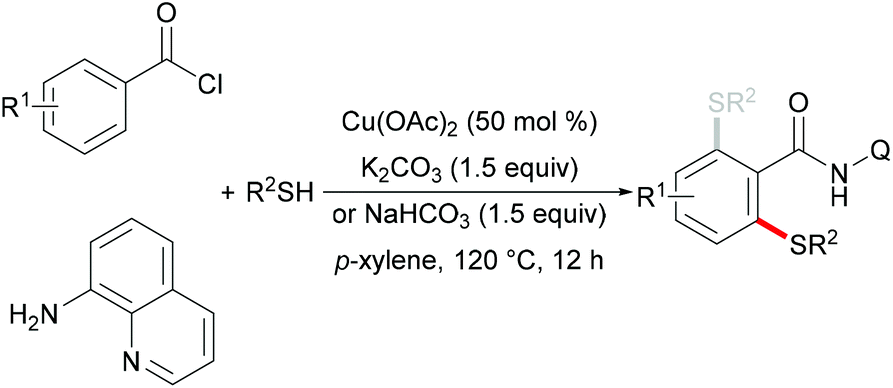DOI:
10.1039/C9QO01497G
(Review Article)
Org. Chem. Front., 2020,
7, 1022-1060
Chelation-assisted transition metal-catalysed C–H chalcogenylations
Received
19th December 2019
, Accepted 10th March 2020
First published on 11th March 2020
Abstract
The development of convenient and mild chalcogenylation reactions for the preparation of unsymmetrical diaryl sulfides and diaryl selenides has received significant attention in recent years due to their prevalence in natural products, organic molecular syntheses, catalysis, drug candidates and functional materials. In contrast with conventional organic transformations which largely rely on the inherent reactivity of functional groups, transition metal-catalysed direct C–H functionalizations have emerged as a powerful strategy that eliminates prefunctionalised starting materials and thus leads to more atom- and step-economical processes. This review summarizes the recent advances in C–S and C–Se formations via transition metal-catalyzed C–H functionalization utilizing directing groups to control the site-selectivity until autumn 2019. Typical examples are listed and mechanistic aspects are discussed in detail.
 Wenbo Ma | Wenbo Ma was born in Hebei province, P. R. China. After he obtained his B.Sc. degree in pharmacy sciences from the North China University of Science and Technology and M.Sc. in medicinal chemistry from the West China School of Pharmacy, Sichuan University, he joined Prof. Dr Lutz Ackermann's research group at the Georg-August University of Göttingen where he obtained his Ph.D. degree in 2015. Afterwards he was appointed as a professor at the Sichuan Industrial Institute of Antibiotics, Chengdu University. His current research interests are focused on medicinal chemistry and methodology study on transition-metal catalyzed C–H functionalization. |
 Nikolaos Kaplaneris | Nikolaos Kaplaneris was born in 1992 in Athens, Greece. He received his bachelor's degree in chemistry from the National and Kapodistrian University of Athens in 2014. He obtained his master's degree in organic chemistry from the same university in 2016 following studies in the area of organocatalysis and photochemistry under the supervision of Prof. Christoforos G. Kokotos. In the same year, he joined the group of Prof. Lutz Ackermann at the Georg-August-Universität Göttingen as a PhD student, working on late-stage peptide diversification and remote functionalization. |
 Xinyue Fang | Xinyue Fang was born in 1996 in Sichuan, P. R. China. She obtained her B.Sc. degree in pharmacy sciences from Chengdu University in 2018. In the same year, she started her M.Sc. studies on transition-metal-catalyzed C–H functionalization under the supervision of Prof. Wenbo Ma at the Sichuan Industrial Institute of Antibiotics, Chengdu University. |
 Linghui Gu | Linghui Gu was born in Sichuan, P. R. China, in 1989. She obtained her B.Sc., M.Sc. and Ph.D. degrees in medicinal chemistry from the West China School of Pharmacy, Sichuan University from 2007 to 2017. She is currently working as an associate investigator in the research group of Prof. Wenbo Ma at the Sichuan Industrial Institute of Antibiotics, Chengdu University. Her current research interests are focused on transition-metal catalyzed C–H functionalization and medicinal chemistry. |
 Ruhuai Mei | Ruhuai Mei received his B.S. in chemistry from Chengdu University of Traditional Chinese Medicine (2010), M.S. from Sichuan University (2013), and Ph.D. from the Georg-August-Universität Göttingen working with Professor Lutz Ackermann in 2017. After one year of postdoctoral work in the same research group, he became a research fellow at the College of Pharmacy and Biological Engineering, Chengdu University. His research interest is focused on oxidative C–H functionalizations. |
 Lutz Ackermann | Lutz Ackermann studied chemistry at the Christian-Albrechts-University Kiel and obtained his Ph.D. in 2001 working with Alois Fürstner at the MPI für Kohlenforschung in Mülheim/Ruhr. He was a postdoctoral fellow with Robert G. Bergman (UC Berkeley) before initiating his independent research in 2003 at the Ludwig-Maximilians-University München, supported within the Emmy Noether Program of the DFG. In 2007, he became a full professor at the Georg-August University of Göttingen, where he served as the Dean of Research and Dean of Chemistry as well as the director of the Wöhler Research Institute for Sustainable Chemistry (WISCh). The development of novel concepts for homogeneous catalysis and their applications in sustainable organic synthesis, late-stage peptide diversification, and molecular imaging are among his main current research interests. |
1. Introduction
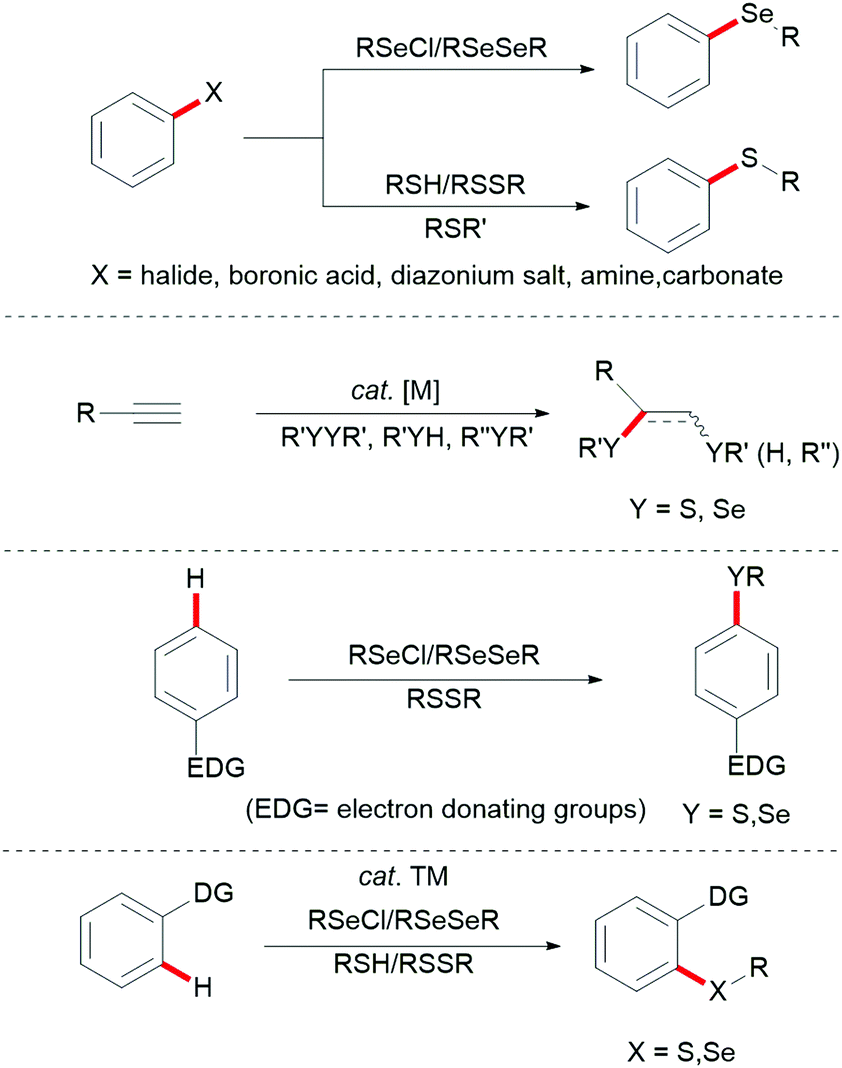
Aryl sulfides and aryl selenides are ubiquitous structural motifs that have been identified as potent drug candidates, bioactive molecules,1–5 and fluorescent probes.6,7 They also play an indispensable role in molecular syntheses8–10 and organocatalysis.11–16 For instance, various sulfur- and selenium-containing molecules have been identified as important therapeutic compounds with bioactivities ranging from antiviral, anti-inflammatory, antibacterial, anticonvulsant, hypoglycemic, human immunodeficiency virus (HIV) protease inhibitor, antioxidative and anticancer activities. Furthermore, they feature likewise significant applications in functional organic materials because the introduction of sulfur or selenium into organic molecules has a profound effect on their physical and electronic properties.17–21 Consequently, the development of new synthetic methods for the assembly of compounds with C–Se and C–S linkages has attracted considerable attention in recent years.22–30 Early examples for C–S and C–Se bond formation largely rely on the inherent reactivity of functional groups, and are generally accomplished by the following three approaches: (a) direct cross-coupling reactions between a metal thiolate and a prefunctionalized aryl substrate, mostly aryl halides, aryl boronic acids, aryl carboxylic acids or aryl diazonium salts;31–46 (b) addition reactions of thiols onto unsaturated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C multiple bonds under free-radical or metal-catalysed conditions;47–56 and (c) direct electrophilic modifications of electron-rich aromatic compounds with thiols or diselenides (Fig. 1).34,57–76
C multiple bonds under free-radical or metal-catalysed conditions;47–56 and (c) direct electrophilic modifications of electron-rich aromatic compounds with thiols or diselenides (Fig. 1).34,57–76
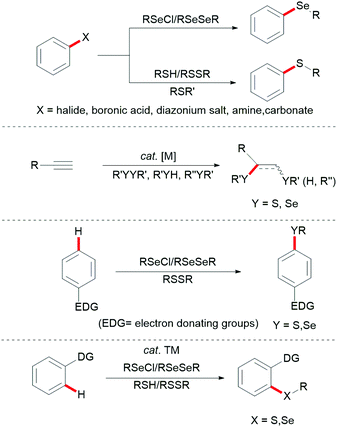 |
| | Fig. 1 Representative C–S and C–Se bond forming reactions. | |
During the past decades, transition metal-catalysed C–H bond activation and functionalization have been established as a powerful tool for the preparation of complex molecules, pharmaceutical agents, natural products and functional materials, thereby providing a streamlined, environmentally sustainable alternative to traditional cross-couplings (Fig. 1d).77–96 This strategy avoids prefunctionalized substrates, thus eliminating the concomitant formation of stoichiometric amounts of undesired by-products. In this context, different transition metal complexes such as palladium, ruthenium and rhodium and Earth abundant first row and transition metals particularly copper, iron, nickel and cobalt have been extensively explored in direct C–H alkenylations, arylations, halogenations, cyanations, annulations, alkylations, aminations and chalcogenylations.77–88,90–98 Chemical approaches for C–S and C–Se bond formation via direct C–H bond functionalization reactions assisted by directing groups to guide the regio-selectivity provided a new insight into the mechanistic understanding of C–S and C–Se coupling reactions and a more effective synthetic route to sulfur- and selenide-containing compounds with high atom economy. In this review, we summarize the recent advances in palladium-, rhodium-, ruthenium-, silver-, cobalt-, copper- and nickel-catalysed C–S and C–Se bond formation via directed C–H activation until autumn 2019.
2. Second row transition metal catalysis
2.1 Palladium catalysis
Palladium catalysis has received more attention since the pioneering report by Suzuki, Negishi and Heck on the coupling reactions which were underlined by the 2010 Nobel Prize.99–101 Palladium can exist in oxidation states ranging from 0 to +4, which allows its complexes to perform a wide range of oxidative transformations.102–104 In the past decades, several classes of organic transformations, including classical cross-couplings, C–H functionalizations, and oxygenations, among others, have thus been widely explored by means of palladium catalysis.105–110
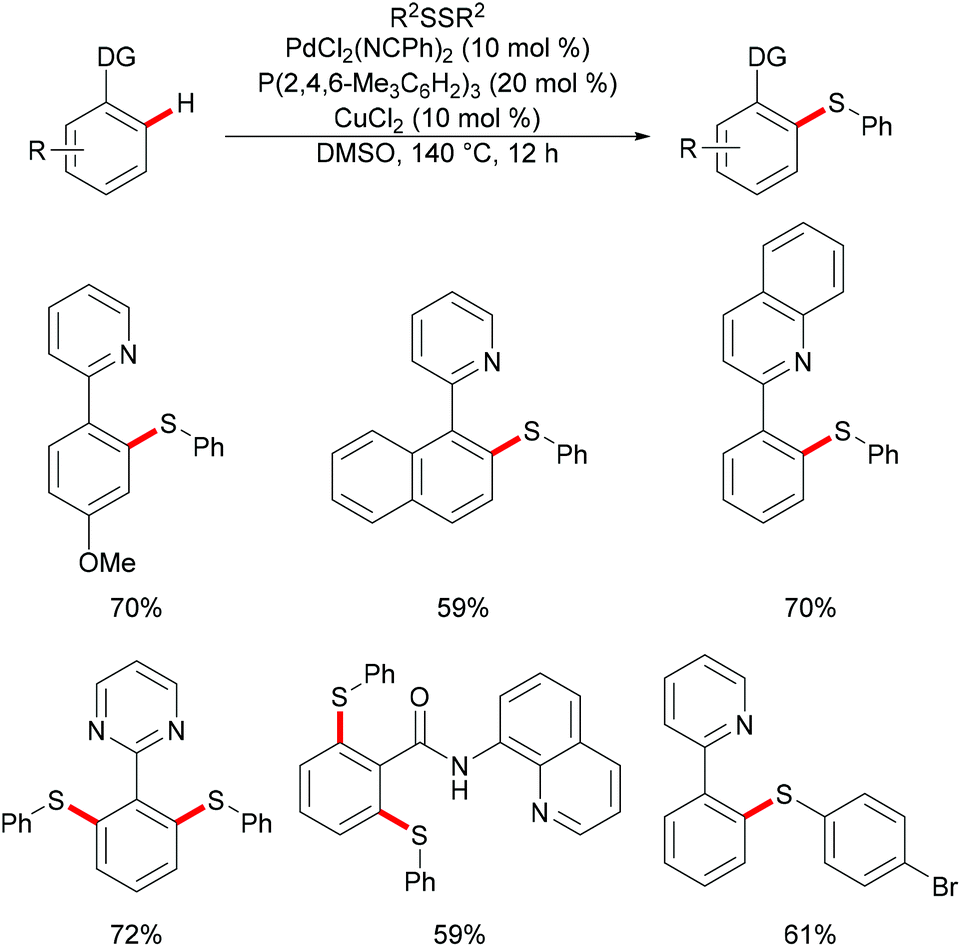
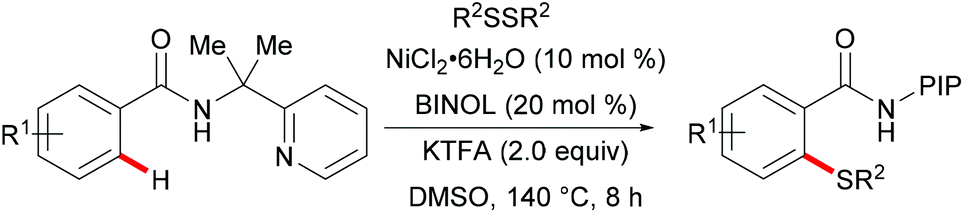
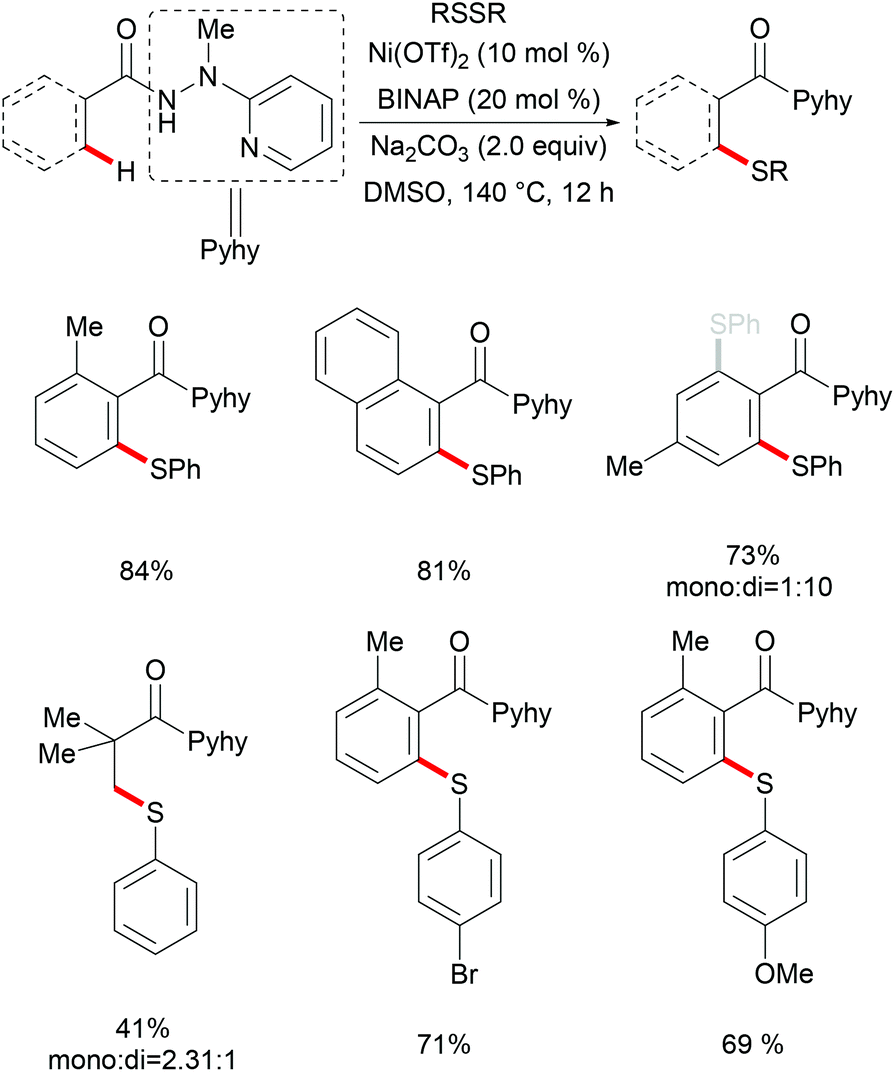
In 2014, Nishihara and Li's group111 reported on the palladium-catalysed C–H thiolation of 2-phenylpyridine with diaryl disulfides in the presence of 20 mol% trimesitylphosphine as a ligand and 10 mol% CuCl2 at 140 °C for 12 h (Scheme 1). 2-Phenylpyridines bearing a wide range of important functional groups, such as ester, methoxy and halides, reacted with diaryl disulfides smoothly to furnish mono-substituted products in moderate to good yields. Moreover, 2-phenylpyrimidine and N-(8-quinolyl)benzamide were converted successfully and resulted in the exclusive formation of dithiolated products. Diaryl disulfides bearing either electron-withdrawing or electron-donating groups were efficiently transformed in this reaction, whereas dialkyl disulfides only gave unsatisfactory yields due to lower reactivity. Importantly, benzenethiol was also a suitable coupling partner under these oxidative conditions and afforded the corresponding product in a good yield. Notably, the ratio of the palladium/phosphine ligand is crucial for this transformation, as no desired product was observed without phosphine ligands. The authors proposed that the ligands might facilitate the dissociation of the palladium dimer to form the active monomeric species. Furthermore, the reaction proceeded efficiently with only 0.6 equivalent of diaryl disulfides, indicating that copper salts could promote the oxidation of aryl thiol to the corresponding disulfide. DMSO was proposed to play a dual role as the solvent and the terminal oxidant, since dimethyl sulfide was observed by GC-MS. The intermolecular and intramolecular kinetic isotope effect (KIE) values of 1.2 and 1.3, respectively, indicated that the C–H bond cleavage is likely not involved in the rate-determining step.
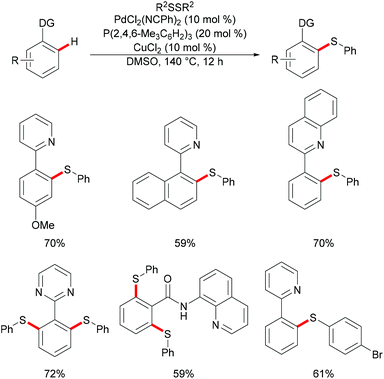 |
| | Scheme 1 Palladium-catalysed direct thiolations of aryl C–H bonds with disulfides. | |
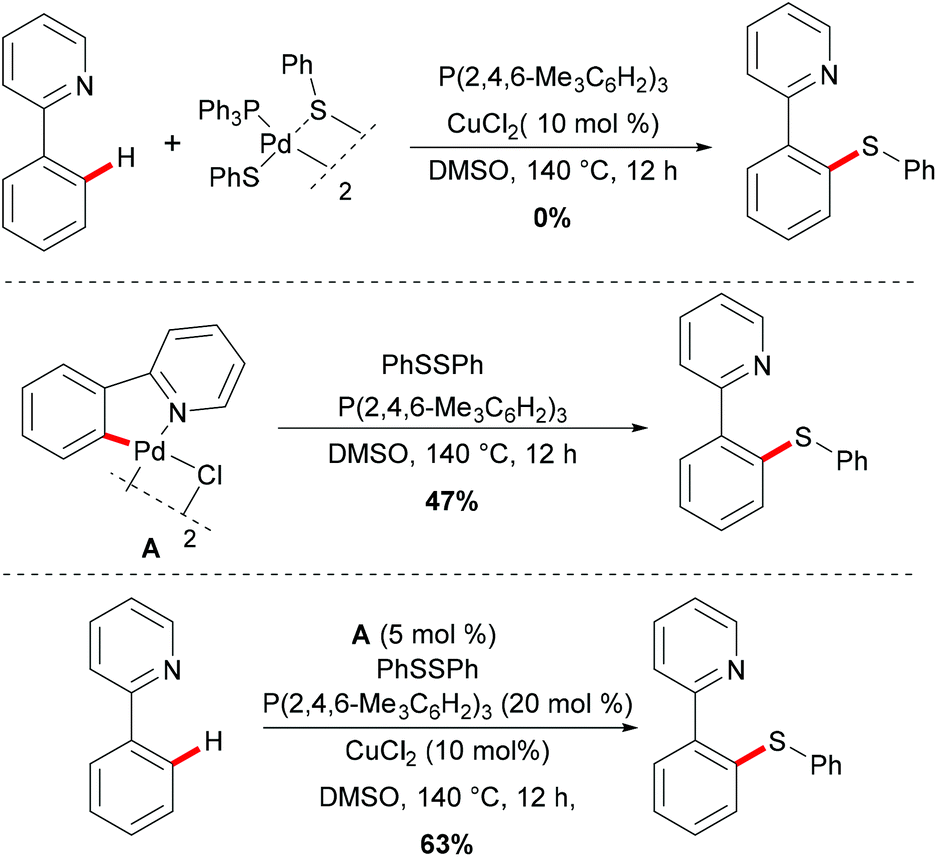
Thereafter, a series of control experiments were carried out with the well-defined five-membered palladacycle or (triphenylphosphine)palladium disulfide dimer as the substrate or catalyst; thus the authors proposed a plausible mechanism involving a Pd(II)/Pd(IV) pathway rather than a Pd(0)/Pd(II) pathway for this thiolation reaction (Scheme 2). Initially, the dimeric palladium catalyst is coordinated by the directing group and achieved the ortho C–H activation of the aryl ring to form a five-membered palladacycle species, followed by an oxidative addition of disulfide to form a palladium(IV) intermediate. Thus, subsequent reductive elimination affords the desired thiolated product and releases the thiolation–palladium complex, which could regenerate the active palladium catalyst that re-enters the catalytic cycle. Finally, the thiol could be oxidized to disulfides by DMSO in the presence of copper salts.
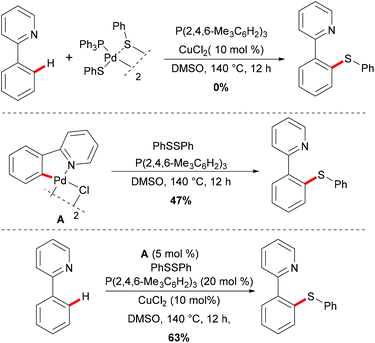 |
| | Scheme 2 Stoichiometric C–H bond thiolations. | |
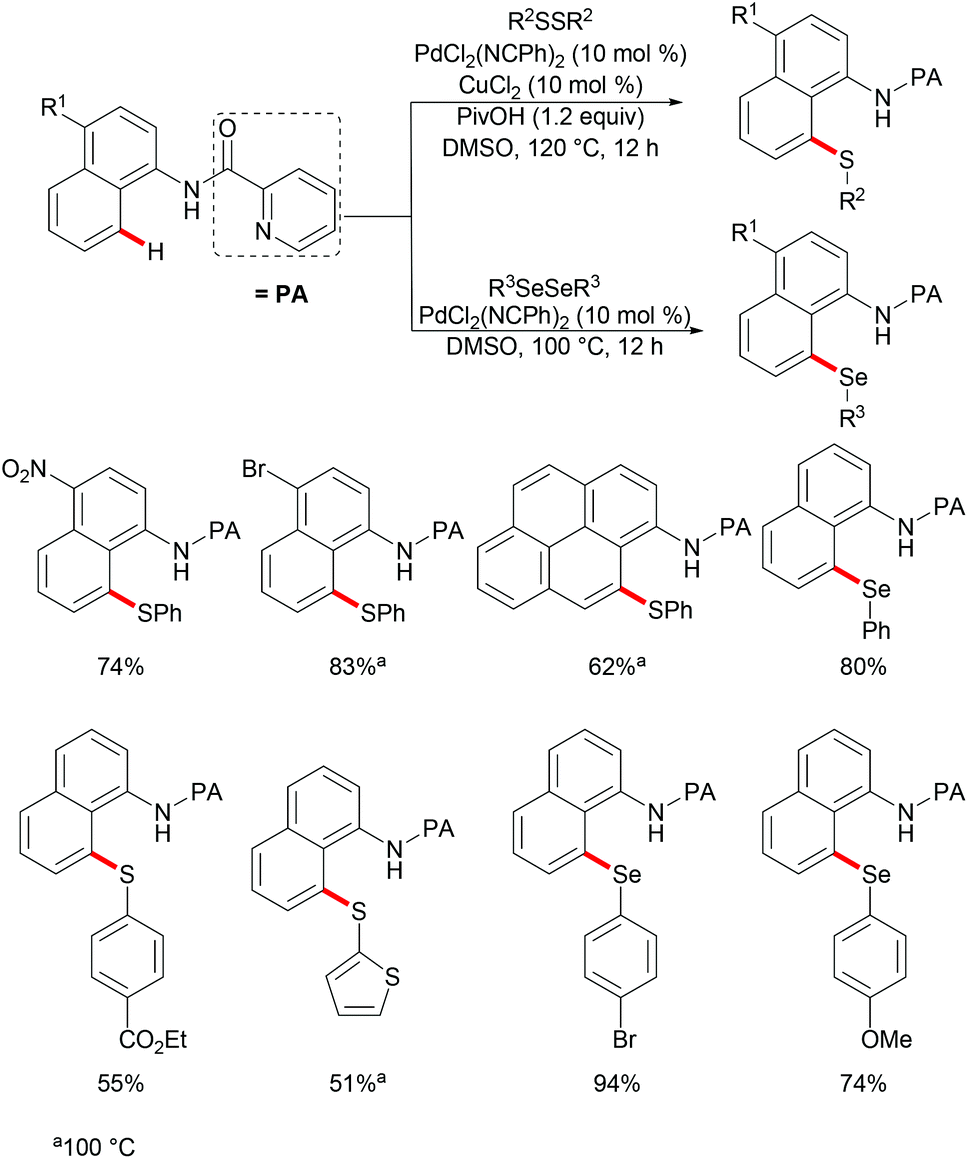
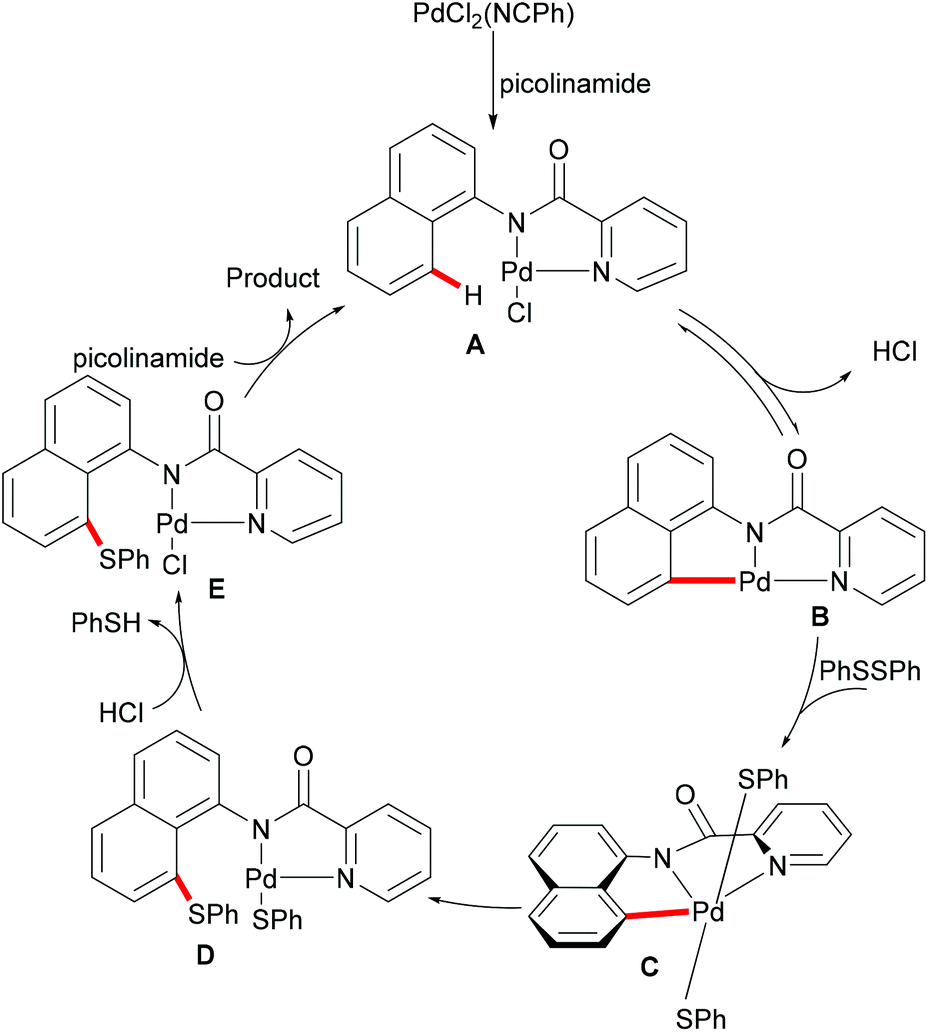
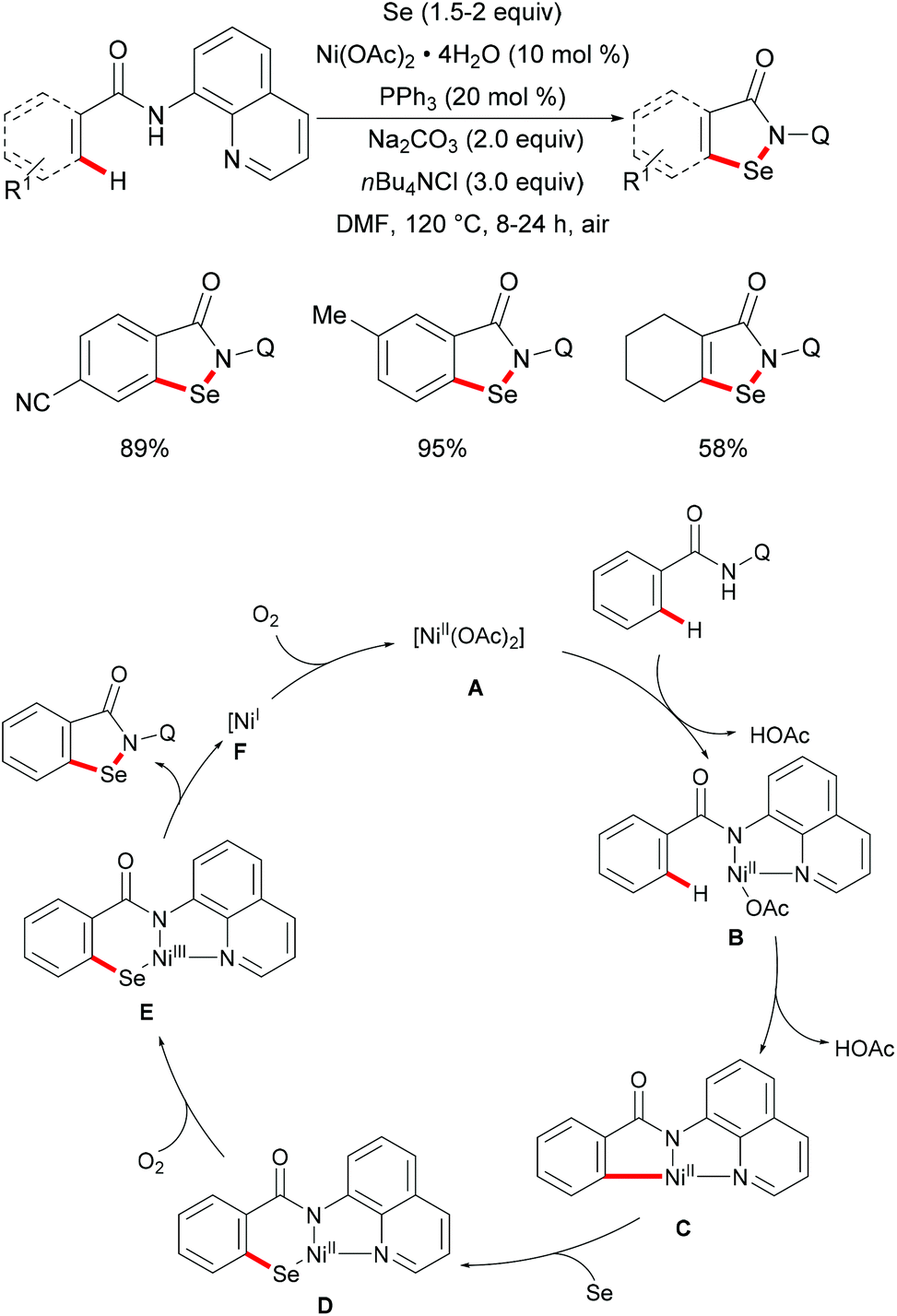
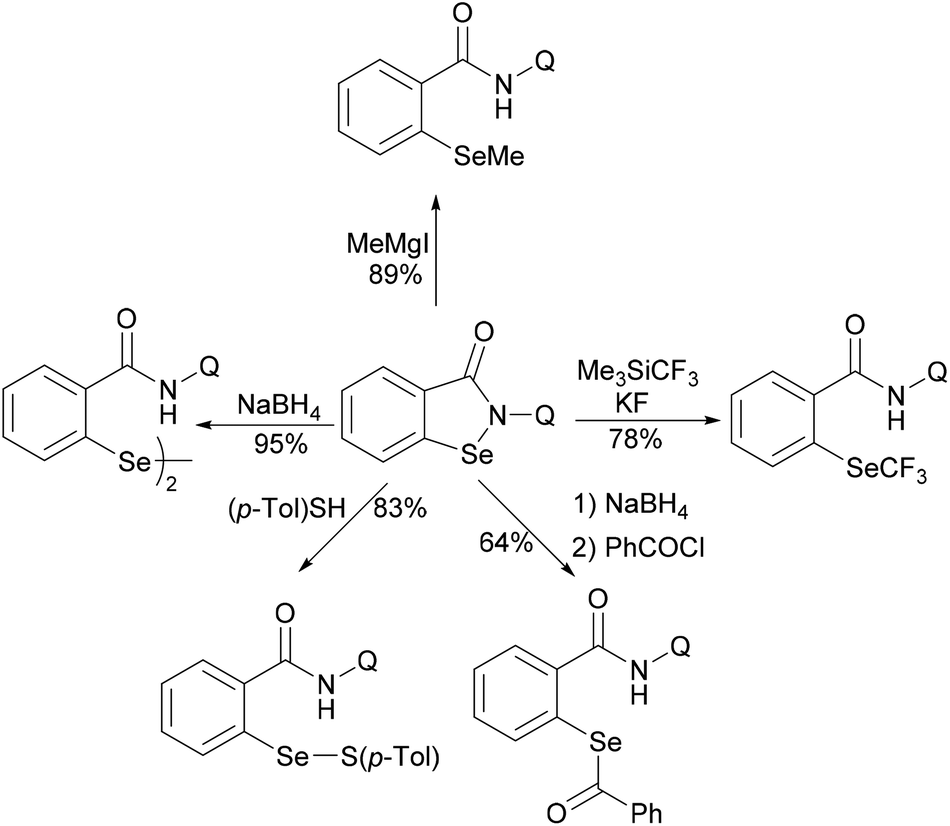
Subsequently, the same group reported on the picolinamide-assisted peri-selective C–H chalcogenylation of naphthylamines under similar reaction conditions (Scheme 3).112 This transformation proved to be successful in the absence of the phosphine ligand. On the basis of the previous observations, showcasing that acid additives could promote C–H functionalizations, the addition of stoichiometric amounts of pivalic acid (PivOH) to the reaction led to significant improvement in the reaction outcome. This robust palladium catalysis tolerated a wide range of substituted naphthylamines and diaryl disulfides, and gave the corresponding products in good to excellent yields. As expected, the palladium catalysis could be expanded to direct selenylation with 0.6 equiv. of diaryl diselanes even in the absence of CuCl2 and PivOH. Importantly, the loading of the catalyst could be reduced to 5 mol% without significant loss of catalytic activity. The picolinamide directing group was easily removed under mild conditions and afforded 8-benzenesulfenyl-naphthylamines, which are important structural motifs in a wide range of functional molecules and pharmaceuticals. Interestingly, when the naphthalene bearing 2-pyridyl and picolinamide directing groups at the same time, peri-thiolation selectively occurred on the 8-position, thus indicating that the bidentate directing group facilitates the formation of the palladacycle due to the bidentate nature of the directing group. Control experiments strongly supported a palladium(II)/palladium(IV) mechanistic pathway (Scheme 4).
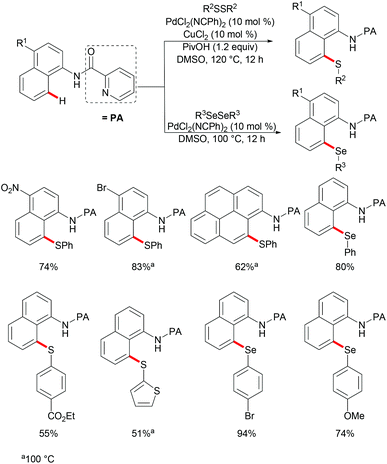 |
| | Scheme 3 Pd-Catalysed peri-selective chalcogenylations of naphthylamines. | |
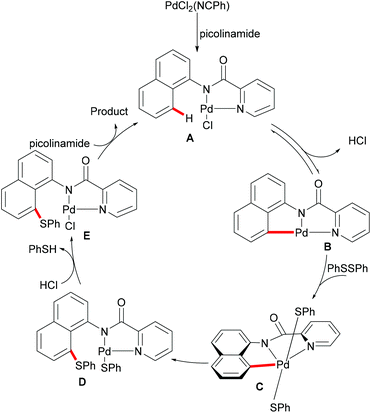 |
| | Scheme 4 Proposed mechanism for the peri-selective chalcogenylations of naphthylamines. | |
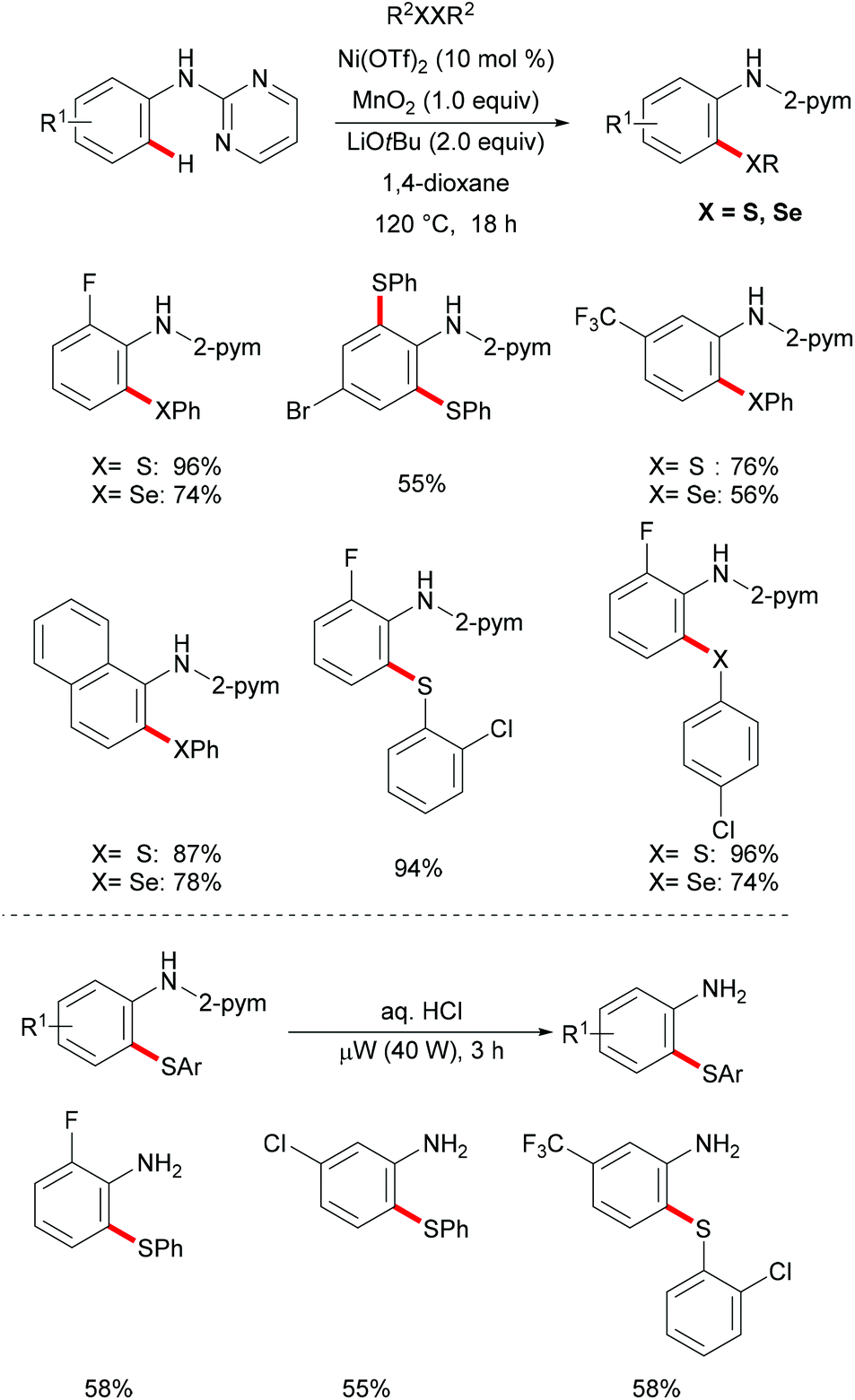
In the same year, Kambe and co-workers independently disclosed a phosphine-ligand-free palladium-catalysed direct C–H chalcogenylation assisted by pyridine or easily removable pyrimidine directing groups.113 This C–H chalcogenylation reaction was compatible with various useful arenes and heteroarenes, including carbazole, 2-phenylpyridine, benzo[h]quinolone, and indole derivatives. Disulfides or diselanes bearing either electron-donating or electron-withdrawing groups smoothly delivered the corresponding products in good to quantitative yields. Notably, dialkyl disulfides also gave the desired products in high yields. Interestingly, when diphenyl disulfides, bis(2,5-dichlorophenyl) disulfides and bis(2,5-dichlorophenyl) diselenides were employed for transformations of indoles, the 2,3-dichalcogenated products were obtained in good yields. These findings imply that the electron-withdrawing nature of the substituent on the chalcogen could accelerate the second chalcogenylation at the 3-position. Finally, detailed mechanistic studies through several control experiments revealed that copper salts could facilitate the transfer of the PhS group to palladium to form a thiolate complex and the bis-six-membered palladacycle was the key intermediate in this reaction. Moreover, the 2-pyrimidyl directing group could be easily removed to provide the thiolated carbazole in high yields (Scheme 5).
