auto-inhibition of the kinase activities. Recently, small molecules binding to this myristoylation pocket, an allosteric site distinct from the ATP pocket, have been shown to overcome the resistance of inhibitors targeting the ATP site. In addition, some other proteins, such as UNC11971 and NOTUM,72,73 could be also targeted through their lipid binding pockets. Taken together, these results strongly imply that a lipid-binding pocket can be “druggable”. There are still some concerns about selectivity issues due to highly conserved fatty acyl binding motifs, and substrate binding site inhibition has also been proposed for some acyltransferases.74 However, specific inhibitors targeting TEADs auto-S-fatty acylation suggest that selectivity could still be achieved.75
Strategies for targeting auto-S-fatty acylation
Inhibitors of ZDHHC family of PATs
It has been proposed that some PAT-mediated catalysis reactions require two steps, and it would be possible to have two strategies to modulate the activities of PATs (Fig. 4). One is to target the first auto-S-fatty acylation step, and the other focuses on the second step of acyl transfer to the substrates. For the second strategy, blocking acyl transfer could be achieved through inhibiting interaction between PATs and substrates. However, the substrate binding site of PATs is less defined for small molecule binding, and the presence of a hydrophobic cavity for lipid binding makes the former strategy more feasible. Although the hydrophobic pockets are conserved among different PATs, there are still significant differences of residues lining inside the pocket, suggesting that selectivity could be achieved. For example, at the end of the cavity of human ZDHHC20, Tyr181 interacts with Ser29 via a hydrogen bond to close off the cavity, whereas Ile182-Phe53 in ZDHHC3 and Ser185-Leu56 in ZDHHC7 act as homologous pair of Tyr181-Ser29 in hDHHC20. Similar to kinase-mediated phosphorylation, a common cofactor (ATP) might bind to pockets of similar shape in different proteins, but selectivity can still be successfully achieved. It is likely that selectivity could also be achieved for lipid-binding pockets of PATs and other auto-S-fatty acylated proteins. For example, TEAD inhibitor (MGH-CP1) does not inhibit the ZDHHC-family of proteins.75 Furthermore, the intrinsic active cysteine in this pocket makes it possible to design covalent modulators, which expands the space for medicinal chemistry manipulation.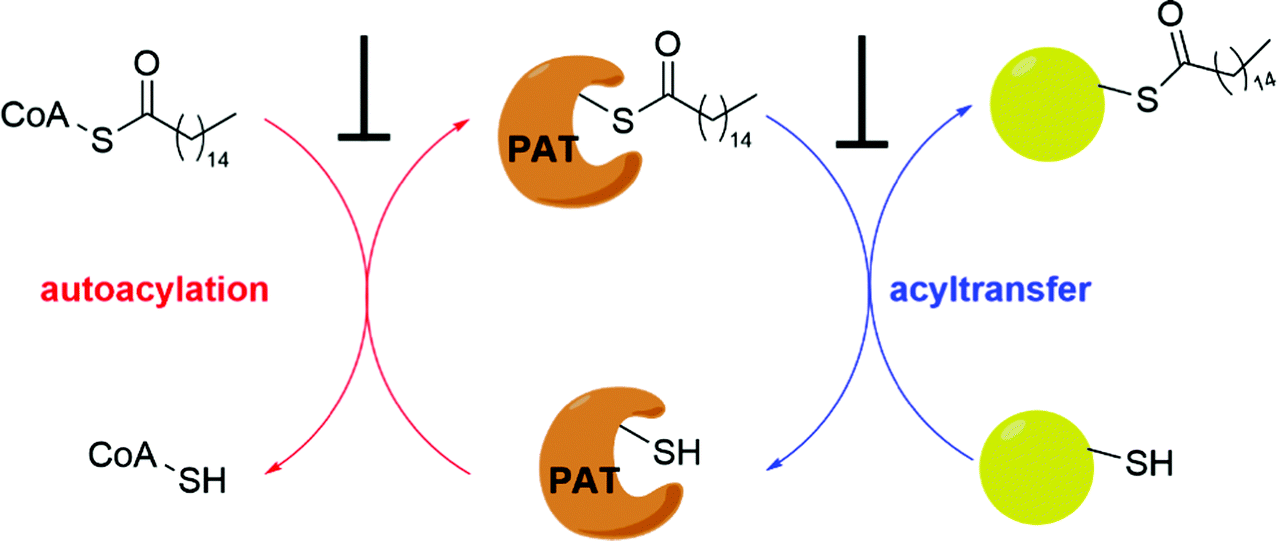 | ||
| Fig. 4 Strategies to inhibit PAT-mediated acylation. | ||
2-BP is a non-specific, pan-inhibitor for PATs auto-S-fatty acylation through formation of an irreversible adduct with PATs (Fig. 5a).76 Under cellular conditions, 2-BP is metabolically converted to 2-BP-CoA,77 which is the metabolic analog of palmitoyl-CoA. Owing to the good leaving capacity of α-bromo of 2-BP, the cysteine at the S-palmitoylation site then reacts with the α-bromo through a nucleophilic reaction, leading to irreversible inhibition of PATs. In addition, 2-BP might also block S-palmitoylation by affecting lipid metabolism to reduce cellular palmitoyl-CoA levels. Global profiling of cellular targets showed that 2-BP also targets other classes of proteins, such as transporter channels, enzymes, and chaperones,77 suggesting 2-BP could be promiscuous. Therefore, it is unsuitable to use 2-BP as an uncontroversial tool to target PATs. Other inhibitors bearing a lipid tail (Fig. 5a), such as cerulenin and tunicamycin, also inhibits activities of PATs in an irreversible manner. However, both cerulenin and tunicamycin have non-specific effects on other processes in cells, including fatty acid biosynthesis78 and N-glycosylation,79 respectively. Such a lack of specificity not only limits their therapeutic applications, but also makes them undesirable as chemical tools to interrogate biological functions of PATs-mediated fatty acylation.
