
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular design and self-assembly of multidomain peptides towards cytocompatible supramolecular cell penetrating nanofibers†
Su Yang and
He Dong *
*
Department of Chemistry and Biochemistry, The University of Texas at Arlington, Arlington, TX 76019, USA. E-mail: he.dong@uta.edu
First published on 10th August 2020
Abstract
The discovery of cell penetrating peptides (CPPs) with unique membrane activity has inspired the design and synthesis of a variety of cell penetrating macromolecules, which offer tremendous opportunity and promise for intracellular delivery of a variety of imaging probes and therapeutics. While cell penetrating macromolecules can be designed and synthesized to have equivalent or even superior cell penetrating activity compared with natural CPPs, most of them suffer from moderate to severe cytotoxicity. Inspired by recent advances in peptide self-assembly and cell penetrating macromolecules, in this work, we demonstrated a new class of peptide assemblies with intrinsic cell penetrating activity and excellent cytocompatibility. Supramolecular assemblies were formed through the self-assembly of de novo designed multidomain peptides (MDPs) with a general sequence of Kx(QW)6Ey in which the numbers of lysine and glutamic acid can be varied to control supramolecular assembly, morphology and cell penetrating activity. Both supramolecular spherical particles and nanofibers exhibit much higher cell penetrating activity than monomeric MDPs while supramolecular nanofibers were found to further enhance the cell penetrating activity of MDPs. In vitro cell uptake results suggested that the supramolecular cell penetrating nanofibers undergo macropinocytosis-mediated internalization and they are capable of escaping from the lysosome to reach the cytoplasm, which highlights their great potential as highly effective intracellular therapeutic delivery vehicles and imaging probes.
Introduction
The discovery of cell penetrating peptides (CPPs) has great impacts on both fundamental and translational biomedical research due to their seeming ability to transverse the cell membrane at will.1–5 The structure-dependent membrane activity of CPPs inspired the design of a range of cell penetrating polymers and liposomes with multivalent presentation of cationic groups, which play important roles in mediating their membrane activity.6,7 Compared to traditional CPPs, these cell penetrating macromolecules show improved stability and tunable pharmacokinetic, however, their cytotoxicity has been a concern.8,9As an alternative for the fabrication of macromolecular structures, peptide self-assembly offers an effective method to generate peptide-based nanomaterials with much higher stability than monomeric peptides, tunable nanostructures, biological activity and good biocompatibility.10–20 In particular, the high aspect ratio peptide nanofibers, which are formed through self-assembly of β-sheet peptides show good stability and high resistance toward proteolysis.21–23 Recently there have been increasing interests in the design of peptide nanofibers for vaccine and gene delivery, both of which often require highly efficient delivery of antigenic and genetic agents into the cytoplasm.12,24 From this perspective, peptide nanofibers with intrinsic cell penetrating activity would be greatly beneficial to the development of peptide-based immuno-and gene therapy.
Inspired by recent advances in peptide self-assembly and cell penetrating macromolecules, in particular self-assembled cell penetrating peptides,25–31 we have developed a new class of peptide assemblies with intrinsic cell penetrating activity, termed supramolecular cell penetrating nanofibers (SCPNs).32–35 SCPNs are generated through the self-assembly of de novo designed cationic multidomain peptides (MDPs) with a general sequence of Kx(QW)6 (K: lysine, Q: glutamine; W: tryptophan). The central (QW)6 domain drives the self-assembly to form “sandwich”-like β-sheet nanofibers while the terminal domain consisting of a variable number of lysine residues drives disassembly due to electrostatic repulsion among the lysine residues. The end products reflect an energetic balance between the attractive forces provided by the (QW)6 domain and the repulsive forces among the lysine residues. Upon self-assembly, MDP nanofibers display a high positive charge density due to the accumulation of the lysine residues at the fiber–solvent interface, and therefore being potentially membrane-active. Although we have fabricated a library of supramolecular peptide assembly using the Kx(QW)6 series as the molecular building block and identified a potent cell penetrating nanofiber based on K10(QW)6,33,35 the sample preparation was tedious. Desalting was often required to induce self-assembly for peptides having a relatively larger number of lysine residues. Although the mechanism for desalting-induced self-assembly has not been fully understood, we presume that parts of the lysine residues were deprotonated upon trifluoroacetate exchange by bicarbonate during the desalting process, thereby the electrostatic repulsion was reduced, and the equilibrium was shifted toward self-assembly. Although desalting is effective to generate SCPNs, the procedure is relatively tedious. In the current work, we aim to apply the self-assembly strategy based on a new set of modularly designed MDPs for the facile generation of cationic supramolecular peptide assemblies and screening their cell penetrating activity.