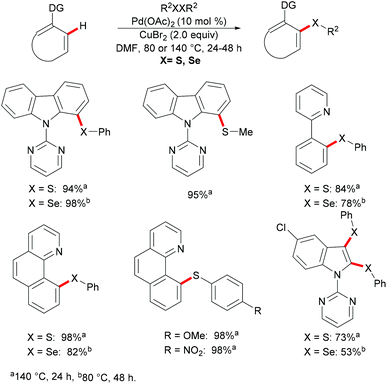 |
| | Scheme 5 Palladium(II)-catalysed intermolecular C–H chalcogenylations of arenes. | |
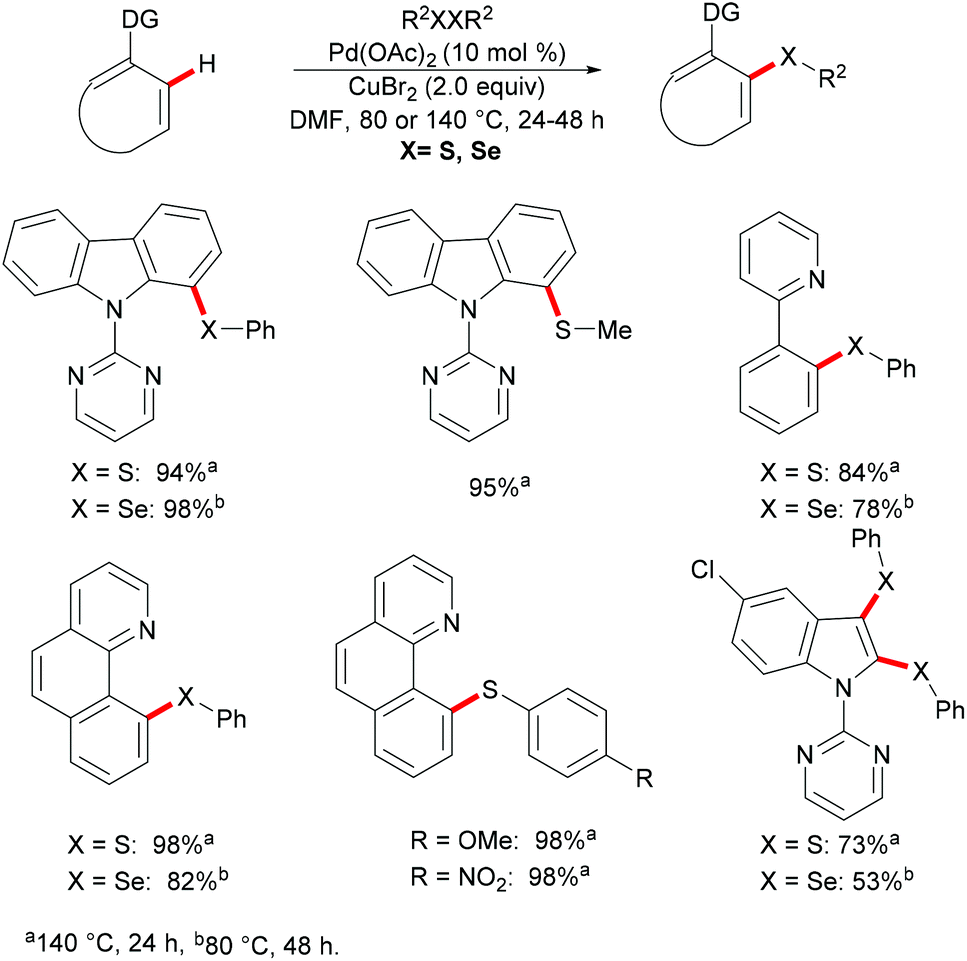
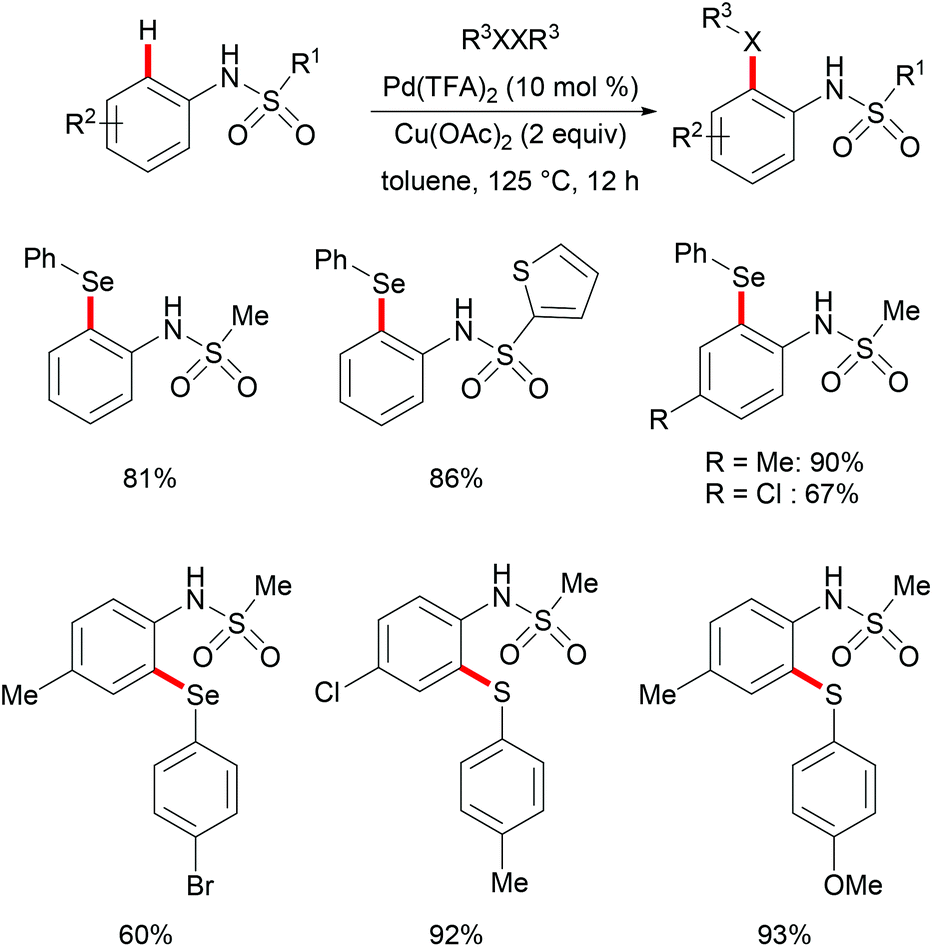
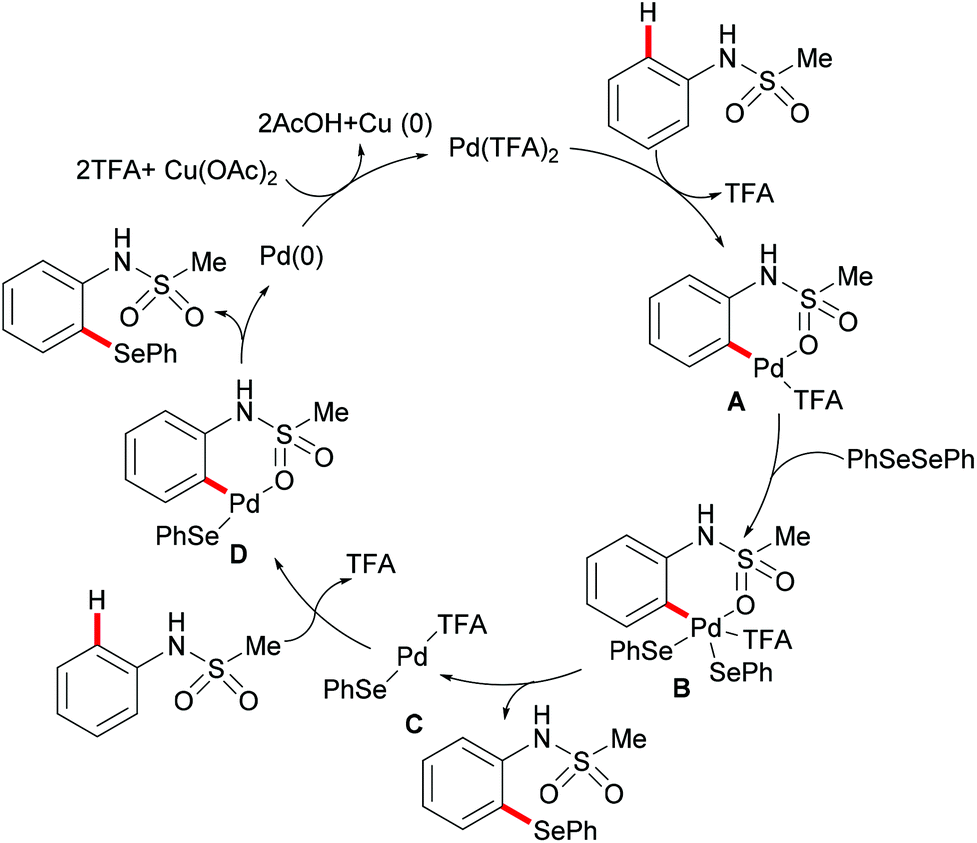
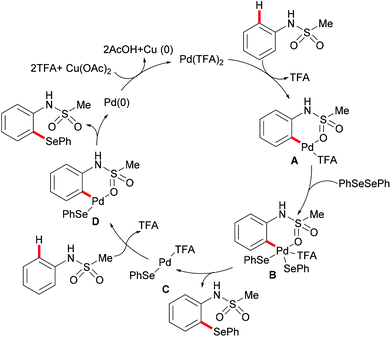
More recently, Ma and coworkers reported the palladium(II)-catalyzed ortho-C–H chalcogenylations of N-arylsulfonamides via weak coordination (Scheme 6). Thus, they found that the combination of 10 mol% Pd(TFA)2 and 2.0 equiv. of Cu(OAc)2 in toluene at 125 °C proved to be optimal for this transformation.114 The reaction tolerated a wide range of valuable electrophilic functional groups in both the arylsulfonamide and disulfide moieties, and delivered selectively mono-chalcogenated products in good yields with excellent chemo- and positional-selectivities, thus providing a novel and step-economical access to diversely selenylated and thiolated sulfonamides. Further mechanistic studies also revealed a palladium(II)/palladium(IV) process, likely initiated by the coordination of the palladium(II) catalyst with the weakly coordinating O-atom of the sulfonamide, followed by ortho-C–H activation to furnish six-membered palladacycle intermediate A. The subsequent oxidative addition of 1,2-diphenyl diselane onto palladacycle species A gives palladium(IV) species B, which subsequently undergoes a reductive elimination to afford the desired product and palladium(II) species C. Then, intermediate C participates in a second C–H activation event to form palladacycle species D. Intermediate D then undergoes reductive elimination generating the target product and a palladium(0) species, which could be converted into the active palladium(II) species in the presence of the external oxidant Cu(OAc)2 (Scheme 7).
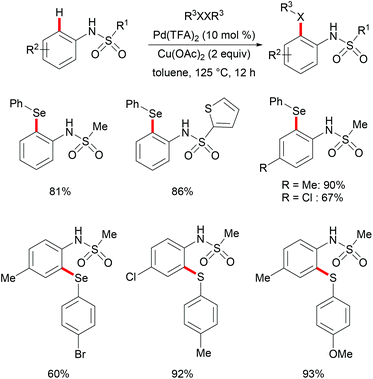 |
| | Scheme 6 Palladium(II)-catalysed ortho-C–H chalcogenylations of N-arylsulfonamides. | |
 |
| | Scheme 7 Proposed mechanism for palladium(II)-catalysed ortho-C–H chalcogenylations of N-arylsulfonamides. | |
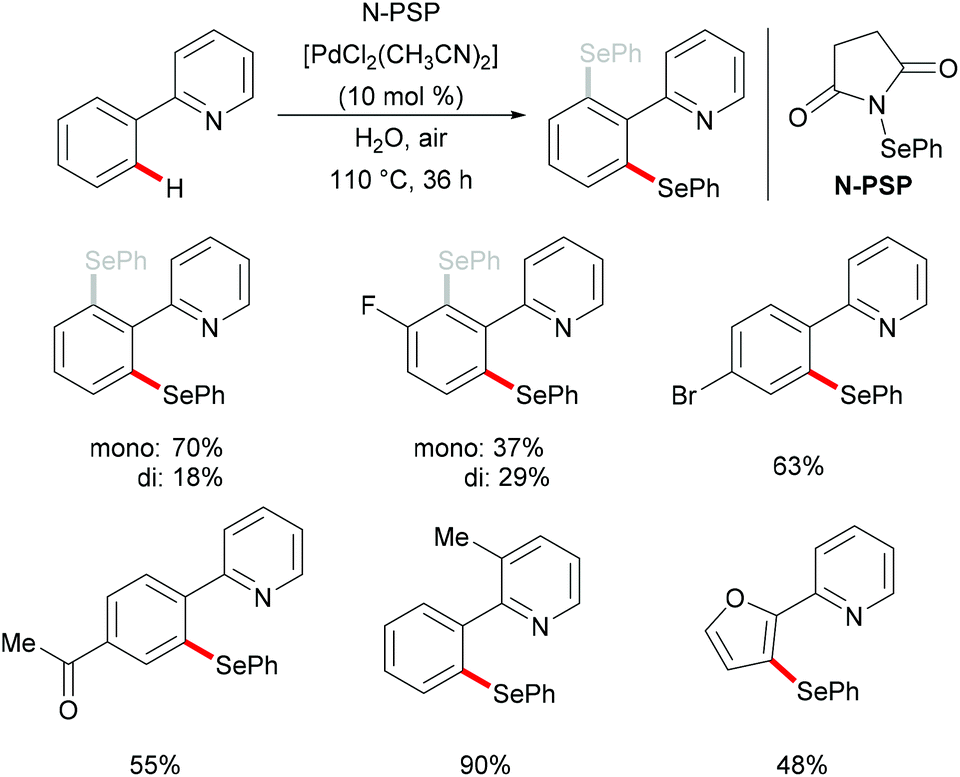
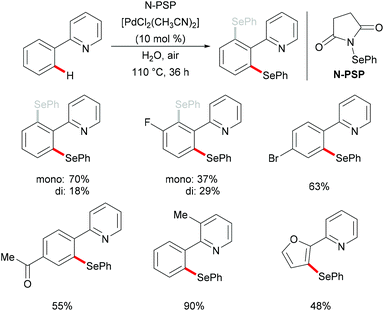
In 2015, Law's and Wong's groups disclosed a highly robust palladium(II)-catalyzed direct C–H phenylselenylation of arenes and heteroarenes with N-(phenylseleno)phthalimide (N-PSP) (Scheme 8).115 Both palladium(0) and palladium(II) catalysts displayed high catalytic efficiency in this reaction. The notable features of this new protocol include wide substrate scope, no use of external oxidants, surfactants, and other additives and water as a green reaction medium. The authors proposed the importance of possible hydrogen bonding between water and the carbonyl group of N-PSP. A variety of electron-donating or electron-withdrawing substituents on the arene were well tolerated and afforded the desired mono- and/or diphenylselenylation products in modest to excellent yields. The Hammett plots were constructed based on the product ratios vs. Hammett constants, indicating that electron-donating groups could facilitate the formation of an active electrophilic palladium complex as a key reaction intermediate. A plausible palladium(II)/palladium(IV) mechanism involving oxidative addition of the chalcogenylation agent to form the palladium(IV) intermediate was proposed based on the preliminary results and the previous reports. Alternatively, other pathways, such as the direct electrophilic aromatic substitution, a process involving Pd–C bond cleavage and C–Se formation sequence or the palladium(II)/palladium(III) catalytic cycle including the formation of bimetallic palladium(III) intermediates, cannot as of yet be ruled out.
 |
| | Scheme 8 Palladium-catalysed direct C–H selenylation of (hetero)arenes in water. | |
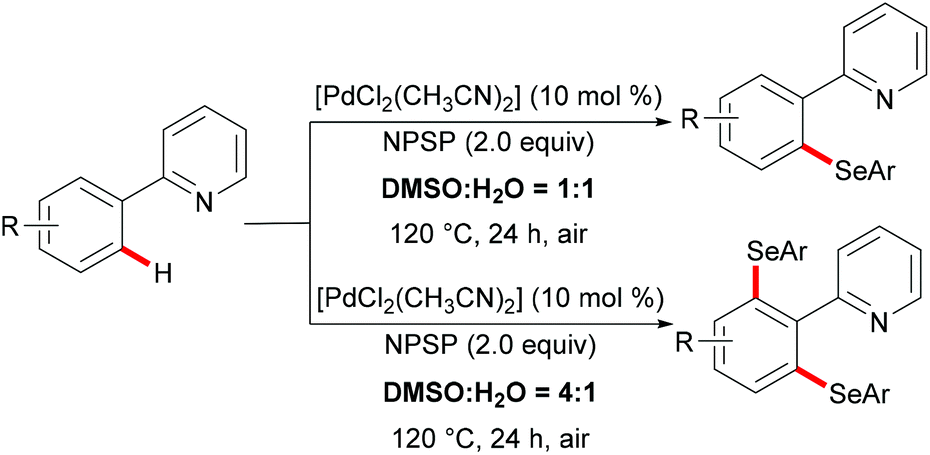
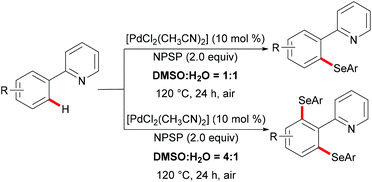
Shortly thereafter, the same groups achieved completely controlled mono- and di-selective selenylations by finely tuning the ratio of DMSO to water (Scheme 9).116 The substrate scope was expanded to include 2-phenoxylpyridines. This new selenylation can be considered as a complementary approach to the previous C–H selenylation (vide supra). Similarly, in contrast with N-(phenylseleno)phthalimide (N-PSP), Zhang and coworkers achieved palladium catalysed selective C–H sulfenylations with N-arylthiobenzamides as both the reactant and the oxidant at the same time (Scheme 10).117 2-Arylpyridines and diaryl disulfides bearing various functional groups were well tolerated under the optimized reaction conditions and afforded the mono-selective products in up to 90% yield. Interestingly, the dithiolation could be achieved by changing the stoichiometry of the substrates to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. Control experiments indicated that the C–H thiolation might involve a palladium(II)/palladium(IV) pathway.
:4. Control experiments indicated that the C–H thiolation might involve a palladium(II)/palladium(IV) pathway.
 |
| | Scheme 9 Palladium(II)-catalysed switchable mono/diselenylation of arenes controlled by the judicious choice of the solvent ratio. | |
 |
| | Scheme 10 Palladium(II)-catalysed selective C–H thiolations of arene with N-arylthiobenzamides. | |
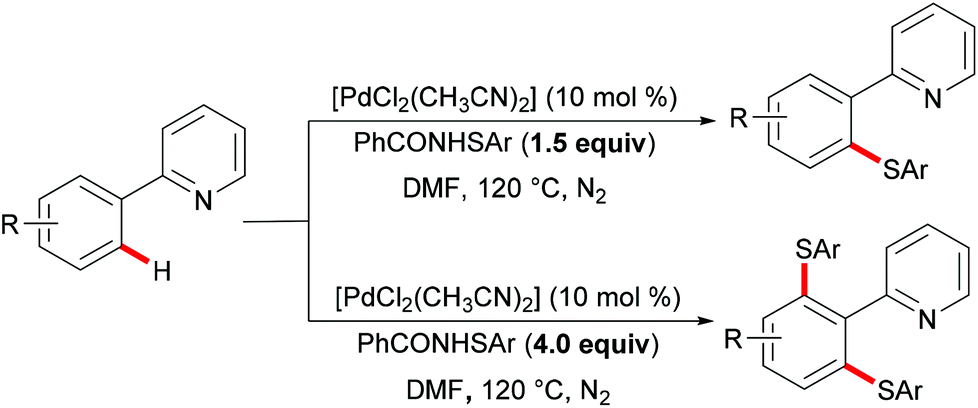
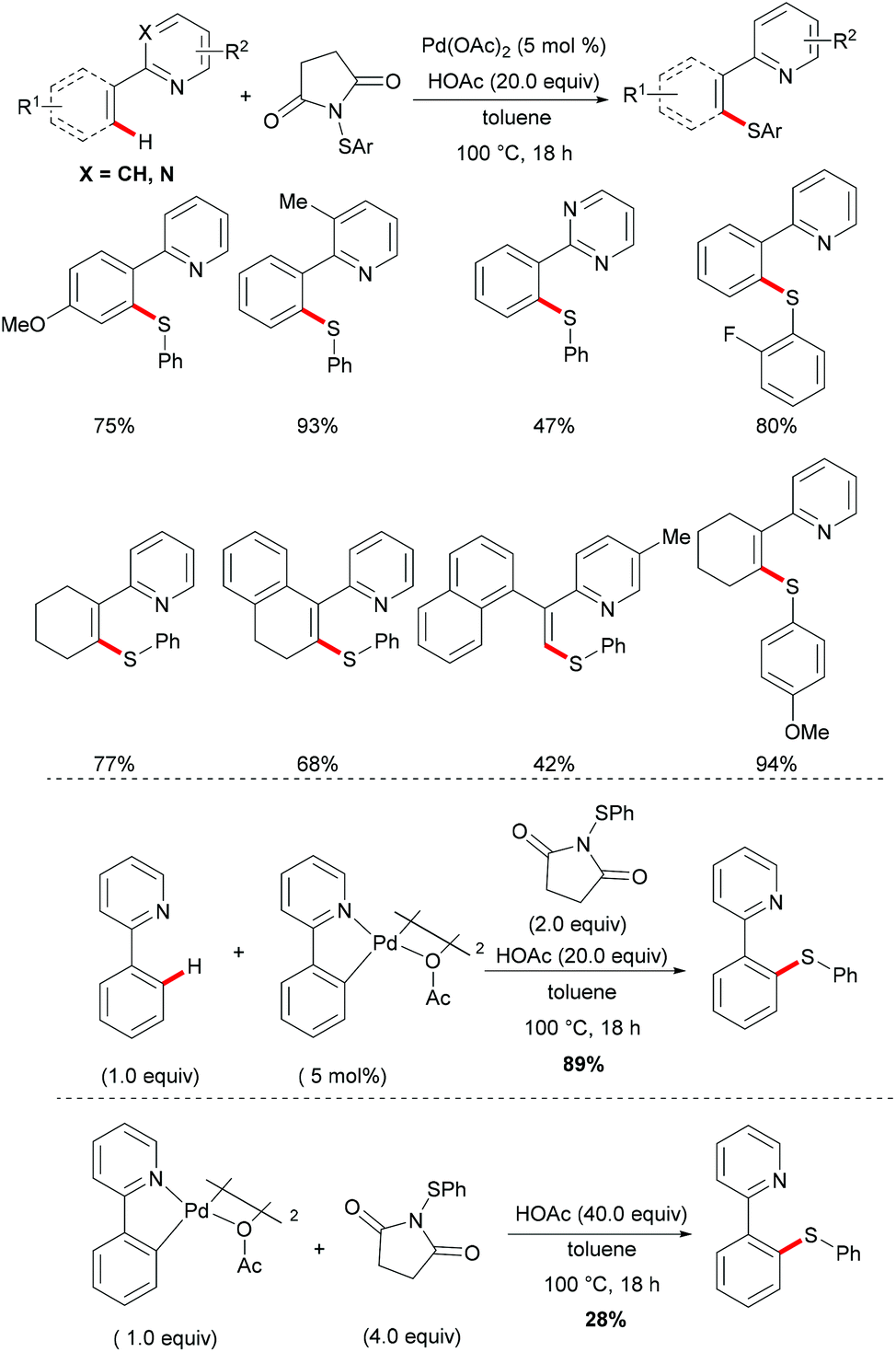
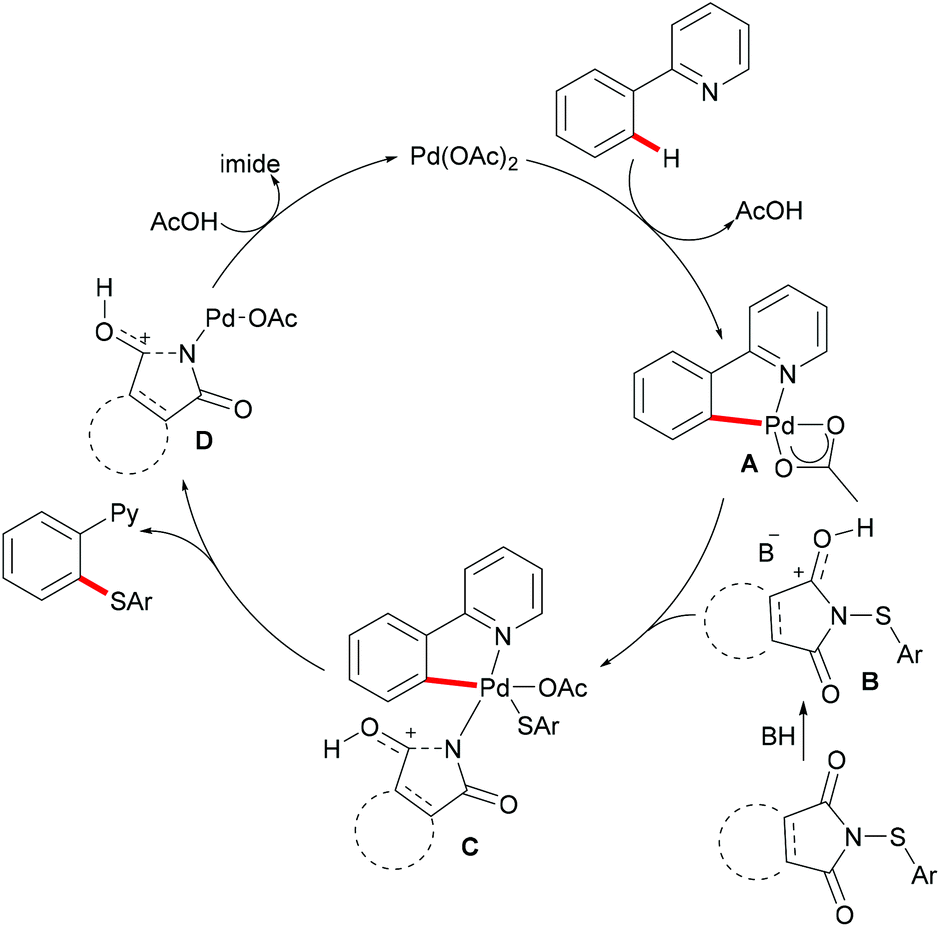
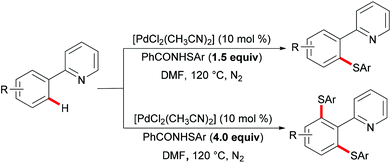
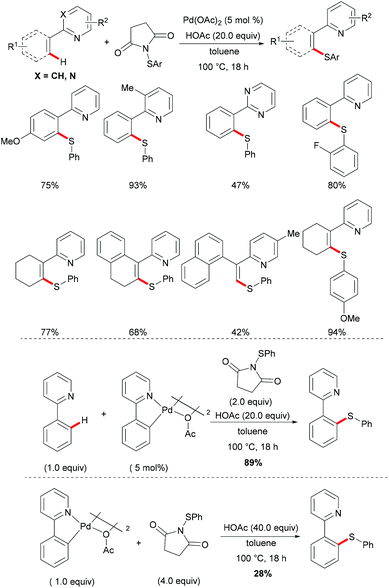
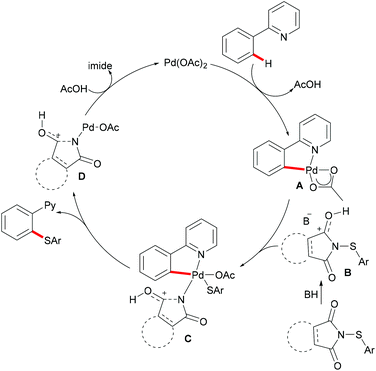
More recently, Anbarasan's group reported on a versatile protocol for palladium-catalysed C–H thiolations and i(a)midation using N-(arylthiol)i(a)mide as a suitable reagent in the presence of either a Brønsted acid or a Lewis acid, respectively.118 This C–H transformation employed N-(arylthiol)i(a)mide as a versatile reagent acting as a thiolating reagent, an amidation reagent and an oxidant in this process (Scheme 11). Moreover, the alkenyl substrates were also smoothly converted under the optimized conditions and afforded the desired products selectively. Notably, the independently prepared five-membered dimeric palladacycle complex displayed high catalytic efficiency and furnished the thiolated product with high efficacy. Moreover, the treatment of a stoichiometric palladacycle with 4.0 equiv. of (phenylthiol)imide and 40.0 equiv. of AcOH afforded the thiolated product in 28% yield. These results indicate that the dimeric palladacycle species may be the resting state of the catalyst and could generate the active monomeric species. Next, variable temperature FTIR and NMR studies of N-(arylthio)imides with acetic acid were conducted to understand the mode of the interaction. The observations of a significant shift in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency in FTIR, a downfield shift of the carbonyl carbon in 13C NMR and no significant change of the aromatic signal in an acidic medium indicated that the imide oxygen atom was protonated by AcOH. Based on the preliminary studies and earlier literature precedence, a plausible mechanism involving a palladium(II)/palladium(IV) process was proposed for the developed transformations (Scheme 12). The reaction is initiated by the coordination of the palladium catalyst by the directing group, promoting the ortho-C–H bond activation to form a five-membered palladacycle, which then undergoes oxidative addition to the N–S reagent, which is activated by AcOH, leading to a palladium(IV) intermediate. The thus formed palladium(IV) species eventually underwent a reductive elimination process to produce the desired thiolated product.
O stretching frequency in FTIR, a downfield shift of the carbonyl carbon in 13C NMR and no significant change of the aromatic signal in an acidic medium indicated that the imide oxygen atom was protonated by AcOH. Based on the preliminary studies and earlier literature precedence, a plausible mechanism involving a palladium(II)/palladium(IV) process was proposed for the developed transformations (Scheme 12). The reaction is initiated by the coordination of the palladium catalyst by the directing group, promoting the ortho-C–H bond activation to form a five-membered palladacycle, which then undergoes oxidative addition to the N–S reagent, which is activated by AcOH, leading to a palladium(IV) intermediate. The thus formed palladium(IV) species eventually underwent a reductive elimination process to produce the desired thiolated product.
 |
| | Scheme 11 Brønsted acid-controlled palladium(II)-catalysed C(sp2)–H thiolations with N-(arylthio)i(a)mides. | |
 |
| | Scheme 12 Proposed mechanism of Brønsted acid-controlled palladium(II)-catalysed C(sp2)–H thiolations. | |
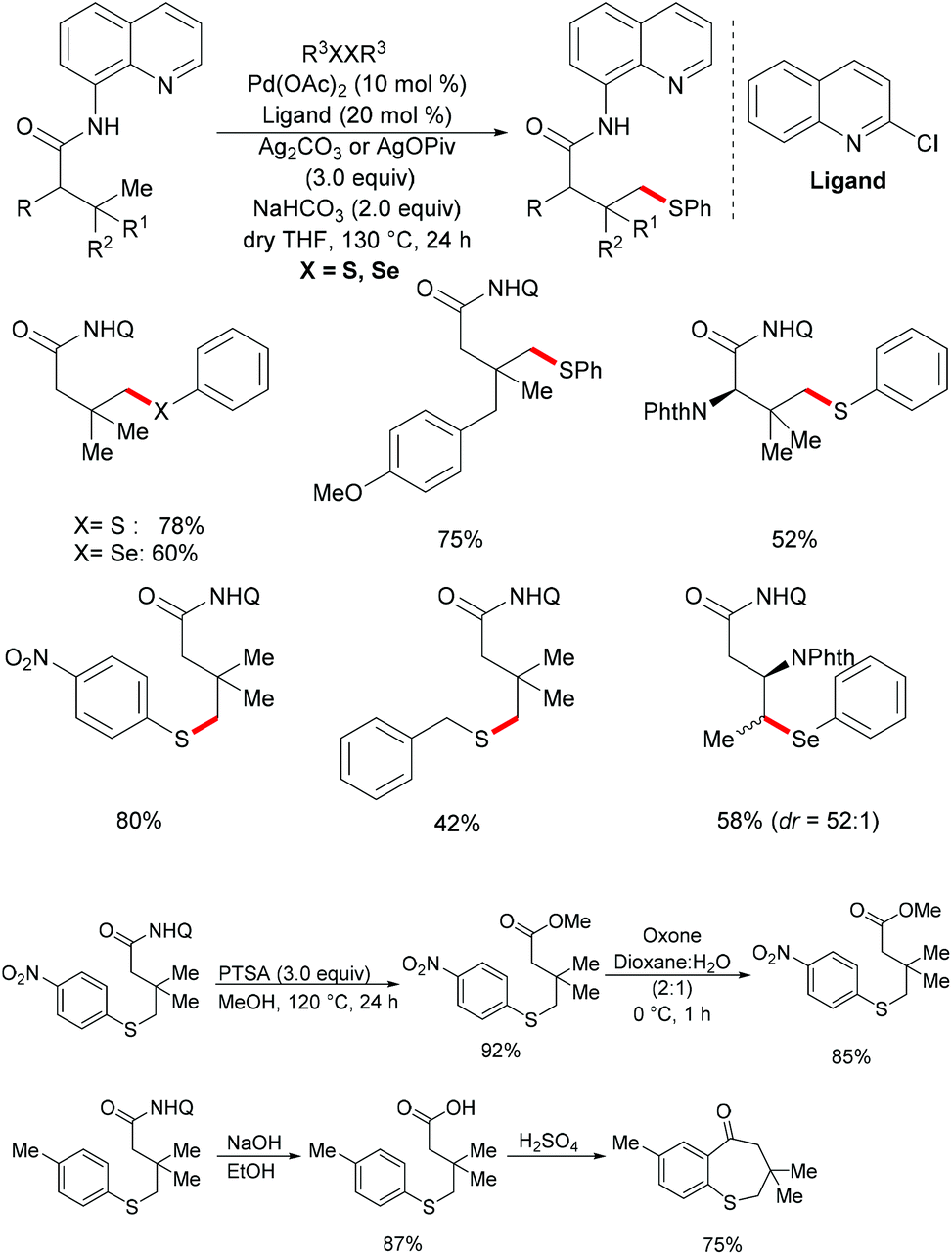
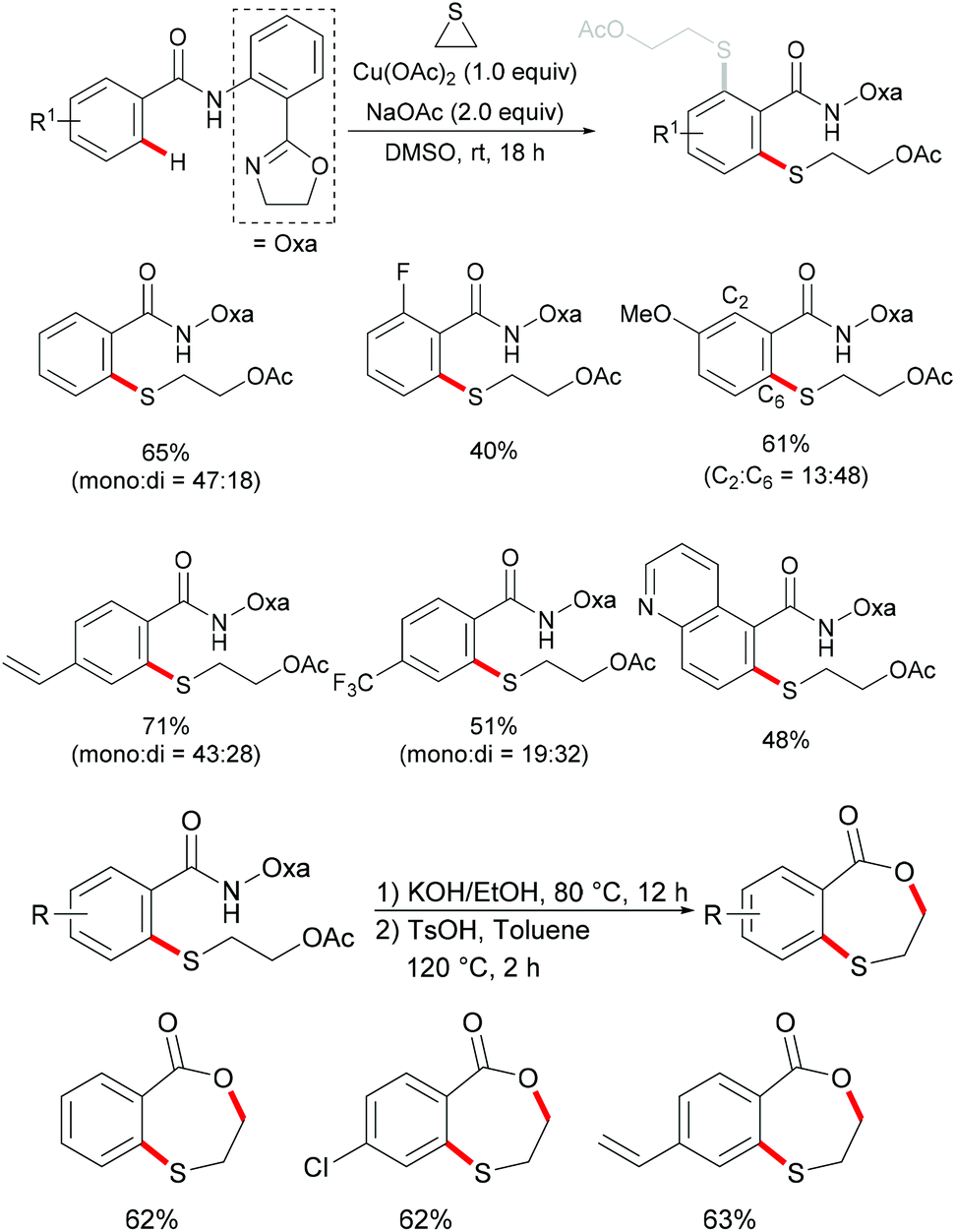
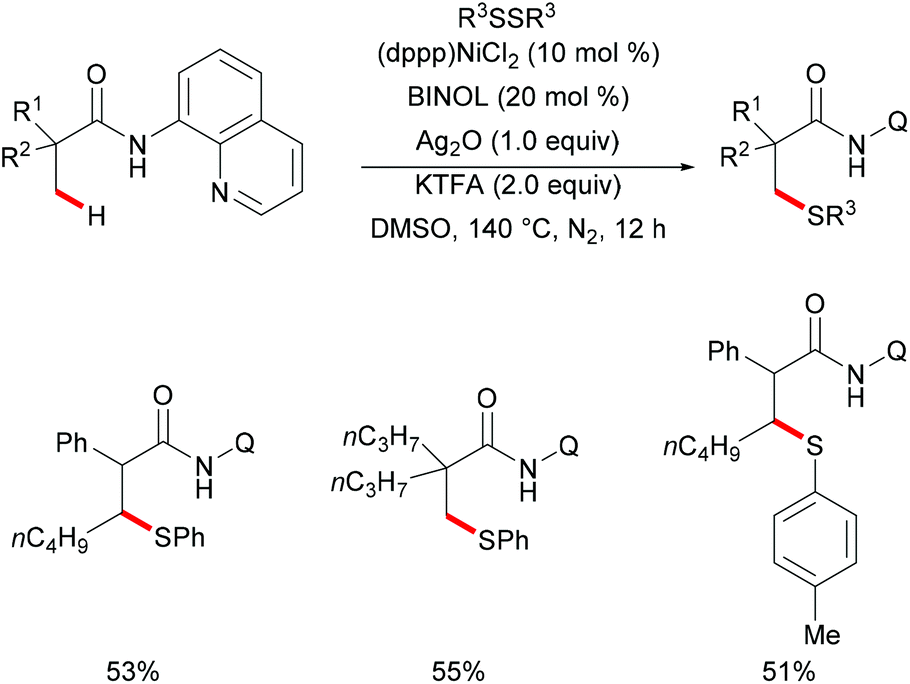
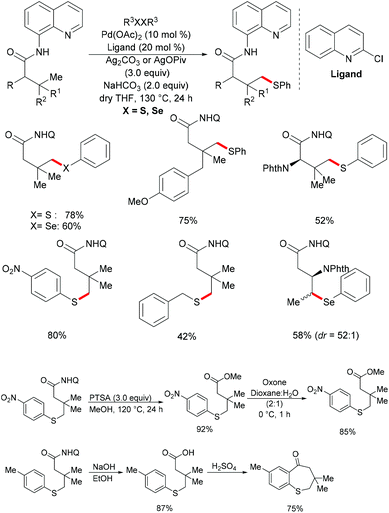
In comparison with the well-established C(sp2)–H bond functionalization processes, the direct chalcogenylation of C(sp3)–H continues to be scarce due to a lack of favourable π-interactions. Despite these challenges, the Maiti group reported a diastereoselective remote γ-C–H chalcogenylation of α-amino acids and aliphatic carboxylic acids via a six-membered palladacycle intermediate (Scheme 13).119 These chalcogenylation reactions smoothly proceeded in the presence 10 mol% Pd(OAc)2, 3.0 equiv. of Ag2CO3, 20 mol% 2-chloroquinoline as a ligand and 2.0 equiv. of NaHCO3 at 130 °C to exclusively provide mono-thiolated products in good yields with high diastereoselectivity. The key to the success was the use of the 2-chloroquinoline ligand, which presumably accelerates and facilitates the regioselective functionalizations of C(sp3)–H and enhances the yield of the desired products. Although secondary C(sp3)–H bonds were also potentially feasible, the primary γ-C–H position was preferentially functionalized presumably due to the steric hindrance of the intermediate metallacycle at the secondary position. Moreover, the sequential hetero bifunctionalization of aliphatic acids was achieved by carrying out γ-arylation as the first step and subsequently thioarylation at the other primary C–H position. It is noteworthy that α-amino acids possessing primary γ-C–H such as L-tert-leucine, L-valine, and L-isoleucine derivatives were also compatible, selectively affording γ-thioarylated products in moderate yields without racemization of the stereogenic α-centre of the amino acids. Diaryl disulfides bearing either electron-donating groups, such as methyl and methoxy, or electron-withdrawing groups, such as chloro and nitro, at the para position reacted smoothly, delivering the thiolated products in satisfactory yields. Importantly, the developed protocol was also compatible with aliphatic disulfides such as dibenzyl disulfide. As expected, diaryl diselenides were also suitable coupling partners with various aliphatic carboxylic acids and amino acids with AgOPiv as the oxidant and provided the γ-selective selenylated products with high diastereoselectivity. Moreover, the established protocol could be expanded to the gram scale, albeit with a slight decrease in the isolated yield. Finally, the directing group was easily removed under either acidic or basic conditions to provide the corresponding methyl ester and carboxylic acid products respectively. In another approach, treatment of the carboxylic acid with conc. H2SO4 gave the biologically relevant benzothiepinone moiety via an intramolecular cyclization process.
 |
| | Scheme 13 Palladium(II)-catalysed diastereoselective γ-C–H chalcogenylations of amino acids and aliphatic carboxylic acids. | |
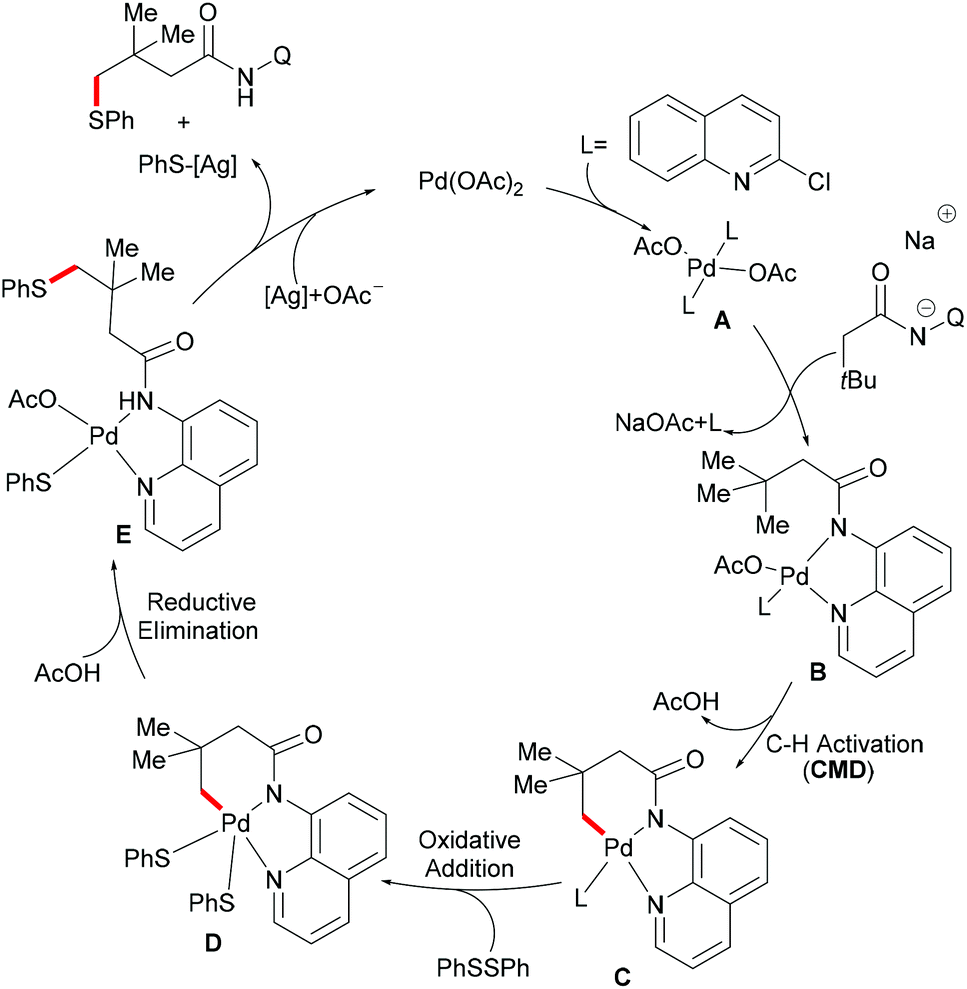
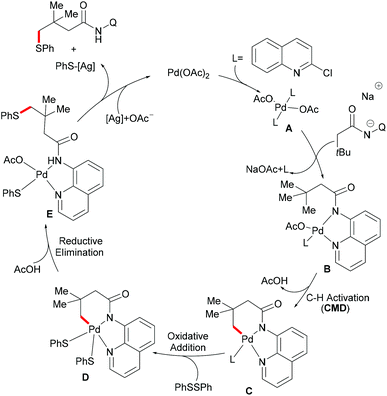
Based on the preliminary mechanistic studies and the previous literature reports, a plausible catalytic cycle was proposed as shown in Scheme 14. Palladium(II) is coordinated by the 2-chloroquinoline ligand to form the pre-catalyst A, which binds with the bidentate amide directing group to give the active catalyst–substrate chelate complex B, followed by selective γ-C–H activation to form a six-membered palladacycle intermediate via the concerted metalation–deprotonation (CMD) pathway.120 Subsequent oxidative addition of disulfide onto the palladium(II) centre provides the palladium(IV) species D that thereafter undergoes a reductive elimination to afford the γ-thioarylated product. Further experimental observations revealed that the C–H activation is reversible and not involved in the rate-determining step.
 |
| | Scheme 14 Plausible catalytic cycle for γ-chalcogenylation. | |
2.2 Rhodium catalysis
As early as 2007, Satoh and Miura's group reported the first rhodium-catalysed chelation assisted C–H activation of arenes.121 Since then the well-characterized [RhCp*Cl2]2 catalysed C–H functionalization with a broader scope of coupling partners, including aromatic or olefin π bonds, strained rings, and electrophilic and nucleophilic reagents has been widely explored and has attracted increasing attention.122–125 In contrast with the well-explored palladium catalyst, the robust [RhCp*Cl2]2 catalysts displayed high activity under mild reaction conditions with broad functional group tolerance due to the uniqueness of the more polarized Rh(III)–C(aryl) bond and the bulky Cp* ligand which could promote favorable steric hindrance during the C–H activation process and enabled the rhodium catalyst to remain thermally stable.