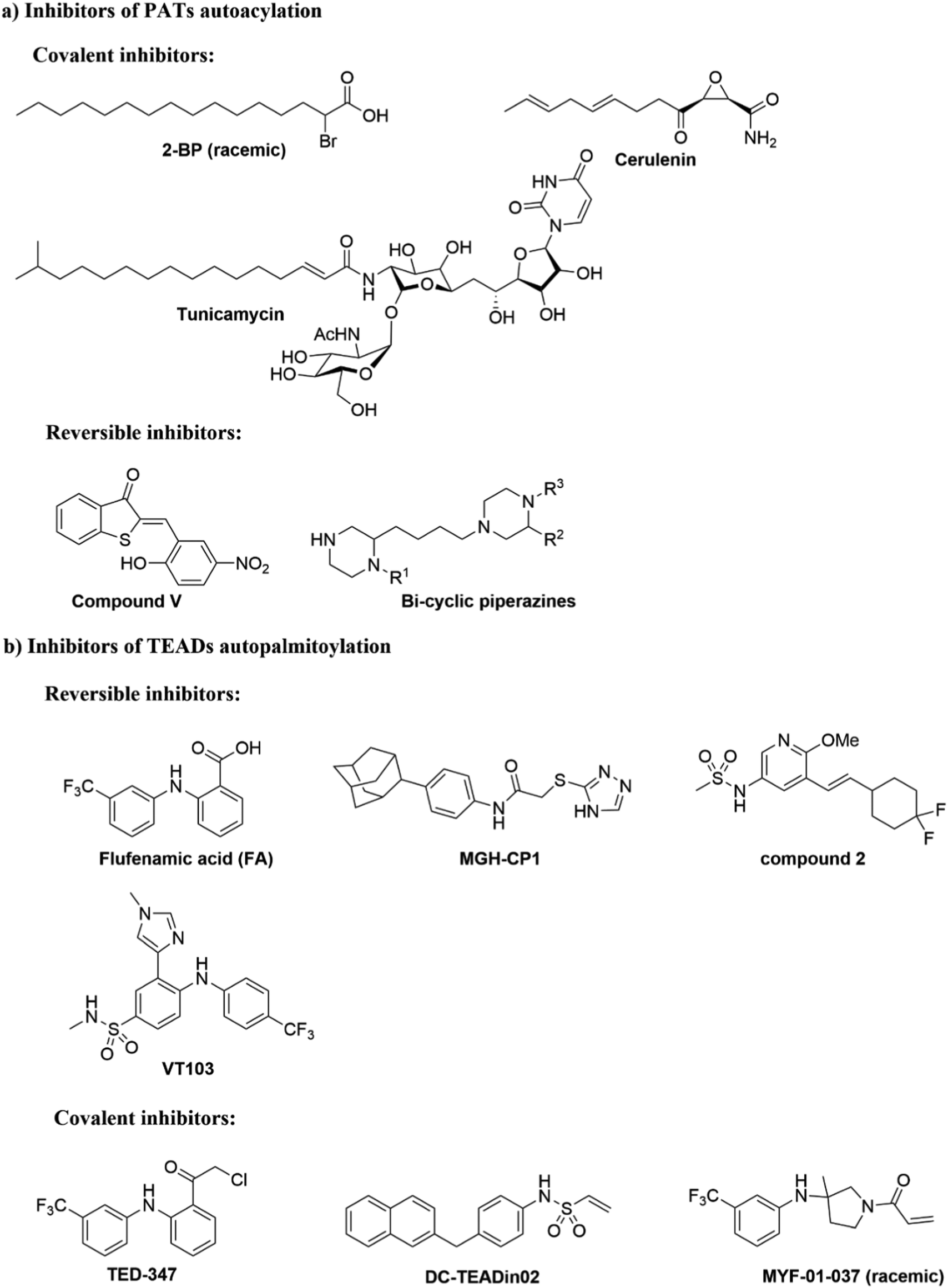 | ||
| Fig. 5 Small molecules targeting autoacylation. (a) Inhibitors of PAT autoacylation. (b) Selected inhibitors of TEAD auto-S-palmitoylation. | ||
Smith and co-workers developed cell-based screening assays to look for inhibitors of PATs.80 From a high throughput screening, they identified five compounds (I–V) as potential inhibitors of S-palmitoylation-related processes. Only compound V (Fig. 5a) was subsequently confirmed to suppress auto-S-fatty acylation of PATs in a reversible manner,81 which is validated with ZDHHC2 in vitro. However, compound V inhibits PAT activities of all four ZDHHCs tested, including yeast pfa3, yeast Erf2/Erf4, human ZDHHC2 and human ZDHHC9/GCP16. Despite these limitations, compound V might serve as a starting point for structure–activity relationship studies to develop more selective and potent PATs inhibitors, if its binding site on ZDHHCs can be determined. To further develop a more sensitive high throughput screening (HTS), Mitchell and co-workers exquisitely explored the detection of CoASH,82 which is released in the auto-S-fatty acylation step. The production of CoASH is monitored via a coupled fluorogenic assay catalyzed by α-ketoglutarate dehydrogenase complex (α-KDH), where α-ketoglutarate is converted to succinyl-CoA along with the formation of fluorescent NADH (Fig. 6). Notably, the coupled fluorogenic assay releases CO2, which makes the fluorescence irreversible. Using this fluorescence assay, Deschenes et al. identified a series of bis-cyclic piperazine-based compounds (Fig. 5a) that inhibit auto-S-fatty acylation of yeast Ras2 PAT, Erf2.83 A fluorescence auto-S-fatty acylation assay validated that the bicyclic compounds acts competitively with BODIPY-C12-CoA for Erf2, while 2-BP is an uncompetitive or mixed inhibitor in this case, implying that these compounds inhibit PAT auto-S-fatty acylation with a different mode-of-action with 2-BP. Some of these compounds indeed restricted the growth of the Ras2-dependent S. cerevisiae strain at the micromolar range. However, the selectivity against different PATs in cells requires further confirmation. Given that Erf2 is the homolog of ZDHHC9, such reports might provide useful information for the discovery of ZDHHC9 inhibitors in the future. While there are other S-palmitoylation inhibitors discovered recently, such as inhibitors of Ras84 and dual leucine-zipper kinase85 (DLK) S-palmitoylation, we will not discuss them in this part due to their obscure mode-of-action.