Results and discussion
Peptide design
To demonstrate the self-assembly strategy for the construction of cell penetrating peptide assemblies, we synthesized a new series of MDPs, Kx(QW)6Ey in which glutamic acids are appended at the opposing end of lysine residues to allow attractive ionic interactions and therefore provide an additional driving force to shift the equilibrium toward self-assembly. Based on our previous work, the supramolecular nanofibers are a prerequisite, but not a sufficient factor for potent cell penetrating activity. The flexibility of the supramolecular charge domain is also critical for improving the membrane activity of supramolecular peptides. For the Kx(QW)6 series which require desalting steps, we found that a minimum of seven lysines seemed to be required to keep a flexible cationic domain to have effective cell membrane interactions because K6(QW)6 did not show appreciable cell penetrating activity.33,35 Therefore, in this work, we intend to keep an excess of lysine residues at or above 7 for the new sequences in order to ensure effective membrane interactions. Specifically, two or three glutamic acids were added to the C-terminus of K10(QW)6 to drive the equilibrium toward self-assembly while keeping sufficient charge domain flexibility. Similarly, K12(QW)6E3 was synthesized in an attempt to promote self-assembly while having a sufficiently long cationic domain upon self-assembly. By having a small library of MDPs with varying numbers of lysine and glutamic acid, we expect to generate self-assembled peptides with different supramolecular nanostructures and intermolecular packing within the assemblies and use them to explore and optimize supramolecular-structure dependent cell penetrating activity. Table 1 shows the full peptide sequences and their abbreviated names which reflect the numbers of lysine and glutamic acid in the sequence. For example, K10-E2 refers to the sequence of K10(QW)6E2. All peptides were synthesized through Fmoc-solid phase peptide synthesis and used without further treatment after HPLC purification.| Abbreviation | Sequences |
|---|---|
| K10 | KKKKKKKKKKQWQWQWQWQWQW |
| K10-E2 | KKKKKKKKKKQWQWQWQWQWQWEE |
| K10-E3 | KKKKKKKKKKQWQWQWQWQWQWEEE |
| K12-E3 | KKKKKKKKKKKKQWQWQWQWQWQWEEE |
Structural determination of self-assembled MDPs
The MDPs were evaluated for their ability to self-assemble in Tris buffer (pH 7.4, 20 mM). Using a previously established fluorescence method,36,37 the critical assembly concentrations (CACs) of all MDPs were determined at 11–12 μM suggesting that all of them can self-assemble (Fig. S3†). To quantitatively determine the oligomerization states of each assembly and the relative abundance of oligomers versus monomers upon equilibrium, we performed sedimentation velocity (SV) experiments using analytical ultracentrifugation (AUC). Samples were prepared in Tris buffer at 20 μM (above their CACs) and incubated for 24 h at 4 °C to drive the equilibrium toward self-assembly. By monitoring the sedimentation profile of each sample in real time, we can obtain a distribution of sedimentation coefficients which can be used to calculate the molecular weight of each species present in each peptide sample. Fig. S4† showed the raw sedimentation scans taken every ∼30 min at 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and the residual plot supplied by SEDFIT software which showed the goodness of fit. As shown by the distribution of sedimentation coefficients (Fig. 1A), we confirmed that the majority (78%) of K10 existed as monomers and 22% formed oligomers consisting of 12 subunits. Adding glutamic acids on K10 increased the percentage of the oligomeric species. For example, both K10 and K10-E2 formed oligomers of a similar size, but the percentage of oligomers in K10-E2 (37%) was much higher than that of K10 (22%) (Fig. 1B). Further increasing the number of glutamic acids drove the equilibrium toward self-assembly and led to the formation of larger assemblies composed of more than 30 subunits (Fig. 1D). The larger scale supramolecular assemblies are more heterogeneous than small oligomers formed by K10 and K10-E2. This observation is consistent with what is commonly found in amyloid-like fibrous peptide assembly.38 It is also worth noting that these assemblies formed by K10-E3 were in equilibrium with dimers rather than monomers as found with K10 and K10-E2. Compared to K10-E3, K12-E3 formed small oligomers in equilibrium with monomers, which was consistent with the sequence-structure design rule for MDP self-assembly (Fig. 1C). Additional glutamic acids are needed to compensate the higher repulsive interactions among the twelve lysine residues in order to drive the equilibrium toward supramolecular assembly. These results showed that by using this small library of MDPs with varying numbers of lysine and glutamic acid, we can generate cationic supramolecular peptide nanostructures with tunable size, morphology and potentially molecular packing in order to probe supramolecular structure-dependent cell penetrating activity.
000 rpm and the residual plot supplied by SEDFIT software which showed the goodness of fit. As shown by the distribution of sedimentation coefficients (Fig. 1A), we confirmed that the majority (78%) of K10 existed as monomers and 22% formed oligomers consisting of 12 subunits. Adding glutamic acids on K10 increased the percentage of the oligomeric species. For example, both K10 and K10-E2 formed oligomers of a similar size, but the percentage of oligomers in K10-E2 (37%) was much higher than that of K10 (22%) (Fig. 1B). Further increasing the number of glutamic acids drove the equilibrium toward self-assembly and led to the formation of larger assemblies composed of more than 30 subunits (Fig. 1D). The larger scale supramolecular assemblies are more heterogeneous than small oligomers formed by K10 and K10-E2. This observation is consistent with what is commonly found in amyloid-like fibrous peptide assembly.38 It is also worth noting that these assemblies formed by K10-E3 were in equilibrium with dimers rather than monomers as found with K10 and K10-E2. Compared to K10-E3, K12-E3 formed small oligomers in equilibrium with monomers, which was consistent with the sequence-structure design rule for MDP self-assembly (Fig. 1C). Additional glutamic acids are needed to compensate the higher repulsive interactions among the twelve lysine residues in order to drive the equilibrium toward supramolecular assembly. These results showed that by using this small library of MDPs with varying numbers of lysine and glutamic acid, we can generate cationic supramolecular peptide nanostructures with tunable size, morphology and potentially molecular packing in order to probe supramolecular structure-dependent cell penetrating activity.