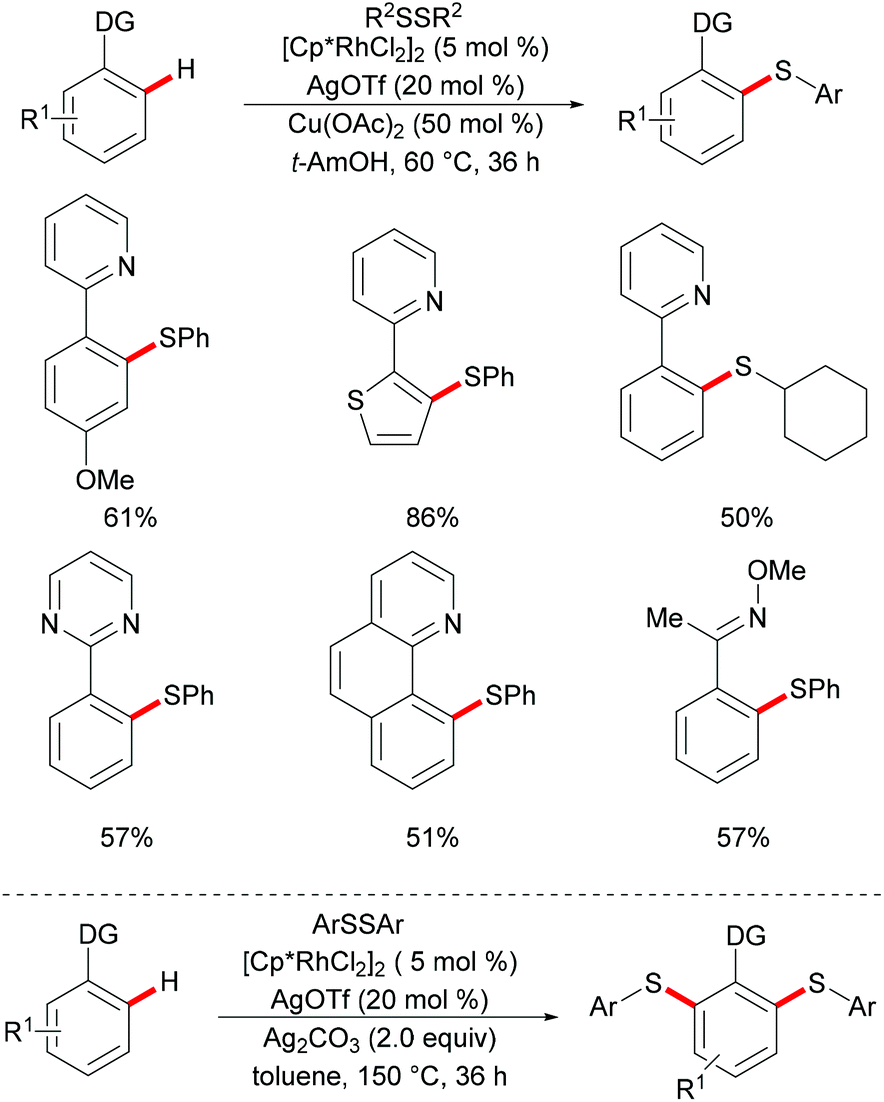
In 2014, Li, Zhou and coworkers reported on the first example of a rhodium-catalysed direct C–H thiolation with disulfides, assisted by the pyridine directing group.126 This protocol proved to be operative under mild reaction conditions with a broad functional group tolerance, hence affording the desired ortho mono-thiolated products in good to excellent yields (Scheme 15). Importantly, dialkyl and dibenzyl disulfides were also compatible and gave the desired products selectively. Notably, numerous different N-containing directing groups, such as ketoximes, pyrimidines and pyrazoles, also served as effective directing groups in the C–H thiolation reaction, thus expanding the scope of this protocol. Notably, this method also allowed for dithiolations by changing the oxidant to Ag2CO3. The intermolecular KIE value of 4 was determined by competition, indicating that C–H bond cleavage is involved in the rate-determining step. Detailed mechanistic studies indicated that a nucleophilic-addition or oxidative addition pathway might be involved in the C–H thiolation (Scheme 16).
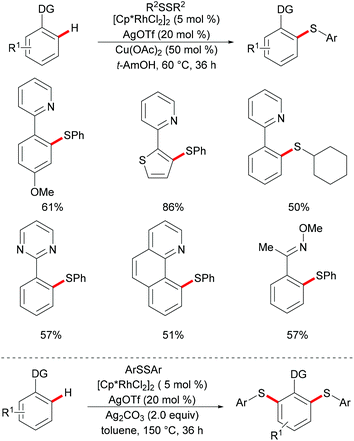 |
| | Scheme 15 Rhodium-catalysed directed thiolations of aromatic C–H bonds. | |
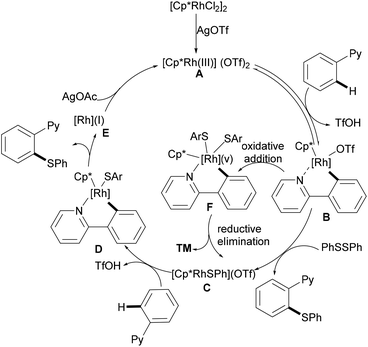 |
| | Scheme 16 Proposed mechanism for rhodium-catalysed directed C–H bond thiolations of arenes. | |
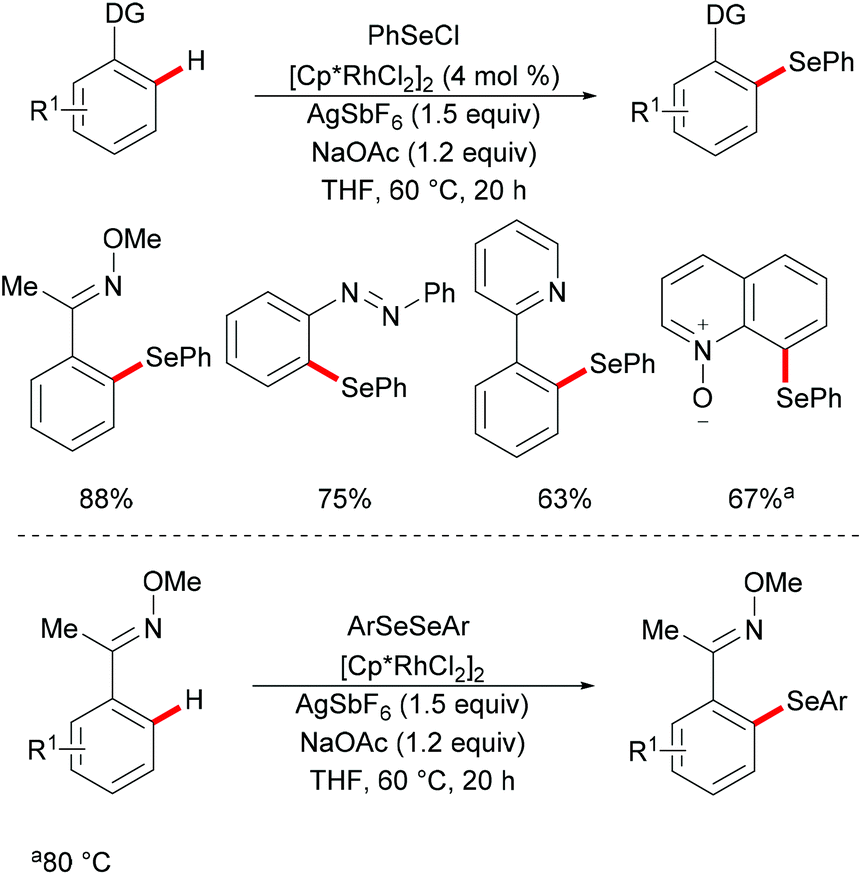
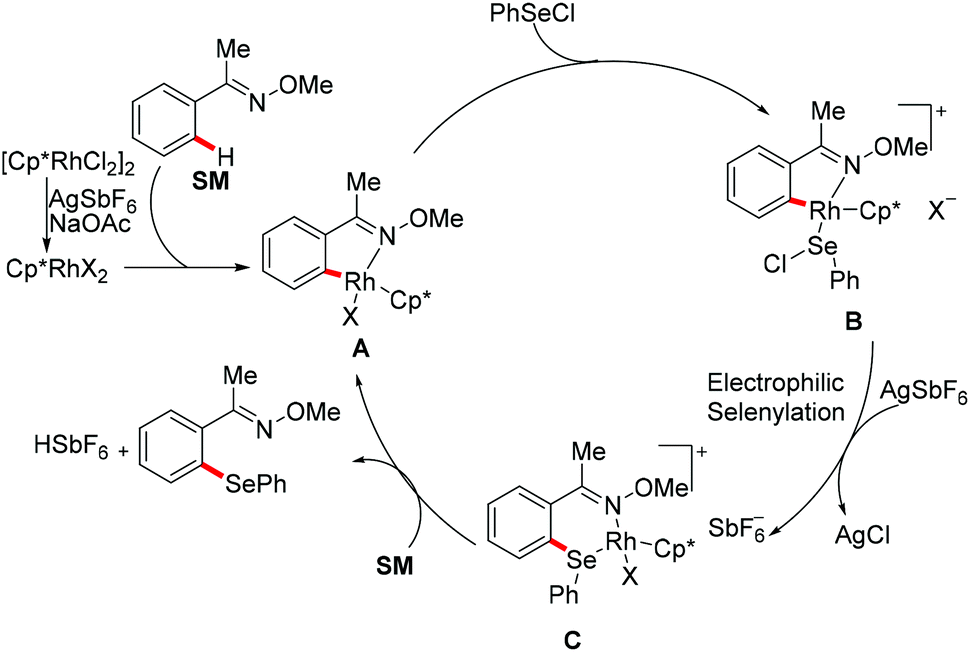
Subsequently, Wan/Li's group reported the rhodium(III)-catalysed direct selenylation of arenes assisted by ketoxime with electrophilic selenenyl chlorides as the selenylated reagents (Scheme 17).127 The silver salt presumably played a dual role in terms of both substrate and catalyst activation. Gratifyingly, arenes bearing oxime, azo, pyridyl, and N-oxide directing groups were also amenable substrates under these reaction conditions and afforded the desired products in good yields. Importantly, the catalyst was highly efficient under mild reaction conditions for a broad range of substrates with excellent functional group tolerance, which provided a new protocol to access unsymmetrical diaryl selenoethers. Moreover, diaryl diselenides were also suitable coupling partners under the same catalytic conditions. A cyclometalated rhodium(III) complex used as the catalyst precursor also afforded the desired product in a moderate yield. On the basis of mechanistic studies and previous literature reports, the authors proposed a plausible catalytic cycle involving an electrophilic substitution pathway for this transformation, as shown in Scheme 18. Initially, the active rhodium catalyst reacts with the substrate to give five-membered rhodacyclic intermediate A through ortho C–H activation of the arene. Next, selenium coordinates to the rhodium catalyst, followed by nucleophilic displacement of Cl by the Rh–C(aryl) bond (electrophilic selenylation) to give five-membered rhodacyclic species B. The subsequent coordination of oxime to intermediate C through another C–H activation process takes place and releases the coupling product along with the regeneration of the active cyclometalated rhodium complex A. However, a rhodium(III)/rhodium(V) pathway involving the Se–Cl bond oxidatively adding to a rhodium(III) species cannot thus far be excluded.
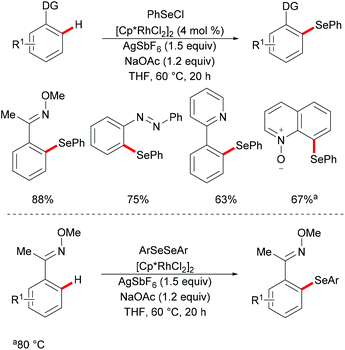 |
| | Scheme 17 Rhodium(III)-catalysed C–H selenylations of arenes with selenenyl chlorides or diselenides. | |
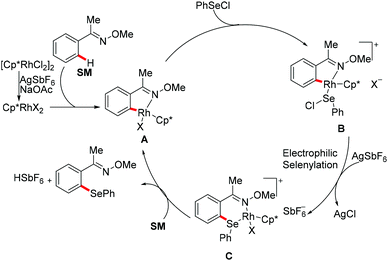 |
| | Scheme 18 Proposed catalytic cycle for the C–H selenylations of arenes with selenenyl chlorides/diselenides. | |
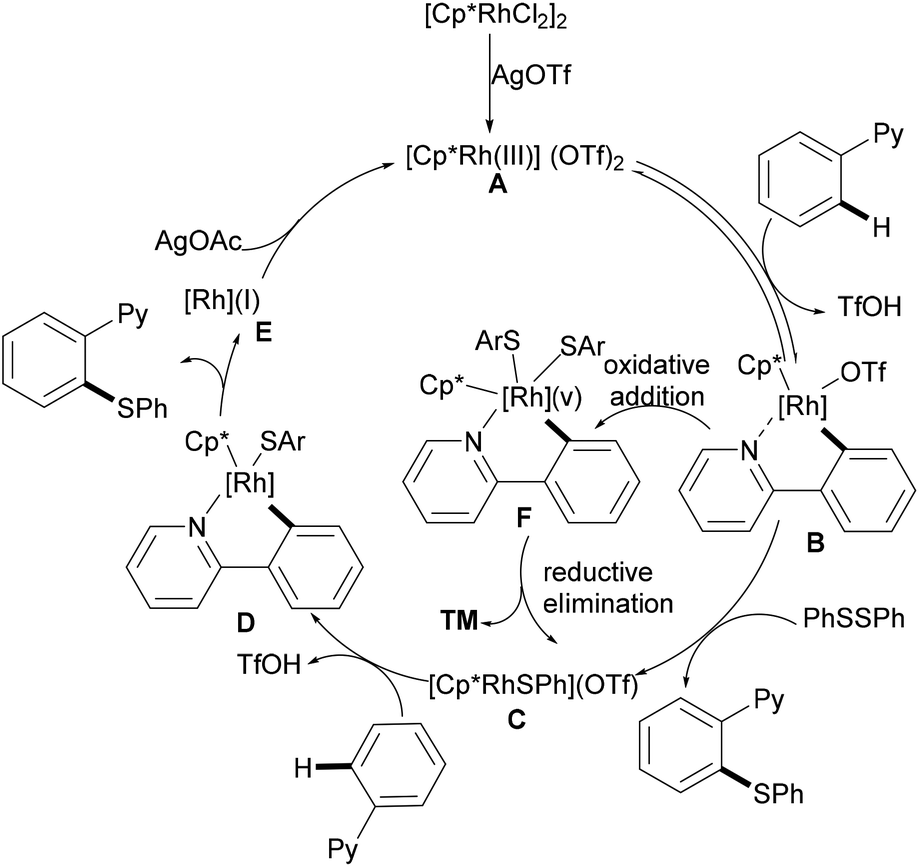
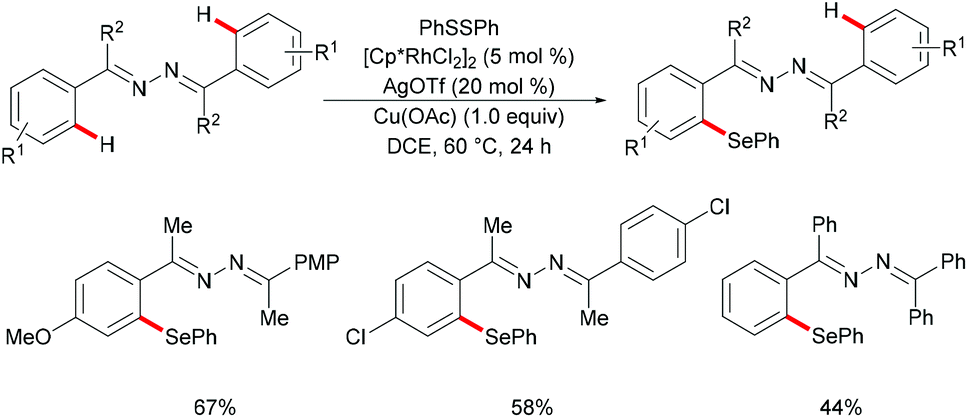
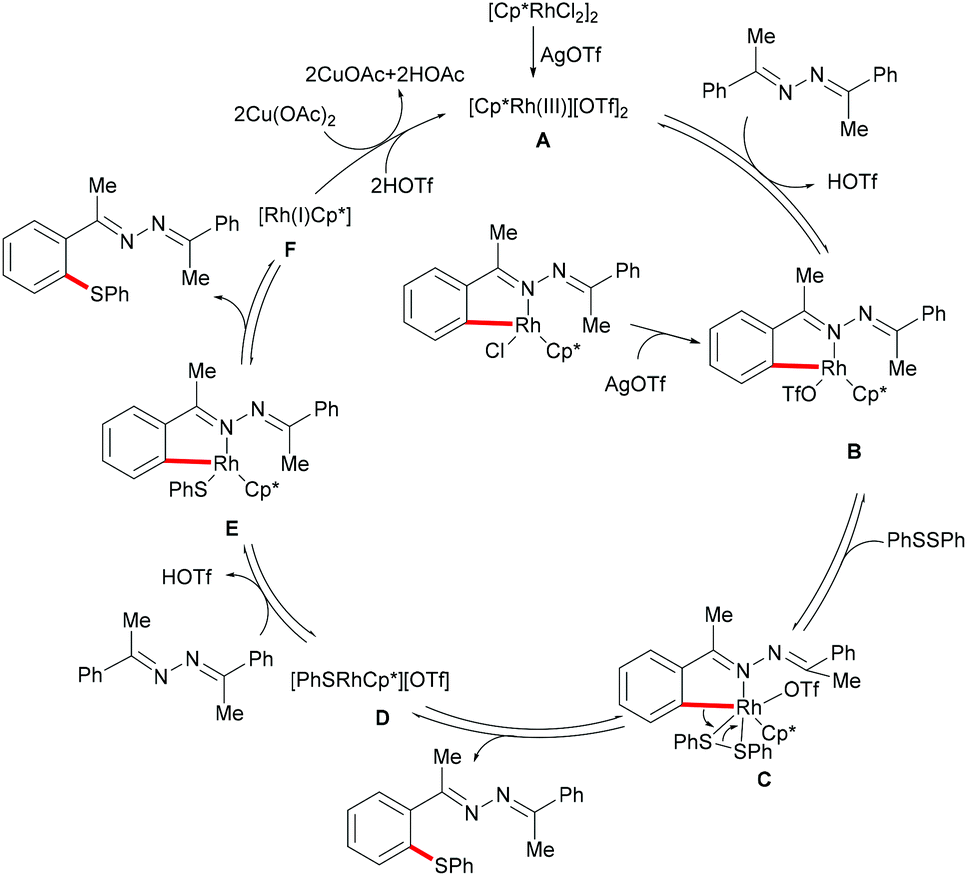
Inspired by the previous reports on the use of CN substructures as viable directing groups, in 2015, Zhu and co-workers succeeded in the direct thiolation of the ortho C–H bond of aryl ketazines with diaryl disulfides catalysed by the in situ generated cationic rhodium species [Cp*RhIII][OTf]2 in the presence of a stoichiometric amount of Cu(OAc)2 as the oxidant in DCE at 60 °C (Scheme 19).128 The authors demonstrated that substrates bearing electron-donating substituents showed better reactivities than substrates decorated with electron-withdrawing groups, giving the mono-thiolated product with excellent regio- and chemo-selectivities. In line with the detailed mechanistic studies with KIE, H/D exchange, substrate competition and catalysis with well-defined rhodium(III) complexes, the authors proposed a plausible mechanism involving the insertion of the S–S bond into the C–Rh bond to form C–S and S–Rh bonds, followed by a coupling reaction to obtain a rhodacycle intermediate, thus releasing the desired product via a reductive elimination event (Scheme 20).
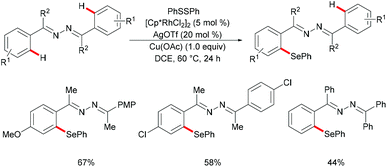 |
| | Scheme 19 Rhodium(III)-catalysed directed ortho-C–H bond functionalization of aromatic ketazines. | |
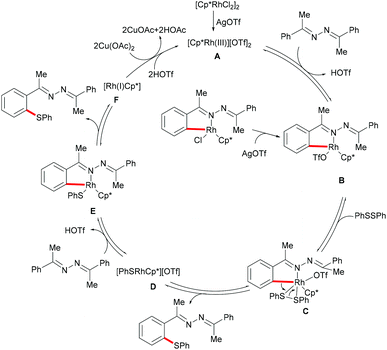 |
| | Scheme 20 Plausible catalytic cycle for C–H thiolations of aromatic ketazines. | |
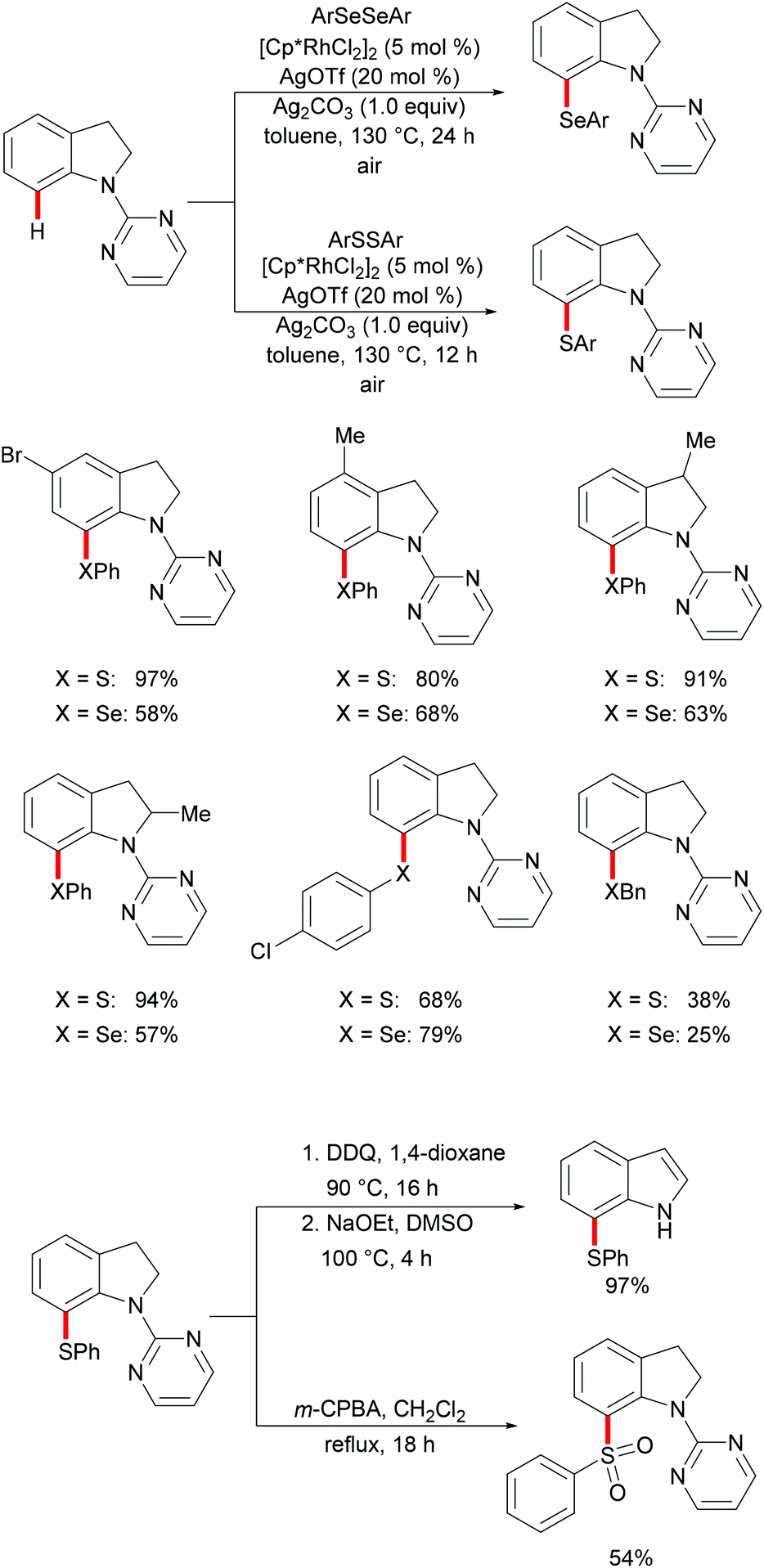
Indoline and indole moieties play an important role as nitrogen-containing heterocycles, while they are omnipresent in natural products, pharmaceuticals and organic dyes. Thus, a variety of methods for the decoration of indoline and indole scaffolds via transition metal-catalysed direct functionalization have been extensively explored in recent years. In 2016, Wang's group reported a rhodium-catalysed selective C7 chalcogenylation of indolines by using pyridimidine as the directing group (Scheme 21).129,130 The additive AgOTf, which presumably abstracts a chloride anion to generate the competent cationic rhodium complex, was indispensable for these transformations. Notably, the obtained indolines could be oxidized to indoles in nearly quantitative yields in the presence of stoichiometric amounts of DDQ. The traceless removal of the pyridimidyl group could be easily performed by treatment with NaOEt in DMSO at 100 °C.131 Furthermore, the thiolated indolines could be selectively oxidized to sulfonyl indolines by treatment with m-CPBA. This approach gave user-friendly access to various chalcogenated indolines and indoles.
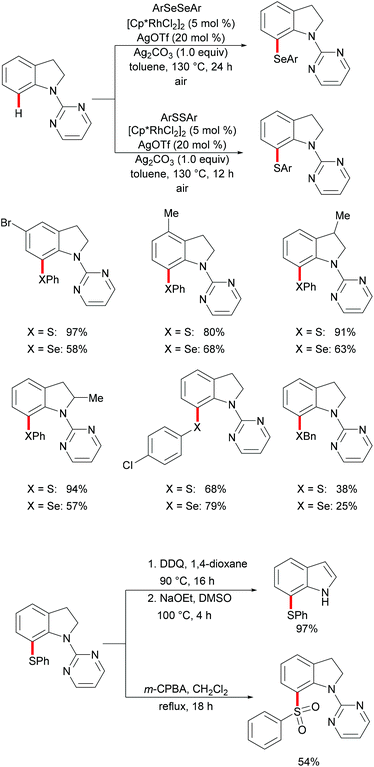 |
| | Scheme 21 Rhodium(III)-catalysed C7-chalcogenylations of indolines. | |
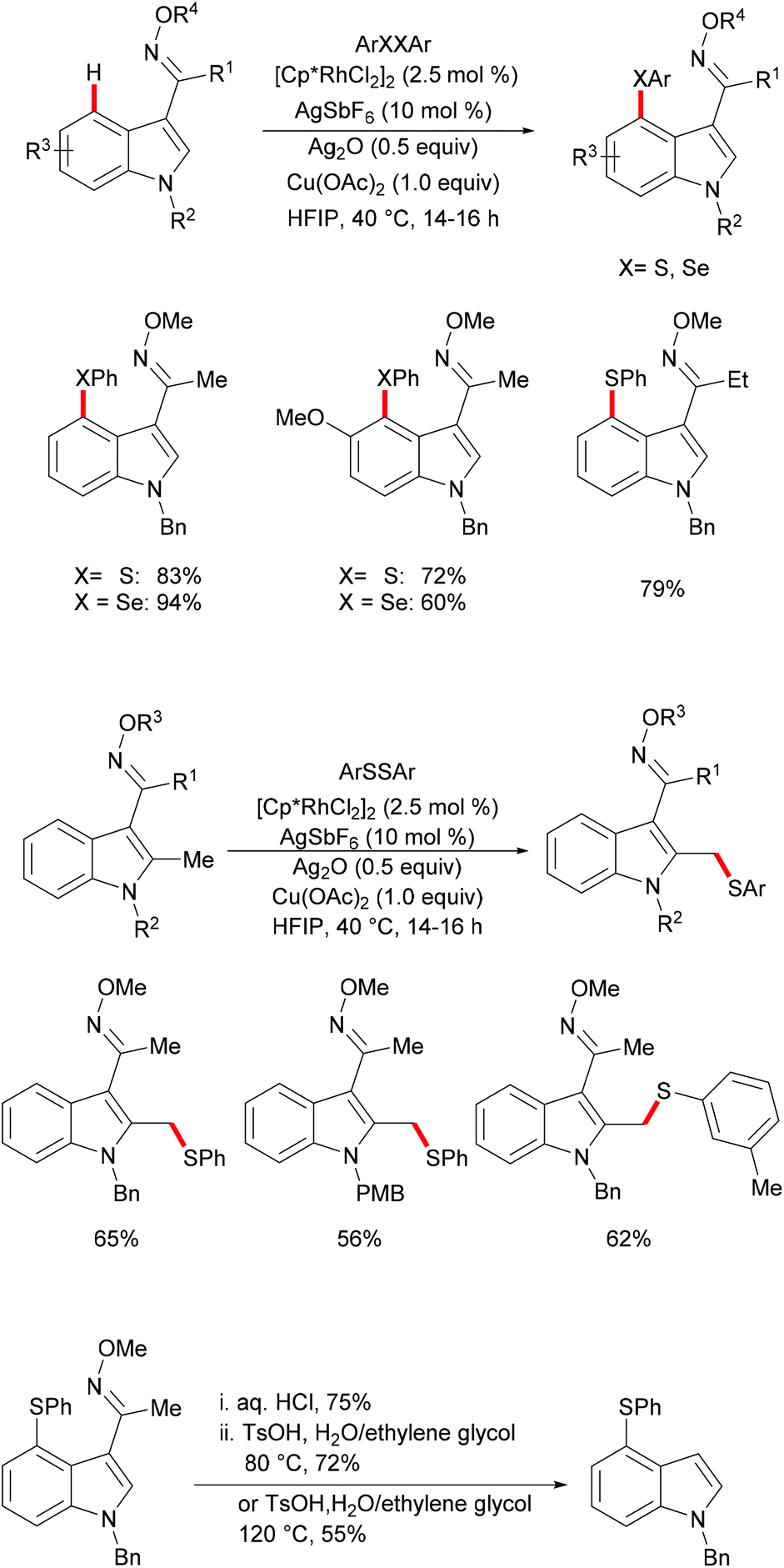
Subsequently, the Samanta group reported on a C-4 selective rhodium-catalyzed C–H chalcogenylation of indoles by employing a removable oxime as the directing group under mild reaction conditions (Scheme 22).132 Interestingly, in the absence of Cu(OAc)2, the reaction did not proceed efficiently, thus indicating that the copper additive is essential for this transformation. Indoles with various protecting groups and electronically and sterically differentiated functional groups at the C5, C6 or C7 position were smoothly transformed, affording the corresponding products in good to excellent yields. Notably, N–H free indole was not compatible in this reaction. Thus, this robust rhodium(III)-catalysed system was suitable for gram-scale synthesis with a slightly decreased yield, using only 1 mol% of the rhodium catalyst. Interestingly, when C-2 methyl indole was employed under the optimized reaction conditions, the thiolation occurred selectively at the methyl position. Finally, in order to demonstrate the synthetic utility of the developed protocol, the oxime directing group was removed in a two-step process, with the corresponding ketone as a key intermediate.
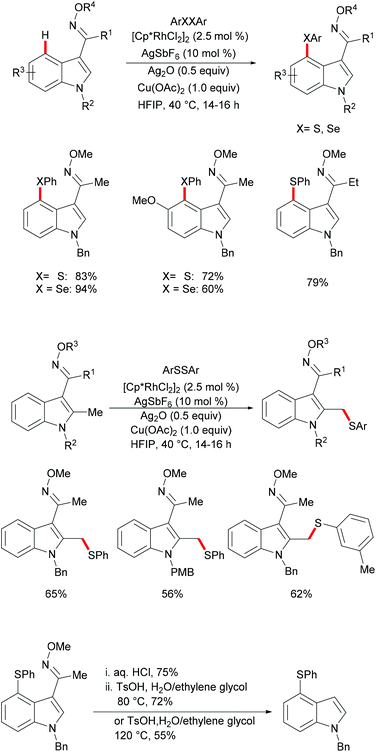 |
| | Scheme 22 Rhodium(III)-catalysed regiocontrolled direct C4 and C2-methyl thiolations of indoles. | |
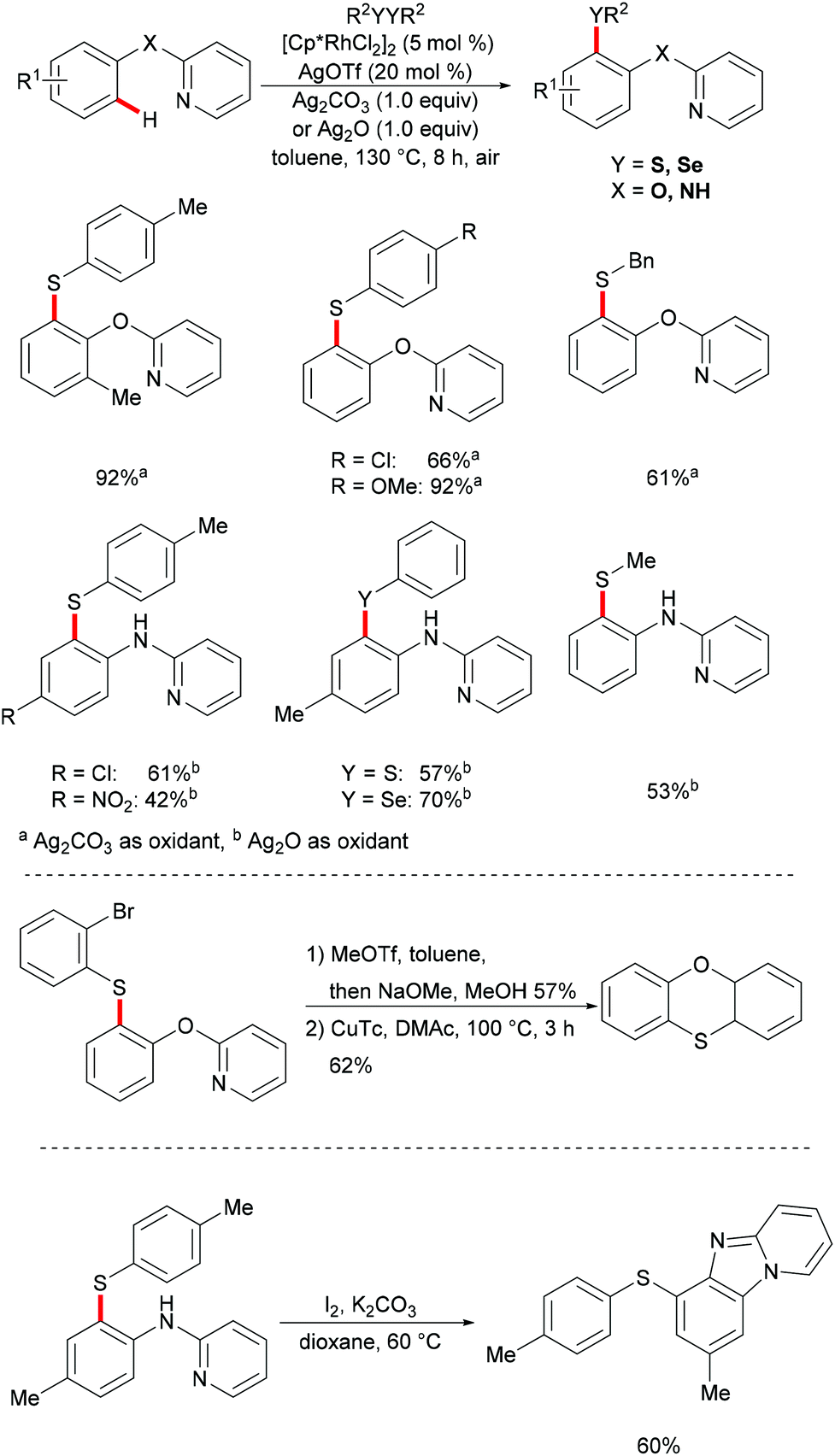
In 2017, the Yang group developed a highly efficient rhodium(III)-catalysed direct C–H chalcogenylation of phenols and anilines assisted by the 2-pyridyl group.133 AgOTf played a key role in this transformation. This protocol showed high levels of functional group tolerance. The directing groups were easily removed, delivering the corresponding 2-(arylthiol)phenol and 2-aminothiophenol in moderate yields. The obtained thiolated products could be further transformed into phenoxathiin and benzimidazole-fused heterocycles via intramolecular C–O cross coupling and intramolecular annulation, respectively. The substrates bearing electron-donating groups were inherently more reactive, indicating that the reaction involves a base-assisted intermolecular electrophilic substitution process (Scheme 23).131,134,135
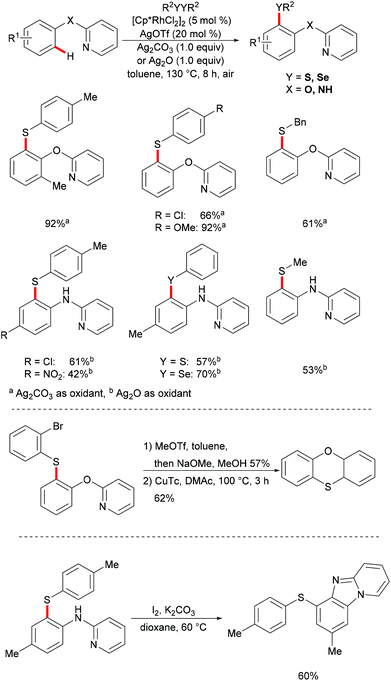 |
| | Scheme 23 Rhodium(III)-catalysed direct ortho-chalcogenylations of phenols and anilines. | |
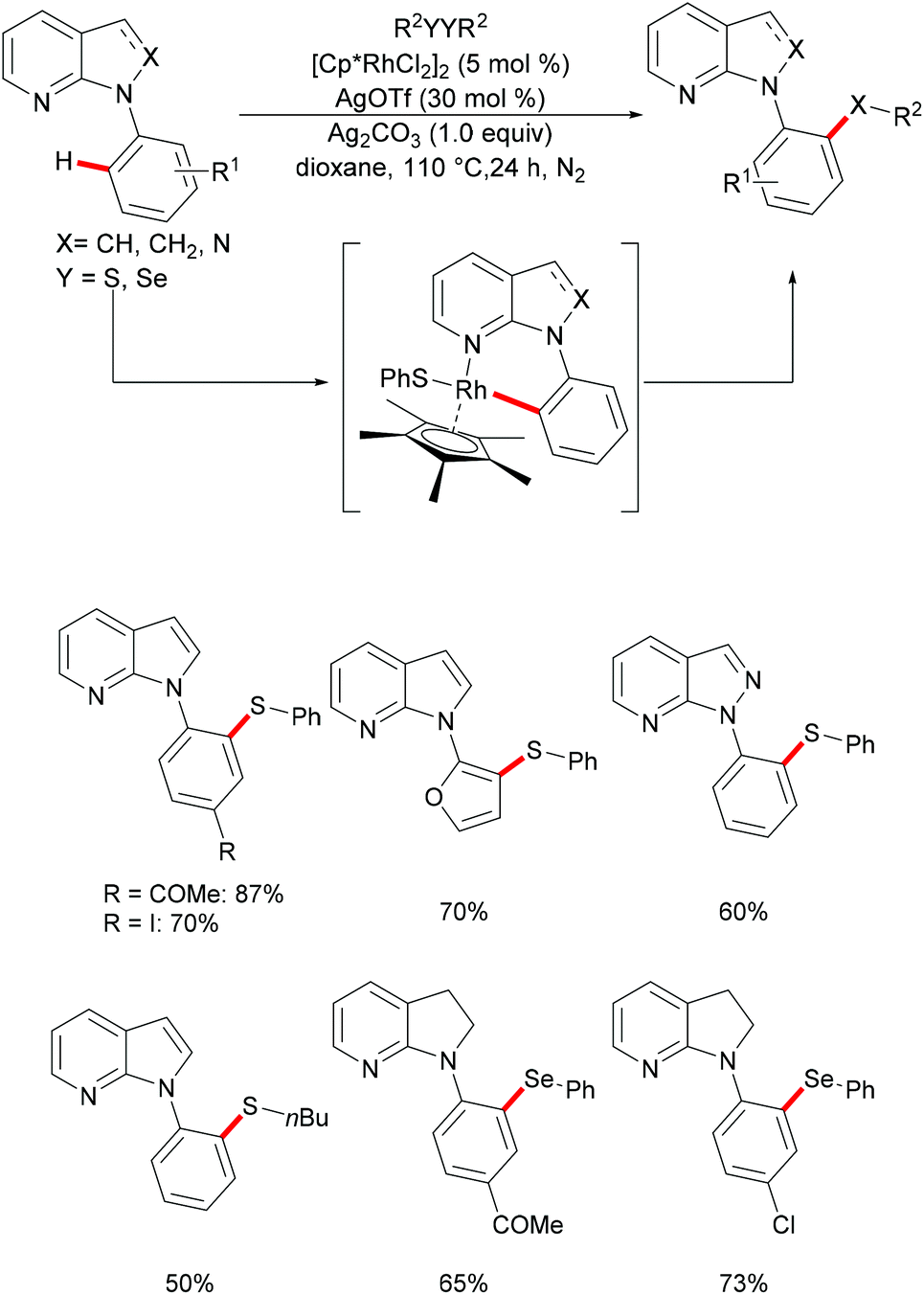
The 7-azaindole motif represents a very important nitrogen-containing heterocyclic scaffold, which is widely found in natural products and marketed pharmaceuticals. In this regard, Deb and coworkers136 reported a selective rhodium-catalysed C–H chalcogenylation of N-(hetero)aryl-7-azaindoles (Scheme 24). This robust rhodium-catalysed system displayed excellent reactivity with broad functional group tolerance. Notably, heteroarenes, such as furans, thiophenes and benzothiophenes, were also converted efficiently and gave the corresponding thiolated products in good yields. This catalyst was not restricted to diaryl disulfides, as dialkyl disulfides were also compatible under the optimised conditions. The independently synthesised well-defined 6-membered rhodium complex reacted with diphenyl disulfide smoothly, thus indicating, presumably, the intermediacy of this rhodacycle.
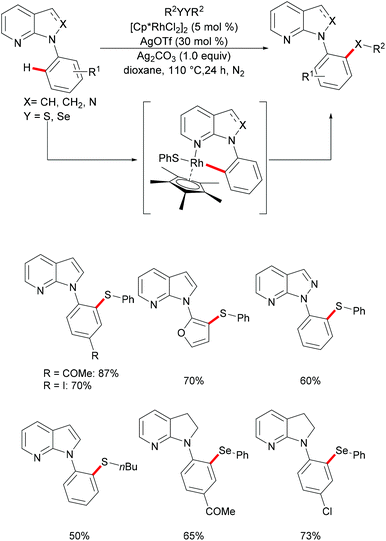 |
| | Scheme 24 Rhodium-catalysed direct and selective ortho C–H chalcogenylations of N-(hetero)aryl-7-azaindoles. | |
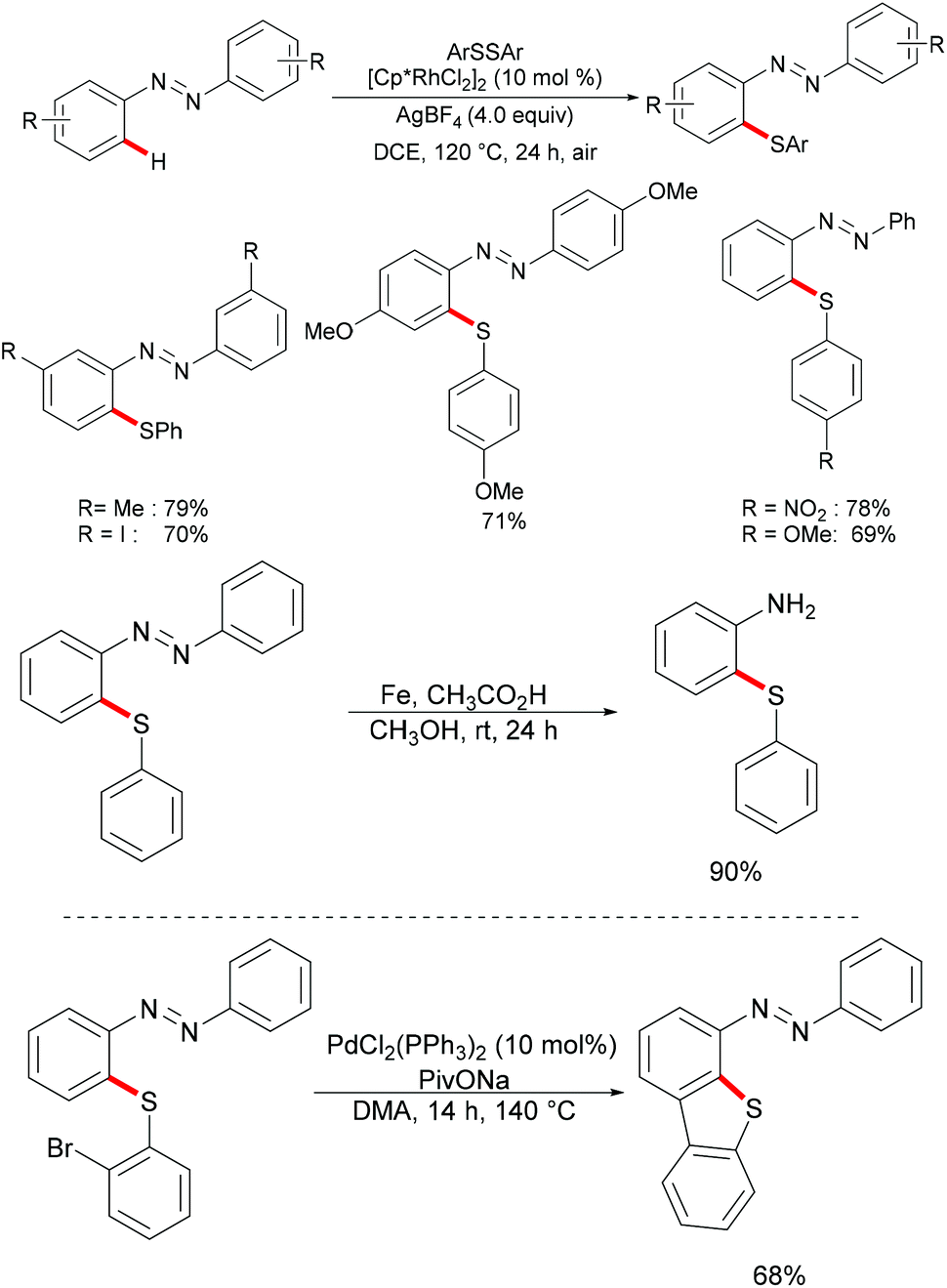
At the same time, the Zhou group reported a rhodium-catalysed C–H thiolation of azobenzenes using AgBF4 as the oxidant (Scheme 25).137 Here, two equivalents of azobenzene substrates were required in order to ensure good yields. A wide range of sensitive functional groups, such as iodine, nitro or ester groups, were well tolerated under the optimised reaction conditions. The substrates bearing electron-donating groups proved to be more reactive and afforded the desired products in good to excellent yields, whereas the substrates bearing electron-withdrawing groups only gave moderate yields. Notably, for meta-substituted azobenzenes, the thiolation selectively occurred at the less sterically hindered position and afforded the corresponding products in good to excellent yields. Further experiments, such as the removal of the directing group under acidic conditions, intermolecular cyclization via C–H activation initiated by oxidative addition into the C–Br bond and oxidation reactions, were carried out under mild reaction conditions, which indicated the unique synthetic utility of the developed protocol. The detailed mechanism was also thoroughly investigated through an H/D exchange experiment, a KIE study and the isolation of a five-membered rhodacycle complex.