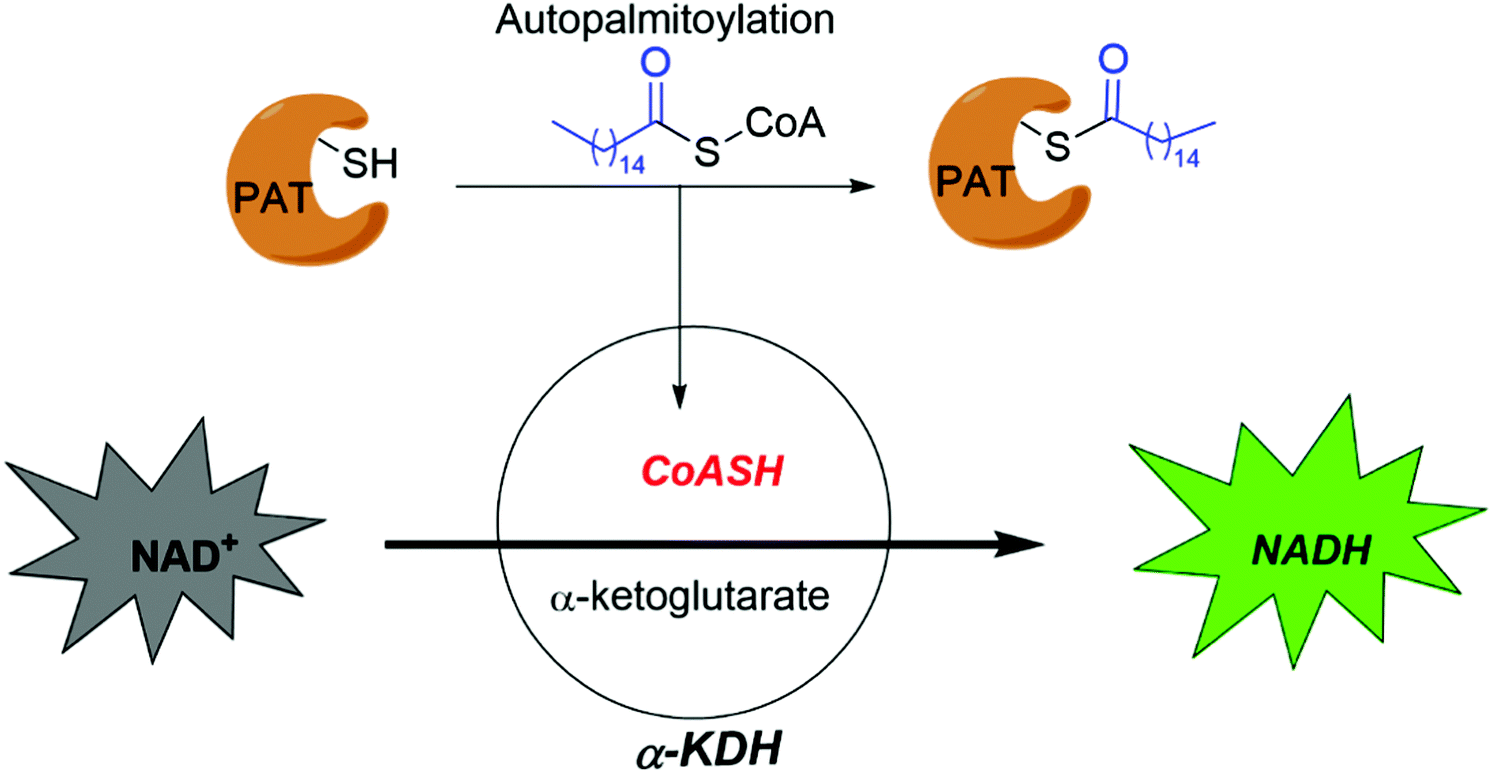 | ||
| Fig. 6 High throughput assay developed by Mitchell et al. | ||
Inhibitors of non-enzymatic auto-S-fatty acylation
The best-characterized example to target auto-S-fatty acylation of non-enzymatic proteins is the development of TEAD inhibitors (Fig. 5b). Deregulation of TEAD–YAP activity is involved in many human cancers.63,64 Developing inhibitors to disturb their interaction has been proposed as a promising strategy for drug discovery. However, given that the interface between TEAD and YAP is shallow and spans a large surface area, developing direct protein–protein interaction inhibitors might be difficult. The crystal structure of TEADs revealed that there is a highly conserved hydrophobic pocket near the active cysteine, where auto-S-fatty acylation occurs to stabilize TEAD structures and facilitate TEAD–YAP association. From a Pharmakon library screening, flufenamic acid (FA) (Fig. 5b) has been shown to bind to TEAD2 YBD.12 The crystal structure of TEAD2 YBD bound to FA indicated that FA primarily binds to the S-palmitoylation pocket, suggesting that this pocket is targetable. However, no significant inhibition of TEAD–YAP binding by FA was detected by Meroueh et al. using the fluorescence polarization (FP) assay.86 To improve the efficacy, they utilized the inherent active cysteine, which provides a strong foundation for covalent inhibitor discovery. In line with this hypothesis, the carboxylic group was replaced with an electrophilic chloromethyl ketone moiety as a warhead to irreversibly inhibit TEAD auto-S-fatty acylation by alkylating the active cysteine residue. TED-347 (Fig. 5b) was reported to form the expected covalent bond within palmitate-binding pocket. After treatment with TED-347 at 10 μM, viability of patient-derived glioblastoma cells, grown as three-dimensional spheroids, was inhibited by 30%. In contrast, more than 100 μM temozolomide, the standard of care for glioblastoma, was required to reach comparable efficiency.Similarly, the Luo group also succeeded by elaborately taking advantage of the active cysteine adjacent to the lipid binding pocket. Molecular docking-based virtual screening was first performed to characterize reversible binders, which are then modified with a reactive warhead. Finally, a series of vinylsulfonamide derivatives were developed to irreversibly inhibit TEAD auto-S-fatty acylation.87 DC-TEADin02 attenuated TEAD4 auto-S-fatty acylation with an EC50 of 197 ± 19 nM in vitro. Likewise, starting from FA, rational installation of a warhead by the Jänne and Gray groups leads to MYF-01-37 (Fig. 5b),88 a covalent inhibitor of TEAD. TEAD–YAP is highly activated in EGFR-mutant non-small cell lung cancer (NSCLC) treated with EGFR (Osimertinib) and MEK inhibitor (Selumetinib), leading to acquired drug resistance. Co-treatment of MYF-01-37, Osimertinib and Selumetinib significantly boosts apoptosis. Recently, we reported that MGH-CP1 (Fig. 5b),75 featured a phenylcarbamothioate scaffold, could reversibly and potently inhibit TEADs auto-S-fatty acylation in vitro with submicromolar IC50. Blockade of the TEAD–YAP complex by MGH-CP1 leads to inhibition of downstream target genes in cells. In an APC-mutant intestine, MGH-CP1 treatment synergized with Last1/2 deletion to efficiently suppress epithelial over-proliferation and induce apoptosis, providing a new approach to treat APC-mutant cancers.