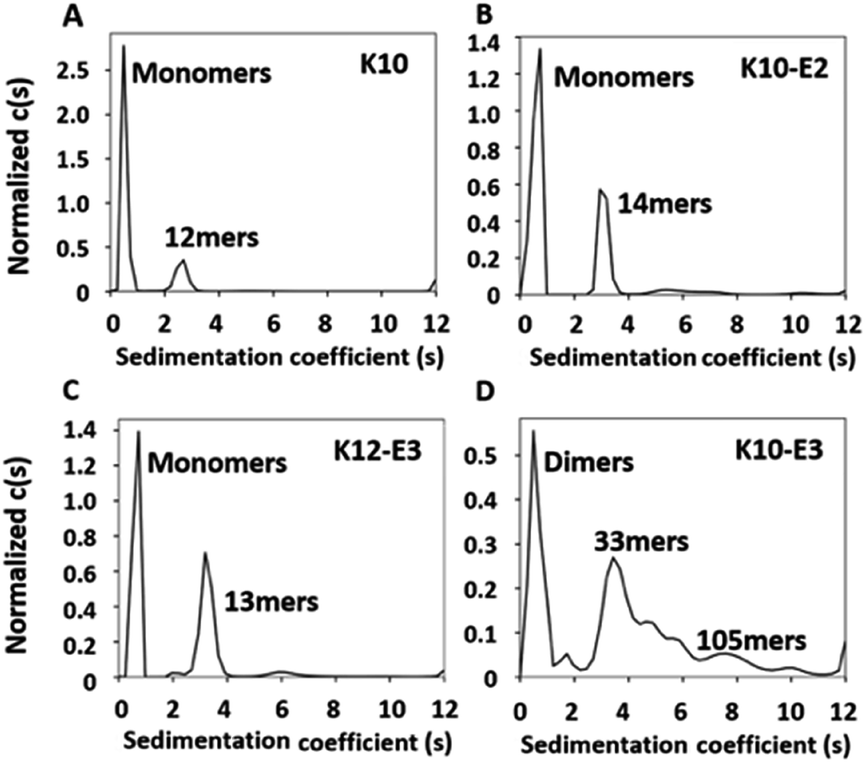 | ||
| Fig. 1 AUC-sedimentation velocity data of (A) K10, (B) K10-E2, (C) K12-E3 and (D) K10-E3 as a semi-quantitative measure of the assembly states. Continuous sedimentation coefficient distribution, c(s) curve, obtained with a regularization procedure. Peptide concentration: 20 μM in Tris buffer (20 mM, pH = 7.4). | ||
The supramolecular structure of peptide assembly was examined by negatively stained transmission electron microscopy (TEM). As shown in Fig. 2A, K10 was invisible under TEM likely due to the fact that K10 largely existed as a monomer. With additional glutamic acids to promote self-assembly, K10-E2 and K12-E3 formed spherical micellar nanostructures while K10-E3 formed nanofibers mixed with a small fraction of micelles (Fig. 2B–D). Based on the AUC and TEM results, we propose that MDPs may follow different self-assembly routes to form either spherical micelles or nanofibers (Scheme 1).