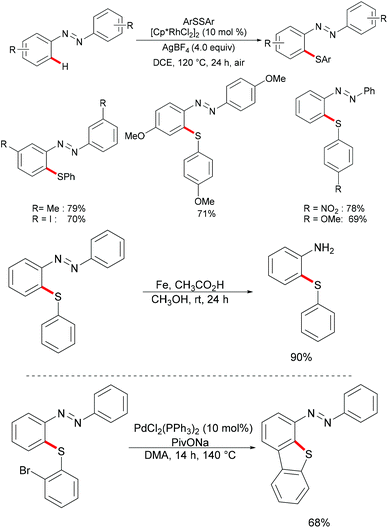 |
| | Scheme 25 Rhodium(III)-catalysed thiolations of azobenzenes. | |
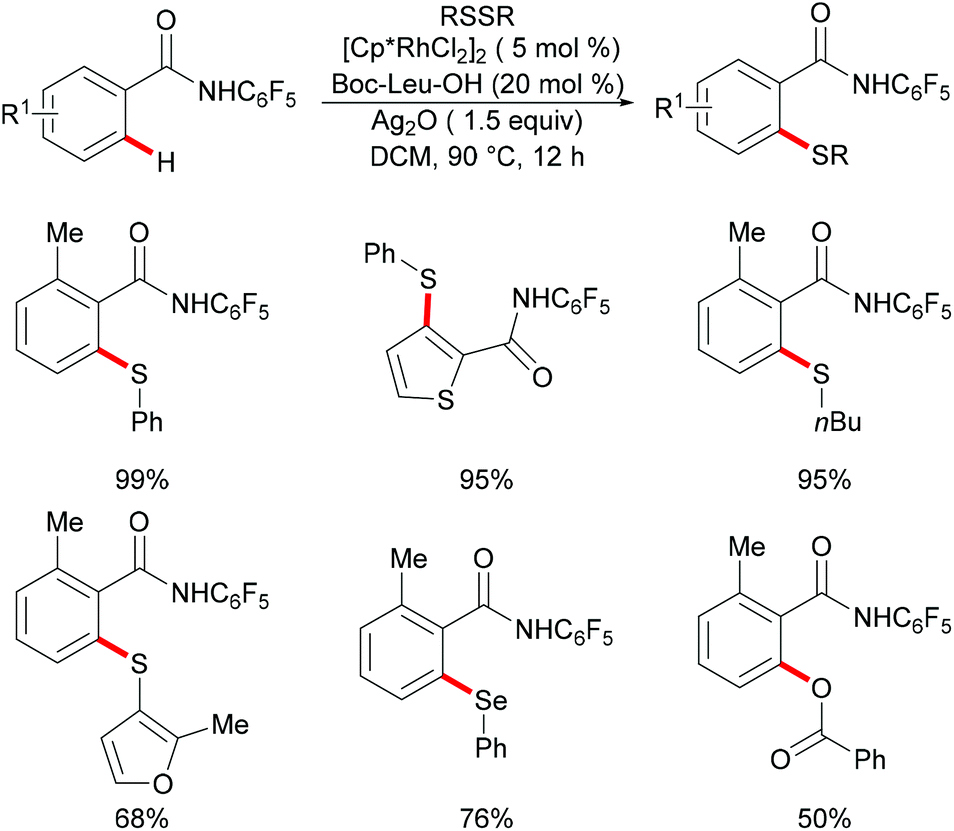
More recently, the Yu and Lu groups realized a ligand-promoted rhodium(III)-catalyzed C–H thiolation of arenes directed by a weakly coordinating amide auxiliary (Scheme 26).138 After an extensive ligand optimization, MPAAs (mono-N-protected amino acids) were found to be the privileged ligands to promote this reaction due to their important roles in stabilizing the catalytic centre. Under the optimized conditions, both diaryl and dialkyl disulfides reacted efficiently with various benzamides giving the corresponding thiolated products in moderate to excellent yields. It is noteworthy that diphenyl diselenides and benzoyl peroxide were also suitable substrates in this transformation.
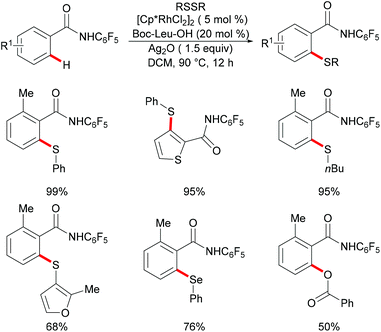 |
| | Scheme 26 MPAA ligand-promoted rhodium-catalysed C–H thiolations of benzamides. | |
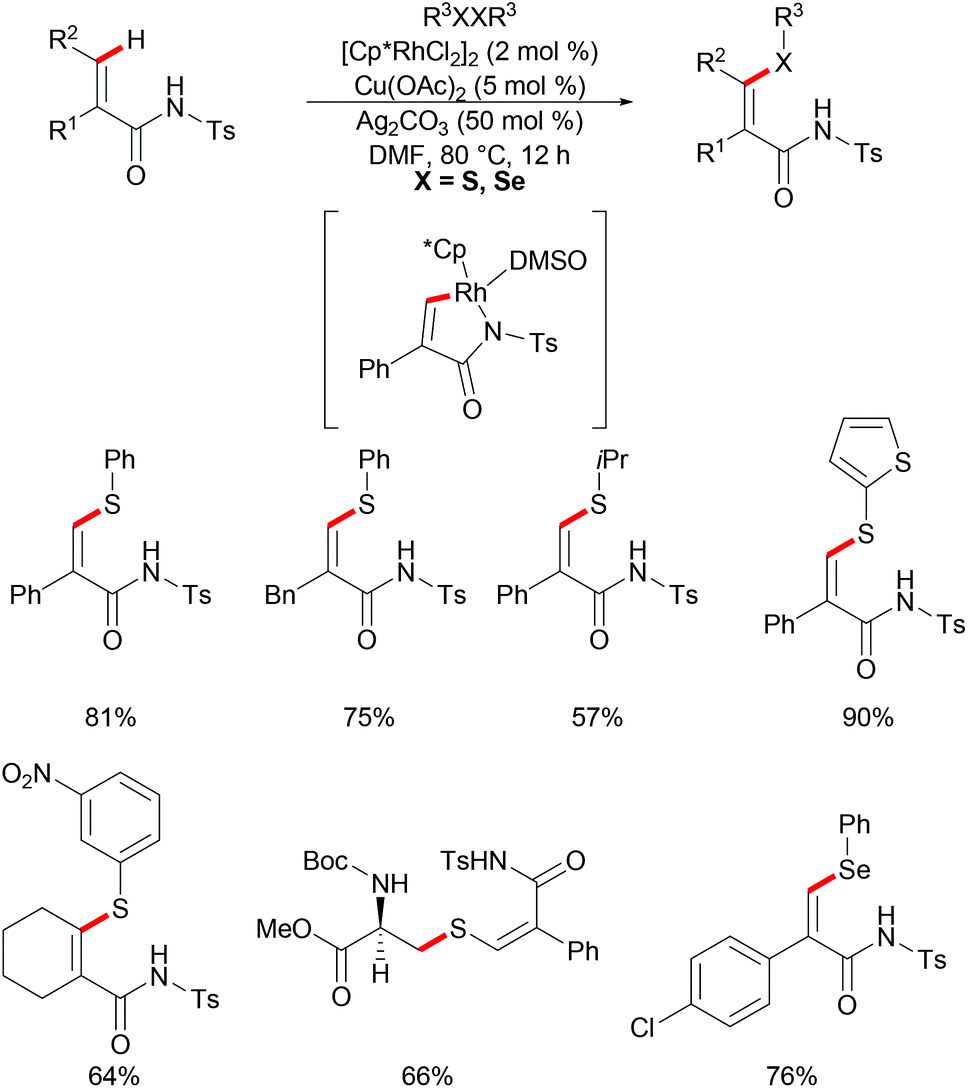
Besides aromatic C–H bonds, olefinic C–H bonds can also participate in rhodium catalysed thiolations. Recently, Ji and coworkers reported on a rhodium-catalysed thiolation of activated alkenyl C(sp2)–H bonds, assisted by N-tosylamide for the preparation of (Z)-β-alkenyl sulphides (Scheme 27).139 Both aryl and alkyl substituted acrylamides, in the α and β positions, reacted with diphenyl disulfides smoothly under relatively mild conditions and gave the corresponding products in good yields in a regio- and stereoselective fashion. Additionally, the internal alkenes were also compatible in this catalytic system, and furnished the desired products in moderate yields. The electronic properties of the substituents on the disulfides did not considerably affect the outcome of the reaction as both electron-donating and electron-withdrawing groups yielded the desired products in moderate to good yields. In addition, heterocyclic and secondary alkyl disulfides were also converted smoothly and gave the thiolated products in moderate to excellent yields, whereas the tertiary alkyl disulfides failed to give acceptable results, presumably due to steric hindrance. Notably, protected cystine was also successfully employed in this method and afforded the alkenylated cysteine derivative in a good yield. Moreover, this reaction also worked with diphenyl diselenides, albeit employing harsher reaction conditions, prolonged reaction time and increased amount of the silver salt. It is noteworthy that the catalyst loading could be reduced to 0.01 mol% with a slight decrease in the yield, albeit by prolonging the reaction time to 48 h, thus indicating the high catalytic efficiency of the developed protocol. Further investigations by KIE studies indicated that the reaction might involve a base-assisted internal electrophilic-type substitution (BIES) pathway. The result of stoichiometric studies strongly suggested the five-membered rhodacycle as a key intermediate in the C–H chalcogenylation process.
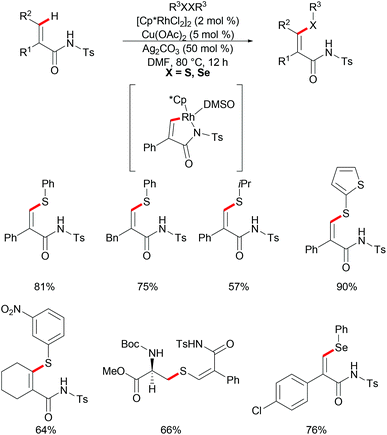 |
| | Scheme 27 Rhodium(III)-catalysed highly regioselective thiolations of N-tosyl acrylamides. | |
2.3 Ruthenium catalysis
The first ruthenium-catalysed C–H functionalization reactions were initially explored by Lewis and Smith140 and further developed by Murai,141 Ackermann,130,142–146 Wang, Bruneau, Dixneuf and Jeganmohan, among others.144,147–150 The cost effective ruthenium catalyst was demonstrated to be robust and capable of forming cyclometalated species via C–H bond cleavage followed by diverse C–H functionalizations, providing a plethora of decorated molecules. Recently, with inspiration from these elegant works, ruthenium(II)-catalysed C–H chalcogenylations of arenes with the assistance of various directing groups (DGs) were widely explored. The notable features of these methods included the use of a cost effective ruthenium catalyst, mild reaction conditions, shorter reaction times, broad substrate scope and the utility in the late-stage chalcogenylations of useful molecules.
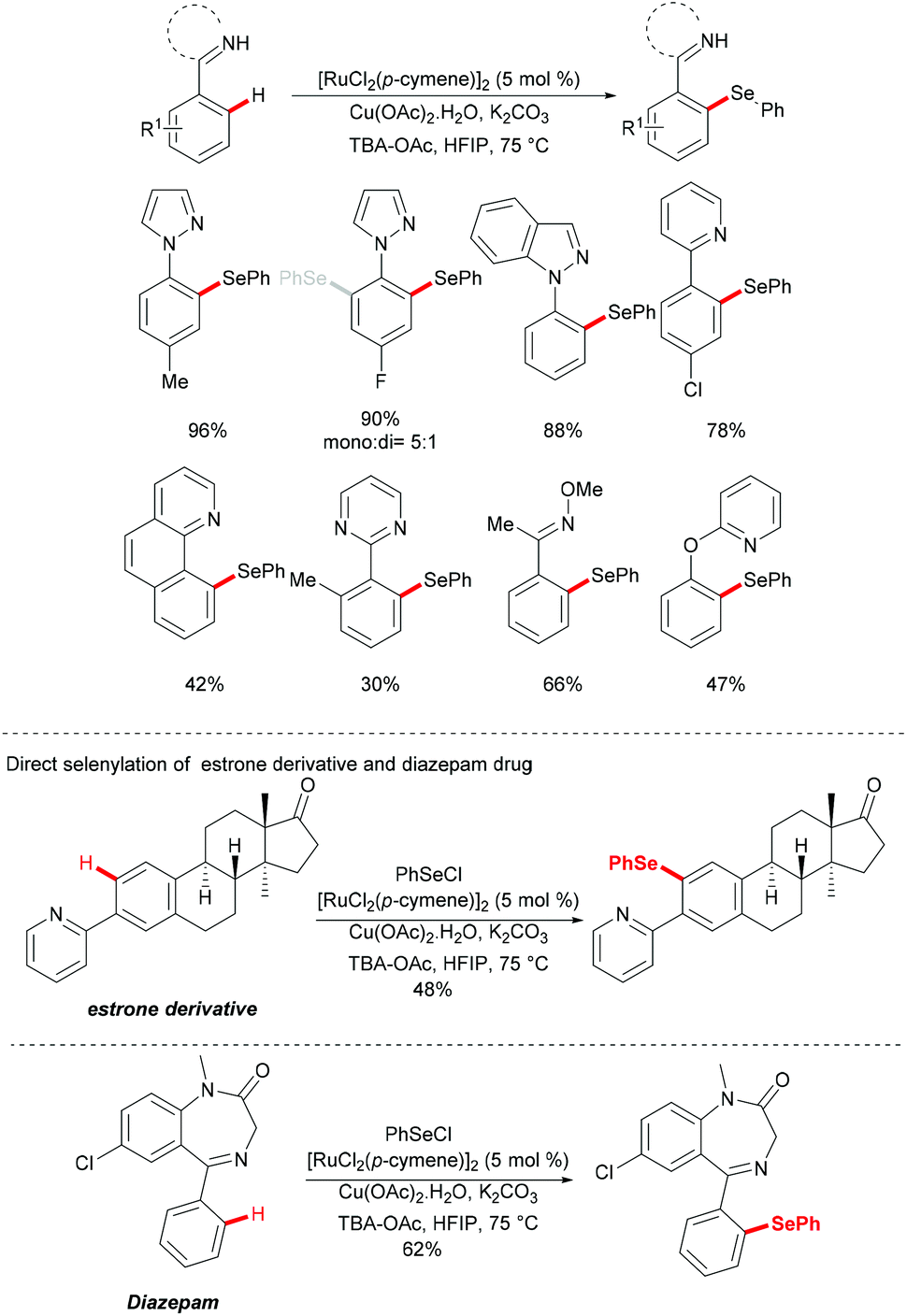
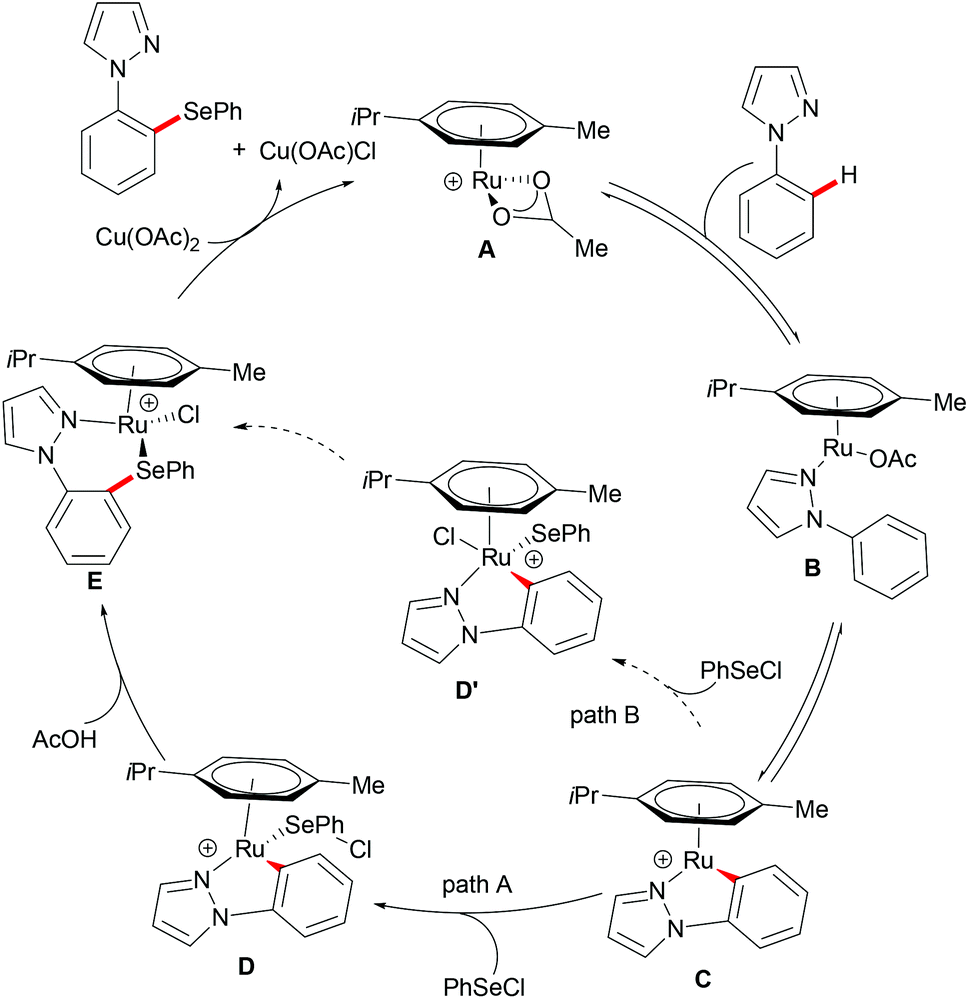
In 2016, Zhang and coworkers reported the ruthenium catalysed C–H selenylation of arenes with selenyl chlorides directed by pyrazoles (Scheme 28).151 This robust [RuCl2(p-cymene)]2/Cu(OAc)2·H2O146 catalyst tolerated a wide range of substrates bearing different functional groups converting them efficiently and furnishing the mono-selective product in an excellent yield. Moreover, upon using substrates bearing an electron-withdrawing substituent at the para-position or an electron-donating substituent at the meta-position, bis-substituted products were also obtained in small quantities. It is noteworthy that pyridine, pyrimidine, benzoquinoline and O-methyl oxime were compatible as the directing groups for this transformation, albeit providing the products in lower yields. Moreover, 2-phenoxylpyridine bearing an oxygen linker could also participate in this selenylation reaction, and afforded the corresponding product in a moderate yield. Importantly, the estrone derivative and the prescription antidepressant drug diazepam were also converted successfully in this reaction, thus indicating the wide utility of the direct C–H selenylated reactions. Detailed mechanistic studies indicated that this ruthenium catalysed C–H selenylation might involve electrophilic substitution or oxidative addition/reductive elimination pathways (Scheme 29).
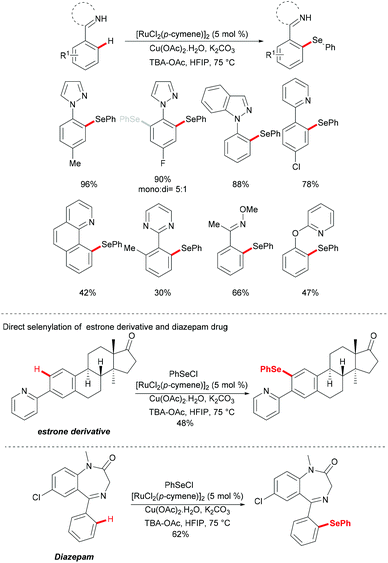 |
| | Scheme 28 Ruthenium-catalysed C–H selenylations of arenes with selenyl chlorides. | |
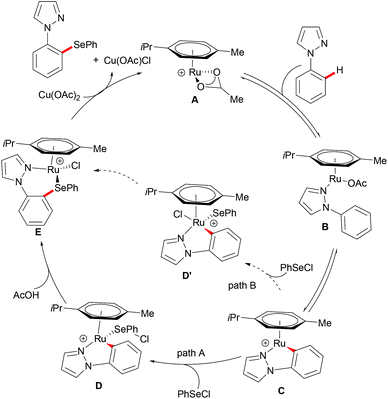 |
| | Scheme 29 Proposed mechanism of the ruthenium-catalysed C–H selenylations with selenyl chlorides. | |
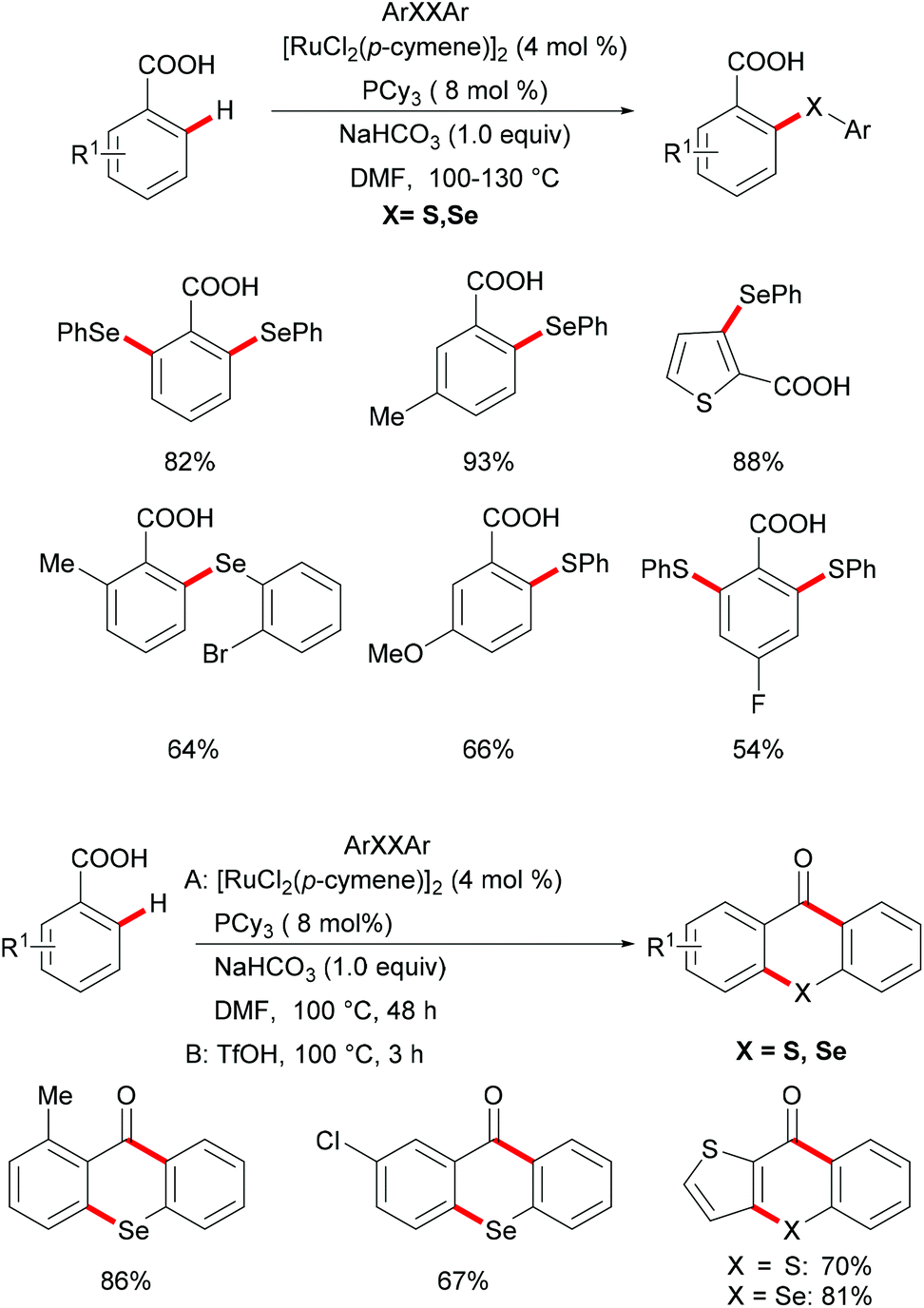
Carboxylic acids are widely found in functional organic molecules and natural products, and they can be easily interconverted into other functional groups or removed in a traceless fashion. Besides, they can also serve as leaving groups in various decarboxylative couplings with the formation of C–C or C–heteroatom bonds. In this context, carboxylate directed transition metal catalyzed C–H functionalizations have been demonstrated by the Yu, Ackermann and Su groups in the last decade.152–163 Inspired by these reports,164,165 in 2017, Baidya's group reported an example of ruthenium catalysed C–H chalcogenylation assisted by weakly coordinating carboxylic acid as the directing group (Scheme 30).166 This method shows good functional group tolerance for both benzoic acid and diaryl diselenide, providing the corresponding mono- or di-chalcogenated products in good yields. Notably, the heteroaryl carboxylic acid was also compatible in this reaction. As expected, diaryl disulfides were also suitable coupling partners in this catalytic system. Finally, the obtained chalcogenated product upon treatment with TfOH at 100 °C afforded the corresponding selenoxanthones and thioxanthone derivatives in high yields via an intramolecular cyclization process, which provides a simple, economical and straightforward synthetic route to chalcogenoxanthones. The kinetic studies based on significant KIE values (kH/kD ≈ 1.89 and 1.74) suggested that the C–H cleavage is likely involved in the rate-determining step.
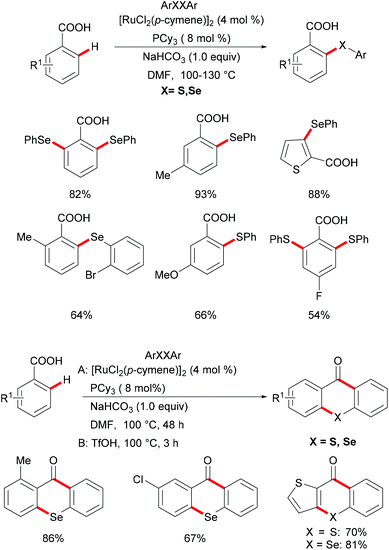 |
| | Scheme 30 Ruthenium(II)-catalysed ortho C–H chalcogenylation of benzoic acids. | |
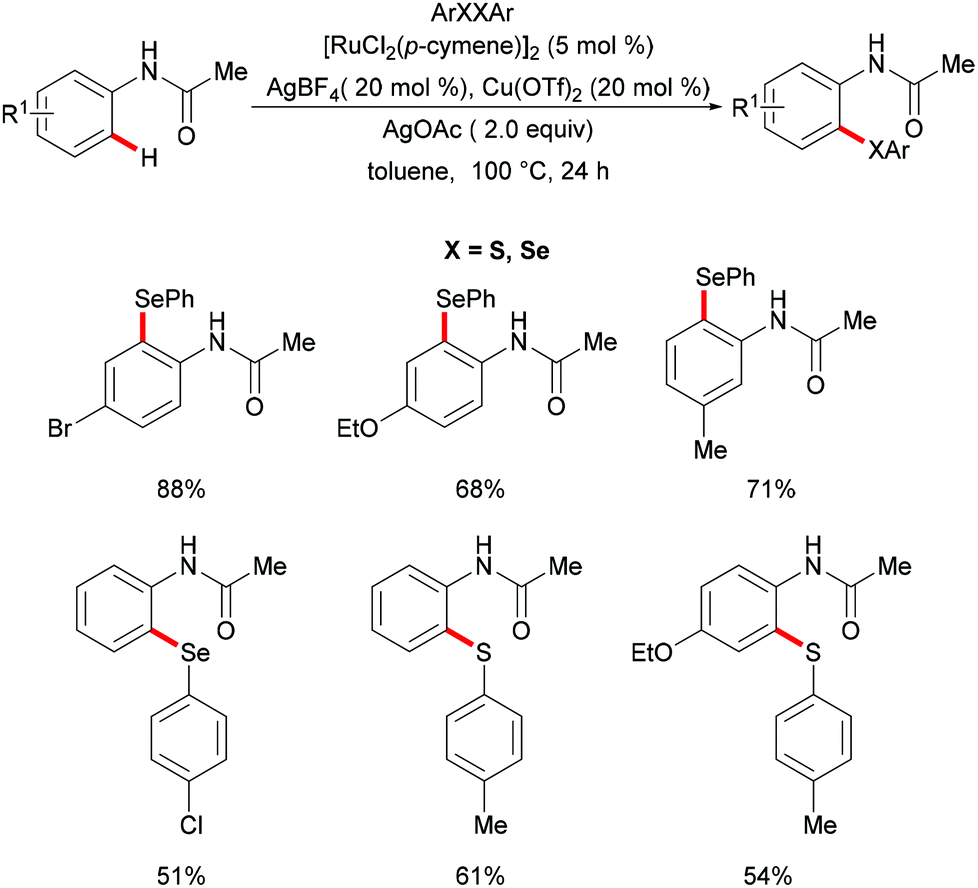
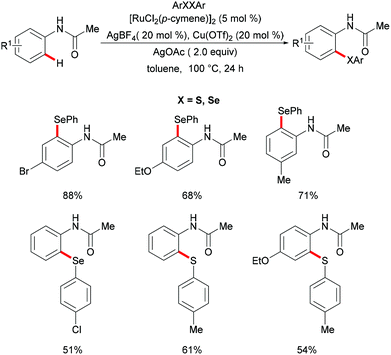
Independently, Ma's group reported ruthenium-catalysed C–H chalcogenylations of synthetically meaningful anilides via weak coordination (Scheme 31). [RuCl2(p-cymene)]2 in combination with AgBF4 and AgOTf as cocatalysts in the presence of Cu(OAc)2·H2O as a base/oxidant in toluene at 100 °C were found to be the optimal conditions for this transformation.167 Anilides bearing either electron-withdrawing or electron-donating groups on the para-position of the arene ring converted efficiently and afforded the corresponding mono-chalcogenated products in high yields. For the meta-substituted anilides, this chalcogenylation occurred at the less sterically hindered position in most cases, while the 3,4-(methylenedioxy)anilides gave the more sterically hindered selenylated product as the major product, due to a secondary directing group effect. Moreover, diaryl disulfides and diaryl diselanes decorated with a wealth of functional groups were also compatible in this reaction. Finally, a BIES C–H activation142 mechanism was proposed based on the intermolecular competition reaction and KIE studies.
 |
| | Scheme 31 Ruthenium(II)-catalysed C–H chalcogenylation of anilides. | |
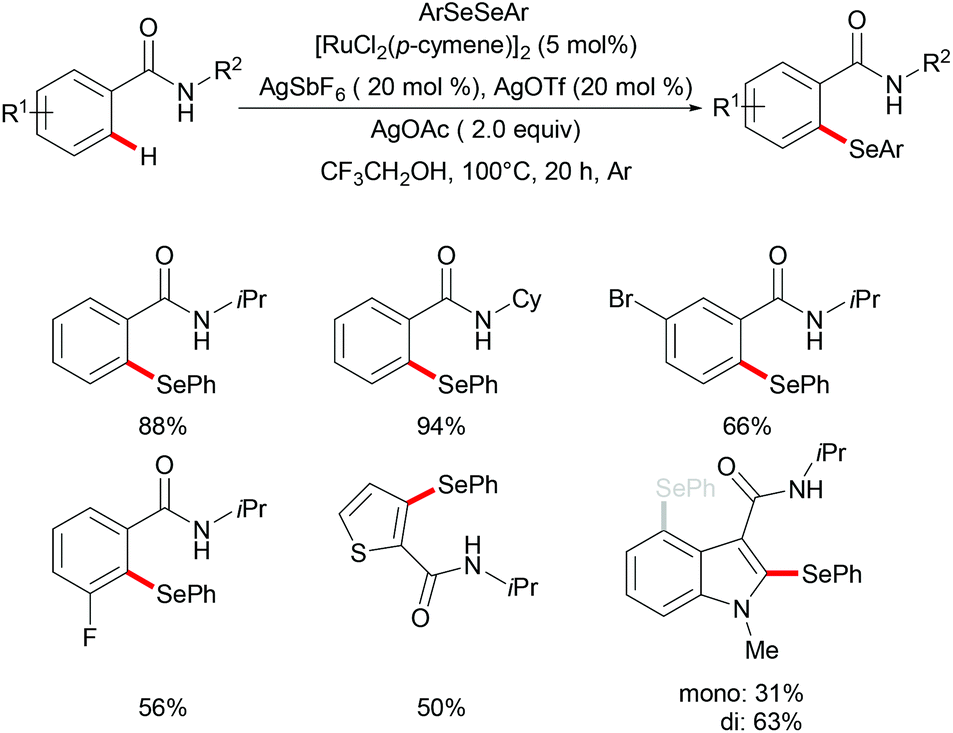
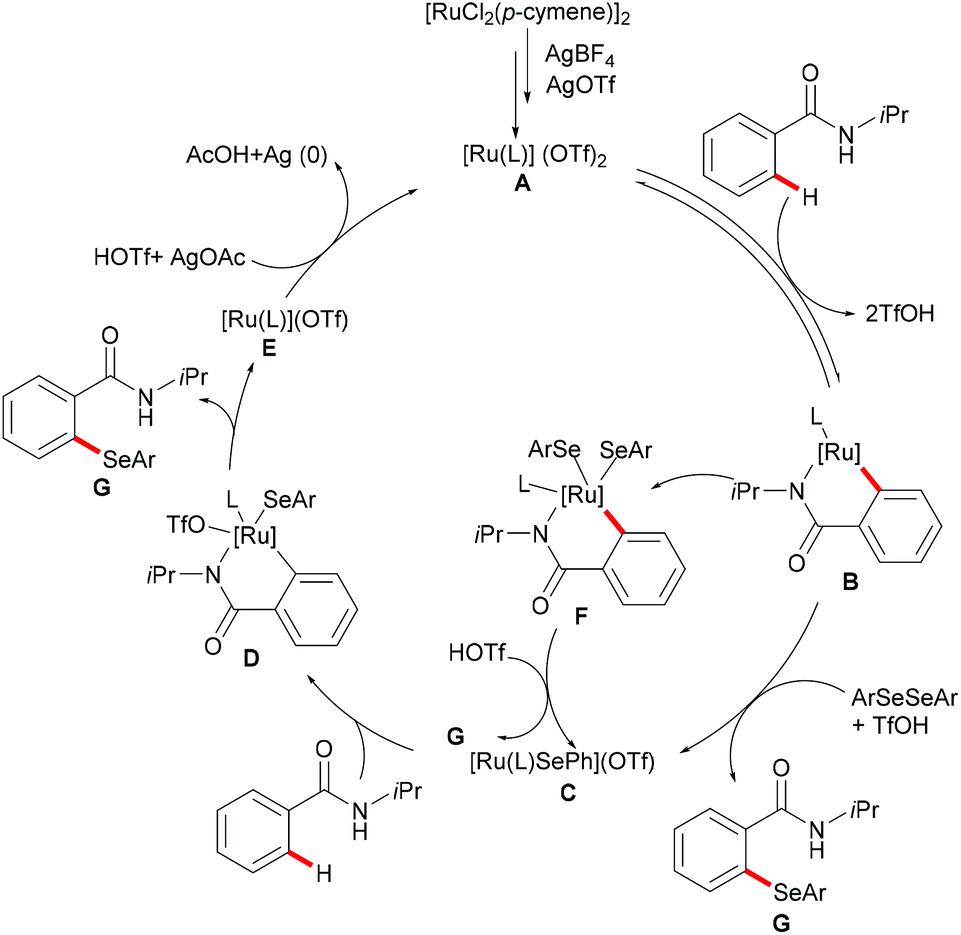
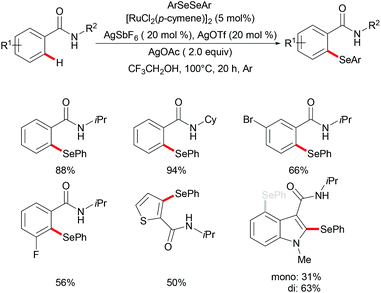
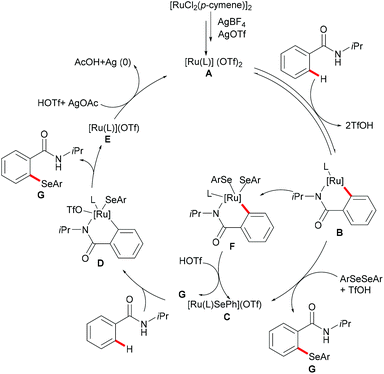
Moreover, the same group achieved ruthenium catalysed C–H selenylation of benzamides under similar reaction conditions, albeit employing AgSbF6 and AgOTf as co-catalysts in CF3CH2OH (Scheme 32).168 The preliminary study showed that increased bulk on the amide substituents was beneficial whereas N-hydroxybenzamide and tertiary amide failed to promote the reaction. Both electron-rich and electron-deficient benzamides converted efficiently to provide the mono-selenylated products in high yields. Notably, for the meta-substituted aromatic amides, the C–H selenylations were largely controlled by steric interactions, thus giving the C5-selective mono-selenylated products. In contrast, meta-fluoro-substituted benzamide exclusively led to the selenylation at the C2 position; the structures were further confirmed through single-crystal X-ray diffraction analysis. Furthermore, this method was applicable to a gram-scale synthesis, which demonstrated the utility of this transformation. Finally, the amide directing group could be further functionalized into a variety of bio-relevant heterocycles and useful functional groups such as carboxylic acid, aldehyde, tetrazole and phosphite under mild reaction conditions. Based on the mechanistic studies and previous literature reports, a plausible catalytic cycle via a BIES pathway was proposed as shown in Scheme 33. First, the activated ruthenium catalyst generated from the [RuCl2(p-cymene)]2 precursor in the presence of AgBF4 and AgOTf coordinated to the directing group and formed five-membered ruthenium(II) species Bvia a reversible C–H bond activation process. The thus formed five-membered cycloruthenated compound B reacted with diaryl diselane to deliver the desired product and intermediate C. Another possible pathway involved the oxidative addition process to give ruthenium(IV) species F, which then underwent reductive elimination to afford the product and ruthenium(II) species C. Intermediate C could participate in a second C–H activation process to form cycloruthenated species D again followed by reductive elimination to generate the target product and ruthenium(I) E, which could be oxidized to an active catalytic ruthenium(II) species in the presence of AgOAc, thus re-entering the catalytic cycle.
 |
| | Scheme 32 Ruthenium-catalysed C–H selenylation of benzamides. | |
 |
| | Scheme 33 Proposed catalytic cycle for the ruthenium-catalysed C–H selenylation of benzamides. | |
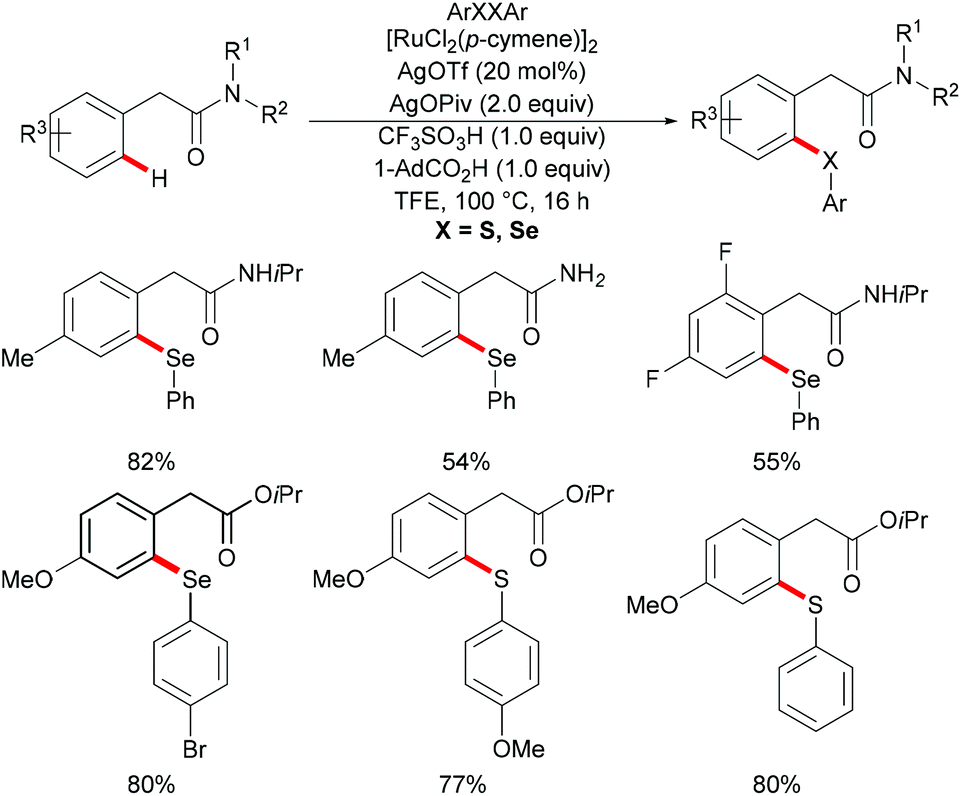
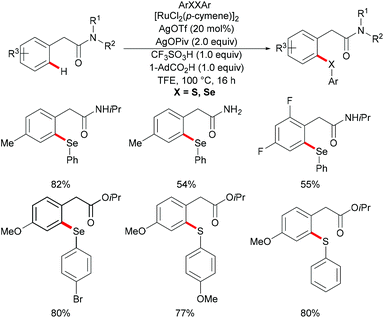
Very recently, Ma's group further extended the ruthenium-catalysed C–H selenylation of aryl acetic amides utilizing a distal weakly coordinating directing group via an unfavorable six-membered metallacycle intermediate (Scheme 34).169 The presence of 1-AdCO2H and TfOH, which could facilitate the cyclometallation and regeneration of the ruthenium catalyst, was found to be crucial for efficient catalysis. Under the optimized reaction conditions, various substituted aryl acetic amides underwent this transformation smoothly to generate the corresponding selenylated products in good yields. Notably, primary and tertiary amides were also compatible under these catalytic conditions and afforded the corresponding selenylated products in moderate yields. Furthermore, aryl acetate ester could also enable this transformation efficiently in the presence of Ag3PO4 as the oxidant; furthermore, the scope of the methodology was not only limited to selenylations, but also expanded to thiolation reactions.
 |
| | Scheme 34 Ruthenium-catalysed C–H selenylation of aryl acetic amides and esters via weak coordination. | |
2.4 Silver catalysis
Silver has a wide range of applications in organic synthesis, ranging from radical to carbene/nitrene transfer reactions.170,171 Silver has been vastly utilized for the construction of saturated nitrogen heterocycles with high levels of positional selectivity.172,173 Despite these advances, silver catalysed CH activation reactions are scarce.174,175
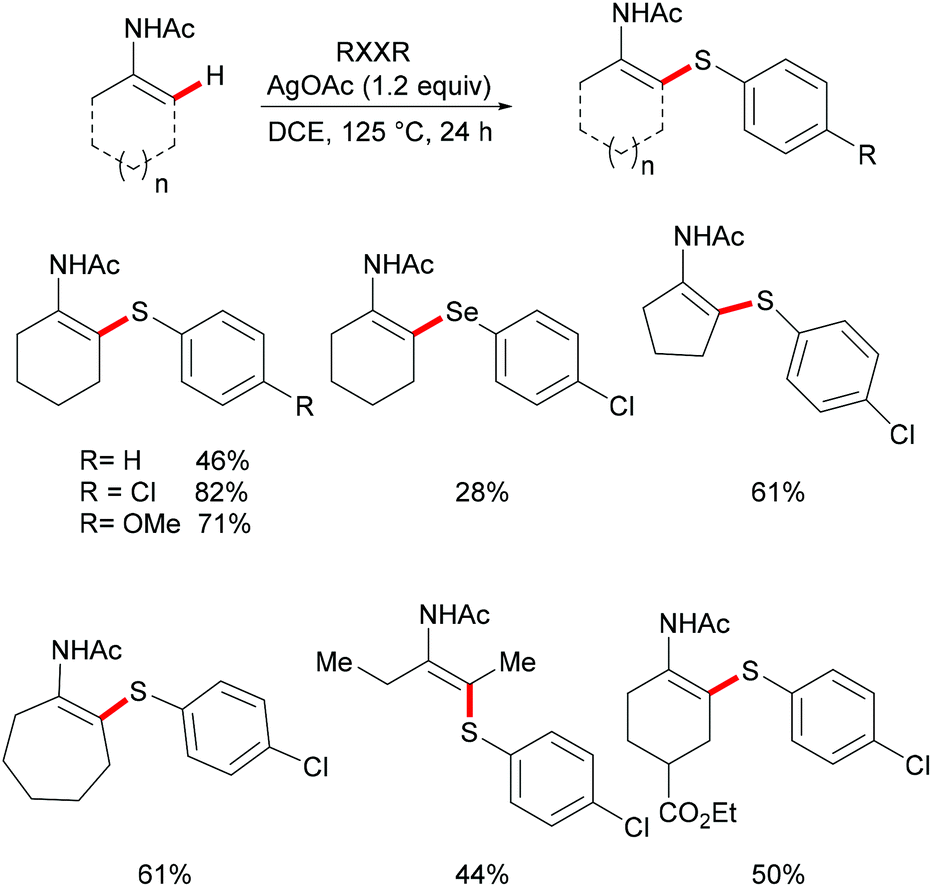
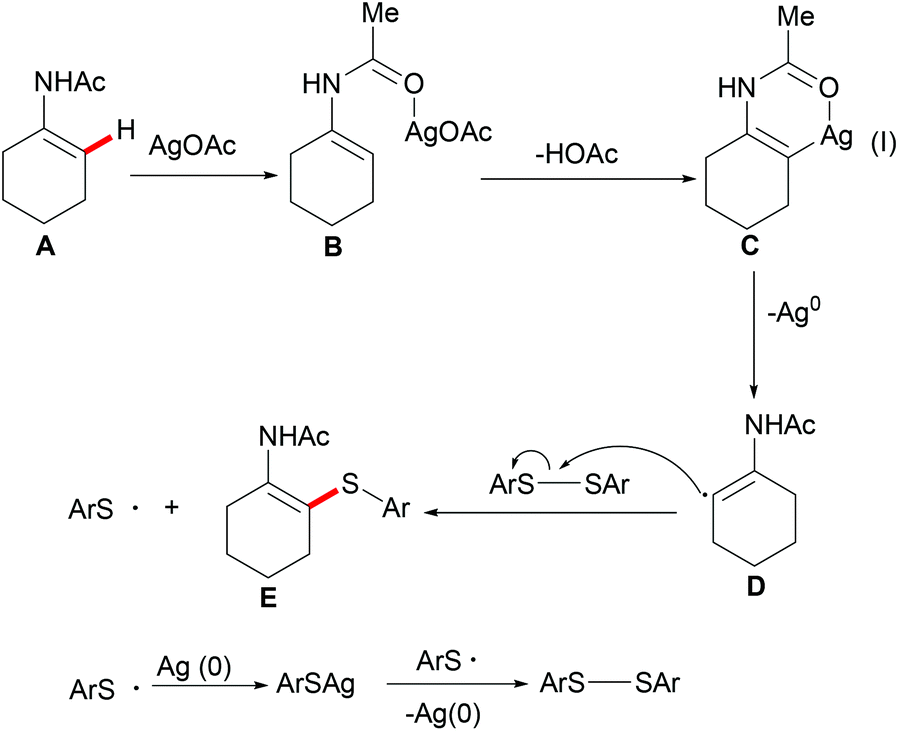
Deng and coworkers explored the Ag-mediated oxidative thiolation of enamides with diaryl disulfides (Scheme 35).176 A variety of substituted diaryl disulfides were effectively converted into the corresponding thiolated products in medium to good yields under the optimized conditions. Dibenzyl disulfide and diphenyl diselenides were also compatible, albeit resulting in lower isolated yields due to incomplete conversion. Both cyclic and linear secondary enamides participated in this alkenyl C(sp2)–H bond thiolation successfully. It is noteworthy that tertiary enamide, such as N-vinyl-2-pyrrolidone, smoothly reacted with diaryl disulfides, and delivered the corresponding (E) thiolated products with high stereoselectivity.