Although inhibition of TEAD S-fatty acylation is thought to disrupt TEAD–YAP association, as demonstrated by small molecules mentioned above, the detailed mode-of-action at the molecular level has not been fully elucidated. Recently, compound 2 (Fig. 5b), discovered by researchers at Genentech, suppressed downstream transcriptional activities of TEADs.89 It is reported that compound 2 disrupts TEAD homeostasis, leading to inhibition of its binding to chromatin, thereby slowing down YAP-dependent tumor growth in a mouse xenograft model. Instead of blocking TEADs–YAP interactions, compound 2 surprisingly surrogates palmitic acid to stabilize the cellular level of exogenous TEADs in HEK293T cells. However, endogenous pan-TEAD levels in several cancer cells did not show significant change after treatment of compound 2. These contradictory results would need further studies to be clarified. In addition, the results showing the association of TEADs and chromatin using in vitro biochemical and cell-based ChIP-seq experiments are also inconsistent. Therefore, the detailed mechanisms of how compound 2 inhibits TEAD are still elusive.90 The therapeutic utilities of TEAD inhibitors are promising with accumulating evidence of TEAD inhibitors in cancer models, including VT103,91 which shows strong antitumor activity in NF2-deficient mesothelioma models in vivo.
Outlook
Non-enzymatic auto-S-fatty acylation has been largely overlooked over the years. It is understandable that enzyme-mediated process is more likely to be regulated and could be directly targeted for drug discovery.92 However, emerging evidence supports that some PATs are also auto-S-fatty acylated as the first step of the catalysis, implying that targeting auto-S-fatty acylation is one potential approach to modulate PATs. In addition, the functions of some important non-enzymatic proteins, such as TEADs and RFX3, are regulated by their auto-S-fatty acylation, and intracellular fatty acyl-CoA levels could directly affect their functions.The crystal structures of auto-S-fatty acylated proteins all revealed a hydrophobic pocket to accommodate lipid binding. It is speculated that such hydrophobic pocket could be a common feature of many auto-S-fatty acylated proteins. This unique structural feature provides a basis for rational drug design. More importantly, the inherent active cysteine near this hydrophobic pocket allows the development of irreversible inhibitors. Although there is still no approved drug targeting auto-S-fatty acylation, small molecule inhibitors targeting auto-S-fatty acylation have been successfully shown to intervene in the functions of the proteins. This approach opens a door to target previously “undruggable” targets to expand the “druggable” proteome. Moreover, these compounds provide novel chemical tools to decipher the functions of protein auto-S-fatty acylation in complex biological networks.
One of the outstanding questions is how we can advance technological methods for an in-depth understanding of auto-S-fatty acylation. In recent years, progress in chemical biology approaches has accelerated profiling of auto-S-fatty acylation, such as identification of new auto-S-fatty acylated proteins. However, our knowledge of how auto-S-fatty acylation is regulated in cell signaling is still lacking. Considering that a large number of non-membrane bound proteins are S-fatty acylated in a biological system, a significant number of these proteins could be “auto-S-fatty acylated” as they are not normally co-localized with the membrane-bound ZDHHC-family of PATs. Therefore, designing new chemical probes with higher specificity and various reactivities would have a significant impact. In addition, a lack of robust and sensitive HTS assays, such as the assays developed for kinases,93 further hinders the discovery of potent inhibitors of auto-S-fatty acylation. Traditional auto-S-fatty acylation assay relies on gel-based methods, which are extremely low throughput and time-consuming. While the throughput has been improved with the enzyme-linked assay by Mitchell et al., it still lacks sufficient sensitivity because of the low fluorescence signal of NADH. Incorporating more sensitive fluorophores or even luminogenic materials will probably increase the detection sensitivity. A high false positive rate also hinders its application. For example, the formation of CoASH might come from hydrolysis of palmitoyl-CoA. In addition, compounds which inactivate the α-KDH-catalyzed reaction will be annotated false-positively as inhibitors of auto-S-fatty acylation. Direct and precise assessment of auto-S-fatty acylation in a more robust, sensitive and high throughput fashion would have a significant impact on drug discovery.
Another outstanding question is how to specifically target auto-S-fatty acylation of an individual PAT. Known inhibitors, such as 2-BP, cerulenin and compound V, are pan-inhibitors and non-specific, and have many off-target effects. It is theoretically feasible to design selective small molecules against each PAT. First, the geometry of the hydrophobic pocket in ZDHHCs is slightly different, evidenced by the fact that ZDHHCs have a preference for lipids with different chain lengths.94 Secondly, the residues in the palmitoyl binding pockets have significant diversity, as evidenced in human ZDHHC20 and zebrafish DHHS15. Current research indicates that some PATs mediate S-palmitoylation of more than one substrate. For example, ZDHHC13 could palmitoylate HTT, as well as proteins in the NF-κB pathway. It is essential to understand the on-target adverse effects of PAT inhibitors. In addition to developing inhibitors of S-fatty acylation, allosteric activation of S-fatty acylation or inhibition of depalmitoylation could be important strategies as well. For example, hypoactivation of auto-S-fatty acylation of HIP14/ZDHHC17 and RFX3 is linked to diseases. Small molecules mimicking the binding and function of palmitate in the pocket might serve as activators of these proteins. Indeed, a small molecule activator of TEADs, which binds to the lipid binding site, has been reported.95 Furthermore, the inhibition of de-fatty acylating enzymes, such as APT1/2 and ABHD proteins, might rescue the loss of S-fatty acylation in disease conditions.