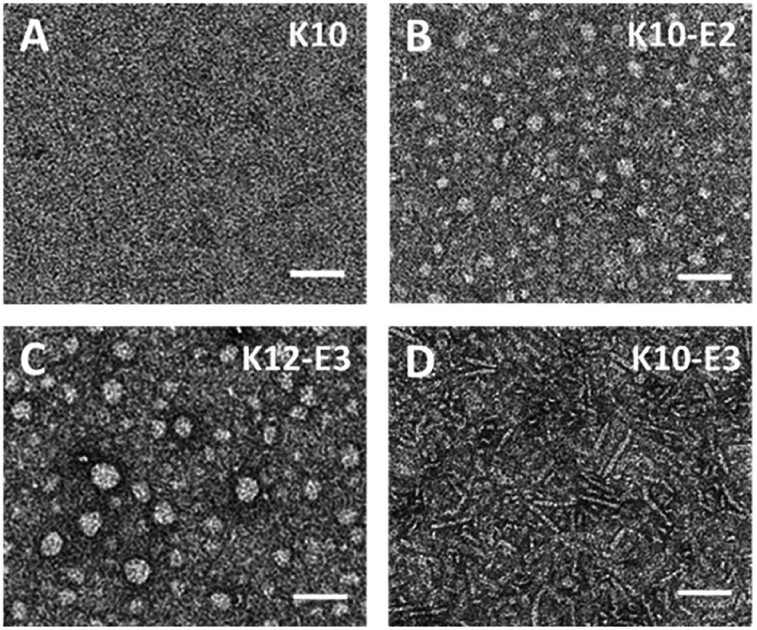 | ||
| Fig. 2 Negatively stained TEM images of (A) K10, (B) K10-E2, (C) K12-E3 and (D) K10-E3. Scale bar = 50 nm. | ||
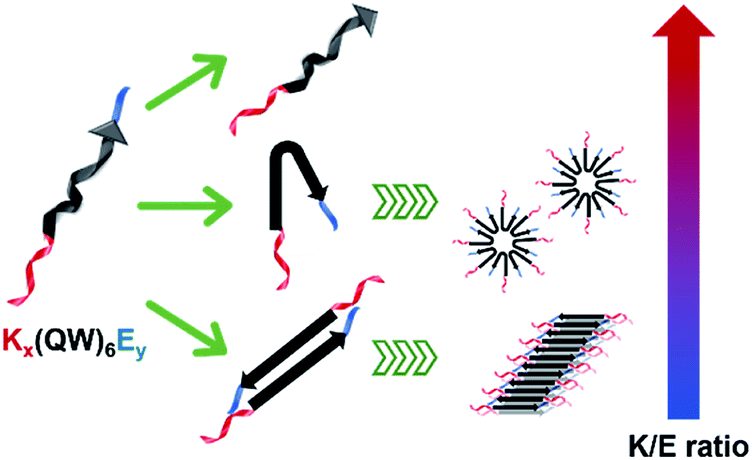 | ||
| Scheme 1 Color-coded schematic representation of Kx(QW)6Ey and their self-assembly routes to form spherical particles and nanofibers. Red: Kx as the cationic domain; black: (QW)6 to drive the supramolecular packing of the β-sheet nanofibers; blue: Ey as the anionic domain. | ||
The self-assembly product is dictated by the energetic balance between the hydrophobic interactions among the (QW) repeating units, the repulsive interactions among the lysine residues and the attractive ionic interactions between lysines and glutamic acids. By varying the numbers of lysine and glutamic acids, we expect to change the energetic balance and therefore the self-assembly pathway and product. For K10-E2 and K12-E3, AUC confirmed the presence of peptide monomers in equilibrium with oligomers (Fig. 1B and C). The monomers are likely to fold into a pseudo β-hairpin conformation as driven by the intramolecular ionic interactions between the lysine and glutamic acids. Although these MDPs were not designed to form a β-hairpin, studies showed that appending oppositely charge amino acids at the peptide termini could drive the formation of β-hairpin conformation.39–41 Indeed, the β-strand conformation of K10-E2 and K12-E3 was confirmed by circular dichroism (CD) spectroscopy (Fig. S5†). These monomers, due to their amphiphilicity can further pack into oligomers and form spherical micelles as observed by TEM. For K10-E3 with an increased portion of glutamic acids with respect to the lysine domain, dimers were found to be in equilibrium with higher ordered assemblies (Fig. 1D). These dimers were possibly formed through combined hydrophobic interaction and intermolecular attractive ionic interactions. The dimers adopted a β-strand conformation as shown by the CD spectroscopy (Fig. S5†), which could further self-assemble into nanofibers as driven by the intermolecular hydrogen bonding. It is important to note that the formation of monomers versus dimers, and micelles versus nanofibers can be achieved by a slight variation of the relative numbers of lysine and glutamic acid in the peptide sequence. This suggests a fine energetic balance exists between the attractive and repulsive interactions on these MDPs, which in turn can be used to manipulate the self-assembly pathways and products.