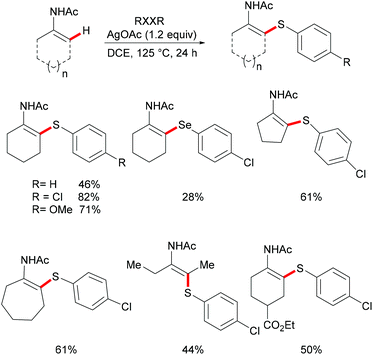 |
| | Scheme 35 Silver-mediated oxidative thiolation of enamides with disulfides. | |
Competition experiments with unsymmetrical diaryl disulfides or electron-rich and electron-deficient disulfides showed that the sulfur connected with the electron-rich aryl group was preferentially transferred. These results indicated that the S–S bond cleavage of diaryl disulfides was unlikely to occur through a nucleophilic attack process. Further investigations with TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl, 1.2 equiv.) as a radical inhibitor in the standard procedure indicated that these thiolation reactions were unlikely to involve a chain radical mechanism. Finally, the authors proposed a plausible nonchain radical mechanism as described in Scheme 36. First, the coordination of the silver catalyst to enamide and subsequent cyclometalation via electrophilic substitution or concerted metalation–deprotonation process afforded the alkenyl silver complex C, which could further transform into the vinyl radical D and the reduced Ag0. The subsequent alkenyl radical D attacked the S–S bond of diaryl disulfide to provide the thiolated product and arylthiol radical Evia a homolytic cleavage process. The arylthiol radical could be eventually terminated by coupling with silver thiophenolate (ArSAg) regenerating the diaryl disulfide and Ag(0) (Scheme 36).
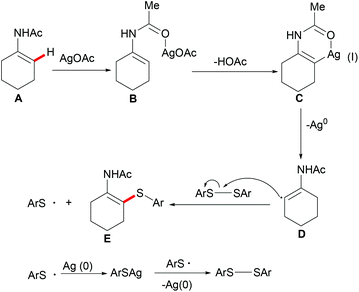 |
| | Scheme 36 Plausible mechanism of the silver-mediated C–H thiolations. | |
3 First row-transition metal catalysis
Over the last few decades, direct C–H functionalizations catalysed by precious 4d and 5d transition metals, such as palladium, rhodium, iridium, platinum and ruthenium, have been greatly explored leading to significant advancements of tools in complex molecular synthesis. These transition metals, which largely feature d8 or d6 electron configuration, exhibited a broad range of reactivities with high efficacy. However, these transition metals are Earth-rare and expensive, which can limit their applications in the industry. Thus, the development of novel catalysts based on inexpensive, less toxic and Earth-abundant first-row transition metals is highly desirable.95,96,177
3.1 Copper catalysis
Copper complexes have been widely used in organic synthesis due to the different oxidation states of copper that are easily accessible, mainly ranging from 0 to +3 that facilitate new bond forming processes by radicals, as well as two-electron transfer pathways via organometallic intermediates.178–180 Due to the low toxicity, high natural abundance in the Earth's crust and therefore cost-efficiency of copper, copper-catalysed reactions have been studied and shaped into valuable tools in the synthetic toolbox of all practitioners of organic synthesis during the last three decades. Thus, after the pioneering work by Ullmann95,181,182 and Goldberg,183 a vast array of transformations has been reported for the efficient construction of C–C and C–Het bonds.184
3.1.1 Thiol as the thiolating agent.
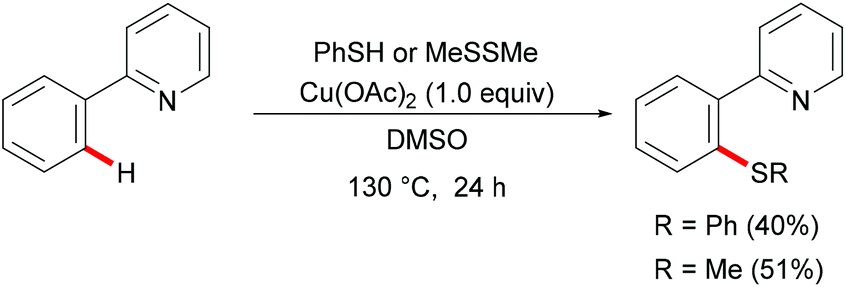
Inspired by these mechanistic and synthetic foundations, Yu's group reported the first copper-mediated C–H thiolations of arenes with thiophenol or dimethyl disulfide assisted by pyridine directing groups in 2006 (Scheme 37). The authors postulated that a single-electron transfer from 2-phenylpyridine to the coordinated copper(II) to form a radical-cation intermediate was the rate-limiting step of this transformation. Although this report only included two examples for C–S bond formation, it provided a new method to access unsymmetric sulfides that enabled further developments in this research area.185
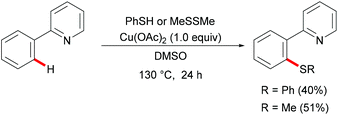 |
| | Scheme 37 Cu(II)-Catalysed C–H thiolations of arenes. | |
In 2014, Liang and coworkers utilized 8-aminoquinoline as the directing group and aryl or aliphatic thiols as thiolating reagents to develop an efficient copper catalysed direct C–H thiolation of aromatic amides (Scheme 38).186 Substrates bearing a broad range of functional groups such as cyano, nitro and halides were converted smoothly to give the mono- or dithiolated products in good yields. Moreover, heterocyclic substrates such as thiophene and pyridine were also compatible in this reaction. Both primary and secondary aliphatic thiols were effective substrates in this catalytic system and afforded the corresponding dithiolated products in synthetically useful yields whereas tBuSH led to a mixture of mono- and di-substituted products. Based on a series of control experiments, the authors proposed that a high-valent Cu(III) intermediate which could be stabilized by bidentate 8-aminoquinoline directing groups might be involved in this thiolation reaction and Ag2CO3 was an efficient oxidant for the oxidation of Cu(II) to Cu(III). Thereafter, Wei and co-workers demonstrated a similar reaction through a one-pot strategy.187 This transformation allowed the direct use of 8-aminoquinoline, benzoyl chlorides and thiophenols, and delivered the desired mono- or di-thiolated products in good yields. Interestingly, these reactions conducted with K2CO3 as the base afforded dithiolated products in good yields, whereas NaHCO3 only gave mono-thiolated products presumably due to the combined effect of weaker basicity and the diminished acidity of the ortho-C–H bond of the monothiolated product (Scheme 39).
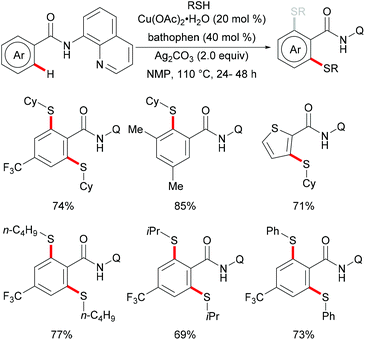 |
| | Scheme 38 Copper(II)-catalysed direct thiolation of aromatic amides with aryl and aliphatic thiols. | |
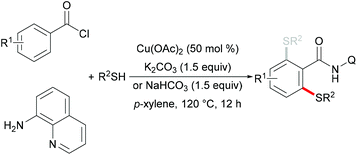 |
| | Scheme 39 Copper-catalysed selective mono- and di-C(sp2)–H thiolations of benzamides by a one-pot strategy. | |
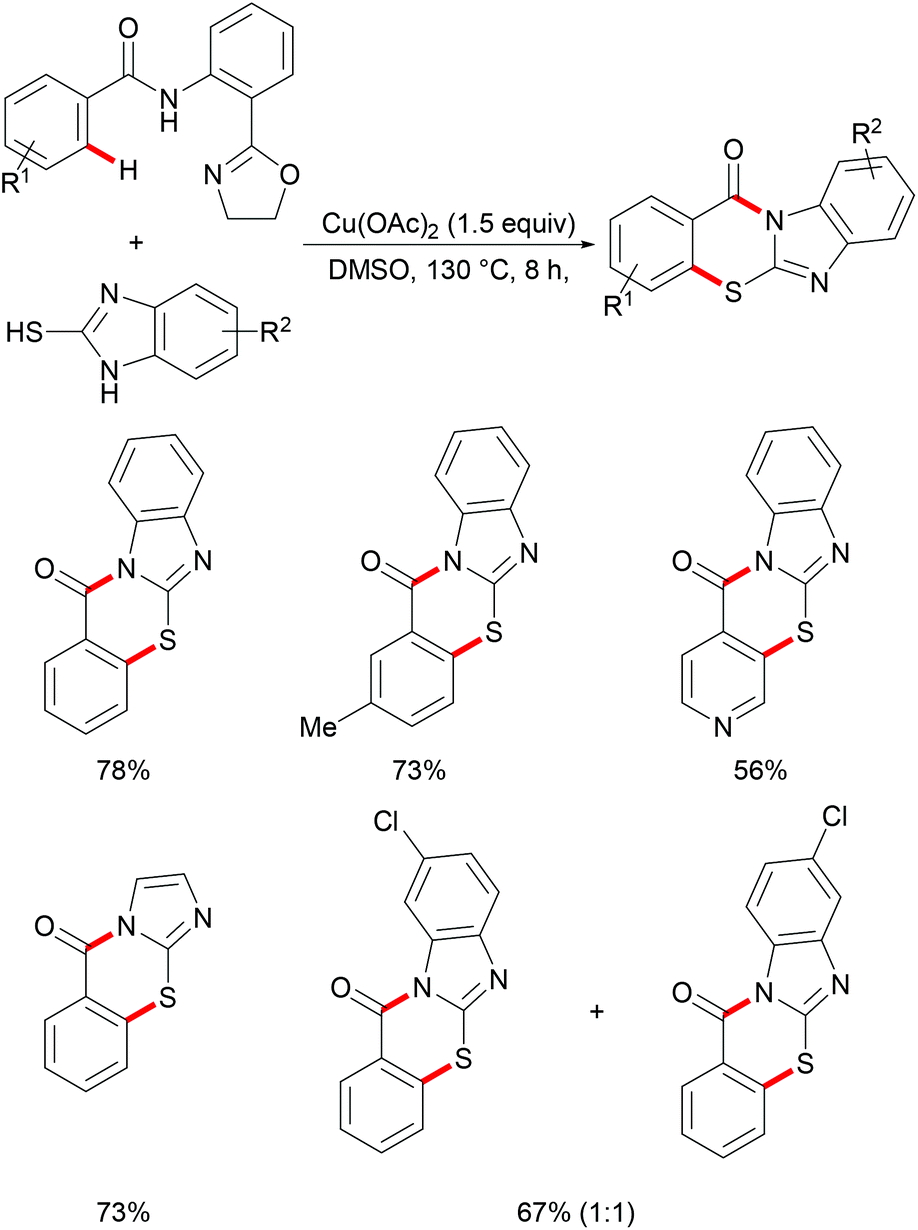
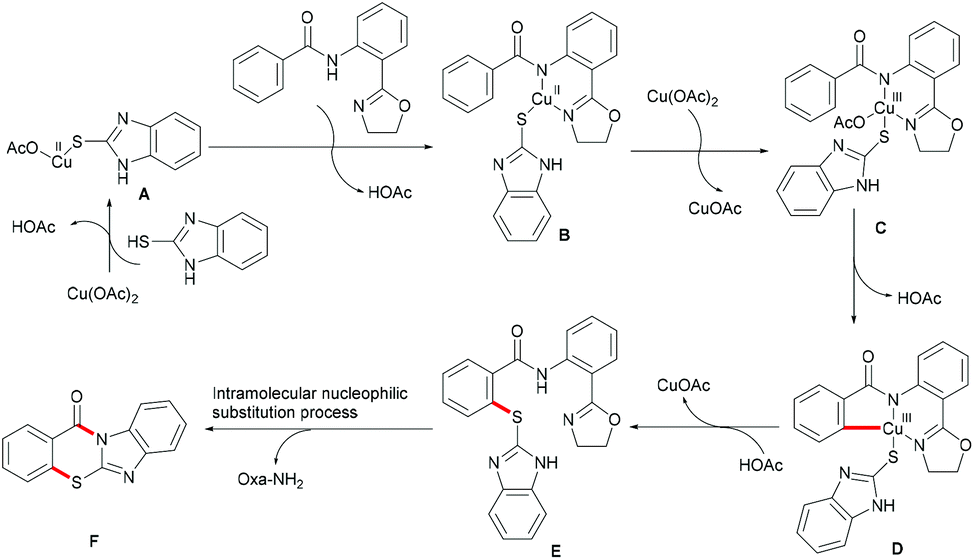
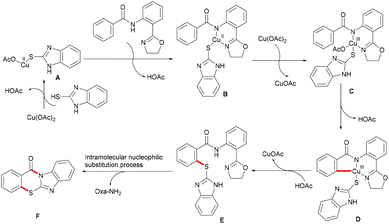
On the basis of this highly facile C–H thiolation protocol by employing thiols as the sulfur source, the synthetic utility of the thus formed thiolated products to enable subsequent transformations was further explored. In 2016, Chen reported on a copper-mediated C–H thiolation of arenes with 2-mercaptobenzimidazole assisted by the removable amide-oxazoline bidentate directing group followed by an intramolecular nucleophilic substitution to provide a series of polycyclic fused thiazinone derivatives (Scheme 40).188 This robust copper catalytic system could tolerate a wide range of functional groups such as acetyl, methoxy, and halides present in benzamides and afforded the corresponding products in good yields. Moreover, heteroaromatic amides, such as pyridine and thiophene derivatives, could also be transformed efficiently and delivered the desired product in a moderate yield. Unfortunately, the unsymmetrical 2-mercaptobenzimidazole reacted with benzamide to provide a mixture of regioisomers due to tautomerism of benzimidalole. Based on the preliminary mechanistic studies, a reasonable catalytic process involving a copper(II)/copper(III) pathway was proposed as shown in Scheme 41. The mechanism was assumed to begin with a ligand exchange with 2-mercaptobenzimidazole followed by coordination of the N,N-bidentate directing group to form 6-membered intermediate B. The thus formed organocopper species could be oxidized by Cu(OAc)2 through a disproportionation process to give Cu(III) complex C and subsequently undergo an acetate-assisted intramolecular C–H cupration to afford fused cyclic Cu(III) D, which then provides the key intermediate E through a reductive elimination process. Finally, intramolecular nucleophilic substitution occurs providing the desired product. Cu(OAc)2 played a dual role as the promoter and the terminal oxidant in this transformation, and thus required more than 1.0 equiv. to ensure successful conversion.
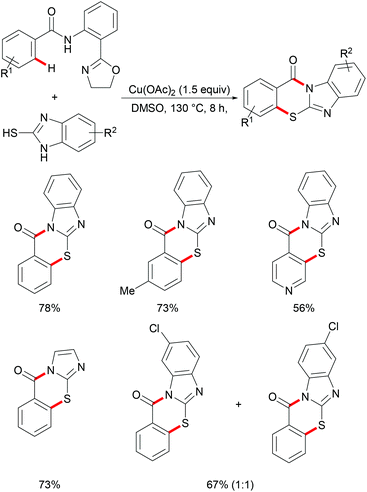 |
| | Scheme 40 Copper-mediated tandem C(sp2)–H thiolation and annulation of benzamides with 2-mercaptoimidazoles. | |
 |
| | Scheme 41 Proposed mechanism for the tandem C–H thiolation and annulation of benzamides with 2-mercaptoimidazoles. | |
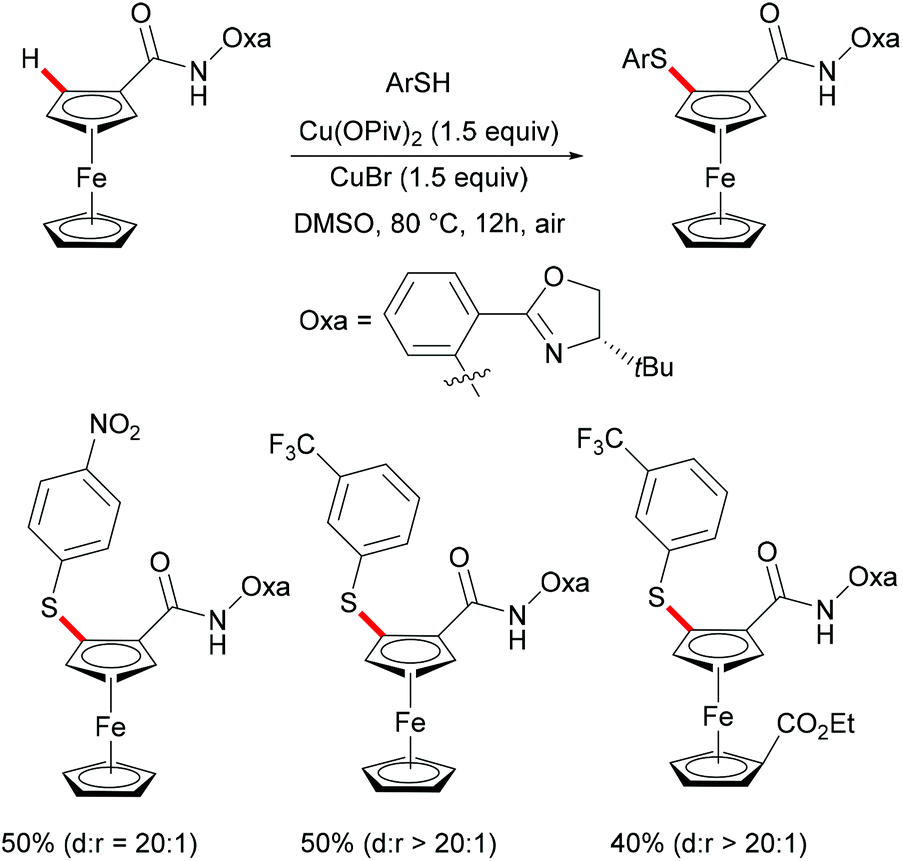
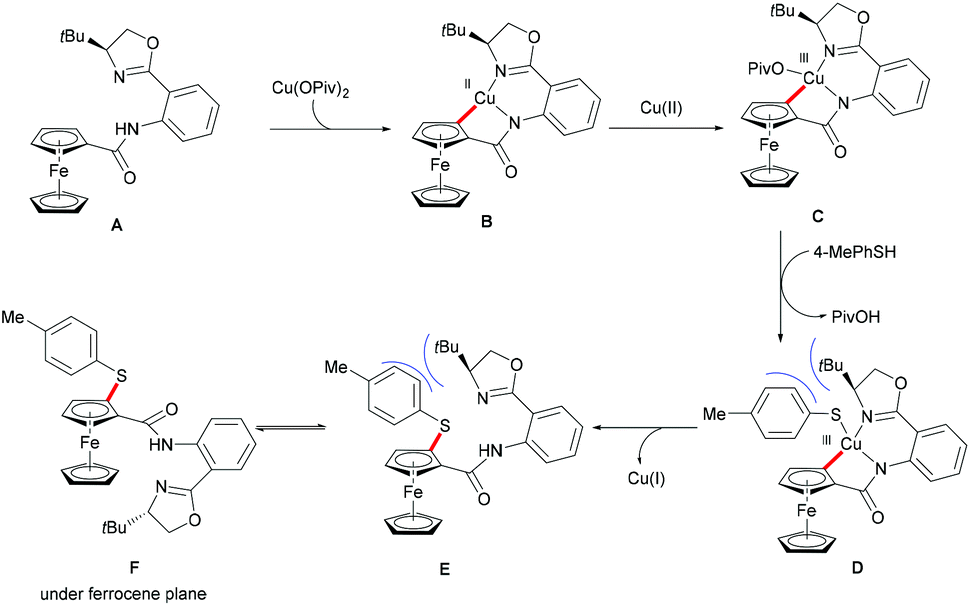
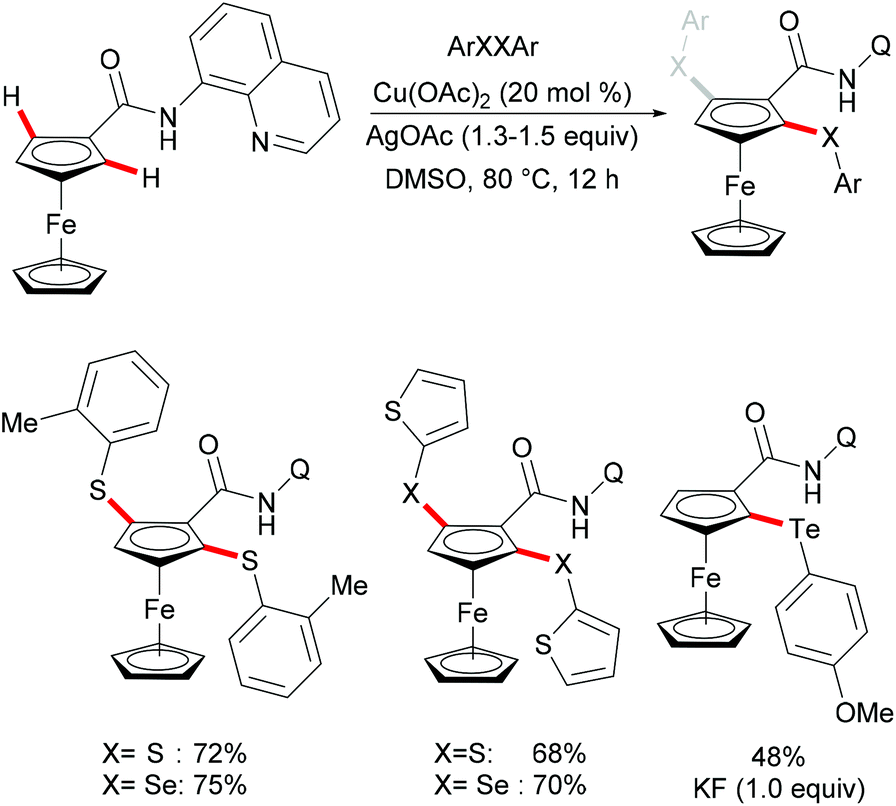
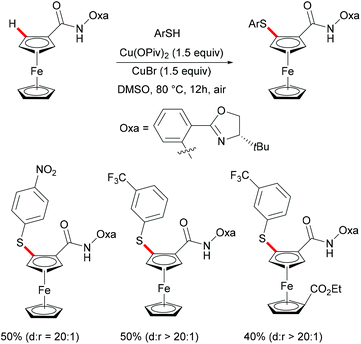
Ferrocene is a useful structural motif and has attracted attention in recent years due to its ability to serve as a backbone of many chiral ligands or catalyst in asymmetric reactions. In this context, Yu and coworkers reported the copper-mediated diastereoselective C–H thiolation of ferrocene by using benzenethiol as the sulfur reagent with the assistance of the amino-oxazoline directing group (Scheme 42).189 After evaluating a series of substituents on oxazoline, the authors demonstrated that the bulky tert-butyl groups above the tethered ferrocene could maintain the desired conformation and therefore increase the diastereoselective ratio. In contrast, the directing group of the obtained mono-thiolated product was forced to the other side to provide an unfavorable conformation for further C–H thiolation, and thus suppressed the overreaction of the mono-thiolated product. Although the mechanism is still unclear to date, the authors proposed a similar copper(II)/copper(III) reaction model for this transformation based also on previous reports (Scheme 43). Initially, the copper coordinates to the N,N-bidentate group to achieve the ortho-C–H activation of the cyclopentadienyl ring, followed by an oxidative process to provide the copper(III) intermediate. After anion exchange, the thus formed species D undergoes reductive elimination releasing the thiolated product and copper(I) complex.
 |
| | Scheme 42 Copper-mediated diastereoselective C–H thiolation of ferrocenes. | |
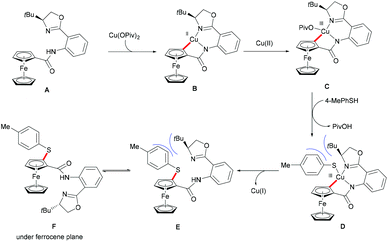 |
| | Scheme 43 Copper-mediated diastereoselective C–H thiolation of ferrocenes. | |
Very recently, the same group further expanded this copper-mediated C–H thiolation directed by the amino-oxazoline group by using ethylene sulfide as the sulfur source.190 This reaction was accomplished with 1.0 equiv. of Cu(OAc)2·H2O at room temperature with a variety of functional groups being tolerated, furnishing mono- and di-thiolated products in moderate to good yields. After a sequential hydrolysis–lactonization process, the thus obtained monothiolated product could be converted into the corresponding seven-membered benzoxathiepinone derivatives in good yields (Scheme 44).
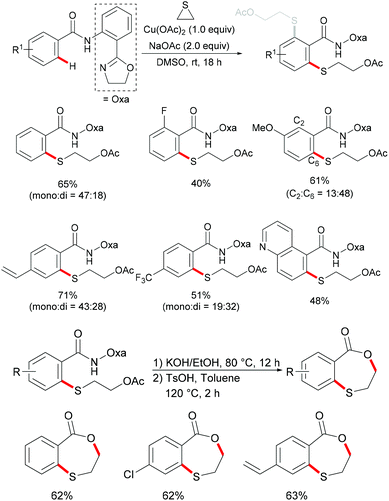 |
| | Scheme 44 Copper-mediated C–H thioetherification of arenes with ethylene sulfide. | |
3.1.2 DMSO as the thiolating reagent.
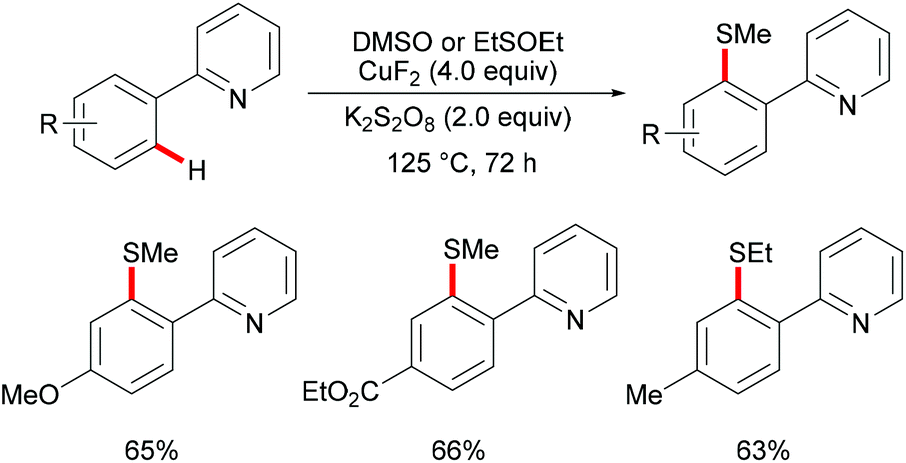
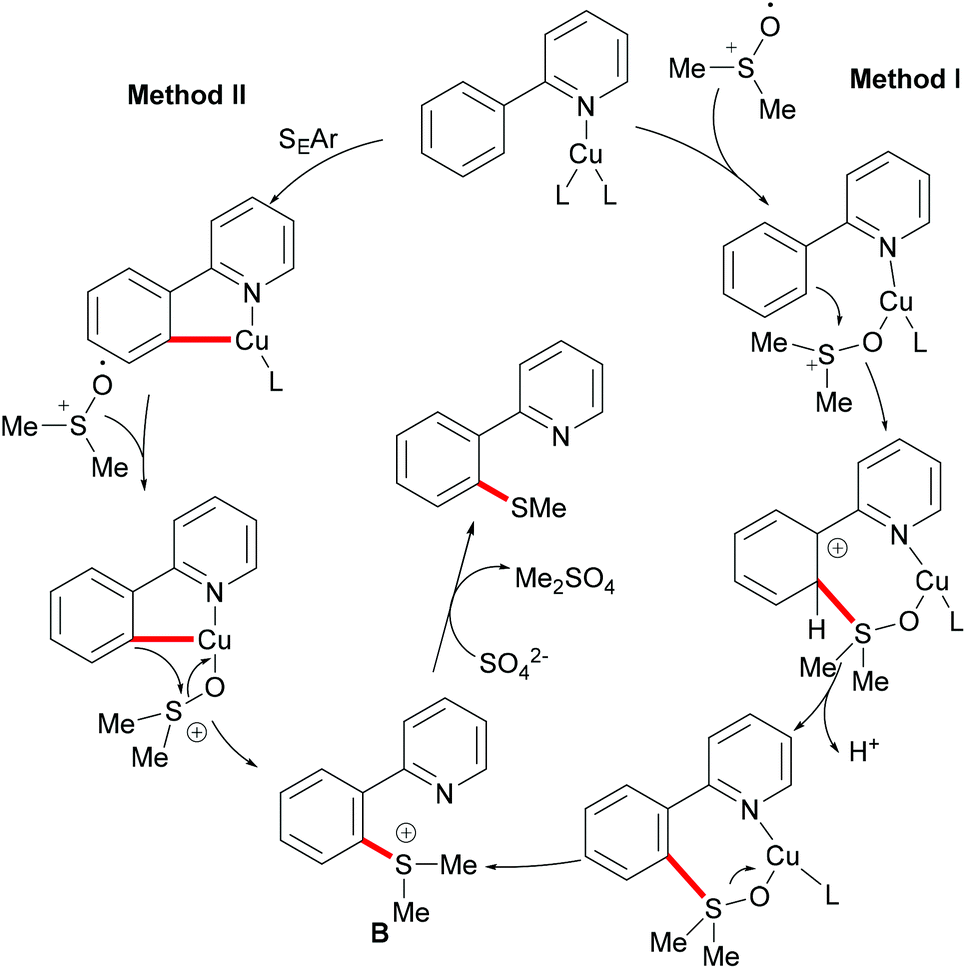
DMSO was also envisioned to be an effective sulfur source under copper catalysis. In this context, the Qing group reported Cu(II) mediated C–H methylthiolations of arenes with DMSO in 2010 (Scheme 45).191 This reaction required an excess of CuF2, acting as a catalyst and an oxidant, extended reaction time and high reaction temperature to ensure full conversion. The substrates bearing either electron-withdrawing or electron-donating groups did not affect the outcome of the reaction and gave the mono-thiolated products in synthetically useful yields along with trace amounts of disubstituted products. Pyrimidine was also an effective directing group for this reaction. Furthermore, diethyl sulfoxide was also a suitable coupling partner and afforded the corresponding products in moderate yields. Finally, the authors proposed two possible pathways using sulfonium ions as key intermediates for this transformation (Scheme 46). Route I involved an intramolecular electrophilic aromatic substitution and then cleavage of the S–O bond with the assistance of CuII. In route II, a metallacycle could be formed from Avia SEAr and then rearranged to give intermediate B. A subsequent methyl-transfer process promoted by a nucleophile would deliver the desired product.
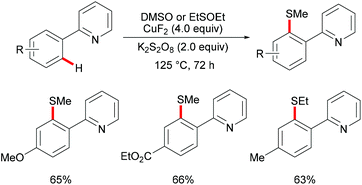 |
| | Scheme 45 Copper(II)-catalysed functionalizations of aryl C–H bonds. | |
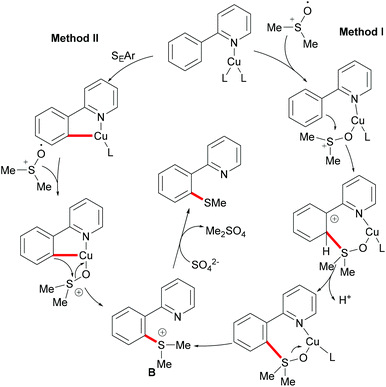 |
| | Scheme 46 Proposed mechanism for copper(II)-catalysed functionalization of aryl C–H bonds. | |
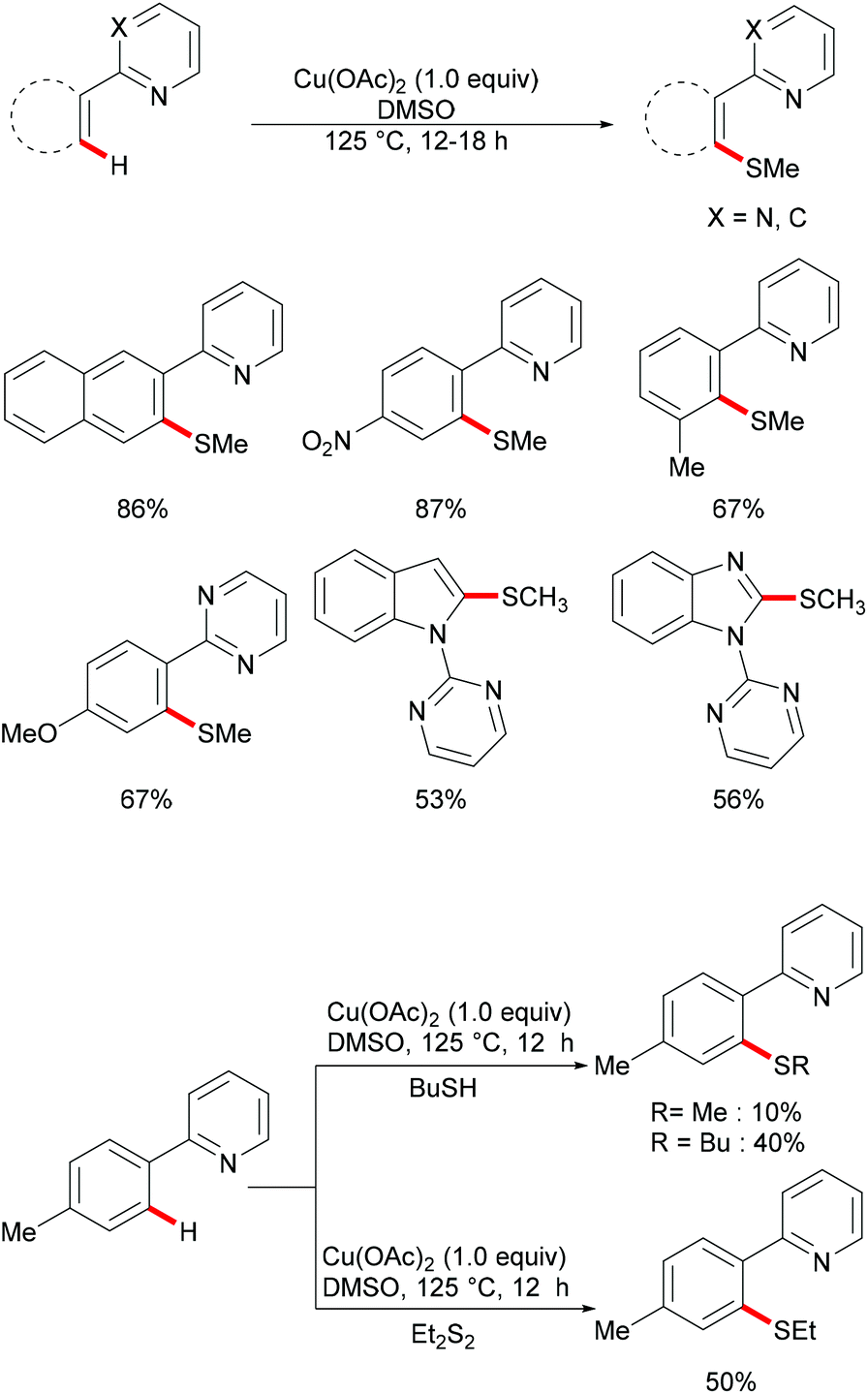
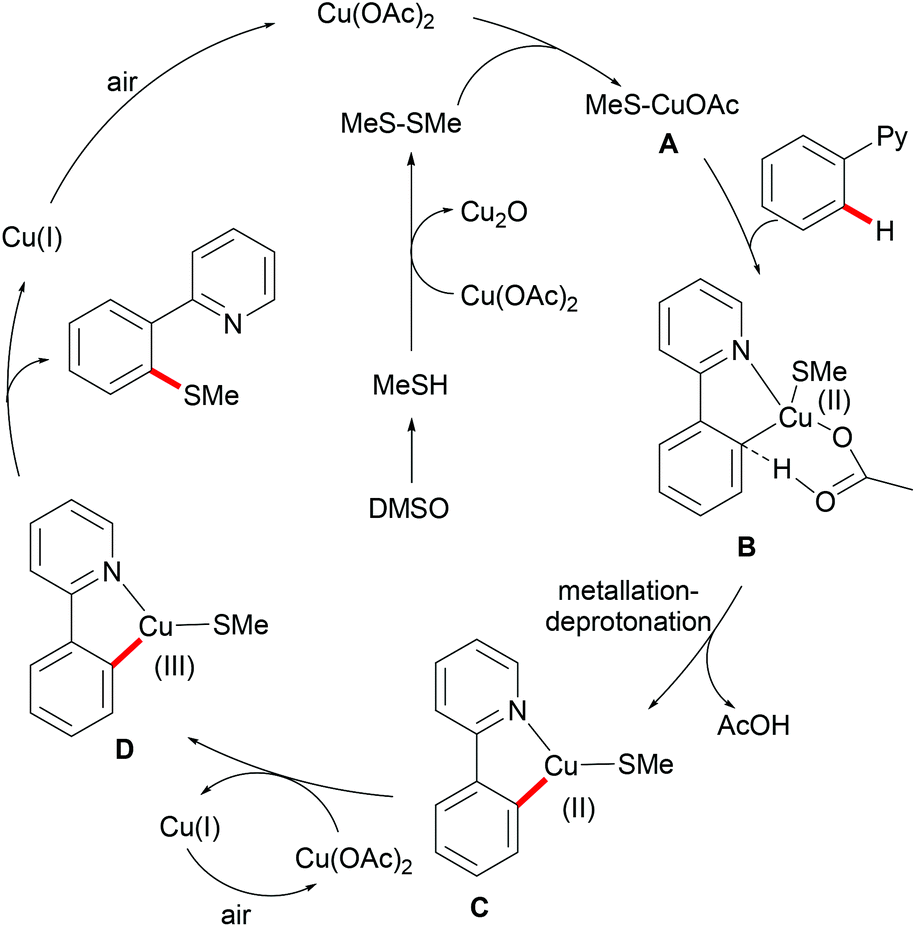
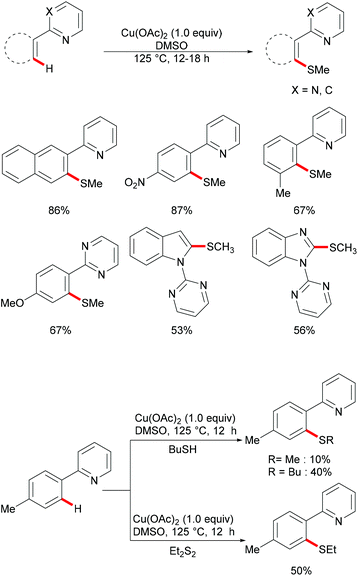
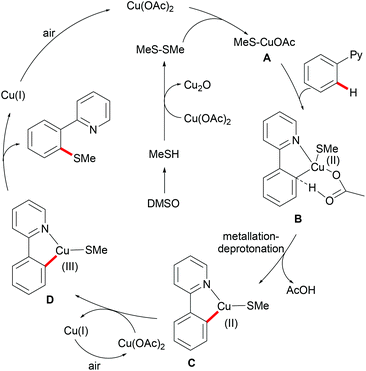
Inspired by this report, Jain disclosed a similar transformation by using 1.0 equiv. of Cu(OAc)2 as the catalyst without any extra oxidant at 125 °C for 12 h (Scheme 47).192 2-Naphthylpyridines displayed a higher reactivity than the phenyl substituted substrates probably due to the lower aromaticity and more localized π-electron density and afforded the thiolated product at the less sterically hindered position with a good yield, whereas the meta-methyl substituted substrates gave the product at the more hindered position. The authors suggested that the copper catalyst could be coordinated to the directing group and the S atom of the formed mono-thiolated product which suppressed the further thiolation process, and thus afforded solely the mono-selective thiolated products. As expected, pyrimidine was also an effective directing group in this transformation. The control experiment conducted with BuSH and Et2S2 as the sulfur source providing the corresponding butylthiolated and ethylthiolated products in moderate yields indicated that disulfides might be the actual thioalkylated reagents. Moreover, the dimethyl disulfide and Cu2O observed through GC-MS and PXRD respectively further supported this hypothesis. On the basis of these results, the authors proposed a reasonable catalytic cycle involving a copper(II)/copper(III) pathway which is shown in Scheme 48.
 |
| | Scheme 47 Copper acetate–DMSO promoted methylthiolation of arenes and heteroarenes. | |
 |
| | Scheme 48 Proposed mechanism for copper acetate–DMSO promoted methylthiolation of arenes and heteroarenes. | |
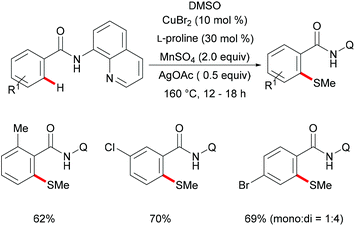
Subsequently, Huang and coworkers demonstrated a highly efficient copper-catalysed direct C–H thiomethylation of arenes with DMSO assisted by the 8-aminoquinoline bidentate directing group (Scheme 49).193 The addition of L-proline as the ligand significantly improved the yield. Benzamides bearing different functional groups on the ortho- or meta-position of the phenyl ring reacted with DMSO smoothly and afforded the monosubstituted products in moderate yields, whereas the unsubstituted or para-substituted substrates resulted in a mixture of mono- and dithiolated products. The control experiment conducted with TEMPO as the radical scavenger indicated that a free radical pathway was not likely operative in this transformation. Finally, the authors proposed a similar copper(II)/copper(III) catalytic cycle as previously described.