Conflicts of interest
X. W. is the inventor of a patent application of TEAD auto-S-palmitoylation inhibitors (WO2017053706A1).Acknowledgements
The authors are supported by grants from NIH (R01CA219814-01A1, R01CA238270, and R01DK107651). Z. T. is partly supported by a dermatology postdoctoral fellowship from Melanoma Research Alliance (MRA).References
- J. Greaves, G. R. Prescott, O. A. Gorleku and L. H. Chamberlain, Mol. Membr. Biol., 2009, 26, 67–79 CrossRef CAS PubMed.
- M. E. Linder and R. J. Deschenes, Nat. Rev. Mol. Cell Biol., 2007, 8, 74–84 CrossRef CAS PubMed.
- D. Morozova and M. Weiss, Biophys. J., 2010, 98, 800–804 CrossRef CAS PubMed.
- X. Guan and C. A. Fierke, Sci. China: Chem., 2011, 54, 1888–1897 CrossRef CAS.
- S. Baekkeskov and J. Kanaani, Mol. Membr. Biol., 2009, 26, 42–54 CrossRef CAS PubMed.
- B. Chavda, J. A. Arnott and S. L. Planey, Expert Opin. Drug Discovery, 2014, 9, 1005–1019 CrossRef CAS.
- S. Tabaczar, A. Czogalla, J. Podkalicka, A. Biernatowska and A. F. Sikorski, Exp. Biol. Med., 2017, 242, 1150–1157 CrossRef CAS.
- R. Harmel and D. Fiedler, Nat. Chem. Biol., 2018, 14, 244–252 CrossRef CAS PubMed.
- N. J. Færgeman and J. Knudsen, Biochem. J., 1997, 323, 1–12 CrossRef PubMed.
- Y. Ohno, A. Kashio, R. Ogata, A. Ishitomi, Y. Yamazaki and A. Kihara, Mol. Biol. Cell, 2012, 23, 4543–4551 CrossRef CAS.
- M. S. Rana, P. Kumar, C. J. Lee, R. Verardi, K. R. Rajashankar and A. Banerjee, Science, 2018, 359, eaao6326 CrossRef PubMed.
- A. V. Pobbati, X. Han, A. W. Hung, S. Weiguang, N. Huda, G. Y. Chen, C. B. Kang, C. S. B. Chia, X. Luo, W. Hong and A. Poulsen, Structure, 2015, 23, 2076–2086 CrossRef CAS.
- P. Chan, X. Han, B. Zheng, M. Deran, J. Yu, G. K. Jarugumilli, H. Deng, D. Pan, X. Luo and X. Wu, Nat. Chem. Biol., 2016, 12, 282–289 CrossRef CAS PubMed.
- A. P. Turnbull, D. Kümmel, B. Prinz, C. Holz, J. Schultchen, C. Lang, F. H. Niesen, K. P. Hofmann, H. Delbrück, J. Behlke, E. C. Müller, E. Jarosch, T. Sommer and U. Heinemann, EMBO J., 2005, 24, 875–884 CrossRef CAS.
- M. Blanc, F. David, L. Abrami, D. Migliozzi, F. Armand, J. Bürgi and F. G. van der Goot, F1000Research, 2015, 4, 261 Search PubMed.
- P. Ko and S. J. Dixon, EMBO Rep., 2018, 19, 1–14 CrossRef PubMed.
- Y. Fukata and M. Fukata, Nat. Rev. Neurosci., 2010, 11, 161–175 CrossRef.
- M. Zaręba-Kozioł, I. Figiel, A. Bartkowiak-Kaczmarek and J. Włodarczyk, Front. Mol. Neurosci., 2018, 11, 1–18 CrossRef.
- Y. G. Kim, J. S. Eun, J. Seo, K. J. Lee, H. S. Lee, I. Hwang, M. Whiteway, M. Sacher and B. H. Oh, Nat. Struct. Mol. Biol., 2005, 12, 38–45 CrossRef.
- D. Kümmel, U. Heinemann and M. Veit, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12701–12706 CrossRef.
- A. F. Roth, J. Wan, A. O. Bailey, B. Sun, J. A. Kuchar, W. N. Green, B. S. Phinney, J. R. Yates and N. G. Davis, Cell, 2006, 125, 1003–1013 CrossRef CAS PubMed.
- B. Chen, J. Niu, J. Kreuzer, B. Zheng, G. K. Jarugumilli, W. Haas and X. Wu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E8403–E8412 CrossRef CAS PubMed.
- M. Berger and M. F. Schmidt, EMBO J., 1984, 3, 713–719 CrossRef CAS PubMed.
- P. J. O’Brien, R. S. Jules, T. S. Reddy, N. G. Bazan and M. Zatz, J. Biol. Chem., 1987, 262, 5210–5215 CrossRef.
- M. Veit, K. Sachs, M. Heckelmann, D. Maretzki, K. P. Hofmann and M. F. G. Schmidt, Biochim. Biophys. Acta, Lipids Lipid Metab., 1998, 1394, 90–98 CrossRef CAS.
- O. A. Bizzozero, J. F. McGarry and M. B. Lees, J. Biol. Chem., 1987, 262, 13550–13557 CrossRef CAS.
- M. Bharadwaj and O. A. Bizzozero, J. Neurochem., 1995, 65, 1805–1815 CrossRef CAS.
- J. A. Duncan and A. G. Gilman, J. Biol. Chem., 1996, 271, 23594–23600 CrossRef CAS.