Evaluation of the cell penetrating activity by in vitro fluorescence cell imaging
In vitro cell uptake experiment was performed to evaluate the cell penetrating activity of different MDPs. For the cell uptake experiment, rather than using 100% of FITC-labeled MDPs, we prepared co-assembled MDPs which consist of a small fraction of FITC to alleviate the potential effect of the bulky, hydrophobic fluorescent moiety on non-specific cell uptake. As demonstrated in our earlier work,42 the co-assembled peptides can be prepared by physically mixing 5% of fluorescein (FITC) labeled MDPs with 95% of non-labeled MDPs in an organic solvent, such as acetonitrile followed by lyophilization and dissolution of MDP powders in Tris buffer. Notably, for the preparation of the co-assembled samples, each peptide was mixed and co-assembled with FITC-labeled K10, rather than using FITC-labeled peptides of their own sequences. Given the consensus (QW)6 domain, FITC-labeled K10 (5% in total) is expected to co-assemble with the other three peptides. The use of a common imaging probe will minimize the variation of fluorescence intensity due to the use of imaging probes with different molecular composition. As a result, the fluorescence intensity observed for cells treated with different peptides is largely attributed to their cell penetrating activities. The co-assembled MDPs were added to HeLa cell culture to reach a final concentration of 20 μM and incubated for 2 h and 24 h for fluorescence imaging and flow cytometry quantification. As shown in Fig. S6,† after 2 h of incubation with HeLa cells, the micelle and fiber forming MDPs showed much higher fluorescence intensity than monomeric K10 although no significant difference was observed among the self-assembling MDPs. It is also noticeable that MDPs mostly localized on the cell membrane upon incubation with cells for 2 h. Fig. 3A–D showed the fluorescence images of HeLa cells upon incubation with different MDPs for 24 h. While K10 still had minimal fluorescence, the intracellular fluorescence was significantly enhanced for micelle and fiber-forming MDPs, suggesting a time-dependent cell penetrating activity. K10-E3 exhibited the highest fluorescence among all MDPs. Flow cytometry was used to qualitatively compare the cell uptake and their cell penetrating activity by measuring the mean fluorescence intensity of HeLa cells incubated with different MDPs for 24 h (Fig. S7†). Consistent with the fluorescence imaging results, HeLa cells treated with K10-E3 showed the highest fluorescence intensity, followed by K10-E2 and K12-E3 which have comparable fluorescence. All self-assembling MDPs demonstrated much higher cell uptake than K10 which mostly formed monomers. Notably, the four peptides have different charge density and therefore different amphiphilicity. To investigate whether amphiphilicity caused the increased cell penetrating activity of K10-E3, we synthesized another multidomain peptide, K7(QW)6 (abbreviated as K7) which has the same net positive charges as K10-E3 but forming different supramolecular structure. Based on the physical characterization results (Fig. S8A–C†), K7 exhibited similar self-assembly behavior as K10-E2 and K12-E3 by forming β-sheet spherical micelles which were composed of 14 mers. Cell uptake imaging showed much less fluorescence (Fig. S8D†) compared to that of K10-E3 (Fig. 3D), further supporting the important role of supramolecular structure on cell penetrating activity.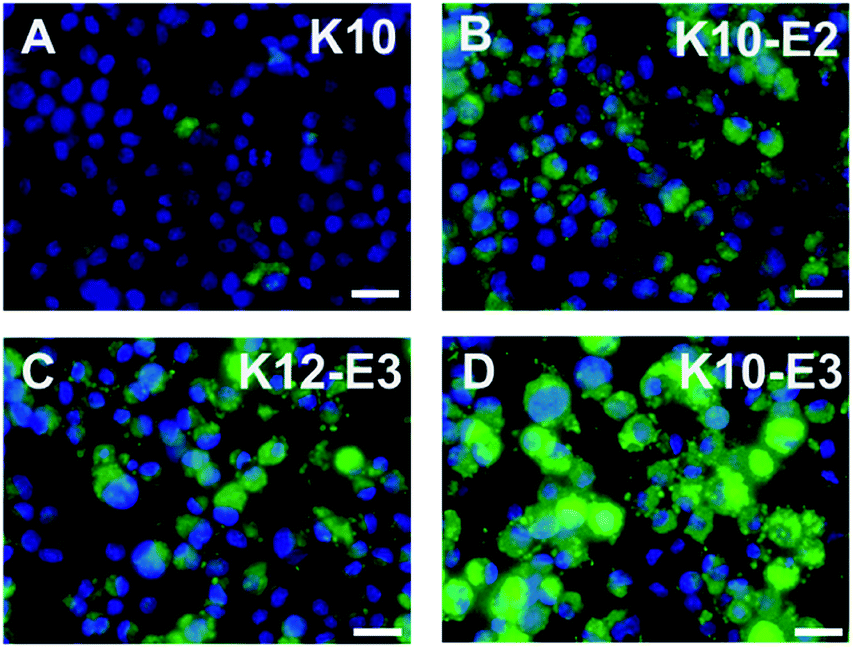 | ||
| Fig. 3 Fluorescence microscopic images of HeLa cells incubated with (A) K10, (B) K10-E2, (C) K12-E3 and (D) K10-E3 for 24 h. Green: FITC labeled peptide, blue: nucleus staining. Scale bar = 25 μm. | ||
Based on the above results, we believe supramolecular assembly can enhance the cell penetrating activity of MDPs. However, we also found that the activity of different self-assembled MDPs varied significantly and seemed to be dependent on the supramolecular structures and possibly the intermolecular packing of MDPs within the assembly. First of all, compared with a spherical micellar structure, a nanofiber can accommodate a larger number of cationic building blocks. This leads to an increase in cationic charge multi-valency to the multivalent interactions with the negatively charged cell membrane. Secondly, in a nanofiber, MDPs are organized into brush-like nanostructures in which the cationic charges are more structurally confined at the fiber–solvent interface. Such an organization could enhance the availability of the cationic clusters for corporative membrane binding. Thirdly, we expect nanofibers are more kinetically stable than spherical micelles because of the highly cooperative non-covalent interactions involved in the formation and stabilization of supramolecular nanofibers. For example, nanofibers are stabilized by the directional hydrogen bonding along the long fiber axis while it does not exist or relatively weak in spherical micelles. These interactions could help improve the kinetic stability of nanofibers in a cellular environment and increase their biological availability. As part of future endeavors, experiments about quantitative kinetic stability measurement will be rationally designed and explored for these supramolecular assemblies.