 |
| | Scheme 49 Copper-catalysed C–H thiomethylation of benzamides with dimethyl sulfoxide. | |
3.1.3 Diselenides and disulfides as chalcogenated reagents.
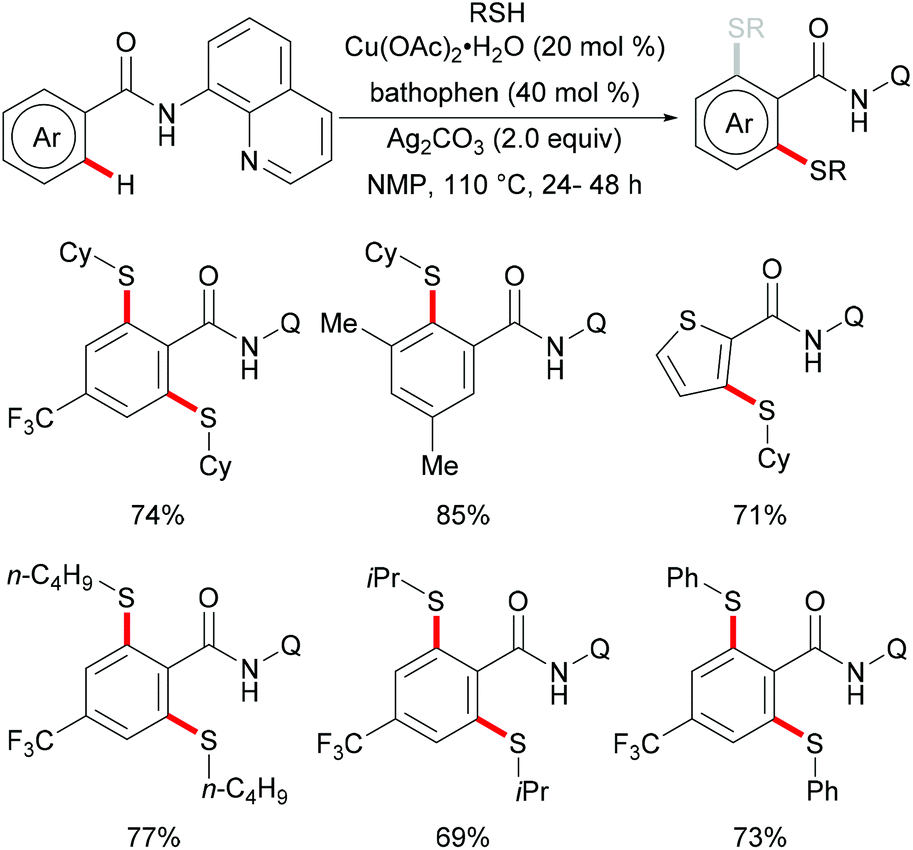
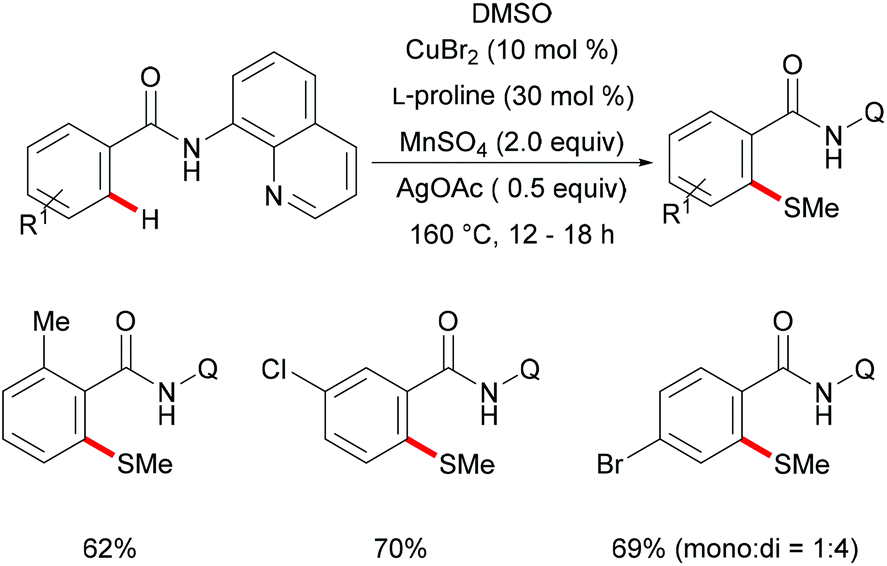
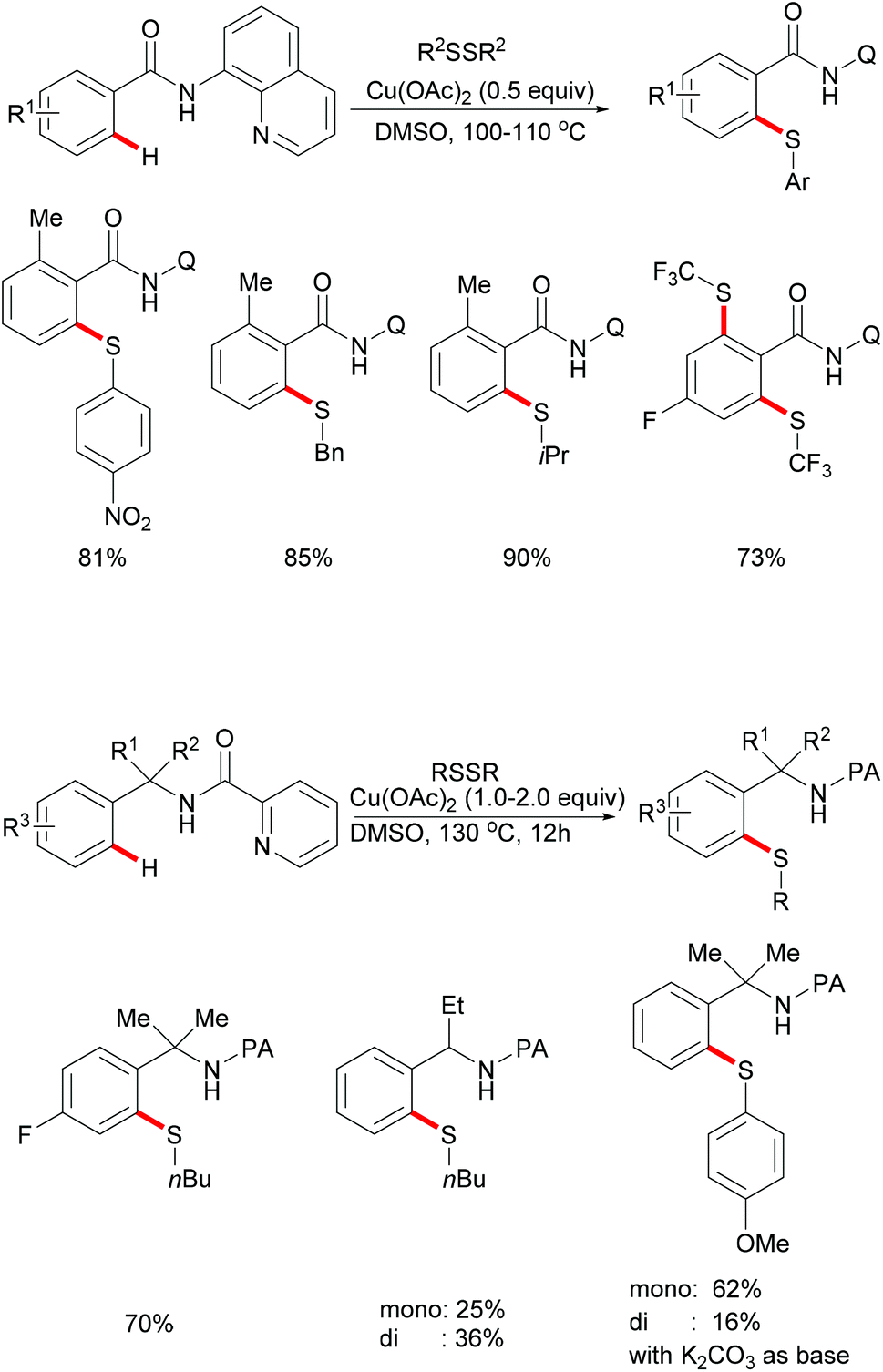
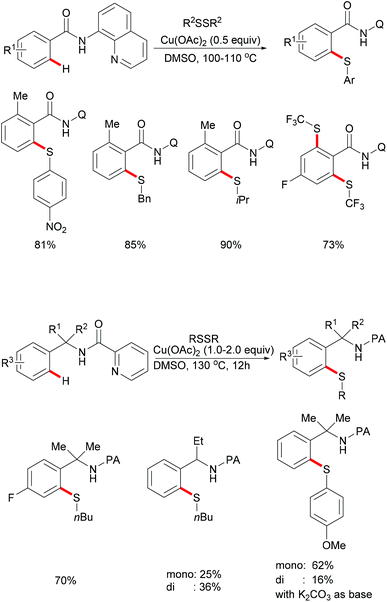
In 2012, Daugulis and coworkers investigated a Cu(OAc)2 mediated C–H thiolation of benzamides by using disulfides as the sulfur reagent with the assistance of the removable 8-aminoquinoline bidentate directing group (Scheme 50).194 DMSO was employed as the ideal solvent due to its potential ability to oxidise thiols to disulfides. Both dialkyl and diaryl disulfides reacted with benzamide substrates smoothly in this reaction and afforded the corresponding mono- or dithiolated products in high yields. Moreover, bis(trifluoromethyl) disulfides were also suitable coupling partners for this transformation. Importantly, benzylamine derivatives decorated with the picolinic acid bidentate directing group were transformed successfully in the presence of a stoichiometric amount of Cu(OAc)2 albeit at a rather high reaction temperature. Control experiments conducted with a reagent grade and ultrapure copper catalyst suggested that catalysis by contaminants was unlikely. Finally, the directing groups were removed through amide N-methylation and subsequent hydrolysis under basic conditions to give thiolated benzoic acid in a good yield.
 |
| | Scheme 50 Copper-promoted thiolation of C(sp2)–H bonds. | |
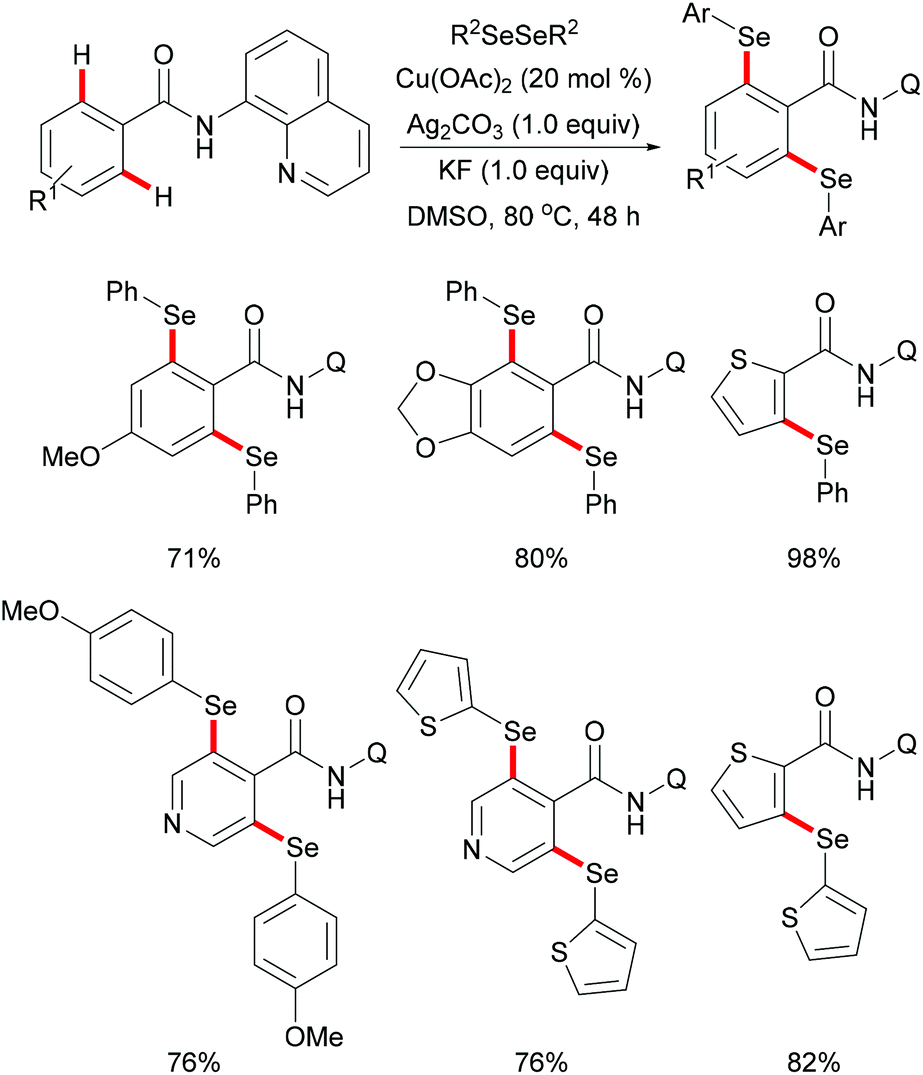
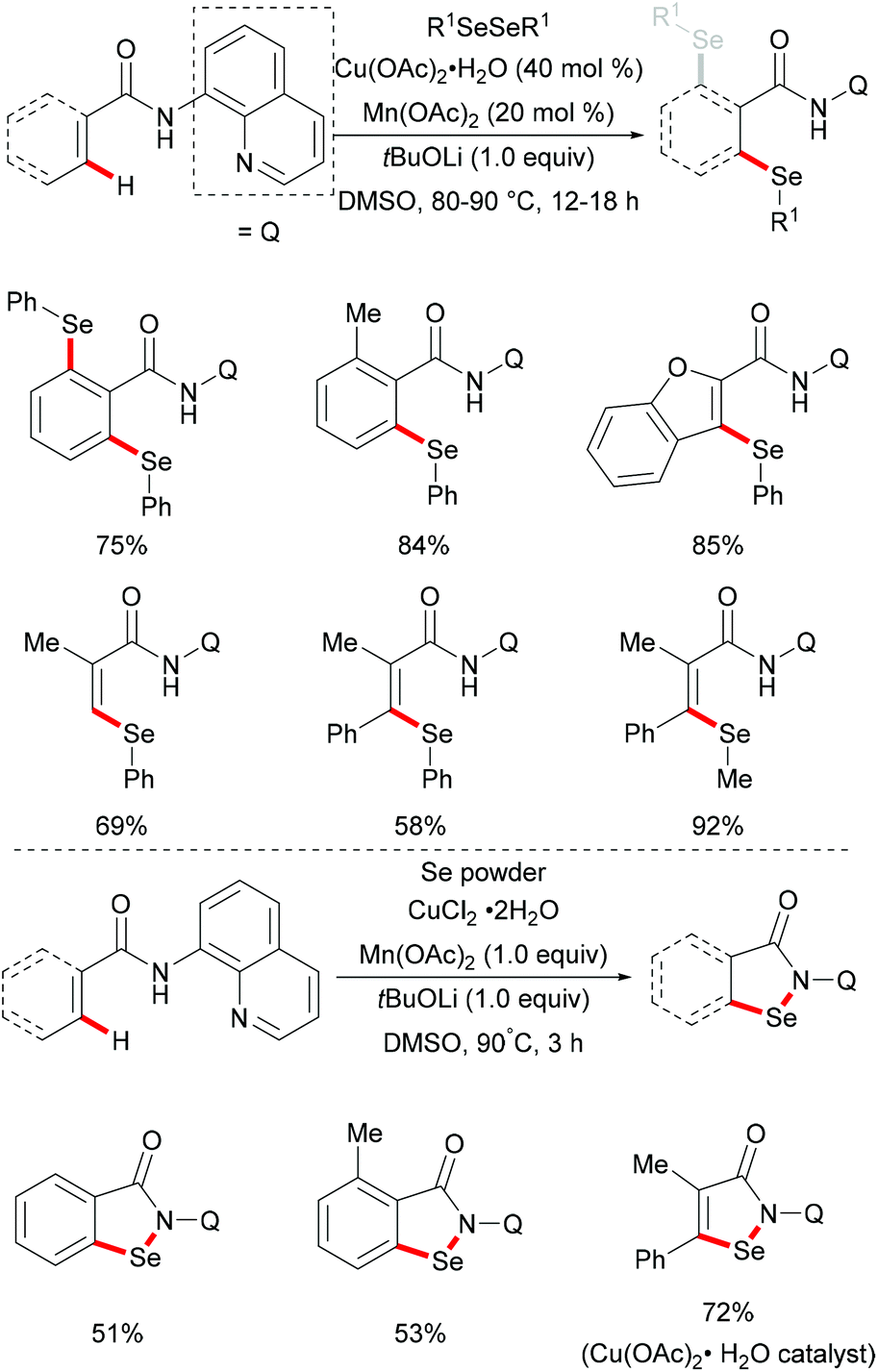
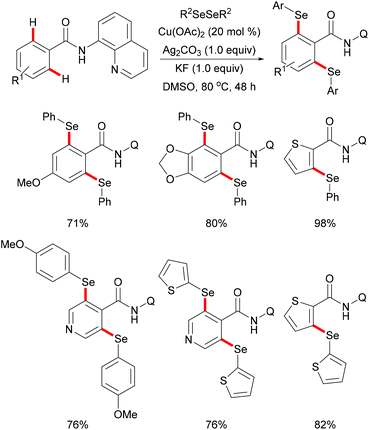
Later, inspired by this report, Baidya's group expanded the substrate scope to include selenylation under comparable reaction conditions (Scheme 51).195 Further investigation showed that this selenylation could also proceed efficiently in the presence of a catalytic amount of Cu(OAc)2 along with Ag2CO3 as an oxidant and KF as an additive, delivering the corresponding mono- or diselenylated products in high yields. The notable features of this reaction included scalability, good functional group tolerance and removal of the directing group. The significant KIE values observed in this reaction indicated that the C–H bond activation might be the rate-determining step. Finally, a plausible mechanism involving the copper(II)/copper(III) pathway for C–H selenylations was proposed based on various control experiments. Initially, the copper catalyst coordinates to the bidentate directing group to achieve the ortho-C–H activation via a base promoted concerted-metalation–deprotonation (CMD) pathway to form the cyclometallated copper(II) intermediate. The subsequent oxidation or disproportionation process to give a copper(III) species, followed by reductive elimination releases the selenylated product and copper(I). Copper(I) could be regenerated to active Cu(II) in the presence of Ag2CO3, re-entering the catalytic cycle (Scheme 52).
 |
| | Scheme 51 Copper-catalysed 8-aminoquinoline-directed selenylation of arene and heteroarene C–H bonds. | |
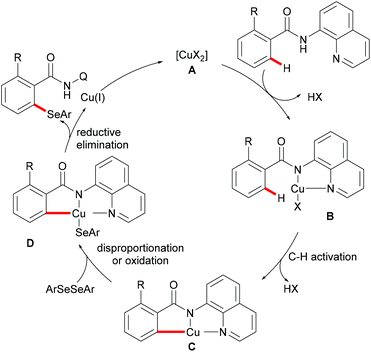 |
| | Scheme 52 Proposed mechanism for 8-aminoquinoline-directed C–H selenylation of arene and heteroarene C–H bonds. | |
More recently, Kumar's group successfully achieved the C–H dichalcogenylations of ferrocene amides with Baidya's protocol (Scheme 53).196 A broad range of functional groups such as methoxy, nitro and halides present in the diaryl disulfide or diaryl diselenide substrates were well tolerated in these reactions. In contrast, dialkyl and dibenzyl disulfides were not compatible. Notably, ditellurides were also suitable substrates in this transformation and afforded the mono-tellurated products in moderate yields. It was supposed that the competing tellurium–copper complexation in the formed mono-tellurated product is favored over the activation of a second C–H bond. Finally, the authors proposed a plausible copper(II)/copper(III) catalytic cycle. However, a free radical process through the homolytic cleavage of dichalcogenides cannot be ruled out at this stage.
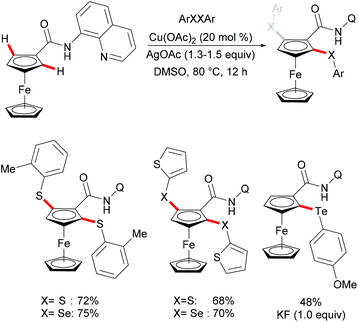 |
| | Scheme 53 Copper-catalysed C–H chalcogenylations of ferroceneamide with aryl disulfides, diselenides, and ditellurides. | |
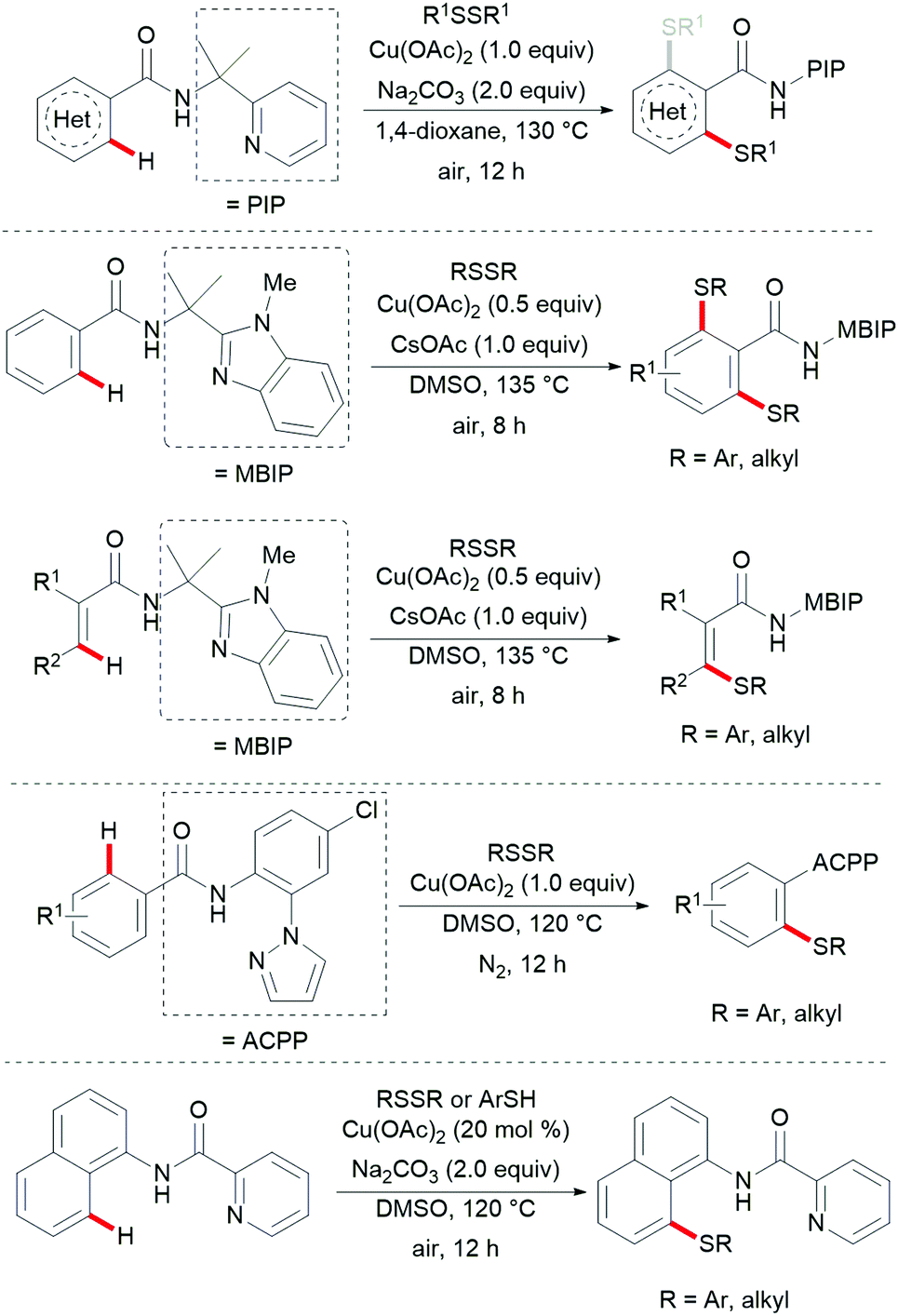
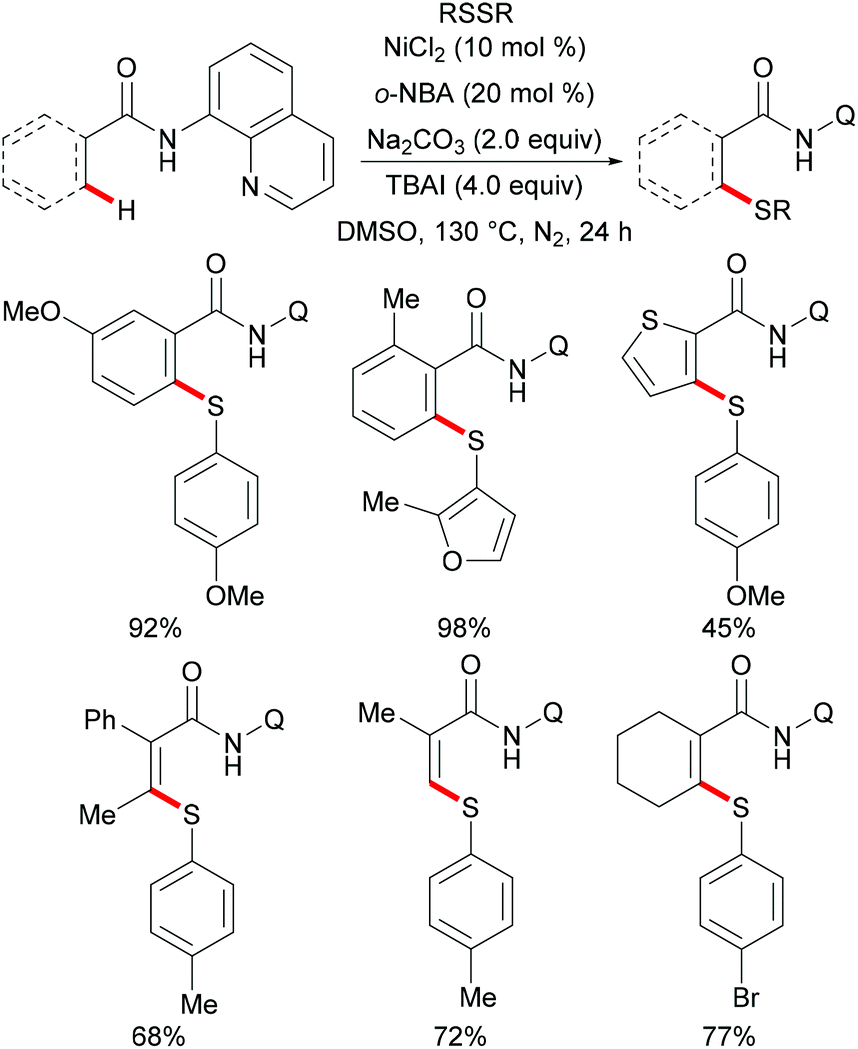
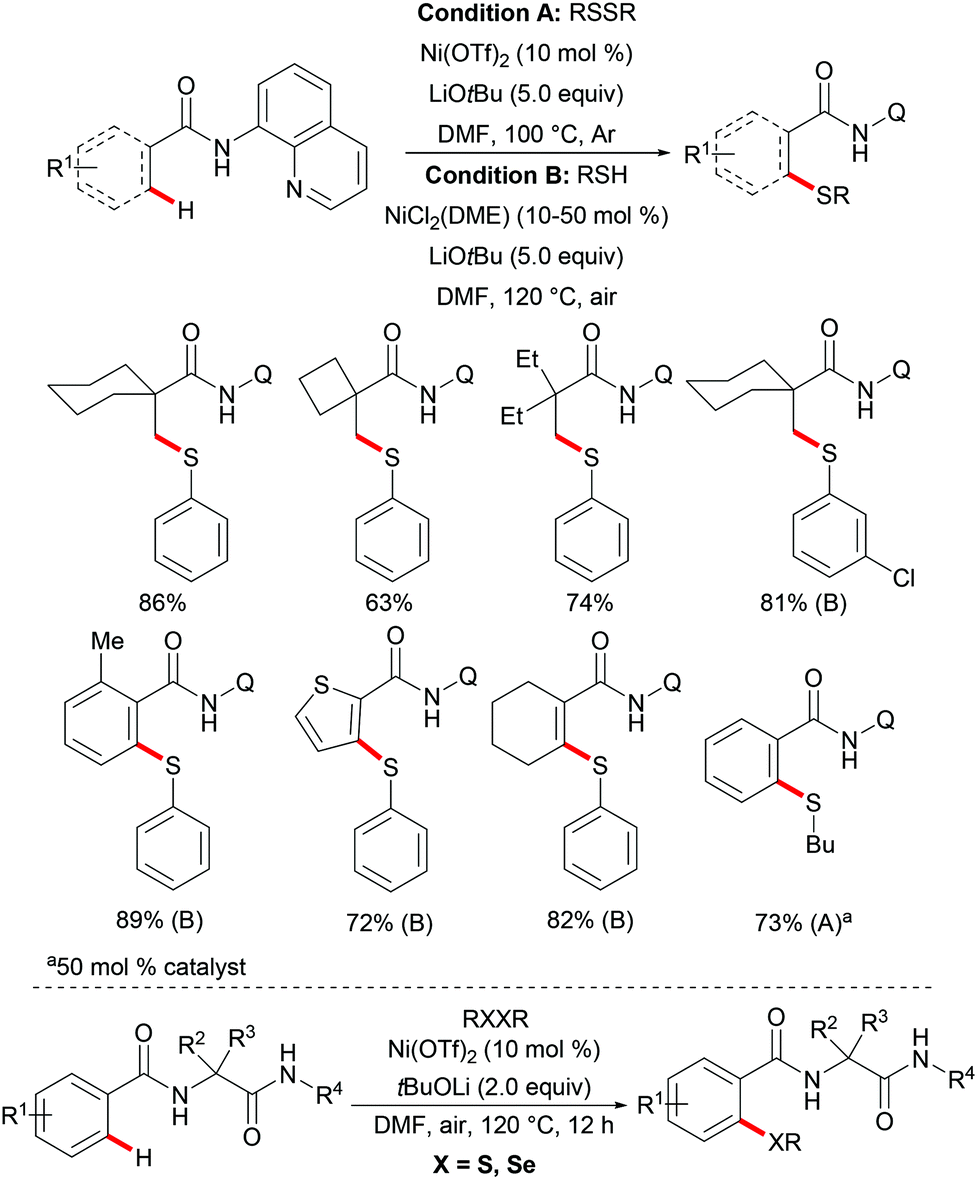
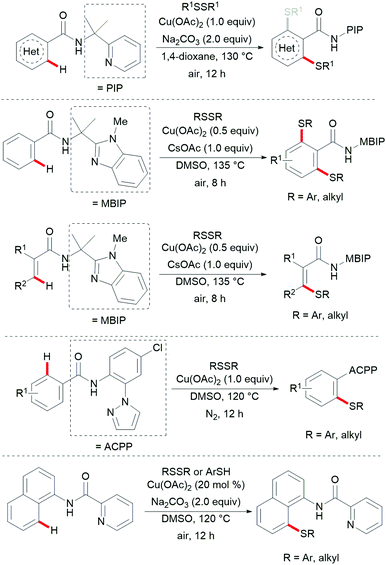
Thereafter, a wide range of bidentate removable directing groups were explored for this copper-catalysed (mediated) C–H chalcogenylation. In 2017, Shi and coworkers reported copper-mediated heteroaryl C–H thiolation with disulfides with the assistance of the PIP bidentate directing group (Scheme 54).197 Typical heterocyclic substrates such as pyridine, thiophene, indole and furan transformed smoothly in the presence of 1.0 equiv. of Cu(OAc)2 and 2.0 equiv. of Na2CO3 without any oxidant and ligands, and delivered the corresponding mono- or di-thiolated products in good to excellent yields. Subsequently, the Song198 and Ji199 groups found that 2-amino alkylbenzimidazole and 2-amino-5-chlorophenyl-1H-pyrazole (ACPP) were also efficient directing groups for C–H thiolations. Benzamides bearing a variety of functional groups reacted with diaryl or dialkyl disulfides smoothly and afforded dithiolated products in moderate to good yields. Importantly, more challenging alkenyl C(sp2)–H substrates such as acrylamide and cycloalkenyl amide were also compatible in this reaction and gave the corresponding mono-thiolated products in good yields. In contrast, the terminal alkenyl substrates gave a mixture of mono- and dithiolated products. Later, Lu and coworkers disclosed an aerobic copper catalyzed peri-selective thiolation of naphthylamines by using picolinamide as the bidentate directing group under similar conditions.200 Detailed mechanistic studies indicated that this reaction proceeded through a copper(II)/copper(III) pathway.
 |
| | Scheme 54 Copper-catalysed C–H thiolations assisted by various directing groups. | |
More recently, Jana and coworkers demonstrated a copper/manganese catalysed C–H selenylation with the assistance of the 8-aminoquinoline directing group (Scheme 55).201 A wide range of functionalized benzamides underwent selenylations with diaryl and dialkyl sulfides, providing the mono- or diselenylated products in high yields. Furthermore, the alkenyl C(sp2)–H acrylamide derivatives could also participate in this reaction and delivered the Z-selective mono-selenylated products. It was noteworthy that selenium powder was also demonstrated to be an effective chalcogen source in the presence of CuCl2 as the catalyst under similar conditions. The considerable intermolecular KIE values (9) implied that the rate-determining step is likely to involve the C–H bond cleavage. Inhibitory effects by radical quenchers such as TEMPO and BHT indicated that a SET pathway may not be involved in this selenation reaction.
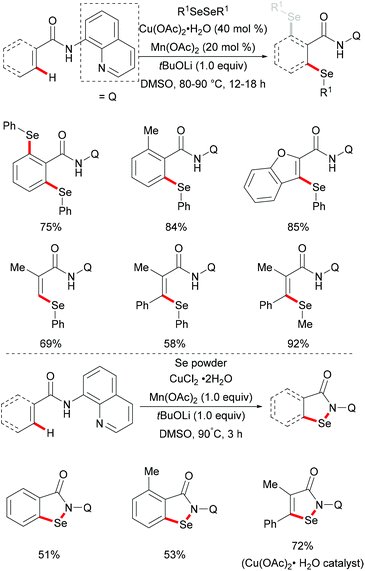 |
| | Scheme 55 Copper/manganese cocatalysed C–H selenylation of arenes, heteroarenes, and alkenes under air. | |
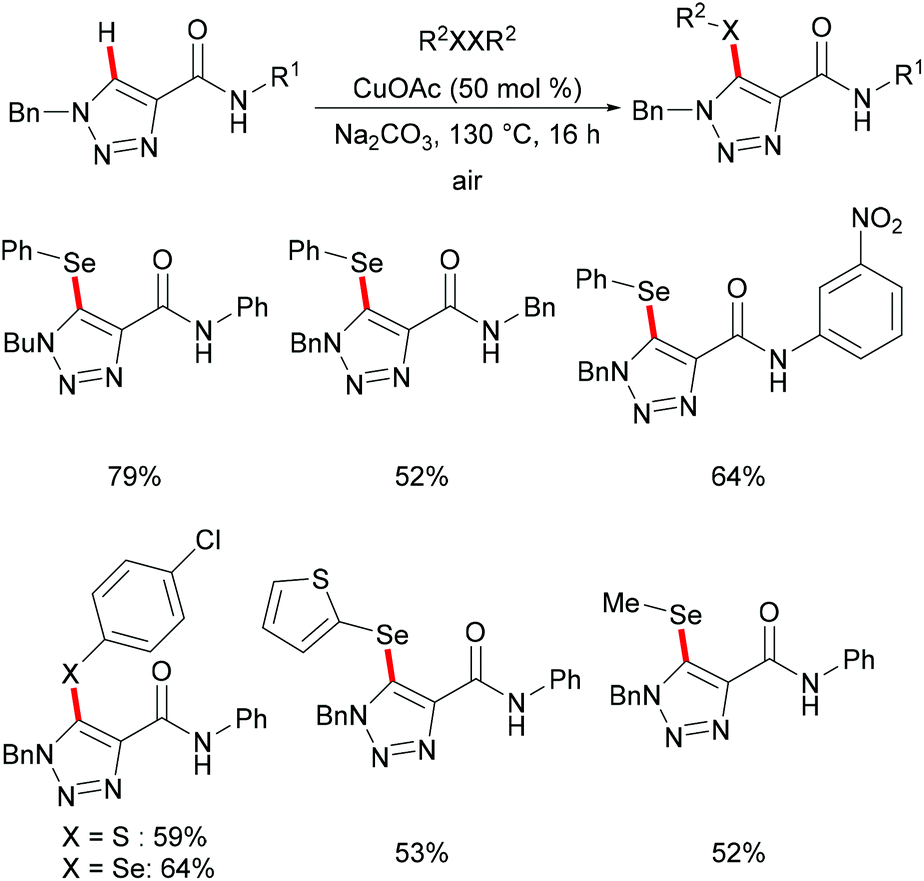
The copper-catalysed C–H chalcogenylations were widely explored with the assistance of a variety of bidentate directing groups due to their versatility in forming the more stable N,N–Cu chelated intermediate which could be easily oxidized to Cu(III) to promote the transformation efficiently. In contrast, the mono-dentate directing groups proved to be elusive. In 2016, the Ackermann group reported copper(I)-catalyzed C–H chalcogenylations of 1,2,3-triazoles by weak O-assistance overriding the strong N,N-bidentate moiety (Scheme 56).202 This strategy tolerated a wide range of functional groups present in both the disulfide and triazole substrates, providing the chalcogenated products in good yields. The well-defined copper(I) selenide PhSeCu reacted with triazole efficiently under air, thus indicating the formation of copper(I) selenides in this manifold.
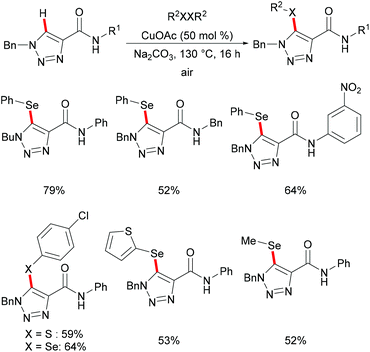 |
| | Scheme 56 Copper-catalysed C–H chalcogenylation with the weak O-assistance. | |
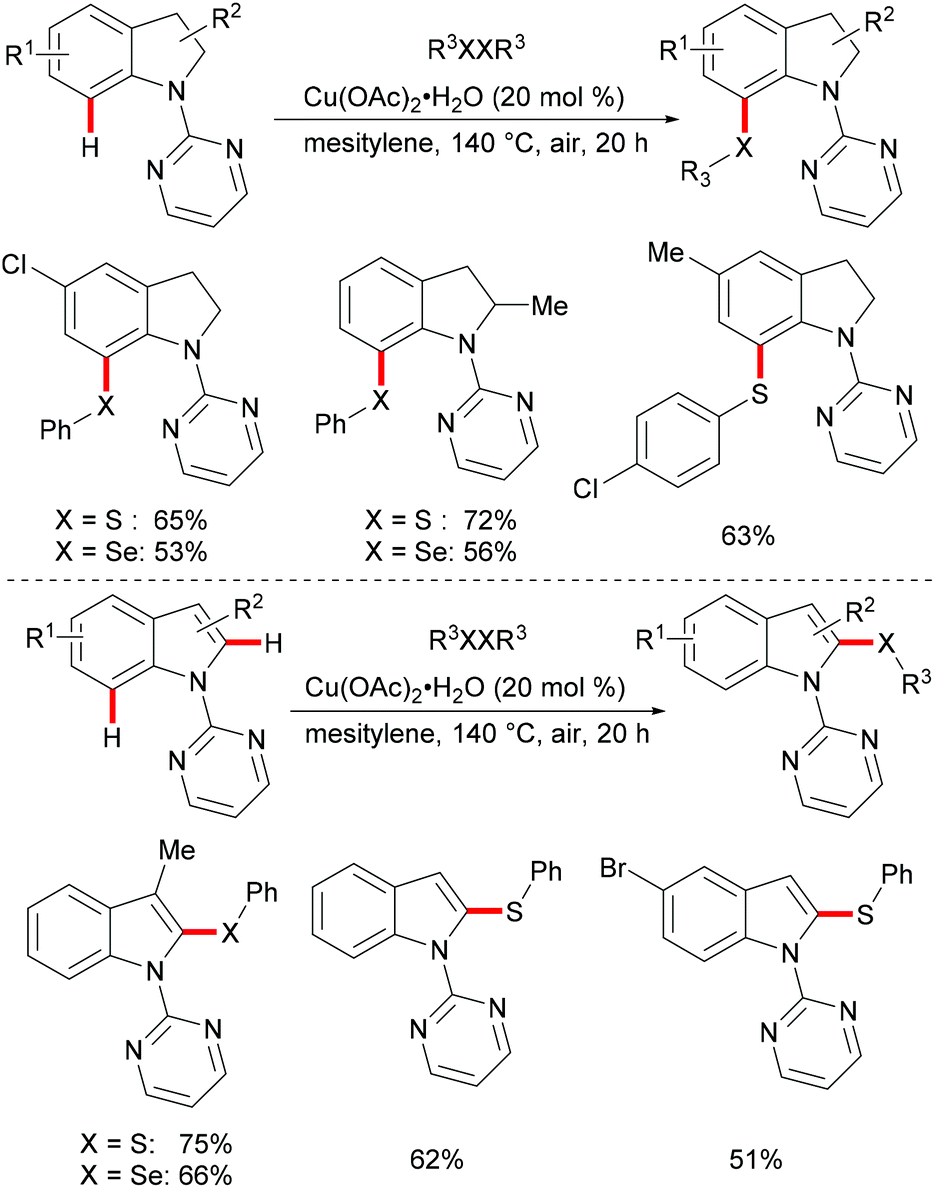
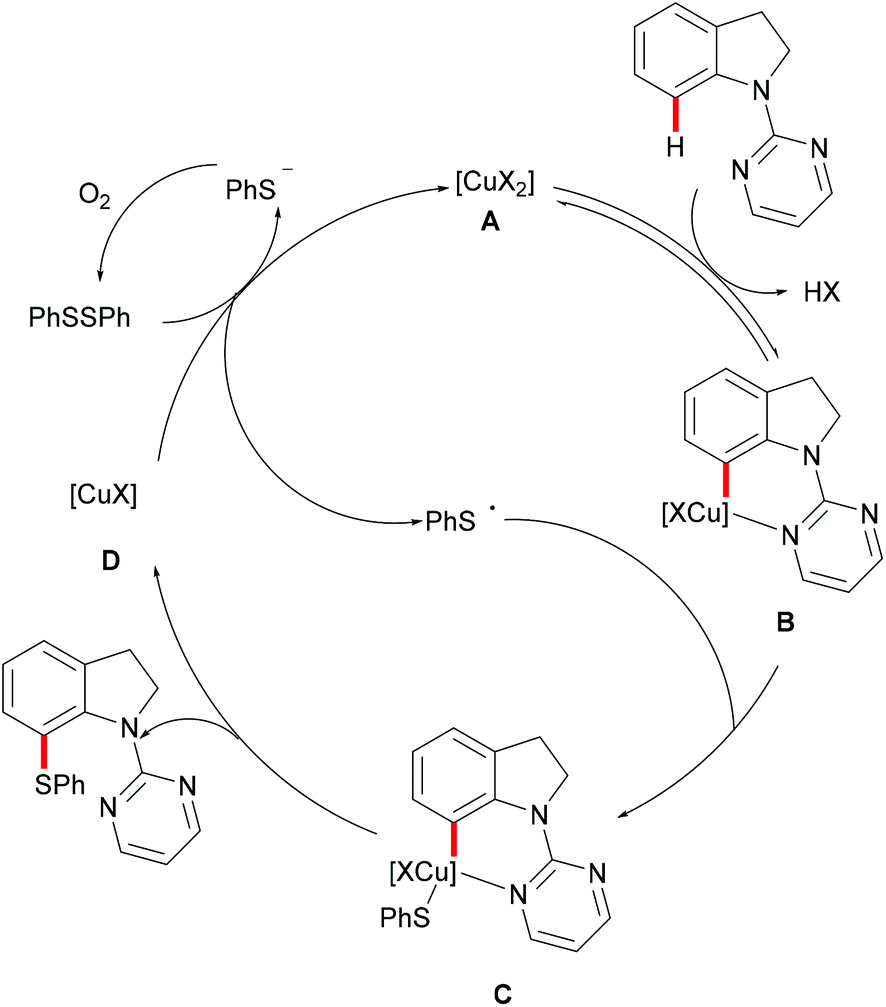
Subsequently, the Ackermann group reported on a copper-catalysed C–H chalcogenylation of indoles and indolines at the C2 and C7 positions, respectively (Scheme 57).203 This protocol featured operational simplicity, broad functional group tolerance and excellent regioselectivity, and the directing group was readily removable. Control experiments conducted with typical radical scavengers significantly suppressed this transformation, thus indicating that a SET pathway might be involved in this copper-catalysed C–H chalcogenylation. Based on the preliminary mechanistic studies and the literature precedents, the authors proposed a copper(II)/copper(III) catalytic cycle. Initially, complexation of the pyrimidine directing group by the copper catalyst took place, followed by C7 C–H activation to form a six-membered cyclometalated copper(II) intermediate which then undergoes an oxidation process by the addition of a sulfenyl radical to generate the copper(III) species. Subsequent reductive elimination delivers the desired product and releases a copper(I) species, which could be reoxidized to copper(II) by the action of disulfide and oxygen (Scheme 58).