- S. Lobo, W. K. Greentree, M. E. Linder and R. J. Deschenes, J. Biol. Chem., 2002, 277, 41268–41273 CrossRef CAS.
- A. F. Roth, Y. Feng, L. Chen and N. G. Davis, J. Cell Biol., 2002, 159, 23–28 CrossRef CAS PubMed.
- D. A. Mitchell, G. Mitchell, Y. Ling, C. Budde and R. J. Deschenes, J. Biol. Chem., 2010, 285, 38104–38114 CrossRef CAS PubMed.
- B. C. Jennings and M. E. Linder, J. Biol. Chem., 2012, 287, 7236–7245 CrossRef CAS PubMed.
- K. Lemonidis, O. A. Gorleku, M. C. Sanchez-Perez, C. Grefen and L. H. Chamberlain, Mol. Biol. Cell, 2014, 25, 3870–3883 CrossRef PubMed.
- M. O. Collins, K. T. Woodley and J. S. Choudhary, Sci. Rep., 2017, 7, 1–14 CrossRef.
- T. Lanyon-Hogg, M. Faronato, R. A. Serwa and E. W. Tate, Trends Biochem. Sci., 2017, 42, 566–581 CrossRef PubMed.
- F. L. Raymond, P. S. Tarpey, S. Edkins, C. Tofts, S. O’Meara, J. Teague, A. Butler, C. Stevens, S. Barthorpe, G. Buck, J. Cole, E. Dicks, K. Gray, K. Halliday, K. Hills, J. Hinton, D. Jones, A. Menzies, J. Perry, K. Raine, R. Shepherd, A. Small, J. Varian, S. Widaa, U. Mallya, J. Moon, Y. Luo, M. Shaw, J. Boyle, B. Kerr, G. Turner, O. Quarrell, T. Cole, D. F. Easton, R. Wooster, M. Bobrow, C. E. Schwartz, J. Gecz, M. R. Stratton and P. A. Futreal, Am. J. Hum. Genet., 2007, 80, 982–987 CrossRef PubMed.
- A. Masurel-Paulet, V. M. Kalscheuer, N. Lebrun, H. Hu, F. Levy, C. Thauvin-Robinet, V. Darmency-Stamboul, S. El Chehadeh, J. Thevenon, S. Chancenotte, M. Ruffier-Bourdet, M. Bonnet, J. M. Pinoit, F. Huet, V. Desportes, J. Chelly and L. Faivre, Am. J. Med. Genet., Part A, 2014, 164, 789–795 CrossRef CAS PubMed.
- J. J. Shimell, B. S. Shah, S. M. Cain, S. Thouta, N. Kuhlmann, I. Tatarnikov, D. B. Jovellar, G. S. Brigidi, J. Kass, A. J. Milnerwood, T. P. Snutch and S. X. Bamji, Cell Rep., 2019, 29, 2422–2437.e8 CrossRef CAS PubMed.
- D. A. Mitchell, L. D. Hamel, K. D. Reddy, L. Farh, L. M. Rettew, P. R. Sanchez and R. J. Deschenes, J. Biol. Chem., 2014, 289, 18582–18592 CrossRef CAS.
- M. Kouskou, D. M. Thomson, R. R. Brett, L. Wheeler, R. J. Tate, J. A. Pratt and L. H. Chamberlain, Exp. Neurol., 2018, 308, 35–46 CrossRef CAS PubMed.
- E. Napoli, G. Song, S. Liu, A. Espejo, C. J. Perez, F. Benavides and C. Giulivi, Sci. Rep., 2017, 7, 1–15 CrossRef CAS.
- C. J. Perez, L. Mecklenburg, J. Jaubert, L. Martinez-Santamaria, B. M. Iritani, A. Espejo, E. Napoli, G. Song, M. Del Río, J. DiGiovanni, C. Giulivi, M. T. Bedford, S. Y. R. Dent, R. D. Wood, D. F. Kusewitt, J. L. Guénet, C. J. Conti and F. Benavides, J. Invest. Dermatol., 2015, 135, 3133–3143 CrossRef CAS PubMed.
- A. N. Saleem, Y. H. Chen, H. J. Baek, Y. W. Hsiao, H. W. Huang, H. J. Kao, K. M. Liu, L. F. Shen, I. wen Song, C. P. D. Tu, J. Y. Wu, T. Kikuchi, M. J. Justice, J. J. Y. Yen and Y. T. Chen, PLoS Genet., 2010, 6, 1–15 Search PubMed.
- I. W. Song, W. R. Li, L. Y. Chen, L. F. Shen, K. M. Liu, J. J. Y. Yen, Y. J. Chen, Y. J. Chen, V. B. Kraus, J. Y. Wu, M. T. Michael Lee and Y. T. Chen, PLoS One, 2014, 9, e92194 CrossRef PubMed.
- L. F. Shen, Y. J. Chen, K. M. Liu, A. N. S. Haddad, I. W. Song, H. Y. Roan, L. Y. Chen, J. J. Y. Yen, Y. J. Chen, J. Y. Wu and Y. T. Chen, Sci. Rep., 2017, 7, 1–14 CrossRef PubMed.
- E. Ponimaskin, G. Dityateva, M. O. Ruonala, M. Fukata, Y. Fukata, F. Kobe, F. S. Wouters, M. Delling, D. S. Bredt, M. Schachner and A. Dityatev, J. Neurosci., 2008, 28, 8897–8907 CrossRef PubMed.
- D. A. Galil, V. Cherkas, N. Ronkina, J. Lafera, M. Gaestel and E. Ponimaskin, Mol. Cell. Biol., 2016, 36, 2208–2225 CrossRef PubMed.
- R. K. Graham, D. E. Ehrnhoefer and M. R. Hayden, Trends Neurosci., 2011, 34, 646–656 CrossRef PubMed.
- N. H. Skotte, S. S. Sanders, R. R. Singaraja, D. E. Ehrnhoefer, K. Vaid, X. Qiu, S. Kannan, C. Verma and M. R. Hayden, Cell Death Differ., 2017, 24, 433–444 CrossRef CAS PubMed.