Evaluation of the cytotoxicity and cell uptake mechanism
Cytotoxicity of cell penetrating macromolecules has been a major concern for their widespread use in biomedical and medical applications. Engineering polymer degradability has been used as an effective method to improve the cytocompatibility of cell penetrating polymers.43–46 The cytotoxicity of these supramolecular peptides was evaluated in HeLa cell culture and the cell viability was quantified by the CCK8 assay after 24 h of incubation of cells with each peptide. As shown in Fig. S9,† all MDPs showed cell viability at >70% up to 80 μM, suggesting good cytocompatibility. The low cytotoxicity is of great interest, which is presumably related to the internalization mechanism employed by these supramolecular assemblies. Endocytosis is a much more preferred route for cell penetrating macromolecules to be internalized because it imposes less physical disruption on the cell membrane.47,48 To investigate the internalization mechanism employed by these supramolecular peptides, we pre-incubated HeLa cells for 2 h using three common endocytosis inhibitors including methyl-β-cyclodextrin (MβCD) for clathrin-mediated endocytosis, filipin III for caveolae-dependent endocytosis, and amiloride for macropinocytosis.49,50 After removing the inhibitors, K10-E3 (the most active cell penetrating assembly) was added to the cell culture and incubated for 24 h for fluorescence imaging. As shown in Fig. 4 (compared with Fig. 3D), while MβCD had no inhibitory effect on the cell uptake of K10-E3, both filipin III and amiloride caused fluorescence reduction of HeLa cells upon K10-E3 treatment, suggesting endocytosis to be the predominant cell uptake mechanism. In particular, significant fluorescence reduction was found for amiloride treated cells, supporting that cell uptake was mostly mediated through macropinocytosis. More interestingly, we observed diffuse intracellular distribution for the internalized K10-E3, rather than the punctate pattern commonly observed for materials trapped within the endosome or lysosome. Indeed, the majority of internalized K10-E3 did not co-localize with the Lysotracker (Fig. S10†), indicating the ability of these supramolecular peptides to escape from the lysosome to reach cytoplasm, which was an important attribute for the design of highly effective intracellular therapeutic delivery vehicles. It is also worth noting that the current work aims to establish a rationale for a fundamental peptide self-assembly mechanism by which cytocompatible SPCNs can be generated. For future practical biomedical and medical applications, we can readily modify these MDPs by introducing various chemical functionalities in response to a range of disease-specific microenvironment to turn on and off the cell penetrating activity. Such efforts would be greatly beneficial for the development of smart SPCNs as disease-specific molecular therapy and imaging agents.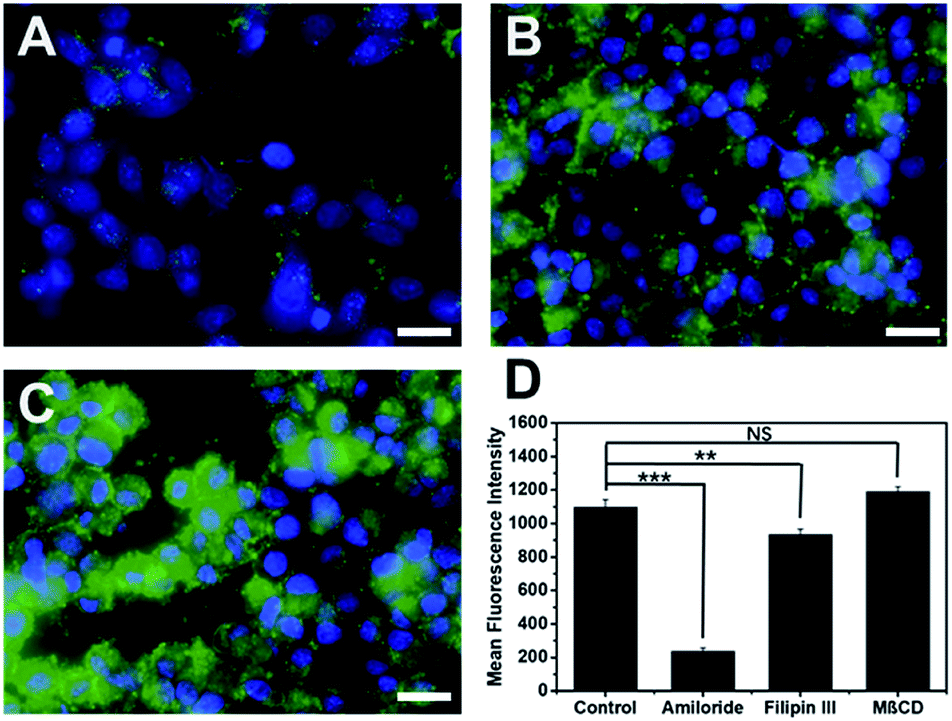 | ||
| Fig. 4 Fluorescence microscopic images of HeLa cells pre-incubated with (A) amiloride, (B) filipin III and (C) MβCD followed by the addition of FITC-labeled K10-E3 for fluorescence cell imaging. Green: FITC labeled K10-E3, blue: nucleus staining. Scale bar = 25 μm. Peptide concentration: 20 μM. (D) Fluorescence intensity of HeLa cells pre-incubated with different endocytosis inhibitors followed by the addition of FITC-labeled K10-E3 as quantified by flow cytometry. Statistically significant differences are indicated by **p < 0.01, ***p < 0.001. | ||
Conclusions