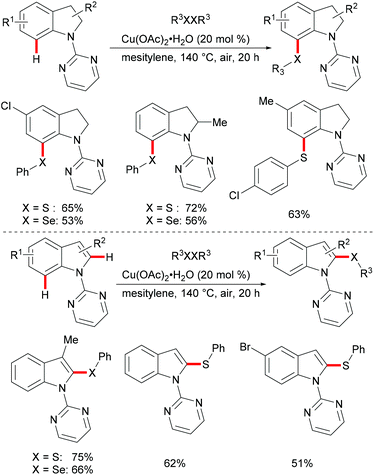 |
| | Scheme 57 Copper-catalysed positional-selective C–H chalcogenylation of indolines and indoles. | |
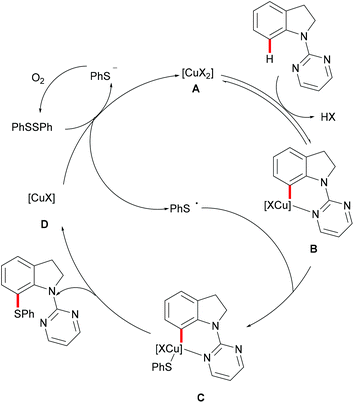 |
| | Scheme 58 Proposed mechanism for the positional-selective C–H chalcogenylation of indolines. | |
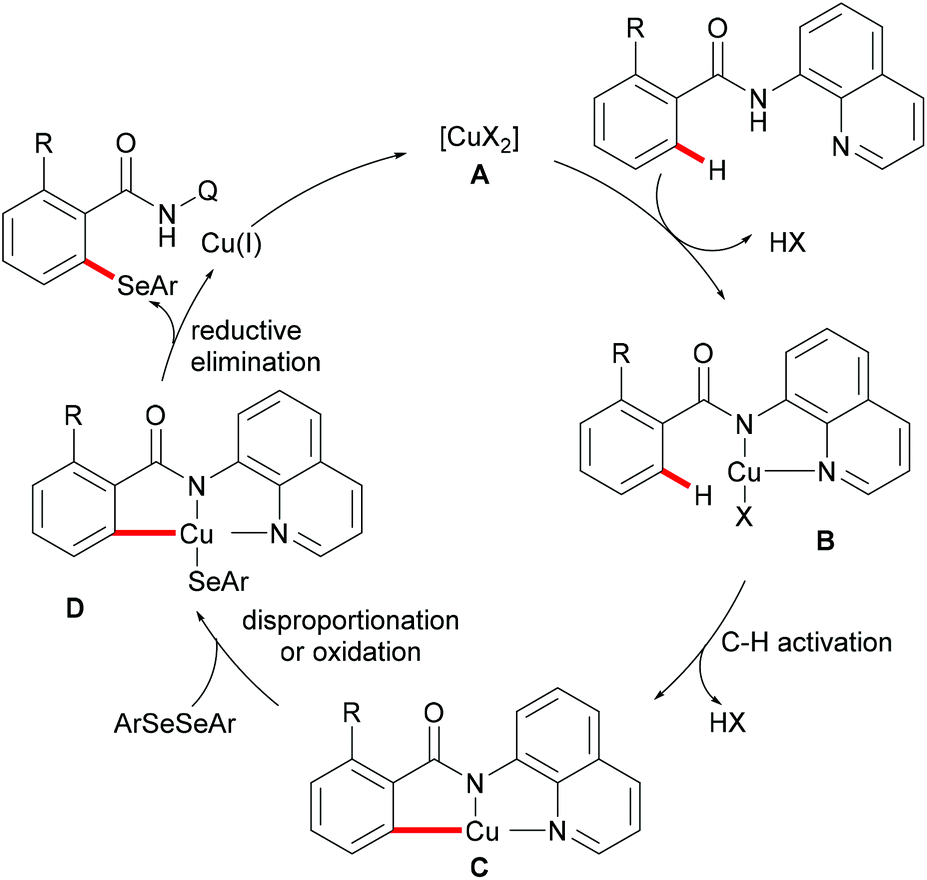
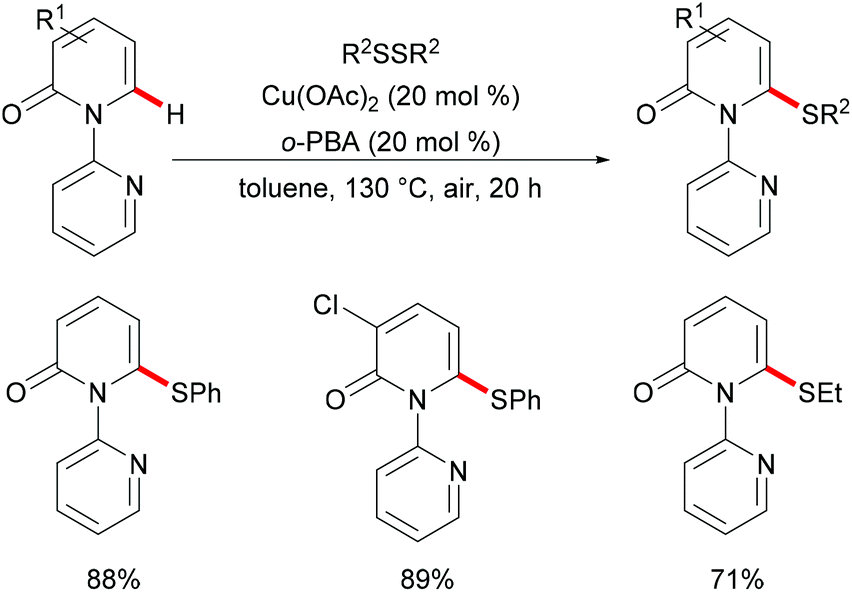
In 2016, Liu's group reported copper(II)-catalysed C6-selective C–H thiolation of 2-pyridones for the preparation of functionalized 6-sulfenylation-2-pyridones.204 In the presence of 20 mol% Cu(OAc)2 as the catalyst and 20 mol% o-PBA (2-biphenylcarboxylic acid) as the additive, a wide range of functional groups were well tolerated under these conditions, providing the corresponding products in good yields. Both the aryl-substituted and the less reactive alkyl disulfides showed acceptable reactivity, furnishing the thiolated products in moderate yields (Scheme 59). Finally, the directing groups could be easily removed by a hydride reduction to give the NH-free thiolated 2-pyridone derivatives. Further mechanistic studies suggested that a free radical process was not likely involved in the copper catalysis.
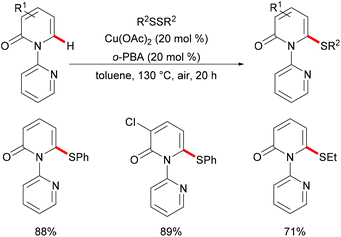 |
| | Scheme 59 Copper(II)-catalysed C6-selective C–H thiolation of 2-pyridones. | |
3.2 Cobalt catalysis
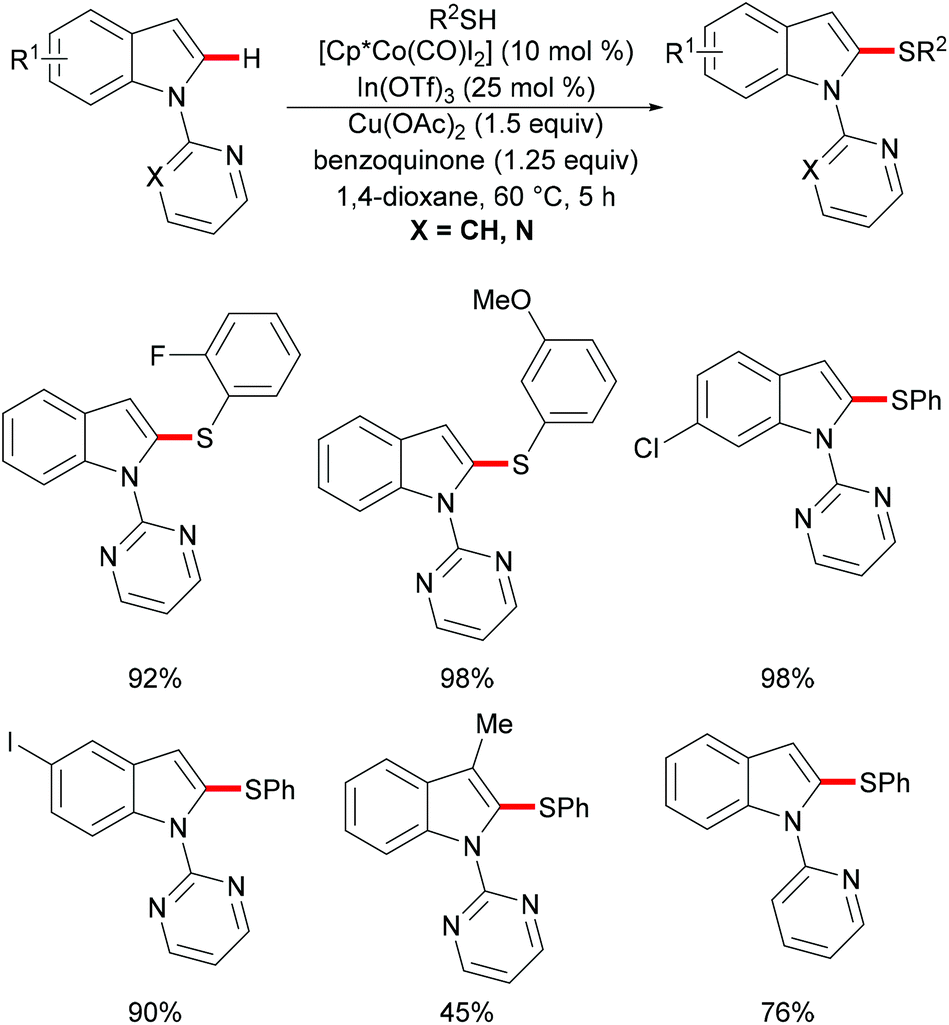
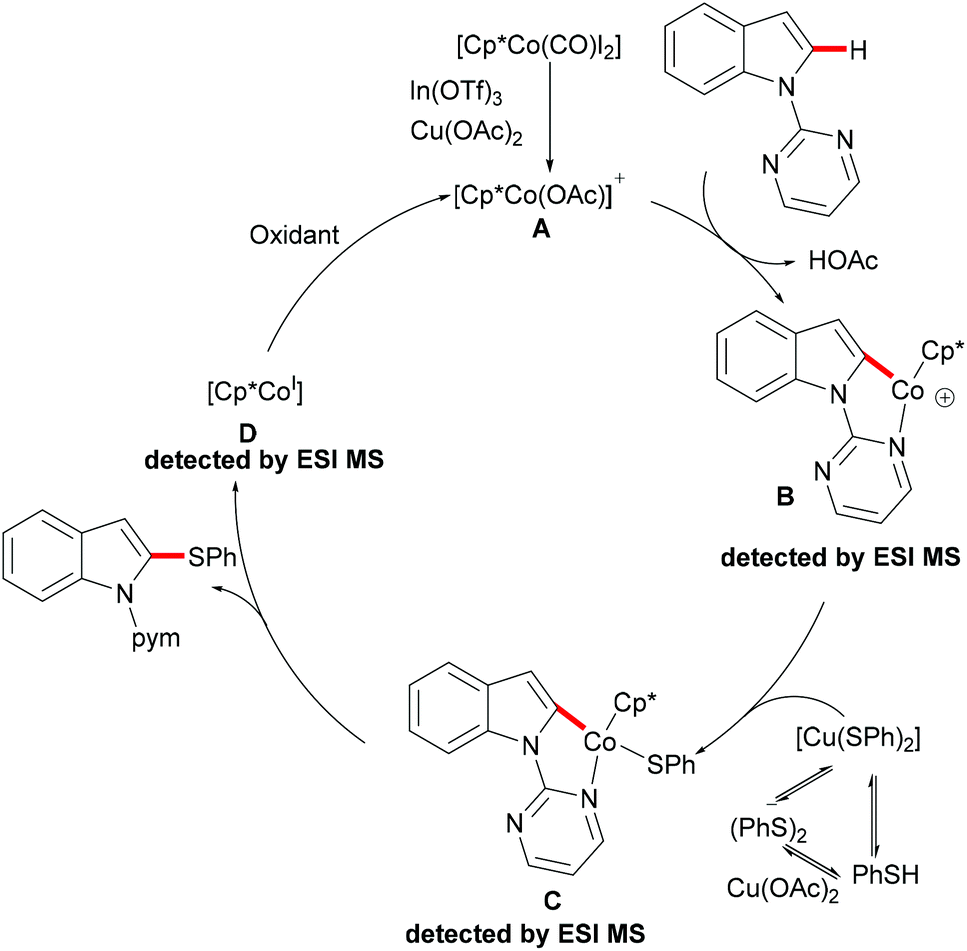
As early as 1955, Murahashi and co-workers reported the first example of a cobalt mediated C–H ortho-carbonylation reaction for the preparation of phthalimidines by the treatment of a Schiff base with Co2(CO)8, which laid the foundation for cobalt catalysed chelation-assisted C–H functionalizations.205–207 In recent years, Yoshikai, Ackermann and Nakamura reported low-valent cobalt-catalysed C–H transformations, including intermolecular hydroarylation of alkynes and alkenes. Pyridine, pyrimidine, and imines were employed as the directing groups for these reactions at room temperature with Grignard reagents as the reducing agent, featuring good yields and high stereoselectivities.208,209 Thereafter, Daugulis and Ackermann reported cobalt catalysed alkyne annulations and alkenylations using the 8-aminoquinoline-derived N,N-bidentate directing group.208,210,211 These reactions were performed under mild reaction conditions which enabled a wide range of substrates to be used. Moreover, the Kanai/Matsunaga group exploited high-valent Cp*CoIII catalysed addition of aryl C–H bonds to polar electrophiles.212 Thus, the cobalt(III) manifold proved to be broadly applicable and enabled C–H cyanations, amidations, amino-carbonylations, halogenations, allylations and annulations.213–216 Inspired by these findings, the Glorius group reported the cobalt-catalysed C–H thiolation of indoles with thiols assisted by pyrimidine or pyridine directing groups through a dehydrogenative cross-coupling (Scheme 60).217 A wide substrate scope was demonstrated and the reaction occurred in all cases with good yields in a regioselective fashion. Control experiments conducted with [Cp*Co(C6H6)][B(C6F5)]2 as the precatalyst resulted in a lower yield without In(OTf)3, thus indicating that In(OTf)3 plays a dual role as a halide abstractor and a promoter of the coordination of thiolates to copper. Additionally, further investigations implied that the copper salt was essential not only for the generation of the active catalyst, but also for the formation of the active thiolation agent. Finally, a plausible mechanism was proposed as shown in Scheme 61 based on control experiments and the previous literature precedents. Initially, the active Co(III) species A was formed by the removal of iodide from [Cp*Co(CO)I2] in the presence of In(OTf)3 and Cu(OAc)2, followed by a directed C–H activation of indole to provide five-membered cobaltacycle B. Next, complex B reacted with an anionic Cu(I) species to deliver cobalt thiolate C, which subsequently underwent a reductive elimination process to release the thiolated product and cobalt(I) species D. Finally, the cobalt(I) species could be reoxidized to regenerate the active catalyst A in the presence of Cu(OAc)2 or benzoquinone. Another alternative mechanism involved a nucleophilic attack process through electrophilic Cu(II) or Cu(III) thiol species on species B or trans-metalation of copper species with indole from B, followed by a reductive elimination process to give the desired product.
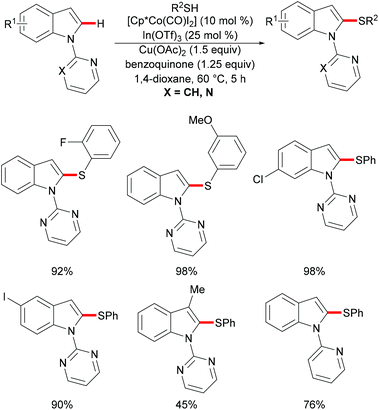 |
| | Scheme 60 Cobalt-catalysed C–H thiolation of indoles with thiols. | |
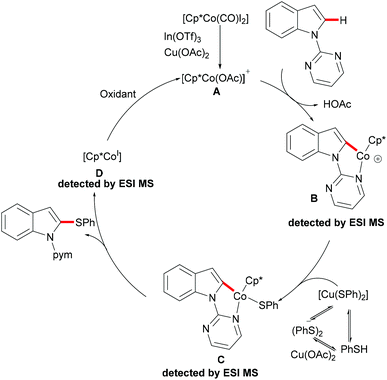 |
| | Scheme 61 Proposed mechanism for the cobalt-catalysed C–H thiolation of indoles with thiols. | |
In 2017, Gui's group reported on a cobalt-catalysed C–H methylthiolation employing inexpensive DMSO as the methylthiolating agent with the assistance of 8-aminoquinoline as the bidentate directing group (Scheme 62).218 The key to the success was the judicious choice of PhCOCl as the additive, while others gave inferior results. Substrates decorated with either electron-withdrawing or electron-donating groups on the aromatic ring were efficiently converted and furnished the mono-selective methylthiolated products with excellent regioselectivity. Diethylsulfoxide could also participate in this reaction and gave the corresponding ethylthiolated products in good yields. Moreover, the acrylamide substrates were also compatible under the optimized conditions and delivered (Z)-(β)-alkenyl sulfides in good yields. As was expected, the amide could be hydrolyzed to carboxylic acid derivates with overall 56% yield in two steps. Finally, a plausible catalytic cycle involving a SET process was proposed and is shown in Scheme 63. The reaction was initiated by the coordination of a cobalt(III) catalyst to the bidentate directing group to form five-membered cobaltacycle intermediate A and the following intramolecular SET pathway gave radical cation intermediate B. In the presence of a silver oxidant, the cobalt(II) complex was oxidized to cobalt(III) intermediate C which subsequently reacted with the methylthio anion generated in situ from DMSO to form radical intermediate D. After an additional intramolecular SET process of species D, the thus formed cation intermediate E underwent a proton elimination pathway from the phenyl ring, and then rearomatised and was transformed into F releasing the final product. The concomitantly released cobalt(II) could be reoxidized to the active cobalt(III) catalyst for a subsequent catalytic cycle.
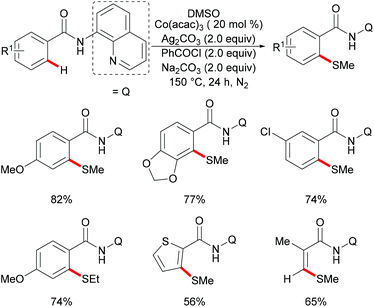 |
| | Scheme 62 Cobalt-catalysed highly mono-selective ortho-methylthiolation of benzamides. | |
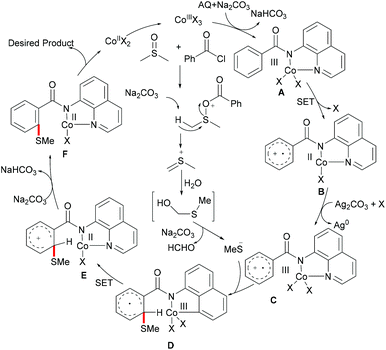 |
| | Scheme 63 Cobalt-catalysed highly mono-selective ortho-methylthiolation of benzamides. | |
More recently, Wang and coworkers disclosed a cobalt-catalysed C–H thiolation assisted by 2-aminopyridine 1-oxide directing groups (Scheme 64).219 This method displayed wide functional group tolerance for both amide substrates and disulfides, and provided the corresponding mono-thiolated products in moderate to good yields. Importantly, this protocol was also applicable for the synthesis of quetiapine, an atypical antipsychotic agent which is an approved drug. Finally, control experiments conducted with typical radical scavengers indicated that this thiolation reaction might involve a free-radical pathway. Based on the preliminary mechanistic investigations and literature precedents, a plausible catalytic cycle involving a radical coupling of the amide substrate with the thioether radical was proposed by the authors as shown in Scheme 65.
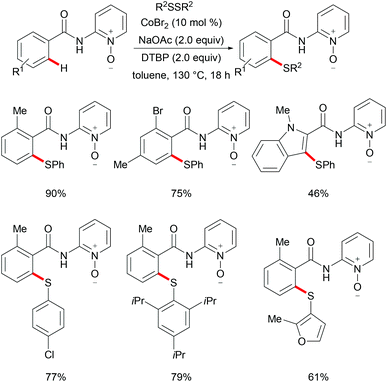 |
| | Scheme 64 Cobalt-catalysed direct C–H thiolation of aromatic amides with disulfides. | |
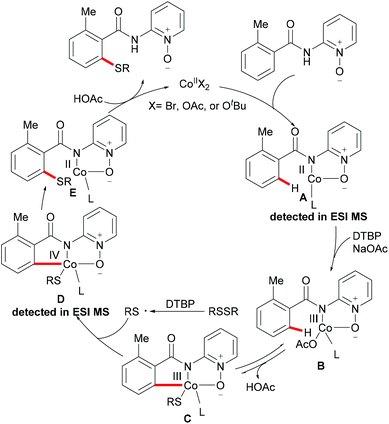 |
| | Scheme 65 Cobalt-catalysed direct C–H thiolation of aromatic amides with disulfides. | |
3.3 Nickel catalysis
Nickel is one of the most well-established 3d transition metals used in catalysis, with a plethora of applications in cross-coupling chemistry.220–223 The wide range of synthetically useful oxidation states that nickel can access produces highly versatile and tuneable catalysts. In addition, nickel has high natural abundance in the Earth's crust and compared with its 4d analogue, palladium, it has a low cost. The first report on stoichiometric C–H activation was published in 1963 by Dubeck, when cyclonickelation occurred from the reaction between azobenzene and nickelocene.224
In 2015, Lu's group reported the nickel-catalysed directed C–H thiolations of arenes assisted by the 2-(pyridine-2yl)isopropylamine (PIP-amine)225–230 removable bidentate directing group (Scheme 66).231 The key to the success was the use of benzoic acid as the additive in these transformations. This methodology showed a high level of functional group tolerance for both benzamide and diaryl disulfide substrates, and delivered the di- or mono-thiolated products in good to excellent yields. Moreover, radical scavenging experiments suggested that this reaction probably involved a SET process. Finally, the authors proposed a plausible nickel(II)/nickel(III) catalytic cycle as shown in Scheme 67. An initially formed five-membered nickel(II) species engaged with the phenyl sulfide radical to give the nickel(III) intermediate that furnished the desired product upon the reductive elimination and protonation process. Shortly afterward, Shi's group independently reported a similar transformation by using BINOL and KTFA as ligands and DMSO as the terminal oxidant.232 In contrast with Lu's work, this reaction might proceed via the nickel(II)/nickel(IV) pathway that involved an oxidative addition process (Scheme 68).
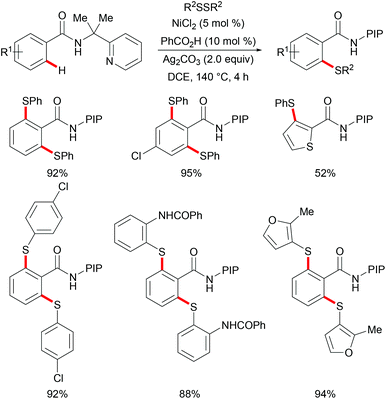 |
| | Scheme 66 Nickel-catalysed and benzoic acid-promoted direct thiolation of unactivated arenes. | |
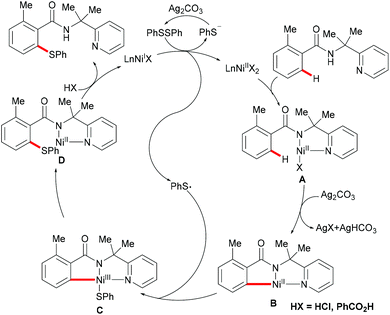 |
| | Scheme 67 Proposed catalytic cycle for the nickel-catalysed direct thiolation of arenes. | |
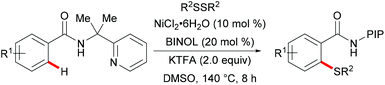 |
| | Scheme 68 Nickel-catalysed thiolation of arenes assisted by the PIP-amine bidentate directing group. | |
Besides the aromatic C(sp2)–H bonds, unactivated C(sp3)–H bonds could also participate in this nickel-catalysed thiolation. Subsequently, the same group expanded the substrate scope to aliphatic carboxamides with the assistance of 8-aminoquinoline directing groups by using the established Ni/BINOL catalytic system (Scheme 69).233 Notably, this transformation selectively took place at β-C(sp3)–H bonds even in the presence of γ-aromatic C(sp2)–H bonds, thus providing an efficient synthetic pathway to decorated β-thiolated aliphatic carboxamides. This new thiolation reaction can be considered as a complementary approach to the previous aromatic C(sp2)–H thiolations. In addition, the control experiment conducted with TEMPO or BHT as representative radical scavengers did not provide evidence for a radical pathway in contrast with the previous report by Lu, thus indicating that the phenyl sulfide radical might not be the key intermediate for this transformation. Finally, upon consideration of the previous reports, the authors proposed a nickel(II)/nickel(IV) catalytic cycle for this thiolation reaction.
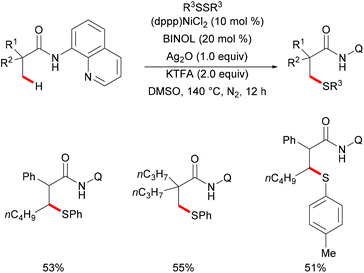 |
| | Scheme 69 Nickel-catalysed direct thiolation of C(sp3)–H bonds assisted by the 8-aminoquinoline directing group. | |
Almost at the same time, Zhang's group concurrently described a direct nickel-catalysed C(sp2)–H thiolation assisted by the 8-aminoquinoline bidentate directing group.234 This transformation was accomplished with stoichiometric quantities of the TBAI additive and o-nitrobenzoic acid ligands at a high reaction temperature with a wide range of functional groups being tolerated (Scheme 70). Notably, not only the aromatic C(sp2)–H bonds, but also the alkenyl C(sp2)–H bonds were compatible under the optimized conditions and selectively afforded (Z)-alkenyl sulfides in good yields. After the preliminary mechanistic study, the authors proposed that a nickel(II)/nickel(IV) pathway might be involved in this thiolation reaction. Shortly afterwards, the more challenging C(sp3)–H thiolation was also achieved by Zhang's and Yin's groups independently with the combination of Ni(OTf)2/Ac-Gly-OH or NiBr2/MesCOOH as catalytic systems.235,236 Importantly, these thiolation reactions preferential occurred at the C(sp3)–H bonds of the β-methyl group over the β-methylene group and the γ-methyl group, thus providing a new approach to access β-thio carboxylic derivates with excellent regioselectivity.
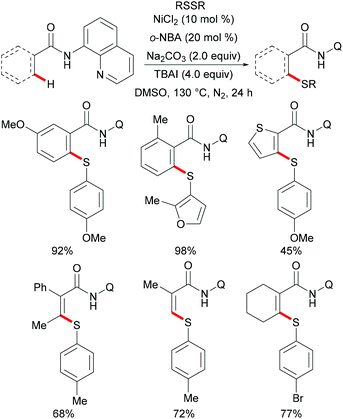 |
| | Scheme 70 Nickel-catalysed direct ortho-thiolation of arenes and alkenes. | |
Independently, Shi's group reported on a nickel-catalysed C(sp3)–H thiolation using disulfide or thiol as the sulfur source with the assistance of the 8-aminoquinoline bidentate directing group (Scheme 71).237 The authors proposed that the Ni–DMF complex formed in the presence of a lithium base was the actual catalyst promoting this transformation. The electron-deficient benzenethiols exhibited better performance and provided the desired products in good yields whereas electron-rich benzenethiols and alkyl thiols only gave unsatisfactory results. Similar to Shi's reports, this thiolation reaction also only occurred at the primary methyl C–H position over methylene, even in the presence of activated benzylic methylene. Additionally, aromatic and alkenyl C(sp2)–H bonds were compatible under similar conditions by employing air-stable NiCl2(DME) as the catalyst precursor. A wide range of functional groups present in both the amide substrates and the disulfides were well tolerated and delivered the corresponding products in good to excellent yields. Notably, this reaction performed equally efficiently at the gram scale, demonstrating the scalability of this transformation. Although the mechanism is not clear, the observation that the thiol/air catalytic system led to an increased reaction rate indicated that these transformations might involve different pathways under these two different conditions. Subsequently, this robust nickel catalytic system could be employed in the C–H thiolations of N-benzoyl-α-amino-acid and dipeptide derivatives.238
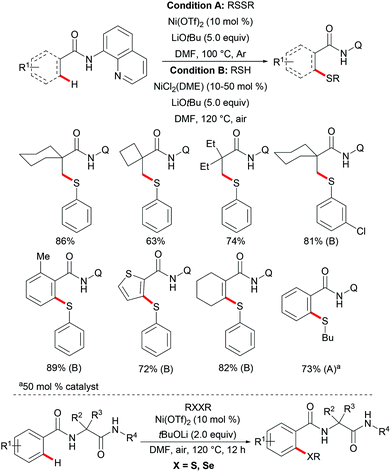 |
| | Scheme 71 Nickel-catalysed directed thiolations of sp2 and sp3 C–H bonds. | |
More recently, Weng and coworkers also achieved the nickel-catalysed C–H thiolation,239 directed by N-(pyridinyl)hydrazine under slightly modified conditions compared to Lu's reports (Scheme 72). This reaction proceeded efficiently using DMSO as the oxidant with a wide range of functional groups being well tolerated. Furthermore, the inherent inactive C(sp3)–H bond also participated in this thiolation smoothly under the optimized conditions. It is noteworthy that 4-methylbenzenethiol was also a suitable coupling partner under the standard conditions presumably due to the oxidation of thiol to disulfide by DMSO. The treatment of the thus obtained thiolated product with SmI2 in the THF/MeOH mixture as the solvent at room temperature furnished the corresponding thiolated benzamide in a moderate yield. Based on a preliminary mechanistic study, the authors postulated a nickel(II)/nickel(IV) catalytic process to be operative in this thiolation reaction.
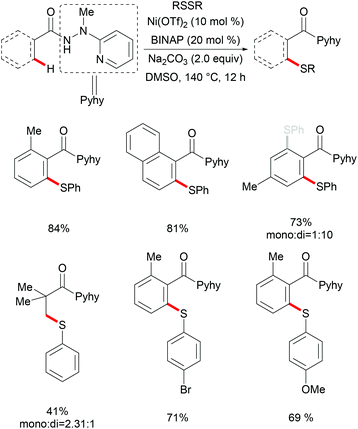 |
| | Scheme 72 Nickel-catalysed direct C–H bond thiolations of acylhydrazines. | |
In 2017, the Nishihara group reported on a nickel-catalysed direct selenylation of 8-quinolyl benzamides with elemental selenium as the selenylating reagent (Scheme 73).240 The reaction proceeded smoothly with Ni(OAc)·4H2O and PPh3 as the catalyst and ligand under air with excellent functional group compatibility. Notably, alkenyl benzamides smoothly reacted with elemental selenium, affording the corresponding isoselenazolone derivatives in moderate yields (Scheme 74). In addition, the thus obtained benzoisoselenazolones could be further transformed, giving rise to valuable organoselenium compounds. Although the detailed mechanism was still unclear at that stage, the authors proposed that the reaction might proceed through an unprecedented single-electron oxidation of the stable nickelacycle(II) species under air.
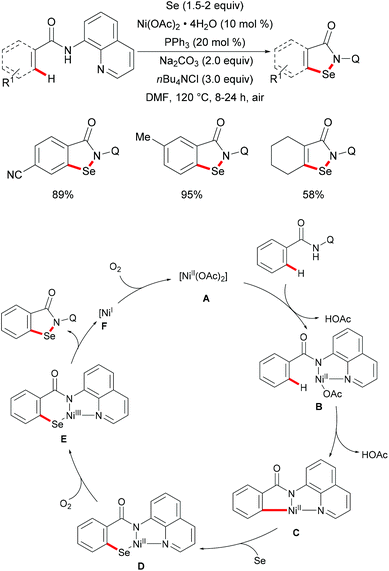 |
| | Scheme 73 Nickel-catalysed direct selenylation of C(sp2)–H bonds with elemental selenium. | |
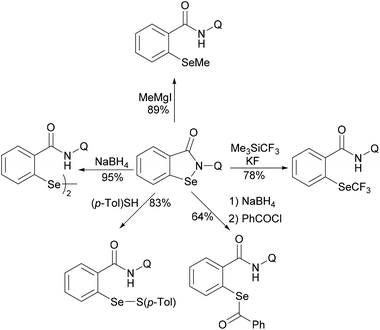 |
| | Scheme 74 Further transformation of benzoisoselenazolone. | |
Although undisputable progress has been achieved in nickel-catalysed C–H thiolations, these established methods were largely limited to electron-deficient benzamide derivatives bearing bidentate directing groups. In this context, Ackermann and coworkers reported on a nickel-catalysed C–H chalcogenylation of anilines with the assistance of the removable monodentate pyrimidine directing group in 2016 (Scheme 75).241 The key to these transformations was the use of MnO2 as the oxidant which could promote the regeneration of disulfides or diselenides. This approach demonstrated excellent functional group tolerance and afforded the corresponding mono- or di-chalcogenated products in good yields. Mechanistic studies showed that this C–H chalcogenylation was totally inhibited by the addition of typical radical scavengers, demonstrating that a radical pathway might be involved. A considerable kinetic isotope effect (KIE) was observed from two independent experiments conducted with deuterium-labelled compounds and the corresponding substrate, suggesting that C–H bond cleavage was the rate-determining step of the reaction. Based on these mechanistic studies, the authors proposed a plausible nickel(II)/nickel(III) catalytic cycle involving oxidation of the free thio radical to form a nickel(III) intermediate, followed by a reductive elimination process (Scheme 76). Finally, the removal of the directing group smoothly occurred in aqueous HCl under microwave irradiation, delivering the desired 2-amino thiophenols in high yields.
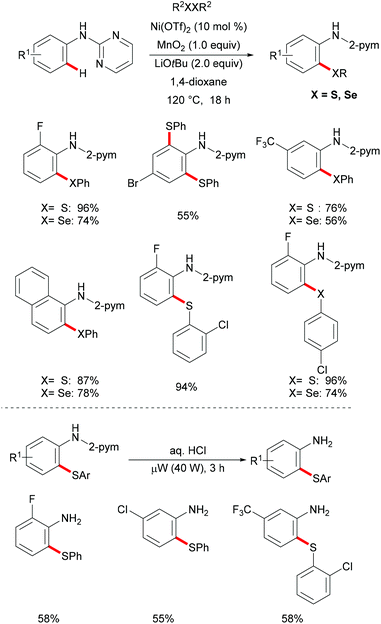 |
| | Scheme 75 Nickel-catalysed C–H chalcogenylations of anilines. | |
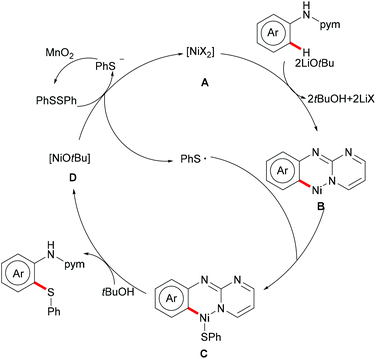 |
| | Scheme 76 Proposed mechanism for nickel-catalysed C–H chalcogenylations of anilines. | |
4. Conclusions
During the past decade, the prevalence of S- and Se-containing molecules in bioactive drugs and functional materials has induced the development of various transition metal-catalysed C–H chalcogenylations by chelation assistance. These step-economical methods have thus emerged as environmentally friendly strategies for the preparation of unsymmetrical sulfides and selenides. Herein, we have summarized the recent advances featuring representative C–H chalcogenylations. Thus, challenging C–H chalcogenylations were accomplished by, among others, precious palladium and ruthenium catalysts, while the recent momentum was gained by the use of less expensive and less toxic Earth-abundant 3d metals. Here, particular advances were realized with versatile nickel, cobalt and copper catalysis manifolds. Despite the indispensable progress, major challenges still need to be overcome. For instance, numerous C–H chalcogenylations were conducted at a relatively high reaction temperature with an excess of metal oxidants or an excess of chalcogenylating reagents to ensure sufficient turnover. Furthermore, the directing groups often required harsh conditions for their removal or were transformed during the reaction, thus considerably limiting the molecular diversity and the application in complex molecule synthesis. Therefore, the development of sustainable strategies for efficient C–S and C–Se bond construction under mild conditions and the design of removable, transformable and transient242–244 directing groups are highly desirable. Moreover, further studies on the mechanistic manifolds of these C–H activations will pave the way for green and efficient routes towards sustainability. We believe that the innovative accomplishments in these areas will provide practical tools for the preparation of valuable structural scaffolds, including 5–7-membered sulphur-containing heterocycles with major potential for drug discovery, molecular labeling, organocatalysis and late-stage diversification, and in materials sciences, among others.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors wish to thank the Sichuan Science and Technology Program (Grant No. 2018JY0247), the Key Laboratory of Medicinal and Edible Plants Resources Development of Sichuan Education Department (Grant No. 10Y201708), the Onassis Foundation (fellowship to NK), and the DFG (Gottfried-Wilhelm-Leibniz award to LA) for generous support.
Notes and references
- E. d. A. dos Santos, E. Hamel, R. Bai, J. C. Burnett, C. S. Tozatti, D. Bogo, R. T. Perdomo, A. M. Antunes, M. M. Marques, M. de F. C. Matos and D. P. de Lima, Synthesis and evaluation of diaryl sulfides and diaryl selenide compounds for antitubulin and cytotoxic activity, Bioorg. Med. Chem. Lett., 2013, 23, 4669–4673 CrossRef PubMed
 .
.
- E. Moreno, D. Plano, I. Lamberto, M. Font, I. Encio, J. A. Palop and C. Sanmartin, Sulfur and selenium derivatives of quinazoline and pyrido[2,3-d]pyrimidine: synthesis and study of their potential cytotoxic activity in vitro, Eur. J. Med. Chem., 2012, 47, 283–298 CrossRef PubMed .
- G. Mugesh, W.-W. du Mont and H. Sies, Chemistry of biologically important synthetic organoselenium compounds, Chem. Rev., 2001, 101, 2125–2179 CrossRef CAS PubMed .
- C. W. Nogueira, G. Zeni and J. B. T. Rocha, Organoselenium and organotellurium compounds: toxicology and pharmacology, Chem. Rev., 2004, 104, 6255–6285 CrossRef CAS PubMed .
- Z. Wen, J. Xu, Z. Wang, H. Qi, Q. Xu, Z. Bai, Q. Zhang, K. Bao, Y. Wu and W. Zhang, 3-(3,4,5-Trimethoxyphenylselenyl)-1H-indoles and their selenoxides as combretastatin A-4 analogs: microwave-assisted synthesis and biological evaluation, Eur. J. Med. Chem., 2015, 90, 184–194 CrossRef CAS PubMed .
- Z. Lou, P. Li and K. Han, Redox-responsive fluorescent probes with different design strategies, Acc. Chem. Res., 2015, 48, 1358–1368 CrossRef CAS PubMed .
- S. T. Manjare, Y. Kim and D. G. Churchill, Selenium- and tellurium-containing fluorescent molecular probes for the detection of biologically important analytes, Acc. Chem. Res., 2014, 47, 2985–2998 CrossRef CAS PubMed .
- S. Kumar, H. B. Singh and G. Wolmershaeuser, Protection against peroxynitrite-mediated nitration reaction by intramolecularly coordinated diorganoselenides, Organometallics, 2006, 25, 382–393 CrossRef CAS .
- T. Wirth, G. Fragale and M. Spichty, Mechanistic course of the asymmetric methoxyselenenylation reaction, J. Am. Chem. Soc., 1998, 120, 3376–3381 CrossRef CAS .
- F. V. Singh and T. Wirth, Selenium and tellurium electrophiles in organic synthesis, Phys. Sci. Rev., 2019, 4 DOI:10.1515/psr-2017-0131 .
- D. M. Browne, O. Niyomura and T. Wirth, Catalytic use of selenium electrophiles in cyclizations, Org. Lett., 2007, 9, 3169–3171 CrossRef CAS PubMed .
- Z. Deng, J. Wei, L. Liao, H. Huang and X. Zhao, Organoselenium-catalyzed, hydroxy-controlled regio- and stereoselective amination of terminal alkenes: efficient synthesis of 3-amino allylic alcohols, Org. Lett., 2015, 17, 1834–1837 CrossRef CAS PubMed .
- R. Guo, J. Huang and X. Zhao, Organoselenium-catalyzed oxidative allylic fluorination with electrophilic N–F reagent, ACS Catal., 2018, 8, 926–930 CrossRef CAS .
- S. Ortgies and A. Breder, Selenium-catalyzed oxidative C(sp2)-H amination of alkenes exemplified in the expedient synthesis of (aza-)Indoles, Org. Lett., 2015, 17, 2748–2751 CrossRef CAS PubMed .
- F. V. Singh and T. Wirth, Selenium reagents as catalysts, Catal. Sci. Technol., 2019, 9, 1073–1091 RSC .
- J. Trenner, C. Depken, T. Weber and A. Breder, Direct oxidative allylic and vinylic amination of alkenes through selenium catalysis, Angew. Chem., Int. Ed., 2013, 52, 8952–8956 CrossRef CAS PubMed .
- D. A. Boyd, Sulfur and its role in modern materials science, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed .
- D. K. Dang, C. Sundaram, Y.-L. T. Ngo, W. M. Choi, J. S. Chung, E. J. Kim and S. H. Hur, Pyromellitic acid-derived highly fluorescent N-doped carbon dots for the sensitive and selective determination of 4-nitrophenol, Dyes Pigm., 2019, 165, 327–334 CrossRef CAS .
- P. Gaur, A. Kumar, G. Dey, R. Kumar, S. Bhattacharyya and S. Ghosh, Selenium incorporated cationic organochalcogen: live cell compatible and highly photostable molecular stain for imaging and localization of intracellular DNA, ACS Appl. Mater. Interfaces, 2016, 8, 10690–10699 CrossRef CAS PubMed .
- G. Wang, Q. Guo, D. Chen, Z. Liu, X. Zheng, A. Xu, S. Yang and G. Ding, Facile and highly effective synthesis of controllable lattice sulfur-doped graphene quantum dots via hydrothermal treatment of durian, ACS Appl. Mater. Interfaces, 2018, 10, 5750–5759 CrossRef CAS PubMed .
- S. Yang, J. Sun, P. He, X. Deng, Z. Wang, C. Hu, G. Ding and X. Xie, Selenium doped graphene quantum dots as an ultrasensitive redox fluorescent switch, Chem. Mater., 2015, 27, 2004–2011 CrossRef CAS .
- I. Beletskaya, Element–element addition to alkynes catalyzed by the group 10 metals, Chem. Rev., 1999, 99, 3435–3461 CrossRef CAS PubMed .
- I. P. Beletskaya and V. P. Ananikov, Transition-metal-catalyzed C–S, C–Se, and C–Te bond formation via cross-coupling and atom-economic addition reactions, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed .
- T. Kondo and T. Mitsudo, Metal-catalyzed carbon–sulfur bond formation, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed .
- C. F. Lee, R. S. Basha and S. S. Badsara, Engineered C–S bond construction, Top. Curr. Chem., 2018, 376, 25 CrossRef PubMed .
- C. F. Lee, Y. C. Liu and S. S. Badsara, Transition-metal-catalyzed C–S bond coupling reaction, Chem. – Asian J., 2014, 9, 706–722 CrossRef CAS PubMed .
- B. J. Li and Z. J. Shi, From C(sp2)–H to C(sp3)–H: systematic studies on transition metal-catalyzed oxidative C–C formation, Chem. Soc. Rev., 2012, 41, 5588–5598 RSC .
- C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. Hor and X. Liu, Recent advances in C-S bond formation via C-H bond functionalization and decarboxylation, Chem. Soc. Rev., 2015, 44, 291–314 RSC .
- N. Ibrahim, M. Alami and S. Messaoudi, Recent Advances in Transition-Metal-Catalyzed Functionalization of 1-Thiosugars, Asian J. Org. Chem., 2018, 7, 2026–2038 CrossRef CAS .
- X. Chen, M. Wang and X. Jiang, C–H functionalization strategies for the construction of thioethers, Acta Phys.-Chim. Sin., 2019, 35, 954–967 Search PubMed .
- Y. Fang, T. Rogge, L. Ackermann, S. Y. Wang and S. J. Ji, Nickel-catalyzed reductive thiolation and selenylation of unactivated alkyl bromides, Nat. Commun., 2018, 9, 2240 CrossRef PubMed .
- S. Fukuzawa, E. Shimizu, Y. Atsuumi, M. Haga and K. Ogata, Copper-catalyzed direct thiolation of benzoxazole with diaryl disulfides and aryl thiols, Tetrahedron Lett., 2009, 50, 2374–2376 CrossRef CAS .
- C. Goux, P. Lhoste and D. Sinou, Synthesis of allyl aryl sulphides by palladium(0)-mediated alkylation of thiols, Tetrahedron Lett., 1992, 33, 8099–8102 CrossRef CAS .
- Z. He, F. Luo, Y. Li and G. Zhu, Copper-catalyzed direct thiolation of xanthines and related heterocycles with disulfides, Tetrahedron Lett., 2013, 54, 5907–5910 CrossRef CAS .
- J. Mao, T. Jia, G. Frensch and P. J. Walsh, Palladium-catalyzed debenzylative cross-coupling of aryl benzyl sulfides with aryl bromides: synthesis of diaryl sulfides, Org. Lett., 2014, 16, 5304–5307 CrossRef CAS PubMed .
- G. Mugesh and H. B. Singh, Heteroatom-directed aromatic lithiation: A versatile route to the synthesis of organochalcogen (Se, Te) compounds, Acc. Chem. Res., 2002, 35, 226–236 CrossRef CAS PubMed .
- S.-I. Murahashi, M. Yamamura, K.-I. Yanagisawa, N. Mita and K. Kondo, Stereoselective synthesis of alkenes and alkenyl sulfides from alkenyl halides using palladium and ruthenium catalysts, J. Org. Chem., 1979, 48, 2408–2418 CrossRef .
- Y. C. Shieh, K. Du, R. S. Basha, Y. J. Xue, B. H. Shih, L. Li and C. F. Lee, Syntheses of thioethers and selenide ethers from anilines, J. Org. Chem., 2019, 84, 6223–6231 CrossRef CAS PubMed .
- R. Sikari, S. Sinha, S. Das, A. Saha, G. Chakraborty, R. Mondal and N. D. Paul, Achieving nickel catalyzed C–S cross-coupling under mild conditions using metal-ligand cooperativity, J. Org. Chem., 2019, 84, 4072–4085 CrossRef CAS PubMed .
- P. Singh, R. Bai, R. Choudhary, M. C. Sharma and S. S. Badsara, Regio- and stereoselective syntheses of allylic thioethers under metal free conditions, RSC Adv., 2017, 7, 30594–30602 RSC .
- A. F. Manuel, Q. Shen and J. F. Hartwig, A general and long-lived catalyst for the palladium-catalyzed coupling of aryl halides with thiols, J. Am. Chem. Soc., 2006, 128, 2180–2181 CrossRef PubMed .
- E. Alvaro and J. F. Hartwig, Resting state and elementary steps of the coupling of aryl halides with thiols catalyzed by alkylbisphosphine complexes of palladium, J. Am. Chem. Soc., 2009, 131, 7858–7868 CrossRef CAS PubMed .
- Y. Li, M. Wang and X. Jiang, Controllable sulfoxidation and sulfenylation with organic thiosulfate salts via dual electron- and energy-transfer photocatalysis, ACS Catal., 2017, 7, 7587–7592 CrossRef CAS .
- D. Ma, Q. Geng, H. Zhang and Y. Jiang, Assembly of substituted phenothiazines by a sequentially controlled CuI/L-proline-catalyzed cascade C–S and C–N bond formation, Angew. Chem., Int. Ed., 2010, 49, 1291–1294 CrossRef CAS PubMed .
- M. Wang, Z. Dai and X. Jiang, Design and application of alpha-ketothioesters as 1,2-dicarbonyl-forming reagents, Nat. Commun., 2019, 10, 2661–2669 CrossRef PubMed .
- Z. Qiao, J. Wei and X. Jiang, Direct cross-coupling access to diverse aromatic sulfide: palladium-catalyzed double C–S bond construction using Na2S2O3 as a sulfurating reagent, Org. Lett., 2014, 16, 1212–1215 CrossRef CAS .
- I. Beletskaya, V. Ananikov and M. Kabeshov, The first example of polymer-supported palladium catalyst for stereo-selective S-S bond addition to terminal alkynes, Synlett, 2005, 2005, 1015–1017 CrossRef .
- J. Chen, S.-X. Su, D.-C. Hu, F.-H. Cui, Y.-L. Xu, Y.-Y. Chen, X.-L. Ma, Y.-M. Pan and Y. Liang, Copper-catalyzed bis- or trifunctionalization of alkynyl carboxylic acids: an efficient route to bis- and tris-selenide alkenes, Asian J. Org. Chem., 2018, 7, 892–896 CrossRef CAS .
- W. Guo, K. Tao, W. Tan, M. Zhao, L. Zheng and X. Fan, Recent advances in photocatalytic C–S/P–S bond formation via the generation of sulfur centered radicals and functionalization, Org. Chem. Front., 2019, 6, 2048–2066 RSC .
- S. Higashimae, T. Tamai, A. Nomoto and A. Ogawa, Selective thiolative lactonization of internal alkynes bearing a hydroxyl group with carbon monoxide and organic disulfides catalyzed by transition-metal complexes, J. Org. Chem., 2015, 80, 7126–7133 CrossRef CAS PubMed .
- T. Ishiyama, K.-I. Nishijima, N. Miyaura and A. Suzuki, Palladium(0)-catalyzed thioboration of terminal alkynes with 9-(alkylthio)-9-borabicyclo[3.3.1]nonane derivatives: stereoselective synthesis of vinyl sulfides via the thioboration-cross-coupling sequence, J. Am. Chem. Soc., 1993, 115, 7219–7225 CrossRef CAS .
- A. Ogawa, H. Kuniyasu, M. Takeba, T. Ikeda, N. Sonoda and T. Hirao, Palladium(0)-catalyzed regio- and stereoselective addition of heteroatom compounds bearing Si–Se, Ge–Se, and Si–Ge bonds to phenylacetylene, J. Organomet. Chem., 1998, 564, 1–4 CrossRef CAS .
- G. Perin, E. J. Lenardao, R. G. Jacob and R. B. Panatieri, Synthesis of vinyl selenides, Chem. Rev., 2009, 109, 1277–1301 CrossRef CAS PubMed .
- T. Tamai, K. Fujiwara, S. Higashimae, A. Nomoto and A. Ogawa, Gold-catalyzed anti-markovnikov selective hydrothiolation of unactivated alkenes, Org. Lett., 2016, 18, 2114–2117 CrossRef CAS PubMed .
- M. Wang, L. Cheng, B. Hong and Z. Wu, Regioselective bis-selenation of allenes catalyzed by palladium complexes: a theoretical study, Organometallics, 2009, 28, 1506–1513 CrossRef CAS .
- L. Shao, Y. Li, J. Lu and X. Jiang, Recent progress in selenium-catalyzed organic reactions, Org. Chem. Front., 2019, 6, 2999–3041 RSC .
- A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Cross-dehydrogenative C–H/S–H coupling reactions, Top. Curr. Chem., 2018, 376, 39 CrossRef PubMed .
- D. Kong, T. Huang, M. Liang, M. Wu and Q. Lin, KIO3-catalyzed cross dehydrogenative coupling reaction: sulfenylation of phenol and arylamine derivatives in water at room temperature, Org. Biomol. Chem., 2019, 17, 830–834 RSC .
- G. Li, G. Zhang, X. Deng, K. Qu, H. Wang, W. Wei and D. Yang, Transition-metal-free KI-catalyzed regioselective sulfenylation of 4-anilinocoumarins using Bunte salts, Org. Biomol. Chem., 2018, 16, 8015–8019 RSC .
- K. Nishino, S. Tsukahara, Y. Ogiwara and N. Sakai, Palladium(II)/copper(II)-catalyzed C–H sulfidation or selenation of arenes leading to unsymmetrical sulfides and selenides, Eur. J. Org. Chem., 2019, 1588–1593 CrossRef CAS .
- S. K. R. Parumala and R. K. Peddinti, Iodine catalyzed cross-dehydrogenative C–S coupling by C(sp2)–H bond activation: direct access to aryl sulfides from aryl thiols, Green Chem., 2015, 17, 4068–4072 RSC .
- C. D. Prasad, S. J. Balkrishna, A. Kumar, B. S. Bhakuni, K. Shrimali, S. Biswas and S. Kumar, Transition-metal-free synthesis of unsymmetrical diaryl chalcogenides from arenes and diaryl dichalcogenides, J. Org. Chem., 2013, 78, 1434–1443 CrossRef CAS PubMed .
- J. Rafique, S. Saba, A. R. Rosario and A. L. Braga, Regioselective, solvent- and metal-free chalcogenation of imidazo[1,2-a]pyridines by employing I2 /DMSO as the catalytic oxidation system, Chem. – Eur. J., 2016, 22, 11854–11862 CrossRef CAS PubMed .
- S. Saba, J. Rafique and A. L. Braga, DMSO/iodine-catalyzed oxidative C–Se/C–S bond formation: a regioselective synthesis of unsymmetrical chalcogenides with nitrogen- or oxygen-containing arenes, Catal. Sci. Technol., 2016, 6, 3087–3098 RSC .
- P. Saravanan and P. Anbarasan, Palladium catalyzed aryl(alkyl)thiolation of unactivated arenes, Org. Lett., 2014, 16, 848–851 CrossRef CAS PubMed .
- H. Wang, Y. Li, Q. Lu, M. Yu, X. Bai, S. Wang, H. Cong, H. Zhang and A. Lei, Oxidation-induced β-selective C–H bond functionalization: thiolation and selenation of N-heterocycles, ACS Catal., 2019, 9, 1888–1894 CrossRef CAS .
- P. Wang, S. Tang, P. Huang and A. Lei, Electrocatalytic oxidant-free dehydrogenative C–H/S–H cross-coupling, Angew. Chem., Int. Ed., 2017, 56, 3009–3013 CrossRef CAS PubMed .
- S. Yi, M. Li, W. Mo, X. Hu, B. Hu, N. Sun, L. Jin and Z. Shen, Metal-free, iodine-catalyzed regioselective sulfenylation of indoles with thiols, Tetrahedron Lett., 2016, 57, 1912–1916 CrossRef CAS .
- T. Zhang, G. Deng, H. Li, B. Liu, Q. Tan and B. Xu, Cyclization of 2-biphenylthiols to dibenzothiophenes under PdCl2/DMSO catalysis, Org. Lett., 2018, 20, 5439–5443 CrossRef CAS PubMed .
- W. Ma, H. Dong, D. Wang and L. Ackermann, Late-stage diversification of non-steroidal anti-inflammatory drugs by transition metal-catalyzed C–H alkenylations, thiolations and selenylations, Adv. Synth. Catal., 2017, 359, 966–973 CrossRef CAS .
- L. Gu, X. Fang, Z. Weng, J. Lin, M. He and W. Ma, PIDA-Promoted Selective C5 C–H Selenylations of Indolines via Weak Interactions, Adv. Synth. Catal., 2019, 361, 4998–5004 CrossRef CAS .
- H. Cao, L. Chen, J. Liu, H. Cai, H. Deng, G. Chen, C. Yan and Y. Chen, Regioselective copper-catalyzed thiolation of imidazo[1,2-a]pyridines: an efficient C–H functionalization strategy for C–S bond formation, RSC Adv., 2015, 5, 22356–22360 RSC .
- A. X. Zhou, X. Y. Liu, K. Yang, S. C. Zhao and Y. M. Liang, Copper-catalyzed direct thiolation of azoles with aliphatic thiols, Org. Biomol. Chem., 2011, 9, 5456–5462 RSC .
- F. Bai, S. Zhang, L. Wei and Y. Liu, Transition-metal-free indole C3 sulfenylation by KIO3 Catalysis, Asian J. Org. Chem., 2018, 7, 371–373 CrossRef CAS .
- F. L. Yang and S. K. Tian, Iodine-catalyzed regioselective sulfenylation of indoles with sulfonyl hydrazides, Angew. Chem., Int. Ed., 2013, 52, 4929–4932 CrossRef CAS PubMed .
- S. Chen, M. Wang and X. Jiang, Pd-catalyzed C–S cyclization via C–H functionalization strategy: access to sulfur-containing benzoheterocyclics, Chin. J. Chem., 2018, 36, 921–924 CrossRef CAS .
- X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Palladium(II)-catalyzed C–H activation/C–C cross-coupling reactions: versatility and practicality, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed .
- L. Ackermann, Carboxylate-assisted transition-metal-catalyzed C–H bond functionalizations: mechanism and scope, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed .
- L. McMurray, F. O'Hara and M. J. Gaunt, Recent developments in natural product synthesis using metal-catalysed C–H bond functionalisation, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC .
- C. S. Yeung and V. M. Dong, Catalytic dehydrogenative cross-coupling: forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed .
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, C–H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed .
- G. Rouquet and N. Chatani, Catalytic functionalization of C(sp2)–H and C(sp3)–H bonds by using bidentate directing groups, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed .
- J. Wencel-Delord and F. Glorius, C–H bond activation enables the rapid construction and late-stage diversification of functional molecules, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed .
- G. Chen and Z.-J. Shi, Privileged strategies for direct transformations of inert aliphatic C–H bonds, Natl. Sci. Rev., 2014, 1, 172–175 CrossRef CAS .
- S. A. Girard, T. Knauber and C. J. Li, The cross-dehydrogenative coupling of C(sp3)–H bonds: a versatile strategy for C–C bond formations, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed .
- M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Recent advances in directed C–H functionalizations using monodentate nitrogen-based directing groups, Org. Chem. Front., 2014, 1, 843–895 RSC .
- Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Transition metal-catalyzed ketone-directed or mediated C–H functionalization, Chem. Soc. Rev., 2015, 44, 7764–7786 RSC .
- Y. Park, Y. Kim and S. Chang, Transition metal-catalyzed C–H amination: scope, mechanism, and applications, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed .
- P. Gandeepan and L. Ackermann, Transient directing groups for transformative C–H activation by synergistic metal catalysis, Chem, 2018, 4, 199–222 CAS .
- J. Kalepu, P. Gandeepan, L. Ackermann and L. T. Pilarski, C4–H indole functionalisation: precedent and prospects, Chem. Sci., 2018, 9, 4203–4216 RSC .
- C. Sambiagio, D. Schonbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnurch, A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC .
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, Electrocatalytic C–H activation, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS .
- A. Tortajada, F. Julia-Hernandez, M. Borjesson, T. Moragas and R. Martin, Transition-metal-catalyzed carboxylation reactions with carbon dioxide, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed .
- Z. Zhang, P. H. Dixneuf and J. F. Soule, Late stage modifications of P-containing ligands using transition-metal-catalysed C–H bond functionalisation, Chem. Commun., 2018, 54, 7265–7280 RSC .
- P. Gandeepan, T. Muller, D. Zell, G. Cera, S. Warratz and L. Ackermann, 3d Transition metals for C–H activation, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed .
- J. B. Peng, F. P. Wu and X. F. Wu, First-row transition-metal-catalyzed carbonylative transformations of carbon electrophiles, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed .
- D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Rhodium catalyzed chelation-assisted C-H bond functionalization reactions, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed .
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Recent advances in transition-metal catalyzed reactions using molecular oxygen as the oxidant, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC .
- R. F. Heck, Palladium-catalyzed reactions of organic halides with olefins, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS .
- N. Miyaura and A. Suzuki, Palladium-catalyzed cross-coupling reactions of organoboron compounds, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS .
- E.-I. Negishi, C. Copéret, S. Ma, S.-Y. Liou and F. Liu, Cyclic carbopalladation. A versatile synthetic methodology for the construction of cyclic organic compounds, Chem. Rev., 1996, 96, 365–394 CrossRef CAS PubMed .
- K. J. Bonney and F. Schoenebeck, Experiment and computation: a combined approach to study the reactivity of palladium complexes in oxidation states 0 to IV, Chem. Soc. Rev., 2014, 43, 6609–6638 RSC .
- M. Kolter, K. Böck, K. Karaghiosoff and K. Koszinowski, Anionic Palladium(0) and Palladium(II) Ate Complexes, Angew. Chem., Int. Ed., 2017, 56, 13244–13248 CrossRef CAS PubMed .
- K. Muñiz, High-oxidation-state palladium catalysis: new reactivity for organic synthesis, Angew. Chem., Int. Ed., 2009, 48, 9412–9423 CrossRef PubMed .
- Q.-A. Chen, Z.-S. Ye, Y. Duan and Y.-G. Zhou, Homogeneous palladium-catalyzed asymmetric hydrogenation, Chem. Soc. Rev., 2013, 42, 497–511 RSC .
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Recent advances in transition-metal catalyzed reactions using molecular oxygen as the oxidant, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC .
- J. J. Topczewski and M. S. Sanford, Carbon–hydrogen (C–H) bond activation at PdIV: a Frontier in C–H functionalization catalysis, Chem. Sci., 2015, 6, 70–76 RSC .
- C.-Y. Wang, J. Derosa and M. R. Biscoe, Configurationally stable, enantioenriched organometallic nucleophiles in stereospecific Pd-catalyzed cross-coupling reactions: an alternative approach to asymmetric synthesis, Chem. Sci., 2015, 6, 5105–5113 RSC .
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed .
- J. Wang and G. Dong, Palladium/norbornene cooperative catalysis, Chem. Rev., 2019, 119, 7478–7528 CrossRef CAS PubMed .
- M. Iwasaki, M. Iyanaga, Y. Tsuchiya, Y. Nishimura, W. Li, Z. Li and Y. Nishihara, Palladium-catalyzed direct thiolation of aryl C–H bonds with disulfides, Chem. – Eur. J., 2014, 20, 2459–2462 CrossRef CAS PubMed .
- M. Iwasaki, W. Kaneshika, Y. Tsuchiya, K. Nakajima and Y. Nishihara, Palladium-catalyzed peri-selective chalcogenation of naphthylamines with diaryl disulfides and diselenides via C–H bond cleavage, J. Org. Chem., 2014, 79, 11330–11338 CrossRef CAS PubMed .
- R. Qiu, V. P. Reddy, T. Iwasaki and N. Kambe, The palladium-catalyzed intermolecular C–H chalcogenation of arenes, J. Org. Chem., 2015, 80, 367–374 CrossRef CAS .
- L. Gu, X. Fang, Z. Weng, Y. Song and W. Ma, Ligand-free palladium(II)-catalyzed ortho-C–H chalcogenations of N-arylsulfonamide via weak coordination, Eur. J. Org. Chem., 2019, 1825–1829 CrossRef CAS .
- W. Jin, P. Zheng, W.-T. Wong and G.-L. Law, Efficient palladium-catalyzed direct C–H phenylselenylation of (hetero)arenes in water, Asian J. Org. Chem., 2015, 4, 875–878 CrossRef CAS .
- W. Jin, P. Zheng, G.-L. Law and W.-T. Wong, Palladium(II)-catalyzed switchable mono-/diselenylation of arenes controlled by solvent effects, J. Organomet. Chem., 2016, 812, 66–73 CrossRef CAS .
- X.-S. Zhang, G. Li, X.-G. Zhang and X.-H. Zhang, Pd(II)-catalyzed selective sulfenylation of arene C–H bonds using N-arylthiobenzamides as thiolation reagent and oxidant, Tetrahedron, 2015, 71, 5458–5464 CrossRef CAS .
- M. Chaitanya and P. Anbarasan, Lewis acid/bronsted acid controlled Pd(II)-catalyzed chemodivergent functionalization of C(sp2)-H bonds with N-(arylthio)i(a)mides, Org. Lett., 2018, 20, 3362–3366 CrossRef CAS PubMed .
- S. Guin, A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, Promoting highly diastereoselective γ-C–H chalcogenation of α-Amino acids and aliphatic carboxylic acids, ACS Catal., 2018, 8, 2664–2669 CrossRef CAS .
- D. Lapointe and K. Fagnou, Overview of the mechanistic work on the concerted metallation–deprotonation pathway, Chem. Lett., 2010, 39, 1118–1126 CrossRef .
- K. Ueura, T. Satoh and M. Miura, Rhodium- and Iridium-Catalyzed Oxidative Coupling of Benzoic Acids with Alkynes via Regioselective C–H Bond Cleavage, J. Org. Chem., 2007, 72, 5362–5367 CrossRef CAS PubMed .
- G. Song, F. Wang and X. Li, C–C, C–O and C–N bond formation via rhodium(III)-catalyzed oxidative C–H activation, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC .
- N. Kuhl, N. Schröder and F. Glorius, Formal SN-type reactions in rhodium(III)-catalyzed C–H bond activation, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS .
- G. Song and X. Li, Substrate activation strategies in rhodium(III)-catalyzed selective functionalization of arenes, Acc. Chem. Res., 2015, 48, 1007–1020 CrossRef CAS PubMed .
- D. A. Colby, R. G. Bergman and J. A. Ellman, Rhodium-catalyzed C–C bond formation via heteroatom-directed C–H bond activation, Chem. Rev., 2010, 110, 624–655 CrossRef CAS .
- Y. Yang, W. Hou, L. Qin, J. Du, H. Feng, B. Zhou and Y. Li, Rhodium-catalyzed directed sulfenylation of arene C–H bonds, Chem. – Eur. J., 2014, 20, 416–420 CrossRef CAS PubMed .
- S. Yu, B. Wan and X. Li, Rh(III)-catalyzed selenylation of arenes with selenenyl chlorides/diselenides via C-H activation, Org. Lett., 2015, 17, 58–61 CrossRef CAS PubMed .
- J. Wen, A. Wu, M. Wang and J. Zhu, Rhodium(III)-catalyzed directed ortho-C-H bond functionalization
of aromatic ketazines via C-S and C-C coupling, J. Org. Chem., 2015, 80, 10457–10463 CrossRef CAS PubMed .
- W. Xie, B. Li and B. Wang, Rh(III)-catalyzed C7-thiolation and selenation of indolines, J. Org. Chem., 2016, 81, 396–403 CrossRef CAS PubMed .
- L. Ackermann and A. V. Lygin, Ruthenium-catalyzed direct C–H bond arylations of heteroarenes, Org. Lett., 2011, 13, 3332–3335 CrossRef CAS PubMed .
- L. Ackermann and A. V. Lygin, Cationic ruthenium(II) catalysts for oxidative C-H/N-H bond functionalizations of anilines with removable directing group: synthesis of indoles in water, Org. Lett., 2012, 14, 764–767 CrossRef CAS PubMed .
- S. Maity, U. Karmakar and R. Samanta, Regiocontrolled direct C4 and C2-methyl thiolation of indoles under rhodium-catalyzed mild conditions, Chem. Commun., 2017, 53, 12197–12200 RSC .
- S. Yang, B. Feng and Y. Yang, Rh(III)-catalyzed direct ortho-chalcogenation of phenols and anilines, J. Org. Chem., 2017, 82, 12430–12438 CrossRef CAS PubMed .
- D. Zell, M. Bursch, V. Muller, S. Grimme and L. Ackermann, Full selectivity control in cobalt(III)-catalyzed C-H alkylations by switching of the C-H activation mechanism, Angew. Chem., Int. Ed., 2017, 56, 10378–10382 CrossRef CAS .
- D. Zell, U. Dhawa, V. Müller, M. Bursch, S. Grimme and L. Ackermann, C–F/C–H activation by manganese(I) catalysis: expedient (per)fluoro-allylations and alkenylations, ACS Catal., 2017, 7, 4209–4213 CrossRef CAS .
- T. K. Vats, A. Mishra and I. Deb, Rhodium-catalyzed direct and selective ortho C–H chalcogenation of N-(hetero)aryl-7-azaindoles, Adv. Synth. Catal., 2018, 360, 2291–2296 CrossRef CAS .
- J. Yang, B. Deng, X. Guo, Z. Li, H. Xiang and X. Zhou, Rhodium(III)-catalyzed thiolation of azobenzenes, Asian J. Org. Chem., 2018, 7, 439–443 CrossRef CAS .
- Y. S. Kang, P. Zhang, M. Y. Li, Y. K. Chen, H. J. Xu, J. Zhao, W. Y. Sun, J. Q. Yu and Y. Lu, Ligand-promoted Rh(III)-catalyzed thiolation of benzamides with a broad disulfide scope, Angew. Chem., Int. Ed., 2019, 58, 9099–9103 CrossRef CAS .
- C. Liu, Y. Fang, S. Y. Wang and S. J. Ji, Highly regioselective Rh(III)-catalyzed thiolation of N-tosyl acrylamides: general access to (Z)-beta-alkenyl sulfides, Org. Lett., 2018, 20, 6112–6116 CrossRef CAS PubMed .
- L. N. Lewis and J. F. Smith, Catalytic C–C bond formation via ortho-metalated complexes, J. Am. Chem. Soc., 1986, 108, 2728–2735 CrossRef CAS .
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins, Nature, 1993, 366, 529–531 CrossRef CAS .
- W. Ma, R. Mei, G. Tenti and L. Ackermann, Ruthenium(II)-catalyzed oxidative C–H alkenylations of sulfonic acids, sulfonyl chlorides and sulfonamides, Chem. – Eur. J., 2014, 20, 15248–15251 CrossRef CAS PubMed .
- W. Ma and L. Ackermann, Ruthenium(II)-catalyzed C–H alkenylations of phenols with removable directing groups, Chem. – Eur. J., 2013, 19, 13925–13928 CrossRef CAS PubMed .
- W. Ma, K. Graczyk and L. Ackermann, Ruthenium-catalyzed alkyne annulations with substituted 1H-pyrazoles by C–H/N–H bond functionalizations, Org. Lett., 2012, 14, 6318–6321 CrossRef CAS PubMed .
- L. Ackermann, L. Wang and A. V. Lygina, Ruthenium-catalyzed aerobic oxidative coupling of alkynes with 2-aryl-substituted pyrroles, Chem. Sci., 2012, 3, 177–180 RSC .
- L. Ackermann, A. V. Lygin and N. Hofmann, Ruthenium-catalyzed oxidative annulation by cleavage of C–H/N–H bonds, Angew. Chem., Int. Ed., 2011, 50, 6379–6382 CrossRef CAS PubMed .
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Ruthenium(II)-catalyzed C–H bond activation and functionalization, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed .
- L. Ackermann, Carboxylate-assisted ruthenium-catalyzed alkyne annulations by C–H/het–H bond functionalizations, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed .
- W. Ma, P. Gandeepan, J. Li and L. Ackermann, Recent advances in positional-selective alkenylations: removable guidance for twofold C–H activation, Org. Chem. Front., 2017, 4, 1435–1467 RSC .
- R. Manikandan and M. Jeganmohan, Recent advances in the ruthenium(II)-catalyzed chelation-assisted C–H olefination of substituted aromatics, alkenes and heteroaromatics with alkenes via the deprotonation pathway, Chem. Commun., 2017, 53, 8931–8947 RSC .
- S. Shu, Z. Fan, Q. Yao and A. Zhang, Ru(II)-catalyzed direct C(sp2)-H activation/selenylation of arenes with selenyl chlorides, J. Org. Chem., 2016, 81, 5263–5269 CrossRef CAS PubMed .
- H. A. Chiong, Q.-N. Pham and O. Daugulis, Two methods for direct ortho-arylation of benzoic acids, J. Am. Chem. Soc., 2007, 129, 9879–9884 CrossRef CAS PubMed .
- S. Mochida, K. Hirano, T. Satoh and M. Miura, Synthesis of stilbene and distyrylbenzene derivatives through rhodium-catalyzed ortho-olefination and decarboxylation of benzoic acids, Org. Lett., 2010, 12, 5776–5779 CrossRef CAS PubMed .
- J. Cornella, M. Righi and I. Larrosa, Carboxylic acids as traceless directing groups for formal meta-selective direct arylation, Angew. Chem., Int. Ed., 2011, 50, 9429–9432 CrossRef CAS PubMed .
- H. X. Dai, G. Li, X. G. Zhang, A. F. Stepan and J. Q. Yu, Pd(II)-catalyzed ortho- or meta-C–H olefination of phenol derivatives, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS PubMed .
- M. Deponti, S. I. Kozhushkov, D. S. Yufit and L. Ackermann, Ruthenium-catalyzed C–H/O–H and C–H/N–H bond functionalizations: oxidative annulations of cyclopropyl-substituted alkynes, Org. Biomol. Chem., 2013, 11, 142–148 RSC .
- H. Gong, H. Zeng, F. Zhou and C. J. Li, Rhodium(I)-catalyzed regiospecific dimerization of aromatic acids: two direct C–H bond activations in water, Angew. Chem., Int. Ed., 2015, 54, 5718–5721 CrossRef CAS PubMed .
- L. Huang, D. Hackenberger and L. J. Goossen, Iridium-catalyzed ortho-arylation of benzoic acids with arenediazonium salts, Angew. Chem., Int. Ed., 2015, 54, 12607–12611 CrossRef CAS PubMed .
- S. Warratz, C. Kornhaass, A. Cajaraville, B. Niepotter, D. Stalke and L. Ackermann, Ruthenium(II)-catalyzed C–H activation/alkyne annulation by weak coordination with O2 as the sole oxidant, Angew. Chem., Int. Ed., 2015, 54, 5513–5517 CrossRef CAS PubMed .
- C. Zhu, Y. Zhang, J. Kan, H. Zhao and W. Su, Ambient-temperature ortho C–H arylation of benzoic acids with aryl iodides with ligand-supported palladium catalyst, Org. Lett., 2015, 17, 3418–3421 CrossRef CAS PubMed .
- A. Bechtoldt, C. Tirler, K. Raghuvanshi, S. Warratz, C. Kornhaass and L. Ackermann, Ruthenium oxidase catalysis for site-selective C–H alkenylations with ambient O2 as the sole oxidant, Angew. Chem., Int. Ed., 2016, 55, 264–267 CrossRef CAS PubMed .
- Y. Liu, Y. Yang, Y. Shi, X. Wang, L. Zhang, Y. Cheng and J. You, Rhodium-Catalyzed Oxidative Coupling of Benzoic Acids with Terminal Alkynes: An Efficient Access to 3-Ylidenephthalides, Organometallics, 2016, 35, 1350–1353 CrossRef CAS .
- M. P. Drapeau and L. J. Goossen, Carboxylic acids as directing groups for C-H bond functionalization, Chem. – Eur. J., 2016, 22, 18654–18677 CrossRef PubMed .
- L. Ackermann and J. Pospech, Ruthenium-catalyzed oxidative C–H bond Alkenylations in water expedient synthesis of annulated lactones, Org. Lett., 2011, 13, 4153–4155 CrossRef CAS PubMed .
- L. Ackermann, J. Pospech, K. Graczyk and K. Rauch, Versatile synthesis of isocoumarins and α-pyrones by ruthenium-catalyzed oxidative C–H/O–H bond cleavages, Org.
Lett., 2012, 14, 930–933 CrossRef CAS PubMed .
- A. Mandal, S. Dana, H. Sahoo, G. S. Grandhi and M. Baidya, Ruthenium(II)-Catalyzed ortho-C-H Chalcogenation of Benzoic Acids via Weak O-Coordination: Synthesis of Chalcogenoxanthones, Org. Lett., 2017, 19, 2430–2433 CrossRef CAS PubMed .
- W. Ma, Z. Weng, T. Rogge, L. Gu, J. Lin, A. Peng, X. Luo, X. Gou and L. Ackermann, Ruthenium(II)-catalyzed C–H chalcogenation of anilides, Adv. Synth. Catal., 2018, 360, 704–710 CrossRef CAS .
- W. Ma, Z. Weng, X. Fang, L. Gu, Y. Song and L. Ackermann, Ruthenium-catalyzed C–H selenylations of benzamides, Eur. J. Org. Chem., 2019, 41–45 CrossRef CAS .
- Z. Weng, X. Fang, M. He, L. Gu, J. Lin, Z. Li and W. Ma, Ruthenium catalyzed C–H selenylations of aryl acetic amides and esters via weak coordination, Org. Lett., 2019, 21, 6310–6314 CrossRef CAS PubMed .
- G. Fang, X. Cong, G. Zanoni, Q. Liu and X. Bi, Silver-based radical reactions: development and insights, Adv. Synth. Catal., 2017, 359, 1422–1502 CrossRef CAS .
- Q.-Z. Zheng and N. Jiao, Ag-catalyzed C–H/C–C bond functionalization, Chem. Soc. Rev., 2016, 45, 4590–4627 RSC .
- M. Álvarez-Corral, M. Muñoz-Dorado and I. Rodríguez-García, Silver-Mediated Synthesis of Heterocycles, Chem. Rev., 2008, 108, 3174–3198 CrossRef PubMed .
- J. M. Alderson, J. R. Corbin and J. M. Schomaker, Tunable, chemo- and site-selective nitrene transfer reactions through the rational design of silver(I) catalysts, Acc. Chem. Res., 2017, 50, 2147–2158 CrossRef CAS PubMed .
- D. Whitaker, J. Burés and I. Larrosa, Ag(I)-catalyzed C–H activation: The role of the Ag(I) salt in Pd/Ag-mediated C–H arylation of electron-deficient arenes, J. Am. Chem. Soc., 2016, 138, 8384–8387 CrossRef CAS PubMed .
- W. Li, D. Yuan, G. Wang, Y. Zhao, J. Xie, S. Li and C. Zhu, Cooperative Au/Ag dual-catalyzed cross-dehydrogenative biaryl coupling: reaction development and mechanistic insight, J. Am. Chem. Soc., 2019, 141, 3187–3197 CrossRef CAS PubMed .
- L. Yang, Q. Wen, F. Xiao and G. J. Deng, Silver-mediated oxidative vinylic C-H bond sulfenylation of enamides with disulfides, Org. Biomol. Chem., 2014, 12, 9519–9523 RSC .
- G. Cera and L. Ackermann, Iron-catalyzed C–H functionalization processes, Top. Curr. Chem., 2016, 374, 191–224 CrossRef PubMed .
- C. Zhang, C. Tang and N. Jiao, Recent advances in copper-catalyzed dehydrogenative functionalization via a single electron transfer (SET) process, Chem. Soc. Rev., 2012, 41, 3464–3484 RSC .
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Aerobic Copper-Catalyzed Organic Reactions, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed .
-
G. Evano and N. Blanchard, Copper-mediated cross-coupling reactions, Wiley, Hoboken, New Jersey, 2014 Search PubMed .
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Copper catalysed Ullmann type chemistry: from mechanistic aspects to modern development, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC .
- F. Ullmann and J. Bielecki, Chem. Ber., 1901, 34, 2174–2185 CrossRef CAS .
- I. Goldberg, Ueber phenylirungen bei gegenwart von kupfer als katalysator, Ber. Dtsch. Chem. Ges., 1906, 39, 1691–1692 CrossRef .
- X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Copper-catalyzed C–H functionalization reactions: efficient synthesis of heterocycles, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed .
- X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, Cu(II)-catalyzed functionalizations of aryl C–H bonds using O2 as an oxidant, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed .
- X.-B. Yan, P. Gao, H.-B. Yang, Y.-X. Li, X.-Y. Liu and Y.-M. Liang, Copper(II)-catalyzed direct thiolation of C–H bonds in aromatic amides with aryl and aliphatic thiols, Tetrahedron, 2014, 70, 8730–8736 CrossRef CAS .
- Y. Liu, M. Huang and L. Wei, Selective mono- and di-C(aryl)–H sulfenylation of benzamides by one-pot assembly of 8-aminoquinoline, benzoyl chlorides, and thiophenols, Asian J. Org. Chem., 2017, 6, 41–43 CrossRef CAS .
- J. Liu, Z. Xue, Z. Zeng, Y.-X. Chen and G. Chen, Copper-mediated tandem C(sp2)-H sulfenylation and annulation of arenes with 2-mercaptoimidazoles: regio- and site-selective access to polycyclic fused imidazo[2,1-b][1,3]thiazinones, Adv. Synth. Catal., 2016, 358, 3694–3699 CrossRef CAS .
- W.-J. Kong, Q. Shao, M.-H. Li, Z.-L. Zhou, H. Xu, H.-X. Dai and J.-Q. Yu, Copper-Mediated Diastereoselective C–H Thiolation of Ferrocenes, Organometallics, 2018, 37, 2832–2836 CrossRef CAS .
- X. Wang, X. Yi, H. Xu and H. X. Dai, Cu-Mediated C-H Thioetherification of Arenes at Room Temperature, Org. Lett., 2019, 21, 5981–5985 CrossRef CAS PubMed .
- L. Chu, X. Yue and F.-L. Qing, Cu(II)-mediated methylthiolation of aryl C–H bonds with DMSO, Org. Lett., 2010, 12, 1644–1647 CrossRef CAS PubMed .
- P. Sharma, S. Rohilla and N. Jain, Copper acetate-DMSO promoted methylthiolation of arenes and heteroarenes, J. Org. Chem., 2015, 80, 4116–4122 CrossRef CAS PubMed .
- X. Cui, X. Liu, X. Wang, W. Tian, D. Wei and G. Huang, Copper-catalyzed ortho-thiomethylation of benzamides via 8-aminoquinoline-assisted C-H activation with dimethyl sulfoxide, ChemistrySelect, 2017, 2, 8607–8611 CrossRef CAS .
- L. D. Tran, I. Popov and O. Daugulis, Copper-promoted sulfenylation of sp2 C–H bonds, J. Am. Chem. Soc., 2012, 134, 18237–18240 CrossRef CAS PubMed .
- A. Mandal, H. Sahoo and M. Baidya, Copper-catalyzed 8-aminoquinoline-directed selenylation of arene and heteroarene C–H bonds, Org. Lett., 2016, 18, 3202–3205 CrossRef CAS PubMed .
- M. Sattar, M. Shareef, K. Patidar and S. Kumar, Copper-catalyzed 8-aminoquinoline assisted aryl chalcogenation of ferroceneamide with aryl disulfides, diselenides, and ditellurides, J. Org. Chem., 2018, 83, 8241–8249 CrossRef CAS PubMed .
- Y. Li, Y.-J. Liu and B.-F. Shi, Copper-mediated thiolation of unactivated heteroaryl C–H bonds with disulfides under ligand- and metal-oxidant-Free conditions, Adv. Synth. Catal., 2017, 359, 4117–4121 CrossRef CAS .
- S.-L. Liu, X.-H. Li, T.-H. Shi, G.-C. Yang, H.-L. Wang, J.-F. Gong and M.-P. Song, Copper-promoted thiolation of C(sp2)–H bonds using a 2-amino alkylbenzimidazole directing group, Eur. J. Org. Chem., 2017, 2280–2289 CrossRef CAS .
- K. Z. Deng, L. L. Zhang, Y. F. Chen, H. X. Xie, X. B. Xu, C. C. Xia and Y. F. Ji, Copper-mediated direct thiolation of aryl C–H bonds with disulfides, Org. Biomol. Chem., 2019, 17, 7055–7065 RSC .
- Y.-S. Xiong, Y. Yu, J. Weng and G. Lu, Copper-catalyzed peri-selective direct sulfenylation of 1-naphthylamines with disulfides, Org. Chem. Front., 2018, 5, 982–989 RSC .
- B. K. Singh, G. Bairy and R. Jana, A general copper/manganese cocatalyzed C–H selenation of arenes, heteroarenes, and alkenes under air, ChemistrySelect, 2017, 2, 9227–9232 CrossRef CAS .
- G. Cera and L. Ackermann, Weak O-assistance outcompeting strong N,N-bidentate directing groups in copper-catalyzed C–H chalcogenation, Chem. – Eur. J., 2016, 22, 8475–8478 CrossRef CAS PubMed .
- P. Gandeepan, J. Koeller and L. Ackermann, Expedient C–H chalcogenation of indolines and indoles by positional-selective copper catalysis, ACS Catal., 2017, 7, 1030–1034 CrossRef CAS .
- P. Peng, J. Wang, C. Li, W. Zhu, H. Jiang and H. Liu, Cu(ii)-catalyzed C6-selective C–H thiolation of 2-pyridones using air as the oxidant, RSC Adv., 2016, 6, 57441–57445 RSC .
- S. Murahashi, Synthesis of phthalimidines from schiff bases and carbon monoxide, J. Am. Chem. Soc., 1955, 77, 6403–6404 CrossRef CAS .
- S. Murahashi and S. Horiie, The reaction of azobenzene and carbon monoxide, J. Am. Chem. Soc., 1956, 78, 4816–4817 CrossRef CAS .
- H.-F. Klein, M. Helwig, M. Karnop, H. König, B. Hammerschmitt, G. Cordier, U. Flörke and H.-J. Haupt, Tris(trimethylphosphine)cobalt(0) Compounds Containing Azaolefinic Ligands-Syntheses and Structures of Benzo[c]cinnoline and Phenylisocyanate Complexes, Z. Naturforsch., 1993, 48b, 785–793 Search PubMed .
- L. Ackermann, Cobalt-catalyzed C–H arylations, benzylations, and alkylations with organic electrophiles and beyond, J. Org. Chem., 2014, 79, 8948–8954 CrossRef CAS PubMed .
- K. Gao and N. Yoshikai, Low-valent cobalt catalysis: new opportunities for C–H functionalization, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS .
- L. Grigorjeva and O. Daugulis, Cobalt-catalyzed, aminoquinoline-directed C(sp2)–H bond alkenylation by alkynes, Angew. Chem., Int. Ed., 2014, 53, 10209–10212 CrossRef CAS PubMed .
- W. Ma and L. Ackermann, Cobalt(II)-catalyzed oxidative C–H alkenylations: regio- and site-selective access to isoindolin-1-one, ACS Catal., 2015, 5, 2822–2825 CrossRef CAS .
- T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, A cationic high-valent Cp*Co(III) complex for the catalytic generation of nucleophilic organometallic species: directed C–H bond activation, Angew. Chem., Int. Ed., 2013, 52, 2207–2211 CrossRef CAS PubMed .
- H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, Pyrroloindolone synthesis via a Cp*Co(III)-catalyzed redox-neutral directed C–H alkenylation/annulation sequence, J. Am. Chem. Soc., 2014, 136, 5424–5431 CrossRef CAS PubMed .
- J. Li and L. Ackermann, Cobalt-catalyzed C-H cyanation of arenes and heteroarenes, Angew. Chem., Int. Ed., 2015, 54, 3635–3638 CrossRef CAS PubMed .
- M. Moselage, J. Li and L. Ackermann, Cobalt-catalyzed C–H activation, ACS Catal., 2015, 6, 498–525 CrossRef .
- B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Air-stable carbonyl(pentamethylcyclopentadienyl)cobalt diiodide complex as a precursor for cationic (pentamethylcyclopentadienyl)cobalt(III) catalysis: application for directed C-2 selective C–H amidation of indoles, Adv. Synth. Catal., 2014, 356, 1491–1495 CrossRef CAS .
- T. Gensch, F. J. R. Klauck and F. Glorius, Cobalt-catalyzed C–H thiolation through dehydrogenative cross-coupling, Angew. Chem., Int. Ed., 2016, 55, 11287–11291 CrossRef CAS PubMed .
- L. Hu, X. Chen, L. Yu, Y. Yu, Z. Tan, G. Zhu and Q. Gui, Highly mono-selective ortho-methylthiolation of benzamides via cobalt-catalyzed sp2 C–H activation, Org. Chem. Front., 2018, 5, 216–221 RSC .
- M. Li and J. J. Wang, Cobalt-catalyzed direct C–H thiolation of aromatic amides with disulfides: application to the synthesis of quetiapine, Org. Lett., 2018, 20, 6490–6493 CrossRef CAS .
- T. Kurahashi and S. Matsubara, Nickel-catalyzed reactions directed toward the formation of heterocycles, Acc. Chem. Res., 2015, 48, 1703–1716 CrossRef CAS PubMed .
- L. N. Cavalcanti and G. A. Molander, Photoredox catalysis in nickel-catalyzed cross-coupling, Top. Curr. Chem., 2016, 374, 39 CrossRef PubMed .
- T. Iwasaki and N. Kambe, Ni-catalyzed C–C couplings using alkyl electrophiles, Top. Curr. Chem., 2016, 374, 66 CrossRef PubMed .
- M. Tobisu and N. Chatani, Nickel-Catalyzed Cross-Coupling Reactions of Unreactive Phenolic Electrophiles via C–O Bond Activation, Top. Curr. Chem., 2016, 374, 41 CrossRef PubMed .
- J. P. Kleiman and M. Dubeck, The preparation of cyclopentadienyl [o-(phenylazo)phenyl]nickel, J. Am. Chem. Soc., 1963, 85, 1544–1545 CrossRef CAS .
- B. Zhan, Y. Liu, F. Hua and B. F. Shi, Nickel-catalyzed ortho-halogenation of unactivated (hetero)aryl C–H bonds with lithium halides using a removable auxiliary, Chem. Commun., 2016, 52, 4934–4937 RSC .
- S. Zhao, B. Liu, B. Zhan, W. Zhang and B. F. Shi, Nickel-Catalyzed Ortho-Arylation of Unactivated (Hetero)aryl C–H Bonds with Arylsilanes Using a Removable Auxiliary, Org. Lett., 2016, 18, 4586–4589 CrossRef CAS PubMed .
- F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S. Zhang and B. F. Shi, Pd(II)-catalyzed alkoxylation of unactivated C(sp3)–H and C(sp2)–H bonds using a removable directing group: efficient synthesis of alkyl ethers, Chem. Sci., 2013, 4, 4187–4192 RSC .
- Y.-H. Liu, Y.-J. Liu, S. Y. Yan and B. F. Shi, Ni(II)-catalyzed dehydrative alkynylation of unactivated (hetero)aryl C–H bonds using oxygen: a user-friendly approach, Chem. Commun., 2015, 51, 11650–11653 RSC .
- Y.-J. Liu, Y.-H. Liu, S.-Y. Yan and B. F. Shi, A sustainable and simple catalytic system for direct alkynylation of C(sp2)–H bonds with low nickel loadings, Chem. Commun., 2015, 51, 6388–6391 RSC .
- B. Li, B. Liu and B. F. Shi, Copper-catalyzed ortho-halogenation of arenes and heteroarenes directed by a removable auxiliary, Chem. Commun., 2015, 51, 5093–5096 RSC .
- K. Yang, Y. Wang, X. Chen, A. A. Kadi, H. K. Fun, H. Sun, Y. Zhang and H. Lu, Nickel-catalyzed and benzoic acid-promoted direct sulfenylation of unactivated arenes, Chem. Commun., 2015, 51, 3582–3585 RSC .
- S. Y. Yan, Y. J. Liu, B. Liu, Y. H. Liu and B. F. Shi, Nickel-catalyzed thiolation of unactivated aryl C–H bonds: efficient access to diverse aryl sulfides, Chem. Commun., 2015, 51, 4069–4072 RSC .
- S. Y. Yan, Y. J. Liu, B. Liu, Y. H. Liu, Z. Z. Zhang and B. F. Shi, Nickel-catalyzed direct thiolation of unactivated C(sp3)–H bonds with disulfides, Chem. Commun., 2015, 51, 7341–7344 RSC .
- C. Lin, D. Li, B. Wang, J. Yao and Y. Zhang, Direct ortho-thiolation of arenes and alkenes by nickel catalysis, Org. Lett., 2015, 17, 1328–1331 CrossRef CAS PubMed .
- C. Lin, W. Yu, J. Yao, B. Wang, Z. Liu and Y. Zhang, Nickel-catalyzed direct thioetherification of beta-C(sp3)-H bonds of aliphatic amides, Org. Lett., 2015, 17, 1340–1343 CrossRef CAS PubMed .
- X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S. F. Yin, Nickel-catalyzed direct thiolation of C(sp3)–H bonds in aliphatic amides, Org. Lett., 2015, 17, 1970–1973 CrossRef CAS PubMed .
- X. Ye, J. L. Petersen and X. Shi, Nickel-catalyzed directed sulfenylation of sp(2) and sp(3) C–H bonds, Chem. Commun., 2015, 51, 7863–7866 RSC .
- F. Gao, W. Zhu, D. Zhang, S. Li, J. Wang and H. Liu, Nickel-catalyzed ortho-C–H thiolation of N-benzoyl alpha-amino acid derivatives, J. Org. Chem., 2016, 81, 9122–9130 CrossRef CAS PubMed .
- J. M. Li, Y. Yu, J. Weng and G. Lu, Nickel-catalyzed direct C–H bond sulfenylation of acylhydrazines, Org. Biomol. Chem., 2018, 16, 6047–6056 RSC .
- M. Iwasaki, N. Miki, Y. Tsuchiya, K. Nakajima and Y. Nishihara, Synthesis of benzoisoselenazolone derivatives by nickel-catalyzed dehydrogenative direct selenation of C(sp2)–H bonds with elemental selenium in air, Org. Lett., 2017, 19, 1092–1095 CrossRef CAS PubMed .
- T. Muller and L. Ackermann, Nickel-catalyzed C–H chalcogenation of anilines, Chem. – Eur. J., 2016, 22, 14151–14154 CrossRef PubMed .
- F. L. Zhang, K. Hong, T. J. Li, H. Park and J. Q. Yu, Functionalization of C(sp3)–H bonds using a transient directing group, Science, 2016, 351, 252–256 CrossRef CAS PubMed .
- S. St John-Campbell, A. J. P. White and J. A. Bull, Single operation palladium catalysed C(sp3)–H functionalisation of tertiary aldehydes: investigations into transient imine directing groups, Chem. Sci., 2017, 8, 4840–4847 RSC .
- F. Zhang and D. R. Spring, Arene C–H functionalisation using a removable/modifiable or a traceless directing group strategy, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC .
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Nikolaos
Kaplaneris
b,
Xinyue
Fang
a,
Linghui
Gu
*a,
Nikolaos
Kaplaneris
b,
Xinyue
Fang
a,
Linghui
Gu