- R. R. Singaraja, K. Huang, S. S. Sanders, A. J. Milnerwood, R. Hines, J. P. Lerch, S. Franciosi, R. C. Drisdel, K. Vaid, F. B. Young, C. Doty, J. Wan, N. Bissada, R. M. Henkelman, W. N. Green, N. G. Davis, L. A. Raymond and M. R. Hayden, Hum. Mol. Genet., 2011, 20, 3899–3909 CrossRef CAS PubMed.
- B. S. Shah, J. J. Shimell and S. X. Bamji, J. Cell Sci., 2019, 132, jcs230052 CrossRef CAS PubMed.
- K. B. Runkle, A. Kharbanda, E. Stypulkowski, X. J. Cao, W. Wang, B. A. Garcia and E. S. Witze, Mol. Cell, 2016, 62, 385–396 CrossRef CAS PubMed.
- B. Chen, B. Zheng, M. Deran, G. K. Jarugumilli, J. Fu, Y. S. Brooks and X. Wu, Nat. Chem. Biol., 2016, 12, 686–693 CrossRef CAS PubMed.
- E. P. Marin, L. Jozsef, A. Di Lorenzo, K. F. Held, A. K. Luciano, J. Melendez, L. M. Milstone, H. Velazquez and W. C. Sessa, Arterioscler., Thromb., Vasc. Biol., 2016, 36, 370–379 CrossRef CAS PubMed.
- S. W. Kim, D. H. Kim, K. S. Park, M. K. Kim, Y. M. Park, S. Muallem, I. So and H. J. Kim, Autophagy, 2019, 15, 327–340 CrossRef CAS PubMed.
- N. Zhang, H. Zhao and L. Zhang, J. Virol., 2019, 93, 1–12 Search PubMed.
- T. M. McMichael, L. Zhang, M. Chemudupati, J. C. Hach, A. D. Kenney, H. C. Hang and J. S. Yount, J. Biol. Chem., 2017, 292, 21517–21526 CrossRef PubMed.
- E. Cho and M. Park, Pharmacol. Res., 2016, 111, 133–151 CrossRef PubMed.
- I. De and S. Sadhukhan, Eur. J. Cell Biol., 2018, 97, 319–338 CrossRef.
- M. R. Gadalla and M. Veit, Expert Opin. Drug Discovery, 2020, 15, 159–177 CrossRef CAS.
- L. Yi and C. Zheng, Crit. Rev. Microbiol., 2021, 47, 34–43 CrossRef CAS PubMed.
- H. Lin, FEBS J., 2021, 7, 1–17 Search PubMed.
- C. D. Gottlieb and M. E. Linder, Biochem. Soc. Trans., 2017, 45, 923–938 CrossRef CAS PubMed.
- C. L. Noland, S. Gierke, P. D. Schnier, J. Murray, W. N. Sandoval, M. Sagolla, A. Dey, R. N. Hannoush, W. J. Fairbrother and C. N. Cunningham, Structure, 2016, 24, 179–186 CrossRef CAS PubMed.
- F. Zanconato, G. Battilana, M. Cordenonsi and S. Piccolo, Curr. Opin. Pharmacol., 2016, 29, 26–33 CrossRef CAS PubMed.
- J. K. Holden and C. N. Cunningham, Cancers, 2018, 10, 81 CrossRef PubMed.
- N. G. Kim and B. M. Gumbiner, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9877–9882 CrossRef CAS PubMed.
- A. M. Waters and P. L. Beales, Pediatr. Nephrol., 2011, 26, 1039–1056 CrossRef PubMed.
- J. F. Reiter and M. R. Leroux, Nat. Rev. Mol. Cell Biol., 2017, 18, 533–547 CrossRef PubMed.
- F. Carofiglio, D. Trisciuzzi, N. Gambacorta, F. Leonetti, A. Stefanachi and O. Nicolotti, Molecules, 2020, 25, 4210 CrossRef PubMed.
- G. Garivet, W. Hofer, A. Konitsiotis, C. Klein, N. Kaiser, T. Mejuch, E. Fansa, R. Alsaabi, A. Wittinghofer, P. I. H. Bastiaens and H. Waldmann, Cell Chem. Biol., 2019, 26, 842–851.e7 CrossRef PubMed.
- R. Brommage, J. Liu, P. Vogel, F. Mseeh, A. Y. Thompson, D. G. Potter, M. K. Shadoan, G. M. Hansen, S. Jeter-Jones, J. Cui, D. Bright, J. P. Bardenhagen, D. D. Doree, S. Movérare-Skrtic, K. H. Nilsson, P. Henning, U. H. Lerner, C. Ohlsson, A. T. Sands, J. E. Tarver, D. R. Powell, B. Zambrowicz and Q. Liu, Bone Res., 2019, 7, 2 CrossRef CAS PubMed.
- W. Mahy, N. J. Willis, Y. Zhao, H. L. Woodward, F. Svensson, J. Sipthorp, L. Vecchia, R. R. Ruza, J. Hillier, S. Kjær, S. Frew, A. Monaghan, M. Bictash, P. C. Salinas, P. Whiting, J. P. Vincent, E. Y. Jones and P. V. Fish, J. Med. Chem., 2020, 63, 12942–12956 CrossRef CAS.
- A. Mousnier, A. S. Bell, D. P. Swieboda, J. Morales-Sanfrutos, I. Pérez-Dorado, J. A. Brannigan, J. Newman, M. Ritzefeld, J. A. Hutton, A. Guedán, A. S. Asfor, S. W. Robinson, I. Hopkins-Navratilova, A. J. Wilkinson, S. L. Johnston, R. J. Leatherbarrow, T. J. Tuthill, R. Solari and E. W. Tate, Nat. Chem., 2018, 10, 599–606 CrossRef CAS.
- Q. Li, Y. Sun, G. K. Jarugumilli, S. Liu, K. Dang, J. L. Cotton, J. Xiol, P. Y. Chan, M. DeRan, L. Ma, R. Li, L. J. Zhu, J. H. Li, A. B. Leiter, Y. T. Ip, F. D. Camargo, X. Luo, R. L. Johnson, X. Wu and J. Mao, Cell Stem Cell, 2020, 26, 675–692.e8 CrossRef CAS PubMed.