In conclusion, supramolecular peptides with different cell penetrating activity were demonstrated using modularly designed MDPs as the molecular building block. The supramolecular nanostructure is mediated by the relative length of the cationic and anion domain on MDPs to control the energetic balance between the attractive and repulsive interactions involved in the self-assembly. AUC and TEM confirmed the formation of nanofibers and spherical micelles for different MDPs and both supramolecular assemblies exhibit much higher cell penetrating activity than monomeric MDPs. It was found that supramolecular nanofibers could further enhance the cell penetrating activity of MDPs while showing excellent cytocompatibility. Through these preliminary findings, we have established a rationale for a peptide self-assembly mechanism by which SPCNs can be generated. Future efforts will focus on expanding the library of MDPs with diverse chemical functionality for further activity optimization as well as the development of disease-responsive SPCNs for targeted molecular imaging and therapy applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Science Foundation (DMR 1824614) and the University of Texas at Arlington. We would like to thank Chad Brautigam and Shih-Chia Tso at the Biophysical core facilities of UT Southwestern Medical School for the excellent technical support and kind discussions on the AUC experiment and data analysis. We appreciate the kind help and discussion with Dr Wei Chen for fluorescence imaging experiments.Notes and references
- M. Zorko and U. Langel, Adv. Drug Delivery Rev., 2005, 57, 529–545 CrossRef CAS PubMed.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2008, 60, 548–558 CrossRef CAS PubMed.
- A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS PubMed.
- M. Green and P. M. Loewenstein, Cell, 1988, 55, 1179–1188 CrossRef CAS PubMed.
- M. C. Morris, J. Depollier, J. Mery, F. Heitz and G. Divita, Nat. Biotechnol., 2001, 19, 1173–1176 CrossRef CAS PubMed.
- L. Yin, Z. Song, K. H. Kim, N. Zheng, H. Tang, H. Lu, N. Gabrielson and J. Cheng, Biomaterials, 2013, 34, 2340–2349 CrossRef CAS PubMed.
- F. Sgolastra, B. M. Deronde, J. M. Sarapas, A. Som and G. N. Tew, Acc. Chem. Res., 2013, 46, 2977–2987 CrossRef CAS PubMed.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS PubMed.
- J. Cai, Y. Yue, D. Rui, Y. Zhang, S. Liu and C. Wu, Macromolecules, 2011, 44, 2050–2057 CrossRef CAS.
- C. A. Hauser and S. Zhang, Chem. Soc. Rev., 2010, 39, 2780–2790 RSC.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- H. Wang, Z. Feng and B. Xu, Theranostics, 2019, 9, 3213–3222 CrossRef CAS PubMed.
- D. M. Raymond and B. L. Nilsson, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- G. A. Hudalla, T. Sun, J. Z. Gasiorowski, H. Han, Y. F. Tian, A. S. Chong and J. H. Collier, Nat. Mater., 2014, 13, 829–836 CrossRef CAS PubMed.
- A. N. Moore and J. D. Hartgerink, Acc. Chem. Res., 2017, 50, 714–722 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- M. C. Branco, D. M. Sigano and J. P. Schneider, Curr. Opin. Chem. Biol., 2011, 15, 427–434 CrossRef CAS PubMed.
- H. Acar, S. Srivastava, E. J. Chung, M. R. Schnorenberg, J. C. Barrett, J. L. LaBelle and M. Tirrell, Adv. Drug Delivery Rev., 2017, 110–111, 65–79 CrossRef CAS PubMed.
- A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- T. J. Deming, Adv. Mater., 1997, 9, 299–311 CrossRef CAS.
- H. Zhang, J. Park, Y. Jiang and K. A. Woodrow, Acta Biomater., 2017, 55, 183–193 CrossRef CAS PubMed.
- M. Yang, D. Xu, L. Jiang, L. Zhang, D. Dustin, R. Lund, L. Liu and H. Dong, Chem. Commun., 2014, 50, 4827–4830 RSC.
- S. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS PubMed.
- J. S. Rudra, T. Sun, K. C. Bird, M. D. Daniels, J. Z. Gasiorowski, A. S. Chong and J. H. Collier, ACS Nano, 2012, 6, 1557–1564 CrossRef CAS PubMed.
- Z. Zhu, D. Tian, P. Gao, K. Wang, Y. Li, X. Shu, J. Zhu and Q. Zhao, J. Am. Chem. Soc., 2018, 140, 17484–17491 CrossRef CAS PubMed.
- Y. Shi, Y. Hu, G. Ochbaum, R. Lin, R. Bitton, H. Cui and H. S. Azevedo, Chem. Commun., 2017, 53, 7037–7040 RSC.
- J. Peng, Q. Yang, Y. Xiao, K. Shi, Q. Liu, Y. Hao, F. Yang, R. Han and Z. Qian, Adv. Funct. Mater., 2019, 29, 1900004 CrossRef.
- H. Jiang, X. Y. Hu, S. Schlesiger, M. Li, E. Zellermann, S. K. Knauer and C. Schmuck, Angew. Chem., Int. Ed. Engl., 2017, 56, 14526–14530 CrossRef CAS PubMed.
- R. Fan, L. Mei, X. Gao, Y. Wang, M. Xiang, Y. Zheng, A. Tong, X. Zhang, B. Han, L. Zhou, P. Mi, C. You, Z. Qian, Y. Wei and G. Guo, Adv. Sci., 2017, 4, 1600285 CrossRef PubMed.
- N. Ashwanikumar, J. S. Plaut, B. Mostofian, S. Patel, P. Kwak, C. Sun, K. McPhail, D. M. Zuckerman, S. C. Esener and G. Sahay, J. Controlled Release, 2018, 282, 76–89 CrossRef CAS PubMed.
- R. Ni and Y. Chau, Angew. Chem., Int. Ed. Engl., 2017, 56, 9356–9360 CrossRef CAS PubMed.
- S. Yang, D. Xu and H. Dong, J. Mater. Chem. B, 2018, 6, 7179–7184 RSC.
- D. Xu, D. S. K. Samways and H. Dong, Bioact. Mater., 2017, 2, 260–268 CrossRef PubMed.
- D. Xu, L. Jiang, L. DeRidder, B. Elmore, M. Bukhari, Q. Wei, D. Samways and H. Dong, Mol. BioSyst., 2016, 12, 2695–2699 RSC.
- D. Xu, D. Dustin, L. Jiang, D. S. Samways and H. Dong, Chem. Commun., 2015, 51, 11757–11760 RSC.
- Y. Chen and M. D. Barkley, Biochemistry, 1998, 37, 9976–9982 CrossRef CAS PubMed.
- D. Xu, Q. Ran, Y. Xiang, J. Linhai, B. M. Smith, F. Bou-Abdallah, R. Lund, Z. Li and H. Dong, RSC Adv., 2016, 6, 15911–15919 RSC.
- S. T. Wang, Y. Lin, R. K. Spencer, M. R. Thomas, A. I. Nguyen, N. Amdursky, E. T. Pashuck, S. C. Skaalure, C. Y. Song, P. A. Parmar, R. M. Morgan, P. Ercius, S. Aloni, R. N. Zuckermann and M. M. Stevens, ACS Nano, 2017, 11, 8579–8589 CrossRef CAS PubMed.
- M. S. Searle, S. R. Griffiths-Jones and H. Skinner-Smith, J. Am. Chem. Soc., 1999, 121, 11615–11620 CrossRef CAS.
- M. Whitney, E. N. Savariar, B. Friedman, R. A. Levin, J. L. Crisp, H. L. Glasgow, R. Lefkowitz, S. R. Adams, P. Steinbach, N. Nashi, Q. T. Nguyen and R. Y. Tsien, Angew. Chem., Int. Ed. Engl., 2013, 52, 325–330 CrossRef CAS PubMed.
- T. Jiang, E. S. Olson, Q. T. Nguyen, M. Roy, P. A. Jennings and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17867–17872 CrossRef CAS PubMed.
- W. Chen, S. Yang, S. Li, J. C. Lang, C. Mao, P. Kroll, L. Tang and H. Dong, ACS Appl. Mater. Interfaces, 2019, 11, 28681–28689 CrossRef CAS PubMed.
- H. Peng, S. Varanasi, D. K. Wang, I. Blakey, F. Rasoul, A. Symons, D. J. T. Hill and A. K. Whittaker, Eur. Polym. J., 2016, 84, 448–464 CrossRef CAS.
- Y. Chandorkar, R. K. Bhagat, G. Madras and B. Basu, Biomacromolecules, 2014, 15, 863–875 CrossRef CAS PubMed.
- A. Bera, A. K. Singh Chandel, C. Uday Kumar and S. K. Jewrajka, J. Mater. Chem. B, 2015, 3, 8548–8557 RSC.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454–14459 CrossRef CAS PubMed.
- W. B. Kauffman, T. Fuselier, J. He and W. C. Wimley, Trends Biochem. Sci., 2015, 40, 749–764 CrossRef CAS PubMed.
- W. B. Kauffman, S. Guha and W. C. Wimley, Nat. Commun., 2018, 9, 2568 CrossRef PubMed.
- K. J. Lim, B. H. Sung, J. R. Shin, Y. W. Lee, D. J. Kim, K. S. Yang and S. C. Kim, PLoS One, 2013, 8, e66084 CrossRef CAS PubMed.
- J. A. Gomez, J. Chen, J. Ngo, D. Hajkova, I. J. Yeh, V. Gama, M. Miyagi and S. Matsuyama, Pharmaceuticals, 2010, 3, 3594–3613 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and additional characterizations. See DOI: 10.1039/d0ra04748a |
| This journal is © The Royal Society of Chemistry 2020 |