- J. M. Draper and C. D. Smith, Mol. Membr. Biol., 2009, 26, 5–13 CrossRef CAS PubMed.
- D. Davda, M. A. El Azzouny, C. T. M. B. Tom, J. L. Hernandez, J. D. Majmudar, R. T. Kennedy and B. R. Martin, ACS Chem. Biol., 2013, 8, 1912–1917 CrossRef CAS PubMed.
- S. Omura, Bacteriol. Rev., 1976, 40, 681–697 CrossRef CAS PubMed.
- D. Duksin and P. Bornstein, J. Biol. Chem., 1977, 252, 955–962 CrossRef CAS.
- C. E. Ducker, L. K. Griffel, R. A. Smith, S. N. Keller, Y. Zhuang, Z. Xia, J. D. Diller and C. D. Smith, Mol. Cancer Ther., 2006, 5, 1647–1659 CrossRef CAS PubMed.
- B. C. Jennings, M. J. Nadolski, Y. Ling, M. B. Baker, M. L. Harrison, R. J. Deschenes and M. E. Linder, J. Lipid Res., 2009, 50, 233–242 CrossRef CAS PubMed.
- L. D. Hamel, R. J. Deschenes and D. A. Mitchell, Anal. Biochem., 2014, 460, 1–8 CrossRef CAS PubMed.
- L. D. Hamel, B. J. Lenhart, D. A. Mitchell, R. G. Santos, M. A. Giulianotti and R. J. Deschenes, Comb. Chem. High Throughput Screening, 2016, 19, 262–274 CrossRef CAS PubMed.
- L. Ganesan, P. Shieh, C. R. Bertozzi and I. Levental, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- D. D. O. Martin, P. S. Kanuparthi, S. M. Holland, S. S. Sanders, H. K. Jeong, M. B. Einarson, M. A. Jacobson and G. M. Thomas, Sci. Rep., 2019, 9, 3632 CrossRef PubMed.
- K. Bum-Erdene, D. Zhou, G. Gonzalez-Gutierrez, M. K. Ghozayel, Y. Si, D. Xu, H. E. Shannon, B. J. Bailey, T. W. Corson, K. E. Pollok, C. D. Wells and S. O. Meroueh, Cell Chem. Biol., 2019, 26, 378–389.e13 CrossRef CAS PubMed.
- W. Lu, J. Wang, Y. Li, H. Tao, H. Xiong, F. Lian, J. Gao, H. Ma, T. Lu, D. Zhang, X. Ye, H. Ding, L. Yue, Y. Zhang, H. Tang, N. Zhang, Y. Yang, H. Jiang, K. Chen, B. Zhou and C. Luo, Eur. J. Med. Chem., 2019, 184, 111767 CrossRef CAS PubMed.
- K. J. Kurppa, Y. Liu, C. To, T. Zhang, M. Fan, A. Vajdi, E. H. Knelson, Y. Xie, K. Lim, P. Cejas, A. Portell, P. H. Lizotte, S. B. Ficarro, S. Li, T. Chen, H. M. Haikala, H. Wang, M. Bahcall, Y. Gao, S. Shalhout, S. Boettcher, B. H. Shin, T. Thai, M. K. Wilkens, M. L. Tillgren, M. Mushajiang, M. Xu, J. Choi, A. A. Bertram, B. L. Ebert, R. Beroukhim, P. Bandopadhayay, M. M. Awad, P. C. Gokhale, P. T. Kirschmeier, J. A. Marto, F. D. Camargo, R. Haq, C. P. Paweletz, K. K. Wong, D. A. Barbie, H. W. Long, N. S. Gray and P. A. Jänne, Cancer Cell., 2020, 37, 104–122.e12 CrossRef CAS PubMed.
- J. K. Holden, J. J. Crawford, C. L. Noland, S. Schmidt, J. R. Zbieg, J. A. Lacap, R. Zang, G. M. Miller, Y. Zhang, P. Beroza, R. Reja, W. Lee, J. Y. K. Tom, R. Fong, M. Steffek, S. Clausen, T. J. Hagenbeek, T. Hu, Z. Zhou, H. C. Shen and C. N. Cunningham, Cell Rep., 2020, 31, 107809 CrossRef CAS PubMed.
- H. Karatas, M. Akbarzadeh, H. Adihou, G. Hahne, A. V. Pobbati, Y. Ng, S. M. Guéret, S. Sievers, A. Pahl, M. Metz, S. Zinken, L. Dötsch, S. Thavam, A. Friese, C. Kang, W. Hong, H. Waldmann, E. Y. Ng, S. M. Guéret, S. Sievers, A. Pahl, M. Metz, L. Dötsch, C. Nowak, S. Thavam, A. Friese, C. Kang, W. Hong and H. Waldmann, J. Med. Chem., 2020, 63, 11972–11989 CrossRef CAS PubMed.
- T. T. Tang, A. W. Konradi, Y. Feng, X. Peng, M. Ma, J. Li, F.-X. Yu, K.-L. Guan and L. Post, Mol. Cancer Ther., 2021, 20, 986–998 CrossRef CAS PubMed.
- P. Wu, T. E. Nielsen and M. H. Clausen, Drug Discovery Today, 2016, 21, 5–10 CrossRef CAS PubMed.
- J. F. Glickman, Assay Guid. Man., 2004, 1–19 Search PubMed.
- J. Greaves, K. R. Munro, S. C. Davidson, M. Riviere, J. Wojno, T. K. Smith, N. C. O. Tomkinson and L. H. Chamberlain, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1365–E1374 CrossRef CAS PubMed.
- A. V. Pobbati, T. Mejuch, S. Chakraborty, H. Karatas, S. R. Bharath, S. M. Guéret, P. A. Goy, G. Hahne, A. Pahl, S. Sievers, E. Guccione, H. Song, H. Waldmann and W. Hong, ACS Chem. Biol., 2019, 14, 2909–2921 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |